DOI:
10.1039/D5RA08633G
(Paper)
RSC Adv., 2026,
16, 5979-5989
Graphene oxide functionalized tannic acid–trimethylolpropane triglycidyl ether nanoparticles: a versatile network nanocatalyst for synthesis of 4-substituted-1,5-benzodiazepine derivatives
Received
8th November 2025
, Accepted 4th January 2026
First published on 28th January 2026
Abstract
This work is described through a one-pot three-component reaction of o-phenylenediamine, and dimedone, under reflux conditions in aqueous ethanol/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, 5 mL) in the presence of GO@tannic acid–TTE. The main advantages of this method are the ease of operation, purification, and the absence of hazardous and expensive solvents and catalysts. Also, this catalyst has the ability to perform this reaction in large quantities on an industrial scale. FT-IR, TGA, XRD, EDX, elemental mapping, TEM, and SEM methods have been used to characterize the prepared nanocatalyst. The catalyst can be reused several times.
:1 v/v, 5 mL) in the presence of GO@tannic acid–TTE. The main advantages of this method are the ease of operation, purification, and the absence of hazardous and expensive solvents and catalysts. Also, this catalyst has the ability to perform this reaction in large quantities on an industrial scale. FT-IR, TGA, XRD, EDX, elemental mapping, TEM, and SEM methods have been used to characterize the prepared nanocatalyst. The catalyst can be reused several times.
1 Introduction
Graphene oxide (GO) has attracted a lot of attention due to its interesting properties. Its versatility and distinctive architecture make it useful for research in many fields. Its outstanding properties enable its application in various fields such as energy, biomedicine, biotechnology, and polymer composites. These interests in graphene oxide are at a stage where one can witness interesting scientific research and related technological developments.1 GO consists of oxygen-containing functional groups, which include carboxyl, carbonyl, epoxy, hydroxyl, lactone, and quinone. These functional groups are polar, which makes the substance strongly hydrophilic and partially soluble in water, causing the solubility to depend on the ratio of conductivity and oxygen. This two-dimensional aromatic structure with oxygenated functional groups exhibits favourable chemical properties, which makes it an environmentally safe substitute for costly metallic catalysts.2
Tannic acid (TA), a type of polyphenol compound, provides a variety of biological benefits.3 Its fundamental properties are biodegradability, as well as antioxidant, antibacterial, antiallergenic, and anticarcinogenic activities.4 The abundance, low cost, and exceptional compatibility make the food tannin beneficial in numerous industries like medical devices, the leather industry, in addition to being used as a food supplement. Its complexing ability was historically exploited for treating diarrhea and other issues of digestion, and was also effective in managing burns and skin ulcers.5 The structure of tannic acid includes a glucose molecule at the centre with esterically bonded gallic acid.6,7 The high concentration of phenolic hydroxyls and ester linkage moieties guarantees high chemical activity because they allow the molecule to undergo covalent and non-covalent interactions like van der Waals, hydrophobic, π–π, and metal–organic coordination.1 Braghiroli et al. showed the synthesis of amine-functionalized TA by reaction with ammonia.8 TA epoxy and TA alkene functionalized were also used in the fabrication of bio-based resins and coatings.9–11 TA is also versatile in that it can complex or cross-link macros through multiple ways, such as hydrogen bonding, ionic interactions, hydrophobic interactions, and metal chelation. TA-based hydrogels have been successfully synthesized by cross-linking with epoxy-functionalized hydrophilic monomers such as trimethylolpropane triglycidyl ether (TTE), and this compound can undergo many chemical changes.12,13 TTE is a chemical reagent owning three reactive epoxy end-groups, which can react with oxygen-containing groups (–COOH and –OH) formed on degraded polymer or networks.14
Multicomponent reactions (MCRs) have attracted significant interest in the synthesis of organic compounds because they can generate target products without the need to separate intermediates in a single operation, thereby reducing reaction time and energy consumption.15–17 Heterocyclic compounds, which contain heteroatoms such as nitrogen, oxygen, or sulfur in their ring structures, are very important in chemistry due to their wide applications in pharmaceuticals, natural products, and materials science.18–20
Benzodiazepines, a significant class of psychotropic medications, have garnered attention for their potent central nervous system depressant effects.21 These compounds interact with the GABAA receptor,22 leading to a range of therapeutic benefits, including sedative, hypnotic, anxiolytic, anticonvulsant, and muscle relaxant properties.23 Generally well-tolerated, benzodiazepines are safe and effective for short-term use across various conditions.24,25 The synthesis of 4-substituted 1,5-benzodiazepine derivatives has been achieved through a three-component reaction involving o-phenylenediamine, dimedone, and aldehydes.26 A review of recent studies shows that environmentally friendly catalysts such as tris(hydrogensulfato)boron [B(HSO4)3]26 and heterogeneous catalyst Fe(OTs)3/SiO2 have significantly advanced green multicomponent methods for the synthesis of benzodiazepine derivatives.27 Current methods for the synthesis of 1,5-benzodiazepine derivatives using green catalysts often require harsh conditions, long reaction times, and large amounts of catalyst. Also, the use of graphene oxide nanostructures in these reactions has been rarely reported. However, developing an efficient synthesis protocol for benzodiazepines remains a worthwhile endeavour. Since GO can be used as a suitable base for functionalizing other spices to prepare an efficient catalyst, in this work, TA-modified TTE was coupled to GO to carry out a one-step reaction of o-phenylenediamine, dimedone, and aldehydes, providing a simple approach to produce benzodiazepine derivatives in high yield and low catalyst loading (Fig. 1).
 |
| | Fig. 1 Synthesis of 4-substituted-1,5-benzodiazepines. | |
2 Experimental
2.1 Materials and methods
All chemicals used in this study were of analytical grade and used without further purification. Graphene oxide was prepared according to a modified Hummers' method. Tannic acid (≥99%), trimethylolpropane triglycidyl ether (≥98%), o-phenylenediamine (≥99%), dimedone (≥98%), and aromatic aldehydes (≥98%) were purchased from Sigma-Aldrich. Ethanol and other solvents were obtained from Merck and used as received. Deionized water was used throughout the experiments. The chemicals used in this work were obtained from Sigma Aldrich Chemical Co. and used with no further purification. The crystal structure of the nanoparticles was determined using X-ray powder diffraction (XRD) (Cu Kα radiation, λ = 1.5405 Å) in the range of 10–80 (2θ) at a speed of 2 min−1 using an X'Pert Pro device from Panalytical. FT-IR spectra were analyzed using an FT-IR spectrometer from Bruker, Germany. The morphology and particle size of the samples were determined using a transmission electron microscope (TEM) using a Leo 912 AB device at an accelerating voltage of 200 kV. Micrometric and FESEM analysis were performed using a ZEISS FESEM device. Thermogravimetric analysis (TGA, PerkinElmer, Pyris 1, USA) was used to investigate the thermal behavior of the samples. NMR analysis was performed using a Bruker instrument at frequencies of 300, 400, and 500 MHz. The reaction progress and purity of the substrates were monitored using thin layer chromatography (TLC) on SILG/UV 254 and 365 silica gel plates.
2.2 Synthesis of GO tannic acid
The method of Hummers was altered in order to obtain graphene oxide nanosheets.28 0.1 g of graphene oxide was sonicated in 10 mL of deionized water for 1 hour. Subsequently, the pH of the solution was adjusted to 4.7 by adding HCl. After that, 0.2 g of tannic acid was added within 1 hour. After heating for 24 hours at 80 °C, in a nitrogen atmosphere, the product was washed with deionized water and ethanol, then dried at 50 °C (Fig. 2).
 |
| | Fig. 2 Synthesis of GO@tannic acid. | |
2.3 Preparation of GO@tannic acid–TTE
As illustrated in Fig. 3, the methodology instructs that in the first step, 0.5 g of GO@tannic acid must be dispersed in 10 mL of deionized water for 20 minutes. Next, a sodium hydroxide (NaOH) solution (0.1 M, 5 mL) was added slowly and stirred for an hour. Following that, 0.9 mL of trimethylolpropane triglycidyl ether (TTE) was incorporated and maintained under stirring at 60 °C for 12 hours. Finally, the resulting product was washed with water and ethanol, dried at room temperature, and stored. Also, to determine the amount of acid in the catalyst according to the literature, 0.1 g of catalyst was added to an aqueous NaCl solution (1 mol L−1, 10 mL) with an initial PH 7.0. The mixture was stirred for 30 min until the pH of the solution decreased to 5.5, which indicates an ion exchange between phenolic protons and sodium ions, and this is equal to a loading of 0.00031 mmol g−1 of phenolic group.
 |
| | Fig. 3 Synthesis of GO@tannic acid–TTE catalyst. | |
2.4 General procedure for synthesis of 4-substituted-1,5-benzodiazepines catalyzed by GO@tannic acid–TTE
To a mixture of o-phenylenediamine (1 mmol, 0.108 g), dimedone (1 mmol, 0.140 g), and aromatic aldehydes (1 mmol; e.g., 4-chlorobenzaldehyde, 0.141 g) in EtOH/H2O (5 mL), GO@tannic acid–TTE (0.05 g) was added, and the mixture was stirred at reflux for 4 h. The progress of the reaction was followed by TLC. Upon completion of the reaction, the catalyst was easily removed. The benzodiazepine product was washed with ethanol. Spectroscopic methods, including FT-IR, 1H, and 13C NMR, were used to characterize the crystalline product obtained.
2.5 Spectra data
2.5.1 3,3-Dimethyl-11-phenyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 483, 526, 622, 699, 742, 835, 1080, 1384, 1571, 1634, 1731, 2853, 2824, 3065, 3421. 1H NMR (300 MHz, DMSO) δ 8.26 (s, 1H), 7.58–6.41 (m, 8H), 4.92 (s, 1H), 4.71 (s, 1H), 2.25 (s, 2H), 2.03 (s, 2H), 1.03 (s, 3H), 0.98 (s, 3H). 13C NMR (75 MHz, DMSO) δ 195.1, 162.8, 144.2, 135.4, 129.4, 128.2, 127.8, 126.9, 123.2, 117.7, 116.6, 115.9, 115.0, 96.2, 50.7, 41.9, 32.8, 32.0, 28.5.
2.5.2 11-(4-Bromophenyl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 486, 537, 712, 744, 826, 1011, 1074, 1384, 1459, 1599, 1630, 2854, 2926, 3086, 3390. 1H NMR (300 MHz, DMSO) δ 8.89 (s, 1H), 8.21–7.06 (m, 8H), 4.93 (s, 1H), 2.25 (s, 2H), 2.02 (s, 3H), 1.08 (s, 3H), 1.00 (s, 3H). 13C NMR (75 MHz, DMSO) δ 195.1, 162.8, 144.2, 135.4, 129.4, 128.2, 127.8, 126.9, 123.2, 117.7, 116.6, 115.9, 115.0, 96.2, 50.7, 41.9, 32.8, 32.0, 28.5.
2.5.3 11-(4-Hydroxyphenyl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 537, 619, 749, 841, 1041, 1100, 1183, 1230, 1295, 1384, 1462, 1509, 1610, 1630, 2874, 2966, 3114, 3165, 3399. 1H NMR (300 MHz, DMSO) δ 9.07 (s, 1H), 8.74 (s, 1H) 7.56–6.53 (m, 8H), 4.88 (s, 1H), 2.35 (s, 2H), 2.02 (s, 2H), 1.03 (s, 3H), 0.99 (s, 3H). 13C NMR (75 MHz, DMSO) δ 195.1, 161.2, 159.6, 152.2, 144.2, 133.9, 129.4, 128.6, 127.8, 122.0, 121.5, 115.9, 114.8, 97.0, 56.5, 50.7, 32.8, 28.4, 19.0.
2.5.4 11-(2-Chlorophenyl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 612, 745, 1050, 1151, 1245, 1312, 1383, 1443, 1527, 1573, 1733, 2957, 3060, 3228, 2205, 3437. 1H NMR (300 MHz, DMSO) δ 9.0 (s, 1H), 7.95–6.68 (m, 8H), 6.01 (s, 1H), 4.65 (s, 1H), 2.39 (s, 2H), 2.01 (s, 2H), 1.05 (s, 3H), 1.03 (s, 3H). 13C NMR (75 MHz, DMSO) δ 194.5, 162.2, 155.6, 149.1, 143.7, 133.2, 132.1, 131.6, 131.2, 130.3, 129.9, 127.4, 116.1, 115.4, 63.1, 54.5, 50.3, 32.3, 31.8, 28.1.
2.5.5 11-(3-Hydroxyphenyl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 612, 746, 889, 1045, 1150, 1244, 1274, 1387, 1458, 1523, 1586, 2872, 2957, 3068, 3224. 1H NMR (300 MHz, DMSO) δ 9.76 (s, 1H), 8.20 (s, 1H), 7.62–6.57 (m, 8H), 4.66 (s, 1H), 2.50 (s, 2H), 2.01 (s, 2H), 1.03 (s, 3H), 0.98 (d, J = 4.2 Hz, 3H). 13C NMR (75 MHz, DMSO) δ 195.0, 158.2, 151.8, 144.2, 131.8, 130.4, 127.9, 117.6, 116.6, 115.9, 113.8, 96.2, 72.9, 63.5, 50.7, 32.8, 32.2, 28.5.
2.5.6 3,3-Dimethyl-11-(3-nitrophenyl)-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 704, 743, 810, 1099, 1277, 1248, 1384, 1426, 1603, 2957, 3063, 3231, 3312, 3400. 1H NMR (300 MHz, DMSO) δ 9.01 (s, 1H), 8.64–6.54 (m, 8H), 5.82 (s, 1H), 3.60 (s, 1H), 2.46 (s, 2H), 2.28 (s, 2H), 1.02 (s, 3H), 0.97 (s, 3H). 13C NMR (75 MHz, DMSO) δ 192.5, 149.4, 148.7, 135.5, 132.9, 131.9, 131.1, 129.8, 124.7, 123.7, 123.2, 121.3, 120.8, 112.3, 86.3, 50.9, 32.1, 28.3, 27.7, 22.2.
2.5.7 11-(3,5-Dichlorophenyl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 465, 778, 1117, 1205, 1270, 1383, 1434, 1462, 1560, 1626, 1663, 1722, 2594, 2660, 2874, 2929, 2958, 3414. 1H NMR (500 MHz, DMSO) δ 9.01 (s, 1H), 7.63–6.37 (m, 7H), 4.68 (s, 1H), 2.39 (s, 3H), 2.00 (s, 2H), 1.01 (s, 3H), 0.96 (s, 3H). 13C NMR (126 MHz, DMSO) δ 194.6, 146.6, 143.7, 134.8, 132.3, 130.5, 128.2, 127.8, 127.4, 127.3, 122.8, 121.0, 117.3, 116.1, 115.5, 114.6, 50.2, 32.3, 31.4, 31.0, 27.9.
2.5.8 11-(3-Hydroxy-4-methoxyphenyl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 611, 746, 1034, 1150, 1273, 1386, 1452, 1503, 1566, 2872, 2957, 3228, 3315. 1H NMR (500 MHz, DMSO) δ 8.40 (s, 1H), 7.78–6.53 (m, 7H), 4.91 (s, 1H), 3.88 (s, 3H), 2.34 (s, 2H), 2.01 (s, 3H), 1.06 (s, 3H), 1.05 (s, 3H). 13C NMR (126 MHz, DMSO) δ 194.8, 160.9, 151.8, 148.5, 147.8, 133.4, 122.7, 121.6, 119.7, 115.6, 115.3, 114.9, 110.3, 96.5, 56.0, 50.2, 32.3, 31.4, 28.5, 18.5.
2.5.9 11-(3-Hydroxy-4-methoxyphenyl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 611, 746, 1034, 1150, 1273, 1386, 1452, 1503, 1566, 2872, 2957, 3228, 3315. 1H NMR (500 MHz, DMSO) δ 8.40 (s, 1H), 7.78–6.53 (m, 7H), 4.91 (s, 1H), 3.88 (s, 3H), 2.34 (s, 2H), 2.01 (s, 3H), 1.06 (s, 3H), 1.05 (s, 3H). 13C NMR (126 MHz, DMSO) δ 194.8, 160.9, 151.8, 148.5, 147.8, 133.4, 122.7, 121.6, 119.7, 115.6, 115.3, 114.9, 110.3, 96.5, 56.0, 50.2, 32.3, 31.4, 28.5, 18.5.
2.5.10 11-(4-Chlorophenyl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one. FT-IR (KBr, cm−1): 471, 748, 1044, 1093, 1151, 1335, 1384, 1531, 1603, 1629, 2872, 2957, 3055, 3414. 1H NMR (400 MHz, DMSO-d6): δ 12.73 (s, 1H), 7.90 (d, J = 7.3 Hz, 2H), 7.63 (s, 2H), 7.52 (d, J = 8.5 Hz, 2H), 7.22 (s, 3H), 6.65 (s, 1H), 5.42 (s, 1H), 3.53 (s, 2H), 2.62 (s, 2H), 1.48 (s, 3H), 1.22 (s, 3H).
3 Results and discussion
3.1 Characterization of GO@tannic acid–TTE catalyst
Fig. 4 shows the FT-IR spectrum of graphene oxide, tannic acid, trimethylolpropane triglycidyl ether, and GO tannic acid@TTE. Graphene oxide FT-IR spectrum reveals a broad peak at 3429 cm−1, indicating hydroxyl groups through the O–H bond's stretching mode. The carboxyl group is evident at 1720 cm−1, while the C–OH group appears at 1356 cm−1. Peaks at 1225 and 1056 cm−1 correspond to C–O–C stretching and C–O vibrational modes. The tannic acid spectrum, featuring a strong absorption band at 3275 cm−1, is attributed to hydroxyl groups of phenols. The spectrum also shows sharp peaks at 2927–2860 cm−1, associated with CH2 and CH3 groups stretching vibrations.29 Tannic acid's aromatic esters are confirmed by carbonyl groups' C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1714 cm−1 and C–O at 1205 cm−1.30 In comparison, after functionalization with trimethylolpropane triglycidyl ether, a strengthened OH peak at 3450 cm−1 due to epoxy ring-opening, with C–O stretching vibration peaks at 1044–1150 cm−1 appeared.31
O stretching at 1714 cm−1 and C–O at 1205 cm−1.30 In comparison, after functionalization with trimethylolpropane triglycidyl ether, a strengthened OH peak at 3450 cm−1 due to epoxy ring-opening, with C–O stretching vibration peaks at 1044–1150 cm−1 appeared.31
 |
| | Fig. 4 The FT-IR comparison of trimethylolpropane triglycidyl ether (TTE) (pink spectrum), tannic acid (brown spectrum), GO (green spectrum), GO@tannic acid (red spectrum), and GO@tannic acid–TTE (blue spectrum) as a new heterogeneous catalyst. | |
3.2 X-ray diffraction (XRD) of GO@tannic acid–TTE
In XRD of GO@tannic acid–TTE, a peak was observed at 2θ 10.6°, corresponding to an interlayer spacing indicating (001) of GO (Fig. 5). One more broad diffraction peak of GO was observed at 26.3°.29 However, only one distinct peak was observed in the XRD pattern of tannic acid, corresponding to the characteristic value of 2θ = 24.4.32 Trimethylolpropane triglycidyl ether, a peak observed at 2θ = 24.6° and 34°.33
 |
| | Fig. 5 The XRD analysis of GO@tannic acid–TTE. | |
3.3 Scanning electron microscopy (SEM)
Scanning electron microscopy was used to determine the morphology of GO tannic acid and the final catalyst (Fig. 6). As shown, the nanoparticles have good integrity with a smooth surface, with irregular and wrinkled flakes. These wrinkles have increased after reaction with trimethylolpropane triglycidyl ether.
 |
| | Fig. 6 SEM images of GO@tannic acid, GO@tannic acid–TTE, and particle size distribution histogram graphs of GO@tannic acid, GO@tannic acid–TTE particles, which were determined. | |
The particle size distribution histogram of GO@tannic acid demonstrates the successful formation of nanoparticles with an average diameter in the nanometer range. The distribution confirms that the synthesized catalyst is predominantly composed of nanoparticles with sizes below 50 nm, indicating effective size control during the synthesis process. Furthermore, the particle size distribution histogram of GO@tannic acid–TTE reveals a dominant particle size in the range of 15–20 nm. The results show that the majority of catalyst nanoparticles are well within the nanoscale regime, with an average diameter of approximately 20 nm. This narrow size distribution and small particle size confirm the successful synthesis of well-dispersed catalyst nanoparticles, with most particles having sizes below 30 nm.
3.4 Transmission electron microscopy (TEM)
To obtain more confirmation about the catalyst structure, a TEM image of the GO tannic acid@TTE was investigated (Fig. 7). TEM images of GO tannic acid@TTE show aggregation that confirms the successful bonding. A clear view of images observed GO tannic acid@TTE particles have more than one layer (a layer with an aggregate of collected nanoparticles) as a catalyst was produced. Also, there were no visible free flakes of GO, and the structure was irregular, branch-like, and bunch-like.
 |
| | Fig. 7 TEM images of GO@tannic acid–TTE. | |
3.5 Energy-dispersive X-ray spectroscopy (EDS) and elemental mapping analysis
A comprehensive EDS analysis revealed the elemental composition of GO@tannic acid–TTE (Fig. 8). The findings indicated the presence of C and O within the catalyst. Furthermore, elemental mapping (Fig. 9) verified the uniform and even distribution of these elements throughout the catalyst's structure.
 |
| | Fig. 8 The EDX spectrum of GO@tannic acid–TTE. | |
 |
| | Fig. 9 Elemental mapping of the C, O, and combination for GO@tannic acid–TTE. | |
3.6 Thermal gravimetric analysis (TGA) of GO@tannic acid and GO@tannic acid@TTE
The TGA of GO@tannic acid and GO@tannic acid–TTE in the 50–850 °C temperature range is shown in Fig. 10. The TGA curve of GO@tannic acid shows an initial weight loss (10%), which takes place in the 50–200 °C temperature range, confirming the evaporation of the solvent adsorbed on the surface. Next, a remarkable weight loss of 42% is observed in the temperature range of 200–500 °C, associated with the decomposition of the organic moieties on the surface of the GO. In the case of GO@tannic acid–TTE, the weight loss of approximately 13% (from room temperature to 200 °C) indicates the amount of evaporation of adsorbed water and bound water. When the temperature rises to 380 °C, the sharp reduction in the amount of GO@tannic acid by 25% is attributed to the decomposition of hydroxyl groups, alkyl, and carboxyl oxygen-containing functional groups on the surface of the GO. Nevertheless, upon increasing the temperature up to 600 °C, the thermal stability is considerably decreased as a result of the decomposition of the catalyst structure. Additionally, increasing the temperature to 500° results in a steep degradation for GO@tannic acid and a step degradation for GO@tannic acid–TTE.
 |
| | Fig. 10 The TGA diagrams of GO@tannic acid and GO@tannic acid–TTE. | |
3.7 Investigating the catalytic activity of GO@tannic acid–TTE for synthesis 4-substituted-1,5-benzodiazepines
Initial investigations focused on different reaction conditions, such as solvent and catalyst amount and temperature (Fig. 11). The data show that without a catalyst, the reaction does not take place. Furthermore, the products were formed when the catalyst was present during the heating process, which showcases the importance of a catalyst in the process. To study the effect of catalyst amounts, the model experiment was done with GO@tannic acid–TTE at different loadings in EtOH/H2O (1:1 v/v, 5 mL) under reflux, as detailed in chart 1. The experiments showed that as the amount of catalyst was increased, the reaction yield also increased until it reached a plateau of 96%. Therefore, based on the yield, 0.05 g was selected as the optimum catalyst amount for further experiments.
 |
| | Fig. 11 Optimisation reaction diagrams. | |
Furthermore, the solvent's effect on the reaction was also tested, including H2O, DMF, n-hexane, EtOH, THF, and even no solvent. Again, 50% aqueous ethanol not only showed to be the most effective solvent, providing the best yield, but also showcased the highest reaction rate and productivity (chart 2, entry 1). Lastly, the temperature's impact on the catalyst performance. As illustrated in chart 3, the product yield experienced a surge to 96% at 90 °C. In terms of reaction duration, the best time was determined to be 4 hours (Scheme 1).
 |
| | Scheme 1 General reaction for the synthesis of 4-substituted-1,5-benzodiazepines in the presence of GO@tannic acid–TTE as a catalyst. | |
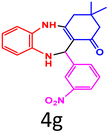
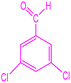
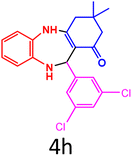
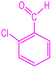
All reactions were carried out under identical conditions and within a fixed reaction time of 4 hours (Table 1). The results demonstrate that both electron-donating and electron-withdrawing substituents on the aromatic ring of aldehydes are well tolerated in this catalytic system, leading to the formation of the desired products with excellent yields. Notably, no significant decrease in reaction efficiency was observed regardless of the electronic nature of the substituents, indicating that the reaction proceeds efficiently for a wide range of aldehydes. The main advantage of this catalytic system, which distinguishes it from other reported methods, is the short reaction time. In order to accurately evaluate the efficiency of the catalyst, the important parameters of turnover number (TON) and turnover frequency (TOF) were investigated. TON represents the total number of moles of product produced by each mole of catalytically active sites before deactivation and is a measure of the overall stability and efficiency of the catalyst, while TOF represents the number of moles of product produced by each mole of catalytic sites per unit time and is an indicator of the catalytic speed and activity of the system. In this study, TON and TOF values were calculated for all products and the results are presented in Table 1.
Table 1 Synthesis of 4-substituted-1,5-benzodiazepines using GO@tannic acid–TTE
| Entry |
Aldehyde |
Product |
Yielda (%) |
Mp (°C) found (lit.) |
TON |
TOF (h−1) |
| Reaction condition: o-phenylenediamine (1 mmol, 0.108 g), dimedone (1 mmol, 0.140 g), aromatic aldehydes, solvent: EtOH/H2O (1:1 v/v, 5 mL), catalyst (0.05 g) at reflux condition and 4 h. 4-Chlorobenzaldehyde (1.021 g), dimedone (0.777 g), o-phenylenediamine (1.296 g), catalyst (0.18 g), time: 5 h. |
| 1 |
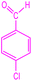 |
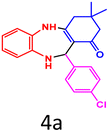 |
93 |
237–238 (ref. 27) |
60000 |
15000 |
| 2 |
 |
 |
88 |
243–245 (ref. 27) |
56800 |
14200 |
| 3 |
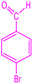 |
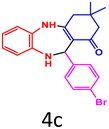 |
91 |
245–246 (ref. 34) |
58700 |
14700 |
| 4 |
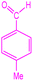 |
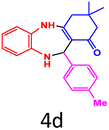 |
90 |
224–227 (ref. 35) |
58100 |
14500 |
| 5 |
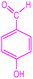 |
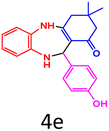 |
90 |
220–223 (ref. 27) |
58100 |
14500 |
| 6 |
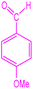 |
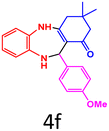 |
88 |
194–196 (ref. 34) |
56800 |
14200 |
| 7 |
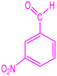 |
 |
90 |
146–148 (ref. 27) |
58100 |
14500 |
| 8 |
 |
 |
89 |
205–207 (ref. 36) |
57400 |
14400 |
| 9 |
 |
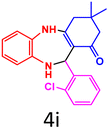 |
92 |
232–233 (ref. 35) |
59400 |
14800 |
| 10 |
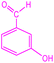 |
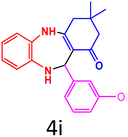 |
90 |
287–289 (ref. 35) |
58100 |
14500 |
| 11 |
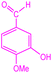 |
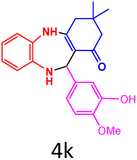 |
89 |
224–226 (ref. 36) |
57400 |
14400 |
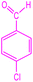
| 12b |
 |
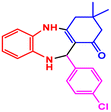 |
80 |
237–238 (ref. 27) |
143000 |
28600 |
3.8 Proposed mechanism for the synthesis of 4-substituted-1,5-benzodiazepines reaction using GO@tannic acid–TTE
The proposed mechanism for the synthesis of 4-substituted-1,5-benzodiazepines is shown in Fig. 12 and also demonstrates the catalytic role of GO@tannic acid–TTE along the reaction pathway. In the first step, the catalyst activates dimedone through hydrogen bonding interactions mediated by the phenolic functional groups. This activation increases the electrophilicity of the carbonyl centers and makes them more susceptible to nucleophilic attack by the aromatic diamine. As a result, intermediate 2 is formed, which is accompanied by the release of a water molecule. The catalyst then subsequently activates the carbonyl aldehyde group, which reacts with the NH2 group of intermediate 2 to form intermediate 3. Finally, intramolecular cyclization of intermediate 3 produces intermediate 4, in which hydroxyl groups on the catalyst facilitate proton transfer during the imine formation, cyclization, and final hydrogen displacement steps, which also contribute to the reaction rate. Once the product is formed, it is easily removed from the catalyst surface, allowing the catalyst to be reused without loss of activity (Fig. 12).37
 |
| | Fig. 12 The proposed mechanism for synthesis 4-substituted-1,5-benzodiazepines, using GO@tannic acid–TTE. | |
3.9 Hot filtration test
A hot filtration experiment was performed to find the natural heterogeneity of GO@tannic acid–TTE. The multicomponent reaction between 4-chlorobenzaldehyde, dimedone, and orthophenylenediamine was carried out by this catalyst under optimal conditions. After 2 h, subsequent reaction stopping, and the obtained yield was 70%. The reaction was repeated to validate the findings. Halfway through this reaction, the catalyst was removed, and the reaction continued without it for the next 2 hours. At this stage, 75% of the final product was produced, confirming the heterogeneous nature of GO@tannic acid–TTE and the occurrence of reactions under heterogeneous conditions and the absence of leaching.
3.10 Reusability potential of GO@tannic acid–TTE results
The recycling of the GO@tannic acid–TTE catalyst was evaluated during the preparation of 4-substituted-1,5-benzodiazepines. The catalyst was simply filtered after each reaction and replaced in subsequent reactions to assess its performance. During the catalyst recycling process over five consecutive cycles, the amount of catalyst used in each cycle was 0.05, 0.043, 0.038, 0.031, and 0.027 grams, respectively. After each reaction was completed and the catalyst was washed and dried, the catalyst used for the next step was weighed and the amounts of starting materials were recalculated for the next reaction. As shown in Fig. 13, the catalyst remained effective for up to 5 consecutive runs, with a 15% decline in activity, which is acceptable. To investigate the stability of the GO@tannic acid–TTE structure after multiple uses, the recovered catalyst (after five runs) was subjected to FTIR, XRD, and SEM spectroscopy. The results of FT-IR, XRD, and SEM analyses indicate that the structure and morphology of the recovered catalyst remained unchanged after five uses (Fig. 14–16).
 |
| | Fig. 13 Recyclability of GO@tannic acid–TTE in the synthesis 4-substituted-1,5-benzodiazepines. | |
 |
| | Fig. 14 Comparison of the FTIR spectra of fresh and reused catalyst after five runs. | |
 |
| | Fig. 15 XRD spectra of fresh and reused catalyst. | |
 |
| | Fig. 16 Reused SEM images of the catalyst. | |
4 Conclusion
In this study, GO@tannic acid–TTE was introduced as a novel, efficient and recyclable heterogeneous catalyst for the synthesis of 4-substituted-1,5-benzodiazepine derivatives. This catalyst was characterized using various techniques including FT-IR, TGA, XRD, TEM and SEM. The introduced catalyst showed high activity in the synthesis of benzodiazepines under mild and green conditions. Also, the reaction was successfully carried out on a gram scale with 80% yield in about 5 h. The findings of this work contribute to the design of stable catalytic systems with biopolymer modification and epoxy crosslinking on graphene-based platforms. The synthesized catalyst is a suitable, cost-effective and environmentally friendly alternative to the old metal-based systems. The catalyst can be reused up to 5 times. Although this study shows acceptable performance of the catalyst, the variety of synthesized products is limited to a specific class of benzodiazepine derivatives and depends on the experimental conditions. Future research should investigate a wider range of derivatives.
Author contributions
Alireza Gharekhani (first author): methodology, validation, research, writing of the initial draft. Professor Maryam Hajjami (corresponding author): funding, supervision, project management, and conceptualization. Reza Hazbavi (co-author): participated in data analysis and manuscript revision.
Conflicts of interest
There are no conflicts to declare.
Data availability
The datasets used and/or analyzed during the present study are available from the corresponding author upon reasonable request.
All data generated or analyzed during this study are included in this published article [and its SI]. Supplementary information is available. See DOI: https://doi.org/10.1039/d5ra08633g.
Acknowledgements
This work was supported by the research facilities of Bu-Ali Sina University, Hamedan, Iran.
References
- C. Sainz-Urruela, S. Vera-López, M. P. San Andrés and A. M. Díez-Pascual, J. Mol. Liq., 2022, 357, 119104 CrossRef CAS.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 Search PubMed.
- M. Hassanisaadi, G. H. S. Bonjar, A. Rahdar, S. Pandey, A. Hosseinipour and R. Abdolshahi, Nanomaterials, 2021, 11, 2033 Search PubMed.
- C. Sainz-Urruela, S. Vera-López, M. P. San Andrés and A. M. Díez-Pascual, Int. J. Mol. Sci., 2021, 22, 3316 Search PubMed.
- J. C.-Y. Lai, H.-Y. Lai, N. K. Rao and S.-F. Ng, J. Ethnopharmacol., 2016, 189, 277–289 CrossRef CAS PubMed.
- R. Lu, X. Zhang, X. Cheng, Y. Zhang, X. Zan and L. Zhang, Front. Chem., 2020, 8, 583484 Search PubMed.
- Z. Gao and I. Zharov, Chem. Mater., 2014, 26, 2030–2037 Search PubMed.
- F. Braghiroli, V. Fierro, A. Pizzi, K. Rode, W. Radke, L. Delmotte, J. Parmentier and A. Celzard, Ind. Crops Prod., 2013, 44, 330–335 CrossRef CAS.
- N. Esmaeili, M. Vafayan, A. Salimi and M. Zohuriaan-Mehr, Thermochim. Acta, 2017, 655, 21–33 Search PubMed.
- R. Liu, J. Zheng, R. Guo, J. Luo, Y. Yuan and X. Liu, Ind. Eng. Chem. Res., 2014, 53, 10835–10840 Search PubMed.
- R. Liu, J. Zhu, J. Luo and X. Liu, Prog. Org. Coat., 2014, 77, 30–37 Search PubMed.
- M. Sahiner, S. Sagbas and B. O. Bitlisli, J. Appl. Polym. Sci., 2015, 132, 41876–41888 CrossRef.
- N. Sahiner, S. Sagbas, M. Sahiner, C. Silan, N. Aktas and M. Turk, Int. J. Biol. Macromol., 2016, 82, 150–159 CrossRef CAS PubMed.
- X. Xu, J. Yu, H. Lu and L. Xue, Pet. Sci. Technol., 2019, 37, 1523–1528 Search PubMed.
- R. Javahershenas, J. Han, M. Kazemi and P. J. Jervis, ChemistryOpen, 2024, 13, e202400185 Search PubMed.
- R. Javahershenas, J. Han, M. Kazemi and P. J. Jervis, ChemistrySelect, 2024, 9, e202401496 Search PubMed.
- R. Javahershenas and S. Nikzat, Ultrason. Sonochem., 2024, 102, 106741 CrossRef CAS PubMed.
- R. Javahershenas and S. Nikzat, RSC Adv., 2023, 13, 16619–16629 Search PubMed.
- R. Javahershenas, H. Mei, M. Koley, V. A. Soloshonok and A. Makarem, Synthesis, 2024, 56, 2445–2461 Search PubMed.
- R. Javahershenas, A. Makarem and K. D. Klika, RSC Adv., 2024, 14, 5547–5565 RSC.
- M. Kurowski, H. Ott and W. Herrmann, Pharmacopsychiatry, 1982, 15, 77–83 Search PubMed.
- P. Nikas, E. Gatta, A. Cupello, M. Di Braccio, G. Grossi, F. Pellistri and M. Robello, Neuroscience, 2013, 243, 158–164 Search PubMed.
- K. T. Olkkola and J. Ahonen, Modern Anesthetics, 2008, pp. 335–360 Search PubMed.
- P. S. Mahadik, G. Senthilkumar, A. S. Powar, D. Devprakash, T. T. Mani and S. A. Gavali, Res. J. Pharm. Technol., 2012, 5, 181–189 Search PubMed.
- L.-Z. Wang, Z.-X. Hua and W.-G. Niu, Chin. J. Org. Chem., 2010, 30, 1664 Search PubMed.
- Z. Karimi-Jaberi and A. Hooshmandpour, Polycyclic Aromat. Compd., 2020, 40, 432–436 Search PubMed.
- S. Tarannum and Z. N. Siddiqui, RSC Adv., 2015, 5, 74242–74250 Search PubMed.
- R. Ghafouri-Nejad, M. Hajjami and R. Nejat, Appl. Organomet. Chem., 2018, 32, e4248 CrossRef.
- A. Gharehkhani and M. Hajjami, Sci. Rep., 2024, 14, 16937 Search PubMed.
- T. Wahyono, D. A. Astuti, I. K. Gede Wiryawan, I. Sugoro and A. Jayanegara, Mater. Sci. Eng., 2019, 546, 042045 Search PubMed.
- D. Zhang, Y. Zhao, Z. Zhang, B. Guo, J. Zhu, Y. Ye and Y. Zhao, J. Appl. Polym. Sci., 2022, 139, 52161 Search PubMed.
- Y. Wei, S. Jiang, J. Li, J. T. Aladejana, T. Zhang, X. Li, Y. Dong and J. Li, Chem. Eng. J., 2023, 458, 141512 Search PubMed.
- A. Sacko, J. F. Nure, H. Nyoni, B. Mamba, T. Nkambule and T. A. Msagati, Water Conserv. Sci. Eng., 2024, 9, 41 CrossRef.
- X. T. Zhu, J. Y. Liu, B. Jiang and S. J. Tu, J. Heterocycl. Chem., 2015, 52, 92–96 Search PubMed.
- H. Naeimi and H. Foroughi, New J. Chem., 2015, 39, 1228–1236 Search PubMed.
- S. Nagaraju, K. Divakar, B. Paplal and D. Kashinath, New J. Chem., 2017, 41, 8993–9001 Search PubMed.
- S. A. Majid, W. A. Khanday and R. Tomar, Biomed. Res. Int., 2012, 2012, 510650 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Reza Hazbavi
* and
Reza Hazbavi








































