DOI:
10.1039/D5RA08480F
(Paper)
RSC Adv., 2026,
16, 5309-5324
Research on the initial corrosion behavior of A100 steel in salt fog-SO2 environment
Received
4th November 2025
, Accepted 7th January 2026
First published on 26th January 2026
Abstract
To investigate the service performance of A100 steel with the chemical composition 23Co14Ni12Cr3Mo in actual ship deck environments, this study innovatively adopts the ASTM G85 “salt spray-SO2 modified salt spray test” to simulate the weakly acidic marine atmosphere containing engine exhaust pollutants. This approach addresses the limitation of domestic studies that primarily rely on neutral salt spray tests following GJB150.11A, which fail to reflect complex service conditions. The corrosion behavior, product composition and electrochemical kinetics of A100 steel were systematically characterized using multiple techniques including SEM, XRD, XPS, FT-IR, EDS, electrochemical workstation and EBSD. Results show that the corrosion process exhibits triphasic kinetics: rapid initial corrosion from 2 to 4 days due to direct contact between the metal and corrosive medium, with charge transfer resistance (Rct) decreasing from 2860 Ω cm2 to 1029 Ω cm2 and corrosion current density (Icorr) increasing from 5.09 µA cm−2 to 7.35 µA cm−2; decelerated degradation from 4 to 8 days by the formation of a dual-layer rust structure consisting of a dense inner layer of Fe3O4 and γ-FeOOH as well as a porous outer layer of Fe2O3, with Rct peaking at 5421 Ω cm2 and Icorr dropping to the minimum of 1.73 µA cm−2; and renewed acceleration from 8 to 10 days caused by the synergistic damage of Cl− and HSO3− to the rust layer, with Rct plummeting to 426 Ω cm2 and Icorr surging to 23.44 µA cm−2. Alloying elements including Cr, Co and Ni regulate corrosion resistance by modifying the rust layer structure and electrochemical properties. Microstructurally, the initial passivation of austenite delays corrosion, while the subsequent dissolution of martensite and the establishment of a dynamic phase equilibrium ultimately expose austenite, accelerating degradation beyond 10 days. This work clarifies the corrosion mechanism of A100 steel under salt spray-SO2 coupling, providing a reliable experimental basis and theoretical support for its safe application in marine aviation equipment.
1 Introduction
In recent years, with the transformation and upgrading of China's maritime strategy, there has been increasing attention on the survival and adaptability of aircraft in marine environments.1 Aircraft that remain on decks or in ship hangars for extended periods are subjected to the corrosive effects of the marine atmosphere. The harsh marine atmospheric environment not only affects aircraft performance but also imposes higher demands on maintenance and upkeep. The new high-strength A100 steel (23Co14Ni12Cr3Mo), known for its excellent toughness, stress-fatigue resistance, and corrosion resistance,2 has been widely used in marine aviation equipment, particularly in landing gear structures.3 However, the high-temperature, high-humidity, and high-salinity characteristic of marine atmospheres make aircraft structures particularly vulnerable to corrosion degradation.4 Research on the corrosion behavior of A100 steel in complex atmospheric environments is of significant importance for the safe operation of aircraft in harsh marine conditions. Currently, both domestic and international research on the corrosion of high-strength steels primarily focuses on corrosion-induced mechanical issues, and China's research in this area is generally aligned with global trends.5,6 With the deepening of research on marine environments, conventional neutral salt spray tests (NSS) and GJB150.11A standards have proven insufficient for effectively assessing the corrosion resistance of materials in marine conditions, as they cannot simulate the weakly acidic environment and Cl−–HSO3− synergistic corrosion induced by SO2 in engine exhaust. In contrast, ASTM G85 can accurately replicate the pollutant composition and dynamic corrosion cycles of actual ship decks, making it more suitable for revealing the service-related corrosion behavior of A100 steel.
Huang Tao7 conducted preliminary research on the rust characteristics and corrosion resistance of high-strength steel in neutral salt solutions. It was found that a dense inner rust layer can effectively prevent the penetration of Cl−, thereby enhancing the material's corrosion resistance.8 Based on Huang Tao's neutral salt spray experiments, Guo Mingxiao and colleagues investigated the corrosion mechanism of SO2 under acidic salt spray conditions.9,10 The study revealed that the synergistic effect of SO2 and Cl− promoted corrosion in the early stages of the experiment, while the SO2/Cl− ratio did not alter the composition of the corrosion products on the carbon steel surface. Additionally, SO2 facilitated the homogenization of carbon steel corrosion. Qian Ang et al.11,12 studied the corrosion and electrochemical characteristics of A100 steel in neutral salt spray environments. Their research indicated that the surface corrosion product layer effectively hindered the penetration and diffusion of the corrosive solution into the substrate, delaying the corrosion process and providing good protection to the substrate. Yang Wendi and Zhao Lianhong13 utilized laser cladding technology to prepare a CoCrNiNb0.1 + xB4C high-entropy alloy coatings on the surface of A100 steel. The results demonstrated that the addition of B4C led to an increase in the coating's microhardness but a reduction in its corrosion resistance. Vishwajeet Bachhar's team demonstrated that Solanum chrysotrichum extract, rich in bioactive phytochemicals, achieves a maximum corrosion inhibition efficiency of 93.38% for mild steel (MS) in 1.0 M HCl at 1.20 g L−1 concentration (following Langmuir isotherm) while exhibiting antioxidant and antibacterial properties, as evidenced by multifaceted characterization and computational analyses.14,15 With the deepening of research on marine environments, conventional neutral salt spray tests and evaluation metrics have proven insufficient for effectively assessing the corrosion resistance of materials in marine conditions, as they significantly differ from the complex deck environments. The deck surface is exposed to a large amount of exhaust fumes emitted by engines during operation, containing gases such as NO2 and SO2, with SO2 posing the greatest threat to the corrosion damage of aircraft structures.16 Currently, domestic studies on the corrosion behavior of A100 steel primarily rely on neutral salt spray tests, conducted in accordance with GJB150.11A “Laboratory Environmental Test Methods for Military Equipment: Salt Spray Test” for relevant testing and verification.17 Compared to neutral salt spray environments, the weakly acidic environment resulting from the combined effects of salt spray and SO2 more closely resembles the actual conditions on ship decks. The U.S. military has extensively applied the “salt spray-SO2” testing method to evaluate the adaptability of materials in marine deck environments, and after decades of development and application, this method has matured.18 The U.S. Department of Defense's “Environmental Engineering Considerations and Laboratory Tests” includes ASTM.G85 “Modified Salt Spray Test Methods, “with the ”salt spray-SO2” testing procedure incorporated into the environmental test method standards MIL-STD-810 versions F and G. The provided data indicate that corrosion exposure tests on ships align with the results of ASTM.G85 “salt spray-SO2” tests.19–21 Diverging from traditional GJB150.11A methodologies, this study adopts the ASTM G85-based modified salt spray test to evaluate A100 steel specimens, aiming to better simulate the service environment of aircraft on ship decks in China. As A100 steel is a primary material for aircraft landing gear, studying it using the salt spray-SO2 corrosion test standard holds significant reference value. This paper investigates the corrosion behavior and electrochemical characteristics of A100 steel under salt spray-SO2 conditions, providing valuable insights for the safe service of this material in harsh deck environments.
2 Experimental
2.1 Experimental materials
The test specimens were fabricated from A100 high-strength alloy steel and machined into cylindrical geometries with dimensions of 6 × 6 mm (Fig. 1). Prior to corrosion testing, all specimens underwent standardized surface treatment to ensure consistent initial conditions: sequentially ground with sandpapers to remove machining defects, ultrasonically cleaned in anhydrous ethanol for 15 minutes to eliminate surface contaminants, and dried with high-purity nitrogen.The heat treatment procedures included: oil quenching at (885 ± 14)°C for 60 minutes, cryogenic treatment at (−73 ± 8) °C for 60 minutes followed by air reversion, and final tempering at (482 ± 3) °C for 6 h with air cooling. Table 1 present the chemical composition and mechanical properties, respectively. The alloy primarily consists of Fe, Co, and Ni with trace amounts of Cr, Mo, and Mn. It exhibits exceptional strength, with a tensile strength of 1970 MPa, and toughness, making it suitable for aircraft landing gear applications.
 |
| | Fig. 1 Sample size and physical image. | |
Table 1 Chemical composition of A100 steel wt%
| C |
Co |
Ni |
Cr |
Mo |
Mn |
Si |
Fe |
| 0.229 |
13.46 |
11.23 |
3.01 |
1.22 |
0.02 |
0.03 |
Bal |
2.2 Salt Spray-SO2 corrosion testing
According to ASTM G85, a HKT 3000 Clima salt spray-SO2 chamber (Kohler Co.) was employed to simulate marine atmospheric conditions. The test parameters strictly corresponded to the actual environmental characteristics of ship decks to ensure simulation authenticity: the exposure zone temperature was maintained at 35 ± 1 °C, with a saturated tower temperature of 47 ± 1 °C. These parameters align with the typical marine atmospheric temperature range of 28–36 °C in coastal navigation areas,14 while accounting for localized surface temperature increases of 3–5 °C caused by engine exhaust. The saturated tower temperature maintained a relative humidity >95%, consistent with the average marine atmospheric humidity of 85–98%.18 The selected SO2 flow rate of 35 cm3 (min m−3) fell within the actual SO2 concentration range of 10–50 cm3 (min m−3) on ship decks,14 specifically simulating peak SO2 exposure from engine exhaust during routine aircraft takeoffs and landings, where SO2 constitutes 60–70% of acidic pollutants.21 The 5 wt% NaCl spray solution matched the salinity of marine atmospheric aerosols (3–6 wt%) in offshore environments,19 reflecting the high-salinity corrosion stress on deck materials. A cyclic protocol comprising 5 hours of salt spray followed by 1 hour of SO2 injection (repeated four times daily, 6-hours cycles) was implemented to reproduce the dynamic alternation of “salt spray deposition–SO2 emission” in practical scenarios: the 5-hours salt spray phase simulated continuous salt exposure during aircraft parking periods, while the 1-hour SO2 injection replicated the periodic SO2 release during daily engine startups (2–3 cycles per day) (Fig. 2).18 This cycling pattern has been validated through U.S. Navy vessel tests, demonstrating a strong correlation (>0.9) with actual corrosion kinetics.20 Six groups of specimens (three replicates each) were extracted at 2, 4, 6, 8, and 10 days for corrosion morphology analysis and electrochemical testing. All data presented in this study are the mean values of three parallel specimens, which helps reduce experimental errors and ensures the accuracy and reproducibility of the results.
 |
| | Fig. 2 Salt spray-SO2 test scheme. | |
2.3 Electrochemical characterization
Electrochemical measurements were conducted using a PARSTAT 4000A workstation (Princeton Applied Research). Post-corrosion specimens were air-dried, welded with central copper leads, and cold-mounted prior to immersion in 5% NaCl at 25 °C. A three-electrode system was employed, consisting of a saturated calomel reference electrode (SCE) and a platinum counter electrode. After a stabilization period of 30 minutes, electrochemical impedance spectroscopy (EIS) was performed over a frequency range from 105 to 10−2 Hz, with a 10 mV amplitude and 30 data points. Potentiodynamic polarization scans were conducted from −0.4 to 0.4 V, utilizing a 0.5 mV step size, a 3-seconds duration per step, and a scan rate of 0.166 mV s−1. The data were analyzed using Zsimpwin and Cview software. Micro-regional electrochemical mapping of a 3 × 3 mm area was performed with a VersaSCAN SKP system (Princeton), employing a 100 µm step size, an 80 µm probe distance, and a 25 µm amplitude.
2.4 Macro/microstructural analysis and corrosion product characterization
Corrosion morphology was examined using an Olympus DSX500 3D microscope and Hitachi S-3400N SEM. Initial low-magnification observations documented the coloration and distribution of the rust layer.XRD analysis (Cu Kα, 40 kV/40 mA, 5–90° range, 2° per min) characterized the composition of the rust layer after 10-days exposure. XPS (Thermo Scientific ESCALAB 250Xi, Al Kα 1486.6 eV, 150 W) determined the elemental oxidation states under ultrahigh vacuum conditions (P < 8 × 10−6 bar), with C 1s (284.8 eV) used as a reference. EDS elemental mapping and FT-IR chemical bonding analysis complemented the phase identification. EBSD (EDAX Velocity Super, 20 kV, 40 nA, 2500 pts per s) elucidated the effects of grain boundary on corrosion progression. The integration of multimodal data ensured analytical reliability.For EBSD characterization, specimens were further polished with 0.05 µm colloidal silica after the initial mirror-finish treatment to eliminate surface stress and oxide films, then ultrasonically cleaned in anhydrous ethanol for 10 minutes and dried with high-purity nitrogen; EBSD tests were performed using an EDAX Velocity Super system with a scanning area of 3 × 3 mm per specimen, a step size of 0.5 µm, and operating parameters of 20 kV, 40 nA, 2500 pts per s, to elucidate the effects of grain boundary on corrosion progression. The integration of multimodal data ensured analytical reliability.
3 Result and analysis
3.1 Corrosion morphology and product analysis
Fig. 3 illustrates the macroscopic surface evolution of A100 steel during salt spray-SO2 testing. After 2-days exposure, numerous dense corrosion pits appear, accompanied by dark brown discoloration and localized reddish-brown corrosion products, along with a loss of metallic luster. On the 4th day, these products develop into patchy rust deposits characterized by powdery detachment, while the dimensions of the pits increase, indicating the formation of iron oxide. Specimens examined the 6th day display extensive reddish-brown outer rust layers overlaying darker inner corrosion products. On the 8th day, a distinct dual-layer rust structure emerges: a reddish-brown outer layer (likely FeOOH/Fe(OH)3) and a compact black inner layer (presumed Fe3O4),22 with surface-deposited NaCl crystals identified via SEM (Fig. 3e). Prolonged exposure until the 10th day results in the formation of lamellar yellow surface layers, indicative of sulfur-containing compounds. Progressive corrosion expansion is evident as color transitions from reddish-brown to yellowish hues across increasingly affected areas. The comparative analysis with neutral salt spray environments2,23 confirms accelerated degradation under combined salt spray-SO2conditions, as evidenced by enhanced surface deterioration at equivalent exposure durations. Furthermore, A100 steel also contains small amounts of metallic elements such as Co, Ni, and Cr, which influence the formation of corrosion products to some extent:24 Cr in A100 steel imparts cation selectivity to γ-FeOOH, preventing the ingress of Cl−; Co can exist in small amounts as CoOOH within FeOOH, increasing the electrochemical impedance of the corrosion product layer; Ni allows the Ecorr of A100 steel to shift in the positive direction, refines the grains of the internal corrosion products, increases their density, promoting the generation of the inner rust layer. These elements collectively improve the initial protective performance of corrosion products, but their regulatory effect on rust layer compactness gradually weakens with the corrosion process under the synergistic erosion of Cl− and HSO3−.
 |
| | Fig. 3 Corrosion morphology of salt spray-SO2 after different time. | |
Fig. 4 presents XRD analysis results of the outer and inner rust layers on A100 steel after 10 days of corrosion. The corrosion products of high-strength steel typically exhibit a dual-layer structure: a loosely adherent outer rust layer and a compact inner layer directly adjacent to the substrate. Following the natural air-drying of corroded specimens, XRD scanning of the upper surface revealed the composition of the outer layer (Fig. 4a). The subsequent removal of superficial rust through ethanol wiping exposed the dense inner layer for characterization (Fig. 4b). A comparative analysis with XRD standard patterns and existing literature2 identified the outer layer as primarily containing Fe2O3, γ-FeOOH, and Fe(OH)3, while the inner layer consisted predominantly of Fe3O4 and γ-FeOOH. These phases correspond to distinct physical properties: the outer layer manifested as a porous reddish-brown substance, contrasting with the compact black morphology of the inner layer.
 |
| | Fig. 4 Analysis results of internal and external rust layer of XRD after salt spray-SO2 corrosion for 10 days. | |
Fig. 5a–c presents XPS spectra of corroded surfaces at varying exposure durations. Reference standards indicate that at 0 days, the Fe 2p spectrum displays unresolved valence states, indicating a predominant metallic iron, while the S 2p spectrum exhibits noisy signals with negligible sulfur content. After 6 and 10 days of corrosion, characteristic Fe 2p1/2 and Fe 2p3/2 peaks appear at 725 eV and 711 eV, respectively. The Fe 2p peaks exhibit corresponding satellite features at higher binding energies – 731.8 eV and 717.7 eV for Fe3+. Peak deconvolution reveals distinct binding energies of 710.8 eV (Fe3+) and 709.6 eV (Fe2+), confirming their coexistence. Quantitative analysis via peak area integration yields the Fe2+/Fe3+ molar ratios: 0.32 ± 0.04 (6 days) and 0.21 ± 0.03 (10 days), indicating an increasing dominance of Fe3+ with prolonged corrosion. This trend aligns with the redox instability of Fe2+, which is readily oxidized to Fe3+ under the weakly acidic salt spray-SO2 environment,25 and correlates with the formation of sulfur-containing corrosion products. The predominance of Fe3+ is consistent with XRD results, which align with the redox instability of Fe2+ and its tendency to oxidize to Fe3+.25 Fig. 5c displays the S 2p spectrum, featuring a prominent single peak at 169 eV, which corresponds to the SO42− reference in the Avantage database. Combined with Fe 2p analysis, this confirms the formation of sulfate through Fe3+–SO42− coordination in corrosion products.26
 |
| | Fig. 5 Fe 2p and S 2p spectra after 10 days of corrosion. | |
Fig. 6 presents SEM observations of specimen surfaces at 0, 6, and 10 days of corrosion exposure (all subfigures with a scale bar of 50.0 µm). The progressive densification of corrosion products is evident, with complete substrate coverage by porous rust layers containing microcracks observed at 6 days (Fig. 6b). This is followed by a reduction in crack dimensions and enhanced in layer compactness at 10 days (Fig. 6c). EDS elemental mapping across three representative regions (Tables 2–4) reveals an increase in sulfur content with prolonged exposure, confirming the incorporation of sulfur compounds. Cross-sectional analysis demonstrates a dominance of iron and oxygen in both rust layers, which can be attributed to redox reactions occurring under salt spray-SO2 conditions. The cathodic oxygen reduction facilitates anodic dissolution of iron, resulting in the formation of Fe2O3 and FeOOH.27 The corrosion mechanism involves two accelerators: chloride ions (0.362 nm diameter) penetrate surface oxides to initiate pitting corrosion,28 while SO2-derived HSO3− acidifies the electrolyte, synergistically enhancing reaction kinetics versus neutral environments.29 Alloying elements exert microstructure-modifying effects: chromium enables γ-FeOOH's cation-selective barrier against chloride ingress; cobalt incorporation as CoOOH increases electrochemical impedance; nickel elevates corrosion potential (Ecorr) while refining grain structure to promote the formation of a dense inner layer.28,30,31
 |
| | Fig. 6 SEM images and EDS analysis of different corrosion cycles. | |
Table 2 The element content on the surface of the specimen after corrosion for 0 days
| Element |
O |
Si |
S |
Cr |
Mn |
Fe |
Co |
Ni |
| Atomic (%) |
0.14 |
0.41 |
0.00 |
2.35 |
0.00 |
73.07 |
13.26 |
9.19 |
| Concentration (%) |
0.04 |
0.21 |
0.00 |
2.18 |
0.00 |
73.05 |
13.99 |
9.65 |
Table 3 The element content on the surface of the specimen after corrosion for 6 days
| Element |
O |
Si |
S |
Cr |
Mn |
Fe |
Co |
Ni |
| Atomic (%) |
45.46 |
0.20 |
24.79 |
0.00 |
0.03 |
24.67 |
3.41 |
1.30 |
| Concentration (%) |
22.81 |
0.18 |
24.93 |
0.00 |
0.05 |
43.21 |
6.30 |
2.40 |
Table 4 The element content on the surface of the specimen after corrosion for 10 days
| Element |
O |
Si |
S |
Cr |
Mn |
Fe |
Co |
Ni |
| Atomic (%) |
12.52 |
0.11 |
28.13 |
0.13 |
0.17 |
48.39 |
5.15 |
5.07 |
| Concentration (%) |
4.52 |
0.18 |
20.34 |
0.16 |
0.22 |
60.95 |
6.84 |
6.72 |
Table 5 Fitting results of electrochemical impedance spectroscopy
| Salt spray time |
Rs (Ω cm2) |
CPE(dl) (µF cm−2) |
nd |
Rf (Ω cm2) |
CPE(f) (µF cm−2) |
nf |
Rct (Ω cm2) |
χ2 |
| 2 days |
8.11 |
187.2 |
0.862 |
20.16 |
13.03 |
0.855 |
2860 |
0.0032 |
| 4 days |
5.97 |
29.46 |
0.569 |
19.63 |
89.26 |
0.756 |
1029 |
0.0041 |
| 6 days |
4.77 |
26.59 |
0.827 |
16.96 |
43.19 |
0.850 |
4263 |
0.0028 |
| 8 days |
3.56 |
75.21 |
0.715 |
27.21 |
55.27 |
0.826 |
5421 |
0.0025 |
| 10 days |
2.14 |
10.81 |
0.892 |
7.56 |
45.62 |
0.762 |
426 |
0.0053 |
Fig. 7 presents the results of Fourier-transform infrared spectroscopy (FTIR) analysis of corrosion products under salt spray-SO2 exposure. The presence of identical infrared absorption bands across various exposure periods demonstrate consistent compositional profiles, primarily containing Fe3O4, γ-FeOOH, and Fe2(SO4)3.1 Characteristic vibrational modes are observed at 605 cm−1 (Fe–O stretching/bending), 832 cm−1 (γ-FeOOH symmetric stretching), and 998 cm−1 (γ-FeOOH antisymmetric vibration). The prominence of the latter among FeOOH polymorphs suggests enhanced corrosion resistance.1 The broad band at 3350 cm−1 corresponds to O–H stretching in Fe2+-bound hydroxyl groups and adsorbed water molecules, whilethe feature at 1635 cm−1 is attributed to H–O–H bending vibrations.32 Notably, the peak at 1096 cm−1 aligns with S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/OSO stretching in SO42−,33 corroborating findings from EDS and XPS (Fig. 4 and 5). The spectral signature at 605 cm−1 further confirms Fe–O–H lattice vibrations in iron oxyhydroxide structures,34 validating sulfate incorporation and the evolution of iron oxide phases during corrosive degradation.
O/OSO stretching in SO42−,33 corroborating findings from EDS and XPS (Fig. 4 and 5). The spectral signature at 605 cm−1 further confirms Fe–O–H lattice vibrations in iron oxyhydroxide structures,34 validating sulfate incorporation and the evolution of iron oxide phases during corrosive degradation.
 |
| | Fig. 7 FT-IR spectra of etching for different days. | |
3.2 Electrochemical test results and analysis
Fig. 8 presents the results of electrochemical impedance spectroscopy (EIS) analysis of A100 steel immersed in 5% NaCl solution at varying corrosion durations. The reduction in capacitive arc radii from day 2 to 4 (Fig. 8a) indicate accelerated reaction kinetics during initial exposure period. The subsequent accumulation of corrosion products between days 6 and 8 leads to the formation of protective rust layers that penetration of HSO3−/Cl− ions, as evidenced by the increasing arc radii. Bode magnitude analysis (Fig. 8b) reveals a decline in corrosion resistance from days 2 to 4, attributed to direct metal–electrolyte interaction prior to the formation of the protective layer. The progressive densification of corrosion products starting from day 6 establishes effective diffusion barriers, reaching the maximum impedance modulus by day 8. However, the degradation of the outer layer initiates a dynamic equilibrium between protective effects and renewed corrosion, resulting in a reduced modulus on day 10 compared to day 4. Characteristic phase angle maxima in the 100–102 Hz range (Fig. 8c) correspond to redox reaction time constants under weak acidic salt spray-SO2 conditions,22 consistent with modulus evolution. The dispersion effects noted in the analysis, attributed to electrode surface heterogeneity and limitations in solution conductivity, necessitate the replacement of ideal capacitors with constant phase element (CPE) components in equivalent circuit modeling.| |
 | (1) |
 |
| | Fig. 8 Ac Impedance Diagram of A100 Steel in 5% NaCl solution under different corrosion time. | |
The equivalent circuit model utilizes the relationship 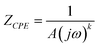 , where A represents the magnitude of capacitive reactance/admittance, j denotes the imaginary unit, ω is the angular frequency, and k signifies the dispersion coefficient. In the context of the rust layer on A100 steel, a five-element equivalent circuit with film components (shown in Fig. 8) was selected to fit the electrochemical impedance spectroscopy data, with detailed parameters provided in Table 5. The circuit components include: Rs (NaCl solution resistance), Rct (charge transfer resistance), CPEdl (comprising double-layer capacitance Cd and dispersion index nd), CPEf (containing film capacitance Cf and dispersion index nf), and Rf (passive film resistance).Rf reflects the structural characteristics of passive films, while Rct corresponds to the resistance encountered during charge transfer through the electrical double layer at the substrate–electrolyte interface. Higher Rct values indicate denser corrosion product layers that prevent corrosive media penetration, thereby enhancing corrosion resistance. Conversely, lower Rct suggests porous rust layers that facilitate charge transfer and accelerate material degradation.35,36
, where A represents the magnitude of capacitive reactance/admittance, j denotes the imaginary unit, ω is the angular frequency, and k signifies the dispersion coefficient. In the context of the rust layer on A100 steel, a five-element equivalent circuit with film components (shown in Fig. 8) was selected to fit the electrochemical impedance spectroscopy data, with detailed parameters provided in Table 5. The circuit components include: Rs (NaCl solution resistance), Rct (charge transfer resistance), CPEdl (comprising double-layer capacitance Cd and dispersion index nd), CPEf (containing film capacitance Cf and dispersion index nf), and Rf (passive film resistance).Rf reflects the structural characteristics of passive films, while Rct corresponds to the resistance encountered during charge transfer through the electrical double layer at the substrate–electrolyte interface. Higher Rct values indicate denser corrosion product layers that prevent corrosive media penetration, thereby enhancing corrosion resistance. Conversely, lower Rct suggests porous rust layers that facilitate charge transfer and accelerate material degradation.35,36
Based on the corrosion characteristics of A100 steel and electrochemical impedance spectroscopy (EIS) features, the five-element equivalent circuit is selected (Fig. 9). The dual-layer rust structure formed during corrosion introduces independent mass transport resistance (Rf) and capacitive response (CPEf) of the corrosion product film, which cannot be fully characterized by simple three-element models accounting only for solution and double-layer effects. Meanwhile, EIS phase angle plots exhibit two distinct time constants, corresponding to the charge transfer at the substrate–electrolyte interface (Rct/CPEdl) and ion diffusion through the rust layer (Rf/CPEf), providing direct experimental evidence for the multi-element model with film components. Additionally, the non-ideal capacitive behavior of the rust layer and electrical double layer (caused by surface roughness, inhomogeneous corrosion products, and adsorbed species) can be accurately described by constant phase element (CPE) instead of ideal capacitors to address the dispersion effect in impedance analysis.
 |
| | Fig. 9 Equivalent circuit diagram. | |
The EIS parameters presented in Table 5 demonstrate distinct stages of corrosion evolution, as evidenced by variations in charge transfer resistance (Rct) and corresponding statistical data. All fitting processes were performed using Zsimpwin software with a five-element equivalent circuit, and the reliability of fitting results was verified by chi-square (χ2) values. During the initial exposure period (2–4 days), a significant decrease in Rct from 2860 to 1029 Ω cm2, was observed, indicating accelerated reaction kinetics due to direct interaction between the bare steel surface and corrosive agents. This initial phase is characterized by minimal formation of protective rust layers, which corresponds with the observed rapid material degradation. Subsequent measurements revealed a notable increase in Rct values, reaching 4263 Ω cm2 by day 6 and peaking at 5421 Ω cm2 on day 8. This upward trend correlates with the progressive development of compact corrosion products that effectively inhibit ionic transport. The maximum Rct value recorded on day 8 confirms the optimal barrier properties achieved by the mature rust layers at this stage. However, a subsequent reduction in Rct to 3815 Ω cm2 by day 10 indicates structural degradation of the protective layer under prolonged exposure to Cl−–HSO3− synergistic effects. This degradation is characterized by microcrack propagation and acidic dissolution processes, which increase the active surface area and consequently facilitate charge transfer processes. The observed trend reversal highlights the dynamic equilibrium between protective film growth and corrosive media penetration, where accumulated structural defects ultimately dominate the corrosion mechanism.37 These quantitative impedance parameters are consistent with microscopic observations (Fig. 3 and 6) and elemental analysis results (Tables 2–4), collectively demonstrating the time-dependent competition between passivation and depassivation processes. The comprehensive data set provides valuable insights into the complex interplay of factors governing the corrosion behavior of the material under investigation.
Fig. 10 shows the polarization curves of A100 steel during corrosion in 5% NaCl solution. The anodic polarization curves exhibit similar characteristics, with the corrosion current increasing as the potential rises, indicating continuous dissolution and corrosion of the specimen. The fitting parameters of the polarization curves are presented in Table 6. The corrosion potential (Ecorr) reflects the thermodynamic tendency of corrosion – a negative shift in Ecorr corresponds to increased corrosion susceptibility, while a more positive Ecorr indicate reduced corrosion activity. βa reflects the kinetics of anodic Fe dissolution with smaller values indicating faster dissolution, while βc relates to cathodic oxygen reduction and reflects changes in cathodic mechanism or mass transport resistance; at 2 days, moderate βa (128.6 ± 8.5 mV dec−1) and βc (112.3 ± 7.9 mV dec−1) indicate unobstructed reactions, 4 days sees βa decrease to 105.4 ± 6.8 mV dec−1 (accelerated anodic dissolution) with stable βc, 6 days shows increased βa (146.2 ± 9.1 mV dec−1) and βc (115.8 ± 8.2 mV dec−1) due to initial rust layer inhibition, 8 days achieves maximum βa (189.5 ± 10.3 mV dec−1) and βc (124.6 ± 9.5 mV dec−1) via dense dual-layer rust protection, and 10d witnesses βa (89.7 ± 5.6 mV dec−1) and βc (96.4 ± 5.8 mV dec−1) plummet as Cl−–HSO3− synergistic attack damages the rust layer, reactivating both anodic and cathodic reactions.
Table 6 Polarization curve fitting result
| Salt spray time |
Ecorr (V) |
Icorr (µA cm−2) |
βa (mV dec−1) |
βc (mV dec−1) |
| 2 days |
−0.663 |
5.093 |
128.6 |
112.3 |
| 4 days |
−0.721 |
7.345 |
105.4 |
108.7 |
| 6 days |
−0.625 |
4.064 |
146.2 |
115.8 |
| 8 days |
−0.530 |
1.726 |
189.5 |
124.6 |
| 10 days |
−0.672 |
23.442 |
89.7 |
96.4 |
 |
| | Fig. 10 Polarization curve of A100 steel in 5% NaCl solution under different corrosion time. | |
Table 6 presents the fitting parameters that illustrate the corrosion evolution of A100 steel. After a 2-days exposure, the corrosion potential (Ecorr) is measured at −0.663 V, with a corrosion current density (Icorr) of 5.093 µA cm−2. By day 4, Ecorr shifts negatively to −0.721 V while Icorr increases to 7.345 µA cm−2, indicating accelerated corrosion activity. On day 6, improved protection is observed as Ecorr rises to −0.625 V and Icorr decreases to 4.064 µA cm−2. Optimal performance is achieved on day 8, with a minimal Icorr of 1.726 µA cm−2 and maximum charge transfer resistance (Rct), which corresponds to dense corrosion product layers that effectively isolate the substrate from the acidic electrolyte. However, by day 10, the degradation of protection results in a negative shift in Ecorr to −0.672 V, with Icorr surging to 23.442 µA cm−2. This acceleration is attributed to accumulated structural damage, an increased active surface area, and compromised rust layers under sustained HSO3−/Cl− attack, despite the slightly elevated Ecorr suggesting a reduced thermodynamic tendency for corrosion.
3.3 Micro electrochemical testing results and analysis
Fig. 11 illustrates the surface potential distribution of A100 steel specimens exposed to salt spray and SO2 corrosion over varying durations, as characterized by scanning Kelvin probe (SKP) measurements. The measured potential (E) profiles were analyzed using Gaussian probability distribution fitting, represented by the following equation:| | |
y = A × exp[−0.5(E − µ)2/σ2]
| (2) |
where A denotes a normalization constant, µ represents the expectation value (mean potential), and σ quantifies the standard deviation (potential distribution dispersion). The fitting results, demonstrated in Fig. 12 and Table 7, reveal systematic evolution of surface electrochemical heterogeneity during corrosion progression.
Table 7 Gaussian fitting result
| Salt spray time per days |
µ mV−1 |
σ |
Maximum potential (mV) |
Minimum potential (mV) |
Potential range (mV) |
| 0 |
−159.46 |
62.83 |
227 |
−294 |
571 |
| 6 |
−62.01 |
85.96 |
95 |
−783 |
878 |
| 10 |
277.53 |
66.89 |
436 |
−282 |
718 |
 |
| | Fig. 11 Scanning Kelvin potential distribution after different time of salt spray-SO2 test. | |
 |
| | Fig. 12 Guassian probability distribution of surface potential of A100 steel at different times. | |
Fig. 11 illustrates the evolution of surface potential characteristics during salt spray-SO2 corrosion. The pristine specimen (0 days) exhibits a homogeneous potential distribution with minimal variation (mean: −159.46 mV, σ = 62.83), reflecting intrinsic micro-galvanic effects between alloying elements with differing electrochemical potentials.38,39 After 6-days exposure, the potential distribution broadens significantly (−783 mV to 95 mV), accompanied by an increased standard deviation (σ = 85.96) and anodic shift (mean: −62.01 mV), indicative of intensified localized corrosion. This transformation results from chloride-induced breakdown of passive films and subsequent micro-galvanic coupling between active substrate regions (anodes) and secondary phase particles/corrosion products (cathodes).40,41 The expanded potential range correlates with accelerated corrosion kinetics due to the cathodic activation of inclusions and microstructural heterogeneities.42,43 By day 10, the surface potential further elevates (mean: 277.53 mV, σ = 66.89) with reduced potential fluctuation, contrasting with electrochemical test trends but aligning with microscopic observations of complete rust coverage. This apparent discrepancy stems from SKP's sensitivity to surface oxide properties, in contrast to electrochemical measurements that reflect bulk charge transfer processes influenced by HSO3−/Cl− penetration and multi-layer corrosion products. It is clarified that the Kelvin potential measured by SKP reflects the surface work function rather than direct corrosion rate, which is dominated by charge transfer kinetics (characterized by EIS and polarization parameters). This explains the discrepancy between SKP and electrochemical trends: SKP is sensitive to surface rust layer integrity, while EIS/polarization tests reflect bulk corrosion involving HSO3−/Cl− penetration. For example, the elevated Kelvin potential at 10d corresponds to sulfur-containing rust (confirmed by XPS/EDS) but does not indicate reduced corrosion activity—consistent with the surged Icorr and dropped Rct in electrochemical tests. The progressive Kelvin potential with exposure duration consistently corresponds with EIS evolution and morphological changes, demonstrating complementary insights from surface-sensitive and bulk electrochemical characterization techniques.
3.4 EBSD results and analysis
Fig. 13 displays inverse pole figures of A100 steel substrate specimens obtained through orientation imaging microscopy at various corrosion durations. The uncorroded specimen presents distinctly visible grain structures. With increasing corrosion time, corrosion products accumulate on the surface, and the grain size distribution shows subtle changes.44,45 Statistical analysis of grain orientations indicates a gradual reduction in low-angle grain boundaries as corrosion advances (Fig. 13b). The grain orientation distribution becomes increasingly heterogeneous, indicating the selective dissolution of specific crystallographic orientations (particularly the (111) orientation marked in Fig. 13a) and structural adjustments related to corrosion-induced phase interactions.44,45
 |
| | Fig. 13 (a) Reverse polarity diagram of A100 steel sample (b) grain orientation statistics. | |
Fig. 14 presents phase diagrams and pole figures of A100 steel after 2, 4, 6, and 10 days of corrosion, with austenite (green) and martensite (red) phases clearly identified. The fraction of the austenite phase initially decreases and then increases during corrosion (Fig. 14a), a phenomenon attributed to the formation of passive films on the surfaces of austenite. Austenite's higher chemical activity and lower corrosion resistance render it more susceptible to attack than martensite, leading to reduced austenite volume fraction in early stages (Table 8). By day 6, accumulated corrosion products form protective passive films on austenite, inhibiting further corrosion,46 while partial dissolution of martensite leads to a decrease in its phase fraction. However, due to the protective effect of the austenite passive film, the austenite volume fraction increased, resulting in a significant elevation of the austenite phase proportion after 10 days of corrosion. The pole figures (Fig. 14b and c) demonstrate weakened characteristic peaks for both phases with prolonged exposure, indicating structural degradation. Initial-stage austenite retains partial orientation features, likely due to preserved passive films, the emergence of new orientation peaks suggest possible recrystallization. Corroded martensite exhibits randomized orientations, reflecting its propensity for brittle fracture propensity during corrosion. These phase-specific evolution patterns reveal that although austenite is initially vulnerable, early passivation temporarily retards corrosion. However, the subsequent dynamic equilibrium between phase dissolution and re-exposure ultimately accelerates degradation by day 10, consistent with electrochemical analyses that indicate a renewed escalation in the corrosion rate.
Table 8 Phase fraction data
| |
2 days |
4 days |
6 days |
10 days |
| Fe(Alpha, Martensite, BCC) |
0.985 |
0.992 |
0.991 |
0.861 |
| Fe(Gamma, Austenite, FCC) |
0.015 |
0.008 |
0.009 |
0.139 |
 |
| | Fig. 14 (a) Phase diagram of A100 steel sample (b and c) austenite and ,artensite pole diagrams of A100 steel sample. | |
Fig. 15a displays grain boundary maps of A100 steel at different corrosion stages, with red, green, and blue lines representing boundaries having misorientation angles below 5°, between 5°–15°, and above 15°, respectively. Progressive corrosion induces significant increases in both grain boundary density and the proportion of high-angle boundaries (Table 9). This phenomenon arises from two concurrent mechanisms: (1) corrosion-induced grain dissolution promotes grain refinement and structural rearrangement, as corroborated by grain size evolution statistics in Fig. 15b; (2) preferential corrosion attack along original grain boundaries transforms initially planar boundaries into irregular configurations, leading to increased high-angle boundary formation.
Table 9 Grain boundary-legend
| |
2 days |
4 days |
6 days |
10 days |
| LAGB |
0.541 |
0.469 |
0.257 |
0.231 |
| HAGB |
0.458 |
0.527 |
0.743 |
0.768 |
| Percentages of LAGB/HAGB |
1.181 |
0.889 |
0.346 |
0.301 |
 |
| | Fig. 15 (a) Grain boundary diagram of A100 steel sample (b) corresponding to grain size of samples with different corrosion times. | |
3.5 Analysis of corrosion behavior mechanism
During salt spray-SO2 testing, a depolarization reaction occurs at the cathode through oxygen participation, constituting a critical step in the corrosion process and typically manifested as localized oxidation features. The anodic reaction predominantly proceeds at the steel substrate/rust layer interface, while cathodic reactions dominate in the outer regions of the inner rust layer. Electrochemical analysis coupled with surface morphological observations reveals that corrosive media penetration to the substrate surface induces localized attack, ultimately forming characteristic corrosion pits.
The synergistic effect of Cl− and HSO3− is not a simple superposition of individual effects but a mutual promotion mechanism: Cl− preferentially adsorbs on the passive film surface, displacing oxygen atoms to form unstable Fe–Cl complexes that break the film and create corrosion channels, while HSO3− derived from SO2 dissolution acidifies the local environment (pH ≈ 4.2–4.8) to accelerate anodic dissolution of Fe and react with Fe3O4 in the inner rust layer to generate porous FeSO4. These channels facilitate HSO3− penetration to the substrate, and the acidic environment further enhances Cl− solubility and mobility, forming a positive feedback loop that rapidly degrades the rust layer. This synergy is verified by EDS/XPS results (Tables 2–4 and Fig. 5) showing coexistence of S and Cl− in corrosion regions, and electrochemical data where the Icorr at 10d (23.442 µA cm−2) is much higher than the sum of corrosion rates induced by single Cl− or SO2 environments.
Fig. 16 illustrates the corrosion mechanism of A100 steel. During the initiation stage, active iron in the substrate acts as an anode, generating Fe2+ through electron loss. This Fe2+ reacts with dissolved oxygen to form FeOOH deposits at pit bases. Concurrently, chloride ions participate in complex reactions with Fe2+, producing corrosive FeCl2·4H2O that accelerates surface degradation. Sulfur dioxide-derived HSO3− further oxidizes to SO42−, forming iron sulfates (Fe2(SO4)3/FeSO4) within corrosion products. Fe(OH)2 decomposition yields FeO, which reacts with oxygen to generate FeOOH, while Fe(OH)3 transforms into stable Fe2O3 and Fe3O4. These oxides gradually develop a dense protective layer that impedes oxygen/chloride diffusion, creating oxygen concentration cells through restricted transport channels and increasing interfacial impedance. The combined effects of protective oxide formation and reduced anodic active area collectively decelerate corrosion rates during later initiation stages through modified electrochemical reaction kinetics.
 |
| | Fig. 16 Analysis of corrosion behavior mechanism. | |
4 Conclusion
This study investigates the early-stage corrosion behavior of A100 steel under salt spray-SO2 conditions through comprehensive characterization and electrochemical analysis, yielding three principal conclusions:
(1) The corrosion process transitions from localized to generalized attack. The inner rust layers primarily consist of dense black Fe3O4 and γ-FeOOH, while outer layers consist of porous reddish-brown Fe2O3. After 10 days of exposure, yellowish outer rust forms, with XPS and FT-IR confirming the incorporation of sulfate and the predominance of Fe3+.
(2) The corrosion follows triphasic kinetics: initial rapid corrosion (0–4 days) with charge transfer resistance (Rct) decreasing from 2860 Ω cm2 to 1029 Ω cm2 and corrosion current density (Icorr) increasing from 5.09 µA cm−2 to 7.35 µA cm−2; decelerated degradation (4–8 days) due to protective rust layer formation, with Rct peaking at 5421 Ω cm2 and Icorr dropping to the minimum of 1.73 µA cm−2; renewed acceleration (8–10 days) induced by Cl−-HSO3− synergy, with Rct plummeting to 426 Ω cm2 and Icorr surging to 23.44 µA cm−2.
(3) Macroscopically, the accumulation of corrosion product (Fe3O4/γ-FeOOH) temporarily inhibits corrosion until structural degradation from Cl−–HSO3− penetration and autocatalytic occluded cell effects reignites propagation. Microstructurally, the initial passivation of austenite delays corrosion, the subsequent dissolution of martensite and the establishment of a dynamic phase equilibrium ultimately expose austenite, accelerating degradation beyond day 10. These findings systematically elucidate the interplay between corrosion product evolution, electrochemical dynamics, and microstructural modifications that govern the degradation mechanisms of A100 steel.
(4) The limitation of this study lies in the simulation of only the synergistic effect between sulfur dioxide (SO2) and chloride ions (Cl−), without considering other pollutants commonly present on ship decks, such as nitrogen oxides (NOx). Future work could focus on expanding the range of pollutants to better replicate real-world environmental conditions, extending the testing duration to investigate long-term corrosion evolution, and incorporating operational loads on landing gear structures to study stress–corrosion coupling behavior.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data that support the findings of this study are included in the main text of the article. The detailed original experimental data, including scanning electron microscopy (SEM) micrographs, X-ray diffraction (XRD) patterns, X-ray photoelectron spectroscopy (XPS) data, Fourier transform infrared spectroscopy (FT-IR) data, energy dispersive spectroscopy (EDS) elemental mapping results, electron backscatter diffraction (EBSD) orientation data, and original datasets of electrochemical tests (electrochemical impedance spectroscopy (EIS), polarization curves, micro-area electrochemical tests, and scanning Kelvin probe (SKP)), are authentic and valid.
Should the original data or additional material data be required to replicate this study or validate the research results, they can be obtained from the corresponding author upon reasonable request.
Acknowledgements
This work was supported by Joint Funds of National Natural Science Foundation of China (No. 52101392), Shandong Provincial Natural Science Foundation of China (No. ZR2024QE253).
References
- Y. L. Chen, Z. Z. Zhang and N. K. Yao, et al., Initial Corrosion Behavior of Non-nitriding and Nitriding 38CrMoAl Steel in Salt Spray Environment, Surf. Technol., 2021, 50(01), 383–394 Search PubMed.
- A. Qian, P. Jin and X. M. Tan, et al., Corrosion and Electrochemical Properties of AerMet100 Steel in Salt Fog, Surf. Technol., 2018, 47(10), 231–239 Search PubMed.
- Z. R. Xian, L. Dong and L. An, et al., Microstructure characterization and mechanical behavior of laser additive manufactured ultrahigh-strength AerMet100 steel, Mater. Sci. Eng., A, 2016, 663, 69–77 CrossRef.
- B. H. Ya, F. D. Chao and L. Hong, et al., Study on the Hydrogen Embrittlement of Aermet100 Using Hydrogen Permeation and SSRT Techniques, Metall. Mater. Trans., 2017, 48, 4046–4057 CrossRef.
- J. K. Guo, J. S. Yu and X. Peng, et al., Study on the Atmospheric Corrosion Behavior of Carbon Steel Using Accelerated Corrosion Test, Surf. Technol., 2014, 43(04), 68–73 CrossRef CAS.
- Z. G. Zhang, J. Y. Zhai and Y. K. Gao, Application Research of Shot Peening Process on 300M Steel Surface, Surf. Technol., 2016, 45(04), 65–68 CrossRef.
- T. Huang, X. P. Chen and X. D. Wang, et al., A Study on the Rust Characteristics and Corrosion Resistance of High Strength Weathering Steels in NaCl Solution, J. Mech. Eng., 2017, 53(20), 45–53 CrossRef.
- Q. Xiang, X. P. Jiang and D. Zhao, et al., Effect of carbonitride treatment temperature on microstructure and corrosion resistance of TA2 titanium alloy, J. Xuzhou Normal Univ., 2021, 39(05), 87–91 Search PubMed.
- M. X. Guo, C. Pan and Z. Y. Wang, et al., A Study on the Initial Corrosion Behavior of Carbon Steel Exposed to a Simulated Coastal-Industrial Atmosphere, Acta Metall. Sin., 2018, 54(01), 65–75 Search PubMed.
- X. X. Song, Research on the Initial Corrosion Behavior of Carbon Steel in Typical Environments of Nuclear Power Plants, University of Science and Technology of China, 2020 Search PubMed.
- A. Qian, X. H. Yang and P. Jin, et al., Micro-zone Electrochemical Behavior of AerMet100 Steel in Salt Spray Environment under Cl−, Equip. Environ. Eng., 2019, 16(10), 88–94 Search PubMed.
- A. Qian, P. Jin and X. M. Tan, et al., Effect of Shot Peening on Surface Integrity and Fatigue Properties of AerMet100 Steel, Surf. Technol., 2019, 48(09), 158–166 Search PubMed.
- W. D. Yang, L. H. Zhao and M. L. Shen, et al., Study on corrosion behavior of CoCrNiNb0.1+B4C high entropy alloy coating on A100 ultra-high strength steel by laser cladding under simulated Marine environment, Surf. Technol., 2024, 1–13 Search PubMed.
- Bachhar, V. Joshi and R. Haldhar, et al., A sustainable triple-action inhibitor: Corrosion protection, antioxidant activity, and antibacterial performance of Solanum chrysotrichum fruit extract, Bioresour. Technol., 2025, 133385 Search PubMed.
- Joshi, V. Bachhar and P. Singh, et al., Rheological and anticorrosion study of Piper chaba extract and coating for mild steel in 2M H2SO4, Colloids Surf., A, 2024, 708, 135989 Search PubMed.
- L. Xu, Y. L. Chen and S. G. Wu, et al., The Study about the Deck Park Environment of Carrier-Based Aircraft and Corrosive State, Aircraft Des., 2016, 36(06), 54–57 Search PubMed.
- S. J. Ketcham, Accelerated Laboratory Corrosion Test for Materials and Finishes Used in Naval Aircraft, Naval Air Development Center, Report No NADC-77252- 3, 1977, 1977 Search PubMed.
- J. Y. Zhu, M. Li and C. G. Cheng, Development and Enlightenment of “Salt Spray-SO2” Test Method for Carrier-based Aircraft of US Navy, Equip. Environ. Eng., 2017, 14(03), 1–6 CAS.
- K. Dhakane, The green fix: environmentally friendly corrosion protection for mass produced metal fasteners, Surf. Coating. Int., 2011,(2), 94 Search PubMed.
- B. D. Sartwell, K. O. Legg, J. Zimmerman, M. Reynolds, J. Gribble, J. Magno and R. Mason, Validation of HVOF Thermal Spray Coatings as a Replacement for Hard Chrome Plating on Hydraulic/Pneumatic Actuators, ESTCP Cost and Performance Report, U.S. Department of Defense, Arlington, VA, 2004 Search PubMed.
- A. Schwartz and E. Beck, Corrosion Performance of AlumiPlate Coated Electrical Connectors with Trivalent Cr Post-Treatment, Naval Air Systems Command, 2007 Search PubMed.
- X. J. Wu, Y. L. Chen and G. X. Bian, et al., Initial Corrosion Behavior of Non-nitriding and Nitriding 38CrMoAl Steel in Salt Spray Environment, Surf. Technol., 2022, 51(05), 234–244 Search PubMed.
- Z. L. Wei, G. X. Bian and A. D. Wang, Effect of Surface Shot Peening on Corrosion Resistance of Aermet100 Steel, Equip. Environ. Eng., 2023, 20(02), 73–81 Search PubMed.
- W. Zhou, W. Lan and S. Zhao, et al., Corrosion Behavior of 42CrMo Steel in Chloride Solution, Surf. Technol., 2017, 46(08), 216–220 Search PubMed.
- C. C. Zhou, C. Li and J. Y. Qian, et al., Adsorption performance of iron oxide red on antimony (V) in printing and dyeing wastewater, J. Zhejiang Univ. (Sci. Ed.), 2022, 49(02), 201–209 CAS.
- D. H. Wang, Z. H. Zhang and P. Li, et al., XPS Study on Hydrogen Sulfide Corrosion Products from Ferric Oxide, Corrosion Sci. Protect. Technol., 2017, 03, 008 Search PubMed.
- Y. Ma, L. Ying and F. Wang, Corrosion of low carbon steel in atmospheric environments of different chloride content, Corros. Sci., 2009, 51, 997–1006 CrossRef CAS.
- Y. L. Chen, A. D. Wang and Y. Zhang, et al., The Synergistic Effect of Cl− and H+ on Initial Corrosion of 2024-T3 Aluminum Alloy, Mater. Rep., 2018, 32(09), 1549–1556 Search PubMed.
- W. Zhou, W. Lan and S. Zhao, et al., Corrosion Behavior of 42CrMo Steel in Chloride Solution, Surf. Technol., 2017, 46(08), 216–220 Search PubMed.
- J. H. Yang, Q. Y. Liu and X. D. Wang, et al., The Progress of Investigation on Weathering Steel and Its Rust Layer, J. Chin. Soc. Corrosion Protect, 2007,(06), 367–372 CAS.
- Y. Xia, F. H. Cao and L. R. Chang, et al., Corrosion Micro-and Macro-electrochemical Behavior of Rusted Carbon Steel and Weathering Steel, Chem. J. Chin. Univ., 2013, 34(05), 1246–1253 CAS.
- X. G. Zheng, W. D. fu and H. Peng, et al., Preparation and characterization of Cu x Zn 1-x S nanodisks for the efficient visible light photocatalytic activity, J. Environ. Chem. Eng., 2017, 6, 9–18 CrossRef.
- L. L. Wu, Generation of Persulfate in Electrolytic System and Treatment of Oilfield Wastewater, Dalian Maritime University, 2023 Search PubMed.
- C. Wei, L. Z. Huai and Z. Jun, et al., Study on the Preparation of A New Composite Coagulant: Poly-Ferric-Titanium-Sulfate and Analysis of FTIR Spectrum and UV-Vis Spectrum, Spectrosc. Spectral Anal., 2016, 36(04), 1038–1043 Search PubMed.
- H. Sheng, C. F. Dong and K. Xiao, et al., Effect of pH on Electrochemical Behavior of 2024-T351 Aluminium Alloy in NaCl Solution, Corrosion Protect, 2013, 34(02), 107–110 CAS.
- D. M. I. Kharitonov, Corrosion Inhibition of Aluminum Alloy AA6063-T5 by Vanadates: Microstructure Characterization and Corrosion Analysis, J. Electrochem. Soc., 2018, 165, C116–C126 CrossRef CAS.
- Y. L. Chen, Z. Z. Zhang and Y. Zhang, et al., Effect of Salt Spray Corrosion Environment on Dynamic Mechanical Properties of 38CrMoAl Steel, Rare Met. Mater. Eng., 2021, 50(06), 2040–2051 CAS.
- Y. L. Chen, H. L. Huang and Y. Zhang, et al., Study on Equivalent Conversion of Galvanic Corrosion Under Different Liquid Film Thickness, Mater. Rep., 2018, 32(09), 1571–1576 Search PubMed.
- G. X. Bian, Y. L. Cheng and Y. Zhang, et al., Equivalent Conversion Coefficient of Aluminum/Titanium Alloy Between Acidic NaCl Solution with Different Concentration and Water Based on Galvanic Corrosion Simulation, Mater. Rep., 2019, 33(16), 2746–2752 Search PubMed.
- B. Losiewicz, M. Popczyk and M. Szklarska, et al., Application of the Scanning Kelvin Probe Technique for Characterization of Corrosion Interfaces, Solid State Phenom., 2015, 228, 369–382 Search PubMed.
- N. E. Benaioun, I. Maafa and A. Florentin, et al., Time dependence of the natural passivation process on AISI 304 in an alkaline medium: Atomic
force microscopy and scanning Kelvin probe force microscopy as additional tools to electrochemical impedance spectroscopy, Appl. Surf. Sci., 2018, 436, 646–652 CrossRef CAS.
- M. Iannuzzi, K. L. Vasanth and G. S. Frankel, Unusual Correlation between SKPFM and Corrosion of Nickel Aluminum Bronzes, J. Electrochem. Soc., 2017, 164, C488–C497 CrossRef CAS.
- A. Nazarov, F. Vucko and D. Thierry, Scanning Kelvin Probe for detection of the hydrogen induced by atmospheric corrosion of ultra-high strength steel, Electrochim. Acta, 2016, 216, 130–139 CrossRef CAS.
- Y. Ma, X. Zhou, Y. Liao, Y. Yi, H. Wu, Z. Wang and W. Huang, Localised corrosion in AA 2099-T83 aluminium-lithium alloy: The role of grain orientation, Corros. Sci., 2016, 107, 41–48 CrossRef CAS.
- Q. Zhou, Z. Zheng and Y. Gao, Abnormal selective dissolution by the partial recrystallization in a plastically deformed austenitic stainless steel, Corros. Sci., 2021, 188, 109548 CrossRef CAS.
- G. Tranchida, M. Clesi, F. Di Franco, F. Di Quarto and M. Santamaria, Electronic properties and corrosion resistance of passive films on austenitic and duplex stainless steels, Electrochim. Acta, 2018, 273, 412–423 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Wenfei Yang*a,
Weijie Fana,
Andong Wanga,
Jia Li
a,
Wenfei Yang*a,
Weijie Fana,
Andong Wanga,
Jia Li








 , where A represents the magnitude of capacitive reactance/admittance, j denotes the imaginary unit, ω is the angular frequency, and k signifies the dispersion coefficient. In the context of the rust layer on A100 steel, a five-element equivalent circuit with film components (shown in Fig. 8) was selected to fit the electrochemical impedance spectroscopy data, with detailed parameters provided in Table 5. The circuit components include: Rs (NaCl solution resistance), Rct (charge transfer resistance), CPEdl (comprising double-layer capacitance Cd and dispersion index nd), CPEf (containing film capacitance Cf and dispersion index nf), and Rf (passive film resistance).Rf reflects the structural characteristics of passive films, while Rct corresponds to the resistance encountered during charge transfer through the electrical double layer at the substrate–electrolyte interface. Higher Rct values indicate denser corrosion product layers that prevent corrosive media penetration, thereby enhancing corrosion resistance. Conversely, lower Rct suggests porous rust layers that facilitate charge transfer and accelerate material degradation.35,36
, where A represents the magnitude of capacitive reactance/admittance, j denotes the imaginary unit, ω is the angular frequency, and k signifies the dispersion coefficient. In the context of the rust layer on A100 steel, a five-element equivalent circuit with film components (shown in Fig. 8) was selected to fit the electrochemical impedance spectroscopy data, with detailed parameters provided in Table 5. The circuit components include: Rs (NaCl solution resistance), Rct (charge transfer resistance), CPEdl (comprising double-layer capacitance Cd and dispersion index nd), CPEf (containing film capacitance Cf and dispersion index nf), and Rf (passive film resistance).Rf reflects the structural characteristics of passive films, while Rct corresponds to the resistance encountered during charge transfer through the electrical double layer at the substrate–electrolyte interface. Higher Rct values indicate denser corrosion product layers that prevent corrosive media penetration, thereby enhancing corrosion resistance. Conversely, lower Rct suggests porous rust layers that facilitate charge transfer and accelerate material degradation.35,36






