DOI:
10.1039/D5RA07375H
(Paper)
RSC Adv., 2026,
16, 15165-15175
The importance of counterion association in the calculated binding constants of Ca2+-aminopolycarboxylate complexes
Received
28th September 2025
, Accepted 6th March 2026
First published on 18th March 2026
Abstract
There exist a number of chemical reactions and processes that have elevated concentrations of metal ions both in solution and as part of a precipitate. To reduce the undesirable effects of metal cations and improve the performance of the active materials in various chemical systems, the application of chelators is required. To this end, different types of chelators have been developed with high affinity towards various metal cations. However, many of the most effective chelators used are limited in biodegradability and could have an impact on environmental pollution. Hence, development of highly efficacious biodegradable alternatives with high affinity for metal cations is an emerging challenge. Electronic structure calculations can assist in predicting new bio-sourced and biodegradable chelators by estimating the binding affinity of metal ion-chelator systems in aqueous solution. In this work, using a set of benchmark aminopolycarboxylate ligands, we calculate binding affinities for a number of chelator-ion systems and investigate the effect of including counterion association on the calculated binding constant. It was found that the system charge neutrality significantly improves the correlation between calculated and experimental values. Furthermore, the influence of conformational flexibility of the chelator structure and pH of the aqueous solution were addressed. The discussed modification in calculation of the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K values considering the counterion association currently serves as a prediction basis and a potential design tool toward new bio-based chelators for applications in industry.
K values considering the counterion association currently serves as a prediction basis and a potential design tool toward new bio-based chelators for applications in industry.
1 Introduction
The use of chelators is required in many industrial processes and consumer applications, e.g. home and personal care products, paper and textile manufacturing, to mitigate the undesirable effects of various metal ions such as Ca2+ or Mg2+.1,2 For example, in home and personal care products, calcium and magnesium cations that are present in tap water (water hardness) can produce scale and reduce the performance of detergents in multiple industrial and domestic processes such as laundering or dishwashing. To reduce water hardness, phosphonates such as (1-hydroxyethane-1,1-diyl)bis(phosphonic acid) (HEDP) and diethylenetriamine penta (methylene phosphonic acid) (DTPMP) may be used. Aminopolycarboxylates (APCs) are another class of industrially relevant chelators, often commercially supplied in the form of their alkali metal salts. Well known examples include ethylenediaminetetraacetic acid (EDTA) and diethylenetriaminepentaacetic acid (DTPA) used as sequestering agents due to their high affinity for Ca2+.3 However, due to the limited biodegradability and other negative environmental effects of these widely used chelators, there is a demand for new, fully renewable and readily biodegradable alternatives.4–7 While some commercial aminopolycarboxylic acids (MGDA, GLDA, EDDS etc.) are biodegradable, their technical performance is lacking compared to phosphonates. Therefore, there is a need for new, renewable, and biodegradable chelators with high affinities for specific metal ions.
Theoretical ex-ante predictions based on the stability constants of complex formation between metal cations and ligands are a valuable tool in identifying the potential of biobased and biodegradable alternatives for commercially available chelators. For chelating agents, the strength of the metal ion-ligand coordination bond in aqueous solution is of importance. The stability of a chelated metal complex is expressed by the stability constant K, which is the equilibrium constant for the formation of a complex in solution. Experimentally there are various methods to determine the stability constant K and for commercially applied chelating agents, these are often known.8–11 Many theoretical studies in this area addressed various approaches to improve the calculated values with respect to the experimental ones. To this end, effective parametrization methods using empirical correction parameters were introduced to correct for omitted energetic contributions to the calculated metal–ligand complexation energies. The defined parametrizations and corrections may closely correlate and be specific to the ligand and metal electronic characteristics as well as structural properties and may vary from one metal–ligand complex to another.12–17 This parameterization was done by introducing either one term energy correction for each metal or an additional correction for each ligand.12,13 To use DFT calculations as a predictive tool for the development of new biobased chelator molecules, a theoretical method is required that produces logK values that are close to the experimental ones in terms of absolute values and trends observed. According to the literature, the calculated logK values so far show large differences with respect to the experimentally measured ones, which is comparable to estimates of molecular acid/base properties such as pKa.18–20 Overestimation of the binding constant has been reported by Laasonen and co-workers. However, the impact of counterions such as Na+ (in addition to the metal cation of interest) association with the chelator-ion system that can physically exist in the experimental environment has not been clearly considered in these studies. In this work we calculate the binding constant values for different benchmark chelators to investigate the effect of including counterion association that significantly improves the correlation between calculated and experimental values without addition of extra parameters and corrections in formulation of the binding constant. According to the literature, it was observed that the formation constants of the Na+ complexes with mono-ligand stoichiometry (ML) linearly increase with the number of charges on the aminopolycarboxylate ligands. This indicates an ionic association between Na+ and anionic carboxylate ligands.21 To validate the effect of such association, we focus on three different ways of calculation of logK, i.e., (i) including implicit water only, (ii) including implicit water + explicit water molecules, (iii) including implicit water only + counterions association. For explicit solvation we included six water molecules forming H-bonds with the ligand anions and for the counterion we added Na+ cations to completely balance the negative charge of the ligand anions. In addition, in this work, the influence of pH and conformational flexibility of the ligand–metal complex on the calculated binding constants is addressed. Isodesmic reactions, where the relative stability of various metal complexes in metal-exchange reactions are calculated, have been widely reported in literature for calculating relative reaction energies due to maximizing the cancellation of errors from the DFT method and from continuum solvent models.22–28 In order to calculate the stability constant values of new biobased chelators with a high degree of confidence, the computational method was validated using a set of known benchmark chelators.
2 Computational details
All geometries have been optimized using the Gaussian 16 package29 and verified to have zero imaginary frequencies. The calculations were carried out with the B3LYP exchange–correlation functional plus D3BJ dispersion correction30–33 and the triple-zeta plus additional polarization function basis set, 6-311G**. The Gibbs free-energies were calculated at 298 K and 1 atm in the solution phase, using the self-consistent continuum approximation (with the default PCM parameterization) of water. The linear transit scan to find the barrier of rotation has been performed using the ADF program package34,35 at BLYP-D3 TZP level of theory starting from and ending at fully optimized geometries.
3 Results and discussion
In Fig. 1A the molecular structures of the aminopolycarboxylic acids considered in this work are shown, while in Fig. 1B a comparison is made between the experimentally measured binding constants (logK) with respect to Ca2+ cation for this series.3 As can be seen in Fig. 1B, there is a clear correlation between several structural characteristics and the measured binding constants. Based on the observed experimental logK values, the most significant contributing factors, that lead to strong chelation, are the number of carboxylate and tertiary amine groups, as well as the length and flexibility of the carbon chains connecting these coordinating groups.36–38 Anionic carboxylate groups cause electronic charge transfer to the Ca2+ and result in strong covalent bond formation (chelation). We note that tertiary amines make stronger coordination than secondary amines, according to the experimental logK values (due to a more available donating lone pair). For example, the chelators with the highest Ca2+ affinity, EDTA and DTPA, have the highest number of tertiary amine (two and three respectively), and carboxylate groups (four and five respectively), as well as very short connecting chains (only one CH2), resulting in conformationally stable five or six membered rings in the complex. One possible theoretical model for obtaining the stability constants of metal ion-chelator complexes is based on the calculated ΔG of the reaction between hydrated calcium cation in the form of an octahedral molecular complex of Ca2+(H2O)6 and the chelator anion to produce the Ca2+ chelator molecular complex and release water molecules (as shown in reaction ii, Scheme 1). In this reaction the chelator anion is considered as an explicitly solvated species with e.g. six water molecules as the first solvation shell that form H-bonds with carboxylate anions. According to the literature, application of explicit and implicit solvation improves the calculated binding constant.39–41
 |
| | Fig. 1 Schematic representation of the experimentally investigated chelators: (A) the structure of the selected benchmark chelators, please note that all the COOH groups are considered deprotonated in this work. (B) The measured logK values (binding constants measured at 25 °C and µ = 0.1 M) in order of decreasing binding affinity vs. Ca2+ based on literature reported data. | |
 |
| | Scheme 1 The reactions used for the calculation of the binding constant of the cation chelation including method (i) only implicit solvation and method (ii) explicit and implicit solvation for ligand molecules. The geometries for this reaction were fully optimized at B3LYP/D3BJ level of theory with 6-311G** basis set. | |
Calculation of logK (K is the equilibrium constant for the reactions in Scheme 1) is based on eqn (1)–(4):
| | |
ΔGreaction = ∑G(product molecules) − ∑G(reactant molecules)
| (1) |
| | |
logKi/ii = −ΔGi/ii/RT(ln10)
| (2) |
| | |
logKcalcd = logKi − 6log[H2O] for reaction i
| (3) |
| | |
logKcalcd = logKii − 12log[H2O] for reaction ii
| (4) |
We note that ΔG(prd.) is the summation of Gibbs free energies of all molecular species on the product side (including H2O molecules) and ΔG(react.) is summation of the Gibbs free energies of all molecular species on the reactant side. In reactions i and ii the ligand anion is considered as a fully deprotonated species with −2 to −5 negative charge (depending on the number of carboxylate groups) in the calculations. The free energies calculated with Gaussian using ideal-gas partition functions are referenced to 1 atm, whereas the experimentally determined K in solution is referenced to a concentration of 1 M. Hence, the free energy of standard-state change must be considered. This term has no effect when the number of moles of reactants and products is the same, however, this is not the case in reactions i and ii due to formation of 6 and 12 H2O molecules, respectively. The calculated conversion constant for ΔG(1 atm → 1 M) is ca. 1.89 kcal mol−1 (details in SI). Furthermore, in order to convert the calculated equilibrium constants by method i and ii (logKi and logKii) into an equilibrium constant that can be better compared with the experimental one, we performed the calculations shown in eqn (3) and (4), which subtract 6log[H2O] and 12log[H2O] from logK of equations i and ii, respectively. The concentration of water is a constant, ∼55.5 M, and therefore log[H2O] is also a constant (∼1.74), which is identical for all cases. Including 1 atm to 1 M correction, the values of logKcalcd were calculated and reported in Tables S8 and S9 in SI, for reaction i and ii, respectively (details in SI).
In solution, reaction ii means that an exchange between water molecules and ligand occurs for the calcium cation. In Fig. 2 we show the calculated logKcalcd values based on the eqn (1)–(4) and reactions i and ii (Scheme 1), for each ligand of Fig. 1A. As can be seen in Fig. 2, the calculated values (logKi) are distributed for some of the chelators (e.g. EDTA, DTPA, EDDM, GLDA) between 40–60 units, and the descending order of the experimental logK that can be observed from EDTA to IDA is not observed for the calculated values and they are distributed randomly. The values of logKii are reduced with respect to logKi, however, some are not realistic (negative values). Hence, the coefficient of determination (R2) between calculated (based on the reaction i and ii) and measured logK values is low. The particularities of the molecular structures of the chelators are not expressed in terms of an impact on the calculated values versus the experimental values. There are two issues that need to be addressed: first, a very large difference in logK values obtained by calculations and experiments and second, a poor correlation between experimental and calculated values is observed. In the next section the influence of ligand electronic structure and negative charge on the calculated logK values in correlation to the experiment are discussed to address these problems.
 |
| | Fig. 2 The calculated logK values based on reaction i and ii vs. the experimentally measured values. The descending order of logKexp from EDTA to IDA is not observed for the calculated ones. | |
To modify logKcalcd values obtained by reactions of Scheme 1, there have been systematic efforts to explore the selectivity of metal binding by DFT approach. These efforts remarkably improved estimation of the metal ion complexation constant with various ligands.12,32–34 However, to the best of our knowledge, association of counterion that is naturally present in the reaction environment was not considered in these attempts, hence, this work.
3.1 Effect of the electronic structure of the ligand on the complexation to Ca2+
In Fig. 3A the variation of the ΔG of complexation between chelators and Ca2+ (with a 1:1 ratio) for each ligand is shown. The ΔG of complexation is the difference between Gibbs free energy of Ca2+-chelator molecular complex and the summation of the Gibbs free energies of free Ca2+ and free chelator, i.e., the free energy of reaction Ca2+ + chelatorn− → Ca-chelator(n−2)−. Variation of the ΔG of reactions i and ii (Scheme 1) is shown in 3B with explicitly hydrated chelators (in orange, method ii) in comparison to the ΔG without explicitly hydrated chelators (in blue, method i) and the ΔG experimental obtained from measured logK values (in black). The negative electronic charge of each ligand is depicted in 3C assuming complete deprotonation of the carboxylic acid groups. Secondary amine and hydroxyl groups are assumed to remain protonated (the lines are there to guide the eyes). Fig. 3B indicates that inclusion of explicit water molecules interacting with the chelator structure (H-Bonds) substantially improves the ΔG values with respect to only implicit solvation (orange vs. blue plots). We note that implicit solvation using water was applied in all calculations. As can be seen in Fig. 3A–C a rather similar pattern for all three graphs is observed, which is in principle similar to the pattern of variation of the negative electronic charge on the ligand anion (3C). This observation indicates that the negative electronic charge on the ligand controls the order of the ΔG of complexation between Ca2+ and chelator as well as the ΔG of reactions in Scheme 1 for the entire ligand series. Hence, the calculated logK values for reaction i and ii are under the influence of the negative electronic charge of the ligand and not the inherent structural characteristics of the ligand. However, as already shown in Fig. 1, the structural particularities of the ligands are determining for the experimental logK and the observed trend of the logKexp. These observations identify that reaction i and ii (and consequently the pattern of the calculations) is under the control of the very strong electrostatic interaction between the entirely negatively charged anion and Ca2+, which overshadows the specific but more subtle effects of chelator structure and functional groups. This indicates that the negative charge on the ligand should be balanced in such a way that it diminishes the very strong electrostatic interaction between metal cation and the ligand anion. This is a valid assumption since a polyanion cannot be solely stabilized by water in a real solution. Accordingly, non-stabilized multiple anionic sites will cause significant electrostatic repulsion and strain in the chelator, while in reality polyanions are dissolved in a stabilizing medium and are always neutralized by counterions.
 |
| | Fig. 3 A) variation of the ΔG of the molecular complex formation between ligands and Ca2+ (the difference between Gibbs free energies of Ca2+⋯chelators molecular complex and free Ca2+ and chelator) no explicit water molecules is considered, (B) variation of the ΔG for the reactions i without explicit water molecules (blue) and with explicitly hydrated ligand (in orange) corresponding to the Scheme 1 and logK values in Fig. 2 in comparison to experimental ΔG values calculated from the available experimental logK values (black), (C) variation of the negative electronic charge of the ligand. In all calculations the implicit solvation using water was included. The lines guide eyes and show the trends. | |
For method i with anionic free ligands, we performed the conformer rotamer ensemble sampling tool (CREST) code,42–45 to find the lowest conformers of the free flexible ligand structures with various dihedral angles. The input structures for CREST were fully optimized geometries of ligands at B3LYP-D3BJ level of theory. Moreover, the ligand structures were calculated with including the explicit water molecules equal to the number of functional groups, i.e. 5H2O for DTPA, 4H2O for EDTA, EDDM, EDDS, HIDS, GLDA, IDS, 3H2O for MGDA and NTA, and 2H2O for, HIDA, IDA and PDA. The results are reported in Table S15 and Fig. S3 in SI. The correlation between calculation and experiment slightly improved with respect to the case with six water molecules for all ligands, i.e., 0.07 units for R2 (0.5779 vs. 0.5002).
3.2 Counterion association
Based on these illustrations, compensation of the negative charge on the ligand is a key step before chelation. To this end, we developed an improved model system by association of the counterions to the chelator anion to realistically overcome the influence of strong electrostatic interaction between Ca2+ and ligand anion. Furthermore, according to the literature, the measurements of logK were performed on the ligands salts.3 Hence, we added extra Na+ cations to the reactions of Scheme 1 and complexed them to the ligand structure to balance the entire negative charge of the ligand before complexation to Ca2+ and reach to a fully neutralized ligand⋯Na+ salt.46 The modified reaction is shown in Scheme 2 (reaction iii). In fact, reaction iii represents an exchange between Na+ and Ca2+ cations with H2O and the chelator. To investigate the direct influence of the Na+ counterion association, explicit water molecules are not included in the chelator molecular structures in reaction iii. As example, Fig. 4 shows the molecular structures of free EDTA anion associated with Na+ and the molecular complex of EDTA-Ca2+-Na+. The advantage of applying reaction iii in the calculation of logK is twofold. First, full neutralization of the negative charge of the anion that compensates the large electrostatic interaction between Ca2+ and ligand (also within the polyanionic ligand). And second, involving the thermodynamic effects of exchange between Ca2+ and Na+ cations with H2O and the ligand in the overall calculated ΔG of the reaction. Full neutralization of the negative charge reduces the electrostatic interactions which is confirmed by Energy Decomposition Analysis (EDA)47–49 results as well as comparison of the complexation energies between Ca2+ and ligandn− versus Ca2+ and Ligandn−Na+(n−2) (reported in Tables S5–S7). Involvement of exchange between cations in a reaction was investigated in previous studies as it maximizes the cancellation of systematic errors inherent to the level of theory and in the calculation of interactions between the continuum solvent and the solute, which can vary significantly with the net electric charge of the solute. As example, calculation of relative energies, thermodynamic constants (pKa values), or the stability of metal complexes using cations exchange were frequently addressed in the literature.24–27
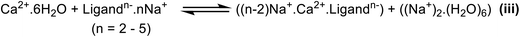 |
| | Scheme 2 The modified reaction used for the calculation of the binding constant (method iii) which contains Na+ counterions (including implicit solvation but no explicit water molecules). We note that (Na+)2·6H2O is a single cluster of two Na+ cations and six water molecules. | |
 |
| | Fig. 4 The optimized molecular structures for EDTA in reaction iii (Scheme 2), i.e., associated with Na+ (left) and EDTA-Ca2+-Na+ (right). No explicit water solvent molecules were included for the chelator (method iii). | |
Fig. 5A shows the calculated ΔG based on reaction iii (Scheme 2) for the ligand series considered in Fig. 1. Fig. 5B shows the calculated values of logK based on reaction iii plotted versus the experimental logK and in comparison to cases i and ii, i.e., without counterion association and only implicit and explicit solvation. The new calculated values (grey line) are now in the same range as the experimental values (4–14) and the obtained correlation (R2) between the calculated logK and the experimental is improved to ca. 0.80 (grey line in Fig. 5B) indicating a rather good agreement between the experimental and theoretical values. Hence, the counterion (Na+) association to the ligand anion significantly improves the calculated logK values.
 |
| | Fig. 5 (A) the calculated free energies for the reaction iii (Scheme 2) considering the Na+ counterion association (correlated to the grey line in B). (B) variation of the calculated vs. experimental logK values for different models, i.e., chelators without explicit water molecules (blue, case i), chelators including explicit water molecules (orange, case ii) and chelators considering the Na+ counter ion (grey, case iii) and no explicit water molecules. | |
Similar to reactions i and ii in reaction iii the ΔG of reaction is the difference between summation of Gibbs free energies of all product molecules and summation of Gibbes free energies of all reactant molecules.
| ΔGiii = G(Na(n−2)CaL) + G(Na2·6H2O) − G(NanL) − G(Ca·6H2O) = ΔG(f,Na(n−2)CaL) − ΔG(f,NanL) |
| ΔGiii = −RTln(K(Na(n−2)CaL)/K(NanL)) |
| | |
log[K(Na(n−2)CaL)] = logKiii + log(K(NanL))
| (5) |
The values of log(K(NanL)), which are considerably small due to the very weak binding interaction with the ligands, were reported in previous experimental investigations in literature.50–55
For a better comparison, the calculated ΔG and logK as well as the measured logK values are reported in Table S1 in SI. The impact of counterion association was examined by addition of K+ cation (instead of Na+) and similar results to Na+ were obtained using EDA and complexation energies (Tables S13 and S14). Consider that continuous solvent models exhibit a strong dependence of their accuracy on the total charge of the solute. This causes larger ligand-Ca2+ electrostatic interactions which yield larger errors and poorer estimations of logK. Since the absolute error of solvation energies (or energies in solution) is much larger for ionic species than for neutral species, as a result of the global charge reduction, the electrostatic interaction with the continuum solvent model is also reduced, thus the free energy in solution (or the solvation energy) of the complexes is reduced.56–61
3.3 Conformational flexibility of the chelator structure
As can be seen in Fig. 5, GLDA and HIDA deviate from the general trend of the obtained ΔG values compared to the rest of the chelators and also the experimental trend, i.e., for GLDA the calculations overestimate the experimental value (11.52 vs. 5.9, respectively) and for HIDA underestimate the experimental value (4.9 vs. 5.3, respectively). To elucidate this deviating behavior, we analyzed the molecular structures of these two chelators in more detail. As shown in Fig. 6, GLDA has a more flexible arm consisting of two methylene (CH2) units, that can be either complexed to the Ca2+ cation or freely associated to a Na+ counterion in the reaction medium. The arrow shows a selected dihedral (C–C bond) angle that, when rotated, results in either a free (associated with Na+) or complexed to Ca2+ molecular structure. The calculated ΔG values (of reaction iii) depicted next to each structure in Fig. 6 (top and middle) demonstrate a large difference in stabilization energies between the two ways of complexation for the GLDA⋯Ca2+ structure, i.e., −15.7 vs. −5.6 kcal mol−1. A linear transit scan around the highlighted dihedral angle results in a 24 kcal mol−1 barrier of rotation starting from the three coordinated molecular complex (flexible arm associated with Na+) to the four coordinated complex. This relatively high rotational barrier indicates that the molecular complex with flexible arm associated with Na+ can be thermodynamically stable in the solution at room temperature. Previous studies also confirmed that in the higher coordination numbers the strain on the ligand back bone forces one or more coordinating atoms (COO−) much farther away.12
 |
| | Fig. 6 The calculated molecular structures of GLDA chelators in various complexation states, i.e., flexible arm is complexed to Ca2+ (top) and associated to Na+ (middle). The lower part shows a linear transit scan of the stabilization energy around the dihedral angle. | |
For a better overview of the flexibility of CH2–CH2 linkage that causes COO− either associated to Na+ or complexed to Ca2+ we performed a dihedral scan starting from the initial optimized geometry at each potential well and slightly changed the dihedral angle in 5° steps to sample more local minima around the optimized geometries. It turned out that three coordinated state (flexible arm associated with Na+ cation) can include several geometries with very close energies (within 1 kcal mol−1) and feasible in solution that can be stabilized through interaction with solvent.62 For the four coordinated state (flexible arm complexed to Ca2+ cation), the variation of the relative energies of rotation of the dihedral angle are larger and the most stable conformer is at least 1.24 kcal mol−1 lower in energy than the others. The results are summarized in Table S3 in the SI. This observation also indicates that the experimentally measured logK value can be an average between many cases (states) between the two depicted extremes in Fig. 6 due to the conformational flexibility in solution, and this can affect the calculated ΔG correlated to reaction iii. Even at room temperature, multiple association/dissociation equilibria exist in which one or more donor groups may temporarily detach from the metal, rotate, and reattach without complete metal–ligand dissociation (one of the factors underlying the stability of polydentate ligands). Furthermore, to study the effect of temperature, the barrier for rotation around the corresponding dihedral angle (shown in the lowest part of Fig. 6) was calculated at four different temperatures that are relevant to a range of industrial processes where chelators are used to control water hardness ions, i.e. 30, 40, 60 and 90 °C (depicted in Fig. S1 in the SI). As example, increasing the temperature from 25 to 90 °C reduces the barrier by 40 percent and increases the chance for a faster transition between three and four coordinated states.
3.4 Effect of pH
In the case of HIDA, the molecule contains a CH2OH group (versus a COO− group in the analogous NTA) that can be deprotonated in basic solution forming an anionic coordination site for complexation to the Ca2+ cation. This results in stronger complexation and lower ΔG of reaction iii as depicted in Fig. 7. However, at a pH lower than the pKa of this OH group, protonation occurs, significantly reducing coordination and hence the logK value. Therefore, for molecules like HIDA, there is a very strong pH effect on logK, that should be included in the comparison with experimental data.
 |
| | Fig. 7 The calculated molecular structures of HIDA chelators in various complexation states, i.e., protonated OH complexed to the Ca2+ (up) and deprotonated O− complexed to Ca2+ (down). | |
A significantly improved value for R2 (0.97) for the logK plot can be obtained by including the influence of partial coordination (flexibility) in GLDA, as well as the pH on the coordination of HIDA (Fig. 8).
 |
| | Fig. 8 (A) The ΔG values of reaction iii, the black dots show the initial values of ΔG for HIDA and GLDA before corrections, i.e., GLDA with four coordination to the Ca2+ and protonated O–H of HIDA. (B) the logK plots including the average between deprotonated (50%) and protonated (50%) complexations for HIDA and the average between three (50%) and four (50%) coordinated states for GLDA. | |
In a realistic reaction solution with a very dynamic situation in the experimental measurements it is possible that a series of transient states are available in between the two extremes for both GLDA and HIDA, which can be sampled by Boltzmann averaging of the calculated energetic states. We note that addition of an anionic counterion such as HCO3− to the reaction iii (Scheme 2) for neutralization of the positive charge of Ca2+ was examined. It turned out the R2 value remains constant, however, the calculated logK values for each ligand slightly decreases (0.4 units). The results are shown in Fig. S2 in SI, logK and ΔG values are reported in Table S2.
In Fig. 9, we have depicted both complexation types, i.e., the product complex in reaction iii (Scheme 2) in the presence of Na+ and without Na+ counterion in reactions Scheme 1. The bond distances between N(tertiary)⋯Ca2+ and O(carboxylate)⋯Ca2+ for all complexes are shown in the figure. The distances between Ca2+ and N/O indicate that all N atoms and carboxylate groups are involved in the complexation. As can be seen in Fig. 9, association of the Na+ counterions to the structure of the molecular complexes results in an increase in the Ca2+⋯N and Ca2+⋯O distances (0.1 Å), which is in line with the weaker complexation (interaction) and less negative ΔG of the reaction iii. Moreover, complexation of oxygen to the Ca2+ has a shorter bond distance (stronger complexation) than nitrogen, which is correlated to hard acid–base (earth alkaline metals and oxygen, respectively) interaction. According to the energy decomposition analysis (EDA), the dominant interaction between Na+-associated ligand and Ca2+ are electrostatic interactions.
 |
| | Fig. 9 The corresponding structures of EDTA, MGDA, and NTA without Na+ association (left) and with Na+ association (right), numbers show the distances in Å. According to the crystallographic measurements, the Ca–O and Ca–N bond distances are 2.408 Å and 2.667 Å, respectively in Ca-EDTA crystal.63 | |
4 Conclusion
We have developed a modified approach for the theoretical calculation of the logK values for the Ca2+ coordination by aminopolycarboxylates. By including charge stabilization in the chelators via Na+ cations association, as well as implicit water solvation in the calculations, conformational flexibility and pH effects, the regression coefficient of the calculated values versus experimentally determined ones for a series of 12 amino carboxylate chelators increased from 0.55 to 0.97. Previous works explained the importance of isodesmic reactions which yield good results for calculating relative energies using DFT and continuum solvation methods.23,26 In this work we highlighted the influence of association of alkali metal cations such as Na+ and K+ neutralizing the negative charge of molecular complex between central metal atom (Ca2+) and multidentate aminocarboxylate ligands. On the one hand, the charge neutralization is an individual physical characteristic that can substantially reduce the electrostatic interaction and hence calculated reaction free energies and logK values toward the experimentally measured binding affinities. On the other hand, when the Na+ or K+ ions are present, the complexation energies of the remaining ligand groups with Ca2+ (which are basically coming from the electrostatic term) are numerically much more similar. This means that the absolute errors in the free energies in solution of each species will be also similar (regardless of the counterion's nature), implying that when calculating differences of free energies between reactants and products, better cancellation of errors will be obtained in free energy of reaction with the presence of Na+ than in free energy of reactions without Na+. A 1:1 ratio of metal–ligand complexes that is included in this work, and previous studies may still be a potential source of deviation from experimental values, i.e. contribution of the other metal:ligand ratios. The discussed modification in calculation of the logK values (considering the counterion association) currently serves as a prediction basis for designing new bio-based chelators for the real application in industry. By analysing the causes of two specific outliers (GLDA and HIDA), we have shown that flexibility of complexation sites of chelators and presence of kinetic barriers for switching between various chelator conformers as well as pH effects can cause significant deviations between calculations and experiments. Full coordination of all available functionalities results in an over-estimation of the calculated logK, due to a thermodynamically stronger complexation between ligand and metal cations. Furthermore, there is the possibility of higher M:L stoichiometry such as ML2, M2L3, etc. that can still improve the average value of the calculated logK with respect to experiment. Understanding the specifics of individual chelator molecules helps in improving the correlations. We are currently expanding the developed methodology to other metal ions, and using modeling to differentiate between predicted logK values of not-yet synthesized novel chelators.
Conflicts of interest
There are no conflicts to declare.
Data availability
The authors declare that the data supporting this article have been included as part of the supplementary information (SI). Supplementary information: includes all xyz coordinates of ligand–Na and ligands–Na–Ca, ΔG and logK for the reactions i, ii and iii, bond distances for ligand–Ca and ligand–Ca–Na, tables with calculated ΔG, relative ΔG and Boltzmann weights of the distribution of molecular structures at three and four Ca2+ coordinated states, plot of variation of the activation barriers vs. temperature for the transition between three-coordinated to four coordinated system, EDA results, conformational search, details of thermodynamic calculation of the logK values. See DOI: https://doi.org/10.1039/d5ra07375h.
Acknowledgements
This work used the Dutch national e-infrastructure with the support of the SURF Cooperative using grant no. EINF-5393. This research was financed by TKI funding from the Topconsortia for Knowledge & Innovation (TKI's) of the Ministry of Agriculture, Fisheries, Food Security and Nature (LVVN, project LWV21.85, Circular Chelates and Ligands with Exceptional Affinities from Natural resources (CCLEAN)) and the companies Archer Daniels Midland Company (ADM) and Bio Detection systems B.V. (BDS).
References
- G. Chauhan, K. K. Pant and K. D. P. Nigam, Environ. Sci.: Processes Impacts, 2015, 17, 12, 10.1039/C4EM00559G
 .
. - G. Chauhan, M. Stein, A. Seidel-Morgenstern, K. K. Pant and K. D. P. Nigam, Chem. Eng. Sci., 2015, 137, 768, DOI:10.1016/j.ces.2015.07.028 .
-
(a) I. S. S. Pinto, I. F. F. Neto and H. M. V. M. Soares, Environ. Sci. Pollut. Res., 2014, 21, 11893, DOI:10.1007/s11356-014-2592-6 ;
(b) R. M. Smith and A. E. Martell, Sci. Total Environ., 1978, 64, 125–147 CrossRef ;
(c) X. C. Liu, L. H. Skibsted, R. Chen and W. Sun, Food Res. Int., 2025, 220, 117128 CrossRef CAS PubMed ;
(d) M. A. Marini, W. J. Evans and R. L. Berger, J. Biochem. Biophys. Methods, 1986, 12, 135–146 CrossRef CAS PubMed ;
(e) Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585–4621 CrossRef CAS PubMed .
- P. W. Jones and D. R. Williams, Int. J. Environ. Anal. Chem., 2001, 81, 73, DOI:10.1080/03067310108044359 .
- K. B. Charkhutian, B. L. Libutti, F. De Cordt and J. S. Ruffini, Process for inhibiting scale and fouling the metal surfaces exposed to an aqueous system, International Patent WO2004013055, 2004 .
- K. B. Charkhutian, B. L. Libutti and M. A. Murphy, Process for inhibiting scale on metal surfaces, International Patent WO2006028917, 2006 .
- J. Richardson, R. Bedinger and J. Wilkins, Methods for reducing scale formation on and removing deposits from heat transfer surfaces, International Patent WO2012068590, 2004 .
- BASF Technical Information Ti/EVD 1418 e - Trilon® M types, 2007.
- AkzoNobel Dissolvine® GL technical brochure, 2010.
- Nippon Shokubai Biodegradable chelating agent: HIDS, https://www.shokubai.co.jp/en/products/functionality/hids.html, accessed in June 2013.
- H. Yvonen and R. Aksela, J. Coord. Chem., 2010, 63, 2013, DOI:10.1080/00958972.2010.496483 .
- J. A. Sillanpää, R. Aksela and K. Laasonen, Phys. Chem. Chem. Phys., 2003, 5, 3382, 10.1039/B303234P .
- H. Pesonen, R. Aksela and K. Laasonen, J. Mol. Struct., 2007, 804, 101, DOI:10.1016/j.theochem.2006.10.013 .
- L. Chen, T. Liu and C. Ma, J. Phys. Chem. A, 2010, 114, 443, DOI:10.1021/jp904296m .
- H. Pesonen, A. Sillanpää, R. Aksela and K. Laasonen, Polymer, 2005, 46, 12641, DOI:10.1016/j.polymer.2005.10.069 .
- B. M. Gindele, K. K. Malaszuk, C. Peter and D. Gebauer, Langmuir, 2022, 38, 14409, DOI:10.1021/acs.langmuir.2c01662 .
- C. Ebenezer and R. V. Solomon, Comments Mod. Chem., 2024, 44(5), 385–459, DOI:10.1080/02603594.2024.2305884 .
- K. S. Alongi and G. C. Shields, Annu. Rep. Comput. Chem., 2010, 6, 113–138, DOI:10.1016/S1574-1400(10)06008-1 .
- S. Pezzola, S. Tarallo, A. Iannini, M. Venanzi, P. Galloni, V. Conte and F. Sabuzi, Molecules, 2022, 27(23), 8590, DOI:10.3390/molecules27238590 .
- A. J. Sanchez and K. Raghavachari, Theor. Chem. Acc., 2023, 142, 86, DOI:10.1007/s00214-023-03024-6 .
- S. Berto, E. Chiavazza, P. Canepa, E. Prenesti and P. G. Daniele, Phys. Chem. Chem. Phys., 2016, 18, 13118–13125, 10.1039/C6CP00192K .
- R. Casasnovas, J. Ortega-Castro, J. Donoso, J. Frau and F. Muñoz, Phys. Chem. Chem. Phys., 2013, 15, 16303–16313, 10.1039/C3CP50840D .
- S. Sastre, R. Casasnovas, F. Muñoz and J. Frau, Phys. Chem. Chem. Phys., 2016, 18, 11202, 10.1039/C5CP07053H .
- A. D. Bochevarov, M. A. Watson, J. R. Greenwood and D. M. Philipp, J. Chem. Theory Comput., 2016, 12, 6001, DOI:10.1021/acs.jctc.6b00805 .
- P. Lian, R. C. Johnston, J. M. Parks and J. C. Smith, J. Phys. Chem. A, 2018, 122, 4366, DOI:10.1021/acs.jpca.8b01751 .
- S. Sastre, R. Casasnovas, F. Muñoz and J. Frau, Theor. Chem. Acc., 2013, 132, 1310–1318 Search PubMed .
- F. Ribeiro Dutra, C. de Souza Silva and R. Custodio, J. Phys. Chem. A, 2021, 125, 65 CrossRef PubMed .
- R. Casasnovas, J. Ortega-Castro, J. Frau, J. Donoso and F. Muñoz, Int. J. Quantum Chem., 2014, 114, 1350, DOI:10.1002/qua.24699 .
- M. J. Frisch and G. W. Trucks, Gaussian 16, Gaussian Inc., Wallingford, CT, 2016 Search PubMed .
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS .
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS PubMed .
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, J. Chem. Phys., 2010, 132, 154104, DOI:10.1063/1.3382344 .
- S. Grimme, J. Comput. Chem., 2006, 27, 1787, DOI:10.1002/jcc.20495 .
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967, DOI:10.1002/jcc.1056 .
- E. J. Baerends, T. Ziegler, ADF 2021, Theoretical Chemistry, Vrije Universiteit Amsterdam: The Netherlands, 2021 Search PubMed .
- S. Heinzman, J. Surfactants Deterg., 1998, 1, 105–108 CrossRef CAS .
- M. M. Crutchfield and J. A. M. OIL, J. Am. Chem. Soc., 1978, 55, 58, DOI:10.1007/BF02673391 .
- H. C. Kemper, et. al, Tenside Surfactants Deterg., 1975, 12, 47 CrossRef CAS .
- O. Gutten, I. Besseova and L. Rulísek, J. Phys. Chem. A, 2011, 115, 11394, DOI:10.1021/jp205442p .
- O. Gutten and L. Rulísek, Phys. Chem. Chem. Phys., 2015, 17, 14393, 10.1039/C4CP04876H .
- O. Gutten and L. R. Rulísek, Inorg. Chem., 2013, 52(18), 10347, DOI:10.1021/ic401037x .
- S. Grimme, J. Chem. Theory Comput., 2019, 155, 2847–2862, DOI:10.1021/acs.jctc.9b00143 .
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192, 10.1039/C9CP06869D .
- S. Spicher, C. Plett, P. Pracht, A. Hansen and S. Grimme, J. Chem. Theory Comput., 2022, 18, 3174–3189, DOI:10.1021/acs.jctc.2c00239 .
- P. Pracht and C. Bannwarth, J. Chem. Theory Comput., 2022, 18, 6370–6385, DOI:10.1021/acs.jctc.2c00578 .
- Critically Selected Stability Constants of Metal Complexes, Version 4.0, ed. A. E. Martell, and R. M. Smith, Texas A & M, University, College Station, TX, 1997 Search PubMed .
- F. M. Bickelhaupt and E. J. Baerends, Rev. Comput. Chem., 2000, 15, 1 CAS .
- F. M. Bickelhaupt and E. J. Baerends, Angew. Chem., Int. Ed., 2003, 42, 35 Search PubMed .
- L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8, e1345, DOI:10.1002/wcms.1345 .
- G. Andereggi, F. Arnauud-Neu, R. Delgado, J. Felcman and K. Popov, Pure Appl. Chem., 2005, 77(8), 1445–1495, DOI:10.1351/pac200577081445 .
- M. Chen and R. S. Reid, Can. J. Chem., 1993, 71, 763–768, DOI:10.1139/v93-100 .
- C. Bretti, K. Majlesi, C. De Stefano and S. Sammartano, J. Chem. Eng. Data, 2016, 61, 1895–1903, DOI:10.1021/acs.jced.6b00063 .
- K. Majlesi, C. Bretti, R. M. Cigala, C. De Stefano and K. Majlesi, Silvio Sammartano, J. Solution Chem., 2018, 47, 528–543, DOI:10.1007/s10953-018-0734-z .
- C. De Stefano, A. Gianguzza, D. Piazzese and S. Sammartano, Anal. Bioanal. Chem., 2003, 375, 956–967, DOI:10.1007/s00216-003-1790-8 .
- C. Bretti, R. M. Cigala, C. De Stefano, G. Lando and S. Sammartano, Fluid Phase Equilib., 2017, 434, 63–73, DOI:10.1016/j.fluid.2016.11.027 .
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2005, 1, 1133 CrossRef CAS PubMed .
- Y. Takano and K. N. Houk, J. Chem. Theory Comput., 2005, 1, 70 CrossRef PubMed .
- A. V. Manerich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef PubMed .
- J. R. Pliego and J. M. Riveros, J. Phys. Chem. B, 2000, 104(21), 5155–5160, DOI:10.1021/jp000041h .
- B. Hanson, M. Smith and P. Li, J. Phys. Chem. B, 2024, 128(48), 11904–11913, DOI:10.1021/acs.jpcb.4c04034 .
- L. C. Kröger, S. Müller, I. Smirnova and K. Leonhard, J. Phys. Chem. A, 2020, 124(20), 4171–4181, DOI:10.1021/acs.jpca.0c01606 .
- M. Heshmat, J. Phys. Chem. A, 2018, 122(40), 7974–7982 CrossRef CAS PubMed .
- B. L. Barnett and V. A. Uchtman, Inorg. Chem., 1979, 18(10), 2674, DOI:10.1021/ic50200a007 .
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Pavlo Kostetskyy
*a,
Pavlo Kostetskyy

![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) log
log







