DOI:
10.1039/D5RA07315D
(Paper)
RSC Adv., 2026,
16, 1802-1819
Spectrochemical, medicinal, and toxicological studies of moxifloxacin and its novel analogs: a quantum chemistry and drug discovery approach
Received
26th September 2025
, Accepted 23rd December 2025
First published on 8th January 2026
Abstract
Moxifloxacin (MOX) is regarded as a fourth-generation fluoroquinolone, demonstrating effectiveness against multidrug-resistant tuberculosis (TB) by inhibiting bacterial DNA gyrase. The therapeutic effectiveness of MOX is negatively influenced by side effects that are dependent on dosage, including heart rate-corrected QT interval prolongation and hepatotoxicity. This study explored the physicochemical, spectral, biological, and pharmacokinetic properties of MOX and its analogues. We incorporated various functional groups such as CH3, NH2, OCF3, NHCONH2, and Cl into the core MOX framework. The geometry was optimized utilizing density functional theory with the B3LYP/6-31g basis set. We conducted geometrical, thermodynamic, molecular orbital, and electrostatic potential analyses to deepen our understanding of their physical and chemical properties. We have obtained the FT-IR and UV-vis spectra and have established correlations with the observed experimental data. The determination of the HOMO–LUMO gap is essential for assessing the chemical reactivity of MOX and its analogs. The methodology of molecular docking was executed, incorporating MOX and its analogs in connection with the targeted protein (PDB ID 5BS8). ADMET prediction was performed to assess absorption, distribution, metabolism, and toxicity, whereas PASS predictions were carried out to examine biological and toxicological properties. MOX13 exhibited a notable HOMO–LUMO gap (3.61 eV), alongside the highest binding affinity (−8.5 kcal mol−1) when compared to all examined analogues. MOX13 exhibits a notably pronounced dipole moment (14.88 debye), alongside an exceptional degree of reactivity. Investigations utilizing molecular dynamics were conducted to assess the stability of receptor–ligand complexes by analyzing RMSD, RMSF, H-bonds, and SASA, suggesting that the ligand would remain bound to its original site.
1 Introduction
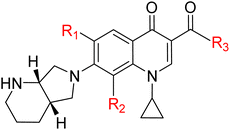
Tuberculosis (TB), caused by the intracellular bacterium Mycobacterium tuberculosis (MTB), persists as an enduring and formidable global public health crisis, exacting a devastating toll on human health, social structures, and economic development, particularly within low- and middle-income countries.1,2 Despite being both preventable and curable, TB continues to claim millions of lives annually, ranking among the leading infectious causes of death worldwide. Recent data underscore the sheer scale of this epidemic, with an estimated 10.8 million new cases and 1.6 million fatalities reported in 2023, highlighting the profound and pervasive nature of this global health emergency.3 The ambitious global targets for TB elimination, such as those outlined in the WHO's End TB Strategy, are continuously undermined by a complex interplay of factors, including the inherent diagnostic challenges, the prolonged and arduous nature of treatment regimens, and most critically, the alarming and escalating emergence and spread of drug-resistant strains.4,5 TB remains a global health crisis, with multidrug-resistant tuberculosis (MDR-TB) or rifampicin-resistant TB (RR-TB) being a significant threat primarily in five key countries; however, the WHO is advancing treatment through new shorter, all-oral regimens including bedaquiline, pretomanid, linezolid, moxifloxacin, delamanid, levofloxacin, clofazimine, and pyrazinamide, alongside enhanced diagnostic guidelines, to combat the disease worldwide.6–10 Quinolones are widely used antibacterials, but their extensive use has led to a steady increase in quinolone-resistant bacterial strains since the 1990s.11 Fluoroquinolones are crucial synthetic antibiotics in TB treatment, particularly for Multi-Drug Resistant M. tuberculosis (MDR-MTB) strains, with ongoing research focusing on structural modifications to enhance their efficiency, tissue penetration, and address emerging bacterial resistance.12 Their potent bactericidal effect is primarily mediated by targeting bacterial DNA gyrase and topoisomerase IV, essential enzymes responsible for regulating DNA topology, including supercoiling, replication, transcription, and DNA repair within the bacterial cell. These enzymes are essential for maintaining the structure and function of the bacterial chromosome.13–17 MOX, a fourth-generation extended-spectrum fluoroquinolone, is a promising second-line agent for drug-resistant TB, including MDR-TB, due to its improved coverage against Gram-positive cocci (including resistant pneumococci) and atypical pathogens, while maintaining good activity against Gram-negative bacteria. It has demonstrated high clinical and bacteriological success rates (≥90%) in trials for various respiratory tract infections, achieving good tissue penetration and requiring no dosage adjustment for elderly or renally/mildly hepatically impaired patients, but its use is associated with gastrointestinal disturbances and QTc prolongation.18–22 Its high potency against MTB, coupled with favorable pharmacokinetic properties such as excellent tissue penetration (including into lung tissue and macrophages, where MTB frequently resides), high oral bioavailability, and a relatively long half-life, contribute significantly to its therapeutic potential and make it a valuable component of current drug-resistant TB regimens.23–28 Despite its advantages, the widespread clinical use of moxifloxacin is not without limitations, including potential dose-dependent adverse effects like hepatotoxicity and a low probability of attaining therapeutic targets. Furthermore, challenges such as a substantial reduction in MOX exposure due to increased clearance and the unconfirmed safety of elevated doses in clinical settings necessitate careful patient monitoring.29,30 MOX, like other fluoroquinolones, exerts its inhibitory effect by interfering with the DNA breakage–reunion cycle of DNA gyrase. Specifically, it stabilizes the transient covalent complex formed between the enzyme and DNA, thereby preventing DNA re-ligation and leading to the accumulation of lethal double-strand breaks within the bacterial genome, ultimately resulting in bacterial cell death.31 The availability of high-resolution structural data, such as the crystal structure of MTB DNA gyrase in complex with DNA (PDB ID 5BS8), has been a transformative development in the field of anti-TB drug discovery. This detailed structural information has revolutionized our understanding of the enzyme's active site, its substrate binding characteristics, and its intricate molecular interactions with existing fluoroquinolone drugs. This provides a precise molecular blueprint for rational drug design strategies.32,33 By elucidating the precise binding interactions between MOX and MTB DNA gyrase, including the identification of key amino acid residues at the binding interface, which are often implicated in resistance, and understanding the conformational changes induced upon ligand binding, computational approaches can be intelligently employed to guide the strategic modification of the MOX scaffold.34 Advanced computational tools and methodologies, such as molecular docking, molecular dynamics simulations, pharmacophore modeling are instrumental in this iterative, multidisciplinary, and highly efficient drug discovery process.35–37 These in silico approaches facilitate the virtual screening of vast chemical libraries, prioritize promising lead compounds, and enable the rational design of novel MOX derivatives with enhanced binding affinity to the target enzyme, improved drug-likeness properties (e.g., solubility, permeability, metabolic stability), and, crucially, a reduced propensity for resistance development by designing compounds that can circumvent or overcome common resistance mutations.38 Furthermore, computational methods allow for early prediction of potential toxicity and metabolic liabilities, significantly de-risking the drug development process and potentially reducing the time and immense cost associated with bringing a new drug to market. In this study, we modified the substituents of MOX at positions 4, 6 and 8 of the quinolone system, as those positions are crucial for bacterial cell penetration, lipophilicity, phototoxicity, anaerobic activity, pharmacokinetic properties.39,40 The overarching objective of this study is to leverage these sophisticated computational methodologies, combined with detailed structural insights from MTB DNA gyrase to design, synthesize, and ultimately evaluate novel MOX derivatives. We hypothesize that by systematically modifying the MOX structure based on a comprehensive understanding of its molecular interactions with MTB DNA gyrase, we can generate compounds that exhibit superior antitubercular activity, improved pharmacokinetic profiles, reduced toxicity, and significantly decreased susceptibility to the emergence of drug resistance. Such improved compounds hold the transformative potential to dramatically shorten treatment regimens, alleviate the severe burden of adverse effects, and ultimately contribute significantly to the global effort to combat the persistent and evolving threat of TB (Table 1).
Table 1 Structural details of MOX and its newly designed analogs
|
| Name |
R1 |
R2 |
R3 |
Molecular formula |
| MOX |
F |
OCH3 |
OH |
C21H24FN3O4 |
| MOX1 |
CH3 |
OCH3 |
OH |
C22H27N3O4 |
| MOX2 |
NH2 |
OCH3 |
OH |
C21H26N4O4 |
| MOX3 |
OCF3 |
OCH3 |
OH |
C22H24F3N3O5 |
| MOX4 |
F |
OCH3 |
CF3 |
C22H23F4N3O3 |
| MOX5 |
F |
OCH3 |
CH2NH2 |
C22H27FN4O3 |
| MOX6 |
F |
OCH3 |
Cl |
C21H23ClFN3O3 |
| MOX7 |
F |
OCH3 |
NH2 |
C21H25FN4O3 |
| MOX8 |
F |
OCH3 |
NHCOCH3 |
C23H27FN4O4 |
| MOX9 |
F |
OCH3 |
NHCONH2 |
C22H26FN5O4 |
| MOX10 |
F |
OCH3 |
OCH3 |
C22H26FN3O4 |
| MOX11 |
F |
CF3 |
OH |
C21H21F4N3O3 |
| MOX12 |
F |
NH2 |
OH |
C20H23FN4O3 |
| MOX13 |
F |
NHCONH2 |
OH |
C21H25FN5O4 |
2 Methods and materials
2.1 Computational details
Quantum mechanical methods focus extensively on the calculation of thermodynamic properties, molecular orbital characteristics, dipole moments, geometries, vibrational frequencies, atomic partial charges, molecular electrostatic potentials, and the interpretation of various types of interactions.41 The initial geometry of MOX was sourced from the PubChem online database (PubChem CID 152946). Gabedit software (version 2.5.0) was utilized to perform molecular activity and conformational analysis based on the predicted AMBER (Assisted Model Building with Energy Refinement) potential values, aiming to identify the most stable conformer with the lowest energy.42 All compounds' structural changes and geometry optimizations were carried out using the Gaussian 16 program, which employs DFT and TD-DFT methods using the B3LYP functional with the 6-31G basis set. The characteristics of frontier molecular orbitals, εHOMO (highest occupied molecular orbital) and εLUMO (lowest unoccupied molecular orbital), were computed using an identical theoretical framework. Subsequently, the HOMO–LUMO energy gap, chemical hardness (η), chemical softness (S), chemical potential (µ), electronegativity (χ), and electrophilicity (ω) were computed employing the Parr and Pearson interpretations of DFT and Koopmans' theorem through the following equation.43
2.2 Protein preparation, docking, and interactions
To prepare the MTB H37Rv receptor, the crystal structure of a topoisomerase II was selected for molecular docking, its three-dimensional structure (PDB ID 5BS8), which had been extracted utilizing X-ray diffraction, and resolution was determined at 2.40 Å, downloaded from the RCSB Protein Data Bank (https://www.rcsb.org/). Initial clean-up involved using Discovery Studio Visualizer 2021 to remove unwanted elements like water molecules, any co-crystallized small molecules, non-protein atoms, and extraneous protein chains. The resulting protein chain was then energy-minimized using the conjugate gradient method in Swiss-PdbViewer (Version 4.1.0) to eliminate unfavorable atomic interactions, especially which exhibit poor interaction of protein atoms. Molecular docking of the synthesized MOX analogs was conducted using PyRx software through Autodock Vina, where the compounds were treated as ligands and the MTB H37Rv receptor as the macromolecule. Blind docking was performed on three different desktops by defining a maximum grid box that encompassed the entire protein with dimensions of 112.42 Å, 57.62 Å, and 75.77 Å along X, Y, and Z axes.44,45 Finally, non-bonded interactions and docking outcomes were analyzed, and the results obtained from the molecular docking experiments were interpreted using Discovery Studio Visualizer 2021.
2.3 Molecular dynamics simulation
Molecular dynamics simulations were carried out applying the GROMACS software package with the CHARMM36 all-atom force field. The system had been set up using a TIP3 standard water model and removed with sodium (Na+) and chloride (Cl−) ions. Throughout the first run, the system completed stabilizing via a two-step equilibration process, firstly at constant volume and temperature (NVT ensemble), and subsequently at constant pressure and temperature (NPT ensemble). The production molecular dynamics simulation was carried out for 100 nanoseconds (ns) under the NPT ensemble, with pressure regulated by a Berendsen thermostat. During the simulation, system coordinates (trajectories) were recorded every 100 ps for later analysis.46 Following the simulation, the remaining trajectories were examined to evaluate the structural stability and dynamics of the system by computing essential properties, such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), the radius of gyration (Rg), solvent-accessible surface area (SASA), and hydrogen bond counts, and a principal component analysis (PCA) was also performed.47
2.4 ADMET and PASS prediction
ADMET (absorption, distribution, metabolism, excretion, and toxicity) and PASS (Prediction of Activity Spectra for Substances) evaluations are essential factors in pharmaceutical development research for predicting the pharmacokinetic behavior and potential biological activity of prospective drug candidates, thereby minimizing late-stage developmental failures. This study employed data on ADMET properties obtained from the admetSAR online server (https://lmmd.ecust.edu.cn/admetsar2).48 We applied the SwissADME tool to evaluate the drug-like properties of the compounds (https://www.swissadme.ch/).49 Additionally, performing early-stage ADME predictions during drug discovery can significantly reduce clinical pharmacokinetic-related failures. The PASS Online service (https://www.way2drug.com/passonline/), which calculates the likelihood of biological activity (Pa) and inactivity (Pi) for every molecule based on structural features, provided the PASS prediction findings for MOX and its analogs. Default thresholds were considered in the analysis; a Pa > 0.5 indicates a high level of activity potential. A simplified entry system for molecular-input lines (SMILES) and structural data files was used in each case to generate the results (https://cactus.nci.nih.gov/translate/).
3 Results and discussion
3.1 Thermodynamic analysis
Thermodynamic properties play a vital role in assessing molecular stability, reactivity, and potential interactions with receptor proteins.50,51 In this study, variations in MOX derivatives considerably impact their thermodynamic parameters, including Gibbs free energy, enthalpy, and entropy. The free energy and enthalpy values for MOX derivatives show distinct trends, indicating the effect of various structural modifications on the molecular stability.52 Among the studies, the parent molecule MOX exhibits a Gibbs free energy of −1378.84 hartree, and MOX6 displays the highest negative free energy value −1763.23 hartree, indicating the most thermodynamically favorable profile, followed by MOX3 −1691.55 hartree (Fig. 1(a)). Besides, derivatives such as MOX1, MOX12, and MOX2 exhibit free energy values of −1318.90 hartree, −1319.71 hartree, and −1334.94 hartree, respectively, displaying less negative free energy values, which suggest reduced thermodynamic stability. The increased negative free energy suggests that binding and interaction occur spontaneously.53,54 Furthermore, the dipole moment serves as a quantitative measure of molecular polarity and provides critical information regarding the nature and strength of intermolecular interactions.55 The dipole moment of MOX (8.37 debye) is found to be the lowest value, whereas MOX13 (14.88 debye) shows the highest value (Fig. 1(c)). The increased dipole moment suggests that they might exhibit stronger binding affinities with receptor proteins through enhanced electrostatic interactions and hydrogen bonding.56 The negative free energy values for these compounds indicate spontaneous binding and interaction with their targets, with larger values correlating with more favorable bonding. For example, MOX6, with a free energy of −1763.23 hartree and a dipole moment of 11.98 debye, is expected to show a strong interaction with the receptor protein, promoting higher stability and activity.57 MOX6 exhibits the most negative free energy, which is the strongest indicator of a spontaneous, high-affinity interaction. This strong thermodynamic favorability is the most crucial determinant of biological potency.58
 |
| | Fig. 1 (a) Free energy, (b) enthalpy, and (c) dipole moment of MOX and its analogs. | |
3.2 Frontier molecular orbital analysis
The frontier molecular orbital (FMO) analysis provides a fundamental understanding of how MOX and its analogs might exhibit their biological activity by linking their electronic structure to their chemical behavior. Molecules interact with biological targets based primarily on their kinetic stability and chemical reactivity, which are quantified by the HOMO–LUMO energy gap, chemical hardness, and softness.59–61 The frontier molecular orbitals, also known as the LUMO and HOMO, are the most significant orbitals in a molecule. The way a molecule interacts with other species is determined by these orbitals. The molecule's kinetic stability and chemical reactivity are described in part by the frontier molecular orbital energy gap. In addition to being highly polarizable, a molecule with a small frontier orbital gap is also known as a soft molecule and is typically linked to low kinetic stability and high chemical reactivity.62 The process of electronic absorption involves the excitation of electrons from HOMO to LUMO. The value of the HOMO–LUMO gap has a direct correlation with both softness and hardness.63 Our research revealed that MOX7 had a high energy gap (4.18 eV) in comparison to the other analogs, while MOX3 had a comparatively small energy gap (3.27 eV) (Fig. 2(a)). Greater chemical stability and decreased reactivity are associated with a wider energy gap, as observed in MOX7, which suggests that the molecule needs more energy for electronic transitions. With hardness and softness values of 2.01 eV and 0.24 eV, respectively, MOX7 is regarded as the hardest among the derivatives. MOX7 is less likely to take part in chemical reactions because of its high hardness, which reflects its resistance to electronic structure deformation. This suggests MOX7 would be less likely to take part in chemical reactions within a biological environment due to its resistance to electron structure deformation, implying a potentially lower or highly selective biological.64 MOX3, on the other hand, exhibited the lowest chemical hardness (1.637) and the highest chemical softness (0.305). This implies that the presence of the OCF3 group makes MOX3 more chemically reactive. The high electronegativity of the three fluorine atoms in OCF3 makes the trifluoromethyl (CF3) group particularly electron-withdrawing, while the oxygen atom itself is highly electronegative. Because of these combined effects of O and CF3, the OCF3 became a potent electron-withdrawing group. The electrical transition from HOMO to LUMO is facilitated by this electron-withdrawing effect, increasing the compound's reactivity.65 Furthermore, the highly electron-withdrawing OCF3 group in MOX3 facilitates the electrical transition, increasing the compound's intrinsic reactivity. According to the Hard and Soft Acids and Bases (HSAB) principle, this softness makes MOX3 more prone to interact with soft biological targets (like certain active sites on enzymes), suggesting it is likely the most chemically reactive analog and could therefore exhibit an enhanced biological response through easier, more potent chemical interaction.66 Thus, the FMO analysis effectively predicts that the more reactive and softer analog, MOX3, will likely possess a greater or different range of biological activity than the stable and hard analog, MOX7.67
 |
| | Fig. 2 (a) HOMO–LUMO energy band gap, and (b) DOS plot of MOX and its analogs. | |
Fig. 2(b) represents the DOS spectra of MOX and its selected analogs, MOX1 and MOX3. From the figure, it is evident that all of them share similar electronic distribution, which in turn indicates that the core molecular framework is preserved. There is a minor variation in the occupied region (−10.0 eV to −5.0 eV), which arises due to structural modification. It also shows comparable electronic excitation behavior, as the conduction-band onset near 2.5 eV remains almost unchanged. These small DOS shifts imply that the structural modification alters the orbital distribution and electronic environments of the molecules, thereby altering their reactivity and potential biological interactions.
3.3 HOMO–LUMO gap and biological activity in MOX analogs
MOX3's smaller energy gap (3.27 eV) suggests it possesses greater chemical reactivity and a higher likelihood of biological interaction compared to MOX7.68 The observed disparity in energy gaps between MOX7 and MOX3 directly implies differences in their kinetic stability and polarizability, with the latter being more prone to forming reactive intermediate.69 This enhanced reactivity for molecules with smaller HOMO–LUMO gaps is often associated with a greater capacity for electron donation and acceptance, crucial for diverse biochemical pathways, and thus can be used to predict the chemical stability and biological activity of molecules.70 The enhanced reactivity, characterized by a smaller HOMO–LUMO gap, also indicates a greater ease with which a molecule can donate or accept electrons during a chemical process, thereby facilitating charge transfer interactions vital for various biological functions.71 Thus, the smaller HOMO–LUMO gap of MOX3 (3.27 eV) compared to MOX7 (4.18 eV) suggests that MOX3 is likely to exhibit higher chemical reactivity and potentially greater biological activity due to its increased electron transfer capabilities and reduced kinetic stability.72 This heightened reactivity, evidenced by a smaller energy gap, often translates into a greater propensity for biological interactions, as the molecule can more readily engage in electron transfer processes critical for various enzymatic reactions and cellular signaling.73 Consequently, a lower energy gap implies a molecule is more “soft” and thus more prone to interaction with biological macromolecules, whereas a higher gap signifies a “harder” molecule with reduced reactivity.74
3.4 Molecular electrostatic potential analysis
Molecular electrostatic potential (MEP) analysis is a crucial technique for exploring how molecules interact and behave chemically, playing an important role in drug development research and predicting chemical reactivity.75 The analysis serves as a vital bridge between the molecular structure of MOX analogs and their anticipated biological activity by mapping the charge distribution across the molecular surface.76 Visualizing the distribution of electrostatic potential across a molecular surface enables the identification of electron-rich and electron-deficient regions, which are essential for predicting noncovalent interactions and optimizing drug design with biological targets like receptors or enzymes.77 The deep red region MEP maps highlight a high electron density area, suitable for electrophilic attack. Whereas intense blue regions indicate areas of electron deficiency, making them highly vulnerable to nucleophilic attack.78 Conversely, green areas suggest regions of nearly zero electrostatic potential, representing electrostatically neutral zones on the surface.79 Among the derivatives studied (Fig. 3), MOX1 showed the most negative electrostatic potential (−9.85 × 10−2 hartree), mainly imputed to the strong electron-releasing influence of the CH3 substituent at the R1 position. The resulting high electron density presents MOX1 as highly susceptible to electrophilic attack, underscoring it as a promising candidate for targeted molecular recognition.80 MOX13 also showed a significant negative value (−9.72 × 10−2 hartree), making it one of the most valuable candidates. In contrast, MOX9 had the lowest negative potential (−7.09 × 10−2 hartree), attributed to the incorporation of the R3 position with an NHCONH2 substituent proximal to the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) within the core structure.81
O) within the core structure.81
 |
| | Fig. 3 MEP map of MOX and its selected analogs. | |
3.5 FT-IR analysis
The important applications of infrared spectroscopy (IR) include confirming functional groups in various chemical compounds and providing information about particles. Spectral characterization was accomplished in this study and then combined with the experimental data of the parent drug MOX (Fig. 4(a)). IR analysis confirms the presence of key groups essential for biological activity, such as N–H (amine/amide) and O–H (hydroxyl groups), and the carbonyl CO moiety.82 These groups are frequently active sites involved in hydrogen bonding and coordination with biological receptors and enzymes. The FT-IR spectral band frequencies were recorded in the 400–4000 cm−1 range.83 The asymmetric stretching band for N–H (primary amine) present in MOX9 was reported at 3743 cm−1, much higher than the theoretically calculated values (3300–3500 cm−1) in this study. All the absorbance of the N–H (secondary amine) functional group present in MOX and its derivatives fluctuated and were found in the higher frequency range. The differences in these frequencies are due to variations in hydrogen bonding and the surrounding chemical environment, which can alter the N–H stretching frequency. The most significant deviation was found (at 3597 cm−1) for N–H present in MOX9. This internal modification affects the 3D shape and electronic accessibility of the functional groups, which are crucial for forming the complementary hydrogen bonds required for high-affinity binding to a biological target. The IR band for C–H present in the cyclic ring of all derivatives and MOX was found in 3084–3087 cm−1. The calculated results are slightly higher than the theoretical values (2950–2850 cm−1).84 Carbonyl and carboxylic acid groups typically exhibit a CO stretching peak around 1700–1740 cm−1. The CO IR stretching is present in the carbonyl of MOX and the carboxylic acid functional group in the range between 1622 and 1697 cm−1. The variation in the IR band arises due to resonance effects and hydrogen bonding, which lowers the stretching frequency, resulting from the modification of the MOX functional group. The O–H vibrations are generally found within the 3600–3620 cm−1 range. The O–H stretching mode associated with the carboxylic acid group in MOX was detected at 3602 cm−1, and all the derivatives that contain the O–H group showed absorption bands between 3602 and 3632 cm−1. The slight differences in these frequencies are due to the hydrogen bonding and the immediate chemical environment surrounding the O–H group, which can cause a minor shift in the stretching frequency.85 By showing specific shifts for certain derivatives (like the high N–H stretching in MOX9), IR spectroscopy provides structural fingerprints that help correlate specific substituent effects with changes in key bond properties, which ultimately links a specific structural modification to a potential change in biological activity.86 The IR analysis confirms that the functional groups critical for biological recognition are present and, more importantly, reveals that their electronic and vibrational properties are modified by the substituents and local environment. These modifications dictate the molecule's final binding characteristics and, consequently, its biological efficacy.
 |
| | Fig. 4 (a) FT-IR, and (b) UV-visible analysis for MOX and some of its selected derivatives. | |
3.6 UV-visible study
TD-DFT methods were applied to analyze the electronic transition characteristics of the molecules.87 The UV-vis analysis contributes significantly to predicting the biological activity of MOX and its analogs by quantifying their electronic transition characteristics, which are direct indicators of their kinetic stability and potential reactivity.88 The simulations provided excitation energies, maximum absorption wavelengths, oscillator strengths, and associated molecular orbital transitions (Table S3). The first singlet excitation (S0 → S1) plays a pivotal role in determining kinetic stability and potential reactive centres of the molecules.89 Among the analogs, MOX4 and MOX11 exhibit broad absorption bands at 619.59 nm and 629.82 nm, with oscillator strengths of 0.0002 and 0.0153, respectively. In a biological setting, these molecules are more likely to undergo chemical changes or interactions, potentially leading to a stronger biological effect or metabolism.90 Their electronic transition is mainly driven by HOMO → LUMO excitation with significant configuration contributions of 0.700 (MOX4) and 0.706 (MOX11). Relatively, the lower excitation energies (2.00 eV for MOX4 and 1.97 eV for MOX11) mean more reactivity and less kinetic stability.91,92 In contrast, MOX7 and MOX3 have higher excitation energies (2.93 eV and 2.90 eV, respectively) and show lower oscillator strengths (0.004 and 0.005), indicating fewer reactive sites and greater kinetic stability, implying they are less prone to spontaneous electronic transitions and, consequently, less likely to participate in certain chemical reactions with biological targets. Thus, the observed electronic properties highlight structural variations as key determinants of the molecule's stability and reactivity.93 By showing a strong correlation between the electronic properties and structural variations, the analysis establishes that the substituents are the key determinants of the molecule's stability and reactivity profile. This is crucial for structure–activity relationship (SAR) studies, allowing researchers to fine-tune substituents to achieve a desired level of reactivity for optimal biological action.
3.7 Binding affinity and interaction analysis
Molecular docking is a computer approach performed to predict the three-dimensional binding orientation of two interacting molecules, playing a vital role in structure-based drug design as well as analysis.94 During the molecular docking simulations, nine distinct binding poses were generated for each ligand. From these, the most favorable pose was selected based on binding affinity and structural alignment criteria. In this study, molecular docking studies were performed to evaluate the binding affinity and nonbonding interactions of MOX and its analogs with the protein (PDB ID 5BS8).95 The binding affinities and key residue interactions are summarized in Fig. 5 and 6. Most analogs displayed favorable docking scores and engaged in significant non-covalent interactions within the active site of the target protein.96 The parent compound MOX exhibited a binding affinity of −7.2 kcal mol−1, interacting with PRO102 through a halogen bond (3.62 Å) and forming π–π T-shaped (PPT) interactions with TRP103. Additionally, alkyl (A) and π–alkyl (PA) interactions were observed with PRO119, LEU312, and HIS280, indicating moderate stability of the ligand–protein complex.97 Among the analogs, MOX13 demonstrated the strongest binding affinity (−8.6 kcal mol−1), attributed to multiple conventional hydrogen bonds with GLY120, ARG98, TRP103, and halogen interactions with ARG98. Particularly, π–alkyl interactions with TRP103 and PRO124, along with carbon bonding with SER104 and PRO119, contributed significantly to the stabilization of the complex. MOX1 also showed strong binding affinity (−7.5 kcal mol−1), forming hydrogen bonds with several key residues, including TRP103, ASP94, SER104, and GLY120, and π-donor hydrogen bonds (PDH) with GLN101. This compound also formed multiple π–alkyl interactions involving ARG98 and TRP103, indicating a robust ligand–receptor interface. Other potent analogs included MOX8 and MOX9 (each with −8.3 kcal mol−1). MOX8 formed hydrogen bonds with GLY117 and PHE116, in addition to π–π T-shaped interactions with TRP103, while MOX9 formed strong hydrogen bonds with ASP122, PRO229, and PRO102, as well as halogen bonds with HIS280. These interactions collectively enhanced the ligand's binding stability.98,99
 |
| | Fig. 5 Binding affinity of MOX and its derivatives with the 5BS8 receptor protein. | |
 |
| | Fig. 6 Superimposed docked conformers selected analogs within the active site of the receptor protein, nonbonding interactions, and hydrogen bond surface area of MOX and some of its derivatives with receptor protein 5BS8. | |
The analog MOX10 demonstrated a notable binding affinity of −7.9 kcal mol−1, establishing multiple hydrogen bonds with TRP103, ARG98, GLN277, and π–alkyl interactions with TRP103 and PRO124. A unique carbon interaction was also noted with SER104 and GLY120, creating flexible non-covalent interaction profiles. Compounds MOX3, MOX4, MOX5, MOX6, MOX7, MOX11, and MOX12 presented moderate binding affinities ranging from −7.2 to −7.8 kcal mol−1, predominantly forming hydrogen bonds, halogen bonds, and π–π interactions. Particularly, MOX4 and MOX5 kept consistent halogen interactions with PRO102 and π–π stacking with TRP103, reinforcing the ligand's affinity through aromatic interactions. In general, conventional hydrogen bonds (H), halogen bonds (HF), alkyl (A), pi–alkyl (PA), π–π T-shaped (PPT), π–σ, π–anion, and carbon bonds (C) contributed significantly to the stabilization of the ligand–protein complexes.100,101 Particularly, residues TRP103, PRO119, HIS280, LEU312, GLY117, and ARG98 were recurrently involved in ligand interactions, indicating their critical role in the binding pocket of the targeted protein. These docking results suggest that several MOX analogs, especially MOX1, MOX8, MOX9, and MOX13, possess improved binding affinities and more extensive interaction networks compared to the parent MOX compound. The diversity and strength of the non-covalent interactions, especially hydrogen and halogen bonds at favorable distances, underscore the potential of these analogs in drug development targeting the 5BS8 protein. In a larger screening (virtual screening), compounds with the most favorable docking scores and optimal interaction profiles are prioritized for further, costly experimental (in vitro and in vivo) biological testing.102 This significantly reduces the time and expense of drug discovery. A better docking score and a greater number/diversity of favorable non-covalent interactions (especially with key active site residues like TRP103, PRO119, HIS280, LEU312, GLY117, and ARG98 in your study) serve as a strong indicator of potentially high biological potency against the targeted protein.
Additionally, in case of resistance, fluoroquinolones are clinically driven by two main mechanisms. First, point mutations within the Quinolone Resistance-Determining Region (QRDR) of the gyrA and gyrB genes (e.g., GyrA D94G or A90V) directly diminish the drug's binding affinity to the enzyme complex. Second, the overexpression of bacterial efflux pumps actively expels the drug from the cell, lowering the intracellular concentration below the effective minimum inhibitory concentration. Designing effective next-generation FQs requires overcoming both structural mutation barriers and efflux system barriers.
Molecular docking experiments confirmed that lead analogs exhibited significantly improved binding to the MTB DNA gyrase structure (PDB ID 5BS8) compared to the parent MOX compound. MOX displayed a binding affinity (ΔG) of −7.2 kcal mol−1. In stark contrast, two designed analogs, MOX9 and MOX13, showed significantly stronger affinity, with values of −8.3 kcal mol−1 and −8.5 kcal mol−1, respectively. This affinity difference of approximately 1.3 kcal mol−1 between MOX and MOX13 implies a substantial theoretical enhancement in inhibitory potency. This fundamental increase in initial binding strength provides the basis for arguing that these analogs are chemically equipped to compensate for the reduction in binding energy typically caused by QRDR mutations. The stronger binding in the wild-type enzyme provides a buffer, allowing the modified drug to maintain efficacy even against mutated enzymes that cause a slight loss of affinity. The parent MOX compound stabilizes its complex primarily through a halogen bond with PRO102 and pi–pi T-shaped (PPT) interactions with TRP103. MOX13, however, achieves its superior affinity (ΔG = −8.5 kcal mol−1) through a robust network involving multiple conventional hydrogen bonds with GLY120, ARG98, and TRP103, alongside halogen interactions with ARG98. The ureido group introduced at the R2 position (C8) of MOX13 facilitates these new hydrogen-bonding anchor points. The engagement of ARG98 via both H-bonding and halogen interactions in MOX13 is a key mechanistic differentiator. Because resistance mutations often cluster adjacent to this region (e.g., near D94), forming strong, novel interactions with neighboring residues like ARG98 and GLY120 suggests that MOX13 operates via a distinct structural stabilizing mechanism. This new interaction network functions as a structural anchor, stabilizing the DNA–enzyme cleavage complex in a manner that is less susceptible to the structural perturbation caused by mutations in the QRDR, thereby providing a clear hypothesis for how MOX13 may bypass resistance conferred by typical gyrase mutations. Beyond target site mutations, efflux pumps represent a crucial mechanism of FQ resistance. Efflux pump substrate recognition is often strongly correlated with a compound's lipophilicity. Structural modification aimed at reducing lipophilicity can thereby diminish a compound's recognition and removal by bacterial efflux systems. MOX13, which features a ureido group (NHCONH2) replacing the methoxy group (OCH3) at the C8 position (R2), exhibits a drastically reduced Consensus log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (0.62) compared to the parent MOX compound (1.85). This substitution introduces substantial polarity, moving MOX13 away from the lipophilicity profile typically favored by efflux pump substrates. This significant reduction in molecular lipophilicity provides a compelling physicochemical strategy for circumventing efflux-mediated resistance, allowing the drug to accumulate intracellularly at concentrations necessary for bactericidal action.
P (0.62) compared to the parent MOX compound (1.85). This substitution introduces substantial polarity, moving MOX13 away from the lipophilicity profile typically favored by efflux pump substrates. This significant reduction in molecular lipophilicity provides a compelling physicochemical strategy for circumventing efflux-mediated resistance, allowing the drug to accumulate intracellularly at concentrations necessary for bactericidal action.
3.8 Molecular dynamics simulation analysis
3.8.1 RMSD analysis. The RMSD analysis describes a clear comparison of the structural stability of four different molecular systems. In such simulations, the RMSD is a critical metric used to quantify the change in the structure of a molecule, typically a protein, over time relative to its initial conformation.103 A low and stable RMSD value indicates that the molecule has settled into a stable three-dimensional shape, whereas high and fluctuating values suggest significant conformational changes and instability.104 The analysis reveals stark differences in the behavior of MOX and its three derivatives, which are likely involved in the protein bound to different ligands labelled MOX, MOX8, MOX9, and MOX13. The most striking result is for the 5BS8_MOX13 complex. This system demonstrates exceptional stability, as its RMSD quickly settles at a very low value of approximately 1 nm and remains consistently flat for the entire duration of the simulation. This behavior is a strong indicator that the ligand MOX13 stabilizes the protein in a specific, favorable conformation. In contrast, the other systems exhibit varying degrees of instability. The 5BS8_MOX complex shows moderate stability, with its RMSD rising and then fluctuating between 2 and 4 nm, suggesting it has found a somewhat stable state but with more fundamental flexibility than the MOX13 complex. The situation deteriorates significantly for the 5BS8_MOX9 system, which displays considerable instability. Its RMSD climbs to high values around 6 nm and fluctuates wildly, indicating that the protein is undergoing continuous and large-scale structural changes without reaching a stable equilibrium. The most unstable system by a clear margin is 5BS8_MOX8, where RMSD soars to over 8 nm and shows extreme fluctuations, which points to a highly disrupted protein structure. This suggests that the MOX8 ligand not only fails to stabilize the protein but may actively be causing it to lose its native fold. Thus, this RMSD analysis provides a comparative measure of how different ligands affect the structural integrity of the 5BS8 protein. The results clearly rank the stability of the complexes as 5BS8_MOX13 being the most stable, followed by 5BS8_MOX, with 5BS8_MOX9 and 5BS8_MOX8 being progressively more unstable. Though 5BS8_MOX13 is more stable than 5BS8_MOX, MOX13 may be selected as the lead compound. The average RMSD value for MOX was 7.49 ± 0.25 nm, indicating substantial structural deviation. In contrast, MOX13 exhibited markedly lower conformational variability (0.76 ± 0.44 nm), while MOX8 (8.56 ± 3.01 nm) and MOX9 (7.62 ± 0.87 nm) showed higher RMSD values, comparable to or exceeding that of MOX.
3.8.2 RMSF analysis. The provided RMSF analysis offered critical insights into the dynamic behavior of the protein 5BS8 when complexed with four different compounds: MOX, MOX8, MOX9, and MOX13.105 The plot visualizes the flexibility of each amino acid residue, where high RMSF peaks signify mobile regions and low-value troughs indicate a stable, rigid structure.106 Across all four analogs, we observe significant flexibility in the N-terminal (residues 10–40) and C-terminal (residues 480–500) regions, as well as in several loop areas (e.g., around residue 125 and 400–440), which are crucial for protein function and interaction. Conversely, the extensive flat regions with low RMSF values correspond to the stable α-helix and β-sheet secondary structures that form the protein's core. When comparing the impact of each compound, a clear difference in the stabilization of the protein emerges. By calculating the average fluctuation across all residues for each complex, we can rank their stability. The analysis reveals that the 5BS8_MOX8 complex is the most stable, exhibiting the lowest average RMSF of 0.169 nm. The following are 5BS8_MOX13 (0.176 nm) and 5BS8_MOX (0.179 nm), with the 5BS8_MOX9 complex being the least stable and most flexible (0.183 nm). Therefore, based on its ability to induce the greatest structural rigidity in the target protein, 5BS8_MOX8 is identified as the best stability compound and the most promising lead compound from this set for further development.
3.8.3 Rg analysis. To evaluate the impact of ligand binding on the overall structural compactness and stability of the protein, the Rg was monitored over a 100 ns MD simulation for each of the four protein–ligand complexes (MOX, MOX8, MOX9, and MOX13). The Rg provides a measure of the mass-weighted root mean square distance of atoms from their common center of mass, serving as a critical indicator of global conformational changes.107 The analysis of the Rg trajectories reveals that all four systems achieved structural stability and convergence after an initial equilibration period of approximately 20 ns. For the remainder of the simulation, the Rg values for each complex remained stable, fluctuating around a consistent average without exhibiting any significant unfolding events or conformational drifts. This indicates that the binding of each ligand resulted in a structurally integral and stable complex over the entire simulation timescale. A quantitative comparison of the average Rg values, calculated over the stable production phase (20–100 ns), highlights distinct differences in protein compactness induced by each ligand. The MOX9 complex displayed the lowest average Rg value of 2.993 nm, signifying that it induced the most compact tertiary structure. Following this were the MOX8 and MOX13 complexes with intermediate average Rg values of 3.013 nm and 3.020 nm, respectively. In contrast, the reference complex, MOX, yielded the highest average Rg of 3.033 nm, indicating a relatively less compact or more expanded conformation compared to the other three ligands. The low standard deviations for all systems confirmed that the observed conformations were not only stable but also maintained a high degree of rigidity. These findings suggest that the chemical nature of the bound ligand directly modulates the global architecture of the protein. The pronounced compactness of the MOX9-bound protein points towards the formation of highly favorable and stabilizing interactions within the binding pocket, which act to draw disparate structural elements of the protein closer together. This induced-fit stabilization is often a hallmark of high-affinity binders. The clear and consistent ranking of compactness −MOX9 > MOX8 > MOX13 > MOX provides compelling evidence of differential structural modulation, offering valuable insights that can be correlated with binding affinity and functional activity to guide further rational drug design efforts (Fig. 7–10).
 |
| | Fig. 7 Results of 100 ns MD simulation of 5BS8 in complex with MOX, MOX8, MOX9, and MOX13 ligands, (a) RMSD values of docked complexes from C-α. The structural changes of the receptor protein were investigated by means of (b) RMSF, (c) the number of hydrogen bonds present between solute and solvent, respectively, and (d) SASA. | |
 |
| | Fig. 8 Results of 100 ns MD simulation of 5BS8 in complex with MOX, MOX8, MOX9, and MOX13 (a) radius of gyration, and (b) volume (nm3). | |
 |
| | Fig. 9 (a–d) The relationship between eigenvalue and percentage of variation in principal component analysis for MOX13, complex with 5BS8. | |
 |
| | Fig. 10 Potential energy and temperature plots from 100 ns molecular dynamics simulation. Number them as (A) temperature plot, and (B) energy file. | |
3.8.4 Hydrogen bond analysis. An analysis of hydrogen bonding within four distinct molecular complexes: MOX, MOX8, MOX9, and MOX13, reveals significant variations in their intermolecular interactions over time, as captured by molecular dynamics simulations.108,109 The investigation, based on data tracking the number of hydrogen bonds at various time points, provides a comparative look into how structural differences between the systems influence their bonding capabilities in complex MOX and MOX9. Hydrogen bonding appears to be a relatively transient and infrequent event. Throughout the simulations, the number of hydrogen bonds in these two complexes generally fluctuates between zero and two, with only brief, sporadic instances of three bonds forming. There is no discernible trend of increasing or decreasing bond formation over time, suggesting a stable but low level of hydrogen-bonding activity. In contrast, complexes MOX8 and MOX13 exhibit a much more dynamic and pronounced hydrogen-bonding profile. Complex MOX13 stands out as having the most robust and sustained hydrogen-bonding network, frequently forming four or five bonds, particularly in the earlier stages of the simulation. Although it experiences fluctuations, it consistently maintains a higher number of bonds compared to the other systems. System MOX8 also shows a high initial propensity for hydrogen bond formation, with several instances of two to five bonds. However, its behavior changes dramatically after about 30 nanoseconds, at which point the number of hydrogen bonds drops significantly and remains low, mostly at zero or one, for the remainder of the simulation. Overall, this comparative analysis highlights that the modifications distinguishing the four systems have a clear impact on their hydrogen-bonding potential. Systems MOX8 and MOX13 demonstrate a greater capacity for forming these crucial intermolecular connections, although with different stability profiles over time. The sustained high level of bonding in MOX13 suggests a particularly favorable conformation for these interactions, while the eventual decline in MOX8 points to a potential conformational change. A deeper examination of the specific molecular structures would be necessary to fully elucidate the underlying reasons for these distinct behaviors.
3.8.5 SASA analysis. Based on the MD simulation data, an analysis of the SASA provides significant insights into the interaction between the MOX protein and three different ligands: MOX8, MOX9, and MOX13. SASA measures the total surface area of a protein that is exposed to the surrounding solvent, and monitoring its changes over time helps characterize ligand binding, protein stability, and conformational shifts.110 The simulation tracks the protein's dynamics over 100 nanoseconds, revealing constant, moment-to-moment fluctuations in SASA, which represent the protein's natural “breathing” and structural flexibility in solutions. When a ligand binds to a protein's surface cavity, or binding pocket, it typically shields a portion of the protein from the solvent. However, the results here show a more complex interaction. The average SASA for the unbound MOX protein was calculated to be 256.78 nm2. Interestingly, upon binding each of the ligands, the total SASA of the complex increased. The average SASA values were 259.03 nm2 for MOX8, 258.11 nm2 for MOX9, and 260.29 nm2 for MOX13. This increase suggests that while the ligands occupy the binding pocket, they induce a larger conformational change elsewhere in the protein, causing other regions to become more exposed to the solvent. The magnitude of this change quantifies the structural impact of each ligand. Compared to the unbound protein, MOX8 caused an average SASA increase of +2.25 nm2, MOX9 an increase of +1.33 nm2, and MOX13 caused the most dramatic increase of +3.51 nm2. Notably, the plot for MOX13 shows its SASA value settling at a new, higher baseline after approximately 40 ns, indicating it has induced a significant and stable structural rearrangement. While a decrease in SASA is classically associated with binding affinity, a large and stable increase like this also signifies a specific and significant interaction that remodels the protein. This analysis has important implications for the protein's “draggability”. The fact that all three ligands induce consistent and stable changes demonstrates that the binding site is “plastic”-it can adapt to accommodate different molecules. This adaptability makes the MOX binding site a promising druggable target. While this SASA analysis is excellent for observing the overall effect, a more detailed per-residue SASA analysis would be the next logical step. Such an analysis would pinpoint which specific amino acid residues are directly involved in the binding event by identifying those whose solvent exposure decreases most significantly upon ligand binding.
3.8.6 PCA analysis. To investigate the primary conformational dynamics of MOX13 during the molecular dynamics simulation, a PCA was performed on the trajectory data. The analysis of the first three principal components (PC1, PC2, and PC3) revealed the most dominant collective motions within the system.111,112 These components were found to contribute 25.19%, 14.08%, and 10.45% of the total variance, respectively. Cumulatively, these first three modes of motion account for 49.72% of the overall structural dynamics, indicating that nearly half of the molecule's flexibility is captured by them. The projection of the simulation snapshots onto these principal components shows a wide distribution of conformations, which signifies that MOX13 is a highly flexible molecule that explores a large conformational space. The clear, directed progression of the conformational clusters, often illustrated by a color gradient, suggests that the system undergoes substantial and well-defined structural changes rather than minor, random fluctuations. Further insight from the scree plot shows a steep initial decline, confirming that a few dominant motions govern the system. However, the fact that seven components are required to capture approximately 70% of the variance suggests that the overall dynamics of MOX13 are complex and involve multiple collective movements. In summary, the PCA reveals that MOX13 is a structurally dynamic molecule characterized by several key, dominant motions that together define its complex behavior.
3.9 ADMET analysis and drug-likeness prediction
An increasingly popular and economical substitute for in vivo drug testing is in silico ADMET screening, which covers absorption, distribution, metabolism, excretion, and toxicity.113 AdmetSAR, used in this study, calculates the ADMET score for eighteen unique attributes on a scale of 0 to 1, with one reflecting optimal performance and zero signifying toxicity or harm. Table 2 outlines the ADMET properties of MOX and its analogs. Human oral bioavailability (HOB) values for all the analogs in this investigation ranged from +0.954 to +1.000, except for MOX1 and MOX2, most of which had greater HOBs than the parent MOX. MOX4, MOX6, and MOX10 can efficiently pass the human intestinal CACO-2 cell monolayer experiment, which assesses their permeability and potential for intestinal absorption. Additionally, a small number of compounds, such as MOX4 to MOX7 and MOX9, showed blood–brain barrier (BBB+) permeability; however, since the current study did not concentrate on brain target receptor therapeutic agents, this may not be a problem. In our study, over 50% of the analogs are not P-glycoprotein (P-gp) inhibitors, although P-gp inhibition can interfere with medication absorption, permeability, and retention.114 Moreover, all the compounds are metabolized by CYP4502C9 isoenzymes and are non-biodegradable. MOX and its modified analogs exhibit weak inhibitory activity against the human ether-a-go-go-related gene (hERG). The inhibition of hERG is associated with long QT syndrome,115 indicating a need for further investigation in this area. This study found that MOX1 and MOX2 exhibit carcinogenic properties, whereas other analogs are non-carcinogenic. With a class III AOT level, all the analogs are relatively low in toxicity and safer for oral administration. All analogs, except MOX12 and MOX13, demonstrate a rat acute toxicity level that exceeds that of MOX. Modified analogs exhibit a higher median lethal dose (LD50) compared to MOX.
Table 2 Drug likeness parameters of MOX and its analogsa
| Compound |
Lipinski |
G/V/E/M |
FC |
CL |
BC |
NHA |
NHD |
BC |
TPSA |
| G = Ghose, V = Veber, E = Egan, M = Muegge, FC = fraction Csp3, CL = consensus logP, NHD = number of H-bond donors, NHA = number of H-bond acceptors, BC = bioavailability score. |
| MOX |
Y (0) |
Y |
0.52 |
1.85 |
0.55 |
6 |
2 |
0.55 |
83.80 |
| MOX1 |
Y (0) |
Y |
0.55 |
1.87 |
0.55 |
5 |
2 |
0.55 |
83.80 |
| MOX2 |
Y (0) |
Y |
0.52 |
1.00 |
0.55 |
5 |
3 |
0.55 |
109.82 |
| MOX3 |
Y (0) |
Y |
0.55 |
2.42 |
0.55 |
9 |
2 |
0.55 |
93.03 |
| MOX4 |
Y (0) |
Y |
0.55 |
3.31 |
0.55 |
8 |
1 |
0.55 |
63.57 |
| MOX5 |
Y (0) |
Y |
0.55 |
1.84 |
0.55 |
6 |
2 |
0.55 |
89.59 |
| MOX6 |
Y (0) |
Y |
0.52 |
2.86 |
0.55 |
5 |
1 |
0.55 |
63.57 |
| MOX7 |
Y (0) |
Y |
0.52 |
1.87 |
0.55 |
5 |
2 |
0.55 |
89.89 |
| MOX8 |
Y (0) |
Y |
0.52 |
2.20 |
0.55 |
6 |
2 |
0.55 |
63.57 |
| MOX9 |
Y (0) |
Y |
0.50 |
1.61 |
0.55 |
6 |
3 |
0.55 |
89.56 |
| MOX10 |
Y (0) |
Y |
0.55 |
2.58 |
0.55 |
6 |
1 |
0.55 |
92.67 |
| MOX11 |
Y (0) |
Y |
0.52 |
2.84 |
0.55 |
8 |
2 |
0.55 |
118.69 |
| MOX12 |
Y (0) |
Y |
0.50 |
1.34 |
0.55 |
5 |
3 |
0.55 |
72.80 |
| MOX13 |
Y (0) |
Y |
0.48 |
0.62 |
0.86 |
6 |
4 |
0.86 |
129.69 |
All MOX analogs (MOX–MOX13) comply with Lipinski's rule of five (zero violations), confirming their potential for oral bioavailability, as evidenced by molecular weight (MW < 500 Da), logP (≤3.31), hydrogen bond donors (HBD ≤ 4), and acceptors (HBA ≤ 9).116 The compounds further satisfy Veber's (rotatable bonds ≤ 10) and Ghose's (molar refractivity 40–130) criteria, with topological polar surface area (TPSA) values (63.57–129.69 Å2) within the optimal range for membrane permeability.117 Particularly, MOX13 exhibits enhanced bioavailability (BC = 0.86) despite its higher TPSA (129.69 Å2), likely due to its balanced logP (0.62) and moderate HBD/HBA counts (4/6).118 The fraction of sp3 carbons (FC = 0.48–0.55) across all analogs suggests favorable saturation, aligning with Muegge's lead-likeness guidelines.119 These results, supported by Molsoft's drug-like scores (>0.55), corroborate the analogs' pharmacokinetic suitability, with MOX–MOX12 meeting all thresholds for intestinal absorption (TPSA < 120 Å2, nRotB ≤ 8) (Table 3).
Table 3 Pharmacokinetic parameters of moxifloxacin and its analogsa
| Name |
Absorption |
Distribution |
Metabolism |
Toxicity |
| HIA |
C2P |
BBB |
P-GpI |
CYP450 2C9 |
hERG |
Carcinogens |
AOT |
Biodegradability |
RAT LD50 (mol kg−1) |
| HIA = human intestinal absorption, C2P = CACO-2 permeability, BBB = blood–brain barrier, p-GpI = p-glycoprotein inhibitor, hERG = human ether-α-go-go related gene, AOT = acute oral toxicity, RAT = rat acute toxicity, I = inhibitor, NI = non-inhibitor, WI = weak inhibitor, NC = non-carcinogen, C = carcinogen, NB = not biodegradable. |
| MOX |
+0.979 |
−0.609 |
−0.960 |
NI (0.756) |
NI (0.774) |
WI (0.809) |
NC (0.903) |
III |
NB (1.000) |
2.327 |
| MOX1 |
+0.954 |
−0.532 |
−0.935 |
NI (0.724) |
NI (0.794) |
WI (0.834) |
C (0.909) |
III |
NB (0.995) |
2.472 |
| MOX2 |
+0.978 |
−0.586 |
−0.907 |
NI (0.851) |
NI (0.859) |
WI (0.859) |
C (0.926) |
III |
NB (0.993) |
2.462 |
| MOX3 |
+0.994 |
−0.593 |
−0.947 |
NI (0.692) |
NI (0.691) |
WI (0.888) |
NC (0.798) |
III |
NB (1.000) |
2.380 |
| MOX4 |
+1.000 |
+0.500 |
+0.640 |
I (0.669) |
NI (0.642) |
WI (0.643) |
NC (0.880) |
III |
NB (1.000) |
2.422 |
| MOX5 |
+1.000 |
−0.548 |
+0.566 |
I (0.523) |
NI (0.758) |
WI (0.659) |
NC (0.863) |
III |
NB (1.000) |
2.462 |
| MOX6 |
+1.000 |
+0.507 |
+0.724 |
I (0.567) |
NI (0.703) |
WI (0.544) |
NC (0.867) |
III |
NB (1.000) |
2.437 |
| MOX7 |
+1.000 |
−0.599 |
+0.599 |
NI (0.584) |
NI (0.667) |
WI (0.936) |
NC (0.855) |
III |
NB (1.000) |
2.525 |
| MOX8 |
+0.997 |
−0.580 |
−0.660 |
I (0.528) |
NI (0.564) |
WI (0.924) |
NC (0.831) |
III |
NB (1.000) |
2.424 |
| MOX9 |
+1.000 |
−0.644 |
+0.550 |
NI (0.627) |
NI (0.631) |
WI (0.814) |
NC (0.852) |
III |
NB (0.997) |
2.414 |
| MOX10 |
+0.996 |
+0.565 |
−0.626 |
I (0.526) |
NI (0.713) |
WI (0.792) |
NC (0.559) |
III |
NB (1.000) |
2.361 |
| MOX11 |
+0.996 |
−0.680 |
−0.886 |
NI (0.837) |
NI (0.810) |
WI (0.828) |
NC (0.859) |
III |
NB (1.000) |
2.330 |
| MOX12 |
+0.995 |
−0.627 |
−0.862 |
NI (0.927) |
NI (0.893) |
WI (0.895) |
NC (0.863) |
III |
NB (1.000) |
2.318 |
| MOX13 |
+0.980 |
−0.705 |
−0.766 |
NI (0.924) |
NI (0.785) |
WI (0.849) |
NC (0.849) |
III |
NB (1.000) |
2.296 |
3.10 PASS prediction
The freely available and widely used PASS online server is a computer-aided approach designed to predict the biological activity spectra (both pharmacological and toxicological) based on their structural formulas for over 4000 types of pharmacotherapeutic effects, modes of action, interactions with the metabolic system, and particular toxicity for drug-like compounds with an average accuracy of more than 95%.120,121 An analysis of the structure–activity connections in the training set, which includes details on the biological activity and structure of over 300000 organic compounds, serves as the basis for the prediction.122 The PASS result is interpreted by calculating the independent probabilities of activity (Pa) and inactivity (Pi), which range from 0 to 1. A Pa larger than 0.7 highly correlates with the anticipated activity and frequently shows the molecule operates as a recognized drug analogue; a Pa between 0.5 and 0.7 denotes a moderate probability of activity, and if Pa is less than 0.5, the compound is likely to be inactive. In this investigation, the probability of activity (Pa) values of MOX and some of its newly created analogs are shown in Table S3, demonstrating that the parent drug MOX has comparatively higher pharmacologic effects than all other analogs. The PASS result shows that MOX has the best antibacterial (0.662), antimicrobial (0.702), and DNA synthesis inhibitor (0.810) activity, indicating that the parent medicines are more capable of acting as antibiotics than other analogs. Except MOX3 (0.697 for AA), MOX11 (0.630 for AA), and MOX10 (0.832 for TI), MOX also has the strongest anti-amyloidogenic activity (0.629) and the ability to inhibit topoisomerase II enzyme (0.820), which plays an important role in DNA replication, repair, and chromosome segregation. The PASS study, on the other hand, shows that most MOX analogs forecast more toxicity than MOX. All the analogs produced negative PASS results for a variety of toxic effects, including stomatitis, torsades de pointes, bradycardia, tachycardia, asthma, hepatitis, QT interval prolongation, hyperglycemia, keratopathy, tremor, and hypertension. However, except for MOX9, nearly all the analogs had higher predicted probabilities for keratopathy (0.700 to 0.947) and asthma (0.707 to 0.939) than MOX (0.730 for AS and 0.726 for KP). In contrast, almost all analogs predicted fewer QT interval prolongations (0.287 to 0.623) and hyperglycemic effects (0.434 to 0.751) than MOX (0.753 for QTIP; 0.800 for Hyp), with MOX10 (0.763 for QTIP) being the only exception. In comparison to all other compounds, MOX9 and MOX13 showed reduced toxicity across most of the criteria that were studied. Finally, PASS results clarify that practically all MOX analogs exhibit lower pharmacologic activity and higher toxicological activity than their parent drug MOX, with only a few newly created analogs outperforming them.
3.11 Structure–activity relationships and pharmacological optimization of quinolone substituents
The intrinsic activity and pharmacological profile of quinolones are highly dependent on substituents located at six important positions: N1 (R1), C5, C6, C7, C8, and the bridgehead atom X (C or N). This position C6, typically bearing a fluorine atom in fluoroquinolones, is critical for achieving potent antibacterial activity. However, the C6 fluorine is also associated with genotoxicity concerns. Therefore, modifying the C6 position is a crucial rational strategy for maintaining or enhancing potency while simultaneously mitigating potential genotoxicity risks, often by substituting fluorine with alternative groups. Substituents at C8 (R8), such as the methoxy group (OCH3) present in MOX, are essential for optimizing the overall molecular configuration. These modifications are known to increase the spectrum of activity against certain strains and are critical for favorable interactions with topoisomerase II. Position C7 typically hosts a bulky heterocyclic moiety (such as the diazabicyclo-octane ring in MOX). The C7 group is paramount for improving tissue penetration and impeding the efficiency of bacterial efflux proteins that compromise intracellular drug concentration.
The modification at R3, which impacts the C7 side chain (the cyclic amine) or an adjacent hydroxyl group near the C3 carboxylic acid, is essential for tuning physicochemical properties necessary for absorption, distribution, and membrane permeability. The strategic modification of R1 and R2 was primarily efficacy-driven, aiming to optimize target binding and overcome resistance mechanisms, while the R3 modifications targeted pharmacokinetic improvements and steric fit at the receptor periphery. The modification of the C8 position (R2) in MOX13 provides a direct answer regarding how to reduce hepatotoxicity. Moxifloxacin contains an 8-methoxy group (OCH3). In MOX13, this is replaced by the highly polar ureido group (NHCONH2). This substitution profoundly affects the compound's physicochemical profile. The consensus logP drops dramatically from 1.85 (MOX) to 0.62 (MOX13), signifying a much lower lipophilicity. Reduced lipophilicity minimizes the risk of the compound accumulating in hepatic tissues and decreases the likelihood of forming reactive lipophilic metabolites, which are often implicated in idiosyncratic Drug-Induced Liver Injury (DILI). Furthermore, the substitution increases the Topological Polar Surface Area (TPSA) to 129.69 Å2 and increases the number of hydrogen bond donors (NHD = 4). This enhanced polarity is predicted to favor rapid hydrophilic clearance pathways, such as enhanced renal excretion or direct conjugation, by passing the slower, oxidative hepatic metabolism pathways that contribute to liver exposure. Consequently, MOX13 shows a lower predicted hepatitis risk (Pa = 0.570) compared to MOX (Pa = 0.621). MOX9, modified at R3, also demonstrated a reduction in predicted hepatitis risk (Pa = 0.500). The R3 modification involves replacing carboxylic acid with an N-carbamoyl carboxamide group, similarly, increasing polarity near that region, and potentially promoting favorable clearance kinetics.
4 Conclusion
The computational investigation into moxifloxacin (MOX) and its novel analogs successfully identified two lead candidates, MOX13 and MOX9, demonstrating superior therapeutic potential against Mycobacterium tuberculosis (MTB) compared to the parent drug. Molecular docking against the MTB DNA gyrase (PDB ID 5BS8) revealed that MOX13 exhibited the strongest binding affinity at −8.5 kcal mol−1, a significant improvement over MOX's −7.2 kcal mol−1. This enhanced affinity in MOX13 stems from a novel, robust network of conventional hydrogen bonds involving residues like GLY120, ARG98, and TRP103, facilitated by the highly polar ureido group (NHCONH2) replacing the methoxy group (OCH3) at the C8 (R2) position. This new interaction mechanism is hypothesized to provide a structural anchor less susceptible to typical Quinolone Resistance-Determining Region (QRDR) mutations. The dynamic stability of the MOX13 complex was confirmed by 100 ns molecular dynamics simulations, showing exceptional stability and the lowest average RMSD value of 0.76 ± 0.44 nm, indicating it holds the protein in a highly favorable conformation throughout the simulation. Concurrently, the analogous modification in MOX13 dramatically reduced its consensus logP to 0.62 from 1.85 (MOX), a reduction in lipophilicity that provides a compelling physicochemical strategy for circumventing efflux-mediated resistance and is linked to a lower predicted Hepatitis risk. Furthermore, ADMET and PASS predictions indicated that both MOX9 and MOX13 maintained the most favorable toxicity profiles, particularly showing a reduced predicted risk of Q–T interval prolongation and hyperglycemic effects compared to the parent MOX. The overall results underscore MOX13 and MOX9 as promising next-generation antitubercular agents with superior efficacy, stability, and enhanced safety characteristics.
Author contributions
Md. Al-Amin: writing – review & editing, writing – original draft, visualization, methodology, investigation. Md. Rakib Hossin Mallik: writing –original draft, visualization, investigation, formal analysis. Md Shohanur Rahman: writing – original draft, methodology, investigation. Abdullah Al Noman: writing – original draft, formal analysis, visualization, investigation. Sarmin Akther Tithy: writing – original draft, methodology, investigation. Omme Samia: writing – original draft. Monir Uzzaman: writing – review & editing, writing – original draft, visualization, validation, supervision, project administration, methodology, investigation, conceptualization.
Conflicts of interest
The authors declared no conflict of interest.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information (SI)]. Supplementary information is available. See DOI: https://doi.org/10.1039/d5ra07315d.
Acknowledgements
We are grateful to the Computer in Chemistry and Medicine Laboratory in Dhaka, Bangladesh, for their important suggestions and support.
References
- S. Singh, Tuberc. Res. Treat., 2024, 2024, 2307742, DOI:10.1155/2024/2307742.
- B. Gilmour and K. A. Alene, Front. Tuberc., 2024, 2, 1487518, DOI:10.3389/ftubr.2024.1487518.
- Global Tuberculosis Report, 2024, https://www.gavi.org/vaccineswork/about Search PubMed.
- C. Lange, D. Chesov, J. Heyckendorf, C. C. Leung, Z. Udwadia and K. Dheda, Respirology, 2018, 23, 656–673, DOI:10.1111/resp.13304.
- C. Lange, K. Dheda, D. Chesov, A. M. Mandalakas, Z. Udwadia and C. R. Horsburgh, Lancet, 2019, 394, 953–966, DOI:10.1016/S0140-6736(19)31882-3.
- L. Guglielmetti, U. Khan, G. E. Velásquez, M. Gouillou, A. Abubakirov, E. Baudin, E. Berikova, C. Berry, M. Bonnet, M. Cellamare, V. Chavan, V. Cox, Z. Dakenova, B. C. de Jong, G. Ferlazzo, A. Karabayev, O. Kirakosyan, N. Kiria, M. Kunda, N. Lachenal, L. Lecca, H. McIlleron, I. Motta, S. M. Toscano, H. Mushtaque, P. Nahid, L. Oyewusi, S. Panda, S. Patil, P. P. J. Phillips, J. Ruiz, N. Salahuddin, E. S. Garavito, K. J. Seung, E. Ticona, L. Trippa, D. E. V. Vasquez, S. Wasserman, M. L. Rich, F. Varaine and C. D. Mitnick, N. Engl. J. Med., 2025, 392, 468–482, DOI:10.1056/nejmoa2400327.
- S. Tiberi, M. J. Vjecha, A. Zumla, J. Galvin, G. B. Migliori and A. Zumla, Int. J. Infect. Dis., 2021, 113, S96–S99, DOI:10.1016/j.ijid.2021.02.067.
- E. A. Gyselen, Health Policy, 2010, 28, 73–74 CrossRef , https://pubmed.ncbi.nlm.nih.gov/23741786/.
- WHO Launches an Update on the Consolidated Guidelines to Diagnose Tuberculosis, https://www.who.int/news/item/17-04-2025-who-launches-an-update-on-the-consolidated-guidelines-to-diagnose-tuberculosis Search PubMed.
- V. Mave, M. Paradkar, F. Conradie, A. Gupta, A. Avihingsanon, G. Meintjes, A. Turkova, K. E. Dooley and R. E. Chaisson, Lancet HIV, 2025, 12, e367–e381, DOI:10.1016/S2352-3018(25)00040-2.
- K. J. Aldred, R. J. Kerns and N. Osheroff, Biochemistry, 2014, 53, 1565–1574, DOI:10.1021/bi5000564.
- A. M. Gutiérrez-Mauricio, J. V. Trujillo-Paez, L. A. Trejo-Martinez, B. Rivas-Santiago, P. Pérez-García, S. Noriega, J. E. López-Ramos, J. Cardoso-Ortiz and A. Rodríguez-Carlos, J. Antibiot., 2025, 2025, 1–18, DOI:10.1038/s41429-025-00839-2.
- I. R. Gilfanov, A. I. Kolesnikova, R. S. Pavelyev, D. V. Sudarikov, P. V. Gribkov, E. S. Izmest'ev, S. A. Rubtsova, S. A. Lisovskaya, O. B. Babaeva, I. Z. Rakhmatullin, R. R. Davletshin, V. V. Klochkov, L. E. Nikitina, A. R. Kayumov and E. Y. Trizna, Chem. Biodivers., 2025, 22, e202402601, DOI:10.1002/cbdv.202402601.
- A. Storper, D. Miller and X. Huo, bioRxiv, 2025, preprint, 2025.06.22.660934, DOI:10.1101/2025.06.22.660934.
- V. Ahlawat, K. Sura, S. Khan, R. Jakhar and M. Dangi, J. Integr. Sci. Technol., 2025, 13, 1112, DOI:10.62110/sciencein.jist.2025.v13.1112.
- P. P. Yılmaz, N. Kulabaş, A. Bozdeveci, S. K. Vagolu, M. Imran, E. Tatar, Ş. Alpay Karaoğlu, D. Sriram, A. A. R. Mahmood and İ. Küçükgüzel, Chem. Biol. Drug Des., 2025, 105, e70126, DOI:10.1111/cbdd.70126.
- M. Salman, P. Sharma, M. Kumar, A. S. Ethayathulla and P. Kaur, Briefings Funct. Genomics, 2023, 22, 180–194, DOI:10.1093/bfgp/elac029.
- V. N. Dahl, O. S. Pedersen, T. Butova, M. Kuzhko, O. Raznatovska, A. Fedorec, V. Vekshyn, V. Hlynenko, N. Yevsiukova, L. Hryshchuk, S. Kornaha, N. Chursina, N. Kondratyuk, L. Markovtsiy, V. Skryp, A. Bogomolov, T. Klymenko, D. Yankovska, M. Ostrovskyy, I. Makoida, D. Levandovska, Y. Shpak, V. Didyk, O. Siomak and D. Butov, Clin. Infect. Dis., 2025, 81, 853–856, DOI:10.1093/cid/ciaf214.
- S. R. Lopes, M. Marçal, N. Fernandes, F. Silva, P. Barbosa, M. Vieira, J. P. Ramos and R. Duarte, Breathe, 2025, 21(1) DOI:10.1183/20734735.0232-2024.
- A. V. Solovyeva, G. V. Volchenkov, O. I. Ponomarenko, T. A. Kuznezova, T. R. Somova, E. V. Belova, S. G. Hinderaker, E. Heldal and S. Keshavjee, Safety, efficacy and feasibility of preventive treatment for drug-resistant tuberculosis with moxifloxacin or bedaquiline, Cold Spring Harbor Laboratory Press, 2025, DOI:10.1101/2025.03.09.25323266.
- F. Schmitz, J. Antimicrob. Chemother., 1998, 41, 481–484, DOI:10.1093/JAC/41.4.481.
- J. A. B. Balfour and H. M. Lamb, Drugs, 2000, 59, 115–139, DOI:10.2165/00003495-200059010-00010.
- T. Gumbo, A. Louie, M. R. Deziel, L. M. Parsons, M. Salfinger and G. L. Drusano, J. Infect. Dis., 2004, 190, 1642–1651, DOI:10.1086/424849.
- S. Shee, S. Singh, A. Tripathi, C. Thakur, T. A. Kumar, M. Das, V. Yadav, S. Kohli, R. S. Rajmani, N. Chandra, H. Chakrapani, K. Drlica and A. Singh, Antimicrob. Agents Chemother., 2022, 66 DOI:10.1128/aac.00592-22.
- A. S. Ginsburg, J. Lee, S. C. Woolwine, J. H. Grosset, F. M. Hamzeh and W. R. Bishai, Antimicrob. Agents Chemother., 2005, 49, 853–856, DOI:10.1128/aac.49.2.853-856.2005.
- M. Fayos, J. T. Silva, M. Fernández-Ruiz, T. Ruiz-Merlo, A. Visentin, C. Loinaz, A. Manrique-Municio, J. M. Caso, J. González-Olmedo, I. Rodríguez-Góncer, F. López-Medrano, C. Lumbreras, J. M. Aguado and R. San-Juan, Transpl. Infect. Dis., 2024, 26, e14382, DOI:10.1111/tid.14382.
- J. A. B. Balfour and L. R. Wiseman, Drugs, 1999, 57, 363–373, DOI:10.2165/00003495-200059010-00010.
- C. A. Debate, C. P. Mathew, J. H. Warner, A. Heyd and D. Church, Respir. Med., 2000, 94, 1029–1037, DOI:10.1053/rmed.2000.0927.
- M. T. Chirehwa, J. E. Resendiz-Galvan, R. Court, M. De Kock, L. Wiesner, N. de Vries, J. Harding, T. Gumbo, R. Warren, G. Maartens, P. Denti and H. McIlleron, Antimicrob. Agents Chemother., 2023, 67 DOI:10.1128/aac.01426-22.
- L. Dridi, J. Tankovic, B. Burghoffer, F. Barbut and J.-C. Petit, Antimicrob. Agents Chemother., 2002, 46, 3418–3421, DOI:10.1128/aac.46.11.3418-3421.2002.
- A. C. Spencer and S. S. Panda, Biomedicines, 2023, 11, 371, DOI:10.3390/biomedicines11020371.
- T. R. Blower, B. H. Williamson, R. J. Kerns and J. M. Berger, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1706–1713, DOI:10.1073/pnas.1525047113.
- Crystal Structure of a Topoisomerase II Complex, 2016, DOI:10.2210/pdb5bs8/pdb.
- B. GL, R. Rajput, M. Gupta, P. Dahiya, J. K. Thakur, R. Bhatnagar and A. Grover, Biochem. J., 2020, 477, 4167–4190, DOI:10.1042/BCJ20200462.
- O. S. Aina, M. O. Rofiu, O. A. Oloba-Whenu, I. A. Olasupo, L. A. Adams and O. B. Familoni, Sci. Afr., 2024, 23, e01985, DOI:10.1016/j.sciaf.2023.e01985.
- S. Zheng, Y. Gu, Y. Gu, Y. Zhao, L. Li, M. Wang, R. Jiang, X. Yu, T. Chen and J. Li, Briefings Bioinf., 2024, 26 DOI:10.1093/bib/bbae696.
- M. M. Matin, M. S. Hasan, M. Uzzaman, M. M. H. Bhuiyan, S. M. Kibria, M. E. Hossain and M. H. O. Roshid, J. Mol. Struct., 2020, 1222, 128821, DOI:10.1016/j.molstruc.2020.128821.
- M. Uzzaman, M. K. Hasan, S. Mahmud, K. Fatema and M. M. Matin, Inform. Med. Unlocked, 2021, 25, 100677, DOI:10.1016/j.imu.2021.100677.
- J. M. Domagala, J. Antimicrob. Chemother., 2018, 685–706, DOI:10.1093/jac/33.4.685.
- D. C. Hooper and G. A. Jacoby, Cold Spring Harb. Perspect. Med., 2016, 6, 1–22, DOI:10.1101/cshperspect.a025320.
- F. Azam, N. H. Alabdullah, H. M. Ehmedat, A. R. Abulifa, I. Taban and S. Upadhyayula, J. Biomol. Struct. Dyn., 2018, 36, 2099–2117, DOI:10.1080/07391102.2017.1338164.
- A. Allouche, J. Comput. Chem., 2012, 32, 174–182, DOI:10.1002/jcc.
- B. Akter, S. Aishee, A. Hridoy, M. M. H. Pulok, M. A. Islam, A. Biswas, A. Patwary, M. Hoque, M. S. Islam, M. N. H. Nirob, F. I. Chowdhury and M. Uzzaman, Chem. Phys. Impact, 2025, 10, 100830, DOI:10.1016/j.chphi.2025.100830.
- S. Dallakyan and A. J. Olson, Methods Mol. Biol., 2015, 1263, 243–250, DOI:10.1007/978-1-4939-2269-7_19.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461, DOI:10.1002/jcc.21334.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718, DOI:10.1002/jcc.20291.
- S. Ghahremanian, M. M. Rashidi, K. Raeisi and D. Toghraie, J. Mol. Liq., 2022, 354, 118901, DOI:10.1016/j.molliq.2022.118901.
- H. Yang, C. Lou, L. Sun, J. Li, Y. Cai, Z. Wang, W. Li, G. Liu and Y. Tang, Bioinformatics, 2019, 35, 1067–1069, DOI:10.1093/bioinformatics/bty707.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- V. A. Adediwura, K. Koirala, H. N. Do, J. Wang and Y. Miao, Expet Opin. Drug Discov., 2024, 19, 671–682, DOI:10.1080/17460441.2024.2349149.
- S. Decherchi and A. Cavalli, Chem. Rev., 2020, 120, 12788–12833, DOI:10.1021/acs.chemrev.0c00534.
- G. Klebe, ChemMedChem, 2015, 10, 229–231, DOI:10.1002/cmdc.201402521.
- I. Muegge and Y. Hu, ACS Med. Chem. Lett., 2023, 14, 244–250, DOI:10.1021/acsmedchemlett.2c00541.
- N. C. Garbett and J. B. Chaires, Expet Opin. Drug Discov., 2012, 7, 299–314, DOI:10.1517/17460441.2012.666235.
- T. Amano, T. Yamazaki and S. Tsuneyuki, Phys. Rev. B, 2024, 110, 165159, DOI:10.1103/PhysRevB.110.165159.
- A. Sultan, J. Sieg, M. Mathea and A. Volkamer, J. Chem. Inf. Model., 2024, 64, 6259–6280, DOI:10.1021/acs.jcim.4c00747.
- Z. Cournia, B. Allen and W. Sherman, J. Chem. Inf. Model., 2017, 57, 2911–2937, DOI:10.1021/acs.jcim.7b00564.
- R. Zahradník, Experientia, 1962, 18(11), 534–536, DOI:10.1007/bf02151616.
- E. A. Eno, H. Louis, P. Ekoja, I. Benjamin, S. A. Adalikwu, M. M. Orosun, T. O. Unimuke, F. C. Asogwa and E. C. Agwamba, J. Indian Chem. Soc., 2022, 99, 100532, DOI:10.1016/j.jics.2022.100532.
- V. S. J. Reeda, J. N. C. Mishma, P. Divya, R. Suja, A. Manikandan, M. Shahid, H. Arora, A. Ali, N. Siddiqui and S. Javed, Spectrosc. Lett., 2025, 58, 57–77, DOI:10.1080/00387010.2024.2401990.
- A. Zochedh, M. Priya, C. Chakaravarthy, A. B. Sultan and T. Kathiresan, Polycycl. Aromat. Compd., 2023, 43, 6516–6548, DOI:10.1080/10406638.2022.2118332.
- B. J. Powell, T. Baruah, N. Bernstein, K. Brake, R. H. McKenzie, P. Meredith and M. R. Pederson, J. Chem. Phys., 2004, 120, 8608–8615, DOI:10.1063/1.1690758.
- S. Saravanan and V. Balachandran, Spectrochim. Acta, Part A, 2014, 120, 351–364, DOI:10.1016/j.saa.2013.10.042.
- A. Martínez, I. A. Ibarra and R. Vargas, PLoS One, 2019, 14, 1–12, DOI:10.1371/journal.pone.0224691.
- A. Mohamed, D. P. Visco, K. Breimaier and D. M. Bastidas, ACS Omega, 2025, 10, 2799–2808, DOI:10.1021/acsomega.4c08626.
- J. Zhang, C. Wang, L. Ji and W. Liu, Chem. Res. Toxicol., 2016, 29, 841–850, DOI:10.1021/acs.chemrestox.6b00018.
- J. Yu, N. Q. Su and W. Yang, JACS Au, 2022, 2, 1383–1394, DOI:10.1021/jacsau.2c00085.
- M. C. Vlasiou, Biointerface Res. Appl. Chem., 2021, 11, 15051–15057, DOI:10.33263/briac116.1505115057.
- K. Abdoulaye, B. A. Lucie, O. Lamoussa, K. Soleymane, B. Kafoumba, K. Abdoulaye, B. A. Lucie, O. Lamoussa, K. Soleymane and B. Kafoumba, J. Mater. Sci. Chem. Eng., 2024, 12, 31–50, DOI:10.4236/msce.2024.123004.
- A. A. Madi, D. Haffar, F. Benghanem, S. Ghedjati, L. Toukal, V. Dorcet and R. Bourzami, J. Mol. Struct., 2021, 1227, 129368, DOI:10.1016/j.molstruc.2020.129368.
- Y. Yıl, Ü. Tarım, B. Dergisi, A. S. Salihu, W. Mohd, N. Hakimi and W. Salleh, Yuzuncu Yil Univ. J. Agric. Sci., 2023, 33, 491–502, DOI:10.29133/yyutbd.1294240.
- K. O. Faloye, S. O. Famuyiwa, K. F. Akinwunmi, C. M. Tata, M. D. Ayoola, E. G. Fakola, O. F. Akinyele and D. T. Ndinteh, Int. J. Sci. Res. Arch., 2022, 7, 456–464, DOI:10.30574/ijsra.2022.7.1.0183.
- Z. Fan, X. Liu, N. Wang, S. Yu, C. Bi, Y. Si, X. Ling, C. Liu, J. Wang and H. Sun, J. Cancer Res. Clin. Oncol., 2024, 150, 253, DOI:10.1007/S00432-024-05784-5.
- Y. T. Hussein, Y. H. Azeez and I. M. Ahmed, J. Turk. Chem. Soc., Sect. A, 2024, 11, 279–290, DOI:10.18596/jotcsa.1246781.
- M. Haritha and C. H. Suresh, WIREs Computational Molecular Science, 2024 DOI:10.1002/wcms.1735.
- A. K. Bhattacharjee, Synth. Lect. Math. Stat., pt F3386, 2025, pp. 123–150, DOI:10.1007/978-3-031-67841-7_7.
- S. R. Gadre, C. H. Suresh and N. Mohan, Molecules, 2021, 26, 3289, DOI:10.3390/molecules26113289.
- F. Vascon, M. Gasparotto, M. Giacomello, L. Cendron, E. Bergantino, F. Filippini and I. Righetto, Comput. Struct. Biotechnol. J., 2020, 18, 1774–1789, DOI:10.1016/j.csbj.2020.06.029.
- C. H. Suresh and S. Anila, Acc. Chem. Res., 2023, 56, 1884–1895, DOI:10.1021/acs.accounts.3c00193.
- D. Shin and Y. Jung, Phys. Chem. Chem. Phys., 2022, 24, 25740–25752, 10.1039/d2cp03244a.
- Z. Wang, W. Wang and H.-B. Li, Molecules, 2023, 28, 3919, DOI:10.3390/molecules28093919.
- R. Tiwari, A. Bijanu, C. S. Sharma, V. Kumar, L. S. Banjara, U. Kumar, K. Kumar, A. K. Meena, Modern Spectroscopic Techniques for Drug Discovery and Environmental Sustainability, 2025, vol. 1AD, pp. 65–96, DOI:10.4018/979-8-3693-7473-3, https://services.igi-global.com/resolvedoi/resolve.aspx?doi=10.4018/979-8-3693-7473-3.ch004.
- M. M. Moreno, P. Garidel, M. Suwalsky, J. Howe and K. Brandenburg, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1296–1303, DOI:10.1016/j.bbamem.2009.01.016.
- E. Ouyang, Z. Hu, J. Zhang, X. Huang, X. Jiang, R. Zhao and H. Yang, Elsevier, 2025, 137583, DOI:10.1016/j.colsurfa.2025.137583.
- H. Yao, J. Liu, S. Xu, Z. Zhu and J. Xu, Expet Opin. Drug Discov., 2017, 12, 121–140, DOI:10.1080/17460441.2016.1272757.
- Z. Chen, H. Luo, A. Gubu, S. Yu, H. Zhang, H. Dai, Y. Zhang, B. Zhang, Y. Ma, A. Lu and G. Zhang, Front. Cell Dev. Biol., 2023, 11, 1091809, DOI:10.3389/fcell.2023.1091809.
- S. K. Pathak, R. Srivastava, A. K. Sachan, O. Prasad, L. Sinha, A. M. Asiri and M. Karabacak, Spectrochim. Acta, Part A, 2015, 135, 283–295, DOI:10.1016/j.saa.2014.06.149.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. A. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protoc., 2014, 9, 1771–1791, DOI:10.1038/nprot.2014.110.
- G. Saielli, Appl. Sci., 2020, 10, 8108, DOI:10.3390/app10228108.
- D. Schauenburg and T. Weil, Adv. Sci., 2024, 11, 1–22, DOI:10.1002/advs.202303396.
- M. Miar, A. Shiroudi, K. Pourshamsian, A. R. Oliaey and F. Hatamjafari, J. Chem. Res., 2021, 45, 147–158, DOI:10.1177/1747519820932091.
- M. Uzzaman, M. K. Hasan, S. Mahmud, A. Yousuf, S. Islam, M. N. Uddin and A. Barua, Inform. Med. Unlocked, 2021, 25, 100706, DOI:10.1016/j.imu.2021.100706.
- J. O. Anhaia-Machado, A. C. G. Soares, C. A. S. de Oliveira Pinto, A. I. Á. Barrera, A. R. Baby and G. H. G. Trossini, Chemistry, 2022, 5, 41–53, DOI:10.3390/chemistry5010004.
- Z. Kocakaya, Y. Sert, M. Kocakaya, G. Ş. Karatoprak, S. İlgün and M. Çadır, J. Mol. Liq., 2025, 417, 126660, DOI:10.1016/j.molliq.2024.126660.
- A. Sagaama, N. Issaoui, O. Al-Dossary, A. S. Kazachenko and M. J. Wojcik, J. King Saud Univ., Sci., 2021, 33, 101606, DOI:10.1016/j.jksus.2021.101606.
- M. Medimagh, N. Issaoui, S. Gatfaoui, A. S. Kazachenko, O. M. Al-Dossary, N. Kumar, H. Marouani and L. G. Bousiakoug, J. King Saud Univ., Sci., 2023, 35, 102645, DOI:10.1016/j.jksus.2023.102645.
- S. Jena, J. Dutta, K. D. Tulsiyan, A. K. Sahu, S. S. Choudhury and H. S. Biswal, Chem. Soc. Rev., 2022, 51, 4261–4286, 10.1039/d2cs00133k.
- S. M. Nashre-ul-Islam, K. K. Borah, F. E. Öztürkkan, M. A. Raza, A. Frontera and D. M. Gil, J. Mol. Struct., 2022, 1268, 133686, DOI:10.1016/j.molstruc.2022.133686.
- A. Anighoro, in Methods in Molecular Biology, Humana, New York, NY, 2020, vol. 2114, pp. 75–86, DOI:10.1007/978-1-0716-0282-9_5.
- S. M. Esther Rubavathy and M. Prakash, Phys. Chem. Chem. Phys., 2025, 27, 1071–1082, 10.1039/d4cp03291h.
- P. Goswami, E. G. Fakola, S. Ghosh, S. Ghosh and S. Das, In silico Pharmacol., 2025, 13, 20, DOI:10.1007/s40203-025-00310-y.
- G. Mandujano-Lázaro, M. F. Torres-Rojas, E. Ramírez-Moreno and L. A. Marchat, Sci. Prog., 2025, 108, 00368504251320313, DOI:10.1177/00368504251320313.
- Y. Maruyama, R. Igarashi, Y. Ushiku and A. Mitsutake, J. Chem. Inf. Model., 2023, 63, 1529–1541, DOI:10.1021/acs.jcim.2c01444.
- W. Schreiner, R. Karch, B. Knapp and N. Ilieva, Comput. Math. Methods Med., 2012, 2012, 173521, DOI:10.1155/2012/173521.
- G. R. C. Pereira, A. N. R. Da Silva, S. S. Do Nascimento and J. F. De Mesquita, J. Cell. Biochem., 2019, 120, 3583–3598, DOI:10.1002/jcb.27636.
- P. Craveur, A. P. Joseph, J. Esque, T. J. Narwani, F. Noël, N. Shinada, M. Goguet, S. Leonard, P. Poulain, O. Bertrand, G. Faure, J. Rebehmed, A. Ghozlane, L. S. Swapna, R. M. Bhaskara, J. Barnoud, S. Téletchéa, V. Jallu, J. Cerny, B. Schneider, C. Etchebest, N. Srinivasan, J.-C. Gelly and A. G. de Brevern, Front. Mol. Biosci., 2015, 2, 140502, DOI:10.3389/fmolb.2015.00020.
- P. Baziyar, B. Seyedalipour, S. Hosseinkhani and E. Nazifi, Iran. J. Biotechnol., 2022, 20, e3178, DOI:10.30498/ijb.2022.310249.3178.
- A. Belhassan, S. Chtita, H. Zaki, M. Alaqarbeh, N. Alsakhen, F. Almohtaseb, T. Lakhlifi and M. Bouachrine, J. Mol. Struct., 2022, 1258, 132652, DOI:10.1016/j.molstruc.2022.132652.
- R. K. Mohapatra, K. Dhama, A. A. El-Arabey, A. K. Sarangi, R. Tiwari, T. B. Emran, M. Azam, S. I. Al-Resayes, M. K. Raval, V. Seidel and M. Abdalla, J. King Saud Univ., Sci., 2021, 33, 101637, DOI:10.1016/j.jksus.2021.101637.
- U. Dasmahapatra, C. K. Kumar, S. Das, P. T. Subramanian, P. Murali, A. E. Isaac, K. Ramanathan, M. M. Balamurali and K. Chanda, Front. Chem., 2022, 10, 991369, DOI:10.3389/fchem.2022.991369.
- S. Rampogu, B. Shaik, J. H. Kim, T. S. Jung, M. W. Ha and K. W. Lee, Heliyon, 2023, 9 DOI:10.1016/j.heliyon.2023.e13324.
- J. Weng, S. Yang, J. Shen, H. Liu, Y. Xu, D. Hao and S. Wang, J. Zhejiang Univ., Sci., B, 2023, 24, 883–895, DOI:10.1631/jzus.b2200634.
- M. Hasan, M. I. Hossain, N. H. Siddiquee, E. Ahmed, M. W. H. Talukder, M. Rahamatolla, T. Nahar, P. R. Paul, M. H. Suhag and M. Uzzaman, PLoS One, 2025, 20, e0326655, DOI:10.1371/journal.pone.0326655.
- M. L. Amin, Drug Target Insights, 2013, 7 DOI:10.4137/dti.s12519.
- M. C. Sanguinetti and M. Tristani-Firouzi, Nature, 2006, 440, 463–469, DOI:10.1038/nature04710.
- C. A. Lipinski, Drug Discov. Today Technol., 2004, 1, 337–341, DOI:10.1016/j.ddtec.2004.11.007.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623, DOI:10.1021/jm020017n.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68, DOI:10.1021/cc9800071.
- I. Muegge, Med. Res. Rev., 2003, 23, 302–321, DOI:10.1002/med.10041.
- A. Lagunin, D. Filimonov and V. Poroikov, Curr. Pharm. Des., 2010, 16, 1703–1717, DOI:10.2174/138161210791164063.
- D. A. Filimonov, A. A. Lagunin, T. A. Gloriozova, A. V. Rudik, D. S. Druzhilovskii, P. V. Pogodin and V. V. Poroikov, Chem. Heterocycl. Compd., 2014, 50, 444–457, DOI:10.1007/S10593-014-1496-1.
- A. Lagunin, A. Stepanchikova, D. Filimonov and V. Poroikov, Bioinformatics, 2000, 16, 747–748, DOI:10.1093/bioinformatics/16.8.747.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ag
*ag










