
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the anti-diabetic potential of bis-Schiff bases of ibuprofen: insights into the in vitro, molecular docking and density functional theory analyses
Muhammad Ayaza,
Aftab Alamb,
Zainabc,
Ahmed A. Elhenawy de,
Imtiaz Ahmadf,
Sajjad Ur Rahmana,
Liaqat Alig,
Abdul Latifa,
Mumtaz Ali*a and
Manzoor Ahmad*a
de,
Imtiaz Ahmadf,
Sajjad Ur Rahmana,
Liaqat Alig,
Abdul Latifa,
Mumtaz Ali*a and
Manzoor Ahmad*a
aDepartment of Chemistry, University of Malakand, P.O. Box 18800, Dir Lower, Khyber Pakhtunkhwa, Pakistan. E-mail: mumtazali@gmail.com; manzoorahmad@uom.edu.pk
bDepartment of Chemistry, Rawalpindi Women University, Rawalpindi, Pakistan
cCollege of Chemistry and Materials Science, Hebei Normal University, Shijiazhuang, China
dChemistry Department, Faculty of Science, Al-Azhar University, Cairo, Egypt
eChemistry Department, Faculty of Science, Al-Baha University, Al-Bahah 65731, Saudi Arabia
fPrograma de Pós-Graduação em Bioquímica e Bioprospecção, Universidade Federal de Pelotas, Campus Universitário Capão do Leão S/N, Pelotas, RS CEP 96010-900, Brazil
gDepartment of Chemistry, University of Mianwali, Mianwali, 42200, Pakistan
First published on 25th February 2026
Abstract
Diabetes mellitus is a chronic metabolic disorder characterized by hyperglycaemia resulting from defects in insulin action or secretion. The synthesized bis-Schiff base derivatives of ibuprofen were evaluated for their in vitro α-amylase and α-glucosidase inhibitory activities using acarbose as a standard. Six compounds (13, 12, 14, 10, 9, and 11) exhibited superior dual inhibitory potency compared to acarbose, with IC50 values ranging from 3.85 ± 0.05 µM to 14.47 ± 0.14 µM (α-amylase) and 4.36 ± 0.12 µM to 16.18 ± 0.12 µM (α-glucosidase), representing 1.1- to 4.2-fold enhanced activity. Compound 13 (3,4,5-trimethoxyphenyl) was the most potent, showing 4.2-fold (α-amylase) and 3.8-fold (α-glucosidase) improvement compared to acarbose. Molecular docking and DFT analyses revealed that compound 13's superior binding affinity (ΔG = −7.033 kcal mol−1 vs. −5.868 kcal mol−1 (acarbose)) arose from optimized π–π stacking with Trp59 and hydrogen bonding with Gln63/Ser108, facilitated by electron-donating methoxy groups. The FMO analysis showed a low HOMO–LUMO gap (ε = 1.840 eV) and a high electrophilicity index (ω = 2.094), correlating with enhanced charge-transfer interactions. Cytotoxicity assessments suggested no toxicity up to 1000 µg mL−1. These findings position ibuprofen-based bis-Schiff bases as promising leads with enhanced potency and potentially improved drug-like properties compared to acarbose.
Introduction
Diabetes mellitus (DM), a chronic metabolic, life-threatening and non-communicable disease mainly caused by reduced insulin secretion, which makes it a worldwide health problem.1 Controlling blood glucose using pharmacological interventions is the main clinical approach for the management of DM, which is rapidly increasing in prevalence and has become a main reason for death worldwide.2,3 DM is also recognized to be a daily life-associated disease that can result in stroke, neuropathy, retinopathy, atherosclerosis, and nephropathy.4 The International Diabetes Federation (IDF)'s approximations show that 529 million individuals received a diabetes diagnosis in 2021,5 while in 2022, the number had increased up to 828 million,6 and by 2030, this number is predicted to increase up to 555 million.7 Type-II DM (T2D) is more common in advanced countries and can be competently treated with α-amylase and α-glucosidase inhibitors that diminish and delay the blood glucose rise occurring after meals.8 α-Amylase and α-glucosidase are the main digestive enzymes used for the metabolism of carbohydrates.9 α-Amylase breaks down complex carbohydrates and starch into disaccharides, which are further hydrolyzed by isomaltase into free glucose, which can then be absorbed. The key digestive isomaltase in human beings is α-D-glucoside glucohydrolase or α-glucosidase, a membrane-bound enzyme present in the epithelial mucosa of the small intestine.10 It is involved in controlling the release of glucose from polysaccharides in the gut and inhibiting its activity. Moderate α-amylase inhibition combined with strong α-glucosidase inhibition has been recommended as an efficient strategy to reduce polysaccharide breakdown and limit glucose release in gastrointestinal tract.11 Several α-amylase and α-glucosidase inhibitors have been made, and their pharmacological properties have been examined.12 Marketed available anti-diabetic drugs like acarbose, miglitol and voglibose block these enzymes in the brush borders of the small intestine, deferring the breakdown of carbohydrates and helping to control post-prandial hyperglycaemia.13–15 However, the long-term usage of these drugs can have some adverse effects, including abdominal pain and diarrhoea.16 Consequently, there is a noteworthy need to develop new and safe α-glucosidase and α-amylase inhibitors having better effectiveness and reduced side effects.17,18In recent decades, azines, also known as bis-Schiff bases or di-imines, have attracted noteworthy attention because of their extensive biological and pharmacological uses.19 These compounds are distinguished by the existence of two carbon–nitrogen (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) double bonds having the general formula RCHN–NHCR and are usually synthesized by the condensation of aliphatic and aromatic aldehydes with primary amines in acidic media, enabling the formation of imine bonds.20 Based on the types of substituents attached to the imine group, azines can exist in asymmetric and symmetric forms, which can have a considerable effect on their biological activity and structural characteristics.21 In addition, azines have been identified as coordination ligands and metal chelators, increasing their potential for enzyme inhibitory activities.22,23 The structural flexibility of azine derivatives allows their further alteration, making them potential candidates for therapeutic interventions and drug development.24
N–) double bonds having the general formula RCHN–NHCR and are usually synthesized by the condensation of aliphatic and aromatic aldehydes with primary amines in acidic media, enabling the formation of imine bonds.20 Based on the types of substituents attached to the imine group, azines can exist in asymmetric and symmetric forms, which can have a considerable effect on their biological activity and structural characteristics.21 In addition, azines have been identified as coordination ligands and metal chelators, increasing their potential for enzyme inhibitory activities.22,23 The structural flexibility of azine derivatives allows their further alteration, making them potential candidates for therapeutic interventions and drug development.24
In recent times, our group has reported the synthesis of bis-Schiff base compounds with diverse biological activities. Notably, bis-Schiff base compounds based on 4-nitroacetophenone are recognized as potent α-glucosidase inhibitors and bis-Schiff bases based on 4-hydroxyacetophenone are reported for their in vitro phosphodiesterase-1 (PDE-1), phosphodiesterase (PDE-3) and anti-diabetic activities.2,22,25,26 Bis-Schiff base compounds derived from ethyl phenyl ketone have shown significant anti-oxidant and anti-urease inhibitory activities,23,27 while flurbiprofen-based bis-Schiff bases have been reported as promising analgesic and anti-inflammatory agents.28 Additionally, ibuprofen-based bis-Schiff bases have been reported to be potential anti-cancer agents.29 Due to the promising biological activities of bis-Schiff base compounds, this research focuses on their in vitro α-amylase and α-glucosidase inhibition activities, and their molecular docking and density functional theory (DFT) analyses are used to provide a systematic understanding.
Results and discussion
Chemistry
In search of potential pharmacologically/biologically active compounds, we, in this study, report the anti-diabetic activities of the synthetic ibuprofen-based bis-Schiff base derivatives 4–15. These compounds were recently synthesized by refluxing ibuprofen with carbonyl diimidazole (CDI) in the tetrahydrofuran (THF) solvent. Then, hydrazine hydrate was added to the mixture and refluxed for 4–5 hours to get the hydrazide (1) of ibuprofen, which was further refluxed with the esterified aldehyde in ethanol to get the Schiff base compound 2. The desired Schiff base compound (2) was further refluxed with hydrazine hydrate in ethanol to get the hydrazide (3) of the Schiff base compound. Finally, a number of aromatic aldehydes were refluxed with the hydrazide Schiff base compound in the ethanol solvent containing a catalytic quantity of acetic acid to get the bis-Schiff base compounds 4–15 (Scheme 1). These compounds were structurally deduced by means of 1H-NMR, 13C-NMR and HR-ESI-MS, and the data of the compounds are available in the reported literature.29 | ||
| Scheme 1 Synthesis of ibuprofen-based bis-Schiff base compounds. | ||
 | ||
| Fig. 1 α-Amylase inhibitory potential of compounds 4–15. | ||
|
|||
|---|---|---|---|
| C. no. | R | α-Amylase | α-Glucosidase |
| IC50 ± SEM (µM) | IC50 ± SEM (µM) | ||
| 4 |  |
89.29 ± 1.44 | 91.52 ± 1.45 |
| 5 |  |
92.43 ± 0.69 | 96.71 ± 1.92 |
| 6 |  |
78.45 ± 0.02 | 82.07 ± 0.79 |
| 7 |  |
85.29 ± 2.32 | 87.42 ± 0.56 |
| 8 |  |
98.05 ± 0.43 | 101.00 ± 0.63 |
| 9 | 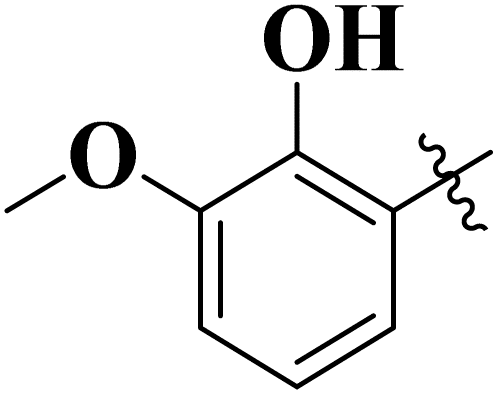 |
12.17 ± 0.14 | 16.60 ± 0.12 |
| 10 |  |
8.66 ± 0.01 | 9.51 ± 0.09 |
| 11 |  |
14.47 ± 0.14 | 16.18 ± 0.12 |
| 12 |  |
5.51 ± 0.04 | 6.11 ± 0.01 |
| 13 |  |
3.85 ± 0.05 | 4.36 ± 0.12 |
| 14 |  |
6.20 ± 0.18 | 8.12 ± 0.14 |
| 15 |  |
32.69 ± 0.16 | 34.22 ± 0.33 |
| Standard | Acarbose | 16.06 ± 0.05 | 16.65 ± 0.07 |

 | ||
| Fig. 2 α-Glucosidase inhibitory potential of compounds 4–15. | ||
Structure–activity relationship (SAR)
To improve understanding, the structure–activity relationship (SAR) was precisely examined by thoroughly assessing the influence of differences in the nature and position of the attached substituents (R) on the benzene ring. These investigations aimed to recognize the main molecular features responsible for biological activities, providing valuable insights into the way precise structural alterations affect the contact of the synthetic compounds with their biological targets. By associating these alterations with the inhibitory potential, the study provides a deeper understanding of the molecular factors crucial for enhancing bioactivity and managing upcoming drug design determinations.The synthesized bis-Schiff base compounds of ibuprofen were screened for their in vitro α-amylase and α-glucosidase inhibitory activities. Amongst the series, compound 13 (IC50 = 3.85 ± 0.05 µM for α-amylase and 4.36 ± 0.12 µM for α-glucosidase) shows powerful inhibitory activity, and the highest activity of this compound could be due to the presence of three electron-donating methoxy groups at meta and para positions. The presence of three methoxy groups on the benzene ring at meta and para positions significantly enhances the electron density of the aromatic ring, enabling powerful hydrogen bonding and π–π interactions with the binding pockets of amino acid residues in the enzymes. According to the comparison of compound 12 (IC50 = 5.51 ± 0.04 µM for α-amylase and 6.11 ± 0.01 µM for α-glucosidase) with 14 (IC50 = 6.20 ± 0.18 µM for α-amylase and 8.12 ± 0.14 µM for α-glucosidase), both derivatives have methoxy groups, but the attachment positions are different. Compared to that of compound 14, the slightly high activity of compound 12 might be attributed to the location of the methoxy substituents at the para and meta positions, instead of the para and ortho positions in compound 14. The positional alteration affects the steric interactions and electronic distributions with the para and meta configurations in compound 12, likely forming superior arrangements and powerful interactions with the active site of enzymes. The condensed steric hindrance and electronic effects in this configuration might ease optimal binding interfaces, leading to the greater activity detected for compound 12. According to the comparison of compound 10 (IC50 = 8.66 ± 0.01 µM for α-amylase and 9.51 ± 0.09 µM for α-glucosidase) with compound 9 (IC50 = 12.17 ± 0.14 µM for α-amylase and 16.60 ± 0.12 µM for α-glucosidase) and compound 11 (IC50 = 14.47 ± 0.14 µM for α-amylase and 16.18 ± 0.12 µM for α-glucosidase), the differences observed in the activities of the compounds might be ascribed to the variations in the potential arrangement of methoxy and hydroxyl groups on the benzene ring. The enhanced inhibitory activity of compound 10 is likely due to the presence of a hydroxyl substituent at ortho and methoxy at para position that increases the steric as well as electronic interfaces with the active site of the enzymes. By shifting the methoxy group from the para to meta position in compound 9, a slight decrease in the activity is observed, and this might be due to weaker hydrogen bonding and less promising electronic effects. However, by changing the position of the hydroxyl substituent from the ortho to meta position in compound 11, a greater decline in the activity is observed. This might be because the hydroxyl group at the ortho position in compound 10 allows powerful intramolecular interfaces and excellent alignment with the binding pockets of the enzyme that are disturbed by changing the position of the hydroxyl group to the meta position in compound 11. These results highlight the crucial role of the attached groups and their positions in controlling the inhibitory activities (Fig. 3). In the case of compounds having electron-withdrawing groups, the activity against α-glucosidase and α-amylase seems to be influenced by the attachment positions of the substituents on the benzene ring. According to the comparison of compound 4 (IC50 = 89.29 ± 1.44 µM for α-amylase and 91.52 ± 1.45 µM for α-glucosidase) with 5 (IC50 = 92.43 ± 0.69 µM for α-amylase and 96.71 ± 1.92 µM for α-glucosidase), both display comparatively lower inhibition. However, the somewhat high activity of compound 4 might be due to the nitro group attached to the benzene ring at the para position. The para position enables improved electron withdrawal by means of resonance, which can increase the interaction of compounds with the active site of the enzymes. In contrast, the nitro group at the meta position of the benzene ring in compound 5 restricts its capability to contribute to resonance, which results in reduced inhibition and weaker electron-withdrawing effects. This shows the significance of the positions of attached substituents in enhancing the binding interactions and electronic environment in compounds having similar structural frameworks.
 | ||
| Fig. 3 The most active α-amylase and α-glucosidase inhibitors in the series. | ||
Cytotoxicity of compounds 13 and 12
The cytotoxic effects of the most active compounds (13 and 12) were evaluated on 3T3 cells by means of the MTT assay. As shown in Fig. 4A and B, neither compound displayed cytotoxic effects within the established concentration range from 0 to 1000 µg mL−1. Cell viability reliably surpassed 90% at all concentrations for both compounds, emphasizing their promising safety profile. These results indicate that the compounds are well-tolerated by 3T3 cells and do not have harmful effects on cellular viability, implying their potential for more biological and therapeutic investigations. | ||
| Fig. 4 Cytotoxicity of the most active compounds against the 3T3 cell line: (A) compound 13. (B) Compound 12. | ||
Molecular docking studies of the compounds against α-amylase
The molecular docking interactions of the most active ibuprofen-based bis-Schiff base derivatives (9–14) with human pancreatic α-amylase (PDB ID: 3BAJ30), a key enzyme in carbohydrate metabolism and a target for anti-diabetic drugs, have been studied. Utilizing docking scores (ΔG), electrostatic (Eele) and solvation (Esol) energies, and solvent-accessible surface areas (SASAs), the binding modes of these active compounds are elucidated and compared with the reference inhibitor, acarbose. Then, we correlate these computational parameters with inhibitory activities in vitro, thereby validating the docking protocol and providing insights into the structure–activity relationships (SARs). Specific interactions, including H-bonding, hydrophobic contacts, and π–π stacking, are examined to understand how substituent variations on the terminal phenyl ring influence binding affinity and biological efficacy (Table 2). In addition, acarbose docks into α-amylase (3BAJ) with ΔG = −5.868 kcal mol−1 and RMSD = 1.323 Å (relative to the crystallographic pose), validating our methodology.| C. no. | ΔG (kcal mol−1) | RMSD (Å) | Eele (kcal mol−1) | Esol (kcal mol−1) | SASA (Å2) | ΔG (kcal mol−1) | RMSD (Å) | Eele (kcal mol−1) | Esol (kcal mol−1) | SASA (Å2) |
|---|---|---|---|---|---|---|---|---|---|---|
| 3BAJ | 5NN8 | |||||||||
| 13 | −7.033 | 1.415 | 60.106 | −49.617 | 10.489 | −6.964 | 1.056 | −46.724 | −39.265 | −7.459 |
| 12 | −6.643 | 1.751 | 63.389 | −32.880 | 30.509 | −5.922 | 1.586 | −7.550 | −36.240 | 28.690 |
| 14 | −7.186 | 7.307 | 39.420 | −33.145 | 6.275 | −6.337 | 1.050 | −1.337 | −26.789 | 25.452 |
| 10 | −7.187 | 1.766 | 53.123 | −44.610 | 8.512 | −6.520 | 1.353 | −92.104 | −32.797 | −59.307 |
| 9 | −6.529 | 1.027 | 42.668 | −22.744 | 19.923 | −6.113 | 1.532 | 100.256 | −34.112 | 134.368 |
| 10 | −6.140 | 1.264 | 48.495 | −30.503 | 17.992 | −6.475 | 1.808 | −38.313 | −44.533 | 6.219 |
| 11 | −6.197 | 1.528 | 240.521 | −170.571 | 69.950 | −5.892 | 1.647 | −29.733 | −53.146 | 42.207 |
| Acarbose | −5.868 | 1.323 | 240.521 | −39.822 | 15.520 | −5.525 | 1.097 | −55.490 | −59.914 | 4.424 |
Generally, a good correlation is observed between the docking scores (ΔG) and the in vitro α-amylase inhibitory activities (IC50 values). Compound 13 (IC50 = 3.85 µM) exhibits a strong docking score of −7.033 kcal mol−1. Compounds 14 (IC50 = 6.20 µM) and 10 (IC50 = 8.66 µM) show the most favourable docking scores (−7.186 and −7.187 kcal mol−1, respectively), suggesting potent binding, which is largely consistent with their excellent in vitro activities. Compounds 12 (IC50 = 5.51 µM, ΔG = −6.643 kcal mol−1) and 9 (IC50 = 12.17 µM, ΔG = −6.529 kcal mol−1) also show good scores, reflective of their activity. In the series, compound 11 (IC50 = 14.47 µM) has the least favourable score among this active set (ΔG = −6.197 kcal mol−1) but still shows better in vitro activity than acarbose. The favourable (more negative) solvation energies (Esol) for most compounds, particularly 13 (−49.617 kcal mol−1) and 10 (−44.610 kcal mol−1), suggest favourable interactions with the solvent-exposed regions of the binding pocket or good desolvation upon binding. Compound 11 presents anomalous Eele (240.522 kcal mol−1) and Esol (−170.571 kcal mol−1) values, alongside a high SASA (69.950 Å2). This might indicate a unique binding mode with substantial electrostatic contributions and significant desolvation penalties or solvent interactions, or potentially an artifact of the scoring. The high SASA implies greater solvent exposure. Fig. 5 reveals key residues within the 3BAJ active site.
 | ||
| Fig. 5 Binding mode of the most active compounds onto the active site of 3BAJ. | ||
The most active compound, 13 (3,4,5-trimethoxyphenyl) with IC50 = 3.85 µM, is stabilized by H-bonds, likely involving its terminal 3,4,5-trimethoxyphenyl ring with Ser108. The phenyl rings of the ibuprofen scaffold is oriented toward the solvent-exposed region, providing lipophilic bulk and facilitating potential hydrophobic interactions with Tyr62, while the central hydrazone linker properly positions the scaffold to interact with the Gln63 backbone. Furthermore, the central phenyl ring extends toward Trp59, forming the primary anchor. The presence of three electron-donating methoxy groups at meta and para positions enhances electron density, favouring polar and van der Waals interactions. Its relatively low SASA (10.489 Å2) suggests that it is well-accommodated within the pocket. Compound 12 (3,4-dimethoxyphenyl), with good activity (IC50 = 5.51 µM), forms H-bonds, likely via its methoxy groups and hydrazide (nitrogens/oxygens), with Glu240 and Lys200. The core structure and terminal ibuprofen phenyl ring are involved in hydrophobic interactions between Trp58 and Trp59 and potentially hydrophobic interactions between His201 and Ile235. The para and meta methoxy groups contribute favourably. Its SASA (30.509 Å2) is higher than that of compound 13, suggesting slightly more solvent exposure. Compound 14 (2,4-dimethoxyphenyl) displays a very good docking score (ΔG = −7.186 kcal mol−1) and activity (IC50 = 6.20 µM). The aromatic systems can engage in π–π stacking with Tyr151 and hydrophobic interactions with Glu149 and Tyr62. The ortho and para methoxy substituents facilitate these interactions. Its low SASA (6.275 Å2) implies a buried pose. Our SAR analysis suggests that the para/meta position is slightly better than the para/ortho position, which aligns with the result that the IC50 value of 12 is slightly better than that of 14, even though 14 has a better docking score. Also, compound 10 (2-hydroxy-4-methoxyphenyl), exhibiting a strong docking score (ΔG = −7.187 kcal mol−1) and activity (IC50 = 8.66 µM), likely forms H-bonds through the oxygen of amide with Lys200, Ala198 and His201. The hydroxyl group at the ortho position and the methoxy at the para position are crucial. π–π stacking with Trp59 and hydrophobic interactions with Gln63 further stabilize its binding. Its low SASA (8.512 Å2) indicates good burial. Compound 9 (2-hydroxy-5-methoxyphenyl) with moderate activity (IC50 = 12.17 µM) interacts with Tyr151, His201, and Lys200 through hydrogen bonds, likely involving its –OH and –OCH3 groups. A potential H-bond with Glu238 is also observed. The shift of the methoxy group from the para (as in compound 10) to meta (relative to the attachment point; here, position 5) position might slightly alter the optimal interaction geometry, as reflected in its higher IC50 value. Compound 11 (3-hydroxy-4-methoxyphenyl) (IC50 = 14.47 µM) forms hydrogen bonds with His201, Lys200, and Glu240 utilizing its amide functionalities. Hydrophobic interactions with Leu165, Leu162, and Trp59 contribute to binding. Acarbose typically binds deep within the α-amylase active site, forming an extensive network of hydrogen bonds with key catalytic residues (Asp197, Glu233, and Asp300) and other polar residues like Gln63, His101, Lys200, His201, His305, and Tyr151. Its inhibitory mechanism involves mimicking the substrate and interacting strongly with the catalytic triad.
Compared to acarbose, the synthesized bis-Schiff bases are structurally distinct and likely adopt the same binding orientations that share interactions with some common residues. Compounds 12, 10, 9, and 11 interact with Lys200 and His201, which are also crucial for acarbose binding, and Gln63 (interacts with 13, 14, and 10) and Trp59 (interacts with 13, 12, 10, and 11) are also implicated in substrate (acarbose) recognition. Glu240 (interacts with 12 and 11) is near the catalytic Glu233. The ibuprofen derivatives primarily rely on a combination of hydrophobic interactions (driven by the isobutylphenyl and other phenyl rings) and specific H-bonds from their hydrazide linkers and terminal substituents. Acarbose, with its multiple –OH groups, forms in a far more extensive H-bond network. The investigated compounds are generally more hydrophobic than acarbose. Generally, the nature and regiochemistry of substituents on the terminal phenyl ring play a pivotal role in modulating inhibitory potency. Generally, electron-donating methoxy and hydroxyl groups enhance activity, with trimethoxy (13) and specific hydroxy–methoxy patterns (2-OH and 4-OCH3 in 10) proving particularly effective. These active compounds demonstrate superior in vitro potency to acarbose, suggesting that they achieve strong enzyme inhibition through a distinct, yet effective, binding mode. The comprehensive framework established by correlating computational binding energies and interaction patterns with biological efficacy offers a robust platform for the rational design and optimization of this class of compounds as novel anti-diabetic agents targeting α-amylase.
Molecular docking of the compounds against α-glucosidase
Molecular docking studies were performed for compounds 9–14 against human lysosomal acid α-glucosidase (PDB ID: 5NN831), a critical enzyme in glycogen metabolism and a target for Pompe disease, with homologous enzymes targeted in diabetes. We aim to elucidate the structural basis of their inhibitory activity by dissecting binding modes, interaction energies (ΔG, Eele, Esol, SASA), and comparing them with the established inhibitor acarbose with ΔG = −5.525 kcal mol−1 (Table 2). Fig. 6 reveals key residues within the 5NN8 active site. Compound 13 (3,4,5-trimethoxyphenyl derivative; ΔG = −6.964 kcal mol−1) forms network π–π stacking interactions with Trp618 and trimethoxyphenyl. Similarly, the terminal phenyl of ibuprofen engages the right-arm aromatic system of compound 13 through π–π stacking or π–alkyl interactions with Trp481 and Met519, respectively. The multiple methoxy groups provide several H-bond acceptor sites. Its highest ΔG score is supported by these multiple interactions. The negative Eele (−46.724 kcal mol−1) suggests favourable electrostatic contributions. For compound 10 (2-hydroxy-4-methoxyphenyl; ΔG = −6.475 kcal mol−1), key H-bonds are observed with Asp518, Trp481, and potentially Ala284 and Asp404. The ortho-hydroxyl and para-methoxy groups are critical for π–π stacking with Trp481. The strong ΔG is well-supported. Favourable Esol (−44.533 kcal mol−1) and Eele (−38.313 kcal mol−1) contribute to its good binding. Compound 14 (2,4-dimethoxyphenyl; ΔG = −6.337 kcal mol−1) forms H-bonds, likely via the carbonyl of amide, with Thr286, and its methoxy groups are involved in hydrophobic interactions with the aromatic residue Trp618, while the phenyl of ibuprofen forms π–π stacking interactions with Trp481. The favorable ΔG together with Eele (−1.337 kcal mol−1) is close to neutral, suggesting hydrophobic and van der Waals interactions are dominant for this pose. | ||
| Fig. 6 Binding mode of the most active compounds onto the active site of 5NN8. | ||
Compound 9 (2-hydroxy-5-methoxyphenyl; ΔG = −6.113 kcal mol−1) engages in important H-bond interactions with His674, Asp616, Thr286, Trp376 and Ala284. Hydrophobic interactions with Leu650 contribute. The moderate ΔG together with a high Eele (100.256 kcal mol−1) suggests significant electrostatic repulsion or an unfavourable charge state in this pose, which might explain its weaker activity despite a seemingly extensive H-bond network. Its very large SASA (134.368 Å2) indicates that it is not well-buried. Compound 12 (3,4-dimethoxyphenyl; ΔG = −5.922 kcal mol−1) forms H-bonds with Met519, Trp481, Asn652, Gly651 and Leu650. Hydrophobic interactions with Leu678 are also present. Its ΔG is less favourable than that of compound 9, even though compound 12 is more active. This could be due to the highly unfavourable Eele of compound 9, whereas compound 12 has a more neutral Eele (−7.550 kcal mol−1). The 3-hydroxy-4-methoxyphenyl substituents in compound 11 (ΔG = −5.525 kcal mol−1) establishes H-bonds with Asp518, Asp282, and Arg527 (common in glucosidase active sites). Hydrophobic interactions with Phe649 are noted. The least favourable ΔG, consistent with its weaker activity. However, it has very favourable Esol (−59.914 kcal mol−1) and Eele (−55.490 kcal mol−1), suggesting strong polar and solvent-mediated interactions, which might partially compensate for a less-optimal van der Waals fit. Acarbose, a well known inhibitor of α-glucosidases. It typically binds in the active site cleft, forming numerous H-bonds with key catalytic residues (Asp and Glu residues acting as nucleophiles and acid/base catalysts) and other polar residues that recognize sugar moieties. It mimics the natural substrate.
Chemical reactivity indices of ibuprofen-based bis-Schiff bases
The chemical reactivity of the most active ibuprofen-based bis-Schiff base derivatives (9, 10, 11, 12, 13, and 14) has been studied using time-dependent density functional theory (TD-DFT) calculations (wB97X-D/6-311G**). Key electronic properties, including HOMO and LUMO energies, are used to derive global reactivity indices such as energy gap (ε), hardness (η), softness (S), electronegativity (χ), and electrophilicity index (ω). These quantum chemical descriptors are correlated with their in vitro inhibitory activities against α-glucosidase and α-amylase, as well as with molecular docking results. The study aims to elucidate how the electronic characteristics of these compounds influence their biological efficacy and interaction with the target enzymes, providing a comprehensive understanding of their structure–activity relationships from both structural and electronic perspectives. The TD-DFT-calculated reactivity indices for compounds 9–14 are presented in Table 3.| C. no. | HOMO | LUMO | ε | η | S | χ | ω |
|---|---|---|---|---|---|---|---|
| 11 | −7.820 | −0.070 | 2.232 | 3.875 | 0.258 | 3.945 | 2.008 |
| 9 | −7.891 | −0.048 | 1.672 | 3.922 | 0.255 | 3.970 | 2.009 |
| 10 | −7.872 | 0.101 | 1.430 | 3.986 | 0.251 | 3.885 | 1.894 |
| 14 | −7.880 | 0.113 | 2.152 | 3.996 | 0.250 | 3.883 | 1.887 |
| 12 | −7.741 | −0.023 | 1.627 | 3.859 | 0.259 | 3.882 | 1.953 |
| 13 | −7.976 | −0.130 | 1.840 | 3.923 | 0.255 | 4.053 | 2.094 |
| Acarbose | −4.942 | −0.934 | 4.008 | 2.004 | 0.499 | 2.938 | 2.153 |
Compound 13, being the most active, exhibits the lowest HOMO and LUMO energies, the highest electronegativity (χ), and the highest electrophilicity index (ω). This suggests that its ability to participate in charge transfer interactions, particularly acting as an electron acceptor (good electrophile), might be a key factor in its superior activity. Compound 12, while having the highest HOMO (less likely to donate electrons), has the lowest hardness (η) and the highest softness (S), indicating that it might be more polarizable and reactive. A smaller energy gap, ε, generally implies higher reactivity. The very active compound 10 has the smallest ε (ε = 1.430). Compound 13 (ε = 1.840, most active) has a moderate ε. Compound 11 (ε = 2.232, less active) has the highest ε. This suggests that lower ε values (indicating easier electronic excitation and higher reactivity) might be beneficial. Hardness is a measure of resistance to deformation or change in electron distribution, while softness is the reciprocal. Softer molecules are generally more reactive. The softest compound, 12 (η = 3.859, S = 0.259), shows good activity. However, the hardest compound, 14 (η = 3.996, S = 0.250), is also quite active. The trend in hardness/softness is not perfectly linear with activity across the whole set but might explain differences between closely related compounds. A higher electronegativity (χ) indicates a greater power to attract electrons. Compound 13 (χ = 4.053, most active) has the highest electronegativity. Compound 9 (χ = 3.970, less active for α-glucosidase) also has high electronegativity. This suggests that high electronegativity is a favourable trait. The electrophilicity index (ω) measures the tendency of a species to accept electrons. A higher ω indicates a better electrophile. The most active compound, 13, for both enzymes has the highest electrophilicity (ω = 2.094). Compounds 11 (ω = 2.008) and 9 (ω = 2.009) also have high ω but are less active. Compounds 10 (ω = 1.894) and 14 (ω = 1.887) have the lowest ω in the series but are very active. The trend in ω is not perfectly linear. While the most active compound (13) has the highest ω, other highly active compounds (10 and 14) have lower ω values. This suggests that while electrophilicity is important, it is not the sole determinant, and an optimal range or interplay with other factors might exist. The ibuprofen derivatives likely rely more on a balance of specific polar interactions (via R-groups and linkers) and broader hydrophobic/π–π interactions, with their electrophilic/nucleophilic character (as described by ω and HOMO/LUMO) playing a role in forming covalent or strong non-covalent charge-transfer interactions if the opportunity arises. The docking studies show that active compounds form hydrogen bonds (polar interactions) and hydrophobic/π–π interactions. The high ω of compound 13 or a low LUMO energy suggests that the compound can readily accept electron density. This could be crucial for interacting with electron-rich (nucleophilic) residues at the enzyme active site (carboxylate side chains of Asp/Glu or lone pairs on His, Ser, or Tyr). If the docking pose places the ligand near such residues, a favourable electrophile–nucleophile interaction could significantly contribute to the binding affinity (ΔG) and, thus, the activity (IC50). The low HOMO energy of compound 13 indicates that while it can donate electrons, it holds them relatively tightly. However, interactions with an electron-deficient (electrophilic) site in the enzyme could still occur. The ε relates to polarizability. Molecules with smaller ε are generally more polarizable and can adapt their electron clouds more easily to interact favourably with the receptor site, potentially leading to stronger induced-fit interactions and better docking scores. Compounds 10 and 13 have relatively smaller ε values and are highly active.
Fig. 7 provides visual representations of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) for a series of active ibuprofen-based bis-Schiff base derivatives (9–14) and the standard inhibitor acarbose. These frontier molecular orbitals (FMOs) are critical in determining a molecule's chemical reactivity, participation in charge-transfer interactions, and, ultimately, biological activity. By examining the spatial distribution of HOMO and LUMO electron densities, we aim to identify regions susceptible to electrophilic and nucleophilic attack, respectively. These insights are correlated with previously discussed DFT-derived reactivity indices, in vitro inhibitory activities against α-glucosidase and α-amylase, and molecular docking results to provide a comprehensive understanding of how electronic delocalization and specific atomic contributions to FMOs influence enzyme–ligand interactions and inhibitory potency. Visualizing the spatial distribution of these FMOs provides invaluable qualitative information on which parts of a molecule are most involved in these electronic processes (Fig. 7). This analysis examines the HOMO and LUMO electron density plots of compounds 9–14 and acarbose, aiming to identify the primary sites of electron donation and acceptance, correlate these distributions with the calculated global reactivity indices (ε, η, ω, etc.), link these electronic features to the specific interactions observed in molecular docking studies with α-glucosidase and α-amylase, and understand how these FMO characteristics contribute to the observed inhibitory activities.
 | ||
| Fig. 7 Visualization of the HOMO and LUMO distributions for ibuprofen-based bis-Schiff bases 9–14. | ||
The HOMO electron density appears to be largely delocalized across the π-conjugated systems of the molecules, particularly involving the central phenoxy acetyl-hydrazone linker and the terminal substituted phenyl ring (the R-group). The ibuprofen's isobutylphenyl moiety also contributes, but often to a lesser extent than the more electron-rich and conjugated central/terminal parts. This suggests that the primary sites for electron donation or electrophilic attack are spread across these conjugated segments. The LUMO density also tends to be delocalized over similar π-conjugated regions, often with significant contributions from the imine bonds (–NCH–) of the hydrazone moieties and the aromatic rings. This indicates that these are the region's are most likely to accept electrons or undergo nucleophilic attack. On the most active compounds, 12 and 13, significant HOMO density is observed on the terminal 3,4,5-/2,4-methoxyphenyl rings and the adjacent hydrazone linker. The methoxy groups, being electron-donating, likely contribute to the high electron density in this region, making them a good electron donor. While the LUMO is strongly localized on the terminal methoxyphenyl rings and the imine bonds. This supports its high electrophilicity index (ω) and its role as a good electron acceptor. The extensive delocalization across this highly substituted ring likely stabilizes the accepted electron. In compound 14 (2,4-dimethoxyphenyl), both FMOs appear delocalized over the entire conjugated system, with significant density on the terminal 2,4-dimethoxyphenyl ring and the imine bonds. The ortho and para methoxy groups influence this distribution. In the HOMO of compound 9 (2-hydroxy-4-methoxyphenyl), significant density is observed on the terminal 2-hydroxy-4-methoxyphenyl ring, with contributions from both the hydroxyl and methoxy groups and extending into the hydrazone linker. The LUMO is also concentrated on the terminal ring system and imine bonds. The presence of the phenolic –OH and methoxy influences the electron distribution. The HOMO and LUMO of compound 9 are similar to those of 10, and FMOs are primarily localized on the terminal substituted phenyl ring and the conjugated linker. The different positions of the methoxy group (5-position vs. 4-position in 10) could subtly alter the precise regions of the highest density and orbital energies. The HOMO and LUMO of compound 11 are delocalized across the terminal ring and conjugated system. The 3-OH and 4-OCH3 patterns dictate the specific regions of electron density. The HOMO density in acarbose appears to be primarily localized on the oxygen atoms of the hydroxyl groups and the glycosidic linkages, as well as on the nitrogen atom of the valienamine-like ring. These are the most electron-rich and nucleophilic sites. The LUMO density is also distributed across the molecule, likely with contributions from regions around the –CN– character of the imine-like moiety, which can stabilize an incoming electron, potentially near the anomeric carbons or the nitrogen atom. Its LUMO is likely higher in energy than that of the more conjugated ibuprofen derivatives. Generally, the LUMO of ligands 9–14 (on the terminal phenyl ring or imine bonds of compounds) overlaps spatially with the HOMO of an electron-rich (nucleophilic) residue in the enzyme's active site (Asp, Glu, His, Ser, or Tyr), and a favourable charge-transfer interaction from the enzyme to the ligand can occur, strengthening binding. This is consistent with the high electrophilicity (ω) of compounds like 13. The docking poses show interactions with such residues. Conversely, if the HOMO of the ligand (on the electron-rich terminal rings) overlaps with the LUMO of an electron-deficient (electrophilic) residue or region in the active site, charge transfer from the ligand to the enzyme can occur. The localization of HOMO density on oxygen and nitrogen atoms (in methoxy, hydroxyl, and hydrazide groups) identifies these as potential H-bond acceptor sites. Similarly, regions of LUMO density near acidic protons could indicate H-bond donor interactions. The FMOs help identify the most electronically active H-bonding atoms. In addition, the delocalization of the HOMO and LUMO across the aromatic rings in compounds 9–14 is fundamental to their ability to engage in π–π stacking interactions with aromatic residues (Trp, Tyr, Phe, and His) in the enzyme active sites, as observed in the docking studies.
Insights into molecular electrostatic potential (MEP) maps
The molecular electrostatic potential (MEP) is a fundamental property that describes the electrostatic interaction energy between a molecule and a positive point charge (a proton) at various points in space around the molecule. It provides a three-dimensional map of the charge distribution, visually identifying regions with high electron density (negative MEP, typically coloured red/orange/yellow) and low electron density (positive MEP, typically coloured blue/purple). Regions with negative MEPs are indicative of lone pairs on heteroatoms or π-electron clouds and are favourable sites for electrophilic attack or hydrogen bond acceptance. Regions with positive MEPs are usually found around hydrogen atoms bonded to electronegative atoms (e.g., O–H and N–H) and are favourable sites for nucleophilic attack or hydrogen bond donation. Understanding the MEP is crucial in drug design as it directly relates to how a ligand interacts with the electrostatic field of a biological receptor, guiding the formation of hydrogen bonds, salt bridges, and other electrostatic interactions that are vital for molecular recognition and binding affinity.4 Negative MEPs (red/orange/yellow regions) are consistently observed around the oxygen atoms of the carbonyl groups (CO) in the ibuprofen moiety and the ester/amide linkages, the oxygen atoms of the –OCH3 and –OH substituents on the terminal phenyl rings, and the nitrogen atoms of the imine (–NCH–) and hydrazide (–NH–N) linkages, particularly if they possess lone pairs not involved in extensive conjugation. These regions are prime candidates for acting as hydrogen bond acceptors or interacting with positively charged/electrophilic sites in the enzyme.
Positive MEPs (blue/purple regions) are typically located around the hydrogen atoms of hydroxyl (–OH) groups on the terminal phenyl rings and the H-atoms of the N–H groups in the hydrazide linkages. These regions can act as H-bond donors or interact with negatively charged/nucleophilic sites in the enzyme. Neutral MEPs (green regions) are predominantly seen over the carbon skeletons of the aromatic rings (phenyl and isobutylphenyl) and aliphatic chains, indicating relatively non-polar, hydrophobic regions. Compounds 12 and 13 show intense negative MEPs around the oxygen atoms of the methoxy groups on the terminal phenyl ring and the carbonyl oxygen of ibuprofen. This highlights their strong hydrogen bond accepting capabilities in these regions. The –NH hydrogens of the hydrazide linkers likely exhibit positive MEPs. The overall molecule has a complex electrostatic landscape, with distinct polar ends (terminal ring and ibuprofen carbonyl) and a more neutral central linker. Compound 14 displays negative MEPs localized on the ortho and para methoxy oxygens. The spatial arrangement of these negative potentials will differ from that in the 3,4-disubstituted 12, potentially influencing the directionality of H-bonds (Fig. 8).
 | ||
| Fig. 8 The MEP for ibuprofen-based bis-Schiff bases 9–14. | ||
For compounds 9 and 10, there is a strong negative MEP around the phenolic oxygen of the –OH group (2-hydroxy-4-methoxyphenyl) and the methoxy oxygen. Significant positive MEPs are observed around the hydrogen of the phenolic –OH group, making it an excellent H-bond donor. This combination of strong H-bond acceptor (oxygens) and donor (hydroxyl H) sites in proximity on the terminal ring is a key feature. Compound 11 shows negative MEPs on the hydroxyl and methoxy oxygens and positive MEPs on the hydroxyl hydrogen. The 3-hydroxy position will present its donor/acceptor capabilities differently than the 2-hydroxy position in 9 and 10. The MEP map of acarbose is dominated by negative MEPs around the numerous oxygen atoms of its multiple hydroxyl groups and glycosidic linkages. This makes acarbose an exceptionally strong hydrogen bond acceptor at many points. The MEP maps directly visualize the H-bond donor (positive MEP) and acceptor (negative MEP) capabilities.
The presence of strongly negative MEP regions on the oxygen atoms of R-group substituents (methoxy and hydroxyl) and linker carbonyls aligns perfectly with the hydrogen bonds observed in docking studies, where these groups act as acceptors with enzyme residues (e.g., Gln, Ser, Tyr, Lys, His, Asp, and Glu). Similarly, positive MEPs on the hydrogens of –OH and –NH match their role as H-bond donors during docking. The overall electrostatic field of the ligand, as depicted by the MEP, can “steer” it into the correct orientation within the enzyme's binding pocket, which itself has a characteristic electrostatic landscape. The overall magnitude and distribution of the MEP are influenced by electronegativity (χ) and electrophilicity (ω). For instance, compound 13, with high χ and ω, shows an intense negative potential around its electronegative oxygen atoms, reflecting its ability to attract and stabilize electron density.
Analytical insights into molecular dynamics (MD) of α-amylase (3BAJ) and α-glucosidase (5NN8)
The intrinsic dynamics have been investigated for α-amylase (PDB ID: 3BAJ) and α-glucosidase (PDB ID: 5NN8) using normal mode analysis (NMA). NMA elucidates the collective motions accessible to these enzymes, providing insights into their flexibility, conformational transitions, and regions susceptible to deformation. Key NMA outputs, including molecular motion visualizations, deformability profiles, β-factor correlations, eigenvalue spectra, variance plots, and covariance matrices, are examined (Fig. 9). These dynamic properties are discussed in the context of ligand binding mechanisms for ibuprofen derivative 13, active site accessibility, and the overall enzyme function. Understanding the inherent flexibility of these enzymes can aid in rationalizing inhibitor binding, identifying allosteric sites, and guiding the design of modulators that leverage protein dynamics. The MD analysis focuses on the NMA results for two key metabolic enzymes: human pancreatic α-amylase (3BAJ) and human lysosomal acid α-glucosidase (5NN8). By examining their dynamic profiles, we identify the flexible and rigid regions within each enzyme, compare their overall dynamic characteristics, relate these dynamics to active site accessibility and potential mechanisms for ligand binding, and understand how intrinsic protein motions influence their catalytic activity and inhibition. | ||
| Fig. 9 Multiple perspectives on the dynamics of the complex, including (a) overall mobility, (b) residue-level deformability, (c) correlation with experimental β-factors, (d) eigenvalue distribution, (e) variance contribution of modes, (f) inter-residue motion correlation, and the (g) underlying elastic network model. | ||
The ribbon diagrams for 13-3BAJ and 13-5NN8, often coloured based on mobility (red/orange for high mobility and blue for low mobility), depict the collective motion described by the lowest-frequency (largest-amplitude) normal modes. The arrows indicate the direction and extent of these global movements (Fig. 9a). These visualizations highlight large-scale conformational changes the enzyme can readily undergo. For instance, they show domain movements, opening/closing of active site clefts, or hinge-bending motions. These are crucial for substrate access, ligand binding, and product release. Observing the nature of this motion provides direct insights into the enzyme's functional dynamics. The motion involves the opening of the active site, and it suggests a mechanism for ligand entry. The global motions (Fig. 9a) reveal how the overall shape of the binding pocket changes, which could affect the binding of elongated ligands like the ibuprofen derivative 13.
The deformability profile (Fig. 9b) shows the normalized displacement (deformability or mobility) of each atom/residue when all modes are considered (or a subset of low-frequency modes). Peaks indicate highly flexible regions, while valleys represent more rigid parts of the protein. The flexible regions correspond to loops; these can be involved in induced-fit mechanisms upon compound 13 binding, which play roles in protein–protein interactions. Rigid regions usually form the structural core or parts of the active site essential for maintaining catalytic geometry. High deformability near the active site (Gln, Ser, Tyr, Lys, His, Asp, and Glu) facilitates ligand entry or conformational changes upon binding. For instance, loops around the active site often show high deformability. The inherent flexibility of MD supports an induced-fit binding mechanism. If they are rigid, the ligand must conform to a pre-existing pocket. The docked ligand fits well into one of the low-energy conformations predicted by NMA, which suggests a conformational selection mechanism, where the ligand binds to a pre-existing favourable state. The significant conformational changes required for binding are represented by higher-energy modes or involve large deformations of specific flexible loops, which points towards an induced-fit mechanism.
The β-factor correlation (Fig. 9c) compares the theoretical β-factors (atomic displacement parameters) calculated from NMA (pink/red) with the experimental β-factors obtained from the PDB crystal structure (gray/black). A good correlation between NMA-derived β-factors and experimental β-factors validates the NMA model, indicating that the calculated collective motions are consistent with the observed atomic fluctuations in the crystal. Peaks in both plots represent regions of high flexibility. Discrepancies can arise from crystal packing effects in the experimental data or limitations of the NMA model (harmonic approximation or absence of a solvent).
The eigenvalue spectra of 13-3BAJ and 13-5NN8 complexes show the eigenvalues of the normal modes (Fig. 9d), typically for the first 20 or so low-frequency modes, often normalized to the first non-trivial mode. Eigenvalues are proportional to the square of the mode frequency; lower eigenvalues correspond to a lower frequency, a larger amplitude, and more collective motions. The distribution of eigenvalues indicates the stiffness of the proteins. A steep rise in eigenvalues suggests that only a few low-frequency modes dominate the large-scale dynamics, while higher-frequency modes contribute to more localized fluctuations. The specific eigenvalue for mode 1 (often the lowest non-trivial) quantifies the ease of the largest collective motion.
The variance plot (cumulative contribution of modes) bar chart in Fig. 9e shows the individual contribution of each of the first ∼20 normal modes to the total variance (mean-square fluctuation) of the protein (purple bars) and the cumulative variance (green bars). This plot demonstrates how many modes are needed to capture a significant portion of the protein's overall dynamics. Typically, a small number of low-frequency modes (the first 10–20) accounts for a large percentage (70–90%) of the total atomic fluctuations. This reinforces the idea that functionally important large-scale motions are often described by just a few collective modes.
The covariance matrix (dynamical cross-correlations) heatmap in Fig. 9f shows the correlation of motion between pairs of residues. Red indicates a positively correlated motion (residues moving in the same direction), blue indicates a negatively correlated (anti-correlated) motion (residues moving in opposite directions), and white indicates an uncorrelated motion. This matrix reveals how motions are coupled across the protein structure. Positive correlations (red blocks/regions) indicate domains or segments that move together as rigid bodies or in a concerted fashion. Negative correlations (blue blocks/regions) suggest hinge regions or parts of the protein that move in the opposite direction, such as the opening/closing of a cleft, where one side moves away as the other moves towards it. This is particularly relevant for active site dynamics. This information identifies the allosteric pathways, where motion in one part of the protein influences another distant part.
The distance fluctuation matrix (Fig. 9g) represents either the connections in an elastic network model (where gray points show residues within a certain cutoff distance) used to a distance fluctuation matrix (where darker gray indicates pairs of atoms whose distance fluctuates more significantly). The diagonal line represents self-correlation.
By comparing the deformability profiles (Fig. 9b), β-factor plots (Fig. 9c), and eigenvalue spectra (Fig. 9d), one can infer the relative overall flexibility. A protein with generally higher deformability peaks, higher β-factors, and lower initial eigenvalues might be considered more flexible. The specific motions, particularly those involving catalytic residues, are essential for the chemical steps of catalysis, including substrate positioning, transition state stabilization, and product release. NMA can help identify modes that contribute to these catalytic events.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values ranging from 5.78 (compound 9) to 6.59 (compound 14). These values are in contrast to the highly hydrophilic acarbose (logP = −2.88). Consequently, the predicted aqueous solubility (logS) is poor for all derivatives (≤−5.49) but excellent for acarbose (−0.733). This aligns with the well-established rule that successful oral drugs typically require a balance, and excessive lipophilicity (logP > 5) is often linked to poor solubility, promiscuous binding, and metabolic instability.32P ∼ 6.0–6.6). Introducing a hydroxyl group (9, 10, and 11) generally reduces enzyme inhibitory potency compared to their methoxy-only counterparts but has a profound impact on ADMET. Hydroxy-substituted compounds (9 and 10) show slightly lower logP (∼5.8) and logD (∼3.6) and significantly lower predicted total clearance (CL: ∼2.0 vs. ∼6.0) compared to the trimethoxy analog 13. This suggests that hydroxylation could be a metabolic detoxification pathway. Crucially, the hydroxyl group is associated with a dramatic increase in predicted carcinogenicity and the activation of stress response pathways (SR-HSE, SR-MMP, and SR-p53), likely introducing reactive or metabolically activated species.
P values ranging from 5.78 (compound 9) to 6.59 (compound 14). These values are in contrast to the highly hydrophilic acarbose (logP = −2.88). Consequently, the predicted aqueous solubility (logS) is poor for all derivatives (≤−5.49) but excellent for acarbose (−0.733). This aligns with the well-established rule that successful oral drugs typically require a balance, and excessive lipophilicity (logP > 5) is often linked to poor solubility, promiscuous binding, and metabolic instability.32P ∼ 6.0–6.6). Introducing a hydroxyl group (9, 10, and 11) generally reduces enzyme inhibitory potency compared to their methoxy-only counterparts but has a profound impact on ADMET. Hydroxy-substituted compounds (9 and 10) show slightly lower logP (∼5.8) and logD (∼3.6) and significantly lower predicted total clearance (CL: ∼2.0 vs. ∼6.0) compared to the trimethoxy analog 13. This suggests that hydroxylation could be a metabolic detoxification pathway. Crucially, the hydroxyl group is associated with a dramatic increase in predicted carcinogenicity and the activation of stress response pathways (SR-HSE, SR-MMP, and SR-p53), likely introducing reactive or metabolically activated species.A comparison of positional isomers (12 vs. 14; 9 vs. 11) shows that while activity differences are subtle, ADMET parameters can vary. For example, 12 (3,4-dimethoxy) has a slightly higher BBB permeant probability and a different CYP substrate profile than 14 (2,4-dimethoxy).
Experimental
General
The chemicals used in the current research work were of analytical grade and purchased from Sigma-Aldrich, TCI, Merck, BDH and Alfa Aesar. The thin-layer chromatographic (TLC) method was utilized on pre-coated aluminium silica gel plates to find out the progress of reactions in the solvent system of ethyl acetate and n-hexane. UV light (254 and 365 nm) was used for visualization of the spots. High-resolution electrospray ionization mass spectrometry (HR-ESI-MS) spectra were recorded on a Waters Quattro Premier XE spectrometer (USA). Moreover, the nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance spectrophotometer working at 150 MHz for 13C-NMR and 600 MHz for 1H-NMR.Synthesis of ibuprofen hydrazide (1)29
The acid of ibuprofen was primarily activated using the coupling reagent 1,1-carbonyldiimidazole (CDI) in a catalytic quantity of triethyl amine (Et3N) in tetrahydrofuran (THF). Consequently, hydrazine hydrate was added to the mixture, and it was refluxed for 3–4 hours while being constantly stirred. The reaction progress was thoroughly confirmed by the TLC method utilizing the solvent system of ethyl acetate and n-hexane (2:3). After the formation of products, the mixture was decanted into cold water; the appeared white precipitates were filtered, washed with cold water, dried in air, recrystallized through ethanol and collected.
Synthesis of the ibuprofen-based Schiff base (2)29
Ibuprofen hydrazide (1) was dissolved in ethanol in a 100 mL round-bottom (RB) flask, and ethyl 2-(4-formylphenoxy)acetate was added to the mixture containing a catalytic amount of acetic acid. The reaction mixture was refluxed for 4–5 hours, and the progress of the reaction was monitored by the TLC method. After completion, the mixture was poured into cold distilled water; the precipitates were formed, filtered, washed and collected. The compound (2) was further dissolved in ethanol, and hydrazine hydrate was added to the reaction mixture. The reaction was refluxed until the formation of Schiff base hydrazide (3). After the confirmation of product formation by TLC, the mixture was transferred to a beaker containing cold water; the appeared precipitates were filtered, washed, dried and collected.Synthesis of bis-Schiff bases (4–15)
The desired Schiff base hydrazide (3) was dissolved in ethanol with a catalytic amount of acetic acid, and different aromatic aldehydes were subsequently added to the mixture. The reaction mixture was stirred and refluxed for 3–4 hours, and TLC was utilized to determine the formation of the product compounds. At the end of the reaction, the mixtures were poured into cold distilled water in a beaker; the formed precipitates were filtered, washed, dried overnight and collected. These compounds were structurally confirmed using 1H-NMR, 13C-NMR and HR-ESI-MS techniques.29In vitro α-amylase and α-glucosidase inhibitory activities
Molecular modelling
logP, TPSA, and solubility) and bioavailability scores. pkCSM (https://biosig.unimelb.edu.au/pkcsm) models were employed to predict human intestinal absorption and other PK parameters. Input structures were prepared as SMILES strings from optimized DFT geometries.Conclusion
This work reports the anti-diabetic (α-amylase and α-glucosidase) activity of the synthesized bis-Schiff bases based on the ibuprofen scaffold. After structural confirmation, these compounds have been tested for their in vitro α-amylase and α-glucosidase inhibitory activities. In the series, six compounds (13, 12, 14, 10, 9 and 11) showed excellent dual inhibitory activities ranging from IC50 = 3.85 ± 0.05 to 14.47 ± 0.14 µM for α-amylase and 4.36 ± 0.12 to 16.18 ± 0.12 µM for α-glucosidase. Similarly, compound 15 showed a significant inhibitory effect, while the remaining compounds were found to be less active. Molecular docking and DFT investigations showed that the most active compounds (9–14) exhibited powerful inhibitory activity against the α-amylase enzyme, supported by a clear correlation amongst the IC50 values and docking scores. Their efficiency is due to the main hydrophobic, π–π interactions and hydrogen bonding with the active site of the enzymes. A robust association among docking scores and IC50 values confirmed the reliability of the computational model. The FMO study further confirmed that electron delocalization and reactive site distribution knowingly affect the binding affinity. These understandings deliver a solid foundation for the rational design of more potent enzyme inhibitors.Author contributions
Conceptualization: Aftab Alam and Manzoor Ahmad; methodology: Muhammad Ayaz and Sajjad Ur Rahman; biological activities: Imtiaz Ahmad; software: Ahmed A. Elhenawy; data curation: Abdul Latif, Zainab and Liaqat Ali; writing – original draft: Aftab Alam and Muhammad Ayaz; and writing – review and editing: Manzoor Ahmad and Mumtaz Ali. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Data availability
Data on the compounds are available in the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ra03358f.Acknowledgements
We are grateful to the Higher Education Commission of Pakistan for financial support under the NRPU research program (no. 14622).References
- P. Seboletswe, G. Kumar, N. Gcabashe, K. Olofinsan, S. Islam, A. Idris and P. Singh, Benzylidenehydrazine Derivatives: Synthesis, Antidiabetic Evaluation, Antioxidation, Mode of Inhibition, DFT and Molecular Docking Studies, Chem. Biodiversity, 2024, e202401556 Search PubMed.
- S. Gul, A. A. Elhenawy, Q. Ali, M. U. Rehman, A. Alam, M. Khan, A. F. AlAsmari and F. Alasmari, Discovering the anti-diabetic potential of thiosemicarbazone derivatives: In vitro α-glucosidase, α-amylase inhibitory activities with molecular docking and DFT investigations, J. Mol. Struct., 2024, 1312, 138671 CrossRef CAS.
- N. Kerru, A. Singh-Pillay, P. Awolade and P. Singh, Current anti-diabetic agents and their molecular targets: A review, Eur. J. Med. Chem., 2018, 152, 436–488 Search PubMed.
- L. M. Aroua, I. S. Alkhaibari, F. M. Alminderej, S. Messaoudi, S. Chigurupati, S. A. Al-mahmoud, A. E. Albadri, A.-H. Emwas and H. A. Mohammed, Synthesis, bioactivity, and molecular docking of pyrazole bearing Schiff-bases as prospective dual alpha-amylase and alpha-glucosidase inhibitors with antioxidant activity, J. Mol. Struct., 2025, 1320, 139291 CrossRef CAS.
- K. L. Ong, L. K. Stafford, S. A. McLaughlin, E. J. Boyko, S. E. Vollset, A. E. Smith, B. E. Dalton, J. Duprey, J. A. Cruz and H. Hagins, Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021, Lancet, 2023, 402(10397), 203–234 CrossRef PubMed.
- B. Zhou, A. W. Rayner, E. W. Gregg, K. E. Sheffer, R. M. Carrillo-Larco, J. E. Bennett, J. E. Shaw, C. J. Paciorek, R. K. Singleton and A. B. Pires, Worldwide trends in diabetes prevalence and treatment from 1990 to 2022: a pooled analysis of 1108 population-representative studies with 141 million participants, Lancet, 2024, 404(10467), 2077–2093 Search PubMed.
- S. Y. Begum, P. M. Imran, A. Kubaib, M. T. Yassin and K. Maniah, Targeted design and synthesis of phenyl-substituted isonicotinyl derivatives with a focus on antimicrobial, antioxidant and antidiabetic properties, J. Mol. Struct., 2025, 1320, 139571 CrossRef CAS.
- S. Agrawal, S. Samanta and S. K. Deshmukh, The antidiabetic potential of endophytic fungi: Future prospects as therapeutic agents, Biotechnol. Appl. Biochem., 2022, 69(3), 1159–1165 CrossRef CAS PubMed.
- B. Konrad, D. Anna, S. Marek, P. Marta, Z. Aleksandra and C. Józefa, The evaluation of dipeptidyl peptidase (DPP)-IV, α-glucosidase and angiotensin converting enzyme (ACE) inhibitory activities of whey proteins hydrolyzed with serine protease isolated from Asian pumpkin (Cucurbita ficifolia), Int. J. Pept. Res. Ther., 2014, 20(4), 483–491 CrossRef CAS PubMed.
- D. Kajaria, J. Tripathi, Y. B. Tripathi and S. Tiwari, In-vitro α amylase and glycosidase inhibitory effect of ethanolic extract of antiasthmatic drug—Shirishadi, J. Adv. Pharm. Technol. Res., 2013, 4(4), 206–209 CrossRef PubMed.
- E. Di Stefano, T. Oliviero and C. C. Udenigwe, Functional significance and structure–activity relationship of food-derived α-glucosidase inhibitors, Curr. Opin. Food Sci., 2018, 20, 7–12 CrossRef.
- R. Khator and V. Monga, Recent advances in the synthesis and medicinal perspective of pyrazole-based α-amylase inhibitors as antidiabetic agents, Future Med. Chem., 2024, 16(2), 173–195 Search PubMed.
- M. Zahir, A. Alam, F. Jan, A. Khan, A. F. AlAsmari, F. Alasmari, M. Khan and A. Al-Harrasi, Synthesis and Multi-Target Inhibition of Bis-Schiff Bases of Barbituric Acid: in Vitro α-glucosidase, α-amylase Inhibitory Activities With Docking and DFT Studies, ChemistrySelect, 2025, 10(1), e202401914 Search PubMed.
- F. Jan, S. Idris, M. Waheed, A. Alam, A. F. AlAsmari, F. Alasmari and M. Khan, Thiosemicarbazone derivatives as potent antidiabetic agents: Synthesis, in vitro, molecular docking and DFT investigations, J. Mol. Struct., 2024, 1311, 138459 CrossRef CAS.
- M. Ali, M. Khan, K. Zaman, A. Wadood, M. Iqbal, A. Alam, S. Shah, A. U. Rehman, M. Yousaf and R. Rafique, Chalcones: as potent α-amylase enzyme inhibitors; synthesis, in vitro, and in silico studies, Med. Chem., 2021, 17(8), 903–912 CrossRef CAS PubMed.
- S. Idris, F. Jan, M. Waheed, A. Alam, M. Ibrahim, A. F. AlAsmari, F. Alasmari, L. Bo and M. Khan, Multifaceted bioactivity of thiosemicarbazide derivatives: Synthesis, characterization, and DFT investigations on inhibition of α-amylase, hydroxyl radical scavenging, and iron chelating activities with molecular docking insights, J. Mol. Struct., 2024, 1304, 137669 CrossRef CAS.
- M. Khan, F. Alam, A. Alam, A. Wadood, S. Shams, M. Ali, S. Shah, A. F. AlAsmari, M. Alharbi and F. Alasmari, Synthesis of Some Novel 4-bromobenzoic Acid Clubbed Hydrazone Schiff Base Derivatives as Potent α-amylase Inhibitors: In vitro and In silico Studies, Lett. Drug Des. Discovery, 2024, 21(15), 3186–3197 CrossRef CAS.
- M. Khan, G. Ahad, A. Alam, S. Ullah, A. Khan, U. Salar, A. Wadood, A. Ajmal, K. M. Khan and S. Perveen, Synthesis of new bis(dimethylamino)benzophenone hydrazone for diabetic management: In-vitro and in-silico approach, Heliyon, 2024, 10(1), e23323 CrossRef CAS PubMed.
- K. M. Khan, M. Khan, M. Ali, M. Taha, S. Rasheed, S. Perveen and M. I. Choudhary, Synthesis of bis-Schiff bases of isatins and their antiglycation activity, Bioorg. Med. Chem., 2009, 17(22), 7795–7801 CrossRef CAS PubMed.
- K. M. Khan, M. Khan, N. Ambreen, F. Rahim, B. Muhammad, S. Ali, S. M. Haider, S. Perveen and M. Choudhary, Bis-Schiff bases of isatins: a new class of antioxidant, J. Pharm. Res., 2011, 4(10), 3402–3404 CAS.
- S. Gul, S. Maab, H. Rafiq, A. Alam, M. U. Rehman, M. Assad, A. F. AlAsmari, F. Alasmari, M. Ibrahim and M. Khan, Exploring Bis-Schiff Bases with Thiobarbiturate Scaffold: In Vitro Urease Inhibition, Antioxidant Properties, and In Silico Studies, Russ. J. Bioorg. Chem., 2024, 50(5), 1627–1638 CrossRef CAS.
- A. Alam, M. Ali, A. Latif, N. U. Rehman, S. Saher, A. Khan, S. Ullah, O. Ullah, S. A. Halim and F. Sani, Novel Bis-Schiff’s base derivatives of 4-nitroacetophenone as potent α-glucosidase agents: Design, synthesis and in silico approach, Bioorg. Chem., 2022, 128, 106058 CrossRef CAS PubMed.
- R. Ahmad, A. Alam, M. Khan, T. Ali, A. A. Elhenawy and M. Ahmad, Antioxidant Activity, Molecular Docking and Quantum Studies of New Bis-Schiff Bases Based on Benzyl Phenyl Ketone Moiety, ChemistrySelect, 2023, 8(35), e202302338 Search PubMed.
- S. Gul, F. Jan, A. Alam, A. Shakoor, A. Khan, A. F. AlAsmari, F. Alasmari, M. Khan and L. Bo, Synthesis, molecular docking and DFT analysis of novel bis-Schiff base derivatives with thiobarbituric acid for α-glucosidase inhibition assessment, Sci. Rep., 2024, 14(1), 3419 CrossRef PubMed.
- A. Alam, S. Gul, Zainab, M. Khan, A. A. Elhenawy, M. S. Islam, M. Ali, S. A. Ali Shah, A. Latif and M. Ahmad, Synthesis of 2,4-dihydroxyacetophenone derivatives as potent PDE-1 and-3 inhibitors: in vitro and in silico insights, Future Med. Chem., 2024, 1–19 Search PubMed.
- S. Gul, A. Alam, M. Assad, A. A. Elhenawy, M. S. Islam, S. A. A. Shah, Z. Parveen, T. A. Shah and M. Ahmad, Exploring the synthesis, molecular structure and biological activities of novel Bis-Schiff base derivatives: A combined theoretical and experimental approach, J. Mol. Struct., 2024, 1306, 137828 CrossRef CAS.
- R. Ahmad, M. Khan, A. Alam, A. A. Elhenawy, A. Qadeer, A. F. AlAsmari, M. Alharbi, F. Alasmari and M. Ahmad, Synthesis, molecular structure and urease inhibitory activity of novel bis-Schiff bases of benzyl phenyl ketone: A combined theoretical and experimental approach, Saudi Pharm. J., 2023, 31(8), 101688 Search PubMed.
- A. Alam, M. Ali, A. Latif, N. U. Rehman, A. J. Shah, I. A. Khan, M. Ayaz, S. U. Rahman, A. Al-Harrasi and M. Ahmad, Discovery of (S)-flurbiprofen-based novel azine derivatives as prostaglandin endoperoxide synthase-II inhibitors: Synthesis, in-vivo analgesic, anti-inflammatory activities, and their molecular docking, Bioorg. Chem., 2023, 141, 106847 CrossRef CAS PubMed.
- M. Ayaz, A. Alam, Zainab, M. Assad, A. Javed, M. S. Islam, H. Rafiq, M. Ali, W. Ahmad and A. Khan, Biooriented synthesis of ibuprofen-clubbed novel bis-Schiff base derivatives as potential hits for malignant glioma: In vitro anticancer activity and in silico approach, ACS Omega, 2023, 8(51), 49228–49243 Search PubMed.
- R. Maurus, A. Begum, L. K. Williams, J. R. Fredriksen, R. Zhang, S. G. Withers and G. D. Brayer, Alternative catalytic anions differentially modulate human α-amylase activity and specificity, Biochemistry, 2008, 47(11), 3332–3344 Search PubMed.
- V. Roig-Zamboni, B. Cobucci-Ponzano, R. Iacono, M. C. Ferrara, S. Germany, Y. Bourne, G. Parenti, M. Moracci and G. Sulzenbacher, Structure of human lysosomal acid α-glucosidase–a guide for the treatment of Pompe disease, Nat. Commun., 2017, 8(1), 1–10 Search PubMed.
- P. D. Leeson and B. Springthorpe, The influence of drug-like concepts on decision-making in medicinal chemistry, Nat. Rev. Drug Discovery, 2007, 6(11), 881–890 CrossRef CAS PubMed.
- J. D. Wessler, L. T. Grip, J. Mendell and R. P. Giugliano, The P-glycoprotein transport system and cardiovascular drugs, J. Am. Coll. Cardiol., 2013, 61(25), 2495–2502 Search PubMed.
- A. J. Scheen, Clinical pharmacokinetics of metformin, Clin. Pharmacokinet., 1996, 30(5), 359–371 Search PubMed.
- U. M. Zanger and M. Schwab, Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation, Pharmacol. Ther., 2013, 138(1), 103–141 Search PubMed.
- A. F. Stepan, D. P. Walker, J. Bauman, D. A. Price, T. A. Baillie, A. S. Kalgutkar and M. D. Aleo, Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States, Chem. Res. Toxicol., 2011, 24(9), 1345–1410 Search PubMed.
- H. Ullah, A. Alam, A. A. Elhenawy, I. Ahmad, A. Latif, M. Ali and M. Ahmad, Exploring the Anti-Diabetic Potential of Bis-Schiff Base Derivatives of Benzimidazole: In-Vitro α-amylase, α-glucosidase Inhibition, Molecular Docking, ADMET and DFT Studies, Comput. Biol. Chem., 2025, 108497 Search PubMed.
- L. Tolosa; M. T. Donato and M. J. Gómez-Lechón, General cytotoxicity assessment by means of the MTT assay, in Protocols in In Vitro Hepatocyte Research, Springer, 2014, pp. 333–348 Search PubMed.
- V. Sahinturk, S. Kacar, D. Vejselova and H. M. Kutlu, Acrylamide exerts its cytotoxicity in NIH/3T3 fibroblast cells by apoptosis, Toxicol. Ind. Health, 2018, 34(7), 481–489 Search PubMed.
- A. Alam, Zainab, A. A. Elhenawy, N. Ur Rehman, M. Shahidul Islam, K. A. Dahlous, F. Talab, S. A. A. Shah, M. Ali and M. Ahmad, Synthesis of Flurbiprofen Based Amide Derivatives as Potential Leads for Diabetic Management: In Vitro α-glucosidase Inhibition, Molecular Docking and DFT Simulation Approach, ChemistrySelect, 2024, 9(17), e202401296 Search PubMed.
- N. Ullah, A. Alam, Zainab, A. A. Elhenawy, S. Naz, M. S. Islam, S. Ahmad, S. A. A. Shah and M. Ahmad, Investigating novel thiophene carbaldehyde based thiazole derivatives as potential hits for diabetic management: synthesis, in vitro and in silico approach, ChemistrySelect, 2024, 9(8), e202304601 Search PubMed.
- Y. Cao, T. Balduf, M. D. Beachy, M. C. Bennett, A. D. Bochevarov, A. Chien, P. A. Dub, K. G. Dyall, J. W. Furness and M. D. Halls, Quantum chemical package Jaguar: A survey of recent developments and unique features, J. Chem. Phys., 2024, 161(5), 052502 Search PubMed.
- T. A. Shah, A. Alam, M. Khan, A. A. Elhenawy, M. Ayaz, M. Ali, A. Latif, S. A. A. Shah and M. Ahmad, Experimental and computational profiling of novel bis-Schiff base derivatives bearing α-naphthalene moiety as potential tyrosinase inhibitors, J. Mol. Struct., 2025, 1321, 139919 CrossRef CAS.
- X.-Q. Yao, L. Skjærven and B. J. Grant, Rapid characterization of allosteric networks with ensemble normal mode analysis, J. Phys. Chem. B, 2016, 120(33), 8276–8288 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |