DOI:
10.1039/D5QM00518C
(Review Article)
Mater. Chem. Front., 2026,
10, 565-602
Advances, practical applications, and future prospects of layered perovskite oxides (LnBaCo2O5+δ) for electrocatalysis reactions
Received
16th July 2025
, Accepted 17th September 2025
First published on 17th September 2025
Abstract
To realize a sustainable and clean society, highly efficient and eco-friendly energy conversion and production technologies have been developed, such as solid oxide cells (SOCs) and water electrolysis. However, most electrocatalytic reactions are normally driven by scarce noble metal-based catalysts. Thus, the exploration of active, stable, and cost-effective electrocatalysts remains an essential challenge. Herein, we summarize a series of layered perovskite oxides (LnBaCo2O5+δ) demonstrating attractive oxygen reduction, oxygen evolution, and hydrogen evolution electrolysis activities. Their variable crystal structures, flexible elemental compositions, and regulated electronic structures endow them with satisfactory activity for electrocatalytic water electrolysis and SOCs (fuel cells and electrolysis cells). This review summarizes the advances in LnBCO-based electrocatalysts and addresses several issues in their development and usage, including structural evolution, fundamental electrical properties, electrocatalytic mechanism and performance, structure–property–performance relationship, activity descriptors, and real merits/demerits. Finally, we outline the opportunities and challenges of layered perovskite oxides for practical applications, encouraging future research on next-generation electrocatalysts applied in an environmentally friendly society.
1. Introduction
High-efficiency energy conversion technologies are very significant to realize an environmentally friendly society, i.e., achieving carbon neutrality before 2060 and peak carbon emissions before 2030. As third-generation fuel cells, solid oxide fuel cells (SOFCs) have been considered as one of the most promising technologies due to their advantages, as follows: (1) high current density and power density; (2) negative anodic and cathodic polarization loss; (3) direct utilization of hydrogen, hydrocarbons, and methanol as fuels without metal catalysts; (4) providing high-quality waste heat and cogeneration of heat and power; (5) high energy conversion efficiency of about 80%; and (6) low noise and pollution emissions (their products are H2O and CO2). Another solution is to convert renewable energy (wind, sunlight, and tides) into electricity. This electricity can be used to produce high-value-added chemicals, such as hydrogen, ammonia, hydrocarbons, and alcohols, via electrocatalysis reactions. Next, these fuels are fed to SOFCs for power generation. The entire layout is clean and environmentally friendly without involving fossil fuels. In particular, hydrogen (H2) with the highest gravimetric energy density among chemical fuels may mitigate the environmental concerns due to its zero emission of carbonaceous species. Both the power generation and electrolysis efficiency are closely related to the performance of electrocatalysts, e.g., electrocatalytic oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER). With the ever-growing demands on fossil fuels and environmental protection, it is necessary to produce green and sustainable energy for next-generation applications.1–4 Water electrolysis driven by sustainable energy is indispensable for the hydrogen economy. John Bockris proposed the hydrogen economy in the 1970s.5 Because the combustion product of hydrogen (H2) is only clean water, and considering its high calorific value (≈282 kJ mol−1), hydrogen energy is an ideal energy carrier for relieving the energy crisis and achieving a carbon-neutral plan.6 Hydrogen fuels are produced via the following routes: (1) steam alcohol reforming; (2) coal gasification; (3) harvesting renewable sources (solar, wind, geothermal, and tidal) to decompose water; and (4) water photolysis/electrolysis.7,8 The produced H2 can be stored and used in the chemical engineering field, such as petroleum, residential life, and fuel cells. More than 95% H2 is produced by the first two routes. However, the high-temperature reforming and gasification processes consume fossil fuels and discharge CO2 pollutant. Therefore, as the reverse reaction of H2 combustion, water electrolysis enables a closed hydrogen cycle with zero carbon emission, customizing sustainable and environmentally friendly developments. The water splitting reaction, H2O → H2 + 1/2O2, is divided into OER at the anode and HER at the cathode, 2O2− – 2e− → O2 and 2H+ + 2e− → H2, respectively. However, the sluggish kinetics at the electrode interfaces (thermodynamic potential = 1.23 V) limit the electrocatalysis efficiency. The state-of-the-art noble metal-based catalysts (Pt/C, RuO2, and IrO2) have been considered as the ideal choices to date, which can act as benchmarks to compare the catalytic activity. However, their scarce reserves, high prices, and relatively natural instability limit their sustainable future commercialization. In this context, earth-abundant noble metal-free electrocatalysts have been designed and explored, such as metals, oxides, (oxy)hydroxides, sulfides, selenides, tellurides, borides, carbides, nitrides, phosphides, and metal–organic frameworks.9–22 Among these materials, perovskite oxides have been considered as low-cost and efficient candidates due to their variable crystal structures, flexible elemental compositions, and regulated electronic structures, providing opportunities and strategies to screen desired electrocatalysts.12–14
The produced H2 is fed to SOFCs, which directly convert chemical potential to electric energy. Representative oxygen-conducting SOFCs (O2−-SOFCs) consist of a porous anode, dense electrolyte, and porous cathode with a sandwich-like configuration, as schematically illustrated in Fig. 1a. The cathode side is fed with an oxidant gas, such as O2 and air. The dissociated O2− ions are transported from the cathode side to the anode side through the dense electrolyte (oxygen–ion conductor). The O2− ions are finally oxidized to H2O or CO2 when reacting with the fuel gas (H2 or CO) at the anode side, while releasing electrons through an external circuit.23–25 Utilizing the reverse reactions, H2O or CO2 can be decomposed into H2/CO fuel and O2 oxidant, i.e., solid oxide electrolysis cells (SOECs) (Fig. 1b).26–28 The gas steam (H2O or CO2) is supplied to the cathode side, and then the dissociated O2− ions are transported to the anode through the electrolyte component, driven by an external voltage (>Nernst voltage). The O2− ions finally evolve into O2 molecules at the anode, accompanied by electron release. As electrochemical devices, SOECs have a number of advantages, as follows: (1) producing hydrocarbon fuel from CO2 or H2O; (2) their reaction process is easy to control by regulating the voltage and reaction temperature; and (3) clean energy sources can be used to drive the process, such as solar, wind, geothermal, tidal, as well as surplus electricity from nuclear and hydroelectric sources. Differing from SOFCs, the high efficiencies of SOECs are attributed to their high operating temperatures. The total energy demand is almost constant from room temperature to 1200 °C for both H2O and CO2 electrolysis. The electrical energy decreases with an increase in temperature, and thus the energy difference is compensated for by increasing the heat supply. Because of the lower cost of heat than electricity, high-temperature electrolysis is more economically favored. Furthermore, increasing the operating temperatures can ensure a sufficient electrode performance from a dynamic perspective. There is a reasonable prospect that the conversion efficiency may exceed 60% for SOECs under desirable operating conditions. To enhance the efficiency of SOECs, studies have been conducted to develop electrode materials. The surface atoms in the cathode and anode act as active sites for the electrocatalytic reactions, the reduction of CO2/H2O and the oxygen evolution, respectively.
 |
| | Fig. 1 Schematic of the operating principles of (a) solid oxide fuel cell (H2 is used as the fuel) and (b) solid oxide electrolysis cell (H2O electrolysis). | |
Nowadays, simple perovskite oxides (ABO3) have been evaluated as potential electrocatalysts for SOFCs, SOECs, and water electrolysis,12–14,29–33 such as La(Sr)Co(Fe)O3−δ (LSCF) and Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF).34,35 The following aspects should be emphasized for an ideal electrocatalyst: (1) sufficient electrical conductivity (ionic, electronic, or mixed conductivity) for the charge transfer reaction; (2) effective active sites for the adsorption, dissociation, and desorption of reactants; (3) excellent long-standing working stability and durability under rigorous practical conditions; and (4) reasonable production cost. In the past two decades, much interest has been paid to a series of layered perovskite oxides, LnBaCo2O5+δ (LnBCO, Ln = lanthanide and Y).36–42 Owing to the difference in ionic radius between Ln3+ and Ba2+, LnBCO crystallizes into A-site cation-ordered structures, following the sequence of 〈Ln–Oδ〉–〈Co–O〉–〈Ba–O〉 along the c-axis direction. Naturally, intrinsic oxygen vacancies are formed in the 〈Ln–Oδ〉 layers, apparently influencing electrocatalytic oxygen-containing reactions. A cubic–tetragonal–orthorhombic phase transition can be identified by regulating the A-site Ln3+ ions, accompanied by oxygen vacancy disorder–order transformation. Both the average cobalt state and oxygen content increase due to an increment in the oxygen-coordinating number of Ln3+, whereupon charge transfer, oxygen surface, and bulk transport processes are improved.37,43 Initially, LnBCO can be used as promising oxygen electrodes for SOFCs, with certain intermediate-temperature ORR activity. It should be noted that their ORR activities are higher than that of the LSCF electrode and comparable to the popular BSCF electrode. In recent years, LnBCO has shown multi-functional electrocatalytic characteristics, e.g., OER and HER activity in alkaline media.44–51 These property diverse properties are attributed to the distinctive structures of layered perovskite with various elemental compositions. LnBCO belongs to a large material library concerning partial cation substitution, anion doping, and elemental stoichiometry, where its A-site comprises lanthanide and alkaline-earth elements, and its B-site almost covers the vast majority of transition metals in the periodic table. Also, its structural and compositional selectivity render a chance for different application demands. More importantly, recent research on perovskite materials unveils some crucial facts for electrocatalysis, such as the correlation between electronic structure and catalytic performance, catalytic mechanisms, and structural evolution. Hence, we deem that layered perovskite oxides (LnBCO) can be employed as perfect electrocatalytic models for strengthening scientific theories.
To date, systematic overviews of “112”-type layered perovskite electrocatalysts have been lacking. Most of the relevant summaries introduce their progress as classified by contained lanthanides (e.g., La, Pr, and Nd-based layered perovskite oxides),36 properties (e.g., electrical conductivity, dilatometric behavior, cathode performance, and chemical stability),36,37 design strategies (e.g., doping, defect engineering, nanostructure, and surface modification),39 and potential applications (e.g., energy and environmental applications).39 However, some scientific questions related to layered perovskite oxides in the electrocatalysis field are rarely summarized, which is required to guide rational prediction and future development directions.
In this review, the advances in a family of layered LnBCO perovskite oxides are systematically discussed toward SOCs and water electrolysis. Firstly, we outline their structural evolution under internal and external conditions, which ensures that LnBCO electrocatalysts exhibit controllable properties. Secondly, we interpret how to regulate their electrical properties (electrical conductivity, surface exchange, and bulk diffusion processes), and then focus on their electrocatalysis mechanism and progress. When exploring the structure–property–performance relationship of the perovskite oxides, their representative activity descriptors (e.g., eg orbital occupancy, O 2p-band center, electronegativity, tolerance factor, charge transfer energy, and Lewis acidity), applicability and inadequacies, are studied. Subsequently, from the perspective of practical applications, their cost, stability, and scalability are compared to that of noble-metal-based catalysts. We highlight their merits and demerits for their rational utilization in definite electrochemical devices and technologies. Finally, their opportunities, challenges, and applicable prospects in the electrocatalysis field are proposed. We hope that this review will contribute to the development of next-generation electrocatalysts for readers.
2. Structural evolution of layered LnBCO perovskite oxides
In the family of layered LnBCO perovskites, Ln3+ and Ba2+ co-occupy the A-site, and Con+ ions are located at the B-site. By reason of the large size difference between Ln3+ (8-fold oxygen coordination) and Ba2+ (12-fold oxygen coordination), the 〈Ln–Oδ〉 layers alternate with the 〈Ba–O〉 layers along the c-axis direction, in which a set number of oxygen vacancies exist in the 〈Ln–Oδ〉 layers. The vacancy mechanism normally governs the oxygen-ionic transport in oxides, and more oxygen vacancies might accelerate electrocatalytic oxygen-containing reactions. Layered LnBCO perovskite oxides can crystallize into variable crystal structures, giving rise to remarkable discrepancies in charge transfer and their electrocatalytic activity. Their structural evolution can be fulfilled via the following strategies: (1) lanthanide modulation; (2) cation or anion doping; and (3) different synthetic routes.
With an increase in the ion radius of Ln3+, the crystal structure transforms from orthorhombic to tetragonal, and to cubic perovskite with order–disorder transition of oxygen vacancies, including orthorhombic structure (space group Pmmm) for Ln = Sm, Eu, and Gd, tetragonal structure (space group P4/mmm) for Ln = Pr and Nd, and cubic structure (space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) for Ln = La.37,43,44 The nonstoichiometry (δ) values are ∼0.5, 0.75, and 1.0 for orthorhombic, tetragonal, and cubic structures (Fig. 2a), respectively. Moreover, the experimental conditions of the synthetic route control the superlattice perovskite structure, such as the surrounding atmosphere, calcining temperature, and annealing time. In the case of larger lanthanides (i.e., from Pr to Tb), doubling of one parameters is observed (an ap × 2ap × 2ap supercell, and ap refers to the lattice parameter of cubic perovskite), whereas in the case of smaller lanthanides (i.e., Ho and Dy), tripling of two parameters is obtained (a 3ap × 3ap × 2ap supercell).41 These crystal data are deduced from air-synthesized perovskite oxides. However, annealing the air-prepared GdBCO (δ = 0.4) in an Ar flow leads to an ap × ap × 2ap superlattice (δ = 0). When reversibly calcining the Ar-annealed GdBCO in an O2-rich atmosphere, the oxygen atoms re-enter the lattice, leading to GdBCO (δ = 0.6) with an ap × 2ap × 2ap supercell. Similarly, annealing of HoBCO (δ = 0.3) in O2 leads to HoBCO (δ = 0.5), and the 3ap × 3ap × 2ap supercell is replaced by an ap × 2ap × 2ap one. In the Mn-doped GdBaCo2−xMnxO5+δ series, GdBaCo1.8Mn0.2O5+δ can be indexed to a tetragonal unit cell of ap × ap × 2ap (P4/mmm). In contrast, a high doping fraction induces a cubic disordered perovskite of GdBaCo0.2Mn1.8O5+δ, and the structures of all the Ar-prepared compounds are similar to that of GdBaCo1.8Mn0.2O5+δ. Under an Ar atmosphere, single-phase GdBaCo2−xMnxO5+δ (0 ≤ x ≤ 2) materials are obtained, while the GdBaCo2O5+δ composition is only obtained in Ar.52 To characterize the A-site cation-ordered structures of layered perovskites, the X-ray diffraction (XRD) technique is employed as well as Rietveld refinement analysis. Multiplet characteristics (peak splitting) are typical for layered perovskite oxides, and the doubling of the unit cell is also confirmed by the small peak at 2θ ≈ 11–12°. It should be noted that this diffraction intensity is very weak for the larger Ln3+ ions, especially Pr3+ and Nd3+. Advanced in situ neutron powder diffraction (NPD) is a more powerful tool to accurately probe crystal structures. The NPD patterns of PrBCO can be fit to a tetragonal layered perovskite structure (P4/mmm), agreeing well with the XRD results.53,54 According to high-resolution transmission electron microscopy (HRTEM) images and selected area electron diffraction (SAED) patterns, we visually observe the supercells of layered perovskite oxides. As studied in PrBa0.95Gd0.05Co2O5+δ, the clear fringes with an interplanar spacing of 0.763 nm are assigned to the (001) plane of tetragonal layered perovskite oxides, and the superlattice diffraction spots are found in the SAED pattern along the [110]P zone axis. Atomic-level energy-dispersive X-ray (EDX) elemental mappings manifest an ap × ap × 2ap supercell, as evidenced by the doubling of the lattice parameter c along the [001]p zone axis.55 The specific crystal structures of perovskite oxides influence their properties. In layered perovskite oxides (LnBCO), oxygen ions migrate through the 〈Ln–Oδ〉 and 〈Co–O〉 planes (ab-plane oriented),56–58 which is conductive to understanding their ionic transport mechanism.
m) for Ln = La.37,43,44 The nonstoichiometry (δ) values are ∼0.5, 0.75, and 1.0 for orthorhombic, tetragonal, and cubic structures (Fig. 2a), respectively. Moreover, the experimental conditions of the synthetic route control the superlattice perovskite structure, such as the surrounding atmosphere, calcining temperature, and annealing time. In the case of larger lanthanides (i.e., from Pr to Tb), doubling of one parameters is observed (an ap × 2ap × 2ap supercell, and ap refers to the lattice parameter of cubic perovskite), whereas in the case of smaller lanthanides (i.e., Ho and Dy), tripling of two parameters is obtained (a 3ap × 3ap × 2ap supercell).41 These crystal data are deduced from air-synthesized perovskite oxides. However, annealing the air-prepared GdBCO (δ = 0.4) in an Ar flow leads to an ap × ap × 2ap superlattice (δ = 0). When reversibly calcining the Ar-annealed GdBCO in an O2-rich atmosphere, the oxygen atoms re-enter the lattice, leading to GdBCO (δ = 0.6) with an ap × 2ap × 2ap supercell. Similarly, annealing of HoBCO (δ = 0.3) in O2 leads to HoBCO (δ = 0.5), and the 3ap × 3ap × 2ap supercell is replaced by an ap × 2ap × 2ap one. In the Mn-doped GdBaCo2−xMnxO5+δ series, GdBaCo1.8Mn0.2O5+δ can be indexed to a tetragonal unit cell of ap × ap × 2ap (P4/mmm). In contrast, a high doping fraction induces a cubic disordered perovskite of GdBaCo0.2Mn1.8O5+δ, and the structures of all the Ar-prepared compounds are similar to that of GdBaCo1.8Mn0.2O5+δ. Under an Ar atmosphere, single-phase GdBaCo2−xMnxO5+δ (0 ≤ x ≤ 2) materials are obtained, while the GdBaCo2O5+δ composition is only obtained in Ar.52 To characterize the A-site cation-ordered structures of layered perovskites, the X-ray diffraction (XRD) technique is employed as well as Rietveld refinement analysis. Multiplet characteristics (peak splitting) are typical for layered perovskite oxides, and the doubling of the unit cell is also confirmed by the small peak at 2θ ≈ 11–12°. It should be noted that this diffraction intensity is very weak for the larger Ln3+ ions, especially Pr3+ and Nd3+. Advanced in situ neutron powder diffraction (NPD) is a more powerful tool to accurately probe crystal structures. The NPD patterns of PrBCO can be fit to a tetragonal layered perovskite structure (P4/mmm), agreeing well with the XRD results.53,54 According to high-resolution transmission electron microscopy (HRTEM) images and selected area electron diffraction (SAED) patterns, we visually observe the supercells of layered perovskite oxides. As studied in PrBa0.95Gd0.05Co2O5+δ, the clear fringes with an interplanar spacing of 0.763 nm are assigned to the (001) plane of tetragonal layered perovskite oxides, and the superlattice diffraction spots are found in the SAED pattern along the [110]P zone axis. Atomic-level energy-dispersive X-ray (EDX) elemental mappings manifest an ap × ap × 2ap supercell, as evidenced by the doubling of the lattice parameter c along the [001]p zone axis.55 The specific crystal structures of perovskite oxides influence their properties. In layered perovskite oxides (LnBCO), oxygen ions migrate through the 〈Ln–Oδ〉 and 〈Co–O〉 planes (ab-plane oriented),56–58 which is conductive to understanding their ionic transport mechanism.
 |
| | Fig. 2 (a) Schematic of the structural evolution of LnBCO perovskite oxides: orthorhombic (Ln = Sm, Eu, and Gd), tetragonal (Ln = Pr and Nd), and cubic (Ln = La) structures with a vacancy ordered-disordered transition. (b) Conductivity (600–800 °C) and (b) k* and D* values (800 °C) of the LnBCO perovskite oxides, along with a structural evolution from orthorhombic to tetragonal, and to cubic structures. The k* and D* data of classic LSFC perovskite oxide are shown in Fig. 2b for comparison. | |
Another method to induce structural evolution in layered perovskite oxides is ionic substitution. Lanthanide/alkaline earth and transition metal ions can be introduced into their A- and B-sites, respectively. Considering the B-site substitution, Zn2+, Ni2+, Fe3+, Sc3+, Mn3+, Zr4+, Nb5+, Ta5+, and Mo6+ have been substituted for Co3+.59–73 Commonly, the B-site Co3+ ions prefer to be replaced by Fe3+ ions due to their similar ionic radii, forming a complete solid solution in a doping fraction range of 0–2.67 At a low-doping level, the solid solutions possess tetragonal layered perovskite structures (P4/mmm), whereas a structural evolution from tetragonal perovskite to cubic perovskite is identified with an increase in the Fe content. As displayed in high-temperature XRD patterns, Fe-doped PrBCO is thermodynamically stable upon heating at 1000 °C, which is beneficial for practical long-term operation.74 Moreover, Fe ion substitution induces a variation in the supercell of layered perovskite oxides. Compared with air-prepared GdBCO (an ap × ap × 2ap supercell), GdBaCo1.4Fe0.6O5+δ shows an ap × 3ap × 2ap superstructure.75 In addition to the Bragg reflections of cubic perovskite, the extra reflections at Gp ± 1/2(001)p and Gp ± 1/3(010)p in the SAED patterns confirm the 3ap × 2ap periodicity. The contrast difference reveals the 2ap and 3ap periodicities along the [001]p and [010]p directions, respectively. The novel perovskite-related supercell of √2ap × √2ap × 10ap is observed in (GdBa)0.8Ca0.4Co0.6Fe1.4O5.41.76 The oxygen content, oxygen-vacancy concentration, and average Co valence state in these supercells can be modulated by controlling the crystal structures, further inuencing electrocatalysis reactions.
A-site alkaline earth ionic substitutions lead to the formation of a highly symmetric cubic structure. After substituting Ca2+ for Ba2+ in PrBCO, a tetragonal–cubic phase transition appears with an increase in the doping fraction, i.e., from a tetragonal structure (x = 0–0.2) to a cubic structure (x = 0.4). Mixed tetragonal and cubic perovskite oxides coexist at x = 0.3.77 Similar structural evolutions are also discovered in Ba-site Sr-doped LnBCO.78–80 Noticeably, these mixed perovskite oxides possess improved electrocatalytic activities arising from the synergistic effects between them.78 We put forward an effective Ca/Sr co-doping strategy, demonstrating a high-performance cubic perovskite oxide (Pr0.94Ba0.6Sr0.2Ca0.2Co2O5+δ) as an oxygen electrode for SOFCs.81 However, in the case of individual Sr doping, tetragonal layered perovskites (NdBa1−xSrxCo2O5+δ and SmBa1−xSrxCo2O5+δ) can be retained even at x = 0.5, which is attributed to the similar ionic radius between Ln3+ and Ca2+.79,80
In general, the structural evolution of LnBCO can be explained from the viewpoint of oxygen content. Taking NdBCO as an example, an orthorhombic layered perovskite oxide (Pmmm) is obtained after calcining at 1200 °C.82 If annealing at 1100 °C in air, NdBCO has a tetragonal ap × ap × 2ap supercell (P4/mmm).83 The lattice oxygen atoms are released with a decrease in oxygen content at elevated temperature. The (212) diffraction peak is split into (142) and (124) peaks, corresponding to an orthorhombic layered perovskite oxide. More lattice oxygen atoms prevent the formation of a vacancy-ordered structure, resulting in tetragonal or cubic perovskite oxides. Surrounding air, O2, and Ar atmospheres also tune the structural evolution by injecting or releasing oxygen atoms. Cation-ordered/disordered perovskite structures can be switched by controlling their annealing temperature. A-site cation-ordered LaBCO is stable at 1000 °C in air, but is transformed into a cation-disordered phase at 1100 °C.84,85 Briefly, structural evolution is an essential and effective strategy for designing perovskite oxide electrocatalysts.
3. Electrical properties of layered LnBCO perovskite oxides
3.1 Electrical conductivity
Layered LnBCO perovskite oxides have been evaluated as potential oxygen electrodes for SOFCs because of their fast charge transfer, oxygen surface exchange, and bulk diffusion kinetics. Their electrical conductivity is ∼200–1400 S cm−1 in the temperature range of 100–800 °C.43 Their relatively high conductivity is responsible for their comparable ORR activities. We have systematically tested the electrical conductivity of layered perovskite oxides (Ln = La, Pr, Nd, Sm, Eu, and Gd).43 At an identical temperature, their conductivity follows the order of cubic > tetragonal > orthorhombic perovskite oxides (Fig. 2b), indicating that the charge transfer reaction is closely related to their structural evolution. Also, their conductivity reflects their metal-like conduction nature at low temperatures. The itinerant electron mechanism might dominate the electron transfer. The itinerant character favors the d–d hopping of electrons, leading to charge fluctuation in the Co 3d band and efficient electron transfer. At high temperatures, the decrease in conductivity is treated by a band model. Given that the conduction band is formed via a three-dimensional Co–O–Co network, the release of lattice oxygen breaks the crystal field of the CoO6 octahedron. The change in the Co d-electron levels results in the creation of trapping levels below the semi-metallic Co d–O p conduction band. Thus, the conduction band becomes narrow and the conductivity is reduced.
To accelerate the charge transfer kinetics, the substitution of Ca2+/Sr2+ for Ba2+ has been considered an effective strategy up to now. Recently, we found that Ca2+/Sr2+ co-doping extremely enhances the electronic conductivity, and improves oxygen surface exchange and bulk diffusion processes.81 The electrical conductivity reaches as high as ∼1100 S cm−1 at 500 °C. The ionic radius of Ca2+ (r = 1.34 Å) and Sr2+ (r = 1.44 Å) is much smaller than that of Ba2+ (r = 1.61 Å) in a 12-fold oxygen-coordinating environment, which relatively matches the ionic radius of Pr3+ (r = 1.126 Å).86 The suitability of ionic size hinders the formation of an A-site cation-ordered structure, with an increase in oxygen content and average Co valence state. More highly oxidative Co4+ species are generated in doped perovskite oxides. As the “3d” hole carriers, Co4+ ions are favorable for electron transfer. After introducing the Ca/Sr dopants, the O–Co–O bond angle is straightened to the ideal 180°, enlarging the bandwidth and Co–O covalency. This is another reason for the increased electrical conductivity of Ca2+/Sr2+-doped LnBCO. In terms of B-site transition metal doping, many choices have been made in LnBCO, such as Zn2+, Ni2+, Sc3+, Fe3+, Mn3+, Nb5+, Ta5+, and Mo6+.59–73 These substitutions inhibit the electronic hopping through the O–Co–O pathway, and the electrical conductivity is reduced. Even though B-site doping is adverse for the charge transfer reaction, a trace of Ta dopants can enhance the electrical conductivity, e.g., ∼234 and 675 S cm−1 for PrBa0.94Co2O5+δ and PrBa0.94Co1.96Ta0.04O5+δ at 700 °C,87 respectively. This enhancement is attributed to the charge carriers provided by the acceptor dopants and stabilized layered perovskite structure. Moreover, cation-defect engineering can also facilitate the charge transfer reaction. As is known, cobalt-based perovskite oxides belong to mixed ionic and electronic conductors (MIECs), in which the charge carriers are electronic holes and oxygen vacancies. Two compensating mechanisms, the generation of extrinsic oxygen vacancies and the oxidation of Co3+ to Co4+, can respond to negatively charged A-site defects. Regarding the oxidation mechanism, enhanced hole carriers (Co4+) lead to an increase in electronic conductivity. An oxygen vacancy-compensated mechanism is observed in A-site-deficient LnBCO (Ln = La, Pr, Nd, and Eu) perovskite oxides.84,88–91 Consequently, the electrical conductivity decreases because the oxygen-ionic mobility is much lower than the electronic mobility in MIECs. The electrical conductivity of layered perovskite oxides is listed for reference in Table 1. Anyway, the charge transfer kinetics is one of the most important aspects for electrocatalysis reactions.
Table 1 The conductivity in air, oxygen surface exchange/chemical diffusion coefficients (k*/D*), and thermal expansion coefficients (TECs) in air of the LnBCO perovskite oxides
|
|
Conductivity (S cm−1) |
k* (cm s−1) |
D* (cm2 s−1) |
TEC (10−6 K−1) |
Ref. |
| LaBaCo2O5+δ |
∼50 (700 °C) |
— |
— |
— |
40
|
| PrBaCo2O5+δ |
∼145 (700 °C) |
— |
— |
— |
40
|
| NdBaCo2O5+δ |
∼170 (700 °C) |
— |
— |
— |
40
|
| SmBaCo2O5+δ |
∼200 (700 °C) |
— |
— |
— |
40
|
| GdBaCo2O5+δ |
∼90 (700 °C) |
— |
— |
— |
40
|
| YBaCo2O5+δ |
∼30 (700 °C) |
— |
— |
— |
40
|
| LaBaCo2O5+δ |
936 (600 °C) |
2.58 × 10−3 (800 °C) |
4.73 × 10−6 (800 °C) |
— |
43
|
| PrBaCo2O5+δ |
853 (600 °C) |
2.58 × 10−3 (800 °C) |
4.82 × 10−6 (800 °C) |
— |
43
|
| NdBaCo2O5+δ |
603 (600 °C) |
2.15 × 10−3 (800 °C) |
3.28 × 10−6 (800 °C) |
— |
43
|
| SmBaCo2O5+δ |
430 (600 °C) |
2.07 × 10−3 (800 °C) |
2.20 × 10−6 (800 °C) |
— |
43
|
| GdBaCo2O5+δ |
360 (600 °C) |
3.05 × 10−4 (800 °C) |
2.93 × 10−6 (800 °C) |
— |
43
|
| EuBaCo2O5+δ |
341 (600 °C) |
6.12 × 10−4 (800 °C) |
2.54 × 10−6 (800 °C) |
— |
43
|
| PrBaCo2O5.5 |
— |
— |
1.9 × 10−8 (500 °C) |
— |
56
|
| PrBaCo2O5+δ |
— |
4.6 × 10−7 (675 °C) |
1.7 × 10−8 (675 °C) |
— |
57
|
| NdBaCo2O5+δ |
— |
∼2.78 × 10−4 (700 °C) |
∼3.7 × 10−5 (700 °C) |
— |
70
|
| NdBaCo2O5+δ |
— |
— |
— |
23.1 (25–1100 °C) |
83
|
| SmBaCo2O5+δ |
— |
— |
— |
20.8 (25–1100 °C) |
83
|
| PrBaCo2O5+δ |
∼2000–500 (25–812 °C) |
6.9 × 10−5 (500 °C) |
3.6 × 10−7 (500 °C) |
— |
95
|
| GdBaCo2O5+δ |
— |
2.8 × 10−7 (575 °C) |
4.8 × 10−10 (575 °C) |
— |
96
|
| LaBaCo2O5+δ |
— |
— |
— |
24.3 (80–900 °C) |
98
|
| NdBaCo2O5+δ |
— |
— |
— |
19.1 (80–900 °C) |
98
|
| SmBaCo2O5+δ |
— |
— |
— |
17.1 (80–900 °C) |
98
|
| GdBaCo2O5+δ |
— |
— |
— |
16.6 (80–900 °C) |
98
|
| YBaCo2O5+δ |
— |
— |
— |
15.8 (80–900 °C) |
98
|
| PrBaCo2O5+δ |
— |
— |
— |
24.03 (100–800 °C) |
161
|
| NdBaCo2O5+δ |
— |
— |
— |
21.5 (80–900 °C) |
162
|
| GdBaCo2O5+δ |
— |
— |
— |
19.9 (80–900 °C) |
162
|
| EuBaCo2O5+δ |
— |
— |
— |
14.9 (100–800 °C) |
163
|
| YBaCo2O5+δ |
— |
— |
— |
14.5 (200–800 °C) |
164
|
| SmBaCo2O5+δ |
— |
3.5 × 10−3 (800 °C) |
5.4× 10−6 (800 °C) |
— |
185
|
3.2 Oxygen surface exchange and bulk diffusion processes
Oxygen surface kinetics is recognized to be a meaningful factor in electrocatalytic ORR, which is widely studied using the electrical conductivity relaxation (ECR) technique. The electrical conductivity of MIECs is proportional to the density of charge carriers. If the charge carrier is either an oxygen vacancy or electron hole, the apparent conductivity is normalized as follows:92| | 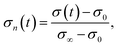 | (1) |
where σ(t), σ0, and σ∞ are the apparent conductivity at time t, the initial equilibrium state, and the new equilibrium state, respectively. The normalized conductivity can be directly expressed by the conductivity associated with oxygen vacancies (σ0) and electron holes (σh). The surface exchange and bulk diffusion processes can be described using Fick's second law under linear kinetic boundary conditions. The oxygen surface exchange coefficient (k*) and chemical diffusion coefficient (D*) are derived from eqn (2)–(4), as follows:93,94| | 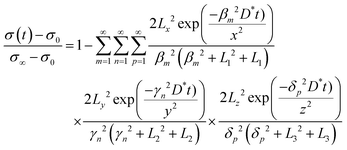 | (2) |
| | βm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) tanβm = Lx; γntanγn = Ly; δptanδp = Lz; tanβm = Lx; γntanγn = Ly; δptanδp = Lz; | (3) |
| | | Lx = x(k*/D*); Ly = y(k*/D*); Lz = z(k*/D*) | (4) |
where x, y, and z represent the length, width, and height, respectively. The parameters βm, γn, and δp. are the non-zero roots. Notably, layered perovskite oxides exhibit fast oxygen surface kinetics,95,96e.g., k* of ∼10−5 cm s−1 and D* of ∼10−7 cm2 s−1 for PrBCO at 500 °C. According to our findings, the k* value of Pr1.1Ba0.9Co2O5+δF0.1 is 2.05 × 10−2 cm s−1 at 800 °C, signifying a valid strategy for improving the oxygen surface process.97 This improvement might be caused by increased oxygen vacancies, as supported by the decreased formation energy of oxygen vacancy according to density functional theory (DFT) computation. Unfortunately, ueliable kinetic parameters are sometimes obtained from the ECR data because the kinetic properties of each specimen are not known beforehand. The experiment noise causes significant uncertainties in the fitted parameters. Thereafter, a generalized ECR (g-ECR) method is developed to promote the accuracy of the fitted kinetic parameters and weaken the uncertainties derived from the experimental noise. The detailed transport modeling and inverse algorithm are discussed in the literature.92 The rationality of g-ECR is verified in a layered perovskite oxide (PrBaCo1.5Fe0.5O5+δ), acquiring more reasonable kinetic parameters than the ECR method with a single measurement.
As a classic intermediate-temperature oxygen electrode for SOFCs, LSCF is characterized by excellent mixed conducting properties and electrocatalytic ORR activity. The electrical conductivity of LSCF is ∼200–300 S cm−1 at 600–900 °C,34 but the maximum value of LaBCO exceeds 1000 S cm−1.43 The convenient charge transfer in layered perovskite oxides might be ascribed to the hopping of d electrons from the high-spin Co3+ to low-spin Co3+ with itinerant electrons. However, the high-spin Co3+ states in LSCF can be stabilized by Fe ions, and thus the thermally activated hopping of d electrons becomes more difficult. The prominent oxygen surface exchange and bulk diffusion rates of LSCF manifest its ORR characteristics, i.e., k* = 1.2 × 10−3 cm s−1 and D* = 6.70 × 10−8 cm2 s−1 at 800 °C, respectively.34 It is noteworthy that the k* and D* values of layered perovskite oxides surpass that of LSCF, e.g., enhanced D* by two orders of magnitude (∼10−6 cm2 s−1) (Fig. 2c).43 The as-collected k* and D* values of layered LnBCO perovskite oxides are listed in Table 1. Their high electrical conductivities, k*, and D* can contribute to the electrocatalytic ORR process.
4. Advances in layered LnBCO perovskite oxides
4.1. Electrocatalytic ORR advances
Layered LnBCO perovskite oxides have been regarded as oxygen electrode materials for SOFCs, with rather good electrocatalytic activity for ORR. At the oxygen electrode interface, four main reaction steps are involved in the entire oxygen reduction process (Fig. 3a), as follows:| | | O2(g) → O2,ads., molecular oxygen adsorption | (5) |
| | | O2,ads → 2Oads., dissociation of adsorbed oxygen | (6) |
| | 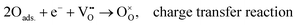 | (7) |
| | 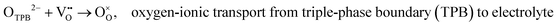 | (8) |
The rate-limiting step can be determined by performing electrochemical impedance spectroscopy at different oxygen partial pressures, permitting the proper design of the oxygen electrode. Layered perovskite oxides (Ln = La, Pr, Nd, Sm, Gd, and Y) have been characterized and evaluated as oxygen permeation membranes and oxygen electrodes.40 The highest oxygen flux (jO2) (∼5.09 × 10−7 mol cm−2 s−1 at 900 °C) and the lowest area-specific resistance (ASR) (∼0.213 Ω cm2 at 600 °C) are achieved in PrBCO. The ORR activities of these oxygen electrodes are ranked as follows: PrBCO > GdBCO > SmBCO > LaBCO > YBCO, corresponding to the unstable layered structure of YBCO. However, different groups reported contradictory trends of electrocatalytic ORR activities.36,40,43 As-synthesized LnBCO materials possess diverse physicochemical characteristics (e.g., crystal structure, composition, oxygen content, and average Co valence state) through different synthesis routes, which profoundly modify their ORR activity. Zhang et al. reported that LnBCO (Ln = La–Gd and Y) possessed an orthorhombic structure (Pmmm), and the detection of impurities in LaBCO.40 We found that the structural symmetries of layered perovskite oxides influence their electrochemical performance.43 A structural evolution occurs through lanthanide modulation, i.e., cubic–tetragonal–orthorhombic phase transition. Cubic LaBCO perovskite oxide has greater ORR activity than tetragonal and orthorhombic layered perovskite oxides. The ASRs are 0.048, 0.054, 0.069, 0.079, 0.089, and 0.096 Ω cm2 for Ln = La, Pr, Nd, Sm, Eu, and Gd at 700 °C, respectively, while LaBCO shows the lowest activation energy (Ea ≈ 104.9 kJ mol−1). As expected, LaBCO has the highest electrical conductivity, k*, and D* among the studied materials. Kim et al. reported similar crystal structures to ours, e.g., LaBCO (cubic), NdBCO (tetragonal), SmBCO (orthorhombic), and GdBCO (orthorhombic).98,99 Their electrochemical performance complies with the order of LaBCO > NdBCO > SmBCO > GdBCO, including electrical conductivity, oxygen-ionic conductivity (σ0), oxygen permeation flux, bulk diffusion coefficient, and peak power density (PPD) in a fuel cell.98,99 A similar performance trend is also corroborated in Ln-deficient Ln0.94BaCo2O5+δ (Ln0.94BCO), and the strong scaling correlation between its structure and performance encourages us to unveil a parameter for predicting its electrocatalytic activity.100 The Goldschmidt tolerance factor (tf) seems to be an effective descriptor for ORR activity. Given that tf is used to predict the geometric stability of perovskite oxides, ABO3-type perovskite oxides (e.g., Ba(Sr)Co(Fe)O3−δ and La(Ba/Sr)Co(Fe)O3−δ), are taken into account to explore the feasibility of tf. The ASRs of a total of 10 perovskite oxides are plotted as a function of tf, revealing a volcano-type change with small deviations from the fitted results using quadratic equations. Resultantly, the La0.94BCO oxygen electrode showed the best ORR activity at tf = 0.992.
 |
| | Fig. 3 The reaction steps of the ORR process at the O2/electrode/electrolyte interfaces. Different ORR pathways for (b) pure electronic conductor oxygen electrode (active TPB zone), (c) MIEC oxygen electrode (active TPB and 2PB zone) and (d) MIEC-electrolyte composite oxygen electrode (expanded TPB length by addition of electrolyte and 2PB zone). | |
To further promote the ORR activity, the most extensive strategy is ion substitution, including A, B, and O-site substitution. At the A-site in LnBCO, Ln3+ or Ba2+ can be replaced by other lanthanide or alkaline earth ions.76–80,101–127 When substituting Ca2+ or Sr2+ for Ba2+, the ORR activity is significantly promoted due to the enhanced electron transfer, as discussed in Section 3.1. Furthermore, Ca2+/Sr2+ co-doping is more effective for improving the ORR activity. We developed a Ca2+/Sr2+ co-doped Pr0.94Ba0.6Sr0.2Ca0.2Co2O5+δ oxygen electrode with the ASR of 0.025 Ω cm2 at 700 °C, approximately reduced by ∼60%.81 This A-site ion substitution is one of the best-performing methods for improving the electrocatalytic properties.
In the case of B-site ion substitution, a variety of transition metals can be considered as dopants, such as Zn, Fe, Mn, Cu, Ni, Sc, Nb, Ta, and Mo.59–73,128–155 It should be noted that A and B-site co-doping is more beneficial to the ORR activity.136,143,156–160 The synergistic effects between two types of dopants have been identified in layered perovskite oxides, LnBa0.5Sr0.5Co1.5Fe0.5O5+δ (Ln = La, Pr, and Nd) and Pr0.8Nd0.2BaCo1.6Fe0.4O5+δ, leading to oxygen vacancy-rich characteristics, fast oxygen surface processes, and excellent durability. A fabricated single cell with an LaBa0.5Sr0.5Co1.5Fe0.5O5+δ-based composite oxygen electrode delivered a high peak power density (PPD) of ∼2200 mW cm−2 at 600 °C,143 suggesting the commercial possibility of this co-doped electrode for SOFCs. However, this family of layered perovskite oxide has a drawback, i.e., thermal expansion coefficients (TECs) of ∼15–24 × 10−6 K−1 (Table 1).83,98,161–164 Their high TECs are ascribed to the configuration transition of Co3+ from low-spin (r = 68.5 pm) to intermediate-spin, and then to high-spin (r = 75 pm) filling states, resulting in an increased volume of CoO6 octahedrons and corresponding lattice expansion. Thereupon, B-site ion substitution can suppress this dilatometric behavior. For example, the TEC value of 30 mol% Ni2+-doped NdBCO decreases from 19.1 × 10−6 K−1 to 16.7 × 10−6 K−1 in the temperature range of 80–900 °C, with an ASR of ∼0.5 Ω cm2 at 700 °C.60 However, in most cases, although Co-site doping restrains high-temperature expansion, it has a negative effect on the ORR activity, for example, in GdBaCo2−xNixO5+δ,61 PrBaCo2−xFexO5+δ,64 PrBaCo2−xNbxO5+δ,68 YBaCo2−xFexO5+δ,71 and YBaCo2−xCuxO5+δ.165
Incorporating the electrolyte components into the electrode backbone can extend the TPB region (electrode–gas–electrolyte), boosting the ORR kinetics at the electrode interfaces.166–193Fig. 3b–d schematically illustrate the possible electrocatalytic ORR zones of pure electronic conductor (PEC), MIEC, and MIEC/electrolyte composite materials, respectively. If a PEC is used as the oxygen electrode, the reaction zone is confined to TPB (Fig. 3b). In the case of an MIEC oxygen electrode, the oxygen reduction process happens not only at TPB but also at the electrode/gas interface, i.e., two-phase boundary (2PB). The MIEC surface can also provide extra active sites for adsorbing molecular O2, and then the oxygen ions migrate from the MIEC to the electrolyte due to oxygen-ionic mobility of MIEC (Fig. 3c). With respect to the composite oxygen electrode (MIEC and electrolyte), the electrochemically active zone is extended due to the expanded TPB length (Fig. 3d). The sum of TPB dominates the electrochemical performance of the oxygen electrode, and thus composite electrocatalysts should be ideal candidates for SOFCs. The fluorite-type oxides Ce0.9Gd0.1O1.95 (GDC) and Ce0.8Sm0.2O1.9 (SDC) are used as dense electrolyte components. Actually, the addition of electrolyte hugely improves the ORR activity of layered perovskite oxides. After adding the GDC electrolyte, the EuBCO/10 wt% GDC oxygen electrode shows an ASR of 0.055 Ω cm2 at 700 °C.166 Moreover, the TEC is lowered to 12.8 × 10−6 K−1, which is compatible with pure GDC electrolyte (11.9 × 10−6 K−1).167 Afterwards, the composite oxygen electrode is made up of Ba-deficient EuBCO and SDC, resulting in a low ASR of 0.028 Ω cm2 at 700 °C.168 In the same way, a single fuel cell with the oxygen electrode (EuBa0.5Sr0.5Co2O5+δ/SDC) can deliver a PPD of ∼1200 mW cm−2 at 700 °C.159 By restructuring the electrolyte and anode components, a PPD of ∼1100 mW cm−2 is achieved in a fuel cell with the configuration of Ni/GDC|GDC| NdBa0.5Sr0.5Co2O5+δ/GDC.170 Besides, elaborately designed oxygen electrodes are explored with simple perovskite oxides, Ruddlesden–Popper oxides, and metals, such as PrBa0.5Sr0.5Co1.5Fe0.5O5+δ/La2NiO4+δ,171,172 PrBCO/LSCF,173 PrBa0.5Sr0.5Co1.5Fe0.5O5+δ/Pr0.9Fe0.7Co0.3O3,174 PrBa0.5Sr0.5Co1.5Fe0.5O5+δ/Ag,175 and PrBa0.8Ca0.2Co2O5+δ/BaCoO3−δ.176 Interestingly, perovskite oxide heterostructures have been successfully developed, e.g., PrBa(Co1−xFex)2O5+δ/Pr0.5Ba0.5Co1−xFexO3−δ and PrBa0.94Co2O5+δ/Pr0.5Ba0.47CoO3−δ.177,178 Tetragonal layered and cubic perovskite oxides provide abundant hetero-interfaces, possessing dramatic oxygen adsorption and dissociation abilities. Benefiting from the high electron transfer efficiency and fast oxygen surface rate, an ASR of 0.026 Ω cm2 is obtained for the heterostructure electrode at 700 °C.178 The ASR can be as low as 0.094 Ω cm2 at 600 °C, which meets the demand of an ideal oxygen electrode for SOFCs (0.1 Ω cm2). The related fuel cell delivers a PPD of 1100 mW cm−2 at 700 °C, along with extremely stable power output over a period of 120 h. Furthermore, we develop BaO nanoparticle-decorated Ln0.94BaCo2O5+δ (e-Ln0.94BCO) perovskite oxides.100 The e-La0.94BCO oxygen electrode exhibits an ASR of 0.03 Ω cm2 at 700 °C, which is reduced by ∼43% relative to pristine La0.94BCO. According to DFT calculations, the interfacial BaO compositions lead to the movement of the bulk O 2p-band center to the Fermi level (EF), meaning a lower energy barrier of oxygen/vacancy hopping and easier oxygen dissociation. Briefly, externally added active compositions improve the ORR activity in some ways, motivating us to continue this work.
Cation-deficient engineering is considered a useful route for designing high-performance perovskite oxide electrocatalysts. Regarding the A-site-deficient status, the Co ions will be partially oxidized to maintain electroneutral balance, which is helpful for the ORR process.88,194,195 The ASRs of the PrBa0.94Co2O5+δ and Pr0.94BaCo2O5+δ oxygen electrodes are 0.042 and 0.11 Ω cm2 at 700 °C, respectively.88,196 Their performance is comparable to that of the popular BSCF electrode (0.094 Ω cm2). In B-site-deficient PrBCO, electron hopping is impeded by the introduced Co defects.197 Owing to the stable valence states of the A-site cations, extrinsic oxygen vacancies are generated in the lattice, facilitating oxygen-ionic transport and oxygen reduction kinetics. These positive impacts on electrochemical performance have been clarified in Nd1−xBaCo2O5+δ,89,90 EuBa1−xCo2O5+δ,91 NdBa1−xCo2−yFeyO5+δ,198 Sm1−xBaCo2O5+δ,199 and Y1−xBaCoCuO5+δ.200 Besides, morphology-controllable perovskite oxides with different shapes also possess excellent ORR activities. Hollow nanofibers of PrBa0.5Sr0.5Co2O5+δ are assembled into a fibrous porous oxygen electrode, with facile channels for gas transport and charge transfer.201 The experimental ASRs and PPDs of LnBCO-based oxygen electrodes are summarized in Table 2.
Table 2 Experimental area-specific resistances (ASRs) and peak power densities (PPDs) of the LnBCO-based cathodes
| Cathode |
ASR (Ω cm2) |
Electrolyte |
PPD (mW cm−2) |
Anode|electrolyte |
Ref. |
|
BZCYYb: BaZr0.1Ce0.7Y0.1Yb0.1O3.
LSGMC: La0.9Sr0.1Ga0.8Mg0.115Co0.085O2.85.
ScSZ: Zr0.9Sc0.1O1.95.
LSGM: La0.9Sr0.1Ga0.8Mg0.2O2.85.
LDC: La-doped CeO2.
PBM: PrBaMn2O5+δ.
BSCZGY: Ba0.5Sr0.5Ce0.6Zr0.2Gd0.1Y0.1.
BZCY: BaCe0.7Y0.2Zr0.1O3.
|
| PrBaCo2O5+δ |
∼0.213 (600 °C) |
SDC |
— |
— |
40
|
| PrBaCo2O5+δ-GDC |
∼0.1 (600 °C) |
SDC |
— |
— |
40
|
| LaBaCo2O5+δ |
0.048 (700 °C) |
GDC |
1212 (750 °C) |
Ni-YSZ|YSZ|GDC |
43
|
| PrBaCo2O5+δ |
0.054 (700 °C) |
GDC |
— |
— |
43
|
| NdBaCo2O5+δ |
0.069 (700 °C) |
GDC |
— |
— |
43
|
| SmBaCo2O5+δ |
0.079 (700 °C) |
GDC |
— |
— |
43
|
| EuBaCo2O5+δ |
0.089 (700 °C) |
GDC |
— |
— |
43
|
| GdBaCo2O5+δ |
0.096 (700 °C) |
GDC |
744 (750 °C) |
Ni-YSZ|YSZ|GDC |
43
|
| PrBa0.95Gd0.05Co2O5+δ |
0.038 (700 °C) |
GDC |
1230 (700 °C) |
Ni-YSZ|YSZ|GDC |
55
|
| PrBa0.9Ca0.1Co1.85Zn0.15O5+δ-BZCYYb |
0.09 (700 °C) |
BZCYYba |
876 (750 °C) |
Ni-BZCYYb|BZCYYb |
59
|
| GdBaCo1.7Ni0.3O5+δ |
0.54 (600 °C) |
GDC |
— |
— |
61
|
| PrBa0.5Sr0.5Co1.9Ni0.1O5+δ |
0.297 (800 °C) |
YSZ |
120 (800 °C) |
Pt|YSZ |
63
|
| PrBaCo1.5Fe0.5O5+δ |
0.091 (700 °C) |
SDC |
— |
— |
64
|
| PrBaCo1.6Fe0.4O5+δ |
0.07 (750 °C) |
SDC |
446.4 (700 °C) |
Ni-SDC|SDC |
65
|
| GdBa0.5Sr0.5Co1.5Fe0.5O5+δ-GDC |
— |
— |
250 (800 °C) |
Ni-YSZ|GDC |
66
|
| YBaCo2O5+δ |
0.11 (700 °C) |
LSGMCb |
873 (800 °C) |
Ni-SDC|SDC|LSGMC |
68
|
| PrBaCo1.5Sc0.5O5+δ |
0.022 (750 °C) |
GDC |
— |
— |
69
|
| NdBaCo1.95Zr0.05O5+δ |
0.024 (700 °C) |
SDC |
1012 (800 °C) |
Ni-ScSZ|ScSZc |
70
|
| PrBaCo1.75Nb0.25O5+δ |
1.034 (600 °C) |
SDC |
— |
— |
71
|
| PrBaCo1.75Ta0.25O5+δ |
0.101 (700 °C) |
BZCY |
755 (700 °C) |
Ni-BZCY|BZCY |
72
|
| PrBa0.8Ca0.2Co2O5+δ-GDC |
0.033 (700 °C) |
LSGMd|LDCe |
460 (700 °C) |
PBMf|LSGM|LDC |
77
|
| Pr0.94Ba0.7Sr0.3Co2O5+δ |
0.031 (700 °C) |
GDC |
1077 (700 °C) |
Ni-YSZ|YSZ|GDC |
78
|
| NdBa0.5Sr0.5Co2O5+δ-GDC |
0.112 (600 °C) |
GDC |
∼1000 (600 °C) |
Ni-GDC|GDC |
79
|
| SmBa0.25Sr0.75Co2O5+δ-GDC |
0.138 (600 °C) |
GDC |
1039 (600 °C) |
Ni-GDC|GDC |
80
|
| Pr0.94Ba0.6Sr0.2Ca0.2Co2O5+δ |
0.025 (700 °C) |
GDC |
1194 (700 °C) |
Ni-YSZ|YSZ|GDC |
81
|
| LaBa0.9Co2O5+δ |
0.118 (600 °C) |
GDC |
— |
— |
84
|
| PrBa0.94Co1.96Ta0.04O5+δ |
0.02 (700 °C) |
GDC |
1050 (700 °C) |
Ni-YSZ|YSZ|GDC |
87
|
| PrBa0.94Co2O5+δ |
0.042 (600 °C) |
GDC |
∼1030 (700 °C) |
Ni-GDC|GDC |
88
|
| Nd0.95BaCo2O5+δ-Bi2O3 |
0.026 (800 °C) |
LSGM |
720 (800 °C) |
Ni-SDC|SDC|LSGM |
90
|
| EuBa0.98Co2O5+δ |
0.107 (700 °C) |
GDC |
505 (700 °C) |
Ni-YSZ|YSZ|GDC |
91
|
| PrBaCo2O5+δ |
0.15 (600 °C) |
GDC |
— |
— |
95
|
| GdBaCo2O5+δ |
∼0.1 (700 °C) |
GDC |
— |
— |
96
|
| Pr1.1Ba0.9Co2O5+δF0.1 |
0.033 (700 °C) |
GDC |
1102 (700 °C) |
Ni-YSZ|YSZ|GDC |
97
|
| LaBaCo2O5+δ-LSGM |
— |
— |
516 (800 °C) |
Ni-GDC|LDC|LSGM |
98
|
| e-La0.94BaCo2O5+δ |
0.03 (700 °C) |
GDC |
1050 (700 °C) |
Ni-YSZ|YSZ|GDC |
100
|
| LaBa0.7Ca0.3Co2O5+δ |
0.033 (700 °C) |
GDC |
1050 (700 °C) |
Ni-YSZ|YSZ|GDC |
101
|
| Pr1.1Ba0.4Sr0.5Co2O5+δ |
0.05 (650 °C) |
GDC |
1506 (750 °C) |
Ni-YSZ|YSZ|GDC |
102
|
| YBaCo2O5+δ |
0.44 (650 °C) |
GDC |
— |
— |
103
|
| Eu0.8Ca0.2BaCo2O5+δ |
0.028 (700 °C) |
GDC |
1210 (700 °C) |
Ni-YSZ|YSZ|GDC |
104
|
| Pr0.94Ba0.7Ca0.3Co2O5+δ |
0.022 (700 °C) |
GDC |
1202 (700 °C) |
Ni-YSZ|YSZ|GDC |
105
|
| Pr0.7Ca0.3Ba0.94Co2O5+δ |
0.027 (700 °C) |
GDC |
1114 (700 °C) |
Ni-YSZ|YSZ|GDC |
106
|
| LaBa0.5Sr0.25Ca0.25Co2O5+δ |
0.075 (800 °C) |
SDC |
662 (800 °C) |
Ni-SDC|SDC |
107
|
| LaBa0.5Sr0.5Co2O5+δ |
0.084 (800 °C) |
SDC |
580 (800 °C) |
Ni-SDC|SDC |
107
|
| LaBa0.8Ca0.2Co2O5+δ |
0.039 (700 °C) |
GDC |
1063 (700 °C) |
Ni-GDC|GDC |
108
|
| LaBa0.5Sr0.5Co2O5+δ |
∼0.25 (800 °C) |
SDC |
— |
— |
109
|
| PrBa0.8Ca0.2Co2O5+δ |
∼0.024 (750 °C) |
SDC |
∼949 (700 °C) |
Ni-BZCYYb|SDC |
110
|
| Pr0.9Ca0.1BaCo2O5+δ |
0.081 (700 °C) |
SDC |
646.5 (800 °C) |
Ni-SDC|SDC |
111
|
| PrBa0.7Ca0.3CoCuO5+δ |
0.052 (650 °C) |
GDC |
2040 (800 °C) |
Ni-YSZ|YSZ|GDC |
112
|
| Pr0.7Y0.3BaCo2O5+δ |
— |
— |
200 (750 °C) |
Ni-GDC|LDC|LSGM |
113
|
| PrBa0.8Ca0.2CoCuO5+δ |
0.051 (800 °C) |
LSGM |
— |
— |
114
|
| NdBa0.75Ca0.25Co2O5+δ |
0.066 (600 °C) |
GDC |
2114 (600 °C) |
Ni-GDC|GDC |
115
|
| Nd0.9La0.1BaCo2O5+δ |
0.083 (700 °C) |
GDC |
1045 (700 °C) |
Ni-YSZ|YSZ|GDC |
116
|
| NdBaCo2O5+δ-GDC |
0.038 (700 °C) |
GDC |
— |
— |
117
|
| SmBa0.8Ca0.2Co2O5+δ |
0.019 (700 °C) |
SDC |
— |
— |
120
|
| Sm0.8Ca0.2BaCo2O5+δ |
0.075 (700 °C) |
SDC |
753 (800 °C) |
Ni-SDC|SDC|LSGM|SDC |
121
|
| SmBa0.5Sr0.5Co2O5+δ |
0.57 (750 °C) |
SDC |
304 (700 °C) |
Ni-SDC|SDC |
122
|
| Gd0.5Pr0.5BaCo2O5+δ |
1.9 (700 °C, wet air) |
BSCZGYg |
— |
— |
123
|
| GdBa0.5Sr0.5Co2O5+δ |
— |
— |
725 (700 °C) |
Ni-SDC|SDC |
124
|
| YBa0.5Sr0.5Co2O5+δ |
— |
— |
468 (800 °C) |
Ni-SDC|SDC|LSGMC |
126
|
| Y0.8Ca0.2BaCo2O5+δ |
0.068 (700 °C) |
LSGM |
— |
— |
127
|
| LaBaCuCoO5+δ |
0.11 (700 °C) |
SDC |
603 (800 °C) |
Ni-SDC|SDC |
128
|
| PrBa0.5Sr0.5Co2O5+δ-GDC |
0.093 (600 °C) |
GDC |
1220 (600 °C) |
Ni-GDC|GDC |
130
|
| PrBaCo2/3Fe2/3Mn2/3O5+δ-SDC |
0.023 (800 °C) |
SDC |
621 (800 °C) |
Ni-SDC|SDC |
131
|
| PrBa0.92CoCuO6−δ |
0.017 (750 °C) |
GDC |
1228 (750 °C) |
Ni-YSZ|YSZ|GDC |
132
|
| PrBa0.8Ca0.2Co1.5Fe0.5O5+δ |
0.08 (600 °C) |
GDC |
1890 (600 °C) |
Ni-GDC|GDC |
133
|
| PrBaCuCoO5+δ |
0.047 (700 °C) |
SDC |
791 (700 °C) |
Ni-SDC|SDC |
135
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ |
0.07 (800 °C) |
LSGM |
697 (850 °C) |
Ni-SDC|SDC|LSGM |
136
|
| PrBaCo2/3Fe2/3Cu2/3O5+δ |
0.038 (800 °C) |
GDC |
659 (800 °C) |
Ni-GDC|GDC |
137
|
| PrBaCo1.6Fe0.2Nb0.2O5+δ–PrBaCo1.6Fe0.2Nb0.2−xO5+δ |
0.079 (750 °C, wet air) |
BZCYYb |
1059 (650 °C) |
Ni-BZCYYb|BZCYYb |
139
|
| PrBa0.8Ca0.2(Co0.95Fe0.05)2O6−δ |
0.017 (700 °C) |
GDC |
1270.1 (700 °C) |
Ni-YSZ|YSZ|GDC |
143
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ-GDC |
∼0.056 (600 °C) |
GDC |
2160 (600 °C) |
Ni-GDC|GDC |
144
|
| NdBa0.5Sr0.5Co2O5+δ-GDC |
0.105 (600 °C) |
GDC |
1477 (600 °C) |
Ni-GDC|GDC |
145
|
| NdBaCo2/3Fe2/3Cu2/3O5+δ |
0.077 (700 °C) |
LSGM |
736 (800 °C) |
Ni-GDC|GDC|LSGM |
146
|
| NdBaCo1.8Sc0.2O5+δ |
0.035 (700 °C) |
SDC |
1188 (800 °C) |
Ni-ScSZ|ScSZ|GDC |
147
|
| NdBa0.9Co1.9Fe0.1O5+δ |
0.14 (700 °C) |
GDC |
— |
— |
148
|
| SmBaCuCoO5+δ |
— |
— |
355 (700 °C) |
Ni-BZCY|BZCY |
150
|
| SmSrCo2O5+δ |
0.073 (700 °C) |
GDC |
— |
— |
151
|
| SmBa0.5Sr0.5CoCuO5+δ |
0.0263 (850 °C) |
LSGM |
857 (850 °C) |
Ni0.9Cu0.1-GDC|LSGM |
152
|
| SmBaCo1.7Ni0.3O5+δ |
0.0464 (800 °C) |
SDC |
536 (800 °C) |
Ni-SDC|SDC |
153
|
| SmBaCo0.5Mn1.5O5+δ |
0.081 (900 °C) |
LSGM |
1060 (900 °C) |
Ni-GDC|LDC|LSGM |
154
|
| GdBaCuCo0.5Fe0.5O5+δ-GDC |
0.118 (750 °C) |
GDC |
— |
— |
155
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ nanofiber |
∼0.025 (750 °C) |
— |
∼2539 (750 °C) |
Ni-YSZ|YSZ|SDC |
156
|
| NdBa0.5Sr0.5Co1.5Fe0.5O5+δ-GDC |
0.263 (800 °C) |
GDC |
886.44 (800 °C) |
Ni-YSZ|YSZ|GDC |
159
|
| Pr0.8Nd0.2BaCo1.6Fe0.4O5+δ |
0.0279 (600 °C) |
GDC |
1345 (600 °C) |
Ni-GDC|GDC |
160
|
| EuBaCo2O5+δ |
0.098 (750 °C) |
GDC |
— |
— |
163
|
| YBaCo2O5+δ–Ag |
2.03 (780 °C) |
YSZ |
— |
— |
164
|
| YBaCo1.4Cu0.6O5+δ |
0.076 (750 °C) |
LSGM |
479 (750 °C) |
Ni-GDC|GDC|LSGM |
165
|
| EuBaCo2O5+δ-GDC |
0.055 (700 °C) |
GDC |
810 (700 °C) |
Ni-GDC|GDC |
166
|
| EuBa0.98Co2O5+δ-SDC |
0.028 (700 °C) |
SDC |
980 (700 °C) |
Ni-YSZ|YSZ|GDC |
168
|
| EuBa0.5Sr0.5Co2O5+δ-SDC |
0.061 (700 °C) |
GDC |
1185 (700 °C) |
Ni-YSZ|YSZ|GDC |
169
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ–La2NiO4+δ |
0.11 (700 °C) |
GDC |
940 (750 °C) |
Ni-YSZ|YSZ|GDC |
171
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ–La2NiO4+δ |
0.51 (700 °C) |
GDC |
710 (700 °C) |
Ni-YSZ|YSZ|GDC |
172
|
| PrBaCo2O5+δ-LSCF |
0.086 (750 °C) |
GDC |
— |
— |
173
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ–Pr0.9Fe0.7Co0.3O3 |
0.09 (700 °C) |
BZCYYb |
1080 (650 °C) |
Ni-BZCYYb|BZCYYb |
174
|
| LaBa0.8Ca0.2Co2O5+δ–BaCoO3−δ |
∼0.03 (750 °C) |
SDC |
940 (750 °C) |
Ni-YSZ|ScSZ|SDCf |
176
|
| PrBaCo1.4Fe0.6O5+δ–Pr0.5Ba0.5Co0.7Fe0.3O3+δ |
0.039 (700 °C) |
BZCYh |
1304 (700 °C) |
Ni-BZCY|BZCY |
177
|
| PrBa0.94Co2O5+δ/Pr0.5Ba0.47CoO3−δ |
∼0.025 (700 °C) |
GDC |
1100 (700 °C) |
Ni-YSZ|YSZ|GDC |
178
|
| PrBaCoFeO5+δ-YSZ |
0.075 (700 °C) |
YSZ |
910 (700 °C) |
Ni-YSZ|YSZ |
180
|
| Pr0.9Y0.1BaCo1.8Ni0.2O6−δ-SDC |
0.11 (700 °C) |
SDC |
— |
— |
181
|
| PrBaCo2O5+δ-SDC |
0.252 (600 °C) |
SDC |
1150 (650 °C) |
Ni-SDC|SDC |
182
|
| NdBaCo2O5+δ-GDC |
0.034 (700 °C) |
GDC |
1000 (700 °C) |
Ni-YSZ|YSZ|GDC |
183
|
| SmBaCo1.7Ni0.3O5+δ-SDC |
0.0272 (800 °C) |
SDC |
628 (800 °C) |
Ni-SDC|SDC |
184
|
| SmBaCo2O5+δ-SDC |
0.159 (700 °C) |
LSGM |
408 (700 °C) |
Ni-GDC|LDC|LSGM |
185
|
| SmBa0.5Sr0.5Co2O5+δ-GDC |
0.013 (700 °C) |
GDC |
1310 (800 °C) |
Ni-YSZ|YSZ|GDC |
186
|
| SmBa0.5Sr0.5Co2O5+δ-SDC |
1.28 (600 °C) |
SDC |
823 (800 °C) |
Ni-SDC|SDC |
187
|
| SmBa0.5Sr0.5Co2O5+δ-GDC |
0.031 (700 °C) |
GDC |
— |
— |
188
|
| SmBa0.5Sr0.5Co2O5+δ-ScSZ |
0.054 (700 °C) |
ScSZ |
1250 (700 °C) |
430L-YSZ|ScSZ |
189
|
| SmBa0.5Sr0.5Co2O5+δ-GDC |
0.019 (700 °C) |
YSZ |
— |
— |
191
|
| PrBaCo2O5+δ-SDC |
0.073 (700 °C) |
LSGM |
758 (800 °C) |
Ni-SDC|SDC|LSGM |
192
|
| PrBaCoFeO5+δ-SDC |
0.037 (800 °C) |
LSGM |
960 (800 °C) |
Ni-SDC|SDC|LSGM |
193
|
| PrBa0.92Co2O5+δ |
0.093 (600 °C) |
GDC |
— |
— |
194
|
| PrBaCo2O5+δ |
0.0594 (700 °C) |
GDC |
— |
— |
195
|
| Pr0.94BaCo2O5+δ |
0.11 (600 °C) |
GDC |
1050 (600 °C) |
Ni-GDC|GDC |
196
|
| PrBaCo1.94O5+δ |
0.059 (700 °C) |
GDC |
889 (650 °C) |
Ni-YSZ|YSZ|GDC |
197
|
| Sm0.95BaCo2O5+δ |
0.038 (750 °C) |
GDC |
— |
— |
199
|
| Y0.93BaCoCuO5+δ |
0.029 (800 °C) |
LSGM |
643 (800 °C) |
Ni-SDC|SDC|LSGM |
200
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ |
— |
— |
986 (800 °C) |
Ni-YSZ|YSZ|GDC |
225
|
4.2. Electrocatalytic OER advances
Compared with the ORR at high temperatures, the OER of perovskite oxides has sluggish reaction kinetics due to the four-proton/electron transfer steps from H2O or OH− to O2 with the HO*, O*, and HOO* intermediates (* represents the surface metal sites), as follows:202| | | O* + H2O → HOO* + H+ + e− | (11) |
Regarding the detailed OER mechanisms, two common pathways are debatable for perovskite oxides, i.e., the adsorbate evolution mechanism (AEM) and the lattice oxygen evolution mechanism (LOEM).13,14 In the AEM procedure, the O2 products are generated from adsorbed H2O or OH−, which proceeds through four-centered proton–electron transfer steps with a series of intermediates (Fig. 4a). The OER activity of electrocatalysts is confined by the different adsorption energies of the intermediates. Regarding the LOEM, the lattice–oxygen atoms of the perovskite oxides are coupled with H2O to produce O2, i.e., lattice–oxygen oxidation (Fig. 4b). Simultaneously, non-concerted proton–electron transfer reactions happen in the LOEM. The LOEM avoids the adsorption scaling relation and overcomes the restriction of a theoretical overpotential (η) of ∼370 mV for the AEM.202 Nonetheless, LOEM-based OER electrocatalysts suffer from operating instability due to the constant generation of oxygen vacancies during the lattice–oxygen oxidation process.
 |
| | Fig. 4 Electrocatalytic mechanisms of the (a) OER AEM, (b) OER LOEM, (c) alkaline HER, and (d) acid HER. | |
The OER investigations of layered perovskite oxides began with PrBCO in alkaline media.47 The structural evolution from disordered to ordered perovskite oxide is determined by the annealing temperature, i.e., disordered (Pr0.5Ba0.5)CoO3−δ, A-site disordered (Pr0.5Ba0.5)2Co2O5.5+δ, A-site ordered PrBaCo2O5+δ, and A-site ordered PrBaCo2O5.5+δ. Almost unchanged OER activity, at the current density (J) = 10 mA cm−2 at ∼1.8 V vs. reversible hydrogen electrode (RHE), is attained in all materials, revealing the negligible effect of the structural bulk phase on OER electrocatalysis. A and B-site co-doping has been performed in NdBCO to improve the OER activity. The overpotentials follow the order of NdBa0.5Sr0.5Co1.5Fe0.5O5+δ (361 mV) < SmBa0.5Sr0.5Co1.5Fe0.5O5+δ (372 mV) < GdBa0.5Sr0.5Co1.5Fe0.5O5+δ (390 mV) at 10 mA cm−2, with the smallest Tafel slope of 88 mV dec−1 for NdBa0.5Sr0.5Co1.5Fe0.5O5+δ.49 Based on the foundation of co-doping, the oxygen defects of PrBa0.5Sr0.5Co1.5Fe0.5O5+δ can be tailored carefully, and it is inferred that excessive oxygen vacancies promote the OH− affinity and reduce the formation energy of O* on the surface.203 We presented a series of EuBa0.5Sr0.5Co2−xFexO5+δ electrocatalysts for OER, gaining the optimal EuBa0.5Sr0.5Co1.6Fe0.4O5+δ one.204 Importantly, its OER activity is higher than that of the commercial IrO2 at η > 460 mV, clearly indicating its potential application at a large J. After that, we designed a composite electrocatalyst of EuBa0.5Sr0.5Co1.6Fe0.4O5+δ/20 wt% RuO2, which outperformed the high-performance RuO2 benchmark. This composite electrode-based electrolyzer delivered a low voltage (U) of 1.47 V for overall water splitting at 10 mA cm−2. DFT computation implies the presence of active Fe and Ru sites for the AEM process. The free energies of the intermediates on the Co, Fe, and Ru sites are calculated for EuBa0.5Sr0.5Co1.6Fe0.4O5+δ and RuO2 at different voltages (U = 0 and 1.23 V). The theoretical η values for the Fe and Ru atoms are 0.43 and 0.78 V, respectively. The results indicate that octahedral Fe atoms are highly active for OER. Conversely, the larger theoretical overpotentials of the Co and Sr atoms (1.73 and 2.01 V) exclude their probabilities as the reaction sites, respectively. In the case of an ideal oxide electrocatalyst, ΔEHOO* and ΔEHO* are predicted to be 3.69 and 1.23 eV, respectively, and ΔEO* is located in the middle at 2.46 eV.202 The octahedral Fe atom has a ΔEO* of 2.60 eV, and the ΔEHOO* and ΔEHO* of the Ru atom are 3.68 and 1.03 eV, respectively, testifying the cooperation between Fe and Ru for water oxidation. Analogous composite electrocatalysts, PrBa0.5Sr0.5Co2O5+δ@FeOOH-20, PrBa0.5Sr0.5Co2O5+δ@CoP, Pr(Ba0.5Sr0.5)0.95Co1.5Fe0.5O5+δ@N-doped graphene, and (PrBa0.8Ca0.2)0.95(Co1.5Fe0.5)0.95Co0.05O5+δ@Co/CoOx, have been explored for alkaline OER.205–208 To find a solution for the low surface areas of perovskite oxides, mesoporous nanofibers (PrBa0.5Sr0.5Co1.5Fe0.5O5+δ and SmBa0.5Sr0.5Co2O5+δ) have been prepared through the electrospinning technique.50,209 Their well-regulated B-site transition metal ratios and high surface areas (∼20 m2 g−1) resulted in a better OER performance than IrO2. Jo et al. found that the reconstruction of perovskite oxides promotes the OER, demonstrating stable and high-efficiency electrocatalysts.210 To expound the function of B-site tuning of perovskite oxides, PrBa0.5Sr0.5Co2−xFexO5+δ with various x values was studied. It was found that the layered perovskite structure and randomly disordered perovskite structure coexisted, altering the intrinsic properties with efficient OER activity and prolonged stability (at 100 mA cm−2 over 2000 h). Oxygen-vacancy defects have been deemed to be crucial for electrocatalytic reactions. Upon treatment in Ar and H2 plasma, oxygen vacancies are generated in PrBa0.5Sr0.5Co1.5Fe0.5O5+δ with tunable concentrations, which are associated with significantly enhanced OER activity.211 The OER properties of layered perovskite oxides are still inferior to that of commercial IrO2 and RuO2 (Table 3), especially operating at a low J (10 mA cm−2). We conclude that the determination of the real active sites is meaningful for the development of OER electrocatalysts, such as surface reconstruction, composition, size, and dimension modulation.
Table 3 Comparison of alkaline OER and HER activities of the LnBCO-based perovskite oxides with that of commercial benchmarks
|
|
OER overpotential (mV) |
Medium |
HER overpotential (mV) |
Medium |
Ref. |
| LaBaCo2O5+δ |
— |
— |
−156 (@10 mA cm−2) |
KOH (1.0 M) |
44
|
| PrBaCo2O5+δ |
— |
— |
−255 (@10 mA cm−2) |
KOH (1.0 M) |
44
|
| NdBaCo2O5+δ |
— |
— |
−389 (@10 mA cm−2) |
KOH (1.0 M) |
44
|
| SmBaCo2O5+δ |
— |
— |
−456 (@10 mA cm−2) |
KOH (1.0 M) |
44
|
| GdBaCo2O5+δ |
— |
— |
−495 (@10 mA cm−2) |
KOH (1.0 M) |
44
|
| EuBaCo2O5+δ |
— |
— |
−551 (@10 mA cm−2) |
KOH (1.0 M) |
44
|
| PrBaCo2O5+δ |
— |
— |
−245 (@10 mA cm−2) |
KOH (0.1 M) |
45
|
| PrBaCo2O5.8 |
— |
— |
−240 (@10 mA cm−2) |
KOH (1.0 M) |
46
|
| PrBaCo2O5+δ |
570 (@10 mA cm−2) |
KOH (1.0 M) |
— |
— |
47
|
| NdBa0.5Sr0.5Co1.5Fe0.5O5+δ |
361 (@10 mA cm−2) |
KOH (0.1 M) |
— |
— |
49
|
| SmBa0.5Sr0.5Co1.5Fe0.5O5+δ |
372 (@10 mA cm−2) |
KOH (0.1 M) |
— |
— |
49
|
| GdBa0.5Sr0.5Co1.5Fe0.5O5+δ |
390 (@10 mA cm−2) |
KOH (0.1 M) |
— |
— |
49
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ nanofiber |
300 (@10 mA cm−2) |
KOH (0.1 M) |
— |
— |
50
|
| GdBa0.6Sr0.4Co2O5+δ |
460 (@1 mA cm−2) |
KOH (0.1 M) |
— |
— |
125
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ–Ag |
340 (@10 mA cm−2) |
KOH (1.0 M) |
∼−120 (@10 mA cm−2) |
KOH (1.0 M) |
175
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ |
313 (@10 mA cm−2) |
KOH (1.0 M) |
— |
— |
203
|
| RuO2 |
290 (@10 mA cm−2) |
KOH (1.0 M) |
— |
— |
204
|
| IrO2 |
400 (@10 mA cm−2) |
KOH (1.0 M) |
— |
— |
204
|
| EuBa0.5Sr0.5Co1.6Fe0.4O5+δ |
420 (@10 mA cm−2) |
KOH (1.0 M) |
— |
— |
204
|
| EuBa0.5Sr0.5Co1.6Fe0.4O5+δ–RuO2 |
290 (@10 mA cm−2) |
KOH (1.0 M) |
— |
— |
204
|
| PrBa0.5Sr0.5Co2O5+δ–FeOOH |
390 (@10 mA cm−2) |
KOH (0.1 M) |
−280 (@10 mA cm−2) |
KOH (0.1 M) |
205
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ–CoP |
378 (@10 mA cm−2) |
KOH (0.1 M) |
— |
— |
206
|
| (PrBa0.5Sr0.5)0.95Co1.5Fe0.5O5+δ-N-doped graphene |
320 (@10 mA cm−2) |
KOH (0.1 M) |
−230 (@10 mA cm−2) |
KOH (0.1 M) |
207
|
| (PrBa0.8Ca0.2)0.95(Co1.5Fe0.5)0.95Co0.05O5+δ@Co/CoOx |
410 (@10 mA cm−2) |
KOH (1.0 M) |
∼−210 (@10 mA cm−2) |
KOH (1.0 M) |
208
|
| SmBa0.5Sr0.5Co2O5+δ nanofiber |
370 (@10 mA cm−2) |
KOH (0.1 M) |
— |
— |
209
|
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ |
470 (@64.36 mA cm−2) |
KOH (1.0 M) |
— |
— |
211
|
| Gd0.5La0.5BaCo2O5+δ |
— |
— |
−240 (@338 mA cm−2) |
KOH (1.0 M) |
237
|
| Pr0.5La0.5BaCo2O5+δ |
— |
— |
−240 (@317 mA cm−2) |
KOH (1.0 M) |
237
|
| LaBaCo2O5+δ |
— |
— |
−233.2 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| Gd0.2La0.8BaCo2O5+δ |
— |
— |
−226.8 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| Gd0.4La0.6BaCo2O5+δ |
— |
— |
−209.8 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| Gd0.5La0.5BaCo2O5+δ |
— |
— |
−210.4 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| Gd0.8La0.2BaCo2O5+δ |
— |
— |
−236.2 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| GdBaCo2O5+δ |
— |
— |
−240.4 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| Pr0.5La0.5BaCo2O5+δ |
— |
— |
−209.9 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| Sm0.5La0.5BaCo2O5+δ |
— |
— |
−227.9 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| Pr0.5Gd0.5BaCo2O5+δ |
— |
— |
−239.9 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| Gd0.5La0.4Sr0.1BaCo2O5+δ |
— |
— |
−229.5 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| PrBa0.5Sr0.5Co2O5+δ |
— |
— |
−242.1 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| Ho0.8Ba0.6Sr0.6Co2O5+δ |
— |
— |
−238 (@10 mA cm−2) |
KOH (1.0 M) |
237
|
| P-doped Pr0.5La0.5BaCo2O5+δ nanofiber |
— |
— |
−208 (@10 mA cm−2) |
KOH (1.0 M) |
238
|
| P-doped Pr0.5La0.5BaCo2O5+δ nanofiber |
— |
— |
−307 (@500 mA cm−2) |
KOH (1.0 M) |
238
|
| Pt/C |
— |
— |
−646 (@1000 mA cm−2) |
KOH (1.0 M) |
240
|
| (LaBa)0.95Co2O5+δ |
— |
— |
∼−215 (@10 mA cm−2) |
KOH (1.0 M) |
240
|
| (LaBa)0.95Co2O5+δ |
— |
— |
−415 (@1000 mA cm−2) |
KOH (1.0 M) |
240
|
| Pt/C |
— |
— |
∼−974 (@2000 mA cm−2) |
KOH (1.0 M) |
241
|
| Pr0.4La0.5Co2O5+δ |
— |
— |
∼−636 (@2000 mA cm−2) |
KOH (1.0 M) |
241
|
| Pt/C |
— |
— |
−35 (@10 mA cm−2) |
KOH (1.0 M) |
242
|
| Pt/C |
— |
— |
−433 (@500 mA cm−2) |
KOH (1.0 M) |
242
|
| PrBa0.94Co2O5+δ/Pr0.5Ba0.47CoO3−δ |
— |
— |
−186 (@10 mA cm−2) |
KOH (1.0 M) |
242
|
| PrBa0.94Co2O5+δ/Pr0.5Ba0.47CoO3−δ |
— |
— |
−364 (@500 mA cm−2) |
KOH (1.0 M) |
242
|
Besides alkaline water electrolysis, H2O or CO2 can be decomposed into H2/O2 or CO/O2via SOECs. ABO3 perovskite oxides have been used as anode materials for oxygen evolution, such as La(Sr)MnO3−δ, LSCF, BSCF, and composite electrocatalysts.3,26,212–220 However, their electrochemical efficiencies are insufficient at high temperatures (≥800 °C). In an 80%H2O/20%H2 atmosphere, the BSCF/SDC anode-based electrolysis cell shows a relatively acceptable J of ∼0.75 A cm−2 at 1.4 V towards H2O decomposition at 850 °C.221 However, the J is merely ∼0.2 A cm−2 at 1.4 V towards the CO2 reduction reaction (CORR) at 800 °C.222 A vanadium-doped BSCF perovskite oxide was developed for the CORR, with a J of ∼0.3 A cm−2 under the same conditions.222 By reason of their superior charge transfer reaction and intrinsic oxygen defects, layered perovskite oxides are considered up-and-coming anode alternatives for the CORR. It should be noted that their efficiencies are much higher than that of simple perovskite oxides, suggesting their more prominent OER activity. A family of layered perovskite oxides (Ln = La, Pr, Nd, Sm, Eu, and Gd) has been assessed as OER electrocatalysts for SOECs. The J values are 1.50, 174, and 1.91 A cm−2 for the GdBCO, NdBCO, and LaBCO anode-based electrolysis cells at 1.8 V towards the CO2RR at 800 °C, respectively.43 The efficiency ordering is the same as their ORR activities, highlighting the relevance of lanthanide modulation with electrocatalytic activity. The doping strategy is carried out to search for high-standard anodes for SOECs. A considerable anode (PrBa0.95Gd0.05Co2O5+δ) is developed by abnormal Ba-site Gd3+ substitution, displaying a large J of 2.63 A cm−2 at 1.8 V towards the CO2RR at 750 °C.58 Given its high-performance LaBCO composition, the J can reach an ultra-high 3.62 A cm−2 for the LaBa0.7Ca0.3Co2O5+δ anode at 1.8 V (CO2RR at 800 °C).102 Yao et al. devised a perovskite oxide anode (PrBaCo0.3Fe1.7O5+δ) by controlling the Co/Fe ratios, possessing a superior OER activity with a J of ∼3.9 A cm2 at 1.8 V (CO2RR at 800 °C).223 The co-doping strategy is still effective in enhancing the oxygen evolution performance. A Ca and Co co-doped PrBa0.8Ca0.2Co1.96Ta0.04O5+δ anode shows robust efficiency, including J = 1.44 A cm−2 at 1.6 V with attractive stability over 100 h for the CO2RR at 800 °C.224 A representative perovskite oxide electrocatalyst, PrBa0.5Sr0.5Co1.5Fe0.5O5+δ, can also be used as the anode material for SOECs. The maximum J of 3.694 A cm−2 is achieved at 2.0 V for water electrolysis at 850 °C, with a stable operation for 120 h with reversibility,225 and this anode stabilizes a long-term J of 1.31 A cm−2 at 1.3 V (800 °C).226 The intrinsic properties can be optimized via dual cation-defect engineering, i.e., balanced oxygen vacancies and oxygen-ionic conductivity. The electrolysis cell with the Pr0.97Ba0.97Co1.5Fe0.5O5+δ/GDC composite anode actualizes a J of 2.61 A cm−2 at 1.5 V for the water electrolysis at 800 °C.227 Layered perovskite oxides are also applied to proton ceramic electrochemical cells. An A-site deficient (PrBa0.8Ca0.2)0.95Co2O6−δ oxygen electrode-based proton electrolysis cell has a J of 0.72 A cm−2 at 1.3 V for water electrolysis at 600 °C.228 It is known that the cathode (fuel electrode) exposed to H2O/CO2 dominates the performance of SOECs. However, our results suggest that the anode also contributes to the performance of SOECs to a certain degree.102 In fact, another series of Fe/Mn-based layered perovskite oxides, LnBa(Fe/Mn)2O5+δ, can serve as cathode materials to absorb the H2O/CO2 reactants,223,229–235e.g., Pr0.95Ba0.95Fe1.6Ni0.2Nb0.2O5+δ.235 Accordingly, future work should aim at developing bifunctional electrodes for ORR and OER, which are used in reversible solid oxide cells (RSOCs).
4.3. Electrocatalytic HER advances
Guiding progress has been made in gaining insight into the electrocatalytic HER mechanism of the water-splitting reaction from experimental and theoretical perspectives. Volmer–Tafel and Volmer–Heyrovsky mechanisms are the two widely accepted reaction pathways (Fig. 4c and d), respectively. In alkaline media, H2O molecules are adsorbed onto the active sites, and then dissociated into H* and OH−. The H* intermediates react with H* or H2O to produce H2:9| | | H2O + e− → H* + OH−, H* + H* → H2 (Volmer–Tafel) | (13) |
| | | H2O + e− → H* + OH−, H* + H2O + e− → OH− + H2 (Volmer–Heyrovsky) | (14) |
Regarding the acid HER, the H+ ions are adsorbed on the active sites, and the H2 products are desorbed from one or two active sites:15| | | H+ + e− → H*, 2H* → H2 (Volmer–Tafel) | (15) |
| | | H+ + e− → H*, H* + H* → H2 (Volmer–Heyrovsky) | (16) |
Layered LnBCO perovskite oxides have assured HER activity in alkaline media. Sun et al. systematically investigated PrBCO electrocatalysts with cubic, tetragonal, and orthorhombic structures, designating the optimal orthorhombic structure (δ ≈ 0.52) with an HER η of 245 mV at 10 mA cm−2 in 0.1 M KOH.45 Constantly progressive reaction steps are involved in the HER process in computational studies, including H2O adsorption on the surface, H2O dissociation into H* and –OH, formation of H* intermediates, and the combination of H* to form H2 (Fig. 5a).236 The RBaCo2O5.5, RBaCo2O5.75, and RBaCo2O6 models are constructed to calculate the free energies of these steps (Fig. 5b), in which a combined interplay between ordered oxygen vacancies (pyramidal high-spin Co3+ sites) and O 2p ligand holes (metallic octahedral intermediate-spin Co4+ sites) is assumed in RBaCo2O5.75. These two active sites produce a near-ideal reaction path to adsorb H2O and release H2, respectively. Experimentally, as-synthesized (Gd0.5La0.5)BaCo2O5.75 is superior to even the commercial Pt/C catalyst in terms of HER activity and stability. The rate-limiting step is determined to be the combination of adsorbed H* to form H2 (Fig. 5c), providing a theoretical basis for hydrogen-evolving electrocatalysis. Moreover, Guan et al. introduced A-site ionic electronegativity (AIE) as an efficient unifying descriptor to predict the HER activities of 13 cobalt-based perovskite oxides,237i.e., LaBaCo2O5.5+δ (Gd0), (Gd0.2La0.8)BaCo2O5.5+δ (Gd0.2), (Gd0.4La0.6)BaCo2O5.5+δ (Gd0.4), (Gd0.5La0.5)BaCo2O5.5+δ (Gd0.5), (Gd0.6La0.4)BaCo2O5.5+δ (Gd0.6), (Gd0.8La0.2)BaCo2O5.5+δ (Gd0.8), GdBaCo2O5.5+δ (Gd1), (Pr0.5La0.5)BaCo2O5.5+δ (Pr0.5La0.5), (Sm0.5La0.5)BaCo2O5.5+δ (Sm0.5La0.5), (Pr0.5Gd0.5)BaCo2O5.5+δ (Pr0.5Gd0.5), PrBa0.5Sr0.5Co2O5.5+δ (PrBa0.5Sr0.5), Ho0.8Ba0.6Sr0.6Co2O5.5+δ (Ho0.8Ba0.6Sr0.6), and Gd0.5La0.4BaSr0.1Co2O5.5+δ (Gd0.5La0.4Sr0.1). Their AIE values endow them with HER activity (η at 10 mA cm−2 and Tafel slope), as shown in the volcano curves. A fully identical layered perovskite oxide, (Gd0.5La0.5)BaCo2O5.5+δ, with an AIE value of ∼2.33, is predicted to have the peak HER activities (Fig. 5d and e). With the aid of X-ray adsorption spectroscopy and DFT computation, this performance can be attributed to the most appropriate electronic states with inductive effects in the perovskite structure, i.e., ∼+3.25 of Co valence state, medium Co–O covalency, band gap (0.14 eV), and O 2p-band position. The charge-transfer energy (Δ) will decrease from 6–7 eV for Co2+ to −2 eV for Co4+ (Fig. 5f), and the Co 3d–Co 2p covalency is enlarged with a shift in the electronic band states close to the EF. However, a perovskite oxide (LnBaCo2O5.75) with a moderate Co valence state (∼+3.25) satisfies the Sabatier principle. When the Co valence is too high (>3.25), the insufficient oxygen vacancies hamper H2O adsorption. If the Co valence is too low, the deficient Co4+ sites are not favorable for desorbing H2. Meanwhile, the O 2p-band center is −2.38 eV for Co3.25+, which is neither too close nor too far from the EF (−2.74 and −2.01 eV for Co3+ and Co3.5+), respectively. In terms of the AIE rule, two perovskite oxides with an AIE value of ∼2.33, Gd0.4La0.6Ba0.4Ca0.6Co2O5.5+δ and Pr0.5La0.5Ba0.5Ca0.5Co2O5.5+δ, have been synthesized for alkaline HER. Their activities are located at the top of the volcano plot and is comparable to that of Gd0.5La0.5BaCo2O5.5+δ. We discovered that the HER performance is related to the structural evolution of layered perovskite oxides.44 At tf ≈ 1.0, cubic perovskite oxide (LaBCO) shows an η of −156 mV at 10 mA cm−2 with a Tafel slope of 64.4 mV dec−1, which is much better than that of reported LnBCO-based electrocatalysts (Table 3). At a large current density (>230 mA cm−2), it even outperforms the Pt/C benchmark. The figure of merit for comparing the intrinsic activities of different electrocatalysts is the turnover frequency (TOF), which is defined as the number of hydrogen molecules evolved per second per active site. The TOF value of LaBCO is 4.1 H2 s−1 at η = 300 mV, significantly surpassing other perovskite oxides and Pt/C. Computational results reveal the rate-limiting step of H2 desorption, and the first-best energy collocation is identified for LaBCO including H2O adsorption (−0.43 eV), H2O dissociation (−0.06 eV), –OH desorption (−0.88 eV), and H2 desorption (1.12 eV). We perceive the necessity of design principles, opening up possibilities for the application of perovskite oxide electrocatalysts. Alternatively, tailored compositions and controlled morphologies ensure exceptional electrocatalytic properties. Li et al. prepared a P-doped Pr0.5La0.5BaCo2O5+δ electrocatalyst with a fiber-like shape, showing an η of −307 mV at 500 mA cm−2 with a Tafel slope of 32.9 mV dec−1.238 This fast hydrogen-evolving action exceeds the commonly used Pt/C. If the H* intermediates are insufficient, the rate-limiting step is the Volmer reaction with a theoretical Tafel slope of 120 mV dec−1. When the Heyrovsky or Tafel reaction is the rate-limiting step, the theoretical Tafel slope is 40 or 30 mV dec−1.239 The smaller value reflects the Volmer–Tafel mechanism with fast dynamics. We explored several perovskite oxide electrocatalysts via cation-defect engineering towards high-rate HER. A-site co-deficient (LaBa)0.95Co2O5+δ merely requires an η of −415 mV at 1000 mA cm−2.240 Most importantly, Pr-deficient Pr0.4La0.5BaCo2O5+δ can be operated under industrial-grade conditions, i.e., 500–2000 mA cm−2, resulting from its enhanced charge transfer and H2 desorption abilities.241 Additionally, layered perovskite oxide-based composite electrocatalysts are also active for the alkaline HER. Hetero-structured Pr0.94BaCo2O5+δ/Pr0.47Ba0.5CoO3−δ (tetragonal/cubic structure) affords a η of −364 mV at 500 mA cm−2.242 Heterostructures have a large surface area, more hydrophilic surface, and increased electrical conductivity arising from in situ exsolved cubic perovskite oxide, while improving the H2O adsorption/dissociation and H2 desorption processes.
 |
| | Fig. 5 (a) Crystal structures of oxygen vacancy-ordered RBaCo2O5.5 and RBaCo2O5.75, as well as RBaCo2O6 without oxygen vacancies. In the ROδ planes, the hollow, grey, and orange circles represent Copyr3+ (with oxygen vacancies), octahedral Co3+ (Cooct3+), and Cooct4+ configurations, respectively. (b) Computational predictions for the alkaline HER processes of H2O adsorption, H2O dissociation, formation of H* intermediates, and subsequent recombination of two H* to form H2 on the surface of RBaCo2O5.75. (c) The calculated free energies of the H2O adsorption (grey), H2O dissociation to adsorbed OH− and H* (blue), OH− desorption (orange), and hydrogen desorption (red) in the alkaline HER on RBaCo2O5.5, RBaCo2O5.75, and RBaCo2O6 surfaces. Reproduced with permission.236 Copyright 2019, Wiley. (d) The HER activity trends of pure-phase cobalt-based perovskite oxides at the overpotential at 10 mA cmoxide−2 as a function of A-site ionic electronegativity. (e) The plots of Tafel slope values as a function of A-site ionic electronegativity (the rate-determining steps estimated from the Tafel slope are shown in pink and purple dashed lines). (f) The charge-transfer models of Co2+, Co3+, and Co4+ in the covalent systems. Reproduced with permission.237 Copyright 2019, Nature. | |
5. Rational design of layered LnBCO perovskite oxides
The flexible elemental compositions and structural evolution of perovskite oxides are attributed to their diverse electronic structures. Their electronic structures significantly influence their physicochemical properties and electrocatalytic performance, which are linear combinations of atomic orbitals. In perovskite oxides, the interaction between their metal and oxygen atoms represents their electronic structures, impacting the adsorption/desorption of surface-bonded reactants and their electrocatalytic activities. Generally, the interaction between the B-site transition metal and O primarily contributes to the electrocatalysis reaction. The transition metal orbitals hybridize with the O 2p orbitals in the BO6 octahedron to produce σ and π orbitals. The σ and π orbitals are formed by the hybridization of dz2/dx2−y2 and dxy/dyz/dxz orbitals with O 2p orbitals, respectively. The σ bonding and π antibonding orbitals are more conventionally called the eg and t2g orbitals for perovskite oxides, respectively. During the electrocatalytic process, B-site transition metal atoms are typically the active sites with BOδ coordination, where the eg and t2g orbitals split into different energy levels. This energy-level splitting is vertical to the adsorption intermediates; thus, the eg states have a stronger overlap with the O 2p orbitals than that of the t2g states. Owing to this reason, the eg-orbital occupancy can approximate the adsorption strength of the intermediates, which correlates the electronic structures with the electrocatalytic properties.
Motivated by the success of the d-band theory as an activity descriptor for metal surfaces, a distinct OER activity design principle, i.e., the near-unity occupancy of the eg orbital of surface transition metal ions, can promote the intrinsic OER activity of perovskite oxides in alkaline media.243 This principle is based on a molecular orbital bonding framework. Because the eg orbital participates in σ-bonding with the anion adsorbates on the surface, the binding ability of oxygen-containing intermediate species at the B-sites affects the OER activity. In the case of La/Ba-based perovskite oxides, plotting their activities (voltage vs. RHE at a surface area-normalized current density of 50 µA cmox−2) as a function of the eg-orbital filling of the surface B-site cation can lead to a volcano plot (Fig. 6a). A highly active OER catalyst, BSCF, is obtained, which is the most popular oxygen electrode for SOFCs and oxygen permeation membranes. Moreover, BSCF has the highest OER activity among the studied perovskite oxides, as predicted by the eg descriptor. The electronic configuration of Co2.8+ in BSCF can be assigned to t52ge∼1.2g given that the Co ions are in the intermediate-spin states on the surface. A low eg-filling in the perovskite oxides is associated with the B-site cation–O bonding, which is too strong for O2 to be desorbed. Instead, high eg-filling prefers weak O2 adsorption. The best OER activity is correlated with a medium eg-filling value of ∼1. Notwithstanding, we take note of the eg occupancy from an ionic model, where the metal atoms are adopted as the active sites, and the participation of active lattice oxygen sites is ignored. Suntivich et al. probed some perovskite oxides with the same eg occupancy, e.g., LaMnO3, LaNiO3, and LaCoO3, but they observed different electrocatalytic activities.244 Consequently, it is difficult to explain these facts using the eg orbital occupancy.
 |
| | Fig. 6 (a) Relation between the OER catalytic activity, defined by the η at 50 µA cmox−2 and occupancy of the eg-symmetry electron of the transition metal (B in ABO3). The data symbols vary with the type of B ions (Cr, red; Mn, orange; Fe, beige; Co, green; Ni, blue; and mixed compounds, purple), where x = 0, 0.25, and 0.5 for Fe. The error bars represent the standard deviations of at least three independent measurements. The dashed volcano line is shown for guidance only. Reproduced with permission.243 Copyright 2011, Science. (b) Activity trend towards oxygen evolution plotted for perovskite oxides. The negative theoretical η is plotted against the standard free energy of the 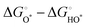 step. The low coverage regime is considered and the calculated values are used to show the activity of each perovskite oxide. The volcano curve is established by using the scaling relation between step. The low coverage regime is considered and the calculated values are used to show the activity of each perovskite oxide. The volcano curve is established by using the scaling relation between 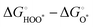 and and 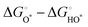 . Reproduced with permission.202 Copyright 2011, Wiley. (c) Experimental ASRs at ∼1000 K vs. the calculated bulk O p-band center of perovskites with simulated composition: 1. Ba0.5Sr0.5Co0.75Fe0.25O3 (BSCF), 2. Sm0.5Sr0.5CoO3 (SSC), 3. PrBaCo2O6 (PBCO), 4. GdBaCo2O6 (GBCO), 5. La0.625Sr0.375Co0.25Fe0.75O3 (LSCF), 6. La0.75Sr.25CoO3 (LSC), 7. LaCoO3 (LCO), 8. La0.75Sr0.25MnO3 (LSM), and 9. LaMnO3 (LMO). The values with vertical errors bars are the average of multiple data values with an error bar equal to the standard deviation in the mean of the ASR. (d) Schematic electronic structure plots illustrating the correlation of the ORR energetics vs. the O p-band center based on a rigid band model. The shaded areas (red and blue in color) represent the occupied transition metal 3d and oxygen 2p states, and the empty areas represent the unoccupied states, respectively. The bold and dotted lines are the Fermi energy level before removing O2− and before adding O2−, respectively. Reproduced with permission.245 Copyright 2011, the Royal Society of Chemistry. (e) Plots of logk* of typical perovskite oxides as a function of calculated O p-band center. The blue symbols are the experimental data. The k* data for the purple symbols are obtained using the reported ASR data. The green diamonds are plotted using predicted logk* values based on the linear fit of the experimental data. Reproduced with permission.246 Copyright 2018, Wiley. (f) Evolution of the iR-corrected potential at 0.5 mAoxide−2versus the O p-band center relative to EF (eV) of (Ln0.5Ba0.5)CoO3−δ with Ln = Pr, Sm, Gd and Ho, for LaCoO3 (LCO), La0.4Sr0.6CoO3−δ (LSC46), Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF82), Ba0.5Sr0.5Co0.4Fe0.6O3−δ (BSCF46) and SrCo0.8Fe0.2O3−δ (SCF82). The error bars represent the standard deviations from at least four independent measurements. Reproduced with permission.247 Copyright 2013, Nature. Experimental plots of (g) logJ and (h) logk* of typical perovskite oxides vs. calculated bulk O p-band enter for PBE. In the order of increasing J value for OER, the materials in (g) correspond to LaCrO3, LaFeO3, LaMnO3, LaCoO3, La0.75Sr0.25CoO3, LaNiO3, GdBaCo2O5.5, SmBaCo2O5.5, La0.5Sr0.5CoO3, and PrBaCo2O5.5. In the order of increasing k* value, the materials in (h) correspond to LaMnO3, LaCoO3, LaFeO3, La0.75Sr0.25CoO3, SrFeO2.75, La0.5Sr0.5CoO3, GdBaCo2O5.5, PrBaCo2O5.5, and Ba0.5Sr0.5Co0.75Fe0.25O2.625. Reproduced with permission.248 Copyright 2019, the American Chemical Society. . Reproduced with permission.202 Copyright 2011, Wiley. (c) Experimental ASRs at ∼1000 K vs. the calculated bulk O p-band center of perovskites with simulated composition: 1. Ba0.5Sr0.5Co0.75Fe0.25O3 (BSCF), 2. Sm0.5Sr0.5CoO3 (SSC), 3. PrBaCo2O6 (PBCO), 4. GdBaCo2O6 (GBCO), 5. La0.625Sr0.375Co0.25Fe0.75O3 (LSCF), 6. La0.75Sr.25CoO3 (LSC), 7. LaCoO3 (LCO), 8. La0.75Sr0.25MnO3 (LSM), and 9. LaMnO3 (LMO). The values with vertical errors bars are the average of multiple data values with an error bar equal to the standard deviation in the mean of the ASR. (d) Schematic electronic structure plots illustrating the correlation of the ORR energetics vs. the O p-band center based on a rigid band model. The shaded areas (red and blue in color) represent the occupied transition metal 3d and oxygen 2p states, and the empty areas represent the unoccupied states, respectively. The bold and dotted lines are the Fermi energy level before removing O2− and before adding O2−, respectively. Reproduced with permission.245 Copyright 2011, the Royal Society of Chemistry. (e) Plots of logk* of typical perovskite oxides as a function of calculated O p-band center. The blue symbols are the experimental data. The k* data for the purple symbols are obtained using the reported ASR data. The green diamonds are plotted using predicted logk* values based on the linear fit of the experimental data. Reproduced with permission.246 Copyright 2018, Wiley. (f) Evolution of the iR-corrected potential at 0.5 mAoxide−2versus the O p-band center relative to EF (eV) of (Ln0.5Ba0.5)CoO3−δ with Ln = Pr, Sm, Gd and Ho, for LaCoO3 (LCO), La0.4Sr0.6CoO3−δ (LSC46), Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF82), Ba0.5Sr0.5Co0.4Fe0.6O3−δ (BSCF46) and SrCo0.8Fe0.2O3−δ (SCF82). The error bars represent the standard deviations from at least four independent measurements. Reproduced with permission.247 Copyright 2013, Nature. Experimental plots of (g) logJ and (h) logk* of typical perovskite oxides vs. calculated bulk O p-band enter for PBE. In the order of increasing J value for OER, the materials in (g) correspond to LaCrO3, LaFeO3, LaMnO3, LaCoO3, La0.75Sr0.25CoO3, LaNiO3, GdBaCo2O5.5, SmBaCo2O5.5, La0.5Sr0.5CoO3, and PrBaCo2O5.5. In the order of increasing k* value, the materials in (h) correspond to LaMnO3, LaCoO3, LaFeO3, La0.75Sr0.25CoO3, SrFeO2.75, La0.5Sr0.5CoO3, GdBaCo2O5.5, PrBaCo2O5.5, and Ba0.5Sr0.5Co0.75Fe0.25O2.625. Reproduced with permission.248 Copyright 2019, the American Chemical Society. | |
The adsorption free energies of the intermediates can directly convey the electrocatalytic activity. Progress in the DFT calculations makes it possible to use them as accurate descriptors. Considering the OER intermediates as O*, HO*, and HOO*, the scaling relationship can be established between the binding energies of these species on the oxide surfaces. Man et al. established the formal scaling relationship between the HO* and HOO* binding energies on rutile, spinel, rock salt, bixbyite, and perovskite oxides.202 The linear correlation implies that HO* and HOO* normally prefer the same type of binding site, and the difference of ΔEHOO* − ΔEHO* is almost kept at a constant of 3.2 eV for all the studied oxides. Given the constant difference between the HO* and HOO* levels, the difference of 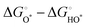 is a unique descriptor for OER activity. The theoretical overpotential (ηOER) under standard conditions is calculated using eqn (17), as follows:
is a unique descriptor for OER activity. The theoretical overpotential (ηOER) under standard conditions is calculated using eqn (17), as follows:
| | 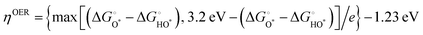 | (17) |
The
ηOER values are plotted as a function of
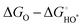
for a series of perovskite oxides, resulting in a universal volcano relationship (
Fig. 6b), in which SrCoO
3 with
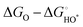
of 1.48 eV is close to the very top of the volcano.
Electronic structure-type descriptors are also proposed for understanding the structure–property–performance relationship of perovskite oxides towards high-temperature ORR. According to Morgan's inference, the experimentally measured ASRs and k* values of the perovskite oxide cathodes for SOFCs are strongly correlated with the DFT-calculated O 2p-band center and oxygen vacancy formation energy, including layered perovskite oxides, PrBCO and GdBCO.245 As shown in Fig. 6c, the log of the ASRs is linearly correlated with the calculated bulk O 2p-band centers, suggesting the effectiveness of the O 2p-band center as a descriptor for ORR activity. The BSCF cathode with the lowest ASR is located at an O 2p-band center of ∼1.5 eV. A united trend is observed in the log of the k* values vs. O 2p-band centers. The O p-band descriptor can be qualitatively understood in terms of a rigid band model, which is used to describe some electron-rich perovskite oxides. Upon the addition of O to the material, electrons move from the EF to the O p-band, and oxygen removal corresponds to the electrons moving in the opposite direction (Fig. 6d). Because the O adsorption/desorption processes govern many aspects of ORR, the O p-band center is considered to be an effective descriptor for ORR and its relevant steps. As correlated between the calculated O 2p-band centers and predicted k* values, 2145 distinct perovskite oxides have been screened as highly active and stable SOFC cathodes via high-throughput DFT computation.246 The screening method qualitatively reproduces the experimental activity, stability, and conduction nature of well-studied cathode materials, among which 52 potential cathode materials with good predicted stability under SOFC operating conditions and predicted k* on par with first-class perovskite oxide cathodes are obtained. The calculated O p-band center is also used as a first principle-based descriptor of k*, which in turn correlates with the ORR activity (Fig. 6e), further verifying the reliability of the O p-band center. Grimaud et al. found that the alkaline OER properties of double perovskite oxides, (Ln0.5Ba0.5)CoO3−δ (Ln = Pr, Sm, Gd, and Ho), can be correlated likewise with the O p-band center.247 The high activity and stability can be explained by having the O p-band center neither too close nor too far from the EF. The η values (0.5 mA cmox−2) of these double perovskite and leading pseudocubic perovskite oxides are well correlated with the computed O p-band centers, as depicted in Fig. 6f. Moving the O p-band center close to the EF from LaCoO3 to (Pr0.5Ba0.5)CoO3 promotes the intrinsic OER activity. However, further lifting the O p-band center gives rise to a decrease in the activity and stability. No visible changes are observed in the perovskite oxides on the left branch during the OER process, whereas rapid amorphization in the near-surface regions occurs for the ones on the right branch, accompanied by leaching of the A-site ions. As a consequence, an active and stable (Pr0.5Ba0.5)CoO3 OER catalyst is obtained upon water oxidation in alkaline solution. However, this prediction largely depends on the DFT computation, given that discrepancies exist among the predictions made using different DFT exchange and correlation functionals and from experiments. Jacobs et al. compared five different DFT exchange and correlation functionals including Perdew–Burke–Ernzerhof (PBE), PBEsol, PBE+U, strongly constrained and appropriately normed functional (SCAN), and Heyd–Scuseria–Ernzerhof (HSE).248 The best correlations for all the measured ORR and OER activities, experimental logJ (1.6 V vs. RHE for OER) and logk*, are made with PBE-level calculations with strong observed linear correlations as a function of bulk O p-band (Fig. 6g and h), respectively. In an account, Shao-Horn et al. reminded us of the potential limitations of this approach in practice.249 A specific limitation of the bulk O p-band center descriptor is that it is more applicable to predicting surface termination-sensitive phenomena. Given that the bulk and surface O p-band center are directly correlated with each other, the bulk descriptor provides a robust trend with surface-sensitive phenomena. More prevalent material databases and machine learning methods offer a chance to explore the descriptor-based mode for discovering and designing electrocatalysts.
Employing soft X-ray emission and absorption spectroscopy, the partial density of states of perovskite oxides are analyzed on an absolute energy scale, and the energy barriers for electron transfer and surface deprotonation are correlated with their OER activity.250,251 The charge-transfer energy is defined as the energy difference between the orbital center of the O 2p orbital and the unoccupied metal 3d orbital,252,253 which plays a pronounced role in adjusting the properties of oxides, e.g., delocalization of electrons and the redox couple of oxides.254–256 According to the plot of J (1.6 V vs. RHE for OER) as a function of change-transfer energy, reducing Δ greatly enhances the OER activity, and the trend passes through all materials from semiconducting to semi-metallic perovskite oxides (Fig. 7a), including layered perovskite oxides (PrBCO, SmBCO, and GdBCO).251 The Fermi level lies in forbidden energies and is poorly defined for semiconducting and insulating oxides. Therefore, the O p-band center is less reliable to describe the OER activity than the charge-transfer energy of all types of perovskite oxides. From semiconducting to semi-metallic perovskite oxides, a decrease in Δ corresponds to an increase in their linear slope relative to their OER activity (Fig. 7a). The change in slope is a typical indicator of a shift in the rate-limiting step. Perovskite oxides with high Δ values have lower activation energies for proton transfer, indicating that the concerted pathway instead becomes the electron-transfer step. When Δ decreases, the perovskite oxides employ a decoupled proton–electron transfer pathway. As Δ decreases further in the case of semi-metallic perovskite oxides, the rate of the OER is limited by a proton-transfer step. It can be seen that a decrease in Δ promotes the electrocatalytic activity, with a change in the OER mechanism from electron-transfer-limited to proton–electron-coupled and proton-transfer-limited reactions. The partial density of states (PDOS) of these 10 perovskite oxides are examined on an absolute energy scale to grasp the function of Δ on physicochemical properties and OER activity. All the perovskite oxides have similar O 2p energy levels, and their Δ values are from the unoccupied 3d-band of their transition metal (Fig. 7b).251 The 3d-band-relevant descriptors (e.g., oxidation state and number of d electrons) should correlate with OER activity because they have a strong effect on the 3d-band position.257 As dictated by Δ, the band positions can decouple the electronic and chemical interactions at the electrode/electrolyte interface for the OER, i.e., electron-transfer kinetics, hydroxide affinity, and adsorbate binding (Fig. 7c and d).251 The electron affinity of the oxide to the electrolyte corresponds to the Schottky barrier for an electron-transfer step. The perovskite oxides with a higher Δ value have a higher Schottky barrier for electron transfer at the electrode/electrolyte interface (Fig. 7c and d). With a decrease in Δ, the unoccupied energy level falls below the OER potential energy without the energy barrier for electron transfer (Fig. 7d). Clearly, semi-metallic perovskite oxides with low energy barriers for electron transfer converge to a Tafel slope of ∼60 mV dec−1. In contrast, semiconducting perovskite oxides with high Δ values have high Tafel slopes of 100–180 mV dec−1.
 |
| | Fig. 7 (a) Correlation between the OER J at 1.6 V vs. RHE and the charge-transfer energy. The charge-transfer energy is defined differently for conductors (open) and semiconductors (closed). Oxide abbreviations: La0.8Sr0.2CoO3−δ (LSCO20), La0.5Sr0.5CoO3−δ (LSCO50), GdBaCo2O5+δ (GBCO), SmBaCo2O5+δ (SBCO), and PrBaCo2O5+δ (PBCO). (b) Electron transfer energy at the electrolyte-oxide interface, hydroxide affinity of the oxide, and oxide metal–oxygen bond strength estimated from X-ray spectroscopic data under rigid band assumption. Schematic of the features used to calculate the electron transfer energy, hydroxide affinity, and charge-transfer energy of the oxides, and trends in the band positions of the perovskite oxides (relative to vacuum) from the lowest to highest OER activity are shown on the right. The arrows are drawn to show a positive difference. The standard thermodynamic sign convention (negative energy is stronger affinity) is used. For conductors, the EF and unoccupied transition metal 3d-O 2p states are identical. The dashed line indicates the O2 and H2O/OH− redox level at pH 13. (c) Surface properties-Tafel slope, pH of zero charge, and adsorbate binding energy of HO* intermediates relative to O* on the transition metal site-relative to the electron transfer barrier, hydroxide affinity, and charge-transfer energy, respectively. (d) Trends in the electron transfer energy, hydroxide affinity of the perovskite oxide surfaces extracted from the band positions as a function of charge-transfer energy, as well as relative adsorbate binding energy of intermediates. Reproduced with permission.251 Copyright 2017, the Royal Society of Chemistry. | |
Currently, a universal descriptor-based approach has not been established to correlate the HER activity. Guan et al. bridged the relationships between OER/HER activities and over ten representative material properties of 12 3d metal-based perovskite oxides (Fe, Co, Ni, and Mn).258 Among these property parameters, the charge-transfer energy can serve as an ideal unifying descriptor, where neither too high nor too low Δ (∼1 eV) is related to the great electrocatalytic activity, fulfilling Sabatier's principle. This principle states that the most efficient electrocatalyst binds the reaction intermediates neither too strongly nor too weakly.250,259 The correlations between macroscopic physicochemical properties (hydrophilicity, aerophobicity, and conductivity) and the OER/HER activities are first studied in 1.0 M KOH solution. The OER/HER η values at 200 µA cmox−2 are used for comparison. The methodologies of significance analysis and regression analysis are applied,260,261 where the degree of correlation between properties and activities can be quantified by the obtained p value. A small p value (≤0.05) means a significant property–activity relationship. The correlation degrees exclude the possibilities of solid–liquid contact angle and electron transfer as universal activity descriptors. However, the solid–gas contact angle can be a potential descriptor. Secondly, the correlations between molecular-level structural properties (bond length, cell volume, and strain) and the OER/HER activities are discussed. The low correlation degrees indicate that these structural features are unsuitable as ideal descriptors. Then, the electronic structural properties of the selected perovskite oxides are correlated with their intrinsic OER/HER activities, such as 3d metal valence state, charge-transfer energy, O 2p-band center, and conduction band gap. The correlations between Δ and OER/HER activities are significant. Systematic experiments and computations untangle that the La0.5Sr0.5CoO3−δ perovskite oxide (Δ ≈ 1 eV) has metal-like high-valence configurations with an LOEM for the OER/HER. However, 12 representative cation-disordered perovskite oxides are considered in this work. Thus, it is hoped that the universality of the Δ descriptor for layered perovskite oxides will be further validated.
To figure out the structure–property–performance relationship, the proper descriptors should be established to guide the design of needed electrocatalysts. As mentioned above, the correlations between the structure/properties of perovskite oxides and their electrochemical performance (ORR, OER, and HER) are discussed with several descriptors, such as eg occupancy, adsorption free energy, O 2p-band center, A-site ionic electronegativity, and charge-transfer energy. Although these parameters are referred to in some studies, they are not rigorously validated across a variety of compositions or experimental conditions. Each is directed against selected materials (specific perovskite series) and has merits and demerits. The accuracy of the proposed descriptors needs to be further verified. The individual proposed descriptor has a limitation in the performance statistics of all perovskite materials. Thus, a unified descriptor is lacking for different perovskite series. Considering the wide physicochemical space of perovskite oxides, machine learning, high-throughput DFT computation, and artificial intelligence represent powerful tools for unveiling their structure–property–performance relationship and searching for new perovskite catalysts.246,262,263 The conventional trial-and-error process for sample synthesis, physico-chemical characterization, and performance testing takes very long. Alternatively, an experimentally proven machine-learning-driven method is demonstrated to accelerate the discovery of efficient air electrodes for SOFCs, where ionic Lewis acid strength (ISA) is adopted as an effective descriptor for the ORR activity of perovskite oxides.262 Four perovskite oxides, Sr0.9Cs0.1Co0.9Nb0.1O3 (SCCN), Ba0.4Sr0.4Cs0.2Co0.6Fe0.3Mo0.1O3 (BSCCFM), Ba0.8Sr0.2Co0.6Fe0.2Nb0.2O3 (BSCFN), and Sr0.6Ba0.2Pr0.2Co0.6Fe0.3Nb0.1O3 (SBPCFN), screened from a huge amount of perovskite compositions (6871 distinct materials), are experimentally synthesized and have superior ORR activities. The overall workflow for the identification of high-performance oxygen electrodes is illustrated in Fig. 8a, including machine learning model training, materials screening, experimental verification, and computational analysis.262 According to the experimental characterization, decreased A-site and increased B-site ISAs in the perovskite oxides can improve their surface oxygen exchange kinetics. Among the four as-synthesized perovskite oxides, SCCN in particular has excellent ORR activity with extremely low ASR of 0.088 Ω cm2 at 550 °C and the lowest Ea (107 kJ mol−1) (Fig. 8b). The Ni/SDC cermet-anode-supported single cell delivers the PPDs of 2.05 W cm−2 at 650 °C (Fig. 8c). According to closed-loop experiments and active learning, a champion four-metal perovskite oxide, Ca0.8Pr0.2Co0.8Fe0.2O3−δ (CPCF), emerged from 10101 perovskite oxide candidates for oxygen-evolving electrocatalysis.263Fig. 8d shows the predicted η values of the ABO3 structures with Ca/Pr at the A-site and Co/Fe at the B-site, namely, CaxPr1−xCoyFe1−yO3. The most promising composition, CPCE, is chosen from the list of candidates. This OER electrocatalyst exhibits an intrinsic η of 391 mV at 10 mA cmoxide−2 (Fig. 8e), which is the lowest value among the four-metal perovskite oxides. However, the prediction of electrocatalytic properties with data-driven machine learning is still in the primary stage, and only a few studies used this technique to predict the ASR, η, and oxygen-ionic conductivity.262–266 Jacobs et al. developed a machine learning model to predict the catalytic properties of perovskite oxides for SOFCs/SOECs, including oxygen surface exchange rate, bulk diffusivity, and ASR. Compared with the models based on ab initio-derived features, these developed models are based on trivial-to-calculate elemental features, which are more accurate and faster.267 A schematic outline of the present work is illustrated in Fig. 9a, in which more mature data-centric approaches can be utilized to discover new materials and understand high-performing materials for SOFCs/SOECs, i.e., electrocatalytic ORR and OER applications. For example, the developed machine leaning model is used to calculate the ASRs of the perovskite oxides, together with calculations of materials cost and stability, screening new promising catalysts from a total of 19072821 materials. To find novel promising perovskite catalysts, the screening criteria are set to be values below a threshold of ASR, cost, and stability, including 1.33/0.21 Ω cm2 (LSCF and BSCF at 500 °C), 133.67 dollars kg−1 (LSCF), and 93.3 meV atom−1 (SCCN at 500 °C), respectively. Fig. 9b–d show the distributions of predicted logASR (500 °C), cost, and stability. It can be seen that 2135396, 2453872, and 1393424 materials separately pass the screening criteria of ASR, cost, and stability, respectively. Finally, 9135 (∼0.05%) materials meet all the screening criteria. From the list of screened materials, the most highly active, cheapest, and most stable candidates are SrCo0.75Nb0.125Ta0.125O3 (logASR = −0.43 Ω cm2), BaFe0.75Cu0.125Zr0.125O3 (1.15 dollars kg−1), and BaFe0.5Co0.25Mo0.25O3 (18.0 meV atom−1), respectively.267 As shown in Fig. 9e, SrZr0.125Nb0.125Co0.625Cu0.125O3 (SZNCCu) (logASR = −0.37 Ω cm2), K0.25Sm0.125Sr0.625Nb0.125Ta0.125Co0.75O3 (KSmSCNT) (logASR = −0.33 Ω cm2), and Bi0.125Sr0.875Y0.125Ni0.125Co0.75O3 (BiSYNC) (logASR = −0.25 Ω cm2) are predicted to outperform high-performing BSCF and SCCN, which are consistent with the experimental ASR values. Furthermore, they have lower Ea values than BSCF, indicating their performance will continue to be utilized <500 °C.267 In summary, to establish a universal electrocatalysis descriptor for perovskite oxides, efforts need to be devoted to all-type perovskite oxides.
 |
| | Fig. 8 (a) The overall workflow diagram: machine learning model training, material screening, experimental verification, and DFT analysis. θ is X-ray incidence angle. (b) Arrhenius-type plots of ASR values of SCCN, BSCCFM, BSCFN, and SPBCFN. (c) Current density–voltage-power density curves of a single cell with the configuration Ni/SDC|SDC|SCCN at 450–650 °C. Reproduced with permission.262 Copyright 2022, Nature. (d) The predicted overpotential at 10 mA cmoxide−2 of CaxPr1−xCoyFe1−yO3 (0 ≤ x, y ≤ 1) oxides according to the precursor mixture ratio of Ca, Pr, Co and Fe. (e) Quantification of the electrochemical properties of CPCF compared to standard catalysts: linear sweep voltammetry curves. The inset shows Tafel slopes. Reproduced with permission.263 Copyright 2023, Nature. | |
 |
| | Fig. 9 (a) Schematic of the present work of using data-centric machine learning approaches for predicting catalytic properties. Violin plots showing the distributions of screened materials: (b) screened ASR, (c) screened materials cost, and (d) screened materials stability. The numbers above each distribution denote the number of materials passing the given screening combination. The high, middle, and low colored ticks denote the maximum, median, and minimum of the distribution, respectively, while the black ticks denote the mean of the distribution. (e) Machine learning-predicted ASR temperature dependence for key materials. The solid lines are machine learning predictions using predicted logASR value at 500 °C together with the machine learning model of predicted ASR barrier to scale the prediction to other temperatures. The error bars are the calibrated one standard deviation error bars from the machine learning model. Data points are experimental ASR values. Reproduced with permission.267 Copyright 2024, Wiley. | |
6. Practical applications of layered LnBCO perovskite oxides
6.1 Electrocatalytic activity and stability
The noble metal-based materials RuO2 (IrO2) and Pt/C have been the most active electrocatalysts for water electrolysis. Electrolyzer devices with an RuO2 (IrO2) and Pt/C couple can act as a standard benchmark. Their overpotentials are recorded at 10 mA cm−2 to compare their OER and HER performances with other catalysts, i.e., acceptable requirement of solar fuel cell applications. The η values of RuO2 and Pt/C reach as low as 290 and −35 mV at 10 mA cm−2 in 1.0 M KOH solution, respectively.204,242 However, their polarization losses increase remarkably with an increase in the current density, such as −433, −646, and −974 mV for Pt/C at 500, 1000, and 2000 mA cm−2, respectively.240–242 During the alkaline HER process, some Pt nanoparticles disappear and tend to agglomerate, corresponding to a Pt dissolution/redeposition mechanism.268 Meanwhile, the surface region of the carbon support becomes amorphous. This surface chemistry modification hampers the anchoring of Pt, which is responsible for the performance degradation of Pt/C. Similarly, Ru (Ir) forms a thermodynamically unstable oxide at high potentials, dissolving according to Ru(Ir)O2 + 2H2O → RuO42− + 4H+ + 2e−.269 In this non-stable stage, Ru(Ir) shows a fast dissolution rate. Alternatively, the degradation of IrO2 during OER may originate from the unstable oxygen anions in its oxide lattice.270
Comparatively, the η values of layered LnBCO perovskite oxides are ∼−120–550 mV for alkaline HER at 10 mA cm−2, while η ≈ 310–570 mV for alkaline OER (Table 3). These activities are still much lower than that of the RuO2 (IrO2) and Pt/C benchmarks (Table 3). In the case of the HER, the computational H* adsorption energy on the Pt (111) surface (ΔGH* = −0.09 eV) is near to the ideal 0 eV,271 which is consistent with Pt being the most efficient HER electrocatalyst. However, this compares with the higher adsorption energy (0.62 eV) at the Co site on the PrBaCo2O6 (100) surface.242 Surprisingly, layered perovskite oxides outperform Pt/C upon a high current density.238,240–242 A-site cation-defect engineering demonstrates a highly active perovskite oxide (Pr0.4La0.5BaCo2O5+δ).241 The HER η is ∼−636 mV at 2000 mA cm−2, whereas η ≈ −974 mV for the commercial Pt/C catalyst. This tendency is in accordance with the OER experiments with the IrO2 benchmark.238,241 Furthermore, it preserves its stability even when operated at 500 mA cm−2 and undergoes a mild degradation rate of ∼0.5 mA cm−2 h−1, whereas Pt/C suffers from severe activity decline of ∼20.7 mA cm−2 h−1. In the ABO3 perovskite oxides, the A-site Ba2+ ions are easily leached from the surface region to the electrolyte due to the ionic bonding characteristic of Ba–O.272 The progressive amorphization breaks the charge transfer and suppresses the electrocatalytic activity. In the case of layered perovskite oxides, no elemental segregations are observed on the surface of Pr0.4La0.5BaCo2O5+δ after a continuous 10 h HER, maintaining a crystalline structure without any amorphous films.240 We infer that A-site Ln3+ ions can aid the stabilization of the Ba–O network. Regarding practical water splitting, the NiFe-LDH‖Pr0.4La0.5BaCo2O5+δ electrolyzer requires a voltage of 2.15 V to drive an ultrahigh J of 2000 mA cm−2, outperforming the IrO2‖Pt/C couple (2.45 V).240 As-reviewed layered perovskite oxides probably solve the problem of low efficiency at high current densities faced by noble metal catalysts. Aside from the electrocatalysts, the practical devices should fully take into account realistic production conditions, such as high current densities, long-term operation, high temperatures, and pressures.273 In laboratory evaluation, the stability test is continued for tens of hours at 10 mA cm−2 at 20–30 °C, and the industrial production is conducted at 200–2000 mA cm−2 under an operating pressure of 0.4–5.0 MPa for thousands of hours. These rigorous conditions probably cause catalyst failure, such as ion leaching, structural evolution, and electrode exfoliation, which cannot be monitored in laboratory-level tests. Therefore, despite the high catalytic performance (activity and stability) actualized in the laboratory, the performance evaluation should be supplied under industrial-grade conditions.
When assessed as oxygen electrodes for SOCs, layered perovskite oxides still possess strong stability at high temperatures. The classic LSCF oxygen electrode has poor durability due to Sr segregation on its surface,274,275 which is caused by the electrostatic attraction of positively charged oxygen vacancies.276 The Sr-enrichment induces the creation of other phases, such as SrCO3 and Sr(OH)2, giving rise to degradation of the electrode performance. Proven by experiments, elemental segregations appear in A-site cation-deficient PrBCO perovskites, forming (PrBa)CoO3 and BaO phases on their surface.100,178,242 Despite in situ exsolution on their surface, these composites influence the ORR activity in a positive way. Exsolved (PrBa)CoO3 perovskite oxides enhance the specific surface areas, electrical conductivity, oxygen adsorption/desorption, and transport abilities.178,242 As an insulator, amorphous BaO nanoparticles can physically adsorb the reaction substances, and then react with surface defects to participate in electrocatalysis.277 Compared to the stability of Ln0.94BaCo2O5+δ, the degradation rates of these oxygen electrode-based fuel cells follow the sequence of Sm0.94BaCo2O5+δ > Pr0.94BaCo2O5+δ > La0.94BaCo2O5+δ.100 After heating treatment, the BaO nanoparticles are in situ exsolved from the bulk. The Ba-segregation on the surface might be associated with the instability of Ln0.94BaCo2O5+δ. Long-term and stepwise tests confirm the steady operation of the LaBCO oxygen electrode in a fuel cell and electrolysis cell, surpassing the NdBCO and GdBCO oxygen electrodes. We deem that the degradation mechanism of layered perovskite oxides is elemental (barium) segregation. Cubic–tetragonal–orthorhombic structural evolution is identified with a decrease in the ionic size of Ln3+, accompanied by a decrease in the Co valence state and increase in oxygen vacancies. These two atomic-scale factors directly affect the catalytic pathways. In Co-containing perovskite oxides, the small polaron hopping mechanism dominates the charge transfer, i.e., the hole carriers of Co4+. Among the components, LaBCO shows the highest conductivity with an average Co valence state of ∼+3.36 and oxygen content of ∼5.86.43 The highest Co valence state corresponds to the best ORR activity of LaBCO, i.e., the highest electronic (hole) conductivity. The performance gradually weakens with a decrease in the ionic size of Ln3+. The general activity follows the sequence of cubic > tetragonal > orthorhombic perovskite oxides. As is known, oxygen-ionic conductivity is key to the ORR activity of MIECs, which is usually controlled by the vacancy mechanism. However, the interstitial oxygen and lattice oxygen mechanisms are also observed in perovskite oxide-like electrocatalysts.278 Although LaBCO has a lower oxygen-vacancy concentration, the oxygen bulk-diffusion process involves hole migration.279 The calculated oxygen-ionic conductivity of LaBCO is 1.65 S cm−1 at 800 °C, which is higher than that of the popular BSCF perovskite oxide (0.96 S cm−1).43,280 Accordingly, the vacancy mechanism is not supposed to be the principal pathway, and the active lattice oxygen mechanism may exist in the electrocatalysis reactions.278 Moreover, the more positive Co4+ sites prefer to adsorb oxygen species due to strong electrostatic affinity. As the initial step, abundant adsorbed oxygen species facilitate the ORR kinetics. A consistent performance trend is also discerned in the LnBCO series for alkaline HER.44 Therefore, advantageous oxygen-ionic conductivity, adequate electronic conductivity, and high average Co valence state play critical roles in the electrocatalytic activity of LnBCO. In all rare earth ions, the ionic radius of La3+ is closest to that of Ba2+. Given the strongest stability of LaBCO, the smaller size mismatch between La3+ and Ba2+ can reduce the segregation level due to the elastic energy difference, enabling more a stable electrode surface.276
In addition to sufficient ORR activity, the reasonable durability of the oxygen electrode is vital in practical SOFC and SOEC devices. If the oxygen electrodes are exposed to ambient air, they inevitably encounter some contaminants, such as ∼1 vol% CO2. Chen et al. reported a Ca-doped PrBCO oxygen electrode (PrBa0.8Ca0.2Co2O5+δ) with excellent ORR activity and strong CO2 tolerance.110 In air with ∼1 vol% CO2, this electrode can retain its ASR value after a 1000 h operation at 750 °C. According to impedance spectroscopy and in situ surface-enhanced Raman spectroscopy, the surface of PrBa0.8Ca0.2Co2O5+δ is more active for oxygen exchange and more robust against CO2 than that of LSCF. As supported by DFT calculations, PrBa0.8Ca0.2Co2O5+δ exhibits much weaker adsorption of CO2 than LSCF (−0.73 eV versus −1.25 eV), suggesting its CO2-tolerant characteristic. A highly active and CO2-tolerant oxygen electrode, Pr0.94Ba0.7Ca0.3Co2O5+δ, has been developed by our group.105 After annealing in air with 10 vol% CO2 for 12 h at 700 °C, the layered perovskite structure is still retained without any secondary phases. In turn, the carbonate of BaCO3 can be detected in undoped Pr0.94BaCo2O5+δ. Several influencing factors contribute to the CO2 tolerance of the oxides, such as average metal–oxygen bond energy (ABE), acidity of cations and defects, and oxygen-vacancy concentration.281–283 The ABE values can be calculated using the following equation:284
| | | 〈ABE〉 = 〈A–O〉 + 〈B–O〉 | (18) |
| | | 〈A–O〉 = (xA/nNA) × (ΔHAnOm − nΔHA − mDO2/2) | (19) |
| | | 〈B–O〉 = (xB/nNB) × (ΔHBnOm − nΔHB − mDO2/2) | (20) |
where
xA(B) is the molar ratio of A(B)-site cation; Δ
HAn(Bn)Om represents the standard molar formation enthalpy of A
n(B
n)O
m; Δ
HA(B) is the sublimation energy of A(B) metal;
NA(B) is the coordination number of the A(B)-site cation; and
DO2 is the dissociated energy of molecular oxygen. A more negative ABE value reflects a stable chemical bond with a strong CO
2-tolerant nature. If a metal cation has high acidity, it is hard to donate electrons to CO
2, which prevents the severe reaction with CO
2. Finally, a lot of oxygen vacancies mean high basicity with convenient reactivity with CO
2. Owing to the higher acidity and smaller radius of Ca
2+ than Ba
2+, Ba-site Ca-doped LnBCO compounds have increased ABE values and decreased oxygen vacancies, signifying their strong character against CO
2.
In summary, layered perovskite oxides exhibit much lower HER (OER) activities than that of the state-of-the-art Pt/C and RuO2 (IrO2) catalysts. However, their performance exceeds noble metal-based catalysts at large current densities, and can be maintained for a long working time. Not evident cation leaching and amorphous film are observed in the as-tested LnBCO electrocatalysts. Compared with Pt/C and RuO2 (IrO2), their intrinsic activity and stability preliminarily meet the requirement of industrial-grade water electrolysis (rapid and large-scale hydrogen production). Next, the studies should be concentrated on water electrolysis under practical conditions. Regarding the applications of SOCs, the electrocatalytic ORR activities of layered perovskite oxides are closely related to the structural evolution. Highly symmetrical cubic perovskite oxides are favorable for the ORR activity and stability, with a higher average Co valence state and mixed ionic and electronic conductivity. More highly oxidative Co4+ species mean more hole carriers, leading to higher electrical conductivity. An active lattice–oxygen mechanism with fast charge transfer rate boosts the ORR kinetics. During long-term operation, the performance degradation originates from the segregation of Ba on the surface. The smaller size mismatch between La3+ and Ba2+ reduces the segregation level, producing a more stable LaBCO oxygen electrode.
6.2 Production costs and scalability
According to the latest data announced by the United States Geological Survey, the reserves of as-proven rare earth and cobalt sources are ∼90 and 11 million tons, respectively. The reserve base of barium is as high as ∼700 million tons in the world. Thus far, the prices of La2O3, Co2O3, and BaO have stabilized at ∼640, 28000, and 1400 dollars ton−1, respectively. Thus, the cost of raw materials for LaBCO electrocatalysts is not more than 20 dollars kg−1. In comparison, the global reserve of platinum is ∼14000 tons, of which 98% is distributed in South Africa and Russia. The reserves of ruthenium and iridium are even scarcer, only ∼5000 and 1600 tons, respectively. At the current mining velocity, the amount of iridium will satisfy 15 years of mining. Thus, this scarcity, coupled with their harsh extraction conditions (high temperatures and high pressures), determines the high costs of noble metal catalysts. At present, the international prices of Pt, Ru, and Ir are ∼42.5, 31.5, and 51.5 dollars g−1, respectively. The progress in large-scale water electrolysis has been limited by the trade-off between the high cost of platinum-group-metal catalysts and the limited performance of earth-abundant metal alternatives. Considering the production costs and industrial-grade efficiency, this review summarizes a family of layered LnBCO perovskite oxides for addressing cost-effective, high-performance, and durable electrocatalysts in sustainable and clean energy fields.
The final stage of hydrogen production is “green hydrogen”, which is produced through the usage of renewable energy sources. For example, relatively mature water electrolysis for hydrogen production can realize complete decarbonization. Depending on their membrane materials, electrolyzers can be divided into alkaline water electrolysis (AWE), proton exchange membrane water electrolysis (PEMWE), anion exchange membrane water electrolysis (AEMWE), and SOEC water electrolysis (SOECWE). KOH electrolyte is used in alkaline water electrolysis cells. Differing from AWE, PEMWE uses a perfluorosulfonic acid proton exchange membrane as a solid electrolyte with good chemical stability, proton conductivity, and gas separation. AEMWE is technology for hydrogen production using an anion exchange membrane, with decreased dependence on noble metals and equipment costs. High-temperature SOECWE uses solid oxides as the electrolyte materials, with a working temperature of 700–1000 °C. The electrochemical performance of electrocatalysts is significantly improved, and the efficiency is higher. The electrodes of SOECs are non-noble metal catalysts. The cathode and the anode use porous metal ceramic (Ni/YSZ) and perovskite oxide, respectively, and the electrolyte uses an oxygen-ionic conductor (YSZ). An all-ceramic solid structure avoids material corrosion problems. According to laboratory-level experiments, LnBCO electrocatalysts are more adapted to AEMWE and SOECWE. In the case of AEMWE, their HER/OER performance (activity and stability) is greater than that of Pt/C and IrO2 (RuO2) catalysts. In high-temperature SOECs, LnBCO-based oxygen electrodes outperform the popular ABO3 perovskite oxides, demonstrating excellent OER activity between 700–800 °C. The electrolysis J can reach ∼2–3 A cm−2 at 1.8 V for the CO2RR at 800 °C, along with satisfactory long-term and cycling stabilities. However, these perovskite oxides are ordinarily prepared at high temperatures (∼1000 °C). The as-prepared electrocatalysts are on a micron scale with irregular shapes, and thus it is difficult to produce a homogeneous electrode ink for AWE and PEMWE. In industrial-grade water electrolysis, the production rate of hydrogen is very rapid, with massive bubbles on the electrode surface. Under extreme production conditions, the perovskite oxide electrocatalysts might be peeled off from the metal substrate. In this context, uniform nanomaterials have an unique merit. We should be dedicated to exploring nanoscale perovskite oxides, while maintaining their high electrochemical performance. In the case of SOECWE application, the practical working environment (high temperature and high humidity) limits the choices and efficiencies of materials for the electrolysis cells, and also restricts large-scale promotion. Presently, the electrochemical test is at the primary experimental stage, and the scalability of these electrocatalysts still has no clear conclusion. Encouraged by the production costs, electrocatalytic activity, and operating stability, we anticipate the expanding the scale of layered perovskite oxides for practical usage.
6.3 Expansion of application fields
We always expect to develop versatile catalysts for diverse catalysis reactions. Among all the types of electrocatalysts, noble metal-based materials (containing Pt, Ru, Ir, Rh, Pd, Au, and Ag) have received widespread attention due to their extraordinary performance in most electrocatalysis reactions, such as OER/HER for water electrolysis, ORR/OER in SOCs and metal–air cells, carbon dioxide reduction reaction (CO2RR), nitrate reduction reaction (NRR), methanol oxidation reaction (MOR), ethanol oxidation reaction (EOR), and formic acid oxidation (FAO).285–287 Furthermore, noble metal materials can also catalyze a variety of industrial non-electrocatalysis reactions, such as Fischer–Tropsch synthesis, CO oxidation/ammonia production, selective hydrogenation/hydroformylation, Suzuki coupling reaction, ethylene epoxidation, and water–gas shift reaction.287
The diversity and flexibility of the elemental components, crystal structures, and electronic structures of layered perovskite oxides bring new opportunities to extend their electrocatalysis scope. Besides the well-investigated ORR for O2−-SOFCs, recent work demonstrates highly active PrBCO-based oxygen electrodes for oxygen-conducting SOECs (O2−-SOECs), proton-conducting SOFCs (H+-SOFCs), and proton-conducting SOECs (H+-SOECs), such as Pr1.5Ba0.5Co2O5+δ, (La0.25Pr0.25Nd0.25Sm0.25)Ba0.5Sr0.5Co1.5Fe0.5O5+δ, PrBa0.5Sr0.5Co1.8Ni0.2O5+δ, PrBa0.5Sr0.5Co1.5Fe0.5O5+δ, and PrBaCo1.9Hf0.1O5+δ.288–292 Regarding extended electrocatalysis reactions, some perovskite oxides have been designed carefully, such as LaMnO3-based perovskite oxides for MOR,293,294 La0.6Sr0.4CoO3 for MOR,295 Eu2FeCoO6 for EOR and benzyl alcohol oxidation reaction (BOR),296 Sr2CuWO6 for CO2RR,297 LaMn0.6Co0.4O3, LaFe0.9Cu0.1O3, and (Ba0.5Sr0.5)0.85Co0.8Fe0.2O3−δ for NO3RR.298 In the reverse water–gas shift reaction, low-temperature CO2 conversion to CO can be catalyzed by the synergistic effect of binary Ni–Fe sites in double perovskite oxides (La2NiFeO6).299 To the best of our knowledge, there is little literature on LnBCO electrocatalysts for extended electrocatalysis and non-electrocatalysis reactions. As reported by Su et al., PrBCO can catalyze the generation of radicals from peroxymonosulfate for the oxidative degradation of organic waste in aqueous solution.300 In the PrBCO catalyst, the easy valence-state change of the B-site Co ions and intrinsic oxygen vacancies could mediate a redox process and be bonded with peroxymonosulfate, respectively. This work puts forward a new catalysis application for layered perovskite oxides. Heterogeneous noble-metal-based catalysts show a superior performance to noble metal-free ones in the above-mentioned reactions, which are adopted as benchmarks for direct performance comparison. Several characteristics of LnBCO make them more suitable for an extended electrocatalysis scope compared with noble-metal-based ones. (1) Their elemental compositions can be tailored vs. facile ion substitution, actualizing specific active sites for catalysis selectivity, e.g., complex MOR and EOR. (2) The synergistic effects of various metal sites in medium-entropy (high-entropy) perovskite oxides might realize multifunctional electrocatalytic activities, e.g., Fe–Co-based LnBCO electrocatalysts for ORR, OER, and HER. (3) Their crystal and electronic structures can be tuned via composition, size, and dimension modulation. Their structural evolution, electron-filled state, Co valence state, and oxygen vacancy defects could be tuned for a variety of electrocatalysis reactions. This goal would be given attention to realize new catalysis properties in layered perovskite oxides.
7. Conclusion and prospects
Over the past two decades, continuous efforts have been devoted to the study of complex oxides (beyond the common transition metal oxides and simple perovskite oxides) for oxygen and hydrogen electrocatalysis, yielding exciting results and stimulating great interest. In this review, we summarize the advances of layered LnBaCo2O5+δ perovskite oxide-based materials as all-in-one electrocatalysts for ORR, OER, and HER, such as Ln = La, Pr, Nd, Sm, Eu, Gd, and Y. In the discussion, we highlight how each crystal structure of layered perovskites is associated with their electrocatalytic properties. The frequently used strategies are introduced to regulate the crystal structures, elemental components, and apparent morphologies, further promoting their electrochemical performance. These approaches comprise cationic/anionic doping, phase-structure tuning (oxygen vacancy and cobalt valence tuning), morphology control, and hybrid composite. To improve their electrocatalytic activity, La/Ba-site and O-site substitutions are more effective than Co-site one. Moreover, creating hybrid composites might be a reliable method for enhancing their performance, leveraging the synergistic effects between different compositions. From a practical viewpoint, layered perovskite oxides can be applied in O2−/H+-SOFCs, O2−/H+-SOECs (H2O/CO2 decomposition), and overall water splitting. However, despite the significant achievements in available OER and HER, we propose the realistic challenges and future prospects. In our opinion, future work should be focused on the following aspects.
7.1 Exploring novel layered perovskite oxides
The comprehensive development of layered perovskite oxides remains a challenge, e.g., electrochemical field. Therefore, it is still necessary to explore novel layered perovskite oxides with improved performances in the future. Several types of electrocatalysts should be designed to address the issues in oxygen and hydrogen electrocatalysis. (1) All types of layered perovskite oxides have been intensively evaluated as oxygen electrodes for SOFCs, i.e., electrocatalytic intermediate/low-temperature ORR. The relevant OER/HER work is much less, and thus exploring high-performance electrocatalysts for the evolution reactions. (2) Nanomaterials have unique physicochemical properties due to the size and quantum effects. Perovskite oxides are typically micron-sized electrocatalysts with low specific surface areas. The large particle sizes of bulk materials hinder the exposure of their active sites, reducing the electrochemical reaction zone. Additionally, the irregular micron-sized particles make it an arduous challenge to obtain a homogeneous catalyst ink, affecting the subsequent deposition on the substrate. Consequently, we need to reduce the calcining temperature/time or develop a more advanced route for their preparation. (3) Regarding oxygen electrode applications, their thermal expansion behaviors should match that of the electrolyte components. The TEC values of common electrolytes are ∼10–12 × 10−6 K−1, and Co-based layered perovskite oxides have a TEC as large as ∼15–20 × 10−6 K−1. To eliminate the dilatometric mismatch, the incorporation of electrolyte components into the electrode backbone is one of the most effective strategies, while maintaining the ORR activity due to extended TPB lengths. Moreover, hybrid materials might exhibit extraordinary OER/HER performances. Thus, future developments should also cover composite electrocatalysts.
7.2 Accurately understanding the structure–property–performance relationship
An accurate understanding of the structure–property–performance relationship in layered perovskite oxides can guide the rational design of ideal electrocatalysts. Although many activity descriptors are correlated with the electrochemical performance of perovskite oxides, their accuracy needs to be further upgraded. In regard to this consideration, some suggestions are recommended, as follows: (1) even if current activity descriptors have made great progress, individual descriptors have monotonicity, limiting the rationalization of the performance trend of different electrocatalyst systems. We should strive to explore a universal descriptor for all oxide electrocatalysts, at least for ABO3 perovskite or layered perovskite oxides. Through high-throughput DFT computation, machine learning, and artificial intelligence, new members of descriptors would be proposed to provide more accurate performance prediction. (2) Structural and electronic features have been discussed for layered perovskite oxides, such as tolerance factor, O 2p-band center, electronegativity, and charge-transfer energy. Each feature has its merits and demerits, which are suitable for specific material systems. According to experimental/theoretical cross-validation, the generalizability of these features can be established beyond selected studies. (3) Transition metals are considered active sites for electrocatalysis reactions. In fact, the precise active sites should be defined by more advanced techniques and DFT computation, such as advanced in situ-electrochemical test/infrared spectroscopy, in situ-TEM, and in situ-X-ray adsorption spectroscopy. These in situ experiments are recognized as reputable techniques to investigate structural evolution and reaction intermediates. The reaction mechanisms and active sites of layered perovskite oxides have not been completely defined. Based on more advanced in situ techniques, dynamic adsorption/desorption on active sites can be identified, with observed structural evolution and intermediate formation. Then, a reasonable reaction route is determined by in situ experimental results and theoretical analysis, understanding the structure–property–performance relationship.
7.3 Prospects of practical applications
Layered perovskite oxides exhibit considerable electrochemical performances, demonstrating their potential application in SOFCs, SOECs, and water electrolysis. Future work should develop the corresponding practical devices, as follows: (1) layered perovskite oxides outperform the noble-metal-based benchmarks at large current densities (500–2000 mA cm−2) towards OER/HER, while exhibiting exceptional operating stability. These properties might allow them to be utilized for industrial-grade water splitting, e.g., ≥1000 mA cm−2 for several thousand hours, at high temperature and high pressure, and under strong alkali corrosion conditions. (2) Future research is advised to develop novel oxygen electrodes for reversible protonic ceramic cells, exerting efficient power generation and CO2/H2O decomposition. (3) The versatility and flexibility of crystal/electronic structures of layered perovskite oxides bring some opportunities for expanding the electrocatalytic scope, e.g., CO2RR, N2RR, electrocatalytic oxidation reaction, and organic synthesis reactions. We deem that this topic would be important if layered perovskite oxides are employed as multifunctional electrocatalysts, effectively addressing the energy and environmental crises.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no competing financial interest.
Data availability
Data for this article is available upon request to the corresponding authors of cited references.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52272197) and Natural Science Foundation of Heilongjiang Province (PL2024E025).
References
- Z.-Y. Yu, Y. Duan, X.-Y. Feng, X. X. Yu, M.-R. Gao and S.-H. Yu, Clean and Affordable Hydrogen Fuel from Alkaline Water Splitting: Past, Recent Progress, and Future Prospects, Adv. Mater., 2021, 33, 2007100 CrossRef CAS.
- A. D. Igalavithana, S. M. You, L. Zhang, J. Shang, J. Lehmann, X. N. Wang, Y.-G. Zhu, D. C. W. Tsang, Y.-K. Park, D. Y. Hou and Y. S. Ok, Progress, Batteries, and Prospects for Achieving a “Hydrogen Society” and Opportunities for Biochar Technology, ACS EST Eng., 2022, 2, 1987–2001 CrossRef CAS.
- Y. Zheng, J. C. Wang, B. Yu, W. Q. Zhang, J. Chen, J. L. Qiao and J. J. Zhang, A Review of High Temperature Co-electrolysis of H2O and CO2 to Produce Sustainable Fuels Using Solid Oxide Electrolysis Cells (SOECs): Advanced Materials and Technology, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- D. S. Dhawale, S. Biswas, G. Kaur and S. Giddey, Challenges and Advancement in Direct Ammonia Solid Oxide Fuel Cells: A Review, Inorg. Chem. Front., 2023, 10, 6176–6192 RSC.
- J. O′. M. Bockris, The Origin of Ideas on a Hydrogen Economy and Its Solution to the Decay of the Environment, Int. J. Hydrogen Energy, 2002, 27, 731–740 CrossRef CAS.
- D. Q. Guan, B. W. Wang, J. G. Zhang, R. Shi, K. Jiao, L. C. Li, Y. Wang, B. Xie, Q. W. Zhang, J. Yu, Y. F. Zhu, Z. P. Shao and M. Ni, Hydrogen Society: from Present to Future, Energy Environ. Sci., 2023, 16, 4926–4943 RSC.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, An Overview of Hydrogen Production Technologies, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- X. X. Zhou and Y. Zhang, Noble Metal-Free Hydrogen Evolution Catalysts for Water Splitting, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- C. L. Hu, L. Zhang and J. L. Gong, Recent Progress Made in the Mechanism Comprehension and Design of Electrocatalysts for Alkaline Water Splitting, Energy Environ. Sci., 2019, 12, 2620–2645 Search PubMed.
- Y. Jiang, H. Fu, Z. Liang, Q. Zhang and Y. P. Du, Rare Earth Oxide Based Electrocatalysts: Synthesis, Properties and Applications, Chem. Soc. Rev., 2024, 53, 714–763 RSC.
- Y. Li, Y. G. Deng, D. Q. Liu, Q. Q. Ji and X. K. Cai, Two Dimensional Oxides for Oxygen Evolution Reactions and Related Device Applications, Mater. Chem. Front., 2024, 8, 880–902 RSC.
- H. N. Sun, J. Dai, W. Zhou and Z. P. Shao, Emerging Strategies for Developing High-Performance Perovskite-Based Materials for Electrochemical Water Splitting, Energy Fuels, 2020, 34, 10547–10567 Search PubMed.
- X. Sun, Y. Yuan, S. Z. Liu, H. Q. Zhao, S. Q. Yao, Y. Y. Sun, M. Y. Zhang, Y. J. Liu and Z. Q. Lin, Recent Advances in Perovskite Oxides for Oxygen Evolution Reaction: Structures, Mechanisms, and Strategies for Performance Enhancement, Adv. Funct. Mater., 2025, 35, 2416705 CrossRef CAS.
- Y. L. Zhu, Z. Tang, L. J. Yuan, B. W. Li, Z. P. Shao and W. L. Guo, Beyond Conventional Structures: Emerging Complex Metal Oxides for Efficient Oxygen and Hydrogen Electrocatalysis, Chem. Soc. Rev., 2025, 54, 1027–1092 RSC.
- J. Zhu, L. S. Hu, P. X. Zhao, L. Y. S. Lee and K.-Y. Wong, Recent Advances in Electrocatalytic Hydrogen Evolution Using Nanoparticles, Chem. Rev., 2020, 120, 851–918 CrossRef CAS.
- N. Han, P. Y. Liu, J. Jiang, L. H. Ai, Z. P. Shao and S. M. Liu, Recent Advances in Nanostructured Metal Nitrides for Water Splitting, J. Mater. Chem. A, 2018, 6, 19912–19933 Search PubMed.
- M. Wang, L. Zhang, Y. J. He and H. W. Zhu, Recent Advances in Transition-Metal-Sulfide-Based Bifunctional Electrocatalysts for Overall Water Splitting, J. Mater. Chem. A, 2021, 9, 5320–5363 RSC.
- W. Zhang, N. Han, Y. H. Dou, X. Zhang, J. S. Luo, S. X. Dou and J. Fransaer, Applications of Cobalt Phosphide-Based Materials in Electrocatalysis, ACS Catal., 2025, 15, 5457–5479 CrossRef CAS.
- S. Perumal, I. Pokhrel, U. Muhammad, X. D. Shao, Y. Han, M. Kim and H. Lee, Recent Advances in Electrochemical Water Splitting Electrocatalysts: Categorization by Parameters and Catalyst Types, ACS Mater. Lett., 2024, 6, 3625–3666 CrossRef CAS.
- M.-I. Jamesh, D. Q. Hu, J. Wang, F. Naz, J. P. Feng, L. Yu, Z. Cai, J. C. Colmenares, D.-J. Lee, P. K. Chu and H.-Y. Hsu, Recent Advances in Noble Metal-Free Electrocatalysts to Achieve Efficient Alkaline Water Splitting, J. Mater. Chem. A, 2024, 12, 11771–11820 RSC.
- A. Hameed, M. Batool, Z. Y. Liu, M. A. Nadeem and R. C. Jin, Layered Double Hydroxide-Derived Nanomaterials for Efficient Electrocatalytic Water Splitting: Recent Progress and Future Perspective, ACS Energy Lett., 2022, 7, 3311–3328 CrossRef CAS.
- R. Abazari, S. Sanati, W. K. Fan, M. Tahir, S. Nayak, K. Parida, M. El-Shahat, R. M. Abdelhameed, D. S. Nesterov, A. M. Kirillov and J. J. Qian, Design and Engineering of MOF/LDH Hybrid Nanocomposites and LDHs Derived from MOF Templates for Electrochemical Energy Conversion/Storage and Environmental Remediation: Mechanism and Future Perspectives, Coordin. Chem. Rev., 2025, 523, 216256 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Materials for Fuel-Cell Technologies, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- S. Park, J. M. Vohs and R. J. Gorte, Direct Oxidation of Hydrocarbons in a Solid-Oxide Fuel Cell, Nature, 2000, 404, 265–267 CrossRef CAS.
- A. J. Jacobson, Materials for Solid Oxide Fuel Cells, Chem. Mater., 2010, 22, 660–674 CrossRef CAS.
- S. E. Wolf, F. E. Winterhalder, V. Vibhu, L. G. J. de Haart, O. Guillon, R.-A. Eichel and N. H. Menzler, Solid Oxide Electrolysis Cells – Current Material Development and Industrial Application, J. Mater. Chem. A, 2023, 11, 17977–18028 RSC.
- H. Liu, M. Yu, X. F. Tong, Q. J. Wang and M. Chen, High Temperature Solid Oxide Electrolysis for Green Hydrogen Production, Chem. Rev., 2024, 124, 10509–10576 CrossRef CAS PubMed.
- J. F. Cao, Y. X. Ji and Z. P. Shao, Nanotechnologies in Ceramic Electrochemical Cells, Chem. Soc. Rev., 2024, 53, 450–501 RSC.
- M. Humayun, Z. S. Li, M. Israr, A. Khan, W. Luo, C. D. Wang and Z. P. Shao, Perovskite Type ABO3 Oxides in Photocatalysis, Electrocatalysis, and Solid Oxide Fuel Cells: State of the Art and Future Prospects, Chem. Mater., 2025, 125, 3165–3241 CAS.
- J. F. Cao, Y. X. Ji and Z. P. Shao, Perovskites for Protonic Ceramic Fuel Cells: A Review, Energy Environ. Sci., 2022, 15, 2200–2232 RSC.
- H. Arandiyan, S. S. Mofarah, C. C. Sorrell, E. Doustkhah, B. Sajjadi, D. Hao, Y. Wang, H. Y. Sun, B.-J. Ni, M. Rezaei, Z. P. Shao and T. Maschmeyer, Defect Engineering of Oxide Perovskites for Catalysis and Energy Storage: Synthesis of Chemistry and Materials Science, Chem. Soc. Rev., 2021, 50, 10116–10211 RSC.
- C. C. Peng, X. Han, S. Mabaleha, P. Kwong, Y. Zheng and X. Y. Xu, Recent Advances in Perovskite Air Electrode Materials for Protonic Solid Oxide Electrochemical Cells, Energy Environ. Sci., 2025, 18, 4555–4595 RSC.
- Y. L. Zhu, Q. Lin, Y. J. Zhong, H. A. Tahini, Z. P. Shao and H. T. Wang, Metal Oxide-Based Materials as an Emerging Family of Hydrogen Evolution Electrocatalysts, Energy Environ. Sci., 2020, 13, 3361–3392 RSC.
- S. P. Jiang, Development of Lanthanum Strontium Cobalt Ferrite Perovskite electrodes of Solid Oxide Fuel Cells – A Review, Int. J. Hydrogen Energy, 2019, 44, 7448–7493 CrossRef CAS.
- Z. P. Shao and S. M. Haile, A-High Performance Cathode for the Next Generation of Solid-Oxide Fuel Cells, Nature, 2004, 431, 170–173 CrossRef CAS.
- R. Pelosato, G. Cordaro, D. Stucchi, C. Cristiani and G. Dotelli, Cobalt Based Layered Perovskites as Cathode Material for Intermediate Temperature Solid Oxide Fuel Cells: A Brief Review, J. Power Sources, 2015, 298, 46–67 CrossRef CAS.
- J.-H. Kim and A. Manthiram, Layered LnBaCo2O5+δ Perovskite Cathodes for Solid Oxide Fuel Cells: An Overview and Perspective, J. Mater. Chem. A, 2015, 3, 24195–24210 RSC.
- I. D. Seymour, A. Chroneos, J. A. Kilner and R. W. Grimes, Defect Processes in Orthorhombic LnBaCo2O5.5 Double Perovskites, Phys. Chem. Chem. Phys., 2011, 13, 15305–15310 RSC.
- X. M. Xu, Y. J. Zhong and Z. P. Shao, Double Perovskites in Catalysis, Electrocatalysis, and Photo(electro)catalysis, Trends Chem., 2019, 1, 410–424 CrossRef CAS.
- K. Zhang, L. Ge, R. Ran, Z. P. Shao and S. M. Liu, Synthesis, Characterization and Evaluation of Cation-Ordered LnBaCo2O5+δ as Materials of Oxygen Permeation Membranes and Cathodes of SOFCs, Acta Mater., 2008, 56, 4876–4889 CrossRef CAS.
- A. Maignan, C. Martin, D. Pelloquin, N. Nguyen and B. Raveau, Structural and Magnetic Studies of Ordered Oxygen-Deficient Perovskites LnBaCo2O5+δ, Closely Related to the “112” Structure, J. Solid State Chem., 1999, 142, 247–260 CrossRef CAS.
- P. S. Anderson, C. A. Kirk, J. Knudsen, I. M. Reaney and A. R. West, Structural Characterisation of REBaCo2O6−δ Phases (RE = Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho), Solid State Sci., 2005, 7, 1149–1156 CrossRef CAS.
- L. Li, Z. W. Dong, T. Xia, X. Li, Q. Li, L. P. Sun, H. Zhao and L. H. Huo, A Series of Bifunctional ReBaCo2O5+δ Perovskite Catalysts towards Intermediate-Temperature Oxygen Reduction Reaction and Oxygen Evolution Reaction, Chem. Eng. J., 2023, 468, 143762 CrossRef CAS.
- Y. N. Dou, Y. Xie, X. F. Hao, T. Xia, Q. Li, J. P. Wang, L. H. Huo and H. Zhao, Addressing Electrocatalytic Activity and Stability of LnBaCo2O5+δ Perovskites for Hydrogen Evolution Reaction by Structural and Electronic Features, Appl. Catal., B, 2021, 297, 120403 CrossRef CAS.
- Q. Sun, Z. Y. Dai, Z. B. Zhang, Z. L. Chen, H. Q. Lin, Y. Gao and D. J. Chen, Double Perovskite PrBaCo2O5.5: An Efficient and Stable Electrocatalyst for Hydrogen Evolution Reaction, J. Power Sources, 2019, 427, 194–200 CrossRef CAS.
- H. Togano, K. Asai, S. Oda, H. Ikeno, S. Kawaguchi, K. Oka, K. Wada, S. Yagi and I. Yamada, Highly Active Hydrogen Evolution Catalysis on Oxygen-Deficient Double-Perovskite Oxide PrBaCo2O6−δ, Mater. Chem. Front., 2020, 4, 1519–1529 RSC.
- F. Gunkel, L. Jin, D. N. Mueller, C. Hausner, D. S. Bick, C.-L. Jia, T. Schneller, I. Valov, R. Waser and R. Dittmann, Ordering and Phase Control in Epitaxial Double-Perovskite Catalysts for the Oxygen Evolution Reaction, ACS Catal., 2017, 7, 7029–7037 CrossRef CAS.
- N.-I. Kim, S.-H. Cho, S. H. Park, Y. J. Lee, R. A. Afzal, J. Yoo, Y.-S. Seo, Y. J. Lee and J.-Y. Park, B-Site Doping Effects of NdBa0.75Ca0.25Co2O5+δ Double Perovskite Catalysts for Oxygen Evolution and Reduction Reactions, J. Mater. Chem. A, 2018, 6, 17807–17818 RSC.
- N.-I. Kim, R. A. Afzal, S. R. Choi, S. W. Lee, D. Ahn, S. Bhattacharjee, S.-C. Lee, J. H. Kim and J.-Y. Park, Highly Active and Durable Nitrogen Doped-Reduced Graphene Oxide/Double Perovskite Bifunctional Hybrid Catalysts, J. Mater. Chem. A, 2017, 5, 13019–13031 RSC.
- Y. F. Bu, O. Gwon, G. Nam, H. Jang, S. Kim, Q. Zhong, J. Cho and G. Kim, A Highly Efficient and Robust Cation Ordered Perovskite Oxide as a Bifunctional Catalyst for Rechargeable Zinc-Air Batteries, ACS Nano, 2017, 11, 11594–11601 CrossRef CAS PubMed.
- Q. X. Liu, F. R. Shen, G. H. Song, T. F. Liu, W. C. Feng, R. T. Li, X. M. Zhang, M. R. Li, L. H. He, X. Zheng, S. S. Yin, G. Z. Yin, Y. F. Song, G. X. Wang and X. H. Bao, Tailoring Ion Ordering in Perovskite Oxide for High-Temperature Oxygen Evolution Reaction, Angew. Chem., Int. Ed., 2023, 62, e202307057 CrossRef CAS PubMed.
- D. Muñoz-Gil, E. Urones-Garrote, D. Pérez-Coll, U. Amador and S. Garcia-Martín, Crystal Structure and Compositional Effects on the Electrical and Electrochemical Properties of GdBaCo2−xMnxO5+δ (0 ≤ x ≤ 2) Oxides for Use as Air Electrodes in Solid Oxide Fuel Cells, J. Mater. Chem. A, 2018, 6, 5452–5460 RSC.
- S. Streule, A. Podlesnyak, E. Pomjakushina, K. Conder, D. Sheptyakov, M. Medarde and J. Mesot, Oxygen Order–Disorder Phase Transition in PrBaCo2O5.48 at High Temperature, Phys. B, 2006, 378–380, 539–540 CrossRef CAS.
- R. A. Cox-Galhotra, A. Huq, J. P. Hodges, C. F. Yu, X. Q. Wang, W. Q. Gong, A. J. Jacobson and S. McIntosh, An In Situ Neutron Diffraction Study of the Crystal Structure of PrBaCo2O5+δ at High Temperature and Controlled Oxygen Partial Pressure, Solid State Ionics, 2013, 249–250, 34–40 CrossRef CAS.
- K. X. Zhang, T. Xia, J. P. Wang, Q. Li, L. P. Sun, L. H. Huo and H. Zhao, Realizing Extraordinary Bifunctional Electrocatalytic Performance of Layered Perovskite through Ba-Site Gd Doping toward Oxygen Reduction and Evolution Reactions, Mater. Today Energy, 2024, 43, 101601 CrossRef CAS.
- I. D. Seymour, A. Tarancón, A. Chroneos, D. Parfitt, J. A. Kilner and R. W. Grimes, Anisotropic Oxygen Diffusion in PrBaCo2O5.5 Double Perovskites, Solid State Ionics, 2012, 216, 41–43 CrossRef CAS.
- M. Burriel, J. Peña-Martínez, R. J. Chater, S. Fearn, A. V. Berenov, S. J. Skinner and J. A. Kilner, Anisotropic Oxygen Ion Diffusion in Layered PrBaCo2O5+δ, Chem. Mater., 2012, 24, 613–621 CrossRef CAS.
- Y.-C. Chen, M. Yashima, J. Peña-Martínez and J. A. Kilner, Experimental Visualization of the Diffusional Pathway of Oxide Ions in a Layered Perovskite-Type Cobaltite PrBaCo2O5+δ, Chem. Mater., 2013, 25, 2638–2641 CrossRef CAS.
- B. Liu, J. J. Yang, D. Yan, L. C. Jia, B. Chi, J. Pu and J. Li, Novel PrBa0.9Ca0.1Co2−xZnxO5+δ Double-Perovskite as an Active Cathode Material for High-Performance Proton-Conducting Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2020, 45, 31009–31016 CrossRef CAS.
- J.-H. Kim and A. Manthiram, Layered NdBaCo2−xNixO5+δ Perovskite Oxides as Cathodes for Intermediate Temperature Solid Oxide Fuel Cells, Electrochim. Acta, 2009, 54, 7551–7557 CrossRef CAS.
- B. Wei, Z. Lü, D. C. Jia, X. Q. Huang, Y. H. Zhang and W. H. Su, Thermal Expansion and Electrochemical Properties of Ni-doped GdBaCo2O5+δ Double-Perovskite Type Oxides, Int. J. Hydrogen Energy, 2010, 35, 3775–3782 CrossRef CAS.
- A. M. Shalamova, Yu. A. Glazyrina and A. Yu, Suntsov, Elevated Electrochemical Activity of Double Perovskites PrBaCo2−xNixO6−δ towards Hydrogen Peroxide Oxidation, J. Electroanal. Chem., 2022, 905, 115959 CrossRef CAS.
- L. Liu, R. S. Guo, S. S. Wang, Y. X. Yang and D. S. Yin, Synthesis and Characterization of PrBa0.5Sr0.5Co2−xNixO5+δ (x = 0.1, 0.2 and 0.3) Cathodes for Intermediate Temperature SOFCs, Ceram. Int., 2014, 40, 16393–16398 CrossRef CAS.
- L. Zhao, J. C. Shen, B. B. He, F. L. Chen and C. R. Xia, Synthesis, Characterization and Evaluation of PrBaCo2−xFexO5+δ as Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2011, 36, 3658–3665 CrossRef CAS.
- J. Zou, J. Park, B. Kwak, H. Yoon and J. Chung, Effect of Fe Doping on PrBaCo2O5+δ as Cathode for Intermediate-Temperature Solid Oxide Fuel Cells, Solid State Ionics, 2012, 206, 112–119 CrossRef CAS.
- C. Kuroda and K. Zheng, and K. Świerczek, Characterization of Novel GdBa0.5Sr0.5Co2−xFexO5+δ Perovskites for Application in IT-SOFC Cells, Int. J. Hydrogen Energy, 2013, 38, 1027–1038 CrossRef CAS.
- N. E. Volkova, L. Y. Gavrilova, V. A. Cherepanov, T. V. Aksenova, V. A. Kolotygin and V. V. Kharton, Synthesis, Crystal Structure and Properties of SmBaCo2−xFexO5+δ, J. Solid State Chem., 2013, 204, 219–223 Search PubMed.
- J. F. Xue, Y. Shen and T. M. He, Double-Perovskites YBaCo2−xFexO5+δ Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2011, 196, 3729–3735 CrossRef CAS.
- X. N. Li, X. N. Jiang, H. X. Xu, Q. L. Xu, L. Jiang, Y. C. Shi and Q. Y. Zhang, Scandium-Doped PrBaCo2−xScxO6−δ Oxides as Cathode Material for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2013, 38, 12035–12042 CrossRef CAS.
- C. Z. Sun, Y. Kong, L. Shao, Q. Zhang, X. Wu, N. Q. Zhang and K. N. Sun, Significant Zirconium Substitution Effect on the Oxygen Reduction Activity of the Cathode Material NdBaCo2O5+δ for Solid Oxide Fuel Cells, ACS Sustainable Chem. Eng., 2019, 7, 11603–11611 CrossRef CAS.
- M. Saccoccio, C. L. Jiang, Y. Gao, D. J. Chen and F. Ciucci, Nb-Substituted PrBaCo2O5+δ as a Cathode for Solid Oxide Fuel Cells: A Systematic Study of Structural, Electrical, and Electrochemical Properties, Int. J. Hydrogen Energy, 2017, 42, 19204–19215 CrossRef CAS.
- D. Wang, Y. P. Xia, H. L. Lv, L. N. Miao., L. Bi and W. Liu, PrBaCo2−xTaxO5+δ Based Composite Materials as Cathodes for Proton-Conducting Solid Oxide Fuel Cells with High CO2 Resistance, Int. J. Hydrogen Energy, 2020, 45, 31017–31026 Search PubMed.
- J. S. Xu, H. D. Cai, G. D. Hao, L. L. Zhang, Z. Y. Song, W. Long, L. Zhang and L. Wang, Characterization of High-Valence Mo-Doped PrBaCo2O5+δ Cathodes for IT-SOFCs, J. Alloys Compd., 2020, 842, 155600 CrossRef CAS.
- D. S. Tsvetkov, I. L. Ivanov, D. A. Malyshkin and A. Y. Zuev, Oxygen Content, Crystal Structure and Chemical Expansion of PrBaCo2−xFexO6−δ Double Perovskites, Dalton Trans., 2014, 43, 11862–11866 RSC.
- D. Muñoz-Gil, D. Pérez-Coll, E. Urones-Garrote, U. Amador and S. Garcia-Martin, Influence of the Synthesis Conditions on the Crystal Structure and Properties of GdBaCo2−xFexO5+δ Oxides as Air-Electrodes for Intermediate Temperature Solid Oxide Fuel Cells, J. Mater. Chem. A, 2017, 5, 12550–12556 RSC.
- X. M. de Irujo-Labalde, D. Muñoz-Gil, E. Urones-Garrote, D. Ávila-Brande and S. García-Martín, Complex Modulation of the Crystal Structure of a Layered Perovskite. A Promising Solid-oxide-Fuel-Cell Cathode, J. Mater. Chem. A, 2016, 4, 10241–10247 Search PubMed.
- C. Lim, A. Jun, H. Jo, K. M. Ok, J. Shin, Y.-W. Ju and G. Kim, Influence of Ca-Doping in Layered Perovskite PrBaCo2O5+δ on the Phase Transition and Cathodic Performance of a Solid Oxide Fuel Cell, J. Mater. Chem. A, 2016, 4, 6479–6486 Search PubMed.
- Z. W. Dong, T. Xia, Q. Li, J. P. Wang, S. Li, L. P. Sun, L. H. Huo and H. Zhao, Addressing the Origin of Highly Catalytic Activity of A-Site Sr-Doped Perovskite Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, Electrochem. Commun., 2022, 140, 107341 Search PubMed.
- S. Yoo, S. Choi, J. Kim, J. Shin and G. Kim, Investigation of Layered Perovskite Type NdBa1−xSrxCo2O5+δ (x = 0, 0.25, 0.5, 0.75, and 1.0) Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, Electrochim. Acta, 2013, 100, 44–50 CrossRef CAS.
- A. Jun, J. Kim, J. Shin and G. Kim, Optimizing of Sr Content in Layered SmBa1−xSrxCo2O5+δ Perovskite Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2012, 37, 18381–18388 CrossRef CAS.
- Z. W. Dong, T. Xia, Q. Li, L. P. Sun, L. H. Huo and H. Zhao, Enabling Extraordinary Oxygen Reduction Reaction Activity of Dual Alkaline Earth-Substituted Perovskite Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, Mater. Res. Bull., 2023, 164, 112245 CrossRef CAS.
- H. T. Gu, H. Chen, L. Gao, Y. F. Zheng, X. F. Zhu and L. C. Guo, Oxygen Reduction Mechanism of NdBaCo2O5+δ Cathode for Intermediate-Temperature Solid Oxide Fuel Cells under Cathodic Polarization, Int. J. Hydrogen Energy, 2009, 34, 2416–2420 CrossRef CAS.
- T. V. Aksenova, L. Yu Gavrilova, A. A. Yaremchenko, V. A. Cherepanov and V. V. Kharton, Oxygen Nonstoichiometry, Thermal Expansion and High-Temperature Electrical Properties of Layered NdBaCo2O5+δ and SmBaCo2O5+δ, Mater. Res. Bull., 2010, 45, 1288–1292 CrossRef CAS.
- S. L. Pang, X. N. Jiang, X. N. Li, H. X. Xu, L. Jiang, Q. L. Xu, Y. C. Shi and Q. Y. Zhang, Structure and Properties of Layered-Perovskite LaBa1−xCo2O5+δ (x = 0-0.15) as Intermediate-Temperature Cathode Material, J. Power Sources, 2013, 240, 54–59 CrossRef CAS.
- A. Olszewska, Z. H. Du, K. Świerczek, H. L. Zhao and B. Dabrowski, Novel ReBaCo1.5Mn0.5O5+δ (Re: La, Pr, Nd, Sm, Gd and Y) Perovskite Oxide: Influence of Manganese Doping on the Crystal Structure, Oxygen Nonstoichiometry, Thermal Expansion, Transport Properties, and Application as a Cathode Material in Solid Oxide Fuel Cells, J. Mater. Chem. A, 2018, 6, 13271–13285 Search PubMed.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr., 1976, A32, 751–767 CrossRef CAS.
- F. F. Lu, T. Xia, Q. Li, L. P. Sun., L. H. Huo and H. Zhao, Ta-Doped PrBa0.94Co2−xTaxO5+δ as Promising Oxygen Electrodes: A Focused Study on Catalytic Oxygen Reduction Reaction Activity, Stability and CO2-Durability, J.
Power Sources, 2019, 417, 42–52 CrossRef CAS.
- J. P. Wang, F. C. Meng, T. Xia, Z. Shi, J. Lian, C. B. Xu, H. Zhao, J.-M. Bassat and J.-C. Grenier, Superior Electrochemical Performance and Oxygen Reduction Kinetics of Layered Perovskite PrBaxCo2O5+δ (x = 0.90-1.0) Oxides as Cathode Materials for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2014, 39, 18392–18404 CrossRef CAS.
- K. H. Yi, L. P. Sun, Q. Li, T. Xia, L. H. Huo, H. Zhao, J. W. Li, Z. Lü, J.-M. Bassat, A. Rougier, S. Fourcade and J.-C. Grenier, Effect of Nd-Deficiency on Electrochemical Properties of NdBaCo2O6−δ Cathode for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2016, 41, 10228–10238 CrossRef CAS.
- X. W. Liu, F. J. Jin, N. Sun, J. X. Li, Y. Shen, F. Wang and J. H. Li, Nd3+-Deficiency Double Perovskite Nd1−xBaCo2O5+δ and Performance Optimization as Cathode Materials for Intermediate-Temperature Solid Oxide Fuel Cells, Ceram. Int., 2021, 47, 33886–33896 Search PubMed.
- S. L. Li, T. Xia, Q. Li, L. P. Sun, L. H. Huo and H. Zhao, A-Site Ba-Deficiency Layered Perovskite EuBa1−xCo2O6−δ Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells: Electrochemical Properties and Oxygen Reduction Reaction Kinetics, Int. J. Hydrogen Energy, 2017, 42, 24412–24425 Search PubMed.
- F. He, Y. M. Jiang, C. L. Ren, G. H. Dong, Y. Gan, M.-J. Lee, R. D. Green and X. J. Xue, Generalized Electrical Conductivity Relaxation Approach to Determine Electrochemical Kinetic Properties for MIECs, Solid State Ionics, 2016, 297, 82–92 CrossRef CAS.
- Y. J. Nie, J. Sunarso, F. L. Liang, W. Zhou, Z. H. Zhu and Z. P. Shao, A Comparative Study of Oxygen Reduction Reaction on Bi- and La-Doped SrFeO3−δ Perovskite Cathodes, J. Electrochem. Soc., 2011, 158, B132–B138 CrossRef.
- Y. H. Li, K. Gerdes, H. Diamond and X. B. Liu, An Improved Method to Increase the Predictive Accuracy of the ECR Technique, Solid State Ionics, 2011, 204–205, 104–110 CrossRef CAS.
- G. Kim, S. Wang, A. J. Jacobson, L. Reimus, P. Brodersen and C. A. Mims, Rapid Oxygen Ion Diffusion and Surface Exchange Kinetics in PrBaCo2O5+x with a Perovskite Related Structure and Ordered A Cations, J. Mater. Chem., 2007, 17, 2500–2505 RSC.
- A. Tarancón, S. J. Skinner, R. J. Chater, F. Hernández-Ramírez and J. A. Kilner, Layered Perovskites as Promising Cathodes for Intermediate Temperature Solid Oxide Fuel Cells, J. Mater. Chem., 2007, 17, 3175–3181 RSC.
- X. Li, T. Xia, Z. W. Dong, J. P. Wang, Q. Li, L. P. Sun, L. H. Huo and H. Zhao, Unveiling Positive Impacts of Fluorine Anion doping on Extraordinary Catalytic Activity of Bifunctional-Layered Double Perovskite Electrodes for Solid Oxide Fuel Cells and Electrolysis Cells, Mater. Today Chem., 2023, 29, 101469 CrossRef CAS.
- J.-H. Kim and A. Manthiram, LnBaCo2O5+δ Oxides as Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, J. Electrochem. Soc., 2008, 155, B385–B390 CrossRef CAS.
- J.-H. Kim, L. Mogni, F. Prado, A. Caneiro, J. A. Alonso and A. Manthiram, High Temperature Crystal Chemistry and Oxygen Permeation Properties of the Mixed Ionic-Electronic Conductors LnBaCo2O5+δ (Ln = lanthanide), J. Electrochem. Soc., 2009, 156, B1376–B1382 CrossRef CAS.
- Z. B. Yang, T. Xia, Z. W. Dong, Q. Li, L. P. Sun, L. H. Huo and H. Zhao, Considerable Oxygen Reduction Activity and Durability of BaO Nanoparticles-Decorated Ln0.94BaCo2O5+δ Electrocatalysts, Sep. Purif. Technol., 2023, 317, 123936 CrossRef CAS.
- H. X. Huo, Z. X. Tang, T. Xia, Y. N. Dou, Q. Li, J. P. Wang, L. P. Sun, L. H. Huo and H. Zhao, Untangling Enhanced Performance Origin of Ca-doped LaBa1−xCaxCo2O5+δ Electrocatalysts toward Intermediate-Temperature Oxygen Reduction/Evolution Reactions, Mater. Sci. Eng., B, 2025, 313, 117940 CrossRef CAS.
- E. H. Hou, X. Li, J. P. Wang and T. Xia, Significantly Improved Electrocatalytic Activity of Pr1.1Ba0.9Co2O5+δ Induced by Ba-Site Sr Doping toward Oxygen Reduction/Evolution Reactions, Mater. Sci. Eng., B, 2025, 317, 118196 CrossRef CAS.
- F. C. Meng, T. Xia, J. P. Wang, Z. Shi, J. Lian, H. Zhao, J.-M. Bassat and J.-C. Grenier, Evaluation of Layered Perovskites YBa1−xSrxCo2O5+δ as Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2014, 39, 4531–4543 CrossRef CAS.
- W. B. Xiang, J. P. Wang, S. L. Li, T. Xia and G. X. Wang, Positive Effects of Calcium-doping on the Cathode Performance of Layered Perovskite Eu1−xCaxBaCo2O5+δ for Intermediate-Temperature Solid Oxide Fuel Cells, J. Alloys Compd., 2019, 801, 220–228 CrossRef CAS.
- X. N. Zhang, T. Xia, Q. Li, L. P. Sun, L. H. Huo and H. Zhao, A Highly Active and CO2-Tolerant Perovskite Cathode for Solid Oxide Fuel Cells Operating below 700 °C, J. Alloys Compd., 2021, 858, 157743 CrossRef CAS.
- Z. J. Nie, J. P. Wang, T. Xia and G. X. Wang, A-Site Ca-Doped Layered Double Perovskite Pr1−xCaxBa0.94Co2O5+δ as High-Performance and Stable Cathode for Intermediate-Temperature Solid Oxide Fuel Cells, J. Alloys Compd., 2022, 905, 164191 CrossRef CAS.
- C. G. Yao, H. X. Zhang, X. J. Liu, J. L. Meng, X. Zhang, F. Z. Meng and J. Meng, Characterization of Layered Double Perovskite LaBa0.5Sr0.25Ca0.25Co2O5+δ as Cathode Material for Intermediate-Temperature Solid Oxide Fuel Cells, J. Solid State Chem., 2018, 265, 72–78 CrossRef CAS.
- H. X. Gu, C. Su, C. Zhou, Y. Liu, Y. Zhang, G. M. Yang, W. Zhou and Z. P. Shao, LaBa0.8Ca0.2Co2O5+δ Cathode with Superior CO2 Resistance and High Oxygen Reduction Activity for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2022, 47, 16214–16221 Search PubMed.
- A. Subardi, K.-Y. Liao and Y.-P. Fu, Oxygen Permeation, Thermal Expansion Behavior and Electrochemical Properties of LaBa0.5Sr0.5Co2O5+δ Cathodes for SOFCs, RSC Adv., 2017, 7, 14487–14495 Search PubMed.
- Y. Chen, S. Yoo, Y. M. Choi, J. H. Kim, Y. Ding, K. Pei, R. Murphy, Y. X. Zhang, B. T. Zhao, W. L. Zhang, H. J. Chen, Y. Chen, W. Yuan, C. H. Yang and M. L. Liu, A Highly Active, CO2-Tolerant Electrode for the Oxygen Reduction Reaction, Energy Environ. Sci., 2018, 11, 2458–2466 RSC.
- D. W. Fu, F. J. Jin and T. M. He, A-Site Calcium-Doped Pr1−xCaxBaCo2O5+δ Double Perovskites as Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2016, 313, 134–141 CrossRef CAS.
- S. L. Pang, J. Xu, Y. J. Su, G. M. Yang, M. Zhu, M. Cui, X. Q. Shen and C. L. Chen, The Role of A-Site Cation Size Mismatch in Tune the Catalytic Activity and Durability of Double Perovskite Oxides, Appl. Catal., B, 2020, 270, 118868 CrossRef CAS.
- H. L. Zhao, Y. Zheng, C. Y. Yang, Y. N. Shen, Z. H. Du and K. Świerczek, Electrochemical Performance of Pr1−xYxBaCo2O5+δ Layered Perovskites as Cathode Materials for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2013, 38, 16365–16372 CrossRef CAS.
- C. G. Yao, J. X. Yang, H. X. Zhang, X. S. Lang and K. D. Cai, Ca-Doped PrBa1−xCaxCoCuO5+δ (x = 0-0.2) as Cathode Materials for Solid Oxide Fuel Cells, Ceram. Int., 2022, 48, 7652–7662 CrossRef CAS.
- S. Yoo, A. Jun, Y.-W. Ju, D. Odkhuu, J. Hyodo, H. Y. Jeong, N. Park, J. Shin, T. Ishihara and G. Kim, Development of Double-Perovskite Compounds as Cathode Materials for Low-Temperature Solid Oxide Fuel Cells, Angew. Chem., Int. Ed., 2014, 53, 1–5 CrossRef.
- A. B. Yu, T. Xia, L. P. Sun, Q. Li, L. H. Huo and H. Zhao, Effects of Rare Earth Doping on Electrochemical Properties of NdBaCo2O6−δ Cathode Materials, J. Alloys Compd., 2020, 837, 155563 CrossRef CAS.
- J. H. Kim and J. T. S. Irvine, Characterization of Layered Perovskite Oxides NdBa1−xSrxCo2O5+δ (x = 0 and 0.5) as Cathode Materials for IT-SOFC, Int. J. Hydrogen Energy, 2012, 37, 5920–5929 CrossRef CAS.
- K. E. Song, S. H. Woo, S. W. Baek, H. Kang, W. S. Choi, J. Y. Park and J. H. Kim, SmBa1−xCaxCo2O5+d Layered Perovskite Cathodes for Intermediate Temperature-Operating Solid Oxide Fuel Cells, Front. Chem., 2021, 8, 628813 CrossRef PubMed.
- H. P. Ding, X. J. Xue, X. Q. Liu and G. Y. Meng, High Performance Layered SmBa0.5Sr0.5Co2O5+δ Cathode for Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2009, 194, 815–817 CrossRef CAS.
- A. M. Asensio, D. Clematis, D. Cademartori, M. P. Carpanese, M. Viviani, C. Carbone and A. Barbucci, Calcium Doping in Double Perovskite SmBa1−xCaxCo2O5+δ to Enhance the Electrochemical Activity of Solid Oxide Cell Reversible Oxygen Electrode, J. Alloys Compd., 2023, 933, 167731 CrossRef CAS.
- X. L. Liu, F. J. Jin, X. W. Liu, N. Sun, J. X. Li, Y. Shen, F. Wang, L. Yang, X. Y. Chu, M. Z. Xu, Y. J. Zhai and J. H. Li, Effect of Calcium Doping on Sm1−xCaxBaCo2O5+δ Cathode Materials for Intermediate-Temperature Solid Oxide Fuel Cells, Electrochim. Acta, 2021, 390, 138830 CrossRef CAS.
- A. Subardi, C.-C. Chen, M.-H. Cheng, W.-K. Chang and Y.-P. Fu, Electrical, Thermal and Electrochemical Properties of SmBa1−xSrxCo2O5+δ Cathode Materials for Intermediate-Temperature Solid Oxide Fuel Cells, Electrochim. Acta, 2016, 204, 118–127 CrossRef CAS.
- K. Singh, A. K. Baral and V. Thangadurai, Electrochemical Studies of Gd0.5Pr0.5BaCo2O5+δ (GPBC) Cathode for Oxide Ion and Proton Conducting Solid Oxide Fuel Cells, Solid State Ionics, 2016, 288, 351–356 CrossRef CAS.
- X. L. Zhang and M. F. Jin, Layered GdBa0.5Sr0.5Co2O5+δ as a Cathode for Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2010, 195, 1076–1078 CrossRef CAS.
- S. S. Pramana, A. Cavallaro, C. Li, A. D. Handoko, K. W. Chan, R. J. Walker, A. Regoutz, J. S. Herrin, B. S. Yeo, D. J. Payne, J. A. Kilner, M. P. Ryan and S. J. Skinner, Crystal Structure and Surface Characteristics of Sr-Doped GdBaCo2O6−δ Double Perovskites: Oxygen Evolution Reaction and Conductivity, J. Mater. Chem. A, 2018, 6, 5335–5345 RSC.
- J. F. Xue, Y. Shen and T. M. He, Performance of Double-Perovskite YBa0.5Sr0.5Co2O5+δ as Cathode Material for Intermediate-Temperature Solid Oxide Fuel
Cells, Int. J. Hydrogen Energy, 2011, 36, 6894–6898 CrossRef CAS.
- Z. H. Du, C. L. Yan, H. L. Zhao, Y. Zhang, C. Y. Yang, S. Yi and Y. Lu, and K. Świerczek, Effective Ca-Doping in Y1−xCaxBaCo2O5+δ Cathode Materials for Intermediate Temperature Solid Oxide Fuel Cells, J. Mater. Chem. A, 2017, 5, 25641–25651 RSC.
- Q. J. Zhou, T. M. He, Q. He and Y. Ji, Electrochemical Performances of LaBaCuFeO5+x and LaBaCuCoO5+x as Potential Cathode Materials for Intermediate-Temperature Solid Oxide Fuel Cells, Electrochem. Commun., 2009, 11, 80–83 CrossRef CAS.
- C.-Y. Yoo, J. H. Joo, H. J. Lee and J. H. Yu, The Effects of Fe-Substitution on the Crystal Structure and Oxygen Permeability of PrBaCo2O5+δ, Mater. Lett., 2013, 108, 65–68 CrossRef CAS.
- S. Park, S. Choi, J. Shin and G. Kim, Tradeoff Optimization of Electrochemical Performance and Thermal Expansion for Co-Based Cathode Material for Intermediate-Temperature Solid Oxide Fuel Cells, Electrochim. Acta, 2014, 125, 683–690 CrossRef CAS.
- F. J. Jin, J. H. Li, Y. Wang, X. Y. Chu, M. Z. Xu, Y. J. Zhai, Y. Zhang, W. H. Fang, P. Zou and T. M. He, Evaluation of Fe and Mn Co-Doped Layered Perovskite PrBaCo2/3Fe2/3Mn1/2O5+δ as a Novel Cathode for Intermediate-Temperature Solid-Oxide Fuel Cell, Ceram. Int., 2018, 44, 22489–22496 CrossRef CAS.
- X. N. Jiang, J. Wang, G. Q. Jia, Z. J. Qie, Y. C. Shi, A. Idrees, Q. Y. Zhang and L. Jiang, Characterization of PrBa0.92CoCuO6−δ as a Potential Cathode Material of Intermediate-Temperature Solid Oxide Fuel Cell, Int. J. Hydrogen Energy, 2017, 42, 6281–6289 CrossRef CAS.
- C. Lim, S. Sengodan, D. Jeong, J. Shin and G. Kim, Investigation of the Fe Doping Effect on the B-Site of the Layered Perovskite PrBa0.8Ca0.2Co2O5+δ for a Promising Cathode Material of the Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2019, 44, 1088–1095 CrossRef CAS.
- L. Li, X. L. Xiong and J. Q. Wang, Atomic-Scale Mechanistic Study of Oxygen Reduction Mechanism for B-Site Doped Pr(Ba,Sr)Co2O5+δ by Density Functional Theory Calculations, Comput. Theor. Chem., 2024, 1241, 114930 CrossRef CAS.
- L. Zhao, Q. Nian, B. B. He, B. Lin, H. P. Ding, S. L. Wang, R. R. Peng, G. Y. Meng and X. Q. Liu, Novel Layered Perovskite Oxide PrBaCuCoO5+δ as a Potential Cathode for Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2010, 195, 453–456 CrossRef CAS.
- L. Jiang, T. Wei, R. Zeng, W.-X. Zhang and Y.-H. Huang, Thermal and Electrochemical Properties of PrBa0.5Sr0.5Co2−xFexO5+δ (x = 0.5, 1.0, 1.5) Cathode Materials for Solid-Oxide Fuel Cells, J. Power Sources, 2013, 232, 279–285 CrossRef CAS.
- F. J. Jin, Y. Shen, R. Wang and T. M. He, Double-Perovskite PrBaCo2/3Fe2/3Cu2/3O5+δ as Cathode Material for Intermediate-Temperature Solid-Oxide Fuel Cells, J. Power Sources, 2013, 234, 244–251 CrossRef CAS.
- Y.-F. Sun, Y.-Q. Zhang, J. Chen, J.-H. Li, Y.-T. Zhu, Y.-M. Zeng, B. S. Amirkhiz, J. Li, B. Hua and J.-L. Luo, New Opportunity for In Situ Exsolution of Metallic Nanoparticles on Perovskite Parent, Nano Lett., 2016, 16, 5303–5309 CrossRef CAS PubMed.
- K. Xu, H. Zhang, Y. S. Xu, F. He, Y. C. Zhou, Y. X. Pan, J. Y. Ma, B. T. Zhao, W. Yuan, Y. Chen and M. L. Liu, An Efficient Steam-Induced Heterostructured Air Electrode for Protonic Ceramic Electrochemical Cells, Adv. Funct. Mater., 2022, 32, 2110998 CrossRef CAS.
- D. Jeong, J. Kim, O. Kwon, C. Lim, S. Sengodan, J. Shin and G. Kim, Scandium Doping Effect on a Layered Perovskite Cathode for Low-Temperature Solid Oxide Fuel Cells (LT-SOFCs), Appl. Sci., 2018, 8, 2217 CrossRef CAS.
- B. V. Politov, A. Yu Suntsov, I. A. Leonidov, M. V. Patrakeev and V. L. Kozhevnikov, High-Temperature Defect Thermodynamics of Nickel Substituted Double-Perovskite Cobaltite PrBaCo2−xNixO6−δ (x = 0.2), J. Alloys Compd., 2017, 727, 778–784 CrossRef CAS.
- A. Y. Suntsov, I. A. Leonidov, M. V. Patrakeev and V. L. Kozhevnikov, Defect Formation in Double Perovskites PrBaCo2−xCuxO5+δ at Elevated Temperatures, Solid State Ionics, 2015, 274, 17–23 CrossRef CAS.
- Y. X. Zhang, J. Z. Cui, Z. Liu, C. Jing, Y. Wang, X. Lin and L. J. Zhang, Rational Design of Two-Layer Fe-Doped PrBa0.8Ca0.2Co2O6−δ Double Perovskite Oxides for High-Performance Fuel Cell Cathodes, J. Phys. Chem. C, 2021, 125, 26448–26459 CrossRef CAS.
- S. Choi, S. Yoo, J. Kim, S. Park, A. Jun, S. Sengodan, J. Kim, J. Shin, H. Y. Jeong, Y. M. Choi, G. Kim and M. L. Liu, Highly Efficient and Robust Cathode Materials for Low-Temperature Solid Oxide Fuel Cells: PrBa0.5Sr0.5Co2−xFexO5+δ, Sci. Rep., 2013, 3, 2426 CrossRef PubMed.
- J. Kim, S. Choi, S. Park, C. Kim, J. Shin and G. Kim, Effect of Mn on the Electrochemical Properties of a Layered Perovskite NdBa0.5Sr0.5Co2−xMnxO5+δ (x = 0, 0.25, and 0.5) for Intermediate-Temperature Solid Oxide Fuel Cells, Electrochim. Acta, 2013, 112, 712–718 CrossRef CAS.
- F. J. Jin, L. Li and T. M. He, NdBaCo2/3Fe2/3Cu2/3O5+δ Double Perovskite as a Novel Cathode Material for CeO2- and LaGaO3-Based Solid Oxide Fuel Cells, J. Power Sources, 2015, 273, 591–599 CrossRef CAS.
- C. Z. Sun, Y. Kong, L. Shao, K. N. Sun and N. Q. Zhang, Probing Oxygen Vacancy Effect on Oxygen Reduction Reaction of the NdBaCo2O5+δ Cathode for Solid Oxide Fuel Cells, J. Power Sources, 2020, 459, 228017 CrossRef CAS.
- G. Cordaro, A. Donazzi, R. Pelosato, L. Mastropasqua, C. Cristiani, I. N. Sora and G. Dotelli, Structural and Electrochemical Characterization of NdBa1−xCo2−yFeyO5+δ as Cathode for Intermediate Temperature Solid Oxide Fuel Cells, J. Electrochem. Soc., 2020, 167, 024502 CrossRef CAS.
- C. Z. Sun, Y. Kong, L. Shao, Q. Zhang, X. Wu, N. Q. Zhang and K. N. Sun, Significant Zirconium Substitution Effect on the Oxygen Reduction Activity of the Cathode Material NdBaCo2O5+δ for Solid Oxide Fuel Cells, ACS Sustainable Chem. Eng., 2019, 7, 11603–11611 CrossRef CAS.
- Z. W. Zhu, Z. T. Tao, L. Bi and W. Liu, Investigation of SmBaCuCoO5+δ Double-Perovskite as Cathode for Proton-Conducting Solid Oxide Fuel Cells, Mater. Res. Bull., 2010, 45, 1771–1774 Search PubMed.
- Y. X. Wang, X. Y. Zhao, S. Q. Lü, X. W. Meng, Y. J. Zhang, B. Yu, X. Y. Li, Y. R. Sui, J. H. Yang, C. W. Fu and Y. Ji, Synthesis and Characterization of SmSrCo2−xMnxO5+δ (x = 0.0, 0.2, 0.4, 0.6, 0.8, 1.0) Cathode Materials for Intermediate-Temperature Solid-Oxide Fuel Cells, Ceram. Int., 2014, 40, 11343–11350 CrossRef CAS.
- B. Wang, G. H. Long, Y. F. Li and Y. Ji, Characterization of SmBa0.5Sr0.5CoCuO5+δ Cathode Based on GDC and LSGM Electrolyte for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2016, 41, 13603–13610 Search PubMed.
- L.-N. Xia, Z.-P. He, X. W. Huang and Y. Yu, Synthesis and Properties of SmBaCo2−xNixO5+δ Perovskite Oxide for IT-SOFC Cathodes, Ceram. Int., 2016, 42, 1272–1280 CrossRef CAS.
- A. Olszewska, Y. Zhang, Z. H. Du, M. Marzec, K. Świerczek, H. L. Zhao and B. Dabrowski, Mn-Rich SmBaCo0.5Mn1.5O5+δ Double Perovskite Cathode Material for SOFCs, Int. J. Hydrogen Energy, 2019, 44, 27587–27599 CrossRef CAS.
- Q. J. Zhou, T. Wei, S. Q. Guo, X. L. Qi, R. F. Ruan, Y. Li, Y. Wu and Q. Liu, Evaluation of GdBaCuCo0.5Fe0.5O5+δ as Cathode Material for Intermediate Temperature Solid Oxide Fuel Cells, Ceram. Int., 2012, 38, 2899–2903 CrossRef CAS.
- C. Gumeci, J. Parrondo, A. M. Hussain, D. Thompson and N. Dale, Praseodymium Based Double-Perovskite Cathode Nanofibers for Intermediate Temperature Solid Oxide Fuel Cells (IT-SOFC), Int. J. Hydrogen Energy, 2021, 46, 31798–31806 CrossRef CAS.
- U. Anjum, T. S. Khan, M. Agarwal and M. Ali Haider, Identifying the Origin of the Limiting Process in a Double Perovskite PrBa0.5Sr0.5Co1.5Fe0.5O5+δ Thin-Film Electrode for Solid Oxide Fuel Cells, ACS Appl. Mater. Interfaces, 2019, 11, 25243–25253 CrossRef CAS.
- I.-M. Hung, D. Mohanty, Y.-R. Lin, S.-W. Lee, C.-J. Tseng and Y.-W. Chen, Synthesis and Electrochemical Performance of PrBa0.5Sr0.5Co1.5Fe0.5O5+δ Double Perovskite Cathode Material for Solid Oxide Fuel Cell, Int. J. Hydrogen Energy, 2025, 145, 371–379 Search PubMed.
- Q. Yang, D. Tian, R. Liu, H. D. Wu, Y. H. Chen, Y. Z. Ding, X. Y. Lu and B. Lin, Exploiting Rare-Earth-Abundant Layered Perovskite Cathodes of LnBa0.5Sr0.5Co1.5Fe0.5O5+δ (Ln = La and Nd) for SOFCs, Int. J. Hydrogen Energy, 2021, 46, 5630–5641 CrossRef CAS.
- I. Jang, H. Lee, R. Tamarany, H. Yoon, C. Kim, S. Kim, C.-W. Lee, T. Song and U. Paik, Tailoring the Ratio of A-Site Cations in Pr1−xNdxBaCo1.6Fe0.4O5+δ to Promote the Higher Oxygen Reduction Reaction Activity for Low-Temperature Solid Oxide Fuel Cells, Chem. Mater., 2020, 32, 3841–3849 CrossRef CAS.
- Y. H. Wan, Y. L. Xing, Y. H. Li, D. M. Huan and C. R. Xia, Thermal Cycling Durability Improved by Doping Fluorine to PrBaCo2O5+δ as Oxygen Reduction Reaction Electrocatalyst in Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2018, 402, 363–372 CrossRef CAS.
- Y. N. Kim, J.-H. Kim and A. Manthiram, Effect of Fe Substitution on the Structure and Properties of LnBaCo2−xFexO5+δ (Ln = Nd and Gd) Cathodes, J. Power Sources, 2010, 195, 6411–6419 Search PubMed.
- Z. Shi, T. Xia, F. C. Meng, J. P. Wang, J. Lian, H. Zhao, J.-M. Bassat, J.-C. Grenier and J. Meng, A Layered Perovskite EuBaCo2O5+δ for Intermediate-Temperature Solid Oxide Fuel Cell Cathode, Fuel Cells, 2014, 14, 979–990 CrossRef CAS.
- Y. Liu, YBaCo2O5+δ as a New Cathode Material for Zirconia-Based Solid Oxide Fuel Cells, J. Alloys Compd., 2009, 477, 860–862 CrossRef CAS.
- Y. J. Zhang, B. Yu, S. Q. Lü, X. W. Meng, X. Y. Zhao, Y. Ji, Y. X. Wang, C. W. Fu, X. Y. Liu, X. Y. Li, Y. R. Sui, J. H. Lang and J. H. Yang, Effect of Cu Doping on YBaCo2O5+δ as Cathode for Intermediate-Temperature Solid Oxide Fuel Cells, Electrochim. Acta, 2014, 134, 107–115 CrossRef CAS.
- Z. Shi, T. Xia, F. C. Meng, J. P. Wang, S. M. Wu, J. Lian, H. Zhao and C. B. Xu, EuBaCo2O5+δ–Ce0.9Gd0.1O2−δ Composite Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells: High Electrochemical Performance and Oxygen Reduction Kinetics, Electrochim. Acta, 2015, 174, 608–614 CrossRef CAS.
- G. Corbel, S. Mestiri and P. Lacorre, Physicochemical Compatibility of CGO Fluorite, LSM and LSCF Perovskite Electrode Materials with La2Mo2O9 Fast Oxide-Ion Conductor, Solid State Sci., 2005, 7, 1216–1224 CrossRef CAS.
- S. L. Li, L. K. Zhang, T. Xia, Q. Li, L. P. Sun, L. H. Huo and H. Zhao, Synergistic Effect Study of EuBa0.98Co2O5+δ–Ce0.8Sm0.2O1.9Composite Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, J. Alloys Compd., 2019, 771, 513–521 CrossRef CAS.
- L. Xing, T. Xia, Q. Li, H. Zhao, L. P. Sun and L.-H. Huo, High-Performance and CO2-Durable Composite Cathodes toward Electrocatalytic Oxygen Reduction: Ce0.8Sm0.2O1.9 Nanoparticle-Decorated Double Perovskite EuBa0.5Sr0.5Co2O5+δ, ACS Sustainable Chem. Eng., 2019, 7, 17907–17918 CrossRef CAS.
- J. Kim, W.-Y. Seo, J. Shin, M. L. Liu and G. Kim, Composite Cathodes Composed of NdBa0.5Sr0.5Co2O5+δ and Ce0.9Gd0.1O1.95 for Intermediate-Temperature Solid Oxide Fuel Cells, J. Mater. Chem. A, 2013, 1, 515–519 Search PubMed.
- J. Li, Q. Zhang, P. Qiu, L. C. Jia, B. Chi, J. Pu and J. Li, A CO2-Tolerant La2NiO4+δ-Coated PrBa0.5Sr0.5Co1.5Fe0.5O5+δ Cathode for Intermediate Temperature Solid Oxide Fuel Cells, J. Power Sources, 2017, 342, 623–628 CrossRef CAS.
- J. Li, P. Qiu, M. Xia, L. C. Jia, B. Chi, J. Pu and J. Li, Microstructure Optimization for High-Performance PrBa0.5Sr0.5Co1.5Fe0.5O5+δ–La2NiO4+δ Core–Shell Cathode of Solid Oxide Fuel Cells, J. Power Sources, 2018, 379, 206–211 CrossRef CAS.
- Y. H. Liu, Z. F. Pan, X. F. Zhao, S. H. Zhong, X. Y. Chen and C. Wang, Performance of PrBaCo2O5+δ Impregnated La0.6Sr0.4Co0.2Fe0.8O3−δ Composites as Potential Cathode Materials for Solid Oxide Fuel Cells, J. Alloys Compd., 2021, 886, 161155 CrossRef CAS.
- H. Zhang, K. Xu, F. He, Y. C. Zhou, K. Sasaki, B. T. Zhao, Y. M. Choi, M. L. Liu and Y. Chen, Surface Regulating of a Double-Perovskite Electrode for Protonic Ceramic Fuel Cells to Enhance Oxygen Reduction Activity and Contaminants Poisoning Tolerance, Adv. Energy Mater., 2022, 12, 2200761 CrossRef CAS.
- A. K. Baral, K. V. Sankar, A. Matatyaho, G. Kushnir and Y. Tsur, Tri-Functional Double Perovskite Oxide Catalysts for Fuel Cells and Electrolyzers, ChemSusChem, 2020, 6, 5671–5682 Search PubMed.
- J. H. Kim, S. Yoo, R. Murphy, Y. Chen, Y. Ding, K. Pei, B. T. Zhao, G. Kim, Y. M. Choi and M. L. Liu, Promotion of Oxygen Reduction Reaction on a Double Perovskite Electrode by a Water-Induced Surface Modification, Energy Environ. Sci., 2021, 14, 1506–1516 RSC.
- M. Fu, X. Lin, L. G. Tan, P. Zhang, H. J. Xie and Z. T. Tao, Self-Assembled Fe-Doped PrBaCo2O5+δ Composite Cathodes with Disorder Transition Region for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2023, 48, 15229–15237 CrossRef CAS.
- F. F. Lu, T. Xia, Q. Li, J. P. Wang, L. H. Huo and H. Zhao, Heterostructured Simple Perovskite Nanorod-Decorated Double Perovskite Cathode for Solid Oxide Fuel Cells: Highly Catalytic Activity, Stability and CO2-Durability for Oxygen Reduction Reaction, Appl. Catal., B, 2019, 249, 19–31 CrossRef CAS.
- S. N. Marshenya, B. V. Politov, D. A. Osinkin, A. Y. Suntsov and V. L. Kozhevnikov, Functional Properties and Electrochemical Performance of Dual-Phase Pr0.9Y0.1BaCo2O6−δ–Ce0.8Sm0.2O1.9 Composite Cathodes, J. Solid State Electrochem., 2018, 22, 1863–1869 Search PubMed.
- S. Choi, J. Shin and G. Kim, The Electrochemical and Thermodynamic Characterization of PrBaCo2−xFexO5+δ (x = 0, 0.5, 1) Infiltrated into Yttria-Stabilized Zirconia Scaffold as Cathodes for Solid Oxide Fuel Cells, J. Power Sources, 2012, 201, 10–17 CrossRef CAS.
- S. N. Marshenya, B. V. Politov, D. A. Osinkin, A. Y. Suntsov, I. A. Leonidov and V. L. Kozhevnikov, Advanced Electrochemical Properties of Pr0.9Y0.1BaCo1.8Ni0.2O6−δ–Ce0.8Sm0.2O1.9 Composite as Cathode Material for IT-SOFCs, J. Alloys Compd., 2019, 779, 712–719 Search PubMed.
- D. J. Chen, R. Ran and Z. P. Shao, Assessment of PrBaCo2O5+δ + Sm0.2Ce0.8O1.9 Composites Prepared by Physical Mixing as Electrodes of Solid Oxide Fuel Cells, J. Power Sources, 2010, 195, 7187–7195 CrossRef CAS.
- L. P. Sun, N. Li, Q. Li, L. H. Huo and H. Zhao, Preparation of NdBaCo2O5+δ–Ce0.9Gd0.1O1.95 Composite Cathode by In Situ Sol-Mixing Method and Its High-Temperature Electrochemical Properties, J. Alloys Compd., 2021, 885, 160901 Search PubMed.
- L.-N. Xia, J. You, Z.-P. He, X.-W. Huang and Y. Yu, Performances of Nickel-Doped SmBaCo2O5+δ-Sm0.2Ce0.8O1.9 Composite Cathodes for IT-SOFC, Int. J. Hydrogen Energy, 2016, 41, 1176–1186 CrossRef CAS.
- Z. H. Du, K. Y. Li, H. L. Zhao, X. Dong, Y. Zhang and K. Świerczek, A SmBaCo2O5+δ Double Perovskite with Epitaxially Grown Sm0.2Ce0.8O2−δ Nanoparticles as a Promising Cathode for Solid Oxide Fuel Cells, J. Mater. Chem. A, 2020, 8, 14162–14170 RSC.
- J. H. Kim, M. Cassidy, J. T. S. Irvine and J. Bae, Electrochemical Investigation of Composite Cathodes with SmBa0.5Sr0.5Co2O5+δ Cathodes for Intermediate Temperature-Operating Solid Oxide Fuel Cell, Chem. Mater., 2010, 22, 883–892 CrossRef CAS.
- A. Subardi and Y.-P. Fu, Electrochemical and Thermal Properties of SmBa0.5Sr0.5Co2O5+δ Cathode Impregnated with Ce0.8Sm0.2O1.9 Nanoparticles for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2017, 42, 24338–24346 CrossRef CAS.
- J. T. S. Irvine, J. Bae, J.-Y. Park, W. S. Choi and J. H. Kim, Electrochemical Properties and Durability of In Situ Composite Cathodes with SmBa0.5Sr0.5Co2O5+δ for Metal Supported Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2017, 42, 1212–1220 CrossRef CAS.
- Y. C. Zhou, D. Han, C. Yuan, M. Q. Liu, T. Chen, S. R. Wang and Z. L. Zhan, Infiltrated SmBa0.5Sr0.5Co2O5+δ Cathodes for Metal-Supported Solid Oxide Fuel Cells, Electrochim. Acta, 2014, 149, 231–236 CrossRef CAS.
- N. S. Tsvetkova, A. Yu Zuev and D. S. Tsvetkov, Investigation of GdBaCo2−xFexO6−δ (x = 0, 0.2) – Ce0.8Sm0.2O2 Composite Cathodes for Intermediate Temperature Solid Oxide Fuel Cells, J. Power Sources, 2013, 243, 403–408 CrossRef CAS.
- J. H. Kim, M. Cassidy, J. T. S. Irvine and J. Bae, Advanced Electrochemical Properties of LnBa0.5Sr0.5Co2O5+δ (Ln = Pr, Sm, and Gd) as Cathode Materials for IT-SOFC, J. Electrochem. Soc., 2009, 156, B682–B689 CrossRef CAS.
- Q. J. Zhou, F. Wang, Y. Shen and T. M. He, Performances of LnBaCo2O5+x–Ce0.8Sm0.2O1.9 Composite Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2010, 195, 2174–2181 CrossRef CAS.
- F. J. Jin, J. Liu, Y. Shen and T. M. He, Improved Electrochemical Performance and Thermal Expansion Compatibility of LnBaCoFeO5+δ-Sm0.2Ce0.8O1.9 (Ln = Pr and Nd) Composite Cathodes for IT-SOFCs, J. Alloys Compd., 2016, 685, 483–491 CrossRef CAS.
- S. L. Pang, X. N. Jiang, X. N. Li, Q. Wang and Z. X. Su, Characterization of Ba-Deficient PrBa1−xCo2O5+δ as Cathode Material for Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2012, 204, 53–59 CrossRef CAS.
- S. L. Pang, W. Z. Wang, T. Chen, Y. G. Wang, K. J. Xu, X. Q. Shen, X. M. Xi and J. W. Fan, The Effect of Potassium on the Properties of PrBa1−xCo2O5+δ (x = 0.00-0.10) Cathodes for Intermediate-Temperature Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2016, 41, 13705–13714 CrossRef CAS.
- F. C. Meng, T. Xia, J. P. Wang, Z. Shi and H. Zhao, Praseodymium-Deficiency Pr0.94BaCo2O6−δ Double Perovskite: A Promising High Performance Cathode Material for Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2015, 293, 741–750 Search PubMed.
- L. K. Zhang, S. L. Li, T. Xia, L. P. Sun, L. H. Huo and H. Zhao, Co-Deficient PrBaCo2−xO6−δ Perovskite as Cathode Materials for Intermediate-Temperature Solid Oxide Fuel Cells: Enhanced Electrochemical Performance and Oxygen Reduction Kinetics, Int. J. Hydrogen Energy, 2018, 43, 3761–3775 CrossRef CAS.
- G. Cordaro, A. Donazzi, R. Pelosato, L. Mastropasqua, C. Cristiani, I. N. Sora and G. Dotelli, Structural and Electrochemical Characterization of NdBa1−xCo2−yFeyO5+δ as Cathode for Intermediate Temperature Solid Oxide Fuel Cells, J. Electrochem. Soc., 2020, 167, 024502 Search PubMed.
- X. N. Jiang, Q. L. Xu, Y. C. Shi, X. N. Li, W. L. Zhou, H. X. Xu and Q. Y. Zhang, Synthesis and Properties of Sm3+-Deficient Sm1−xBaCo2O5+δ Perovskite Oxides as Cathode Materials, Int. J. Hydrogen Energy, 2014, 39, 10817–10823 CrossRef CAS.
- S. Q. Lü, X. W. Meng, X. M. Fu, M. H. Liu, Y. R. Sui, Y. L. Chen, J. Cao, Y. F. Sun, Y. Ji and L. L. Yang, The Evolution of Structure and Electrochemical Properties of Y-Site Deficiency Y1−xBaCoCuO5+δ Cathode for Solid Oxide Fuel Cells, Int. J. Hydrogen Energy, 2020, 45, 23227–23236 Search PubMed.
- Y. Chen, Y. F. Bu, B. T. Zhao, Y. X. Zhang, D. Ding, R. Z. Hu, T. Wei, B. Rainwater, Y. Ding, F. L. Chen, C. H. Yang, J. Liu and M. L. Liu, A Durable, High-Performance Hollow-Nanofiber Cathode for Intermediate-Temperature Fuel Cells, Nano Energy, 2016, 26, 90–99 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- Y. M. Zhu, L. Zhang, B. T. Zhao, H. J. Chen, X. Liu, R. Zhao, X. W. Wang, J. Liu, Y. Chen and M. L. Liu, Improving the Activity for Oxygen Evolution Reaction by Tailoring Oxygen Defects in Double Perovskite Oxides, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- X. Wang, Y. N. Dou, Y. Xie, J. P. Wang, T. Xia, L. H. Huo and H. Zhao, A-Site Cation-Ordering Layered Perovskite EuBa0.5Sr0.5Co2−xFexO5+δ as Highly Active and Durable Electrocatalysts for Oxygen Evolution Reaction, ACS Omega, 2020, 5, 12501–12515 CrossRef CAS.
- Z. H. Zhang, B. B. He, L. J. Chen, H. W. Wang, R. Wang, L. Zhao and Y. S. Gong, Boosting Overall Water Splitting via FeOOH Nanoflake-Decorated PrBa0.5Sr0.5Co2O5+δ Nanorods, ACS Appl. Mater. Interfaces, 2018, 10, 38032–38041 CrossRef CAS.
- Y.-Q. Zhang, H.-B. Tao, Z. Chen, M. Li, Y.-F. Sun, B. Hua and J.-L. Luo,
In Situ Grown Cobalt Phosphide (CoP) on Perovskite Nanofibers as an Optimized Trifunctional Electrocatalyst for Zn–Air Batteries and Overall Water Splitting, J. Mater. Chem. A, 2019, 7, 26607–26617 RSC.
- Y. F. Bu, H. Jang, O. Gwon, S. H. Kim, S. H. Joo, G. Nam, S. Kim, Y. Qin, Q. Zhong, S. K. Kwak, J. Cho and G. Kim, Synergistic Interaction of Perovskite Oxides and N-Doped Graphene in Versatile Electrocatalyst, J. Mater. Chem. A, 2019, 7, 2048–2054 RSC.
- B. Hua, M. Li, Y.-F. Sun, Y.-Q. Zhang, N. Yan, J. Chen, T. Thundat, J. Li and J.-L. Luo, A Coupling for Success: Controlled Growth of Co/CoOx Nanoshoots on Perovskite Mesoporous Nanofibres as High-Performance Trifunctional Electrocatalysts in Alkaline Condition, Nano Energy, 2017, 32, 247–254 CrossRef CAS.
- H. J. Zhang, D. Q. Guan, X. C. Gao, J. Yu, G. Chen, W. Zhou and Z. P. Shao, Morphology, Crystal Structure and Electronic State One-Step Co-Tuning Strategy towards Developing Superior Perovskite Electrocatalysts for Water Oxidation, J. Mater. Chem. A, 2019, 7, 19228–19233 Search PubMed.
- H. Jo, Y. J. Yang, A. Seong, D. Jeong, J. Kim, S. H. Joo, Y. J. Kim, L. J. Zhang, Z. Liu, J.-Q. Wang, S. K. Kwak and G. Kim, Promotion of the Oxygen Evolution Reaction via the Reconstructed Active phases of Perovskite Oxide, J. Mater. Chem. A, 2022, 10, 2271–2279 RSC.
- Y. M. Zhu, X. Zhong, S. G. Jin, H. J. Chen, Z. Y. He, Q. Y. Liu and Y. Chen, Oxygen Defect Engineering in Double Perovskite Oxides for Effective Water Oxidation, J. Mater. Chem. A, 2020, 8, 10957–10965 RSC.
- S. D. Ebbesen, S. H. Jensen, A. Hauch and M. B. Mogensen, High Temperature Electrolysis in Alkaline Cells, Solid Proton Conducting Cells, and Solid Oxide Cells, Chem. Rev., 2014, 114, 10697–10734 CrossRef CAS PubMed.
- X. J. Hou, Y. Jiang, K. Y. Wei, C. R. Jiang, T.-C. Jen, Y. L. Yao, X. Y. Liu, J. J. Ma and J. T. S. Irvine, Syngas Production from CO2 and H2O via Solid-Oxide Electrolyzer Cells: Fundamentals, Materials, Degradation, Operating Conditions, and Applications, Chem. Rev., 2024, 124, 5119–5166 CrossRef CAS PubMed.
- I. Jang, J. S. A. Carneiro, J. O. Crawford, Y. J. Cho, S. Parvin, D. A. Gonzalez-Casamachin, J. Baltrusaitis, R. P. Lively and E. Nikolla, Electrocatalysis in Solid Oxide Fuel Cells and Electrolyzers, Chem. Rev., 2024, 124, 8233–8306 Search PubMed.
- X. M. Xu, C. Su and Z. P. Shao, Fundamental Understanding and Applications of Ba0.5Sr0.5Co0.8Fe0.2O3−δ Perovskite in Energy Storage and Conversion: Past, Present, and Future, Energy Fuels, 2021, 35, 13585–13609 CrossRef CAS.
- M. Pidburtnyi, B. Zanca, C. Coppex, S. Jimenez-Villegas and V. Thangadurai, A Review on Perovskite-Type LaFeO3 Based Electrodes for CO2 Reduction in Solid Oxide Electrolysis Cells: Current Understanding of Structure-Functional Property Relationships, Chem. Mater., 2021, 33, 4249–4268 CrossRef CAS.
- E. Hernández, F. Baiutti, A. Morata, M. Torrell and A. Tarancón, Infiltrated Mesoporous Oxygen Electrodes for High Temperature Co-Electrolysis of H2O and CO2 in Solid Oxide Electrolysis Cells, J. Mater. Chem. A, 2018, 6, 9699–9707 Search PubMed.
- S. Dey, R. Saravanan, S. Hati, S. Goswami, A. Suresh, D. Jaiswal-Nagar, M. Ghosh, S. Paul, A. Bhattachaya, M. Mukhopadhyay and J. Mukhopadhyay, Influence of Intrinsic Spin Ordering in La0.6Sr0.4Co0.8Fe0.2O3−δ and Ba0.6Sr0.4Co0.8Fe0.2O3−δ towards Electrocatalysis of Oxygen Redox Reaction in Solid Oxide Cell, RSC Adv., 2024, 14, 30590–30605 RSC.
- W. Q. Zhang, B. Yu and J. M. Xu, Investigation of Single SOEC with BSCF Anode and SDC Barrier Layer, Int. J. Hydrogen Energy, 2012, 37, 837–842 Search PubMed.
- Y. F. Song, X. M. Zhang, K. Xie, G. X. Wang and X. H. Bao, High-Temperature CO2 Electrolysis in Solid Oxide Electrolysis Cells: Developments, Challenges, and Prospects, Adv. Mater., 2019, 31, 1902033 Search PubMed.
- B. Yu, W. Q. Zhang, J. M. Xu and J. Chen, Microstructural Characterization and Electrochemical Properties of Ba0.5Sr0.5Co0.8Fe0.2O3−δ and Its Application for Anode of SOEC, Int. J. Hydrogen Energy, 2008, 33, 6873–6877 Search PubMed.
- Q. X. Liu, Y. F. Song, R. T. Li, H. F. Lv, W. C. Feng, Y. X. Shen, X. M. Zhang, G. X. Wang and X. H. Bao, A Vanadium-Doped BSCF Perovskite for CO2 Electrolysis in Solid Oxide Electrolysis Cells, Int. J. Hydrogen Energy, 2021, 46, 19814–19821 CrossRef CAS.
- B. Y. Yao, Y. N. Dou, T. Xia, T. Song, Q. Li, H. Zhao and L. H. Huo, Bifunctional Fe/Co-Containing PrBaFe2−xCoxO5+δ Electrode: Enhanced Catalytic Properties through Modulating the Ratio of Fe/Co Cations, Mater. Sci. Eng., 2025, 314, 118034 Search PubMed.
- S. X. Wang, X. X. Zhang, Y. Chen, F. Fan, C. Jiang, Y. K. Xiang, X. Xiao, Y. Fang, A. N. Alodhayb, J. H. Li, J. J. Huang, Y. F. Sun and Z. Chen, Dual-Doping Strategy for Lowering the Thermal Expansion Coefficient and Promoting the Catalytic Activity in Perovskite Cobaltate Air Electrodes for Solid Oxide Cells, Small, 2025, 21, 2410672 CrossRef CAS.
- Y. F. Tian, J. Li, Y. Y. Liu, J. J. Yang, B. Liu, L. C. Jia, J. H. Jiang, B. Chi, J. Pu and J. Li, Preparation and Properties of PrBa0.5Sr0.5Co1.5Fe0.5O5+δ as Novel Oxygen Electrode for Reversible Solid Oxide Electrochemical Cell, Int. J. Hydrogen Energy, 2018, 43, 12603–12609 CrossRef CAS.
- A. Jun, J. Kim, J. Shin and G. Kim, Achieving High Efficiency and Eliminating Degradation in Solid Oxide Electrochemical Cells Using High Oxygen-Capacity Perovskite, Angew. Chem., Int. Ed., 2016, 55, 12512–12515 CrossRef CAS PubMed.
- X. R. Huang, F. Y. Yu, Y. Yang, P. Dai, Y. Tang, J. Jiang, K. Y. Feng, M. Y. Zhang, Y. F. Tian and B. Chi, Enhancing Electrochemical Performance of Reversible Solid Oxide Cells with Double Element-Deficient Pr0.97Ba0.97Co1.5Fe0.5O5+δ Oxygen Electrodes, Energy Fuels, 2024, 38, 21432–21440 CrossRef CAS.
- W. Tang, H. P. Ding, W. J. Bian, W. Wu, W. Li, X. B. Liu, J. Y. Gomez, C. Y. R. Vera, M. Zhou and D. Ding, Understanding of A-Site Deficiency in Layered Perovskites: Promotion of Dual Reaction Kinetics for Water Oxidation and Oxygen Reduction in Protonic Ceramic Electrochemical Cells, J. Mater. Chem. A, 2020, 8, 14600–14608 RSC.
- T. H. Shin, J.-H. Myung, M. Verbraeken, G. Kim and J. T. S. Irvine, Oxygen Deficient Layered Double Perovskite as an Active Cathode for CO2 Electrolysis Using a Solid Oxide Conductor, Faraday Discuss., 2015, 182, 227–239 Search PubMed.
- X. Y. Wang, H. B. Hu, C. Y. Xie, Y. F. Wang, H. W. Li and X. F. Ding,
In Situ Construction of a Double Perovskite Heterostructure with Exsolved FeNi3 Alloy Nanoparticles for CO2 Electrolysis in Solid Oxide Electrolysis Cells, J. Mater. Chem. A, 2024, 12, 11701–11709 RSC.
- Y. Song, H. Kim, J.-H. Jang, W. J. Bai, C. C. Ye, J. M. Gu and Y. F. Bu, Pt3Ni Alloy Nanoparticle ELectro-Catalysts with Unique Core–Shell Structure on Oxygen-Deficient Layered Perovskite for Solid Oxide Cells, Adv. Energy Mater., 2023, 13, 2302384 CrossRef CAS.
- Y. J. Wang, H. B. Hu, Z. Y. Zhao, H. S. Zheng and X. F. Ding, Enhancing the CO2 Adsorption of the Cobalt-Free Layered Perovskite Cathode for Solid-Oxide Electrolysis Cells Grains Excellent Stability under High Voltages via Oxygen-Defect Adjustment, ACS Appl. Mater. Interfaces, 2024, 16, 33548–33558 CrossRef CAS.
- G. D. Li, Y. J. Gou, X. J. Cheng, Z. Bai, R. Z. Ren, C. M. Xu, J. S. Qiao, W. Sun, Z. H. Wang and K. N. Sun, Enhanced Electrochemical Performance of the Fe-Based Layered Perovskite Oxygen Electrode for Reversible Solid Oxide Cells, ACS Appl. Mater. Interfaces, 2021, 13, 34282–34291 Search PubMed.
- A. W. Sun, J. Yang, Y. Liu, Z. P. Gao, S. Liu, G. M. Yang and H. R. Xu, Enhancing Solid Oxide Electrolysis Cell Cathode performance through Defect and Bond Engineering: A Study on K-Doped PrBaMnO5+δ Perovskites, J. Power Sources, 2025, 627, 235807 CrossRef CAS.
- M. J. Ma, X. X. Yang, C. M. Xu, R. Z. Ren, J. S. Qiao, W. Sun, Z. H. Wang and K. N. Sun, Constructing Highly Active Alloy-Perovskite Interfaces for Efficient Electrochemical CO2 Reduction Reaction, Sep. Purif. Technol., 2022, 296, 121411 CrossRef CAS.
- D. Q. Guan, J. Zhou, Z. W. Hu, W. Zhou, X. M. Xu, Y. J. Zhong, B. Liu, Y. H. Chen, M. G. Xu, H.-J. Lin, C.-T. Chen, J.-Q. Wang and Z. P. Shao, Searching General Sufficient-and-Necessary Conditions for Ultrafast Hydrogen-Evolving Electrocatalysis, Adv. Funct. Mater., 2019, 29, 1900704 CrossRef.
- D. Q. Guan, J. Zhou, Y.-C. Huang, C.-L. Dong, J.-Q. Wang, W. Zhou and Z. P. Shao, Screening Highly Active Perovskites for Hydrogen-Evolving Reaction via Unifying Ionic Electronegativity Descriptor, Nat. Commun., 2019, 10, 3755 CrossRef PubMed.
- S. Li, T. Xia, Y. N. Dou, Y. Xie, J. P. Wang, H. Zhao and L. H. Huo, Phosphatizing Engineering of Perovskite Oxide Nanofibers for Hydrogen Evolution Reaction to Achieve Extraordinary Electrocatalytic Performance, Adv. Funct. Mater., 2022, 32, 2112164 Search PubMed.
- E. Skúlason, V. Tripkovic, M. E. Björketun, S. Gudmundsdóttir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jónsson and J. K. Nørskov, Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations, J. Phys. Chem. C, 2010, 114, 18182–18197 Search PubMed.
- X. X. Yang, T. Xia, Q. Li, J. P. Wang, L. P. Sun, L. H. Huo and H. Zhao, Achieving Highly Active A-Site Cation-Deficient Perovskite Electrocatalysts Operated at Large Current Density toward Hydrogen Evolution Reaction, Sep. Purif. Technol., 2024, 339, 126715 CrossRef CAS.
- K. Q. Li, T. Xia, R. P. Deng, Y. N. Dou, J. P. Wang, Q. Li, L. P. Sun, L. H. Huo and H. Zhao, Tuning A-Site Cation Deficiency in Pr0.5La0.5BaCo2O5+δ Perovskite to Realize Large-Scale Hydrogen Evolution at 2000 mA cm−2, Small, 2024, 20, 2400760 CrossRef CAS.
- Y. Y. Liu, Y. N. Dou, S. Li, T. Xia, Y. Xie, Y. Wang, W. Zhang, J. P. Wang, L. H. Huo and H. Zhao, Synergistic Interaction of Double/Simple Perovskite Heterostructure for Efficient Hydrogen Evolution Reaction at High Current Density, Small Methods, 2021, 5, 2000701 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles, Science, 2011, 334, 1383–1385 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Design Principles for Oxygen-Reduction Activity on Perovskite Oxide Catalysts for Fuel Cells and Metal-Air Batteries, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- Y.-L. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn and D. Morgan, Prediction of Solid Oxide Fuel Cell Cathode Activity with First-Principles Descriptors, Energy Environ. Sci., 2011, 4, 3966–3970 RSC.
- R. Jacobs, T. Mayeshiba, J. Booske and D. Morgan, Material Discovery and Design Principles for Stable, High Activity Perovskite Cathodes for Solid Oxide Fuel Cells, Adv. Energy Mater., 2018, 8, 1702708 CrossRef.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Double Perovskites as a Family of Highly Active Catalysts for Oxygen Evolution in Alkaline Solution, Nat. Commun., 2013, 4, 2439 CrossRef.
- R. Jacobs, J. Hwang, Y. Shao-Horn and D. Morgan, Assessing Correlations of Perovskite Catalytic Performance with Electronic Structure Descriptors, Chem. Mater., 2019, 31, 785–797 CrossRef CAS.
- L. Giordano, K. Akkiraju, R. Jacobs, D. Vivona, D. Morgan and Y. Shao-Horn, Electronic Structure-Based Descriptors for Oxide Properties and Functions, Acc. Chem. Res., 2022, 55, 298–308 CrossRef CAS PubMed.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Perovskites in Catalysis and Electrocatalysis, Science, 2017, 358, 751–756 Search PubMed.
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. L. Yang and Y. Shao-Horn, Charge-Transfer-Energy-Dependent Oxygen Evolution Reaction Mechanisms for Perovskite Oxides, Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- A. Fujimori, A. E. Bocquet, T. Saitoh and T. Mizokawa, Electronic Structure of 3d Transition Metal Compounds: Systematic Chemical Trends and Multiplet Effects, J. Electron Spectrosc. Relat. Phenom., 1993, 62, 141–152 CrossRef CAS.
- A. E. Bocquet, T. Mizokawa, T. Saitoh, H. Namatame and A. Fujimori, Electronic Structure of 3d-Transition-Metal Compounds by Analysis of the 2p Core-Level Photoemission Spectra, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 3771–3784 CrossRef CAS PubMed.
- J. Zaanen, G. A. Sawatzky and J. W. Allen, Band Gaps and Electronic Structure of Transition-Metal Compounds, Phys. Rev. Lett., 1985, 55, 418–421 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Challenge for Rechargeable Li Batteries, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. B. Goodenough, Covalency Criterion for Localized vs Collective Electrons in Oxides with the Perovskite Structure, J. Appl. Phys., 1966, 37, 1415–1422 CrossRef CAS.
- F. Calle-Vallejo, N. G. Inoglu, H.-Y. Su, J. I. Martínez, I. C. Man, M. T. M. Koper, J. R. Kitchin and J. Rossmeisl, Number of Outer Electrons as Descriptor for Adsorption Processes on Transition Metals and Their Oxides, Chem. Sci., 2013, 4, 1245–1249 RSC.
- D. Q. Guan, H. Y. Xu, Q. W. Zhang, Y.-C. Huang, C. L. Shi, Y.-C. Chang, X. M. Xu, J. Y. Tang, Y. X. Gu, C.-W. Pao, S.-C. Haw, J.-M. Chen, Z. W. Hu, M. Ni and Z. P. Shao, Identifying a Universal Activity Descriptor and a Unifying Mechanism Concept on Perovskite Oxides for Green Hydrogen Production, Adv. Mater., 2023, 35, 2305074 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design, Science, 2017, 355, eaad4998 Search PubMed.
- L. Held and M. Ott, On p-Values and Bayes Factors, Annu. Rev. Stat. Appl., 2018, 5, 393–419 CrossRef.
- N. Fumo and M. A. R. Biswas, Regression Analysis for Prediction of Residential Energy Consumption, Renewable Sustainable Energy Rev., 2015, 47, 332 CrossRef.
- S. Zhai, H. P. Xie, P. Cui, D. Q. Guan, J. Wang, S. Y. Zhao, B. Chen, Y. F. Song, Z. P. Shao and M. Ni, A Combined Ionic Lewis Acid Descriptor and Machine-Learning Approach to Prediction of Efficient Oxygen Reduction Electrodes for Ceramic Fuel Cells, Nat. Energy, 2022, 7, 866–875 CrossRef CAS.
- J. Moon, W. Beker, M. Siek, J. Kim, H. S. Lee, T. Hyeon and B. A. Grzybowski, Active Learning Guides Discovery of A Champion Four-Metal Perovskite Oxide for Oxygen Evolution Electrocatalysis, Nat. Mater., 2024, 23, 108–115 CrossRef CAS PubMed.
- P. C. Xu, T. Lu, X. B. Ji, M. J. Li and W. C. Lu, Machine Learning Combined with Weighted Voting Regression and Proactive Searching Progress to Discover ABO3−δ Perovskites with High Oxide Ionic Conductivity, J. Phys. Chem. C, 2023, 127, 17096–17108 CrossRef CAS.
- Y. Zhang and X. J. Xu, Modeling Oxygen Ionic Conductivities of ABO3 Perovskites through Machine Learning, Chem. Phys., 2022, 558, 111511 CrossRef CAS.
- H. L. Xin, Catalyst Design with Machine Learning, Nat. Energy, 2022, 7, 790–791 Search PubMed.
- R. Jacobs, J. Liu, H. Abernathy and D. Morgan, Machine Learning Design of Perovskite Catalytic Properties, Adv. Energy Mater., 2024, 14, 2303684 CrossRef CAS.
- A. Zadick, L. Dubau, N. Sergent, G. Berthomé and M. Chatenet, Huge Instability of Pt/C Catalysts in Alkaline Medium, ACS Catal., 2015, 5, 4819–4824 CrossRef CAS.
- M. Schalenbach, O. Kasian, M. Ledendecker, F. D. Speck, A. M. Mingers, K. J. J. Mayrhofer and S. Cherevko, The Electrochemical Dissolution of Noble Metals in Alkaline Media, Electrocatalysis, 2018, 9, 153–161 CrossRef CAS.
- O. Kasian, S. Geiger, T. Li, J.-P. Grote, K. Schweinar, S. Y. Zhang, C. Scheu, D. Raabe, S. Cherevko, B. Gault and K. J. J. Mayrhofer, Energy Environ. Sci., 2019, 12, 3548–3555 RSC.
- J. K. Nøskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y.-L. Lee, A. Grimaud and Y. Shao-Horn, Influence of Oxygen Evolution during Water Oxidation on the Surface
of Perovskite Oxide Catalysts, J. Phys. Chem. Lett., 2012, 3, 3264–3270 CrossRef CAS.
- Y. N. Wang, L. N. Wang, K. X. Zhang, J. Y. Xu, Q. N. Wu, Z. B. Xie, W. An, X. Liang and X. X. Zou, Electrocatalytic Water Splitting over Perovskite Oxide Catalysts, Chin. J. Catal., 2023, 50, 109–125 CrossRef CAS.
- M. L. Liu, M. E. Lynch, K. Blinn, F. M. Alamgir and Y. M. Choi, Rational SOFC Material Design: New Advances and Tools, Mater. Today, 2011, 14, 534–546 Search PubMed.
- H. P. Ding, A. V. Virkar, M. L. Liu and F. Liu, Suppression of Sr Surface Segregation in La1−xSrxCo1−yFeyO3−δ: A First Principles Study, Phys. Chem. Chem. Phys., 2013, 15, 489–496 RSC.
- W. Lee, J. W. Han, Y. Chen, Z. H. Cai and B. Yildiz, Cation Size Mismatch and Charge Interactions Drive Dopant Segregation at the Surfaces of Manganite Perovskites, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS.
- Y. F. Song, W. Wang, L. Ge, X. M. Xu, Z. B. Zhang, P. S. B. Julião, W. Zhou and Z. P. Shao, Rational Design of a Water-Storable Hierarchical Architecture Decorated with Amorphous Barium Oxide and Nickel Nanoparticles as a Solid Oxide Fuel Cell Anode with Excellent Sulfur Tolerance, Adv. Sci., 2017, 4, 1700337 Search PubMed.
- Y. Huan, S. X. Chen, R. Zeng, T. Wei, D. H. Dong, X. Hu and Y. H. Huang, Intrinsic Effects of Ruddlesden–Popper-Based Bifunctional Catalysts for High-Temperature Oxygen Reduction and Evolution, Adv. Energy Mater., 2019, 9, 19011573 Search PubMed.
- R. Z. Ren, Z. H. Wang, C. M. Xu, W. Sun, J. S. Qiao, D. W. Rooney and K. N. Sun, Tuning the Defects of the Triple Conducting Oxide BaCo0.4Fe0.4Zr0.1Y0.1O3−δ Perovskite toward Enhanced Cathode Activity of Protonic Ceramic Fuel Cells, J. Mater. Chem. A, 2019, 7, 18365–18372 Search PubMed.
- D. J. Chen and Z. P. Shao, Surface Exchange and Bulk Diffusion Properties of Ba0.5Sr0.5Co0.8Fe0.2O3−δ Mixed conductor, Int. J. Hydrogen Energy, 2011, 36, 6948–6956 CrossRef CAS.
- Y. L. Zhu, J. Sunarso, W. Zhou and Z. P. Shao, Probing CO2 Reaction Mechanisms and Effects on the SrNb0.1Co0.9−xFexO3−δ Cathodes for Solid Oxide Fuel Cells, Appl. Catal., B, 2015, 172–173, 52–57 CrossRef CAS.
- N. C. Jeong, J. S. Lee, E. L. Tae, Y. J. Lee and K. B. Yoon, Acidity Scale for Metal Oxides and Sanderson's Electronegativities of Lanthanide Elements, Angew. Chem., Int. Ed., 2008, 47, 10128–10132 CrossRef CAS PubMed.
- B. Gu, J. Sunarso, Y. Zhang, Y. F. Song, G. M. Yang, W. Zhou, Z. P. Shao and A. High, Performance Composite Cathode with Enhanced CO2 Resistance for Low and Intermediate-Temperature Solid Oxide Fuel Cells, J. Power Sources, 2018, 405, 124–131 Search PubMed.
- R. J. H. Voorhoeve, J. P. Remeika and L. E. Trimble, Defect Chemistry and Catalysis in Oxidation and Reduction over Perovskite-Type Oxides, Ann. N. Y. Acad. Sci., 1976, 272, 3–21 Search PubMed.
- H.-J. Yin, J.-H. Zhou and Y.-W. Zhang, Shaping Well-Defined Noble-Metal-Based Nanostructures for Fabricating High-Performance Electrocatalysts: Advances and Perspectives, Inorg. Chem. Front., 2019, 6, 2582–2618 Search PubMed.
- T.-H. Yang, J. Ahm, S. Shi, P. Wang, R. Q. Gao and D. Qin, Noble-Metal Nanoframes and Their Catalytic Applications, Chem. Rev., 2021, 121, 796–833 CrossRef CAS.
- Y. F. Shi, Z. H. Lyu, M. Zhao, R. H. Chen, Q. N. Nguyen and Y. N. Xia, Noble-Metal Nanocrystals with Controlled Shapes for Catalytic and Electrocatalytic
Applications, Chem. Rev., 2021, 121, 649–735 CrossRef CAS.
- L. N. Yu, X. Y. Hu, Y. G. Guo, S. W. Zhang, J. C. Yu, G. Zou, X. M. Zhang, M. R. Li, Z. T. Ma, X. S. Zheng, Y. F. Song, M. L. Liu, G. X. Wang and X. H. Bao, Breaking the Ion Ordering in the Perovskite Anode for Enhanced High-Temperature Oxygen Evolution Reaction Activity, J. Am. Chem. Soc., 2025, 147, 27000–27010 CrossRef CAS PubMed.
- X. Han, K. X. Li, Q. Shao, W. J. Zhang, T. Li, Y. Gao, P. Z. Feng, Z. Lv and Y. H. Ling, A Durable and Highly Active Oxygen Electrode for Solid Oxide Cells: New Insight into Segregation Suppression of Layered Perovskite, Adv. Mater., 2025, 37, 2502068 CrossRef CAS PubMed.
- J. Yun, H. Shin, S. Kim, B. Seong, S. Lee, K. Kim, S. H. Choi and S. Choi, Highly Active Air Electrode with Enhanced Proton Conduction via Isovalent Doping in a Layered Perovskite for Reversible Protonic Ceramic Cells, Adv. Funct. Mater., 2025, 35, 2508758 CrossRef CAS.
- M. Choi, D. Kim, T. K. Lee, J. Lee, H. S. Yoo and W. Lee, Interface Engineering to Operate Reversible Protonic Ceramic Electrochemical Cells Below 500 °C, Adv. Energy Mater., 2024, 15, 2400124 CrossRef.
- X. Y. Hu, Y. C. Zhou, Z. Y. Luo, H. Y. Li, N. Shi, Z. J. Liu, W. L. Zhang, W. N. Wang, Y. Ding and M. L. Liu, Data-Driven Discovery of Electrode Materials for Protonic Ceramic Cells, Energy Environ. Sci., 2024, 17, 9335–9345 RSC.
- B.-S. Jiang, R. Chang and Y.-C. Lin, Partial Oxidation of Ethanol to Acetaldehyde over LaMnO3-Based Perovskite: A Kinetic Study, Ind. Eng. Chem. Res., 2013, 52, 37–42 Search PubMed.
- X. L. Wang, F. Zhou, Y. F. Gao, Y. H. Li, B. X. Cheng, T. Murayama, T. Ishida, M. Y. Lin and G. L. Xu, Revealing the Role of Mn–O Bonds in Electrocatalytic Methanol Oxidation to Value-Added Formate in LaMnO3, ACS Sustainable Chem. Eng., 2025, 13, 4845–4856 Search PubMed.
- K. A. R. Rao, J. Hwang, L. Giordano, X. R. Wang, E. Crumlin, D. S. Weinberger and Y. Shao-Horn, Regulating Oxygen Activity of Perovskites to Promote Activity and Selectivity for Methanol Oxidation to Formaldehyde, ACS Catal., 2024, 14, 7649–7663 CrossRef.
- S. Ganguly, R. Datta, C. Loha and S. Ghosh, Enhancing Trifunctional Electrocatalytic Activities via Lanthanide Modulation in Fe–Co-Based Double Perovskite Oxides, Energy Fuels, 2024, 38, 13196–13205 Search PubMed.
- J. W. Zhu, Y. Zhang, Z. T. Chen, Z. B. Zhang, X. Z. Tian, M. H. Huang, X. D. Bai, X. Wang, Y. F. Zhu and H. Q. Jiang, Superexchange-Stabilized Long-Distance Cu Sites in Rock-Salt-Ordered Double Perovskite Oxides for CO2 Electromethanation, Nat. Commun., 2024, 15, 1565 CrossRef CAS PubMed.
- A. A. Bayode, O. T. Ore, E. A. Nnamani, B. Sotunde, D. T. Koto, E. I. Unuabonah, B. Helmreich and M. O. Omorogie, Perovskite Oxides: Syntheses and Perspectives on Their Application for Nitrate Reduction, ACS Omega, 2024, 9, 19770–19785 Search PubMed.
- H. S. Lim, Y. Kim, D. Kang, M. Lee, A. Jo and J. W. Lee, Fundamental Aspects of Enhancing Low-Temperature CO2 Splitting to CO on a Double La2NiFeO6 Perovskite, ACS Catal., 2021, 11, 12220–12231 Search PubMed.
- C. Su, X. G. Duan, J. Miao, Y. J. Zhong, W. Zhou, S.
B. Wang and Z. P. Shao, Mixed Conducting Perovskite Materials as Superior Catalysts for Fast Aqueous-Phase Advanced Oxidation: A Mechanistic Study, ACS Catal., 2017, 7, 388–397 CrossRef CAS.
|
| This journal is © the Partner Organisations 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Ziwei
Dong
a,
Yingnan
Dou
a,
Qiang
Li
*a,
Ziwei
Dong
a,
Yingnan
Dou
a,
Qiang
Li










 step. The low coverage regime is considered and the calculated values are used to show the activity of each perovskite oxide. The volcano curve is established by using the scaling relation between
step. The low coverage regime is considered and the calculated values are used to show the activity of each perovskite oxide. The volcano curve is established by using the scaling relation between  and
and  . Reproduced with permission.202 Copyright 2011, Wiley. (c) Experimental ASRs at ∼1000 K vs. the calculated bulk O p-band center of perovskites with simulated composition: 1. Ba0.5Sr0.5Co0.75Fe0.25O3 (BSCF), 2. Sm0.5Sr0.5CoO3 (SSC), 3. PrBaCo2O6 (PBCO), 4. GdBaCo2O6 (GBCO), 5. La0.625Sr0.375Co0.25Fe0.75O3 (LSCF), 6. La0.75Sr.25CoO3 (LSC), 7. LaCoO3 (LCO), 8. La0.75Sr0.25MnO3 (LSM), and 9. LaMnO3 (LMO). The values with vertical errors bars are the average of multiple data values with an error bar equal to the standard deviation in the mean of the ASR. (d) Schematic electronic structure plots illustrating the correlation of the ORR energetics vs. the O p-band center based on a rigid band model. The shaded areas (red and blue in color) represent the occupied transition metal 3d and oxygen 2p states, and the empty areas represent the unoccupied states, respectively. The bold and dotted lines are the Fermi energy level before removing O2− and before adding O2−, respectively. Reproduced with permission.245 Copyright 2011, the Royal Society of Chemistry. (e) Plots of log
. Reproduced with permission.202 Copyright 2011, Wiley. (c) Experimental ASRs at ∼1000 K vs. the calculated bulk O p-band center of perovskites with simulated composition: 1. Ba0.5Sr0.5Co0.75Fe0.25O3 (BSCF), 2. Sm0.5Sr0.5CoO3 (SSC), 3. PrBaCo2O6 (PBCO), 4. GdBaCo2O6 (GBCO), 5. La0.625Sr0.375Co0.25Fe0.75O3 (LSCF), 6. La0.75Sr.25CoO3 (LSC), 7. LaCoO3 (LCO), 8. La0.75Sr0.25MnO3 (LSM), and 9. LaMnO3 (LMO). The values with vertical errors bars are the average of multiple data values with an error bar equal to the standard deviation in the mean of the ASR. (d) Schematic electronic structure plots illustrating the correlation of the ORR energetics vs. the O p-band center based on a rigid band model. The shaded areas (red and blue in color) represent the occupied transition metal 3d and oxygen 2p states, and the empty areas represent the unoccupied states, respectively. The bold and dotted lines are the Fermi energy level before removing O2− and before adding O2−, respectively. Reproduced with permission.245 Copyright 2011, the Royal Society of Chemistry. (e) Plots of log is a unique descriptor for OER activity. The theoretical overpotential (ηOER) under standard conditions is calculated using eqn (17), as follows:
is a unique descriptor for OER activity. The theoretical overpotential (ηOER) under standard conditions is calculated using eqn (17), as follows:
 for a series of perovskite oxides, resulting in a universal volcano relationship (Fig. 6b), in which SrCoO3 with
for a series of perovskite oxides, resulting in a universal volcano relationship (Fig. 6b), in which SrCoO3 with  of 1.48 eV is close to the very top of the volcano.
of 1.48 eV is close to the very top of the volcano.


