
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrosilanes. From laboratory curiosity to semiconductors
Daniel Griess
ab,
Madeleine Heurix
ab,
Matthias Paris
ab,
Philomena Vatter
ab and
Michael Haas
 *ab
*ab
aInstitute of Inorganic Chemistry, Graz University of Technology, Stremayrgasse 9/IV, 8010 Graz, Austria. E-mail: michael.haas@tugraz.at
bChristian Doppler Laboratory for New Semiconductor Materials based on Functionalized Hydrosilanes, Stremayrgasse 9/IV, 8010 Graz, Austria
First published on 24th April 2026
Abstract
Hydrosilanes represent a versatile class of silicon compounds that bridge fundamental main-group chemistry with technologically relevant materials science. Since the early discovery of monosilane in the nineteenth century, the chemistry of silicon hydrides has evolved from a laboratory curiosity into a cornerstone of modern semiconductor processing and silicon materials synthesis. This review provides a comprehensive overview of hydrosilane chemistry, focusing on three central aspects: (i) synthetic approaches to mono- and oligohydrosilanes, (ii) their functionalization through silanide intermediates and related transformations, and (iii) their application as molecular precursors for silicon-based materials and deposition technologies.
 Daniel Griess | Daniel Griess MSc obtained his Master's degree in Chemistry in 2024 under the supervision of Prof. Rolf Breinbauer. He is currently a Ph.D. student in Haas’ group at the Institute of Inorganic Chemistry at Graz University of Technology (Austria). His research interests cover mainly the synthesis of aminosilanes. |
 Madeleine Heurix | DI Madeleine Heurix obtained her Master's degree in Chemistry in 2024 under the supervision of Dr Michael Haas. She is currently a Ph.D. student in Haas’ group at the Institute of Inorganic Chemistry at Graz University of Technology (Austria). Her research interests cover mainly the synthesis of oligohydrogermanes and silylgermanes. |
 Matthias Paris | DI Matthias Paris obtained his Master's degree in Chemistry in 2024 under the supervision of Dr Michael Haas. He is currently a Ph.D. student in Haas’ group at the Institute of Inorganic Chemistry at Graz University of Technology (Austria). His research interests cover mainly the synthesis of silanides. |
 Philomena Vatter | Philomena Vatter BSc obtained her Bachelor's degree in Chemistry in 2024 from Karl Franzens University Graz with thesis supervison from Prof. Tremel at JGU Mainz. She is currently a Master's student in Haas’ group at the Institute of Inorganic Chemistry at Graz University of Technology (Austria). Her research interests cover mainly the liquid phase deposition of functionalized hydrosilanes. |
 Michael Haas | Prof. Michael Haas received his Ph.D. under the supervision of Prof. H. Stueger. He subsequently carried out postdoctoral research at Monash University (Australia) in the group of Prof. C. Jones. He is currently a group leader at Graz University of Technology (Austria). His research interests include the enolate chemistry of heavier carbon homologues, silane chemistry, liquid-phase deposition of silicon heterostructures, and the design of new group 14 photoinitiators. |
Introduction
Hydrosilanes are a class of silicon-based compounds that have gained significant attention due to their unique chemical reactivity and broad applications in material science and nanotechnology. Although silicon chemistry shares some conceptual similarities with the more extensively explored realm of carbon chemistry, it boasts its own distinct characteristics and complexities. Unlike their carbon-based counterparts, hydrosilanes exhibit distinctive properties owing to the weaker Si–Si bonds and the polar nature of Si–H bonds. These properties make hydrosilanes highly reactive and suitable for various applications, including silicon deposition, semiconductor processing, hydrogen storage, and the development of next-generation optoelectronic materials. Their role in forming advanced silicon-based nanostructures has also sparked interest in fields such as catalysis and biomedical research.Recent advances in hydrosilane chemistry have focused on improving their synthetic accessibility, stability, and processability. Methods such as reductive coupling, dehydrogenative coupling, plasma synthesis, and chloride-induced disproportionation have been explored to optimize their production. Additionally, applications in printable electronics, thin-film solar cells, and silicon-based nanostructures have further driven research in this field. Several reviews and reference works have summarized aspects of hydrosilane chemistry over the past decades. Early comprehensive treatments can be found in classical sources such as the Gmelin Handbook of Inorganic Chemistry, as well as survey articles and encyclopedia entries published during the 1980s and 1990s.1,2 These works primarily focused on the fundamental properties, preparation, and industrial relevance of simple silanes. Later contributions,3 including chapters in Comprehensive Inorganic Chemistry II,4 expanded on these topics but remained largely centered on established synthetic methodologies and basic silicon hydride chemistry.
More recently, a review by Gerwig, Böhme, and Friebel has revisited the field with emphasis on hydrosilanes; however, this work mainly addresses hydrosilanes themselves and does not cover the broader chemistry of higher hydrosilanes, their functionalization, and their emerging roles as precursors for modern deposition technologies.5 Therefore, despite the availability of these valuable contributions, a comprehensive and updated overview connecting classical hydrosilane chemistry with recent developments in functionalization and materials applications remains highly desirable.
Consequently, this review summarizes the broad field of hydrosilanes in terms of their synthesis (i), their functionalization (ii) and, if investigated, their applicability with respect to several deposition techniques (iii).
This review does not deal with hydrosilanes, which have only one hydride R3SiH (e.g.: (Me3Si)3SiH, Ph3SiH, or HPh2SiSiPh2H etc.), as in these cases the organic substituents largely dominate the chemical reactivity. Furthermore, molecular species of the type R2SiH2 (e.g.: (Me3Si)2SiH2, Ph2SiH2, or Et2SiH2 etc.) are likewise excluded for the same reason. However, R2SiH2 units within higher silicon hydrides and Si–Si bonded systems are implicitly included in this review. For reviews about hydrosilylation the reader is referred to two very recent publications.6 For a summary of inorganic tetrylenes the reader is referred to the excellent review of Rivard.7
Historical development
The classical approach is the reaction of silicides with diluted acids. The pioneers in this field were F. Wöhler and H. Buff, who synthesized SiH4 by the reaction of diluted acids with aluminium silicides.8 In 1902, H. Moissan and S. Smiles identified Si2H6 as another product formed by this reaction.9 Between 1919 and 1921 A. Stock and K. Somieski pioneered the field of organosilanes and aminosilanes with the synthesis of methylsilane10 and aminosilanes with trisilylamine.11 In the following years they showed that also higher homologs of the series SinH2n+2 were formed.10 G. Fritz was able to obtain the first silylphosphane in 1953 by pyrolysis of PH3 and SiH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 and in 1955 B. J. Aylett, H. J. Emeléus and A. G. Maddock were able to obtain evidence of the existence of trisilylphosphane.13 The next milestone in the synthesis of hydrosilanes was achieved by F. Fehér and co-workers, who used the same methodology and implemented procedures for a targeted isolation up to Si8H18 on a preparative scale. His scientific life is excellently portrayed in M. Baudler's reminiscence.14 M. A. Ring and D. M. Ritter were the first to report the synthesis of silyl potassium KSiH3.15 E. Amberger and H. Boeters were the first to isolate and report the properties of trisilylphosphane in 1962.16
12 and in 1955 B. J. Aylett, H. J. Emeléus and A. G. Maddock were able to obtain evidence of the existence of trisilylphosphane.13 The next milestone in the synthesis of hydrosilanes was achieved by F. Fehér and co-workers, who used the same methodology and implemented procedures for a targeted isolation up to Si8H18 on a preparative scale. His scientific life is excellently portrayed in M. Baudler's reminiscence.14 M. A. Ring and D. M. Ritter were the first to report the synthesis of silyl potassium KSiH3.15 E. Amberger and H. Boeters were the first to isolate and report the properties of trisilylphosphane in 1962.16
A change in the paradigm was the targeted synthesis of the first cyclic hydrosilane derivatives by E. Hengge and co-workers in 1973.17
In 1993 W. Sundermeyer reported the synthesis of branched silylgermanes, demonstrating the structural diversity accessible in mixed silicon–germanium hydride systems.18
13 years later T. Shimoda and co-workers demonstrated that these higher silanes have an application as precursors for liquid phase deposition (LPD).19 This landmark development resulted in a boom of this chemistry with many groups in academia and industry working on the implementation of solution-based deposition in several industrial processes. In 2015, M. Wagner and co-workers reported the synthesis of silafullerens, expanding the structural landscape of silicon-based cluster chemistry.20
In 2017 H. Stueger and co-workers published the synthesis of 2,2,3,3,4,4-hexasilylpentasilane, which is to date the highest isolated hydrosilane on preparative scale.21 Recently our group pioneered the use of functionalized hydrosilanes as precursors for LPD.22 In summary this can be seen as a showcase example of an ideal workflow in science. Starting from the very fundamental studies to the several applications. In Fig. 1 the historical development of this research topic is summarized.
 | ||
| Fig. 1 Historical development. | ||
Silane synthesis
Hydrolysis of silicides
The hydrolytic decomposition of silicides with aqueous acids was first observed by Wöhler and Buff in 1857, marking the first documented formation of a silicon- and hydrogen-containing compound.8 Subsequently, in 1902, Moissan and Smiles were the first to characterize SiH4 along with higher hydrosilanes following the decomposition of magnesium silicide with aqueous HCl.9 This discovery laid the foundation for the use of silicide hydrolysis as a commonly applied method for silane synthesis. The reaction produces monosilane (SiH4) alongside disilane (Si2H6) and minor amounts of higher hydrosilanes. The yield and product distribution are highly dependent on factors like purity of Mg2Si, the solvent and acid used, and overall reaction conditions. Stock implemented extensive studies on this topic and successfully isolated and characterized higher silanes, including tetrasilane (Si4H10), pentasilane (Si5H12) and hexasilane (Si6H14) (see Scheme 1). The application of preparative gas chromatography further enabled the separation and identification of both linear and branches silanes up to octasilane (Si8H18).23–25 Fehér expanded upon these studies by investigating the hydrolysis of Mg2Si using sulfuric and phosphoric acid, leading to the formation and identification of silanes up to pentadecasilane (Si15H32). Moreover, distinct linear silanes up to heptasilane (Si7H16), as well as branched species such as 2-silyltrisilane, 2-silyltetrasilane and 2-silylpentasilane, were isolated and characterized.26 | ||
| Scheme 1 Synthesis of monosilane and oligohydrosilanes with product distribution according to the work of Stock et al. | ||
Johnson reported that using ammonium bromide (NH4Br) in liquid ammonia instead of aqueous HCl increased silane yields to 80–90%.27
Hydrogenation of chlorosilanes
The hydrogenation of chlorosilanes with mild reducing agents such as lithium aluminium hydride (LiAlH4) or diisobutylaluminium hydride (i-Bu2AlH) is a well-established method for synthesizing hydrosilanes on a laboratory scale (see Scheme 2). However, this approach is limited by the availability of chlorosilane precursors and side reactions involving Si–Si bond cleavage (especially LiAlH4 results in the cleavage of silyl groups). | ||
| Scheme 2 Formation of oligohydrosilanes via the hydrogenation of chlorosilanes. | ||
Finholt et al. first reported this reaction in 1947, demonstrating the formation of disilane (Si2H6) via the reduction of hexachlorosilane (Si2Cl6) with LiAlH4 (see Scheme 2). The yield of hydrosilanes varies significantly depending to reaction conditions and chlorosilane employed.28 In 1973, Höfler reported the formation of neopentasilane, short NPS, (neo-Si5H12) via the hydrogenation of dodecachloroneopentasilane (neo-Si5Cl12) with LiAlH4, though low yields were observed due to Si–Si bond cleavage. The use of i-Bu2AlH instead of LiAlH4 improved selectivity and increased product yields.29
The Litz–Ring process (see Scheme 3), while primarily employed for the reduction of tetrachlorosilane (SiCl4) to monosilane using alkali metal hydrides regenerated in situ by molten-salt electrolysis, has also been investigated for related hydride-mediated silicon transformations. The process enables continuous, high-yield production of ultra-pure monosilane and was demonstrated at pilot scale with strong industrial relevance for low-cost, high-purity silicon production for semiconductors.30
 | ||
| Scheme 3 Simplified Litz–Ring process. | ||
Hengge et al. reported the synthesis of the cyclic hydrosilanes, cyclopentasilane (cy-Si5H10) and cyclohexasilane (cy-Si6H12) via the hydrogenation of their respective cyclic chlorosilanes using LiAlH4, achieving excellent yields (see Scheme 4).31
 | ||
| Scheme 4 Hydrogenation of cyclic chlorosilanes. | ||
In 2013, Kroke and co-workers reported the successful crystallization of cyclopentasilane, enabling its structural characterization by X-ray diffraction (see Fig. 2).32
 | ||
| Fig. 2 Crystal structure of Si5H10 adapted from original literature by Kroke and co-workers. | ||
An alternative approach involves the reduction of [Si6Cl14]2− complexes, which are generated from HSiCl3 through a redistribution reaction, facilitated by alkylated amines such as tetraethylenediamine (TEEDA) or pentaethyldiethylenetriamine (PEDETA) and the presence of a base like ethyldiisopropylamine (EDIPA). The resulting hexacoordinate species can then be hydrogenated with LiAlH4, yielding cy-Si6H12 (see Scheme 5).33 Wagner and co-workers have further explored the rich amine-mediated chemistry of chlorosilanes and related silicon cluster species; a comprehensive overview of this chemistry can be found in their review.34
 | ||
| Scheme 5 Synthesis of cyclohexasilane via the reduction of the [Si6Cl14]2− complexes. | ||
The first synthesis of an unsubstituted bicyclic halosilane, bi(cylclopentasilanyl), was reported by Stueger, Lassacher and Hengge in 1995.35 The key intermediate bis(nonachlorocyclopentasilanyl) is obtained either by Hg-mediated coupling of nonachlorocyclopentasilane using (t-Bu)2Hg in n-heptane or by catalytic phenyl abstraction from the perphenylated bicyclic compound with HCl/AlCl3, both routes affording Si10Cl18 in good yields. Subsequent hydride reduction with LiAlH4 at −40 °C selectively converts the Si–Cl functionalities into Si–H bonds, yielding the unsubstituted bicylic Si10H18 (see Scheme 6). This reduction is accompanied by the partial cleavage of the central Si–Si bond, leading to approximately 15% cyclopentasilane (cy-Si5H10) as a side product.
 | ||
| Scheme 6 Synthesis of bicyclic hydrosilane. Yields of the final product not reported since it could not be separated from the side-product (cy-Si5H10). | ||
Disproportionation of chlorosilanes
The disproportionation is the main large-scale method used nowadays for the production of monosilane (SiH4). Trichlorosilane (HSiCl3) undergoes disproportionation in the presence of a catalyst (e.g. AlCl3, ZnCl2, BCl3, FeCl3). The reaction leads to the formation of SiH4 in high purity alongside SiCl4. The process is continuous and operates in a closed-loop system, allowing recycling of SiCl4 (see Scheme 7).36 | ||
| Scheme 7 Large-scale production of SiH4. | ||
Direct synthesis of Si and H2
It is also possible to synthesize monosilane and also higher hydrosilanes from silicon powder and hydrogen in the presence of catalytic amounts of H2S or various sulfides (see Scheme 8). The product distribution is controlled by the H2 pressure. However, this method requires high temperatures and pressures, making it less practical compared to other methodologies.37 | ||
| Scheme 8 Reaction of Si-powder and hydrogen at high temperatures. *Product distribution and total yield when CuS is used as the catalyst. | ||
Catalytic dehydrogenative coupling
The formation of Si–Si bonds from monosilane (SiH4) can be achieved through dehydrogenative coupling reactions, in which primary and secondary organomonosilanes undergo hydrogen abstraction in the presence of a catalyst (see Scheme 9). Such reactions typically require transition metal complexes from group 4 to 10 which work as catalysts.38 | ||
| Scheme 9 Schematic equation for dehydrogenative coupling of silanes. | ||
Harrod was the first to report the catalytic formation of higher hydrosilanes from SiH4 in 1988.39 Building on this work, Okumura et al. demonstrated the synthesis of disilane (Si2H6) and trisilane (Si3H8) via catalytic condensation of SiH4 using platinum, rhodium and ruthenium-based complexes.40 Moreover, n-tetrasilane was afforded via catalytic conversion of SiH4, Si2H6 and Si3H8.41 However, a significant challenge associated with these approaches is the concurrent formation of insoluble polymeric byproducts, which can negatively impact selectivity and complicate product isolation.
Wurtz-type coupling
A Wurtz-type approach to hydrosilane synthesis was introduced by Craig et al. in 1962, involving the reaction of iodosilane (H3SiI) with a liquid sodium/mercury amalgam at room temperature (see Scheme 10).42 | ||
| Scheme 10 Synthesis of disilane (Si2H6) via Wurtz-type coupling. | ||
This method resulted in the formation of disilane with a reported yield of 67%.42 However, the broader application of this method remains limited, as alkali metals tend to also react with Si–H bonds.
An alternative route to higher hydrosilanes involves the nucleophilic substitution of halosilanes with silanide anions at low temperatures (see Scheme 11).43,44 A detailed discussion on the synthesis of silanides follows in Functionalization with group 1 (synthesis of silanides).
 | ||
| Scheme 11 Formation of mono-, di-, and trisilane via Wurtz-type coupling. | ||
This method not only enables the formation of linear oligomers but also the synthesis of branched silanes, such as neopentasilane (neo-Si5H12) and neohexasilane (Si(SiH3)3(Si2H5)) (see Scheme 12). These branched species can be obtained through the reaction of phenylchlorosilane with a mixture of different potassiumsilanides (KSiH(SiH3)2, KSi(SiH3)3 and KSi(SiH3)2(Si2H5)) followed by sequential bromination and reduction of the resulting phenyl-substituted oligosilanes using HBr and LiAlH4, respectively. Nevertheless, the separation and isolation of the individual products was not reported.45
 | ||
| Scheme 12 Formation of branched silanes in a 3-step synthesis. | ||
Building on Fehér's work on silanide synthesis (see Functionalization with group 1 (synthesis of silanides)), Sundermeyer and co-workers investigated the protonation of higher silanides (KSinH2n+1) with phenylsulfonic acid (PhSO3H) resulting in a mixture of various branched hydrosilanes with the general formula (SiH4−n(SiH3)n).46,47
However, they noted that a selective synthesis of higher silanides could not be achieved under the investigated conditions, consequently the reactions were leading to mixtures or less well-defined products.
Stueger et al. demonstrated that LiSi(SiH3)3 (prepared from isolated neopentasilane) reacts selectively with SiCl2Ph2 to form the branched nonasilane (SiH3)3SiSiPh2Si(SiH3)3. Subsequent dephenylation using triflic acid, followed by hydrogenation with i-Bu2AlH, resulted in the formation of 2,2,4,4-tetrasilylpentasilane in good overall yield (see Scheme 13).48,49
 | ||
| Scheme 13 Synthesis of (SiH3)3SiSiPh2Si(SiH3)3 and subsequent treatment with TfOH and i-BuAlH. | ||
In addition, an alternative synthesis of 2,2,4,4-tetrasilylpentasilane was achieved independently by two groups. While Dow Corning employed borosilicate glass surfaces to promote the conversion of neopentasilane,50 the groups led by Stueger and Haas, as well as Evonik, demonstrated that 2,2,4,4-tetrasilylpentasilane also forms in the presence of a Lewis acid (see Scheme 14).51
 | ||
| Scheme 14 Alternative synthesis of 2,2,4,4-tetrasilylpentasilane. | ||
In addition, Haas et al. turned to an alternative method inspired by earlier work of Gilman and Harrell.52 In this approach, 0.5 equiv. of dibromoethane (BrCH2CH2Br) were added to LiSi(SiH3)3 at −80 °C, leading the formation of 2,2,3,3-tetrasilyltetrasilane (a branched octasilane) shown in Scheme 15 accompanied by minor amounts of 2,2,3,3,4,4-hexasilylpentasilane (a branched undecasilane). This reaction is driven by an initial halogen–metal exchange, forming BrSi(SiH3)3 as key intermediate.21
 | ||
| Scheme 15 Synthesis of branched octasilane with minor amounts of branched undecasilane. | ||
Moreover, Stueger and co-workers successfully obtained crystals of the branched undecasilane appropriate for X-ray diffractometry (see Fig. 3).21
 | ||
| Fig. 3 Crystal structure from 2,2,3,3,4,4-hexasilylpentasilane (a branched undecasilane). Reproduced from Haas et al.21 with permission from Angewandte Chemie International Edition, © 2017. | ||
Impact of energy on SiH4
An alternative route to higher hydrosilanes involves energy-induced transformation of monosilane, such as by pyrolysis, photolysis, or electrical discharge (see Scheme 16). These approaches rely on the homolytic cleavage of Si–H and Si–Si bonds, promoting the stepwise build-up of longer-chained silanes. | ||
| Scheme 16 Impact of energy on SiH4. Total yield and product distribution depends on the energy source and reaction conditions. | ||
The most straightforward method employs thermal energy. As early as 1950, Fritz demonstrated that disilane could be synthesized through the pyrolysis of monosilane. Continued pyrolysis of disilane leads to the formation of trisilane, while further thermal treatment of trisilane results in the generation of tetrasilane, indicating that this process enables chain elongation through successive decomposition and recombination steps.53,54
In addition to thermal activation, monosilane can also be exposed to silent electrical discharge within an ozonizer. This method yields a mixture of disilane, trisilane, and smaller amounts of higher homologues.2,55 A further photochemical approach involves the irradiation of SiH4 with ultraviolet light. When mercury is employed as a photosensitizer, a distribution of di-, tri-, and higher silanes can be generated.56
Silafullerenes
A major conceptual advance in silicon hydride chemistry was achieved with the realization of silafullerenes, more precisely endohedral silafullerenes (see Fig. 4), by Wagner and co-workers in 2015–2021.20,57 Building on earlier theoretical predictions that halide ions could template silicon cages, they reported the synthesis of the dodecahedral silicon cluster [Cl@Si20H20]−, the silicon analogue of the smallest fullerene C20H20 (see Scheme 17). | ||
| Fig. 4 Crystal structures from silafullerenes by Wagner et al. Reproduced from ref. 57 with permission from Journal of American Chemical Society, © 2021. | ||
 | ||
| Scheme 17 Formation of silafullerenes by Wagner et al. | ||
The synthesis proceeds via controlled stepwise reduction of perchlorinated precursor, ultimately converting all exohedral Si–Cl bonds into Si–H functionalities while preserving an encapsulated chloride ion at the centre of the cage. Although formally ionic compounds and therefore isolated as a salt with a counter-cation, the cluster is structurally and chemically dominated by twenty-terminal Si–H bonds. As such, these silafullerenes represent a unique class of molecular hydrosilanes, bridging classical neutral hydrosilanes and anionic silicon clusters, and they offer a hydrogen-terminated silicon surface that is amenable to further functionalization while being stabilized by the endohedral Cl− template.
Silane derivatization
Furthermore, alkali metal silanides with the general formula MSixHy (M = Li, Na, K, Rb, Cs) serve as versatile precursors for subsequent functionalization, enabling the incorporation of diverse heteroatoms while preserving the Si–H functionality. The scope and methodology of these transformations are discussed in detail in this review.
In 1961, Ring and Ritter reported the formation of potassium silanide (KSiH3) by reducing monosilane with potassium metal in DME. Reaching 75–100% consumption of silane required about two months (see Scheme 18, path a). A Na/K alloy greatly accelerated the process, achieving 92% SiH4 consumption in 14 days (see Scheme 18, path b). They also showed that KH reacts with disilane to give quantitative conversion within one day, whereas lithium hydride (LiH) affords only 12% conversion after 48 hours (see Scheme 18, path c). Although isolated yields were not determined directly, subsequent reactions of KSiH3 proceeded in essentially quantitative yield.15
 | ||
| Scheme 18 Synthesis of MSiH3 via reduction (path a–c). No yields were reported. | ||
Hagenmuller and Pouchard prepared sodium silanide (NaSiH3) by introducing SiH4 into a finely dispersed suspension of Na in DME. Under these conditions, both reactants partially (approx. 20%) dissolve within 6 hours in equimolar proportions, affording a yellowish solution. However, the resulting NaSiH3 solution is unstable and undergoes decomposition under the synthesis conditions.61
In 1966 Kennedy et al. reported a significant improvement in the preparation of potassium silanide by reacting disilane with potassium sand at −78 °C, achieving complete conversion to KSiH3 within 24–48 h.43
Subsequent studies by Amberger and Römer demonstrated that using a K/Na alloy (5:1 w/w) in DME or diglyme affords full conversion to KSiH3 at room temperature within 3 days. In contrast, THF and 2,2-dimethyl-1,3-dioxolane give slower reactions. While triglyme accelerates the conversion further, the resulting KSiH3 solutions are less stable than those obtained in DME or diglyme. Notably, the addition of diethyl ether (Et2O) to solutions of KSiH3 induces precipitation of a white, crystalline solid that retains its reactivity under N2 or vacuum and can be fully redissolved thereafter.62
Cradock, Gibbon, and van Dyke later showed that hexamethylphosphoramide (HMPA) enables complete conversion to NaSiH3 and KSiH3 at 10 °C within just 15 minutes. However, isolation of the free silanides from HMPA proved unsuccessful due to the high boiling point of the solvent and the high solubility of the products, which prevented precipitation upon addition of nonpolar solvents (see Scheme 19).63
 | ||
| Scheme 19 Synthesis of MSiH3 in HMPA. Isolation of the silanide failed. | ||
Amberger, Römer, and Layer undertook a detailed study of alkali-metal reactivity in DME and diglyme, establishing a clear increase in rate down group 1 (Na < K < Rb < Cs). Relative to sodium, caesium reduced the time required to reach full conversion by up to 92%. Under otherwise comparable conditions, diglyme generally afforded faster conversions than DME (see Scheme 20, path a and b). Mixed-metal systems offered additional benefits: an Rb/Na alloy shortened reaction times relative to rubidium alone (see Scheme 20, path c). By contrast, an Na/Hg amalgam was distinctly sluggish, reaching only 17% conversion after 23 days (see Scheme 20, path d). Isolation of discrete MSiH3 (M = Na, K, Rb, Cs) was not attempted; instead, the reactive solutions were directly subjected to subsequent functionalization.64
 | ||
| Scheme 20 Synthesis of MSiH3 by Amberger, Römer, and Layer (path a–d). No yields were reported. | ||
Bürger and Eujen were the first in 1974 to study the generation of higher silanides in more detail. They used K or KSiH3 and reacted it with varying equivalents of silanes with the formula SinH2n+1 (n = 1–3) and were able to prove via nuclear magnetic resonance (NMR) spectroscopy the existence of KSiHn(SiH3)3−n (n = 1–3). When reacting KSiH3 with an excess of SiH4 they observe next to KSiH3 the formation of KSi2H5 (see Scheme 21, path a). On the other hand, upon the reaction of K with di- or trisilane, or KSiH3 with trisilane they report the formation of the higher silanides KSiHn(SiH3)3−n (n = 1–3) (see Scheme 21, path b). Furthermore, they note that when an excess of trisilane is used the main product is KSi(SiH3)3 (see Scheme 21, path c), and an excess of K leads to cleavage of Si–Si-bonds. Finally, when they reacted the formed silanides with HCl, they always observe SiH4 and Si2H6 next to small amounts of the expected silanes SinH2n+2.65
 | ||
| Scheme 21 Synthesis of higher silanides, isolated yields no reported (path a–c). | ||
In the same year Fehér and Freund extended the generation of higher alkali-metal silanides using DME as solvent. Reaction of KSiH3 with disilane furnished KSi2H5 cleanly. With equimolar trisilane, NMR spectroscopy revealed a product ensemble comprising KSiH3, KSi2H5, KSiH(SiH3)2 and KSi(SiH3)3, with KSiH(SiH3)2 as the major species. In line with the observations of Bürger and Eujen, employing 3–4 equivalents of trisilane yielded exclusively KSi(SiH3)3 (see Scheme 22).65,66 Isolation of the individual silanides for yield determination was not attempted; instead, the in situ solutions were directly subjected to electrophilic functionalization (see Scheme 51).66
 | ||
| Scheme 22 Synthesis of higher silanides by Fehér and Freund, yields were not determined. | ||
Fehér, Betzen, and Krancher (1981) achieved the first isolation of solvent-free potassium silanide KSiH3. Concentration of freshly prepared KSiH3 solutions (generated using a Na/K alloy, 1:3 w/w) led to crystallization; subsequent high-vacuum drying afforded analytically pure KSiH3 in 63.5% isolated yield. They further quantified the solvent dependence of KSiH3 solubility, finding a clear increase with donor atom count (triglyme > diglyme > DME), consistent with enhanced cation coordination. In benzene, addition of 18-crown-6 (18-c-6) increased solubility linearly with crown concentration. A temperature-dependent study in DME (10–51 °C) revealed an inverse temperature dependence, with the highest solubility at 10 °C.67
In a subsequent report, Fehér and Krancher showed that replacing the Na/K with an ultrasonically prepared K dispersion reduced the reaction time by up to 9 h. Moreover, adding 17% (v/v) benzene to 0.8–0.9 M DME solutions of KSiH3 accelerated recrystallization to the solvent-free KSiH3. The isolated yields were essentially unchanged: 64.4% using the Na/K dispersion and 64.9% with the K dispersion.68
Fehér and Krancher (1983)69 then examined the formation of higher silanides from various hydrosilane precursors, targeting the homologous series KSiHn(SiH3)3−n (n = 1–3) over a range of silane stoichiometries (see Tables 1–3). “Reaction time” was defined as the point at which the analyzed composition became invariant; longer times were generally required at higher silane loadings. Transient higher silanides – namely KSi5H11 and KSi6H13 – were detected at early stages but disappeared upon extended reaction, while increasing the silane-to-metal ratio favored formation of higher silanides overall. Product ratios were established by quenching with a fivefold excess of benzyl chloride followed by GC analysis of the resulting products. No H2 evolution during the build-up reaction was observed; rather, SiH4 was the only gaseous byproduct. In addition to the targeted silanides and SiH4, the reactions produced KH and a potassium-containing silicon hydride polymer, (K0.09SiH1.19)n. On the basis of these observations, a mechanism (see Scheme 23) was proposed in which stepwise nucleophilic substitutions at silicon generate KH and higher silanes as intermediates; these silanes undergo competing silylation and metalation. Because the intermediate silanes (other than SiH4) re-enter the reaction network, the sequence converges to mixtures of KSiHn(SiH3)3−n, SiH4, and the potassium containing polymeric silicon hydride.69
 | ||
| Scheme 23 Proposed mechanism by Fehér and Krancher towards the formation of higher silanides. | ||
| Molar ratio x | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Reaction time [h] | 0.25 | 0.5 | 6 | 24 | 72 |
| KH | Trace | Trace | 4.8 | 3.9 | 1.5 |
| KSiH3 | 33.3 | Trace | Trace | — | — |
| KSi2H5 | 63.9 | 6.5 | Trace | — | — |
| KSiH(SiH3)2 | 2.8 | 93.5 | 68.8 | 22.2 | 5.0 |
| KSi(SiH3)3 | — | Trace | 26.4 | 73.9 | 93.5 |
| Molar ratio x | 0.5 | 1.0 | 1.5 | 2.0 | 2.5 | 3.0 |
|---|---|---|---|---|---|---|
| Reaction time [h] | 0.25 | 0.33 | 24 | 48 | 72 | 72 |
| KH | — | — | 2.7 | 3.9 | 2.0 | — |
| KSiH3 | 42.6 | 2.5 | — | — | — | — |
| KSi2H5 | 50.8 | 61.5 | Trace | Trace | — | — |
| KSiH(SiH3)2 | 6.6 | 34.5 | 91.0 | 22.3 | 2.5 | 2.2 |
| KSi(SiH3)3 | — | 1.5 | 6.3 | 73.8 | 95.5 | 97.8 |
| Reaction | KSiH3 + 2A | KSiH3 + 2B | KSiH3 + 2C | |||
|---|---|---|---|---|---|---|
| Reaction time [h] | 1 | 72 | 1 | 72 | 24 | 48 |
| KH | 2.2 | 3.6 | 1.8 | 4.2 | 7.1 | 1.8 |
| KSiH3 | Trace | — | Trace | — | — | — |
| KSi2H5 | Trace | — | Trace | — | — | — |
| KSiH(SiH3)2 | 82.6 | 1.5 | 85.9 | 1.7 | 66.9 | 3.4 |
| KSi(SiH3)3 | 15.2 | 94.9 | 12.3 | 94.1 | 26.0 | 94.8 |
Fieselmann and Dickson showed that adding 18-c-6 to the reaction of SiH4 with potassium in glyme markedly accelerates formation of [K(18-c-6)SiH3], reducing the reaction time to hours (see Scheme 24). Under these conditions the silanide precipitates as a white solid; after 3 h, monitoring of hydrogen evolution indicated that 87% of the SiH4 had been consumed. The resulting silanide was subsequently subjected to electrophilic functionalization (see Scheme 48).70
 | ||
| Scheme 24 Formation of KSiH3 complexed with 18-c-6, no yields reported. | ||
F. Fehér, Krancher, and M. Fehér (1991) revisited the Na/SiH4 system and established that reactions of SiH4 with sodium in ether solvents invariably yield mixtures of sodium silanides NaSiH3−n(SiH3)n (n = 0–3), rather than exclusively NaSiH3 (see Scheme 25, path a). Product distributions determined by benzyl chloride trapping followed by GC analysis, together with direct NMR characterization of the silanide solutions, contradict earlier claims by Hagenmuller and Pouchard of sole NaSiH3 formation.61,71 The sodium silanides proved markedly more stable in solution than previously suggested, showing no detectable degradation over weeks at room temperature. In DME, the steady-state composition contained approximately twice as much NaSiH3 as NaSi2H5; in diglyme, the ratio increased to about 4.5:1. Minor NaSiH(SiH3)2 was consistently observed, with only trace amounts of NaSi(SiH3)3. Substantial NaH formation was detected only at −30 °C, and the overall conversion of SiH4 to sodium silanides at room temperature was 54–60%. Further investigations examined the effects of used sodium, reaction vessel size, solvent volume, reaction time, and temperature; full details are provided in the original publication. Attempts to isolate discrete sodium silanides by concentrating the solutions promoted redistribution toward higher silanides (see Table 4), whereas evaporation to dryness gave only NaH and polysilanes, indicating complete silanide decomposition (see Scheme 25, path b). The related reaction of KSiH3 with excess of SiH4 in DME exhibited a pronounced concentration dependence: dilute solutions favored the distribution KSi2H5 > KSiH3 > KSiH(SiH3)2, whereas more concentrated mixtures reacted approximately twice as fast but afforded KSiH3 > KSi2H5 > KSiH(SiH3)2 > KSi(SiH3)3 (see Scheme 25, path c).71
 | ||
| Scheme 25 a) Monosilane reacting with Na or K forming different silanides MSiHn(SiH3)3−n (n = 0–3). (b) Attempted isolation of formed silanides leading to degradation. (c) Concentration dependency of formed silanides. | ||
| Silanide | Composition of the starting solution [mol%] | Composition of the solution after concentrating it [mol%] |
|---|---|---|
| NaSiH3 | 65 | 25.7 |
| NaSi2H5 | 30.4 | 16.1 |
| NaH(SiH3)2 | 4.6 | 30.3 |
| NaSi(SiH3)3 | Trace | 27.9 |
Lobreyer, Oeler, and Sundermeyer (1991) designed a purpose-built reactor (design details in Fig. 5) that enabled complete consumption of 15 g of potassium to form KSiH3 at 65 °C in only 105 minutes in diglyme and 150 minutes in DME. The yield of KSiH3 was quantitative with respect to potassium.46,72 This represented a substantial acceleration over the fastest previously reported procedure, which required 11–12 hours to consume 15 g of potassium.68
 | ||
| Fig. 5 Reactor design developed by Lobreyer, Oeler, and Sundermeyer. (a) Stirrer bearing, (b) gas-sparging (hollow) stirrer, (c) vortex breaker, (d) G4 frit, (e) gas inlet, (f) temperature probe. Reproduced from ref. 46 with permission from European Chemical Society. © 1991. | ||
In a follow-up study, Lobreyer et al. attempted to prepare NaSiH3 in diglyme using the same reactor. Instead, they consistently obtained mixtures of sodium silanides NaSiHn(SiH3)3−n (n = 0–3). Spectroscopic analysis after functionalization with p-toluenesulfonic acid (PTSA) indicated approximately 50% conversion to NaSi(SiH3)3. Analogously, direct reaction of potassium metal with SiH4 in the specialized reactor furnished higher potassium silanides KSiH3−n(SiH3)n (n = 0–3) in approx. 90% yield relative to the potassium input, as determined by titration. For both K and Na, extended reaction times at 100 °C afford greater quantities of K/NaSiHn(SiH3)3−n (n = 0, 1); concentrating the solution by solvent removal likewise promotes formation of higher silanides. Using K at 70 °C yields exclusively KSiH3 with no evidence of the build-up reaction. The resulting solutions were subsequently engaged in follow-up transformations with C-, P-, and Sn-based electrophiles (see Schemes 53, 129 and 73).47
Stueger et al. accessed iso-tetrasilanides by the reaction of methyllithium (MeLi) with neopentasilane.73 Later Lainer et al. succeeded in the isolation of iso-tetrasilanide through the addition of tetramethylethylenediamine (TMEDA) (see Scheme 26, path a). Full removal of solvent leads to decomposition, but the isotetrasilanide precipitates from the reaction solution, enabling isolation.
 | ||
| Scheme 26 Synthesis of isotetrasilanides via different routes (path a–c). Yields according to NMR-spectroscopy are quantitative. | ||
Analogously, neopentasilane with MOtBu (M = Li, Na, K, Rb, Cs) in THF, and with LiN(iPr)2 in Et2O (see Scheme 26, path b and c) also led to the isotetrasilanides.48,74 For M = K, identical outcomes were obtained in Et2O and DME as in THF.48 Attempts at isolation or recrystallization led to yellow, insoluble polymers; however, ethereal solutions were stable for several hours and could be employed for downstream functionalization (see Scheme 55). Formation of iso-tetrasilanides was quantitative as determined via NMR-spectroscopy.48
In 2015, Leich, Spaniol, and Okuda introduced a concise, high-yield route to the α-polymorphs of potassium silanide, α-KSiH3 and α-KSiD3, via hydrogenolysis/deuterolysis of the triphenylsilyl complex [K(Me6TREN)SiPh3] (Me6TREN = tris[2-(dimethylamino)ethyl]amine), affording isolated yields of up to 98% (see Scheme 27).75
 | ||
| Scheme 27 Generation of silanide through hydrogenolysis/deuterolysis. | ||
Schuhknecht et al. broadened the hydrogenolysis strategy by using the chelating ligand 1,4,7,10-tetramethyl-1,4,7,10-tetraaminocyclododecane (Me4TACD). The triphenylsilyl precursors [(L)MSiPh3]n (M = Li, Na, K, Rb, Cs; L = Me4TACD) underwent hydrogenolysis with H2 (Scheme 28, path a) to give the corresponding hydridosilanide complexes [(L)MSiH3]m in yields up to 93% for M = Na, K, and Rb. In the lithium system, only small amounts (<10%) of [(L)LiSiH3]m could be isolated; extensive ligand decomposition was observed alongside neutral silanes Hn+1SiPh3−n (n = 0–3). For cesium, hydrogenolysis afforded the ligand-free, polymeric trihydridosilanide [CsSiH3]∞.
 | ||
| Scheme 28 Generation of silanides through a hydrogenolysis reaction (path a) and a redistribution route (path b). | ||
Alternatively, [(L)MSiH3]m (M = Li, Na, K, Rb) can be prepared via a redistribution route (Scheme 28, path b): catalytic amounts of [(L)MSiPh3]n promote scrambling of HSiPh3 in solution to generate H2SiPh2 and SiH4 in situ; subsequent reaction of SiH4 furnishes the hydridosilanide complexes. This indirect route is particularly advantageous for M = Li, substantially improving the final yield relative to direct hydrogenolysis.76
Lainer et al. prepared the magnesium silanide Mg[Si(SiH3)3]2 in low yield by salt metathesis of the isotetrasilanide LiSi(SiH3)3 with MgBr2 (see Scheme 29, path a); however, the product is unstable and decomposes over several days at room temperature. In contrast, reaction of LiSi(SiH3)3 with [(ArylNacNac)MgI(OEt2)] afforded the corresponding ligand-supported magnesium silanide [(ArylNacNac)MgSi(SiH3)3] (Aryl = 2,6-diisopropylphenyl (Dipp), 2,4,6-trimethylphenyl (Mes)) in good yields (see Scheme 29, path b). This complex remains stable after solvent removal, when stored at low temperature. Single-crystal X-ray diffraction of [(DippNacNac)MgSi(SiH3)3]·Et2O (see Fig. 6) provided the first crystallographic characterization of a higher silanides.74
 | ||
| Scheme 29 Synthesis of magnesium isotetrasilanides by Lainer et al. (path a and b). | ||
 | ||
| Fig. 6 Crystal structure of the magnesium isotetrasilanide [(DippNacNac)MgSi(SiH3)3]·Et2O from ref. 74 published by wiley under CC-BY 4.0. © 2022. | ||
Functionalization with group 3 (synthesis of silyltetrels)
Amberger and Römer employed KSiH3 to access a range of silyl boranes via salt metathesis with chloroboranes. While several new SiH3–BR2 derivatives were obtained (see Scheme 30, path a–c), reactions with ClB(n-Bu)2, Cl2BNMe2, BCl3, and HBCl2 did not yield isolable target compounds (see Scheme 30, path d). From these results they concluded that isolable silyl boranes require compensation of the electron deficiency at boron by neighbouring donor substituents capable of pi donation (e.g., amino groups), whereas purely inductive electron release from alkyl substituents is insufficient; consistent with this, H3SiB(n-Bu)2 could not be isolated.62
 | ||
| Scheme 30 Synthesis of silylboranes using SiH3K (path a–d). | ||
Gaines and Iorns prepared the silylborane µ-H3SiB5H8 in 80% isolated yield by reacting LiB5H8 with H3SiCl, representing the first fully hydrogenated silicon–borane compound (see Scheme 31, path a). Stirring µ-H3SiB5H8 with diethyl ether at ambient temperature for 8 h afforded 2-H3SiB5H8 (see Scheme 31, path b).80 In a subsequent study, heating of 2-H3SiB5H8 to 150 °C for 13 h gave 1-H3SiB5H8, identified as the most stable isomer (see Scheme 31, path c).80,81 Geisler and Norman selectively halogenated the silyl substituent in silyl-substituted pentaboranes using BCl3, BBr3, and HBr (see Scheme 32, path a–c). Under otherwise comparable conditions, BCl3-mediated chlorination of µ-H3SiB5H8 required longer reaction times than chlorination of 2-H3SiB5H8 (Scheme 32, path a and c).82
 | ||
| Scheme 31 Generation of µ-silylpentaborane and its isomerization reactions (path a–c). | ||
 | ||
| Scheme 32 Halogenation reactions of 2- and µ-silylpentaborane (path a–c). | ||
Geisler, Soice, and Norman investigated BCl3-mediated halogenation of the silyl substituent in silylpentaboranes (see Scheme 33, path a).83 For 1-H3SiB5H8, the halogenation rate was indistinguishable from that of 2-H3SiB5H8, whereas the µ isomer required higher temperatures and longer reaction times to reach full conversion (see Scheme 33, path c and d).82,83 They also synthesized µ-disilylpentaborane by treating LiB5H8 with Si2H5Cl (see Scheme 33, path b). The µ isomer rearranged to the 2 isomer upon stirring in Et2O at room temperature for 4 h, and to the 1 isomer only under more forcing conditions (120 °C, 14 h) (see Scheme 33, path c and d). This behaviour parallels that of the corresponding monosilylpentaboranes; in both series, the thermodynamic stability follows µ ≪ 2 < 1.80,81,83
 | ||
| Scheme 33 Halogenation of 1-silylpentaborane and generation of disilylpentaborane and its isomers (path a–d). | ||
Lainer et al. synthesized the lithium hypersilyl borate [(H3Si)3SiBH3]Li in a single step by treating the isotetrasilanide LiSi(SiH3)3 with Me2S·BH3. The obtained salt is notably robust, remaining unchanged for extended periods as a solid or in solution at ambient temperature (see Scheme 34). Attempts to access cationic or neutral derivatives by hydride abstraction with B(C6F5)3 or with Me3N·HCl were unsuccessful; in both cases, uncharacterized polysilanes formed. The structure of the new hypersilyl borate was confirmed by single-crystal X-ray diffraction (see Fig. 7).84
 | ||
| Scheme 34 Synthesis of lithium hypersilyl borate and attempted hydride abstraction reactions. | ||
 | ||
| Fig. 7 Crystal structure of hypersilyl borate [(H3Si)3SiBH3]Li from ref. 84 published by Wiley under CC-BY 4.0. © 2024. | ||
 | ||
| Scheme 35 Generation of trisilylalane by Hagenmuller and Pouchard (path a) and monosilylalane by Semenenko and Taisumov (path b). For path (a) no yields were reported. | ||
 | ||
| Scheme 36 Synthesis of silylalane by Amberger and Römer. | ||
Functionalization with group 4 (synthesis of silyltetres)
For the synthesis of organosilanes two prevalent methods are used in the literature, either the reduction of silyl halides or via transformations of inorganic silanes.
Reduction of silyl halides is the most commonly used approach for the synthesis of alkylsilylhydrides. Most frequently LiAlH4 is used for the reduction, though other common reductants like lithium hydride, sodium hydride, triethyl aluminium (AlEt3) and diisobutylaluminium hydride can be employed as well (see Scheme 37).28,91
 | ||
| Scheme 37 Synthesis of organosilanes via reduction with LiAlH4. The yields for R = Me, vinyl, n-Bu and i-Bu were only given as range of 80 to 90% by Tannenbaum et al. For R = Pr, no detailed yield was given. | ||
Consequently using di- to tetrabromosilylmethane with LiAlH4 allows for the synthesis of the respective silylmethane.92–94 When bromosilylmethanes were treated in standard solvents such, as n-Bu2O, the yields were very poor, however, using tetraline in combination with benzyltriethylammoniumchloride (TEBAC), excellent yields were achieved (see Scheme 38, path b).93
 | ||
| Scheme 38 Synthesis of C(SiH3)4 starting from C(SiH2Ph)4 (path a) and synthesis of CH4–n(SiH3)n (path b). | ||
Schmidbaur and co-workers were the first to report tetrasilylmethane (C(SiH3)4). The usual choice of a polar solvent was replaced with a two-phase system with phase transfer catalyst, in order to supress the cleavage of Si–C bonds, but the sideproduct trisilylmethane (HC(SiH3)3) was still observed (see Scheme 38, path a).94,95 Furthermore they were able to obtain 2,2-disilylpropane from 2,2-dibromosilylpropane Me2C(SiH2Br)2.96
While commonly Et2O is used as the solvent of choice for the synthesis of lower boiling silanes, Kozhevnikov and co-workers were able to obtain methylsilane (MeSiH3) in excellent yield, using n-Bu2O as solvent (see Scheme 39, path a).97 Bellama and co-workers prepared disilylmethane (H2C(SiH3)2), via reduction of bistrichlorosilylmethane (H2C(SiCl3)2), in 50% yield (see Scheme 39, path b).98
 | ||
| Scheme 39 Synthesis of MeSiH3 (path a) and H2C(SiH3)2 (path b) via reduction with LiAlH4. | ||
A representative example of metal-hydride reduction of alkylsilyl halides is the conversion of 1,1,3,3-tetrachloro-1,3-disilabutane to 1,3-disilabutane. The tetrachloro precursor is prepared in 41% yield by reacting HCl with elemental Si and MeSiCl2CH2Cl using Cu based catalysts,99 followed by reduction with LiAlH4 to afford 1,3-disilabutane (see Scheme 40).100
 | ||
| Scheme 40 Synthesis of 1,3-disilabutane by reduction with LiAlH4. No yield and experimental details were reported for the LiAlH4 reduction. | ||
Fritz and co-workers showed that treatment of chlorinated C-spiro-linked 2,4-disilacyclobutanes with LiAlH4 results in the cleavage of the four membered rings (see Schemes 41 and 42, path b). In contrast, when they employed i-Bu2AlH no cleavage was observed, and the hydrogenated C-spiro-linked 2,4-disilacyclobutanes were obtained (see Schemes 41 and 42, path a).101
 | ||
| Scheme 41 Example of the hydration of a C-spiro linked 2,4-disilacyclobutane with LiAlH4 (path b) and i-Bu2AlH (path a). No yield for the LiAlH4 reaction was reported. | ||
 | ||
| Scheme 42 Hydration of a C-spiro linked 2,4-disilacyclobutane with LiAlH4 (path b) and i-Bu2AlH (path a). No yield for the LiAlH4 reaction was reported. | ||
Pyrolysis of the methylchlorosilanes methyltrichlorosilane (MeSiCl3), dichlorodimethylsilane (Me2SiCl2) and chlorotrimethylsilane (Me3SiCl) at 700 °C afford mixtures of linear and cyclic alkylchlorosilanes, for example, 1,3-disilapropane. The chlorine containing pyrolysis products were then reduced with LiAlH4, though isolation of pure compounds is problematic.102 Furthermore, pyrolysis of a mixture of SiH4 and ethylene leads to formation of mixtures of the alkylsilanes ethylsilane (EtSiH3), diethylsilane (Et2SiH2), triethylsilane (Et3SiH), though no yields were reported.103 The copyrolysis of disilane with methylsilane (MeSiH3) afforded methyldisilane (MeH2SiSiH3), with dimethylsilane (Me2SiH2) it led to formation of 1,1-dimethyldisilane (Me2SiHSiH3) and with trimethylsilane (Me3SiH) it resulted in the formation of 1,1,1-trimethyldisilane (Me3SiSiH3), though again no isolation was performed.104
Furthermore, freshly prepared bis- and tristrifluoromethanesulfonatosilylmethanes are effective precursors for di- and trisilylmethanes. Although the formation of these intermediates proceeds in essentially quantitative yield, they undergo decomposition and isomerization at ambient temperature; therefore, immediate reduction is required to achieve optimal yields (see Scheme 43).105
 | ||
| Scheme 43 Synthesis of di- and trisilylmethane via reduction.105 | ||
Westermark et al. employed LiAlH4 in combination with aluminium chloride (AlCl3) to reduce alkyltriethoxysilanes to the respective alkylsilanes in good yields (see Scheme 44).106
 | ||
| Scheme 44 Reduction of alkyltriethoxysilanes to afford alkylsilanes. | ||
Trialkylstannanes can be used together with Lewis-base catalysts for the partial and full reduction of alkylsilylhalides to alkylhydrosilanes. Usually a distribution of products is obtained, depending on the catalyst used, the stannane used and the molar ratio of stannane to silane.107–109 When using tributyltin hydride (Bu3SnH) and catalytic amounts of triphenylphosphine (PPh3), methylchlorodisilanes show significant Si–Si bond cleavage; in contrast, no cleavage is observed when the analogous methylbromodisilanes are used (see Scheme 45).107
 | ||
| Scheme 45 Reduction of methylbromodisilanes with Bu3SnH and PPh3. No specific yields were reported for the individual compounds, but yields were given as 80% to quantitative yields for the reductions. | ||
One of the first reported synthesis of monoalkylsilanes is the synthesis of MeSiH3 from H3SiCl, by Stock and Somieski in 1919. But no information about the purity of the obtained MeSiH3 is given (see Scheme 46).10
 | ||
| Scheme 46 Synthesis of MeSiH3 via reaction of H3SiCl with ZnMe2. | ||
Another approach is the direct hydrosilylation of unsaturated substrates, though early work demonstrated that direct addition of SiH4 to alkenes or alkynes generally affords product mixtures with low yields under both thermal and photochemical conditions.110 The use of catalysts, for example alkali metal aluminates111 and LiAlH4112 promotes more selective addition of SiH4 to alkenes, significantly improving the selectivity. The best results were achieved by employing Pt(PPh3)4 as catalyst (see Scheme 47).113
 | ||
| Scheme 47 Synthesis of alkylsilanes via addition of SiH4 to an alkene. | ||
SiH4 was also employed as feedstock for nucleophilic substitution via silyl anions. Conversion of SiH4 into KSiH3 allows for subsequent reaction with organic halides, for example MeSiH3 was obtained by reaction of methyliodide (MeI) with KSiH3 (see Scheme 48, path a).114 Ritter and Ring obtained MeSiH3 in excellent yields by treating KSiH3 with excess methyl chloride (MeCl) (see Scheme 48, path b).15 Fieselmann and Dickson improved the yields by using potassium(18-crown-6) silanide ([K(18-c-6)]SiH3) and demonstrated the synthesis of H2C(SiH3)2 (see Scheme 48, path c).70 Similar to Na- and KSiH3, RbSiH3 and CsSiH3 are suitable for the reaction with MeI as well.64
 | ||
| Scheme 48 Synthesis of alkylsilanes by reaction of KSiH3 with MeX (X = Cl, I) (path a–c). | ||
Nucleophilic substitution between KSiH3 and bromomethylsilane (MeSiH2Br) only leads to formation of methyldisilane in poor yields of 5%, a more suitable approach is the reaction between a monohalodisilane with methyllithium (MeLi) (see Scheme 49, path a and b).92
 | ||
| Scheme 49 Synthesis of methyldisilane (path a and b). | ||
Alkali metal silanides derived from SiH4 can be converted to alkylsilanes in hexamethylphosphoramide upon treatment with the corresponding alkyl halides, typically in good yields (see Scheme 50).63
 | ||
| Scheme 50 Synthesis of methyl- and ethylsilane from K- or NaSiH3. No detailed procedures with exact amounts of reagents were given for R = Me, Et. | ||
Treatment of mixtures of potassium silanides with ethyl chloride (EtCl) afforded the corresponding ethylsilanes in approximately 25% overall yield, notably individual product yields were not reported (see Scheme 51).66
 | ||
| Scheme 51 Synthesis of ethyl substituted silanes from potassium silanides with ethylchloride. No specific yield for the individual compounds was given. | ||
Furthermore, treatment of mixtures of higher potassium silanides with phenyl chlorosilane (PhH2SiCl) afforded the corresponding phenylsilanes in approximately 20% overall yield, notably no specific yield was given for the individual products (see Scheme 52).45
 | ||
| Scheme 52 Synthesis of phenylisotetra-, phenylneopenta-, and phenylneohexasilane from the respective potassium silanide and phenylchlorosilane. No specific yield for the individual compounds was assigned. | ||
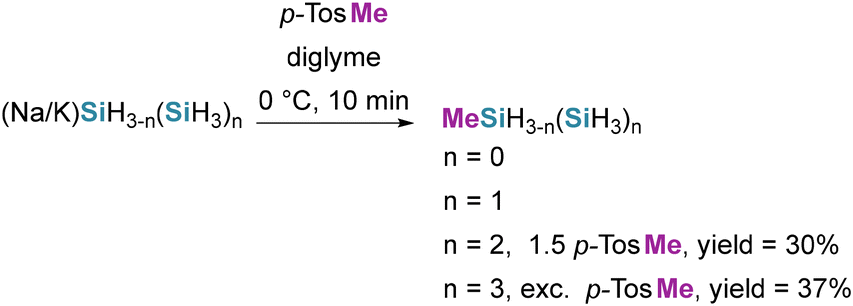
Sundermeyer and co-workers accessed the methylsubstituted silanes MeSiH3, methyldisilane (MeSiH2SiH3), 2-methyltrisilane (SiH3SiHMeSiH3) and MeSi(SiH3)3 via the buildup reaction between SiH4 and K in diglyme, and subsequent alkylation with methyl p-toluenesulfonate (p-TosMe) (see Scheme 53).47,115
 | ||
| Scheme 53 Synthesis of methylsilanes from silanide mixtures. While products are formed with n = 0–3, the isolated yield was only given for n = 2 and n = 3. | ||
Stueger and co-workers employed lithium and potassium isotetrasilanides to access methylisotetrasilane (MeSi(SiH3)3) and a range of alkyl- and phenyl-substituted neopentasilanes. Treatment of solutions of lithium isotetrasilanide (LiSi(SiH3)3) with methyl p-toluenesulfonate afforded methylisotetrasilane in 36% yield, while using triphenylchlorosilane (ClSiPh3) as the electrophile furnished triphenylsilylneopentasilane (SiPh3Si(SiH3)3) in 50% yield (see Scheme 54, path a and b). When dimethylphenylchlorosilane (ClSiMe2Ph) or phenylchlorosilane (ClSiH2Ph) were employed, both the corresponding monosubstituted neopentasilanes and the disubstituted derivatives were obtained (see Scheme 55, path a and b). To suppress disubstitution, the authors avoided an excess of potassium silanide by adding the silanide slowly to the electrophile.48
 | ||
| Scheme 54 Synthesis of methylisotetrasilane (path a) and triphenylsilylneopentasilane (path b) from lithiumisotetrasilanide. | ||
 | ||
| Scheme 55 Synthesis of dimethylphenyl- and phenylneopentasilane from potassiumisotetrasilanide, with concomitant formation of the respective disubstituted product (path a and b). The yield for disubstituted bisdimethylphenylneopentasilane was not reported. | ||
Stueger, Haas and co-workers obtained several phenyl-substituted organosilanes in good yields by treating lithium isotetrasilanide with phenylchlorosilanes as electrophiles. When dichlorophenylsilane (Cl2HSiPh) and 1,2-dichloro-1,2-tetrakisphenyl-disilane were employed as electrophiles, the corresponding branched nonasilane and branched decasilane were isolated as pure compounds, however, the silane byproducts were not identified (see Scheme 56, path a and b). A crystal X-ray structure was determined for (SiH3)3Si(SiPh2)2Si(SiH3)3 (see Fig. 8).49
 | ||
| Scheme 56 Synthesis of phenylsubstituted nona- and decasilanes from lithium isotetrasilanide (path a and b). Both compounds were isolated as pure compounds, but during the reaction undefined silanes emerged as byproducts. | ||
 | ||
| Fig. 8 Crystal structure of (SiH3)3Si(SiPh2)2Si(SiH3)3, adapted from Stueger and Haas et al.49 with permission from American Chemical Society © 2019. | ||
In contrast, when dichlorodiphenylsilane (Cl2SiPh2) was used as electrophile the corresponding diphenylnonasilane, and two cyclic phenylsilane byproducts were obtained in low yields (see Scheme 57). A crystal structure from (SiH3)3SiPh2(SiH3)3 was obtained (see Fig. 9).49
 | ||
| Scheme 57 Synthesis of phenylsubstituted organosilanes with dichlorodiphenylsilane. | ||
 | ||
| Fig. 9 Crystal structure of (SiH3)3SiPh2(SiH3)3, adapted from Stueger and Haas et al.49 with permission from American Chemical Society © 2019. | ||
Moreover, treatment of (H3Si)3Si(SiPh2)2Si(SiH3)3 with triflic acid gave the partially hydrogenated derivative (H3Si)3Si(SiPhH)2Si(SiH3)3, this compound was not isolated but was used in situ to prepare the fully hydrogenated product (H3Si)3Si(SiH2)2Si(SiH3)3 (see Scheme 58).49
 | ||
| Scheme 58 Synthesis of (H3Si)3Si(SiPhH)2Si(SiH3)3. The compound was not isolated and reacted further to (H3Si)3Si(SiH2)2Si(SiH3)3. | ||
Haas and co-workers converted dodecamethoxyneopentasilane to the corresponding potassium silanide using potassium t-butoxide (KOt-Bu) (see Scheme 59, path a). The subsequent treatment of the silanide with electrophiles, such as MeI resulted in the formation of the corresponding nonamethoxyisotetrasilanes (see Scheme 59, path b). Reduction of these derivatives with a slight excess of i-Bu2AlH afforded the corresponding organosilanes in good yields (see Scheme 59, path c).22,116 Applying the same strategy to a potassium disilanide delivered the cyclic 1,1,4,4-tetrakis(silyl)octamethyl-cyclohexasilane (see Scheme 60). A crystal structure of this compound was also obtained (see Fig. 10).22
 | ||
| Scheme 59 Synthesis of organosilanes from potassium silanides via reaction with electrophiles and subsequent reduction with i-Bu2AlH (path a–c). | ||
 | ||
| Scheme 60 Synthesis of organohexasilane compounds from a potassiumdisilanide. | ||
 | ||
| Fig. 10 Crystal structure of 1,1,4,4-tetrakis(silyl)octamethylcyclohexasilane. Adapted from Haas et al.22 published by American Chemical Society under CC-BY 4.0. | ||
Silyl halides are viable substrates for Grignard based synthesis of alkylsilanes, though only in moderate yields. As an example, H3SiBr reacts with various Grignard reagents to afford the respective alkylsilane (see Scheme 61).117
 | ||
| Scheme 61 Synthesis of alkylsilanes from silyl halides and Grignard reagents. No specific molar ratios or temperatures were given for the synthesis. | ||
Moreover, halomethylsilanes, such as, bromo- and chlorosilylmethane are suitable precursors to silylmethyl Grignard reagents (XMgCH2SiH3), which subsequently react with halosilanes to furnish silyl-substituted methanes, exemplified by CH2(SiH3)2 (see Scheme 62). However, neither bromo- nor chloromethylsilane reacts with magnesium under standard conditions, the corresponding Grignard reagent is obtained only when activated magnesium is generated by reducing magnesium chloride (MgCl2) with K in THF under reflux. They stated that they obtained 1,2-disilylethane (H3SiEtSiH3) in 26% yield from the preparation of H3SiCH2MgBr due to Wurtz-coupling. Further examples of this synthesis approach are given in Table 5.118
 | ||
| Scheme 62 Example synthesis of alkylsilanes from silylmethylgrignard compounds. | ||
| Grignard reagent | Halosilane | Product | Yield [%] |
|---|---|---|---|
| Yields marked with asterisk are crude yields. | |||
| Me3SiCH2MgCl | H3SiBr | Me3SiCH2SiH3 | 86 |
| Me2SiHCH2MgCl | H3SiBr | Me2SiHCH2SiH3 | 20–30* |
| MeSiH2CH2MgCl | H3SiBr | MeSiH2CH2SiH3 | 92 |
| H3SiCH2MgBr | H3SiBr | CH2(SiH3)2 | 91 |
| H3SiCH2MgBr | Me2SiHCl | H3SiCH2SiHMe2 | 30* |
| H3SiCH2MgBr | Me3SiCl | H3SiCH2SiMe3 | 85 |
Dimethyltitanocene (Me2TiCp2) is able to catalyze redistribution reactions of diethoxymethylsilane ((EtO)2MeSiH) affording triethoxymethylsilane ((EtO)3MeSi) and methylsilane in excellent yields.119 Other catalysts like sodium ethoxide are used for the redistribution as well (see Scheme 63).120
 | ||
| Scheme 63 Catalytic redistribution of (EtO)2MeSiH. | ||
Ozonizer-type silent electrical discharge tubes were also employed in the synthesis of methyldisilane from a mixture of Me2O and SiH4, though this approach is not synthetically useful due to the low yield of 4% and the poor selectivity.121
Treatment of silanes with organolithium reagents provides an alternative route to organosilanes. Bolduc and Ring reported that condensing disilane into a solution of ethyllithium (LiEt) in benzene (C6H6), furnishes ethyldisilane (EtH2Si–SiH3) in 37% yield by the following reaction at room temperature (see Scheme 64).122
 | ||
| Scheme 64 Synthesis of ethyldisilane from disilane and ethyllithium. | ||
This approach can be expanded to larger silanes such as tri-, n-tetra- and n-pentasilane as well. When solutions of the respective silanes in n-hexane are treated with n-butyllithium (n-BuLi) solutions, the respective n-butylsilane is afforded (see Scheme 65, path a and b). Disubstituted dibutylsilanes are observed as well and are the main product of the reaction when 2 equivalents of n-BuLi are used. Additionally, a solid polysilane is formed as well. While it was mentioned that SiH4 and disilane are formed in the reaction with trisilane as well, it was not indicated if these silanes are observed with tetra- and pentasilane as well.123
 | ||
| Scheme 65 Synthesis of butylsilanes from tri-, tetra- and pentasilane (path a and b). No specific yields for the disubstituted n-butylsilanes with 1 equivalent n-BuLi and no individual yields for the monosubstituted yields were given. | ||
The most basic member of the Si–Ge hydride family is monosilylgermane (H3SiGeH3). Its first targeted preparation was reported by Cox and Varma in 1964, who achieved the formation of H3SiGeH3 in approximately 20% yield via the nucleophilic substitution of chlorogermane with potassium silanide (see Scheme 66). This study represents the earliest attempt to generate a Si–Ge single-bonded hydride under controlled conditions.125
 | ||
| Scheme 66 Synthesis of silylgermane (H3GeSiH3). | ||
In the same year, Phillips and co-workers identified H3SiGeH3 as a minor product during silent electrical discharge experiments, although the hydride was obtained only in trace amounts (∼2%).126
An alternative approach to the silylation of germyl anions employs silyltriflate (H3Si-OTf) or silylnonaflate (H3Si-ONf) as highly electrophilic silylating agents (see Scheme 67). This approach was demonstrated by Sundermeyer and co-workers (1994) and later expanded by Kouvetakis and co-workers (2005). Using these reagents, H3SiGeH3 can be obtained in moderate yields ranging from 22–35%, representing a significant improvement over earlier methods based on halide replacement.46,127
 | ||
| Scheme 67 Synthesis of silylgermane (H3GeSiH3) after treatment of KGeH3 with SiH3-OTf and SiH3-ONf. | ||
Sundermeyer and co-workers also demonstrated the synthesis of branched silylgermane with the general formula (SiH3)nGeH4−n (n = 2–4). In their studies, a mixture of branched silylsilanides (SiH3)3−nSiNa (n = 0–2) was first generated by reacting sodium with monosilane in diglyme. Subsequent treatment of this anion mixture with monogermane induced a rearrangement in which the germanium atom migrated into the central position of the silyl framework, forming branched silylgermanides. Finally, silylation with H3SiONf afforded the corresponding silylgermanes (see Scheme 68). However, the individual products could not be separated, and yields were therefore only estimated by gas chromatography, limiting the characterization of the mixture.18
 | ||
| Scheme 68 3-Step-synthesis of silylgermanes. *No isolated yields; only determined via gaschromatography. | ||
With a similar procedure, Ritter et al. synthesized the different silylgermanes with the general formular (H3Ge)nSiH4−n. They also employed silyltriflates (HnSi(OTf)4−n) and silylnonaflates (HnSi(ONf)4−n), enabling the selective synthesis and isolation of the individual products (Scheme 69).127
 | ||
| Scheme 69 Synthesis of germylsilanes. | ||
More recently, Stueger et al. introduced a complementary and highly selective approach to silyl-germanes based on nucleophilic substitution reactions of well-defined silanide reagents.73 Using lithium tris(silyl)silanide, LiSi(SiH3)3, the previously unknown germaisotetrasilane Ph3GeSi(SiH3)3 could be synthesized in high yield on a multigram scale by reaction with Ph3GeCl (see Scheme 70). The resulting Ph3Ge-substituted silyl-germanes serve as versatile intermediates for further derivatization. Selective treatment with methyllithium induces cleavage of a single Si–Si bond while preserving the Si–Ge linkage, generating lithium silanide species that can be reacted with other chlorosilanes or chlorogermanes. This strategy enables the efficient synthesis of higher substituted silyl-germanes containing one or two Ph3Ge groups in excellent yields. In contrast, attempts to access hydrogen rich silylgermanes via stepwise dephenylation at the germanium center using triflic acid followed by hydride reduction were significantly less selective.
 | ||
| Scheme 70 Synthesis of different branched germasilicon hydrides. | ||
It was also possible to obtain single crystals suitable for X-ray diffraction analysis (Fig. 11 and 12).
 | ||
| Fig. 11 Crystal structure of Si(GePh3)(SiH3)3. Reproduced from ref. 73 with permission from American Chemical Society, © 2016. | ||
 | ||
| Fig. 12 Crystal structure of Si(GePh3)2(SiH3)2. Reproduced from Stueger et al.73 with permission from American Chemical Society, ©2016. | ||
Mixed silylgermanes with preformed Si–Ge bonds were reported by Wagner and co-workers as part of a two-step synthesis strategy toward single-source precursors for Si–Ge materials.128 In a first step, dichlorogermanes (R2GeCl2, R = Ph, n-Bu) were reacted with hexachlorodisilane in the presence of catalytic [n-Bu4N]Cl, generating bis(trichlorosilyl)-germanes Cl3Si–GeR2–SiCl3 in high yields. The reaction proceeds via in situ formation of the nucleophilic [SiCl3]− species, enabling efficient Si–Ge bond formation under mild conditions. Subsequent hydride reduction of the SiCl3 groups with LiAlH4 afforded the corresponding hydrosilanes H3Si–GeR3–SiH3 in good to excellent yields (see Scheme 71).
 | ||
| Scheme 71 Formation of germylsilanes with the general formula H3Si–(GR)m–SiH3. | ||
Moreover, Wagner and co-workers were successful in gaining crystals for X-ray diffractometry of intermediates and products (see Fig. 13). This modular approach provides convenient access to well-defined silylgermanes while avoiding preformed Si–Cl bonds and suppressing scrambling reactions typically encountered for fully hydride-substituted Si–Ge compounds.
 | ||
| Fig. 13 Crystal structures of Cl3Si–(GePh2)2–SiCl3 and H3Si–(GePh2)2–SiH3. Reproduced from Wagner and co-workers128 with permission from American Chemical Society, ©2022. | ||
The simplest hydridic silylstannane, H3Si–SnH3, has been experimentally synthesized only under highly controlled conditions. The first targeted synthesis was reported by Wiberg and co-workers, who obtained the compound via low-temperature hydride reduction of prefunctionalized Si–Sn acetates or chlorides using LiAlH4, generating ethereal solutions of the mixed hydride (see Scheme 72).129
 | ||
| Scheme 72 Formation of silylstannane (H3SnSiH3) via the treatment of chloro-, or acetatesilylstannane with LiAlH4. No yields reported. | ||
However, even under these conditions, H3Si–SnH3 was found to be stable only in highly diluted solutions below −80 °C, while enrichment or warming led to rapid decomposition into silane, metallic tin and hydrogen. Complementary matrix-isolation IR studies later provided spectroscopic evidence for hydridic Si–Sn species formed by insertion of atomic tin into silane, yielding HSnSiH3 complexes stabilized in inert argon matrices at cryogenic temperatures.130 Together, these studies demonstrate while hydridic silylstannanes can be generated and spectroscopically characterized, their extreme thermal lability fundamentally limits their isolation and practical application. Sundermeyer and co-workers were successful in generating silyl stannides with the general formular NaSn(SiH3)nH3−n via the reaction of NaSi(SiH3)nH3−n and SnH4 at −40 °C (see Scheme 73).47 In contrast to the corresponding silylgermanides systems (see Scheme 68), no further follow-up chemistry or reactivity studies were reported.
 | ||
| Scheme 73 Generation of silylstannides. | ||
Functionalization with group 5 (synthesis of silylpnictogens)
A widely used route to silylamines involves the reduction of silylamino halides. Although several hydride reagents are effective, LiAlH4 is most commonly employed and usually Cl is the halide of choice. The employed silylamino halides are typically prepared by reacting silyl halides with the corresponding lithium amides,135,136 or via reaction of the respective amine with the silylhalide,137 followed by hydride reduction to afford the target silylamines. A general reaction is given in Scheme 74, path a, several examples can be found in Scheme 74, path b to g.135–139
 | ||
| Scheme 74 General synthesis of silylamines from chlorosilanes and example synthesis (path a–g). | ||
Another widely used reductant for aminohalosilanes is diisobutylaluminium hydride. For example, reduction of diisopropylaminopentachlorodisilane with i-Bu2AlH affords the partially halogenated diisopropylaminodichlorodisilane in 83% yield (see Scheme 75).140
 | ||
| Scheme 75 Example synthesis of partially halogenated aminochlorosilanes using i-Bu2AlH. | ||
An alternative, synthetically straightforward route exploits nucleophilic substitution at halogenated silanes by the corresponding amines, typically generating the ammonium halide of the amine as a byproduct.141–143 Stock and Somieski were able to obtain trisilylamine from the reaction between ammonia (NH3) and H3SiCl, notably if an excess of NH3 was used the reaction was unselective, while excess H3SiCl leads to the selective formation of trisilylamine.11,144,145 They reasoned that the reaction proceeds stepwise with silylamine and disilylamine (HN(SiH3)2) as intermediates, but could not isolate them and they observed the formation of a polymeric solid and SiH4 (see Scheme 76).144 Later Aylett and Hakim showed that disilylamine decomposes to trisilylamine and NH3 and in the presence of NH3 disilylamine decomposes to SiH4 and a solid polymer.146
 | ||
| Scheme 76 Synthesis of trisilylamine from H3SiCl and NH3, according to Stock. No yield was reported. | ||
Burg and Kuljian adapted Stocks and Somieskis synthesis by slowly mixing gaseous ammonia, from below, into H3SiCl, resulting in the formation of trisilylamine in 80% yield (see Scheme 77). Notably they mentioned that slow addition of ammonia is necessary to obtain high yields and good selectivity.147 When ammonia and H3SiCl were condensed into a flask the reaction afforded trisilylamine in low yields.148
 | ||
| Scheme 77 Synthesis of trisilylamine with gaseous ammonia and H3SiCl, according to Burg. | ||
Emeléus and Miller already employed this method in 1939 for the synthesis of dimethylsilylamine (Me2NSiH3), ethyldislylamine (EtN(SiH3)2) and methyldisilylamine (MeNSiH3) (see Scheme 78, path a and b), though they only identified them and gave no yields.141 For instance, monochlorodisilane (H3SiSiH2Cl) undergoes substitution with diisopropylamine (i-Pr2NH) to furnish diisopropylaminodisilane (H3Si–SiH2Ni–Pr2), concomitant with formation of diisopropylammonium chloride (i-Pr2NH2Cl) (see Scheme 78, path c).143 Both isopropylamine (i-PrNH2) and t-butylamine (t-BuNH2) react with H3SiCl to afford the corresponding disilylamines in good yields (see Scheme 78, path b),142 while monochlorodisilane reacts with diisopropylamine to give diisopropylaminodisilane.149 Burg and Kuljian prepared methyldisilylamine (MeN(SiH3)2) from methylamine (MeNH2) and ammonia in 85% yield (see Scheme 78, path b).147 Bromodisilane (H3Si–SiH2Br) reacts with dimethylamine (Me2NH) to afford dimethylaminodisilane (H3Si–SiH2NMe2) in 8% yield (see Scheme 78, path d).150
 | ||
| Scheme 78 Example synthesis of silylamines from halosilanes and amines (path a–d). When no yields are given for the product, no yield was reported. | ||
Furthermore, disilylamine reacts with H3SiI to afford trisilylamine in 67% yield, when a slight excess of H3SiI is used to afford trisilylamine the yield increases to 87%, though no experimental details were given (see Scheme 79, path a).146,151 Similarly, anhydrous hydrazine reacts with H3SiI to give tetrasilylhydrazine in 40% yield, which is a strong reducing agent, but explodes in contact with air (see Scheme 79, path b).152
 | ||
| Scheme 79 Synthesis of trisilylamine (path a) and tetrasilylhydrazine (path b) using H3SiI. | ||
Ward and MacDiarmid treated iododisilane with NH3 to afford trisdisilylamine in 64% yield (see Scheme 80).153
 | ||
| Scheme 80 Synthesis of tridislanylamine from monoiododisilane and NH3. | ||
Diaminosilylamines, such as t-BuN(SiH2NMe2)2, can be obtained from dichlorosilane (H2SiCl2) in a two-step synthesis in good yields. H2SiCl2 reacts with the respective primary amine, either in excess154 or under use of a aiding base like trimethylamine (NMe3),155 for example t-BuNH2 to obtain a dichlorosilylamine, which subsequently reacts with an amine to afford the respective diaminosilylamine (see Scheme 81).154,155
 | ||
| Scheme 81 Synthesis of diaminosilylamines from H2SiCl2 and the respective amines. | ||
Wells and Schaeffer investigated the reaction of trisilylamine with ammonia by condensing both reagents into a flask and allowing them to react in the liquid phase. The reaction afforded N,N′,N″-trisilylcyclotrisilazane only in trace amounts, however, multiple runs provided sufficient material for characterization. They reasoned that ammonia catalyses the elimination of SiH4 from trisilylamine (see Scheme 82).148
 | ||
| Scheme 82 Reaction between trisilylamine and ammonia. | ||
Various azapolysilanes can be prepared by reacting diisopropylaminodisilane with primary amines, such as t-BuNH2. Alternatively by the reaction of chlorodisilane with primary amines in the presence of triethylamine as a base, or by the treatment of 1,2-dichlorodisilane (ClH2Si–SiH2Cl) with primary amines and triethylamine (NEt3) (see Scheme 83).156
 | ||
| Scheme 83 Example synthesis of silylamines from diisopropylaminodisilane, monochlorodisilane and 1,2-chlorodisilane. No isolated yields were given. | ||
Furthermore, alkalimetal amides, such as sodium amide, are suitable reagents to form silylamines via reaction with silylhalides. After formation of the respective sodium amide a silylhalide is added affording the formation of silylamines, this approach allows for the synthesis of diisopropylaminosilane (i-Pr2NSiH3) (see Scheme 84).157
 | ||
| Scheme 84 Synthesis of silylamines from alkalimetal amides and silylhalides. No isolated yields were given. | ||
Schmidbaur and co-workers accessed 1,1- and 1,2-diaminodisilanes from solutions of 1,2-disilanediylditriflate. The solution of 1,2-disilanediylditriflate is afforded from di-p-tolyldisilane and triflic acid and reacts with solutions of the respective amine and triethylamine to afford the 1,2-diaminodisilanes (see Scheme 85, path a and b). Notably, in the case of diethylamine (HNEt2) the 1,1-substituted diethylaminodisilane is also formed (see Scheme 85, path a), whereas the 1,1-diisopropylaminodisilane is not observed (see Scheme 85, path b).158
 | ||
| Scheme 85 Synthesis of diaminodisilanes from disilanediylditriflate using secondary amines (path a and b). | ||
When primary amines are employed, the reaction furnishes six-membered cyclic aminosilanes containing two disilyl units, rather than the 1,1- and 1,2-substituted linear diaminodisilanes obtained with secondary amines (see Scheme 86, path a). When 1,4-difunctional diamines are used instead, the reaction yields five-membered cyclic compounds bearing an exocyclic silyl substituent (see Scheme 86, path b).158
 | ||
| Scheme 86 Synthesis of cyclic silylamines from disilanediylditriflate. (path a and b). | ||
Dehydrogenative coupling offers an attractive, atom-economical route to silylamines. A variety of catalysts promote the coupling of amines with hydrosilanes, including the Lewis acid trispentafluorophenylborane (B(C6F5)3),159 transition metal systems such as triruthenium dodecacarbonyl (Ru3(CO)12),160 main-group reagents such as di-n-butylmagnesium (n-Bu2Mg),161 Mn based catalysts162 and Zn based catalysts.163 Sanchez et al. reported the dehydrogenative coupling of amino- and diaminosilanes derived from disilane and trisilane using ruthenium on carbon (Ru/C) as the catalyst, although only non-isolated yields were provided (see Scheme 87, path a and b).164
 | ||
| Scheme 87 Synthesis of aminodisilane (path a) and aminotrisilane (path b) using Ru/C as catalyst. Only non-isolated yields were reported. Several R groups can be employed. | ||
SiH4 can be dehydrogenatively coupled with diisopropylamine using di-n-butylmagnesium n-Bu2Mg as catalyst, as shown by Maddock et al. (see Scheme 88).159,161
 | ||
| Scheme 88 Dehydrogenative coupling of SiH4 and diisopropylamine, using n-Bu2Mg. | ||
Moreover, Rekken employed Zn-based catalysis to achieve dehydrogenative coupling, affording diisopropylaminodisilane and bisdiisopropylaminodisilane. They further demonstrated that Zn-based catalysis is applicable to SiH4, albeit with low conversions. No isolated yields were reported, the product compositions were determined by GC-FID, and diisopropylamine was used in large excess (see Scheme 89).163
 | ||
| Scheme 89 Dehydrogenative coupling of disilane with diisopropylamine, using Zn based catalysts. Only conversions were reported. | ||
Using Zn(NTf2)2 as a catalyst enabled the dehydrogenative coupling to afford diisopropylaminoneopentasilane, however, only crude product conversions determined by GC-FID were reported (see Scheme 90).163
 | ||
| Scheme 90 Dehydrogenative coupling of neopentasilane with Zn(NTf2)2 and diisopropylamine. Only conversions were reported. | ||
In 2022, Trovitch and co-workers demonstrated that the dehydrogenative coupling of SiH4 with primary and secondary amines proceeds using the manganese-based catalyst [(2,6-iPr2PhBDI)Mn(μ-H)]2.162 With primary or secondary amines, di- and trisubstituted aminosilanes are obtained, and the steric demand of the amine strongly influences the reaction time. Diamines and triamines furnish polycarbosilazanes, whereas ammonia affords perhydropolysilazane (see Scheme 91).162
 | ||
| Scheme 91 Synthesis of aminosilanes from SiH4 via Mn based catalysis. | ||
A different approach to obtain aminosilanes is the amine exchange pathway. Various aminosilanes are accessible via transamination with amines at room temperature or elevated temperature. The educts of choice for transaminations in the literature are diisopropylaminosilanes. For example, diisopropylaminosilane forms disilylamine with NH3 and subsequently forms trisilylamine (see Scheme 92).159
 | ||
| Scheme 92 Synthesis of di- and trisilylamine, via transamination of diisopropylamine. No detailed ratios of the reagents were given; therefore, the overall equation is given. | ||
Aylett and Hakim reported the synthesis of disilylamine in 75% yield by treating diphenylaminosilane (Ph2NSiH3) in toluene at low temperature with ammonia (see Scheme 93).151
 | ||
| Scheme 93 Synthesis of disilylamine via transamination of Ph2NSiH3 with NH3. | ||
Transamination of diisopropylaminosilane proceeds with a broad range of secondary amines; with primary amines, the corresponding disilylamines are obtained, while polyfunctional amines undergo silylation at multiple amino groups (see Scheme 94, path a to c).143
 | ||
| Scheme 94 Example transaminations of diisopropylaminosilane (path a–c). | ||
This methodology extends to larger aminosilane frameworks, for example, Zhou et al. employed diisopropylaminodisilane in transamination reactions to access a range of monosubstituted as well as bissubstituted (disilanyl)amines, although only GC-MS conversions were reported and no isolated yields (see Scheme 95 and Table 6).165
 | ||
| Scheme 95 Transamination of diisopropylaminosilane. For conversions see Table 6. R = organic rest, R′ = H or organic rest, see Table 6. | ||
| Amine (2 eq.) | GC conversion [%] | |
|---|---|---|
| A | B | |
| Diethyl | 38 | — |
| Methyl | 69 | 22 |
| Ethyl | 73 | 23 |
| Propyl | 62 | 21 |
| Butyl | 71 | 25 |
| 2-Aminobutane | 96 | 1 |
| Pentylamine | 51 | 13 |
| 2-Aminopentane | 95 | 1 |
| 1,2-Dimethylpropyl | 98 | — |
| t-Pentyl | 83 | — |
| Cyclopentyl | 84 | 12 |
| Cyclohexyl | 92 | 5 |
| Aniline | 74 | — |
| o-Toluidine | 38 | — |
Using an NH3:diisopropylaminodisilane ratio of 1:3 gives 99% conversion to bisdisilanylamine; with an additional equivalent of diisopropylaminodisilane, trisdisilanylamine is obtained.165 Furthermore, Rekken et al. obtained trisdisilanylamine by thermal degradation of bisdisilanylamine at 110 °C (see Scheme 96, path a to c).166
 | ||
| Scheme 96 Transamination of diisopropylaminosilane affording bis- and trisdisilanylamine and thermal degradation of bisdisilanylamine (path a–c). No isolated yields were reported. For the thermal redistribution of bisdisilanylamine only the general equation is given. | ||
Silyl iso-cyanide (H3SiNC) was previously prepared but wrongfully assigned as silyl cyanide (H3SiCN) and no detailed analysis of its properties was carried out.167 MacDiarmid was able to obtain and analyze both silyl iso-cyanide and silyl iso-thiocyanate (H3SiNCS). The treatment of silver cyanide (AgNC) with H3SiI at room temperature afforded silyl iso-cyanide in 90% yield and by reaction with mercuric cyanide at elevated temperatures, though no yield was reported (see Scheme 97, path a). Silyl iso-thiocyanate was afforded via the reaction between H3SiI and silver thiocyanate (AgNCS) in 66% yield (see Scheme 97, path b).168 Furthermore, the reaction between trisilylamine and cyanic acid (HOCN) affords silyl iso-cyanate (H3SiNCO) in 95% yield, and with thiocyanic acid (HSCN), it leads to the formation of silyl iso-thiocyanate in 58% yield (see Scheme 97, path c and d).169
 | ||
| Scheme 97 Synthesis of silyl iso-cyanide, silyl iso-cyanate and silyl iso-thiocyanate from H3SiI (path a and b) and trisilylamine (path c and d). | ||
Gaseous silylphosphane (H3SiPH2) reacts readily with cyanic acid as well as thiocyanic acid affording silyl iso-cyanate or silyl iso-thiocyanate in yields greater than 80% (see Scheme 98, path a and b).170
 | ||
| Scheme 98 Synthesis of silyl iso-cyanate (path a) and silyl iso-thiocyanate (path b). No amounts of the used reagents were given. | ||
Disilylsulfide (S(SiH3)2) is a viable feedstock for the synthesis of silyl i-thiocyanate. In combination with silver thiocyanate (AgNCS), it afforded H3SiNCS in 86% yield (see Scheme 99). When instead cyanogen chloride was employed, the reaction led to a mixture of disilylsulfide, silyl cyanide, sulfur, and silyl i-thiocyanate, but the mixture proved to be inseparable.171
 | ||
| Scheme 99 Synthesis of silyl i-thiocyanate from disilylsulfide. The amount of AgNCS used was not reported. | ||
Ebsworth and Mays reported the synthesis of silyl i-cyanate via treatment of silvercyanate (AgOCN) and powdered glass with iodosilane vapor with an overall yield of 20% (see Scheme 100).172
 | ||
| Scheme 100 Synthesis of silyl i-cyanate from iodosilane and silvercyanate. | ||
Silylamines such as N(SiH3)3 and N(H2Si–SiH3)3 serve as precursors to silylaminoboranes. Trisilylamine reacts quantitatively at low temperature with boron trihalides BX3 (X = Cl, F) to afford the corresponding disilylaminohaloboranes.147,173 Witz and Sujishi showed that boron trifluoride (BF3) forms adducts with trisilylamine, methyldisilylamine, and dimethylsilylamine, these adducts decompose at room temperature to give silyl fluoride and the corresponding silylaminoborane (see Scheme 101, path a and b).173
 | ||
| Scheme 101 Synthesis of bissilylaminodichloro- and bissilylaminodifluoroborane (path a and b). | ||
Furthermore, trisilylamine reacts readily with bromodiborane (B2H5Br) at low temperature to afford disilylaminoborane in quantitative yield; subsequent treatment with diborane furnishes bissilylaminodiborane in 80% yield (see Scheme 102, path a and b).147 The same methodology extends to methyldisilylamine, but instead of the desired methylsilylaminoborane it yields methylsilylaminodiborane in 20% yield, accompanied by formation of H3SiBr, SiH4 and B2H6 (see Scheme 102, path c). The yield was improved to 44% by adding 1.3 equivalents of diborane, an effect attributed to reduced disproportionation of the intermediate methylsilylaminoborane (see Scheme 102, path c).147
 | ||
| Scheme 102 Reactions between trisilylamine (path a and b) and methyldisilylamine (path c and d) with bromodiborane and subsequent reaction of the formed silylamnioboranes with diborane. | ||
MacDiarmid and Abedini formed the corresponding adduct of trisdisilanylamine with BF3 at low temperature; upon heating, the adduct decomposed to afford bisdisilanylamino-fluoroborane and fluorodisilane, although only the isolated yield of the fluorodisilane was reported (see Scheme 103).150
 | ||
| Scheme 103 Synthesis of F2BN(SiH2SiH3)2. In the second reaction only the yield for fluorodisilane was reported. | ||
Norman and Scantlin reported that boranes catalyze the condensation of trisilylamine and methyldisilylamine to give the corresponding silazanes with simultaneous liberation of SiH4, in good yields (see Scheme 104, path a and b). When B2H6 or B5H9 is used, the reaction affords H2Si(N(SiH3)2)2 with only minor formation of higher condensation products Si7N3H16. In contrast 1- and 2-BrB5H8 catalyze much faster reactions that are more difficult to control, leading to increased formation of Si7N3H16.174
 | ||
| Scheme 104 Borane catalyzed condensation of trisilylamine (path a) and methyldisilylamine (path b). Path (a) no isolation was performed under catalysis by 1- and 2-BrB5H8. | ||
Silyl azide (H3SiN3) can be prepared by reacting trisilylamine with hydrazoic acid (HN3) in n-Bu2O, however, it is unstable at room temperature and slowly decomposes with evolution of SiH4 (see Scheme 105, path a).175 Alternatively, treatment of H3SiBr with tributyltinazide (Bu3SnN3) affords silylazide in 71% yield (see Scheme 105, path b).176 A further route involves treating H3SiPH2 with a dilute solution of hydrazoic acid, though no yield was reported (see Scheme 105, path c).170
 | ||
| Scheme 105 Synthesis of silylazide (path a–c). No yield or experimental details were reported for the reaction of H3SiPH2 with hydrazoic acid. | ||
Fritz and Berkenhoff investigated the reaction between silylphosphane and NH3 and concluded that at −80 °C silylamine forms and subsequently converts to polymeric solids of the type [SiH2NH]x. To support this interpretation, they also examined the reaction of H3SiCl and NH3, which led to the same solids, although the proposed intermediates could not be directly observed (see Scheme 106).177
 | ||
| Scheme 106 Ammonolysis of H3SiPH2 according to Fritz. No details about the amounts of H3SiPH2 and NH3 as well as reaction time were given. For the following reaction of the formed H3SiNH2 only the general formula is given. | ||
Norman and Jolly further investigated the H3SiPH2 and NH3 system. They found that depending on reaction time, temperature, and ratio of H3SiPH2 to NH3, a complex product mixture is formed. At −78 °C to −63 °C and a ratio above 6:1 H3SiPH2:NH3 mainly PH3 and HN(SiH3)2 are formed in a 2:1 ratio, with small amounts of N(SiH3)3 and SiH4 from subsequent reactions.178 When the reaction was carried out with a ratio of H3SiPH2:NH3 below 6:1 or at higher temperatures, only a complex product mixture was observed, notably small amounts of the silazanes SiH2(NHSiH3)2 and (SiH3)2NSiH2NHSiH3 were formed. They also could not find direct evidence for the formation of H3SiNH2.178
Glidewell further examined the reactions between H3SiPH2 and amines and imines. In the gas phase, methylamine and diethylamine react with PH3 to give the corresponding silylamines in quantitative yield. In contrast, methylamine does not react with gaseous H3SiPH2 and with condensed H3SiPH2 only a white polymeric solid is obtained (see Scheme 107).170
 | ||
| Scheme 107 Synthesis of silylamines from H3SiPH2 with amines. The amounts of reagents used were not given. | ||
Treatment of cyanamide (CH2N2) with H3SiPH2 or with disilylsulfide affords disilylcarbodiimide (C(NSiH3)2) in excellent yields (see Scheme 108, path a and b). However, the yield of the S(SiH3)2 reaction was not explicitly reported. In contrast, trisilylphosphane (P(SiH3)3) gives the carbodiimide only in poor yield with concomitant decomposition of P(SiH3)3, and SiH3PMe2 undergoes complete decomposition under the reaction conditions.170
 | ||
| Scheme 108 Synthesis of disilylcarbodiimide (path a and b). No experimental details were given, therefore only the general formula is given. | ||
Passing H3SiI vapor over powdered glass wool and silver cyanamide (Ag2CN2) led to the formation of disilylcarbodiimide in 20% yield, with SiH4, and HCN as side products. Neither using PbCN2, or H3SiBr with Ag2CN2 improves the yields and results in 10–20% yield of disilylcarbodiimide. Notably, H3SiI and undiluted Ag2CN2 led to an explosion.179 When H3SiI vapor is diluted with N2, the yield of the reaction is increased to up to 60%.180 While it was first unclear if disilylcyanamide NCN(SiH3)2 or disilylcarbodiimide was formed during the reaction, later IR spectra studies180 and gas phase electron diffraction181 showed that disilylcarbodiimide is formed (see Scheme 109).
 | ||
| Scheme 109 Synthesis of disilylcarbodiimide from halosilanes and Ag2CN2. | ||
Schmidbaur and Schuh employed NaH and NaNH2 in the presence of 18-c-6 as a phase-transfer catalyst to convert 1,4-disilabutane and n-tetrasilane into their corresponding aminosilanes (see Scheme 110, path a and b). While 1,4-disilabutane reacts with diethylamine using the NaNH2/18-c-6 system in pentane to afford the aminosilanes with evolution of H2, n-tetrasilane undergoes cleavage, giving mixtures of smaller aminosilanes and higher silanes. They found that NaH alone does not catalyze these reactions. When pyrrole was used with n-tetrasilane instead of diethylamine, the main product was Si(NC4H4)4, accompanied by formation of trisilane.182
 | ||
| Scheme 110 Reactions of 1,4-disilabutane (path a) and n-tetrasilane (path b) with amines under NaNH2 catalysis. | ||
Perhydropolysilazanes such as bisdisilylaminosilane are obtainable from trisilylamine with a suitable catalyst, such as B(C6F5)3 or PdCl2 (for more examples see Scheme 111 and Table 7). Though it is not stated if the product obtained is pure and no number average molecular weight Mn for the products was given. When the reaction is carried out with increased catalyst loading, Mn increases and the ratio of SiH2:SiH3 of the products increases as well.183
 | ||
| Scheme 111 Synthesis of bis(disilylamino)silane and perhydropolysilazanes. Yields for path a are given in Table 7. No yields or molecular weight distributions were given for path b. | ||
| Catalyst | Catalyst loading [mol%] | Cocatalyst [mol%] | T [°C] | t [h] | Yield [%] |
|---|---|---|---|---|---|
| Pt/C | 5 | NEt3 31% | 100 | 18 | 48 |
| Pt/C | 5 | NMe2Et 25% | 100 | 4 | 36 |
| Pt/C | 5 | NMe2Et 25% | 100 | 22 | 46 |
| Pt/C | 0.4 | NMe2Et 25% | 100 | 4 | 31 |
| Pt/C | 0.4 | NMe2Et 25% | 105 | 25 | 39 |
| [RuCl2((AMPY) (DPPB))] | 0.4 | — | 100 | 18 | 10 |
| [RuCl2((AMPY) (DPPB))] | 0.4 | NEt3 30% | 100 | 18 | 83 |
| [RhCl(PPh3)3] | 0.4 | NMe2Et 25% | 100 | 1 | 86 |
| [(R,R)-teth-TsDpenRuCl] | 0.6 | NMe2Et 25% | 100 | 1.5 | 83 |
| Ru3(CO)12 | 1.1 | NMe2Et 25% | 80 | 1 | 80 |
The composition of silylphosphane precursors is especially relevant for semiconductor applications. Silylphosphanes consisting only of P, Si, and H are attractive for semiconductor processing because they are carbon- and halogen-free and decompose to benign H2, reducing impurity incorporation that degrades carrier lifetimes and interface quality.185,186 Their preformed Si–P bonds make them single-source reagents that deliver Si and P together, enabling in situ n-type doping of Si/Ge and the growth of Si–P materials with improved dopant incorporation efficiency and spatial uniformity compared with separate feeds.186,187
The first reported silylphosphane H3SiPH2 was prepared by Fritz, in 1952, via thermal decomposition of equimolar amounts of SiH4 and PH3, proceeding through a radical mechanism.12,53,188 Furthermore, under the reaction conditions, less volatile species such as H2Si(PH2)2 and SiP2 are generated.189,190 This pathway enabled the first investigations into the properties of silylphosphanes but was problematic for the synthesis of H3SiPH2 in a preparative scale, due to the further decomposition of H3SiPH2 towards SiP2 during the reaction (see Scheme 112).190
 | ||
| Scheme 112 Synthesis of H3SiPH2 via pyrolysis of SiH4/PH3 mixtures. No yields were reported. | ||
Sabherwal and Burg adapted the thermal decomposition conditions to a decreased temperature of 300 °C in the presence of trace amounts of I2, leading to a significantly increased yield of 61% for H3SiPH2.191 The enhanced yield was attributed to a lower degree of product degradation, due to the lower temperature and to the formation of H3SiI, which reacts with PH3 to give H3SiPH2 and HI. The formed HI then reacts with SiH4 to regenerate H3SI.191 Another early report of silylphosphanes was made by Aylett et al. in 1955, who demonstrated that H3SiI reacts with white phosphorous to afford PI2SiH3 with various side products, most notably P(SiH3)3, which they could not characterize fully. They further showed that H3SiPH2 can be formed via reaction of NMe3SiH3I with PH3.13
In addition to the thermal route photochemistry was also investigated to yield silylphosphanes. Blazejowski obtained small quantities of H3SiPH2 by IR irradiation of a PH3/SiH4 mixture, photosensitized by SiF4,192 later by irradiation of a PH3/SiH4 mixture,193 and by photolysis of PH3/SiH4 mixtures at 147 nm.194 The formation of H3SiPH2 and hydrosilanes such as disilane and trisilane is rationalized by the generation of silylene groups that insert into Si–H and P–H bonds.192,193 Another alternative approach towards silylphosphanes such as H3SiPH2 employed electrical discharge methods. Mixtures of SiH4 and PH3 form silylphosphanes in an ozonizer-type silent electrical discharge tube.195,196 Using this setup Jolly and Gokhale obtained HP(SiH3)2 from a mixture of H3SiPH2 and SiH4.196,197 Furthermore ozonizer-type silent electrical discharge tubes have also facilitated the preparation of H3SiSiH2PH2 from disilane and PH3. However the silent electrical discharge approach has proven to be unsuitable for larger scales, due to the low yields and poor selectivity (see Scheme 113, path a and b).196,197
 | ||
| Scheme 113 Synthesis of HP(SiH3)2 (path a) and Si2H5PH2 (path b) via silent electrical discharge. No yields for P2H4 and H3SiPH2 were given in the reaction of disilane. | ||
Halide mediated exchange reactions between silylphosphane and halodisilanes were also investigated in order to obtain silylphosphanes. Drake et al. obtained disilanylphosphane (H3SiSiH2PH2) via exchange reaction between H3SiPH2 and disilylhalides (see Scheme 114).198,199
 | ||
| Scheme 114 Synthesis of H3SiSiH2PH2 from H3SiPH2 and H3SiSiH2X. They noted that all compounds are present in equimolar amounts at the equilibrium. | ||
They further employed SiH2Cl2 and SiHCl2SiH2Cl with H3SiPH2 to synthesize the chlorinated silylphosphanes SiH2ClPH2 and SiHCl2SiH2PH2 (see Scheme 115, path a and b). Whereas 1,1-dichlorodisilane does not react with H3SiPH2, 1,2-dichlordisilane undergoes facile conversion to 1,2-diphosphinodisilane via the intermediate SiH2ClSiH2PH2 (see Scheme 115, path c). Furthermore 1,1,2-trichlorodisilane affords 1,1-dichloro-2-phosphinodisilane.199
 | ||
| Scheme 115 Synthesis of silylphosphanes and chlorosilylphosphanes from H3SiPH2 with chlorosilanes (path a–d). No yields were given for the reaction of H3SiPH2 with 1,1,2-trichlorodisilane. | ||
Silylphosphane synthesis can also be achieved through nucleophilic substitution of silyl halides by alkali metal phosphides. Amberger and Boeters demonstrated that the direct reaction of bromosilane with potassium dihydrophosphide (KPH2) does not afford H2PSiH3, but instead leads to the synthesis of P(SiH3)3 (see Scheme 116).200
 | ||
| Scheme 116 Synthesis of P(SiH3)3 from KPH2 and H3SiBr. | ||
A sequence of transmetalation steps results in the formation of P(SiH3)3 steps involving mono- and disilylated intermediates (see Scheme 117).16,200–202
 | ||
| Scheme 117 Formation of P(SiH3)3 from H3SiBr through the intermediates H2PSiH3 and HP(SiH3)3. | ||
Glidewell and Sheldrick demonstrated that the intermediate mono- and disilylsubstituted silylphosphanes can be obtained as well, when using an excess of potassium dihydrophosphide with H3SiBr, followed by acidification of the non-volatile products with hydrogen sulfide.202
Lewis acid driven redistribution reactions also lead to the formation silylphosphanes. MacDiarmid and Russ demonstrated that H3SiPH2 forms a Lewis adduct with BF3, which subsequently decomposes at low temperatures to a mixture of compounds containing P(SiH3)3. Both pathways (a and b) proceed to comparable extents (see Scheme 118).203
 | ||
| Scheme 118 Decomposition pathways of H3SiPH2·BF3. Both reactions a and b occur equally. | ||
Further investigations showed that boranes promote the redistribution of silylphosphanes and their mixtures into trisilylphosphanes.203–206 In the first step of these redistribution reactions the silylphosphanes form Lewis acid–base complexes (see Scheme 119, path a) and, depending on the used borane, then undergo redistribution into trisilylphosphanes (see Scheme 119, path b).204 The best yields are obtained when using an excess of BF3 with 83% formation of P(SiH3)3 and small amounts of HP(SiH3)2 and 90% for P(SiH2SiH3)3 with small amounts of HP(SiH3)2 as side product.204,206 BCl3 induces only limited redistribution, while BBr3 predominantly led to the formation of H3SiBr and BH3 complexes only redistributed in trace amounts.204 Mixed trisilylphosphanes are obtained when mixtures of H3SiPH2 and H3SiH2SiPH2 are subjected to these conditions.204
 | ||
| Scheme 119 Formation of Lewis acid–base complexes of boranes and silylphosphanes and subsequent BF3 promoted redistribution reaction, forming trisilylphosphanes (path a and b). No yields were given for the BX3 and diborane adducts. | ||
When disilanylphosphane is converted to borane adducts and subsequently heated, redistribution to trisilylphosphanes is not observed, instead SiH4 and a polymeric substance is formed (see Scheme 120, path a). Only when 9 equivalents of disilanylphosphane were employed could trisdisilanylphosphane be detected, though no yields were given (see Scheme 120, path b).205
 | ||
| Scheme 120 Formation of the borane adduct of H2PSiH2SiH3 (path a) and thermal degradation into trisdisilanylphosphane (path b). No isolated yields were given. | ||
In 2010 Tice et al. reported a solvent free approach in which P(SnMe3)3 reacts with H3SiBr to afford P(SiH3)3 in 98% yield (see Scheme 121).207
 | ||
| Scheme 121 Synthesis of P(SiH3)3 from P(SnMe3)3. | ||
Recently Li et al. employed exchange reactions between P(SiMe3)3 and an excess of monochlorosilanes to generate the corresponding trisilylphosphanes (see Scheme 122).208
 | ||
| Scheme 122 Synthesis of trisilylphosphanes via exchange reaction of P(SiMe3)3 with monochlorosilanes. | ||
The direct reaction between metalated phosphanes and silylhalides was also thoroughly investigated. Lithium tetrakisdihydrogenphosphidoaluminate was prepared by Finholt et al. in 1963,209 which proved as a useful reagent for phosphanylation. In contrast to reactions of silylhalides with KPH216,200–202 the transmetalation sequence leading to the formation of P(SiH3)3 is not observed when metal tetrakisdihydrogenphosphidoaluminates are employed, giving access to H3SiPH2 (see Scheme 123, path a and b).210,211
 | ||
| Scheme 123 Synthesis of silylphosphanes from metal tetrakis(dihydrogenphosphido)aluminates (path a and b). | ||
Use of LiAl(PH2)4 as phosphanylation agent enables the synthesis of diverse silylphosphanes from silylhalides, notably they were able to use SiH2Br2 to obtain SiH2(PH2)2 and SiHBr3 for SiH(PH2)3 respectively.210,212,213 Norman and co-workers showed that analogous methodology employing sodium tetrakisdi-hydrogenphosphidoaluminate (NaAl(PH2)4) enables access to several silylphosphanes, as well as germylphosphanes.210,212 The reaction of NaAl(PH2)4 with H3SiBr has proven to be a viable synthetic approach for the phosphanylation of silicon halides, allowing the synthesis of silylphosphanes such as H2PSiH3.211 In 1994 Becker et al. successfully employed silyltriflate (CF3SO2OSiH3) as silylation agent for NaAl(PH2)4 to obtain H3SiPH2 in 60% yield (see Scheme 124).214
 | ||
| Scheme 124 Synthesis of H3SiPH2 from CF3SO2OSiH3. No yield for silyltriflate was given, as it is freshly prepared and used immediately without isolation. | ||
Lithium dialkylphosphides are useful for preparing metalated silylphosphanes via reaction with H3SiPH2 (see Scheme 125, path a and b). Solutions of the monometalated silylphosphanes with concentrations above 0.1 mol l−1 undergo disproportionation at room temperature in apolar solvents, establishing an equilibrium between H3SiPHLi and LiP(SiH3)2 (see Scheme 125, path b).215 This behavior prevents the direct synthesis of disilylphosphanes from monometalated H3SiPHLi by subsequent reaction with silylhalides, as trisilylphosphanes will be formed as well.215 Precipitation of LiPH2 after removing most of the solvent and adding benzene drives the equilibrium towards LiP(SiH3)2 and allows for recrystallization in Et2O.215
 | ||
| Scheme 125 Synthesis of metalated silylphosphanes from H3SiPH2 (path a and b) and disproportionation equilibrium of H3SiPHLi. No isolated yield was given for the metalated silylphosphanes but for the associated byproducts. | ||
The monometalated H3SiPHLi can be treated with AlCl3 in diglyme to form LiAl(PHSiH3)4, which reacts with H3SiBr to afford HP(SiH3)2. Owing to the disproportionation of H3SiPHLi into LiP(SiH3)2 and LiPH2, the initially formed LiAl(PHSiH3)4 also contains LiAlP(SiH3)2 and LiAlPH2, which in turn yield H3SiPH2 and P(SiH3)3 upon reaction with H3SiBr. Additionally, HP(SiH3)2 undergoes dismutation to monosilyl- and trisilylphosphane, leading to the approximately equimolar formation of mono-, di-, and trisilylphosphanes. By quickly separating the disilylphosphanes the dismutation can be circumvented. The dismutation is only observed with PH containing silylphosphides (see Scheme 126).215
 | ||
| Scheme 126 Synthesis HP(SiH3)2 from LiAl(PHSiH3)4 and subsequent disproportionation into H3SiPH2 and P(SiH3)3. | ||
Drake and Anderson converted P(SiH3)3 with LiAlH4 to the disilylphosphinoaluminate ion LiAlH[P(SiH3)2]3, which further reacts with halides, such as, monobromodisilane H3SiSiH2Br to give P(SiH3)2SiH2SiH3 (see Scheme 127, path a).216 H3SiPEt2 derived from H3SiBr undergoes H/Li exchange with LiPEt2 to form H2Si(PEt2)2 and LiH. Subsequent reactions of H2Si(PEt2)2 with 2 equivalents of LiPEt2 provides HSi(PEt2)3 (see Scheme 127, path b).217
 | ||
| Scheme 127 Path (a) synthesis of disilylphosphinoaluminate and subsequent reactions with halides. Path (b) synthesis of H3SiP(Et)2, H2Si(P(Et)2)2 and HSi(P(Et)2)3 from H3SiBr. No isolated yields were given. | ||
HSi(PEt2)3 is interesting in the context of silylphosphanes, as it reacts with LiPEt2 to give LiSi(PEt2)3, which reacts with H3SiBr, resulting in the formation of H3SiSiH(PEt2)2, but was only isolated with sideproducts (see Scheme 128).217 Metalation of silylphosphanes using n-BuLi leads to cleavage of the Si–P bond.215,217
 | ||
| Scheme 128 Synthesis of H3SiSiH(PEt2)2 from LiSi(PEt2)3. The formation of H3SiSiH(PEt2)3 was confirmed but isolation as not possible due to degradation during distillation. | ||
Silylphosphides as interesting reagents for further functionalization were obtained either via metalation of silylphosphanes,218,219 or by employing build up reactions of silanes.115 In 1973, Cradock et al. obtained LiP(SiH3)2 from P(SiH3)3 in 90% yield by metalation with MeLi (see Scheme 129, path a).218,219 In 1994 Sundermeyer and co-workers accessed KPHSiH3 and KP(SiH3)2 via the buildup reaction between SiH4 and K in diglyme and subsequent reaction with PH3 (see Scheme 129, path b).47 LiP(SiH3)2 can be synthesized through the dismutation of H3SiPH2 and LiPHCH3 (see Scheme 125, path b), and isolated as [(TMEDA)2Li][P(SiH3)2] (see Scheme 129, path c). A crystal structure is shown in Fig. 14.214
 | ||
| Scheme 129 Synthesis of LiP(SiH3)2 (path a), KPHSiH3, KP(SiH3)2 (path b) and [Tmeda2Li][P(SiH3)2] (path c). | ||
 | ||
| Fig. 14 Crystal structure of the anion (a) and cation (b) of [(TMEDA)2Li][P(SiH3)2]. For better visibility the anion and cation are not shown in the same scale. Adapted from G. Becker, B. Eschbach, D. Käshammer, et al.214 with permission from John Wiley and Sons © 1994. | ||
The smallest member of the silylarsane family is silylarsane (H3SiAsH2) and was prepared by Jolly and Drake in 1962. Similar to the synthesis of H3SiPH2, using an equimolar mixture of SiH4 and arsine (AsH3) in ozonizer-type silent electrical discharge tube yields H3SiAsH2, as well as disilanylarsane (H3SiH2SiAsH2) (see Scheme 130).195 Notably disilane as well as trisilane are formed as sideproducts.221
 | ||
| Scheme 130 Synthesis of H3SiAsH2 and Si2AsH7 from a SiH4/AsH3 mixture. No experimental details were given. | ||
When H3SiAsH2 and B2H6 are condensed into a vessel they react upon warming to 0 °C by forming a solid as well as disilylarsane (HAs(SiH3)2) and trisilylarsane (As(SiH3)3) (see Scheme 131).221 Reactions between H3SiAsH2 and bromodiborane (B2H5Br) only afford the formation of bromosilane and a arsine containing polymer.221 H3SiAsH2 forms the 1:1 adduct with BX3 (X = Cl, Br), but H3SiAsH2BX3 decomposes at low temperatures forming H3SiX.221
 | ||
| Scheme 131 Synthesis of HAs(SiH3)2 and As(SiH3)3 from H3SiAsH2 and B2H6. | ||
Aylett et al. showed in 1955, that monoiodosilane reacts with arsenic to afford diiodosilylarsane (AsI2SiH3) with various side products, most notably trisilylarsane, additionally they showed that trimethylarsane (AsMe3) undergoes reaction with iodosilane, giving access to trisilylarsane.13
Arsenides show similar reactivity towards silyl halides as phosphides. Trisilylarsane can be accessed through nucleophilic substitution of silyl halides by alkali metal arsenides. Amberger and Boeters used bromosilane with potassium dihydroarsenide (KAsH2) to afford As(SiH3)3 (see Scheme 132).200 The same reported method of Glidewell and Sheldrick, in which H3SiPH2 and HP(SiH3)2 can be obtained from the reaction of KPH2 with H3SiBr via acidification with H2S also works for potassium arsenide KAsH2, but leads mostly to silylarsine in 54% yield.202 The formation of As(SiH3)3 from KAsH2 proceeds via a series of transmetalation steps, similar to the respective synthesis of trisilylphosphane P(SiH3)3 (see Scheme 117).202
 | ||
| Scheme 132 Synthesis of As(SiH3)3 from KAsH2 and H3SiBr.200 | ||
Metalated silylarsanes can react with silyl halides to form the respective silylarsanes. Lithium tetrakisdihydrogenarsido-aluminate (LiAl(AsH2)4) reacts with silylhalides H3SiX (X = Br, I) to afford silylarsine, though monochlorosilane only led to formation of H3SiAsH2 in trace amounts (see Scheme 133).222,223
 | ||
| Scheme 133 Synthesis of H3SiAsH2 from LiAl(AsH2)4 and silylhalides. | ||
Anderson and Drake were able to obtain disilylarsane HAs(SiH3)2 by treating LiAl(AsH2)4 with bromosilane. After removal of the volatiles the remaining solid was treated with dihydrogen selenide (H2Se), resulting in the formation of HAs(SiH3)2.202 Furthermore the treatment of lithium tetrasilylarsenide (LiAs(SiH3)4) with bromodisilane led to the formation of disilanylarsane (H3SiH2SiAsH2) in 59% yield (see Scheme 134).223
 | ||
| Scheme 134 Synthesis of H3SiH2SiAsH2 from LiAl(AsH2)4 and H3SiSiH2Br. | ||
Disilanylarsane can also be obtained from the exchange reaction with monohalodisilanes H3SiH2SiX (X = Cl, Br) at low temperatures. The yields for H3SiH2SiAsH2 can be brought to 30%, by stepwise removal of the formed silyl halide H3SiX (see Scheme 135).198
 | ||
| Scheme 135 Synthesis of H3SiSiH2AsH2 via exchange reaction from H3SiSiH2X (X = Cl, Br) and H3SiAsH2. | ||
Lithium disilylarsenide (LiAs(SiH3)2) can be obtained from trisilylarsane in 90% yield by metalation with MeLi, as shown by Cradock et al. in 1973.218,219
Similar to trisilylphosphanes, trisilylarsanes are also available through an exchange reaction between tristrimethylsilylarsane (As(SiMe3)3) and monohalosilanes, though only the reaction with chlorotrisilane is reported (see Scheme 136).208
 | ||
| Scheme 136 Synthesis of As(SiH2SiH2SiH3)3 via exchange reaction from H3SiH2SiH2SiCl and As(SiMe3)3. | ||
The nucleophilic substitution of phosphides and arsenides with silyl halides gave access to their respective trisilyl compound, but similar reactions with antimonides and stibanes did not lead to the desired trisilylstibane (Sb(SiH3)3).200,225 Amberger and Boeters instead accessed Sb(SiH3)3 via the salt methathesis reaction of lithium antimonide (Li3Sb) with H3SiBr (see Scheme 137).200,225
 | ||
| Scheme 137 Synthesis of Sb(SiH3)3 via salt metathesis reaction of Li3Sb and H3SiBr. | ||
Functionalization with group 6 (synthesis of silylchalcogenes)
In 1917, Stock, Somieski, and Wintgen reported the first isolation of O(SiH3)2. Their method involved shaking H3SiBr with degassed H2O, allowing the mixture to stand, and then purifying the product by fractional distillation (see Scheme 138). They proposed that hydrolysis generates H3SiOH as intermediate, which rapidly self-condenses to give O(SiH3)2 with loss of water.228
 | ||
| Scheme 138 Synthesis of O(SiH3)2 via hydrolysis of SiH3Br. | ||
In a follow-up study Stock and Somieski isolated O(Si2H5)2 by reacting Si2H5Br (which was contaminated with Si2HnBr6−n, n = 0–6) with degassed H2O, and then extracting it with benzene (see Scheme 139). They failed in isolating the pure compound and always had some H2O next to O(Si2H5)2. The higher silyl bromides present in the educt as contaminations did not interfere with the reaction, since their resulting siloxanes polymerized and then precipitated as a white solid.145
 | ||
| Scheme 139 Hydrolysis of Si2H5Br, contaminated with higher bromosilanes, leading to O(Si2H5)2 and polymeric siloxanes. Yields of the siloxane were not determined. | ||
Emeléus, MacDiarmid, and Maddock attempted the reduction of O(SiCl3)2 with LiAlH4, but were unable to prepare O(SiH3)2 via this route. Instead, they obtained O(SiH3)2 in 37% yield by passing H2 through boiling O(SiCl3)2, then directing the saturated gas stream over aluminium foil at 500 °C (see Scheme 140, path a). They also generated O(SiH3)2 via hydrolysis of SiH3I, S(SiH3)2, and Se(SiH3)2 (see Scheme 140, path b and c). For the Se(SiH3)2 route, the yield could not be determined because H2Se could not be separated from the silylether.229
 | ||
| Scheme 140 Synthesis of O(SiH3)2 from O(SiCl3)2 (path a), SiH3I (path b), S(SiH3)2 and Se(SiH3)2 (path c). Yields of Se(SiH3)2 hydrolysis reaction not determined. | ||
Kriner, MacDiarmid, and Evers investigated reactions of O(SiH3)2 with aluminum halides. Treatment with Me4Al2Br2 afforded Me4Al2(OSiH3)2 (see Scheme 141), which is stable at −78 °C but decomposes at room temperature to give SiH4 and a non-volatile viscous residue. In contrast, reactions of O(SiH3)2 with Al2X6 (X = Cl, Br, I) yielded only the corresponding halosilanes; no aluminum-containing products could be isolated.230
 | ||
| Scheme 141 Synthesis of Me4Al2(OSiH3)2 from Me4Al2Br2 and O(SiH3)2. | ||
Ward and MacDiarmid isolated pure O(Si2H5)2 by hydrolysing purified Si2H5I. Purification of O(Si2H5)2 required an extensive protocol: multiple fractional distillations, drying over P2O5 (noting that prolonged contact causes degradation of the silylether and formation of PH3), followed by repeated distillations. The final product was obtained in 96% yield (see Scheme 142).231
 | ||
| Scheme 142 Hydrolysis of O(Si2H5)2 from Si2H5I. | ||
Sternbach and MacDiarmid (1961) investigated routes to H3SiOCH3. Reacting SiH4 with methanol (MeOH) at room temperature gave only unreacted SiH4, H2, and a complex mixture of methoxysilanes. With copper powder as a promoter, low yields of H2Si(OCH3)2 and HSi(OCH3)3 were isolated (see Scheme 143, path a). Direct reaction of SiH3I with methanol was highly vigorous, leading mainly to polymeric material; only traces of H3SiOCH3 were detected in the volatile fraction (see Scheme 143, path b). In contrast, treating the amine adduct SiH3I·N(Me)3 with methanol furnished MeOSiH3 in 28% yield (see Scheme 143, path c). They further showed that MeOSiH3 reacts with BF3 to give SiH3F and MeOBF2 (see Scheme 143, path d).232
 | ||
| Scheme 143 Synthesis of alkoxysilanes (path a–c) and reaction of H3SiOMe with BF3 (path d). | ||
Onyszchuk found that O(SiH3)2 reacts with BF3 and BCl3 to give H3SiOBX2 (X = F, Cl), but both products are highly unstable. The fluorinated derivative decomposed during isolation, even at low temperature, and could not be isolated. The chloro analogue decomposes more slowly and was successfully isolated at −64 °C; several subsequent distillations at −112 °C were used to remove H3SiCl formed in the process (see Scheme 144).233
 | ||
| Scheme 144 Reactions of O(SiH3)2 with BF3 (path a) and BCl3 (path b). | ||
Van Dyke and MacDiarmid prepared the unsymmetrical silylether H3SiOSi2H5 by two routes: path (a) hydrolysis of a mixture containing SiH3I and Si2H5Br; path (b) equilibration between O(SiH3)2 and O(Si2H5)2 (see Scheme 145).234
 | ||
| Scheme 145 Synthesis of the unsymmetrical silylether H3SiOSi2H5 via hydrolysis (path a) and equilibration reactivity of silylethers (path b). *Yield of product mixture. | ||
Van Dyke examined the reactions of O(SiH3)2 with phosphorus halides PX3 (X = F, Cl, Br). No reaction was observed with PF3 or PCl3. In contrast, PBr3 caused H2 evolution and formation of a yellow solid; the product mixture contained, in addition to unreacted starting materials, SiH3Br and (H3SiO)nSiH4−n (n = 2, 3) (see Scheme 146, path a). These products were inseparable, so no yields were reported. The absence of SiH4 formation and the appearance of a yellow, presumably polymeric phosphorus hydride (PH)x led Van Dyke to propose the transformation shown in (Scheme 146, path b).235
 | ||
| Scheme 146 a) Reaction of O(SiH3)2 with PBr3 and (b) the proposed reaction mechanism, no yields were reported. | ||
Glidewell and Rankin prepared the aryl silylether PhOSiH3 in nearly quantitative yield by reacting S(SiH3)2 with phenol (PhOH) (see Scheme 147).236
 | ||
| Scheme 147 Synthesis of PhOSiH3 using S(SiH3)2. | ||
Ebsworth and Thompson prepared silyl esters RCO2SiH3 (R = CF3, H, Me) by protonolysis of trisilylamine (N(SiH3)3) with the corresponding carboxylic acids (see Scheme 148, path a). Side products depended on R: with R = CF3 they observed traces of monosilane; with R = H they detected SiH4, H3SiOSiH3, and traces of CO2. They next examined the reaction of silver trifluoroacetate (F3CC(O)OAg) with silyl halides H3SiX (X = Cl, Br, I) (see Scheme 148, path b). SiH3Cl gave the trifluoroacetate silyl ester in low yield, SiH3Br improved the yield to about 60%, and with SiH3I isolation of the ester failed. Finally, they attempted the analogous synthesis with silver formate (HC(O)OAg). With SiH3I, substantial iodine was liberated along with non-condensable gases; the mixture contained disiloxane, formic acid, and SiH3I bearing only small amounts of silyl formate (HC(O)OSiH3). With H3SiBr only traces of silyl formate were obtained, and warming the reaction tube to 0 °C caused it to explode (see Scheme 148, path c).169
 | ||
| Scheme 148 Synthesis towards silyl esters using N(SiH3)3 (path a) and SiH3X (X = Cl, Br, I) (path b and c). Yield of reaction between H3SiI and silver trifluoroacetate not reported. | ||
Cradock et al. reported that treatment of O(SiH3)2 with MeLi liberates MeSiH3 in approximately 85% of the theoretical amount, forming Li(OSiH3) (see Scheme 149). They then attempted to react solutions containing Li(OSiH3) with different electrophiles, but never observed the expected product.219
 | ||
| Scheme 149 Synthesis of Li(OSiH3), no reaction details reported and yields were not determined. | ||
Drake, Henderson, and Hemmings synthesized the silyl esters H3SiOC(O)CX3 (X = F, Cl) by adapting the Ebsworth–Thompson protocol, replacing N(SiH3)3 with a bis(silyl)carbodiimide [C(NSiH3)2] as the silylating agent (see Scheme 150).169,237 This provided a complementary route to the trihaloacetate esters.
 | ||
| Scheme 150 Synthesis of silyl esters using C(NSiH3)2. | ||
Glidewell prepared mixed silyl ethers ROSiH3 (R = Me, Et, i-Pr, t-Bu) by reacting the corresponding lithium tetraaluminates with SiH3Br (see Scheme 151, path a). The phenyl silyl ether PhOSiH3 was obtained from PhONa using either SiH3Br or silyl trifluoroacetate (Scheme 151, path b). He further showed that silylphosphine can serve as an SiH3-transfer reagent: 2,2,2-trifluoroethanol was smoothly converted to CF3CH2OSiH3 (a transformation that failed under the tetraaluminate/SiH3Br conditions), and benzyl alcohol gave BnOSiH3 selectively (Scheme 151, path b and c).238
 | ||
| Scheme 151 Synthesis towards mixed silyl ethers using SiH3Br and tetraaluminates, SiH3R (R = Br, OC(O)CF3) and PhONa, and H2PSiH3 with ROH (R = F3CCH2, Bn). Treatment of mixed silylethers ROSiH3 (R = Et, i-Pr, t-Bu, F3CCH2) with HI (path a–c). Yield of the reactions between lithium aluminate and H3SiBr, and silylphosphine and BnOH were not determined. | ||
Fehér, Fischer, and Skrodzki found that UV irradiation (medium-pressure Hg lamp) of trisilane in the presence of acetone affords a mixture of alkoxysilanes (silylethers) (see Scheme 152, path a). They note that the substitution pattern can be tuned by conditions: short irradiation times with an excess of silane favour predominantly monosubstituted alkoxysilanes, whereas excess ketone and prolonged irradiation shift the distribution toward highly alkoxylated silanes. They further showed that the formed isopropoxysilanes undergo acid-promoted hydrolysis with dilute, non-oxidizing acids to give the corresponding bis-silyl ethers (disiloxanes), in high yields (see Scheme 152, path b).239
 | ||
| Scheme 152 UV irradiation of trisilane in the presence of acetone leading to different mixed silylethers (path a) and the hydrolysis of formed silylethers with aqueous HCl (path b). | ||
Lobreyer, Oeler, and Sundermeyer (1991) reported two routes towards silyl nonaflate. Reaction of silver nonaflate with SiH3Cl afforded low yields (see Scheme 153, path a), whereas an alternative approach employing phenylsilane with perfluorobutanesulfonic acid gave significantly improved isolated yields (see Scheme 153, path b). The resulting silyl nonaflate was then used in further functionalization reactions (Scheme 67).46
 | ||
| Scheme 153 Formation of silyl nonaflate using silver nonaflate with SiH3Cl (path a) and perbutanesulfonic acid with phenylsilane (PhSiH3) (path b). | ||
Cradock et al. generated (TfOSiH2)2 in situ by protonolysis of bis(p-tolyl)disilane with triflic acid (TfOH) (see Scheme 154). The disilyl di(triflate) was not isolated; instead, it was used directly for subsequent functionalization (Scheme 86).158
 | ||
| Scheme 154 Synthesis towards disilyl di(triflate) using (p-tolyl-SiH2)2 and triflic acid. Isolated yields not reported. | ||
Emeléus, MacDiarmid, and Maddock prepared disilyl sulfide S(SiH3)2 by passing SiH3I vapour three times through a tube packed with HgS. Subsequent reaction of S(SiH3)2 with H2S at room temperature for 31 days furnished SiH3SH (see Scheme 155).229
 | ||
| Scheme 155 Synthesis of S(SiH3)2 using SiH3I and HgS, reaction of S(SiH3)2 with H2S forming SiH3SH. | ||
Downs and Ebsworth prepared F3CSSiH3 by passing H3SiI vapor over Hg(SCF3)2 deposited on glass wool. The initially obtained material partially decomposed to give SiH3F and CSF2; repeated fractional distillation removed these volatile by products and furnished pure F3CSSiH3 (see Scheme 156).241
 | ||
| Scheme 156 Reaction between SiH3I and Hg(SCF3)2 forming F3CSSiH3. | ||
Sternbach and MacDiarmid synthesized MeSSiH3 by reacting SiH3I·NMe3 with MeSH at 0 °C (see Scheme 157).242
 | ||
| Scheme 157 Formation of MeSSiH3 by reacting SiH3I·NMe3 with MeSH. | ||
Ward and MacDiarmid prepared S(Si2H5)2 by passing Si2H5I vapor over excess HgS at room temperature (see Scheme 158).153
 | ||
| Scheme 158 Synthesis of S(Si2H5)2 by the reaction of Si2H5I with HgS. | ||
Schmeißer and Frouzanfar attempted to prepare monosubstituted thiosilanes (RS)SiH3 by LiAlH4 reduction of RS-SiCl3, but instead obtained di-, tri-, and tetrasubstituted products. They attributed this outcome to AlCl3 formed in situ, which promotes disproportionation of thiolato groups. The product distribution depends on the substituent R: with n-alkyl groups, (RS)3SiH formed together with SiH4 (see Scheme 159, path a); with a branched alkyl group (i-Bu), (RS)2SiH2 and SiH4 were obtained (see Scheme 159, path b); and with phenyl, a mixture of tri- and tetrasubstituted thiosilanes accompanied by SiH4 (Scheme 159, path c). Monosubstituted thiosilanes were obtained only when employing lead mercaptides in aliphatic or aromatic solvents, which furnished isolable (RS)SiH3 (Scheme 159, path d).243
 | ||
| Scheme 159 Synthesis of mono-, di-, tri- and tetrasubstituted thiosilanes starting either from trichlorosilanes and LiAlH4 (path a–c), or from SiH3I and lead mercaptides (path d). | ||
Glidewell and Rankin prepared PhSSiH3 by salt metathesis of PhSK with SiH3Br. They also examined the reactivity of PhSK toward boron halides: no reaction was observed with BF3, whereas BCl3 afforded a white 1:1 adduct at low temperature that dissociated on warming to ambient temperature (see Scheme 160).236
 | ||
| Scheme 160 Formation of PhSSiH3 and reaction of PhSSiH3 with BCl3. | ||
Glidewell later obtained disilyl sulfide S(SiH3)2 in excellent yields by salt metathesis of Li2S with SiH3Br (see Scheme 161). Alternatively, treatment of SiH3Br with Me3N·H2S also furnished S(SiH3)2.171
 | ||
| Scheme 161 Synthesis of disilylsulfide by Glidewell. | ||
Ebsworth, Glidewell, and Sheldrick monitored the reaction of trisilylphosphine P(SiH3)3 with elemental sulfur in CS2 in an NMR tube over an extended period. After 3 weeks, NMR spectroscopy indicated 16% formation of S(SiH3)2; after 2 months, conversion to S(SiH3)2 was essentially complete. In contrast, P(SiH3)3 showed no detectable reaction with H2S under analogous conditions (see Scheme 162).244
 | ||
| Scheme 162 Synthesis of disilylsulfide by reacting trissilylphosphane with sulfur. | ||
Drake and Riddle subjected gas mixtures to silent electrical discharge and observed broad product formation. For an equimolar SiH4/H2S mixture, in addition to higher silanes, S(SiH3)2 and H3SiSH were detected. Using SiH4/MeSH, the products included S(SiH3)2, H3SiSMe, Me2S and Me2S2, along with higher silanes. Finally, a SiH4/GeH4/H2S mixture afforded H3SiSGeH3, H3SiSH, H3GeSH, S(SiH3)2, S(GeH3)2, higher silanes and germanes, as well as mixed silylgermanes (see Scheme 163).245
 | ||
| Scheme 163 Silent electrical discharge reactions of different SiH4 mixtures. Yields of individual products were not reported. | ||
Glidewell obtained MeSSiH3 by reacting LiAl(SMe)4 with H3SiBr (see Scheme 164). Treatment of Li2S with H3SiBr furnished the expected S(SiH3)2, but it could not be isolated, contrary to his previous work were isolation was successful (Scheme 161).171,238
 | ||
| Scheme 164 Synthesis of silylsulfides starting from alkali sulfides. No yields for S(SiH3)2 reported. | ||
Anderson and Drake prepared a range of thiosilanes by using the thiolato-aluminate LiAl(SMe)4 as nucleophilic RS-source. Treatment of LiAl(SMe)4 with H3SiBr or H5Si2Br furnished the corresponding MeSSiH3 and MeSSi2H5, respectively (see Scheme 165), providing a convenient route to both silyl- and disilanyl-substituted thiosilanes.246
 | ||
| Scheme 165 Synthesis of silylsulfides using the thiolato-aluminate LiAl(SMe)4. | ||
Cradock, Ebsworth, and Jessep obtained S(SiH3)2 in quantitative yield by treating N(SiH3)3 with H2S, also forming the ammonium salt NH3·HSSiH3. The same ammonium salt was also obtained quantitatively by combining HSSiH3 with NH3 (see Scheme 166, path a). Using partially substituted amines derived from N(SiH3)3, they similarly prepared NMe2H·HSSiH3 and NMeH2·HSSiH3 (see Scheme 166, path b). They then explored the electrophile scope of NH3·HSSiH3. MeI afforded MeSSiH3, and Me3SiCl gave Me3SiSSiH3, as expected (see Scheme 166, path c). Reaction with H3GeBr produced H3GeSSiH3 together with the follow-up products S(GeH3)2 and S(SiH3)2 (see Scheme 166, path d). With acetyl chloride, using an excess led predominantly to SiH3Cl, whereas sub-equimolar MeC(O)Cl furnished MeC(O)SSiH3, which isomerized in solution to MeC(S)OSiH3 with an isomer ratio of at least 7:1 in favor of MeC(S)OSiH3 (see Scheme 166, path e).247
 | ||
| Scheme 166 Generation of disilylsulfide and the ammonium salt HSSiH3·NH3 and reactivity studies of the ammonium salt (path a–e). Yields of the reactions between the ammonium salt and H3GeBr and acetylchloride not reported. | ||
Cradock et al. reported that treatment of S(SiH3)2 with MeLi liberates MeSiH3 in approximately 90% of the theoretical amount, forming Li(SSiH3) (see Scheme 167). A solution of Li(SSiH3) can then be quenched with Me3SiCl to give the expected (Me3Si)S(SiH3).248
 | ||
| Scheme 167 Generation of Li(SSiH3) and its reactivity with Me3SiCl. No yields were reported. | ||
Finch and Van Dyke generated H3GeSSiH3 by treating S(SiH3)2 with H3GeCl, but the product disproportionates to S(GeH3)2 and S(SiH3)2, preventing isolation of pure H3GeSSiH3 (see Scheme 168). S(GeH3)2 can be removed readily, but H3GeSSiH3 and S(SiH3)2 have very similar volatilities and could not be separated by distillation.249 This behaviour mirrors the disproportionation reaction observed by Cradock, Ebsworth, and Jessep when NH3·HSSiH3 was reacted with GeH3Br, which likewise furnished H3GeSSiH3 together with S(GeH3)2 and S(SiH3)2 (see Scheme 166, path d).247
 | ||
| Scheme 168 Synthesis of H3GeSSiH3 and its disproportionation towards digermyl- and disilylsulfide, no yields were reported. | ||
Cradock et al. published follow-up chemistry on Li(SSiH3), reacting it with Me3SiCl yields Me3SiSSiH3 (see Scheme 169, path a). Reaction with MeI results in the expected MeSSiH3 formation; acetylchloride giving the thioester MeC(O)SSiH3 and with Me2SiCl2 the corresponding Me2Si(SSiH3)2 (see Scheme 169, path b and c). They also reported several unsuccessful attempts that did not deliver the targeted S-substituted products (see Scheme 169, path d). Reactions with MeSiCl3, SiCl4, PBrF2, PF3, HgCl2, SnCl2, and BCl3 mainly produced S(SiH3)2, SiH3F, or SiH4. Finally, chalcogen exchange experiments showed that reacting Li(SSiH3) with Se(SiH3)2, or alternatively Li(SeSiH3) with S(SiH3)2, converged in both cases to Li(SeSiH3) and S(SiH3)2 (see Scheme 169, path e).219
 | ||
| Scheme 169 Reactivity of Li(SSiH3) with different electrophiles (path a–e). Yields of MeC(O)SSiH3 and Me2Si(SSiH3)2 were not reported. | ||
Haas and Vongher isolated the cyclic trimer (SSiH2)3 in 1978. Their route began with diiodosilane (SiH2I2) and HgS at elevated temperature, followed by filtration and solvent removal to give an oligomeric mixture. After leaving the oligomeric mixture exposed to light for about one week, this mixture converted into a gummy-like, polymeric solid. Vacuum depolymerization at 210 °C for 2 h furnished (SSiH2)3 (see Scheme 170), which is stable at −80 °C but slowly polymerizes at 20 °C over several days. Thermolysis in the presence of Al2S3 showed no change up to 170 °C; above this temperature the trimer decomposes to give mainly H3SiSH and S(SiH3)2, with traces of H2S.250
 | ||
| Scheme 170 Synthesis of the cyclic (SSiH2)3 by reacting HgS with SiH2I2. | ||
Haas and Hitze reported new routes to cyclotrisilathiane (SSiH2)3 in 1984. When Li(SSiH3) was combined with SiH2Cl2, the expected H2Si(SSiH3)2 was not obtained; instead, its decomposition products S(SiH3)2 and (SSiH2)3 were formed (see Scheme 171, path a). The analogous reaction with SiH2I2 gave similar outcomes. Notably, (SSiH2)3 prepared from SiH2Cl2 was more stable than material obtained from SiH2I2, which the authors attributed to iodide catalyzing polymerization. Alternatively, reacting S(SiH3)2 directly with SiH2Cl2 furnished cyclotrisilathiane and, in parallel, H2Si(SSiH3)2, which they succeeded in isolating (see Scheme 171, path b). Using the purified H2Si(SSiH3)2, they conducted a thermal stability study and determined its degradation pathway leading to cyclotrisilathiane (see Scheme 171, path c).251
 | ||
| Scheme 171 Synthesis of the cyclotrisilathiane via different routes (path a–c). Yields for H2ClSiSSiH3 were not reported. | ||
Haas, Süllentrup, and Krüger generated the new six-membered cyclic disilathiane (SSi2H4)2 by two routes. By combining (H2SiCl)2 with S(SiH3)2 (see Scheme 172, path a), or by reacting Li(SSiH3) with one equivalent of (ClSiH2)2, wherein two molecules of the intermediate (ClH5Si2)S(SiH3) undergo self-condensation with elimination of SiH3Cl to close the ring (see Scheme 172, path b).252
 | ||
| Scheme 172 Two pathways towards the cyclic disilathiane (SSi2H4)2 by Haas, Süllentrup and Krüger (path a and b). | ||
A thermolysis study at 90 °C for 1 h showed that, alongside the recovered ring, S(Si2H5)2 is the principal degradation product (Scheme 173, path a). They also accessed linear disilathianes of the type (H3Si)nS(Si2H5)2−n (n = 0, 1) by treating S(SiH3)2 with two or one equivalent(s) of Si2H5Cl, respectively (Scheme 173, path b and c).252
 | ||
| Scheme 173 Thermolysis of disilathiane (SSi2H4)2 (path a) and generation of the unsymmetrical silylsulfide (path b) and disilylsulfide (path c). No yields reported for H3SiS(Si2H5). | ||
In 1955, Emeléus, MacDiarmid, and Maddock attempted to synthesize Se(SiH3)2 by reacting SiH3I with either Se or HgSe over extended periods, but both approaches failed. By contrast, using Ag2Se led to rapid formation of Se(SiH3)2 within about 30 minutes (see Scheme 174).229
 | ||
| Scheme 174 Synthesis of disilylselenide using Ag2Se and SiH3I. | ||
Ebsworth, Emeléus, and Welcman prepared perfluoroalkyl silyl selenides RSeSiH3 (R = CF3, C3F7) by reacting H3SiI with Hg(SeR)2 (see Scheme 175).254
 | ||
| Scheme 175 Synthesis of mixed silylselenides. | ||
Contrary to the H2S case, Ebsworth, Glidewell, and Sheldrick reported that P(SiH3)3 reacts with H2Se to furnish the expected Se(SiH3)2 (see Scheme 176).244
 | ||
| Scheme 176 Generation of disilylselenide through the reaction of trisilylphosphide and H2Se. | ||
Cradock and Ebsworth obtained Se(SiH3)2 in quantitative yield by reacting Li2Se with H3SiBr (see Scheme 177), providing a straightforward halide/selenide metathesis.255
 | ||
| Scheme 177 Synthesis of disilylselenide using lithiumselenide and SiH3Br. | ||
Drake and Riddle applied silent electrical discharge to equimolar SiH4/H2Se and observed, in addition to higher silanes, the formation of Se(SiH3)2 and H3SiSeH (see Scheme 178). With ternary SiH4/GeH4/H2Se mixtures, the product spectrum expanded to include H3SiSeGeH3, H3ESeH and Se(EH3)2 (E = Si, Ge), along with higher silanes, higher germanes, and silylgermanes.245
 | ||
| Scheme 178 Silent electrical of SiH4/H2Se and SiH4/GeH4/H2Se mixtures. Yields of individual products were not reported. | ||
Anderson and Drake synthesized MeSeSiH3 by combining H3SiBr with LiAl(SeMe)4 (see Scheme 179), directly analogous to the sulfur congener prepared from LiAl(SMe)4 (Scheme 165).246
 | ||
| Scheme 179 Synthesis of the mixed MeSeSiH3 selenide using LiAl(SeMe)4 and SiH3Br. | ||
Cradock, Ebsworth, and Jessep showed that N(SiH3)3 reacts with H2Se to give Se(SiH3)2 together with the ammonium selenosilane NH3·HSeSiH3 (see Scheme 180, path a). Replacing NH3 with partially substituted amines such as HNMeSiH3 or Me2NSiH3 resulted in the formation of the corresponding salts HNRR′·HSeSiH3 (R, R′ = H, Me) (see Scheme 180, path b). The NH3·HSeSiH3 adduct was examined in detail: methylation with MeI delivered MeSeSiH3; treatment with Me3SiCl afforded Me3SiSeSiH3, which slowly disproportionated at room temperature to (Me3Si)2Se and Se(SiH3)2 (see Scheme 180, path c); and exposure to PF2Br produced a complex mixture containing PF3, SiH3F, unreacted PF2Br, H2Se, Se(SiH3)2, and SiH3·NH·PF2. Finally, reaction with acetyl chloride yielded a mixture of Se(SiH3)2, diacetyl selenide (Se[(O)CMe]2), and the desired silylselenoacetate MeC(O)SeSiH3, which could not be isolated; nonetheless, the isomer ratio at room temperature was determined to be approximately 2.5:1, with MeC(O)SeSiH3 as the major isomer (see Scheme 180, path d).247
 | ||
| Scheme 180 Generation of disilylselenide and the ammonium salt HSeSiH3·NH3 and reactivity studies of the ammonium salt (path a–d). Yields of the reaction between the ammonium salt and Me3SiCl and acetylchloride not reported. | ||
Cradock et al. reported that treating Se(SiH3)2 with MeLi evolves methylsilane (MeSiH3) in approximately 90% of the theoretical amount and forms Li(SeSiH3). The resulting solution of Li(SeSiH3) can then be quenched with Me3SiCl to afford the expected Me3SiSeSiH3 (see Scheme 181).248
 | ||
| Scheme 181 Generation of Li(SeSiH3) by the reaction of disilylselenide and MeLi and its reaction with Me3SiCl. Yields of products were not determined. | ||
Barker, Drake, and Hemmings showed that LiAl(SeMe)4 reacts with H3SiBr to give H3SiSeMe and with H2SiBr2 to give H2Si(SeMe)2; both products were successfully isolated, and in each case only traces of SiH4 were detected as a byproduct (see Scheme 182).256
 | ||
| Scheme 182 Generation of mixed silylselenides using LiAl(SeMe)4. | ||
Cradock et al. showed that Li(SeSiH3) reacts cleanly with Me3SiCl to furnish Me3SiSeSiH3 (see Scheme 183, path a). If NMe3 is present prior to quenching, the Lewis adduct Me3SiSeSiH3·NMe3 is formed, subsequent treatment with BF3 releases the free selenide (with formation of NMe3·BF3) (see Scheme 183, path b). Acylation with acetyl chloride affords MeC(O)SeSiH3, which isomerizes in solution and exists in equilibrium with MeC(Se)OSiH3 (see Scheme 183, path c). Extending this electrophile scope gave divergent outcomes. With MeC(O)2O or MeCO2H, the main product is again the acetyl silyl selenide MeC(O)SeSiH3. In contrast, CF3CO2H reacts mainly to the silyl trifluoroacetate CF3C(O)OSiH3, while the acyl halides CF3C(O)Br and CClH2C(O)Cl lead predominantly to Se(SiH3)2. No reaction is observed with NMe3 alone under the same conditions (see Scheme 183, path d).219
 | ||
| Scheme 183 Reactivity scope of Li(SeSiH3) (path a–d). | ||
Drake and Hemmings synthesized phenylselenosilanes by two complementary routes: reaction of Li(SePh) with H3SiBr to give PhSeSiH3, and reaction of the selenoaluminate LiAl(SePh)4 with H2SiBr2 to furnish H2Si(SePh)2 (see Scheme 184).257
 | ||
| Scheme 184 Generation of phenylselenosilanes. | ||
Drake, Glavinčevski, and Hemmings prepared Se(SiH3)2 by reacting H3SiI with the selenoaluminate LiAl(SeH)4, which serves as an efficient selenide-transfer reagent (see Scheme 185).258
 | ||
| Scheme 185 Synthesis of disilylselenide using selenoaluminate and SiH3I. | ||
Haas and Hitze were the first to obtain the cyclic selenosilane (SeSiH2)3 by heating HgSe with H2SiI2 to 120 °C. The initial reaction produced oligomeric mixtures, which upon depolymerization under vacuum at 140 °C, followed by recrystallization in benzene furnished crystalline (SeSiH2)3 (see Scheme 186, path a). The crystals are unstable at 20 °C and revert to an oligomeric mixture within 1–2 hours. Attempts to reach the same product via Na2Se and H2SiI2 in benzene at 20 °C led to decomposition into iodine and selenium; performing the reaction in ether gave the same outcome, with decomposition occurring above −40 °C. Reactions between SiH2I2 and K6[HgSe4] behaved similarly (see Scheme 186, path b). When Ag2Se was combined with H2SiI2, no reaction was observed at 20 °C, and upon refluxing the mixture, SiH3I and Se(SiH3)2 were obtained instead of (SeSiH2)3 (see Scheme 186, path c).259
 | ||
| Scheme 186 Attempted reactions towards the cyclic selenosilane (SeSiH2)3 and its successful generation using HgSe and SiH2I2 (path a–c). | ||
Haas, Süllentrup, and Krüger generated the new cyclic compound (SeSi2H4)2 by reacting (SiH2Cl)2 with Se(SiH3)2, with elimination of H3SiCl (see Scheme 187, path a). Thermolysis of this ring at 90 °C for 1 h gave Se(Si2H5)2 as the principal degradation product together with Si2H6 (see Scheme 187, path b). They also obtained the new linear disilaselenides (H3Si)nSe(Si2H5)2−n (n = 0, 1) by reacting Se(SiH3)2 with one or two equivalents of Si2H5Cl (see Scheme 187, path c and d).252
 | ||
| Scheme 187 Synthesis of the cyclic disilylselenide (SeSi2H4)2 via different pathways (path a) and generation of mixed and symmetrical disilylselenides (path b–d). | ||
Bürger and Goetze were the first in 1967 to synthesize Te(SiH3)2 by reacting Li2Te with SiH3I in tetralin and heating the mixture at 70 °C for 3 days (see Scheme 188).261
 | ||
| Scheme 188 Synthesis of Te(SiH3)2 using TeLi2 and SiH3I in tetralin. | ||
Two years later, Cradock, Ebsworth, and Rankin obtained Te(SiH3)2 by reacting SiH3Br with Li2Te in Me2O at low temperature (see Scheme 189).255
 | ||
| Scheme 189 Synthesis of Te(SiH3)2 using TeLi2 and SiH3Br. | ||
Anderson and Drake prepared MeTeSiH3 by reacting the telluroaluminate LiAl(TeMe)4 with SiH3Br, but the product was obtained only in low yield (see Scheme 190).246
 | ||
| Scheme 190 Synthesis of the mixed MeTeSiH3 using LiAl(TeMe)4 and SiH3Br. | ||
Drake and Hemmings attempted to prepare RTeSiH3 (R = Me, Ph) by reacting LiTeR with SiH3Br, but they were unable to isolate the desired products; instead, they observed Te(SiH3)2 and traces of silane among the volatile species alongside intractable polymeric material. In contrast, an exchange route employing RTeMe3Si (R = Me, Ph) with SiH3Br or SiH3I, eliminating Me3SiBr or Me3SiI, results in a successful RTeSiH3 formation (see Scheme 191, path a). They also found that MeTeSiH3 decomposes after prolonged storage or upon laser excitation to give H2Si(TeMe)2 and SiH4 (see Scheme 191, path b).262
 | ||
| Scheme 191 Attempted and successful reaction towards RTeSiH3 (path a) and decomposition products of MeTeSiH3 (path b). | ||
Drake, Glavinčevski, and Hemmings later synthesized Te(SiH3)2 by reacting SiH3I with an excess of Li2Te at low temperatures in Me2O (see Scheme 192).258
 | ||
| Scheme 192 Generation of disilyltelluride using SiH3I and TeLi2 in ether. | ||
Functionalization with group 7 (synthesis of silylhalogenes)
Halohydrosilanes represent a central class of silicon compounds in which Si–X bonds (X = F, Cl, Br, I) replace one or more Si–H or Si–Si functionalities. The strong polarity and thermodynamic stability of Si–halogen bonds significantly influence their chemical behaviour: halohydrosilanes are highly sensitive to hydrolysis, readily undergo redistribution reactions, and frequently serve as activated intermediates in silicon bond construction. From a synthetic perspective, they occupy a strategic position between elemental silicon and hydrosilanes, acting both as precursors to reduce silicon species and electrophilic building blocks for tailored silicon frameworks.Industrial and technological interest in halohydrosilanes is driven largely by thin-film deposition processes. In chemical vapor deposition and liquid phase deposition or solution-based routes, halohydrosilanes provide a controlled silicon delivery, variable reactivity, and favourable volatility. Chlorinated and fluorinated silanes, in particular, are widely exploited for semiconductor fabrication, surface passivation, and the formation of silicon-containing coatings. Their decomposition pathways allow precise regulation of film growth kinetics, impurity profiles, and microstructure – critical parameters for semiconductor devices, photovoltaics, and protective layers. Furthermore, halohydrosilanes serve as key intermediates in the preparation of higher hydrosilanes and functional silicon oligomers, linking fundamental synthesis with applied materials chemistry.263
Synthetic access to halohydrosilanes generally follows several recurring strategies (see Scheme 193).
 | ||
| Scheme 193 Method (i)–(v) for the formation of different halohydrosilanes. | ||
Structurally, halohydrosilanes can be devided into acyclic perhalogenated, cyclic compounds and mixed derivatives. The following sections summarize the principal compound classes and their synthetic approaches, organized according to the halogen substituent – beginning with fluorohydrosilanes and proceeding through chlorosilanes and bromohydrosilanes to iodohydrosilanes – reflecting their sequence in the periodic table.
Monofluorosilane (H3SiF) has been prepared primarily through electrophilic fluorination or Lewis-acid-induced Si–X bond cleavage reactions. Early work by Emeléus and Maddock demonstrated that H3SiF can be obtained in approximately 79% yield by fluorination of chlorosilane with antimony trifluoride at room temperature (see Scheme 194, path a), although competing oxidative fluorination and disproportionation limited product stability and purity.265 A significant cleaner and higher-yielding approach was later introduced by Onyszchuk, who showed that boron trifluoride induces efficient Si–O bond cleavage in disiloxane, affording H3SiF in up to 85% isolated yield under solvent-free conditions (see Scheme 194, path b).233 Related studies by Sternbach and MacDiarmid further established that boron trifluoride readily converts alkoxysilanes into H3SiF, confirming the general applicability of BF3-mediated fluorination pathways, although this route was not optimized for preparative yields (see Scheme 194, path c).232 In addition, Ebsworth and Mays reported the formation of H3SiF from the reaction of silyl isocyanate with boron trifluoride at −78 °C, an observation made in the course of probing the reactivity of silylisocyanate rather than as dedicated synthetic approach to H3SiF (see Scheme 194, path d).172 Treatment of PhOSiH3 with HF affords H3SiF in excellent yields (see Scheme 194, path e).236 The most common method for the synthesis of fluorohydrooligosilanes is the co-condensation of SiF2 with B2H6 at a cool copper surface (method iv). The reactive SiF2 intermediate is generated in situ by passing SiF4 over elemental silicon at high temperatures. Under these conditions, a mixture of F3Si–SiH3, F3Si–SiF2H and H2Si–(SiF3)2 is formed (see Scheme 195). Although isolated yields were not determined, spectroscopic analyses indicated that H2Si–(SiF3)2 is formed in relatively higher amounts, while F3Si–SiH3 and F3Si–SiF2H are produced in comparable portions. This species were separated by high vacuum, low temperature fractional condensation.266
 | ||
| Scheme 194 Methods for the synthesis of monofluorosilane (path a–e). For the reactions of H3SiOMe + BF3 (path c) and SiH3·NCO + BF3 (path d) no yields reported. | ||
 | ||
| Scheme 195 Formation of different fluorohydrosilanes from Si2F with B2H6; no yields reported. | ||
F3Si–SiH3 is also formed as a byproduct during the synthesis of SiF2HPH2 and SiF3PH2 via the condensation of SiF2 with PH3 (method iv) (see Scheme 196).267
 | ||
| Scheme 196 Formation of F3Si–SiH3 as a byproduct during the condensation of SiF2 and PH3; no yields reported. | ||
Reactions of SiF2 with protic reagents (such as HBr) typically generate initially formed fluorinated bromodisilanes that are thermally unstable and undergo rapid secondary transformation. Scheme 197 shows that the reaction with HBr first produces 1-bromo-1,1,2,2-tetrafluorodisilane, which decomposes quickly to a mixture of different mixed halohydrosilanes. Subsequent treatment of the resulting mixture with excess SbF3 converts it efficiently into F3Si–SiF2H.268
 | ||
| Scheme 197 Treatment of SiF2 with HBr yielding in the unstable BrF2Si–SiF2H. Subsequent treatment of decomposition products with SbF3 affording F3Si–SiF2H. No yields reported. | ||
A similar pattern is observed with H2S: the initially formed silanethiol F2HSi–SiF2SH decomposes within minutes, again yielding F3Si–SiF2H (see Scheme 198).269
 | ||
| Scheme 198 Reaction of SiF2 with H2S. Decomposition at room temperature affords F3Si–SiF2H. No yields reported. | ||
Direct fluorination routes of hydrosilanes (method i) have not been reported, but indirect access is possible through selective hydrogenation of mixed halodisilanes (method v). In compounds containing both, fluorine and chlorine or bromine substituents, the heavier halides can be selectively reduced with Me3SnH without cleaving Si–F bonds. Using this strategy, F3Si–SiH3 can be obtained in high yields from the corresponding mixed halo precursors (see Scheme 199).270
 | ||
| Scheme 199 Hydrogenation of mixed halodisilane yielding in F3Si–SiH3. | ||
In contrast, partially fluorinated bromodisilanes do not undergo clean hydrogenation. Instead, F/Br redistribution occurs prior to reduction, producing mixtures of fluorinated disilanes. Stronger hydride reagent led to even less selective outcomes, promoting Si–Si bond cleavage and partial reduction of Si–F bonds. Together, these observations highlight the delicate balance between halogen redistribution, bond stability and reductive pathways in fluorinated disilane chemistry.270
FH2Si–SiH3 was obtained by fluorination of dichlorodisilanes or dichlorotrisilane with SbF3 (method iii) (see Scheme 200).271,272
 | ||
| Scheme 200 Fluorination of oligohydrosilanes with SbF3. | ||
ZnF2 is another well-established fluorinating agent and has been used to prepare F2HSi–SiHF2 and FH2Si–(SiH2)2–SiH2F from their corresponding chlorinated or brominated precursors (method iii) (see Scheme 201).273,274
 | ||
| Scheme 201 Fluorination of chloro-, and bromooligohydrosilanes with ZnF2. | ||
In addition, cleavage of Si–N bonds in N(Si2H5)3 by BF3 provides another pathway to FH2Si–SiH3 (see Scheme 202).150
 | ||
| Scheme 202 Formation of FH2Si–SiH3 via the reaction of N(H5Si2)3 and BF3. | ||
Moreover, it is possible to fluorinate different methoxyoligosilanes to the corresponding branched fluorohydrosilanes with BF3 (see Scheme 203).275
 | ||
| Scheme 203 Fluorination of branched methoxysilanes with BF3. | ||
Finally, fluorohydrosilanes can be accessed via an electrical discharge approach. In this method, various monofluorosilanes were converted into F2HSi–SiH3, FH2Si–SiH2F, F2HSi–SiHF2, F3Si–SiF2H, F2HSi–Si2H5 and F3Si–Si2H5. However, the resulting product mixtures proved difficult to impossible to separate.271,276
Already in 1919 Stock and Somieski reported the synthesis of monochlorosilane (H3SiCl).10 They treated monosilane with hydrogen chloride and catalytic amounts of AlCl3 (method i) (see Scheme 204). Heating this mixture to 100 °C for 30 hours afforded ClSiH3 in 50–55% yield, with dichlorosilane as byproduct (ratio ClSiH3:Cl2SiH2 = 4:1). More recent studies showed, that the yields of ClSiH3 can be increased to 76% when a zeolite catalysator (Na-ZSM-5) is used.279
 | ||
| Scheme 204 Methods for the synthesis of monochlorosilane (path a–d). | ||
Hollandsworth et al. showed, that monosilane can also be chlorinated with silver chloride to afford monochlorosilane (see Scheme 204).280 When phenylsilane (PhSiH3) is used as starting material, H3SiCl could be obtained in yields of 78% (method ii) (see Scheme 204, path c).281 In addition, Glidewell and Rankin showed, that treatment of PhOSiH3 with HCl affords H3SiCl (see Scheme 204, path d).236
Starting in the 1960s, synthetic efforts towards chlorooligohydrosilanes increasingly focused on the chlorination (method i) of smaller hydrosilanes such as disilane, trisilane and tetrasilane.
Early studies by Drake et al. and van Dyke et al. established boron trichloride as an efficient chlorination reagent for these substrates (see Scheme 205, path a).271,276,282,283 In analogy to observations made of monosilane, hydrogen chloride284 and silverchloride285,286 were subsequently shown to be suitable chlorinations agents for disilane as well (see Scheme 205, path b and c). Beyond these systems, tetrachlorostannane was demonstrated to chlorinate di-, tri-, and tetrasilane efficiently, thereby broadening the scope of applicable chlorination reagents (see Scheme 205, path d).287 Finally, direct halogenation using molecular chlorine was reported by Fehér and co-workers, providing a more straightforward, albeit harsher, approach to the chlorination of disilane (see Scheme 205, path e).288
 | ||
| Scheme 205 Chlorination of oligohydrosilanes using different chlorination agents (path a–e). | ||
However, all of the mentioned methods for direct halogenation of oligohydrosilanes yield complex product mixtures that often require time consuming separation or further derivatization before reliable analysis is possible.271,276,284,286–289 Another challenge is the tendency of higher silanes to undergo isomerization.283
Van Dyke, and MacDiarmid prepared the silylethers H3SiOSi2H5 and H3SiOSi2H5 (see Scheme 145). In subsequent reactions, both O(Si2H5)2 and H3SiOSi2H5 reacted with BCl3 to give Si2H5Cl as the main product (see Scheme 206).234
 | ||
| Scheme 206 Reaction of silylethers with BCl3. | ||
In 2012 Stueger et al. demonstrated the selective chlorination of neopentasilane with 3.5 equivalents SnCl4 to afford tetra(chlorosilyl)silane (see Scheme 207 and Fig. 15).290 However, a significant drawback of employing SnCl4 as halogenating reagent is the formation of large quantities of SnCl2, which are difficult to remove completely, particularly on a preparative scale. To address limitation, the authors also examined an alternative protocol based on gaseous HCl in presence of catalytic AlCl3 and obtained the selective formation of tetra(chlorosilyl)silane (85%), accompanied only by minor amounts of 1,2,3-trichloroneopentasilane as byproduct (15%) in a total yield of more than 60%.291
 | ||
| Scheme 207 Synthesis of branched chlorooligohydrosilanes. | ||
 | ||
| Fig. 15 Crystal structure of Si(SiH2Cl)4 reproduced from Stueger et al.48 with permission from American Chemical Society, © 2012. | ||
An additional example for the synthesis of branched chlorohydrosilanes, is the preparation of HSi(SiCl3)3. Höfler et al. reported two efficient synthetic approaches to this compound. The first involves the reaction of dodecachloroneopentasilane (Si(SiCl3)4) with HCl, eliminating SiCl4, affording the target product in 70% yield (see Scheme 208). The second approach is based on the chlorination of HSi[Si(OMe)3]3 with nine equivalents of BCl3, providing HSi(SiCl3)3.292
 | ||
| Scheme 208 Two methods for the synthesis of hexachlorotrisilane. | ||
The synthesis of an even higher branched chlorooligohydrosilane was demonstrated by Christopoulos et al. First, they generated phenylnonasilane via the reaction of the trissilyllithium anion and 0.5 equivalents dichlorophenylsilane. Subsequent treatment with triflic acid affords the triflate substited nonasilane, which can be chlorinated with an excess of lithiumchloride and the chlorononasilane can be obtained in yields of 81% (see Scheme 209).49
 | ||
| Scheme 209 Synthesis of chlorononasilane starting from neopentasilane. | ||
An alternative method for the synthesis of chlorohydrosilanes is the partial hydration of perchlorooligosilanes using hydrating reagents, such as LiAlH4 or Bu3SnH (method v). But also these approach were found to yield in unselective product mixtures.107,109
A more attractive strategy involves the controlled partial hydrogenation (method v) with substoichiometric amounts of i-Bu2AlH (see Scheme 210). This methodology enables access to broader range of chlorohydrooligosilanes under comparatively mild conditions. i-Bu2AlH promotes selective Si–Cl hydrogenation in both linear and branched chlorooligosilanes without inducing Si–Si bond cleavage.291
 | ||
| Scheme 210 Synthesis of chlorooligosilanes via partial hydrogenation. | ||
An additional method to prevent the complete hydrogenation of oligochlorosilanes with LiAlH4 is the implementation of N(SiMe3)2 or N(SiMe2Ph)2 substituents as protecting groups into the molecule (see Scheme 211). After the hydrogenation, these groups can easily be cleaved off with HCl.139
 | ||
| Scheme 211 Synthesis of chlorodisilanes using N(SiMe3)2 (path a) and N(SiMe2Ph)2 (path b) as protecting groups. No reported yields of ClH2Si–SiH2Cl. | ||
In contrast to hydrosilanes, aryl-substituted silanes (ArmSinH2n+2−m) are excellent starting materials for the selective synthesis of chlorohydrosilanes (ClmSinH2n+2−m, n = 2–7, m = 1–9) (Scheme 212). The aryl substituents can be cleanly replaced by chlorine upon treatment with liquefied hydrogen chloride or with HCl solutions in benzene, enabling controlled halogen incorporation with good selectivity (method ii).252,273,274,293,294
 | ||
| Scheme 212 Synthesis of chlorooligosilanes via dearylation. | ||
To suppress equilibrium of the chlorinated products,295 Uhlig et al. proposed a stepwise dephenylation strategy in which triflic acid is first used to remove the organic substituents, followed by either hydride reduction with LiAlH4 or chlorination with Et3NHCl (see Scheme 213). Using Ph3Si–SiH3 as precursor, this route enables the selective formation of HCl2Si–SiH3 and Cl3Si–SiH3, respectively.296
 | ||
| Scheme 213 Multi-step synthesis of chlorodisilanes by Uhlig et al. | ||
An analogous electrical discharge strategy for the formation of fluorohydrosilanes, has also been applied for chlorohydrosilanes. Under comparable conditions, monochlorosilane undergoes coupling reactions to give mixtures of chloro-substituted di- and oligosilanes. Similar to the fluorinated systems, these discharge processes produce complex product distributions, and separation of the individual components is challenging.276
Monobromosilane can be synthesized by reacting monosilane with various bromination agents, including HBr117 (see Scheme 214, path a), SnBr4298 (see Scheme 214, path b), and AgBr280 (see Scheme 214, path c) (method i). A more efficient approach involves the conversion of phenylsilane with hydrogen bromide and catalytic amounts of AlBr3 (see Scheme 214, path d) (method ii).281,299
 | ||
| Scheme 214 Different methods for the synthesis of BrSiH3 (path a–d). No reported yields for the bromination of SiH4 with HBr (path a). | ||
As already shown in the section Siloxanes, H3SiBr is also formed at the reaction of O(SiH3)2 with PBr3 (see Scheme 146, path a).
Similar to the preparation of chlorooligohydrogermanes, different bromooligohydrosilanes can be generated through the bromination of oligohydrosilanes (method i) using reagents like BBr3271,272 (see Scheme 215, path a) HBr117,286 (see Scheme 215, path b), SnBr4298 (see Scheme 215, path c), AgBr280 and Br2288,300 (see Scheme 215, path d). These transformations typically proceed through non-selective halogen exchange leading to complex mixtures of mono- and oligobrominated silanes rather than single, well-defined products.
 | ||
| Scheme 215 Methods for the synthesis of bromooligosilanes (path a–d). *Yield was not isolated. **Yield of the product mixture. | ||
Also similar to the synthesis of chlorohydrosilanes, is the possibility of electrophilic cleavage of silicon–aryl bonds with HBr (method ii) (see Scheme 216).274,294,301
 | ||
| Scheme 216 Synthesis of bromooligosilanes via dearylation. | ||
Maddock and co-workers first reported the preparation of monoiodosilane in 1939, describing its formation via the reaction of monosilane and hydrogen iodide in the presence of catalytic amounts of AlI3 (method i) (Scheme 217, path a).167,303 An alternative efficient method towards monoiodosilane employs phenylsilane as the starting material (method ii) (Scheme 217, path b).281,304 Ward et al. further demonstrated that H3SiI can be generated via the chlorination of phenylchlorosilane and subsequent hydration to form the chlorophenylsilane, which upon treatment with hydrogen iodide afforded monoiodosilane in yields of up to 65% (Scheme 217, path c).299 Across a range of silyl ethers, treatment with HI afforded H3SiI in quantitative yield and the corresponding alcohol (ROH) as byproduct (Scheme 217, path d).238 It is also possible to convert PhOSiH3 to H3SiI by treatment with HI (Scheme 217, path e).236
 | ||
| Scheme 217 Methods for the synthesis of ISiH3 (path a–e). | ||
In 1960, Ward and MacDiamid extended these studies to iodohydrooligosilanes.231 In their work, disilane was reacted with hydrogen iodide in the presence of catalytic amounts aluminium iodide, leading to the formation of iododisilane IH2Si–SiH3 (method i) (see Scheme 218).
 | ||
| Scheme 218 Iodation of disilane with HI yielding in IH2Si–SiH3. | ||
Approximately a decade later Fehér et al. carried out intense studies to the synthesis of different iodooligohydrosilanes following an alternative approach applied to di-, tri- and tetrasilane, in which iodide serves as iodation reagent (see Scheme 219).305,306 These reactions consistently produced mixtures of mono- and diiodinated silanes. Nevertheless, individual isomers of iodinated di-, tri-, and tetrasilanes could be separated and identified by gas chromatographic methods.
 | ||
| Scheme 219 Reaction of di-, tri- and n-tetrasilane with iodide. *Yield of the monoiodotetrasilane was not reported. | ||
Nearly twenty years afterward, Hassler and co-workers reported significant progress in the synthesis of iodooligohydrosilanes. Analogous to strategies established for chloro- and bromohydrosilanes, their work demonstrated that electrophilic cleavage of silicon-aryl bonds with hydrogen iodine enables the selective introduction of iodine substituents (see Scheme 220). Using this approach, a broad range of iodohydrosilans was prepared in high efficiency.294,307
 | ||
| Scheme 220 Synthesis of iodooligosilanes via dearylation. | ||
Cyclic halohydrosilanes
Nonahalocylcopentasilanes (HSi5X9) and octahalocyclo-pentasilanes (1,3-H2Si5X8) (X = Cl, Br, I) can be prepared via dephenylation of the corresponding phenyl-substituted precursors HSi5Ph9 and H2SiPh8 (see Scheme 221).35,308 | ||
| Scheme 221 Synthesis of cyclic halohydrosilanes. | ||
Partially chlorinated cyclopentasilanes were reported by Roewer and co-workers through the stepwise hydride reduction of decachlorocyclopentasilane using Me3SnH (see Scheme 222). The reaction proceeds without detectable formation of SiHCl moieties. The major product observed was assigned to 1,1,3,3,-tetrachlorocyclopentasilane, while 1,1-dihydrooctachlorocyclopentasilane was not detected. Structural assignments were based on 29Si NMR spectroscopic data; the intermediates were not isolated.107
 | ||
| Scheme 222 Synthesis of cyclic chlorohydrosilanes. | ||
Stueger et al. reported the partial hydrogenation of perchlorinated cyclopentasilane in 2012 in a three-step synthesis (see Scheme 223).290 The final yield of the product (Si5HCl9) could not be determined since it could not be separated from cyclopentasilane which was formed as byproduct.
 | ||
| Scheme 223 Synthesis of nonachlorocyclopentasilane by Stueger et al. No yield of the final product reported. | ||
Mixed halohydrosilanes
The bromination of Si–H bonds using BBr3 was employed to generate chlorobromohydrosilanes. Thus, ClH2Si–SiH3 and Cl2HSi–SiH3 were converted into HBrClSi–SiH3 and BrCl2Si–SiH3, respectively (see Scheme 224). These products were detected by GC; however, no isolation was reported.271 | ||
| Scheme 224 Synthesis of mixed halodisilanes; no yields reported. | ||
Mixed halohydrosilanes of the general formula Si2XmYnH6−m−n (X = F, Y = Br) are formed via co-condensation of SiF2 with HBr. Upon warming the reaction mixture to room temperature, its composition changes, although the mixed halohydrosilanes BrF2Si–SiF2H, F3Si–SiFHBr and F3Si–SiHBr2 could be isolated but no yields were reported (see Scheme 225). The compounds HBrFSi–SiF2Br and HBr2Si–SiF2Br were identified in the final reaction mixture by 19F NMR spectroscopy, but were not isolated. Subsequent chlorination of BrF2Si–SiF2H and F3Si–SiHBr2 with SnCl4 afforded F3Si–SiHCl2 and ClF2Si–SiHF2. Yields of the isolated products have not been reported.270,309
 | ||
| Scheme 225 Synthesis of mixed halodisilanes; no yields reported. | ||
Key physical and chemical properties for the central hydrosilanes
In Table 8 the key physical and chemical properties for the central hydrosilanes are depicted.| Silane | m.p. [°C] | b.p. [°C] | Physical/state | Solubility | Safety |
|---|---|---|---|---|---|
| SiH4 | −185 | −112 | Gas | Slightly in all organic and inorganic solvents | Pyrophoric |
| Si2H6 | −133 | −15 | Gas | All organic and inorganic solvents | Pyrophoric |
| Si3H8 | −115 | 53 | Liquid | All organic and inorganic solvents | Pyrophoric |
| n-Si4H10 | −89.9 | 108 | Liquid | All organic and inorganic solvents | Pyrophoric |
| iso-Si4H10 | −99.4 | 102 | Liquid | All organic and inorganic solvents | Pyrophoric |
| n-Si5H12 | −72.2 | 153 | Liquid | All organic and inorganic solvents | Pyrophoric |
| neo-Si5H12 | −57.8 | 134 | Liquid | All organic and inorganic solvents | Pyrophoric |
| cyclo-Si5H10 | −10.5 | 194 | Liquid | All organic and inorganic solvents | Pyrophoric |
| n-Si6H14 | −44.7 | 194 | Liquid | All organic and inorganic solvents | Pyrophoric |
| cyclo-Si6H12 | 16.5 | 226 | Liquid | All organic and inorganic solvents | Pyrophoric |
| n-Si7H16 | −30.1 | 227 | Liquid | All organic and inorganic solvents | Pyrophoric |
| Si8H18 | 28 | x | Oil | All organic and inorganic solvents | Pyrophoric |
| N(SiH3)3 | −106 | 52 | Liquid | All organic and inorganic solvents | Pyrophoric |
| N(SiH2SiH3)3 | −97 | 176 | Liquid | All organic and inorganic solvents | Pyrophoric |
| Et2NSiH2SiH2NEt2 | x | 23/0.05 Torr | Liquid | All organic and inorganic solvents | Pyrophoric |
| SiH3SiH(NEt2)2 | x | 23/0.2 Torr | Liquid | All organic and inorganic solvents | Pyrophoric |
| iPr2NSiH2SiH2NiPr2 | x | 68/0.05 Torr | Liquid | All organic and inorganic solvents | Pyrophoric |
| H3SiPH2 | <135 | 12.7 | Gas | All organic and inorganic solvents | Pyrophori, toxic |
| P(SiH3)3 | −73 | 114 | Liquid | All organic and inorganic solvents | Pyrophori, toxic |
| O(SiH3)2 | −144 | −15.2 | Gas | All organic and inorganic solvents | Not pyrophoric |
| S(SiH3)2 | −70.0 | 58.8 | Liquid | All organic and inorganic solvents | Pyrophori, toxic |
| Se(SiH3)2 | −68.0 | 85.2 | Liquid | All organic and inorganic solvents | Toxic |
| Te(SiH3)2 | x | 49/50 Torr | Liquid | All organic and inorganic solvents | Toxic |
Deposition methods
Silicon (Si) is the second most abundant element in the Earth's crust constituting approximately 28%.310 As a semiconductor, silicon plays a crucial part in the electronics industry. Depending on deposition method and process conditions silicon thin films can exhibit monocrystalline, polycrystalline or amorphous structures, which significantly influence the electronic properties and application potential. Monocrystalline silicon (c-Si) possesses a long-range ordered crystal lattice and high charge carrier mobility, making it the preferred material for microelectronic devices. Polycrystalline silicon (poly-Si) consists of crystalline grains separated by grain boundaries, which reduce carrier mobility but allow deposition at lower temperatures, enabling its use in thin film transistors (TFTs) and photovoltaic devices. Amorphous silicon (a-Si) lacks long-range order and exhibits a high density of dangling bonds.311–313 The structural motifs are summarized in Fig. 16. | ||
| Fig. 16 Structural motifs of (a) monocrystalline silicon, (b) polycrystalline silicon and (c) amorphous silicon. Dangling bonds marked as dotted line.314 | ||
Early on, a-Si attracted scientific interest as a semiconductor, however, it exhibited significant drawbacks with respect to electrical conductivity. At the beginning of the 1970s, new deposition methods were developed, that enabled the preparation of amorphous silicon thin-films with improved electronic properties through the incorporating of hydrogen, resulting in hydrogenated amorphous silicon (a-Si:H).315 As shown in Fig. 17, passivation of dangling bonds with hydrogen leads to a drastic reduction in defect density, thereby improving conductivity and carrier mobility. Furthermore, effective p- and n-type doping of a-Si:H can be achieved by incorporating appropriate dopant species, such as diborane or phosphine, during deposition, as dopant atoms are no longer compensated by dangling bond defects.316
 | ||
| Fig. 17 (a) Schematic passivation of dangling bonds by hydrogen and (b) schematic illustration of density of states in a-Si and a-Si:H. Graphic adapted from Vora.317 | ||
Since then, several different deposition techniques have been employed to produce silicon thin films. These silicon thin films have a wide range of applications, including thin film transistors, flexible large area displays, and photovoltaics.318,319
Chemical vapor deposition
Chemical vapor deposition describes the deposition of a solid component on the surface of a substrate from the vapor phase by means of a chemical reaction. The term was first introduced by J. M. Blocher in 1960 to distinguish the process from other deposition methods, that do not involve chemical reactions such as sputtering.320 During the CVD process, gaseous reactant species are supplied and transported into a reaction chamber, as schematically illustrated in Fig. 18. Upon entering the reactor, the reactant gases are conveyed toward the substrate by mass transport and may either diffuse directly through the boundary layer to adsorb onto the heated substrate surface or participate in gas-phase reactions, forming intermediate species and gaseous byproducts. These intermediates can subsequently reach the substrate by diffusion and adsorption. On the substrate surface, adsorbed species undergo surface diffusion and heterogeneous chemical reactions, ultimately leading to thin-film growth. Any unreacted gaseous precursors or byproducts are subsequently desorbed from the surface and removed from the chamber in the gas phase.312,320–323 | ||
| Fig. 18 Schematic illustration of the CVD process. Graphic adapted from Sun et al.323 and Katsui et al.322 | ||
A variety of CVD processes and techniques exist, including thermal CVD (TCVD), plasma-enhanced CVD (PECVD), photo-assisted CVD (PACVD), and hot-wire CVD (HWCVD). Depending on the deposition conditions such as temperature, pressure, and plasma environment, CVD can yield silicon films with different microstructures, including amorphous, microcrystalline, polycrystalline, or epitaxial silicon. The various CVD techniques therefore differ not only in deposition rate and defect concentration but also in the structural properties of the resulting films. A special form of CVD is atomic layer epitaxy (ALE), which enables the growth of monoatomic layers on a substrate surface. In the following section, these different methods are described in more detail.315
Thermal chemical vapor deposition
The most common sub-technique of CVD is the thermal CVD. In this process, thermal energy is used to activate the chemical reactions. The required energy can be supplied by several different methods, such as radio-frequency heating, thermal radiation or resistance heating. A typical reactor configuration for TCVD, including gas delivery, substrate heating, and exhaust, is schematically illustrated in Fig. 19. | ||
| Fig. 19 Scheme of a typical reactor configuration for TCVD. Graphic adapted from Nnadozie et al.324 | ||
The industrial implementation of silicon chemical vapor deposition emerged in the early 1960s at Bell Telephone Laboratories, where Theuerer demonstrated controlled epitaxial growth of single-crystalline silicon via hydrogen reduction of SiCl4.325 This chlorosilane-based approach enabled precise control over film thickness, doping, and conductivity type, establishing CVD as a technologically viable route for device-grade epitaxial layers. However, these processes required elevated substrate temperatures typically exceeding 1100 °C and involved kinetically demanding Si–Cl bond cleavage with concomitant HCl elimination. These limitations motivated the exploration of hydrosilanes as thermodynamically more labile alternatives for thermal silicon deposition. Lewis and co-workers demonstrated that monosilane undergoes efficient thermal decomposition to silicon and hydrogen, enabling high-purity silicon deposition at temperatures substantially lower than those required for chlorosilane reduction.326 Subsequent kinetic studies by Joyce and Bradley revealed two distinct growth regimes during silane-based epitaxy: a transport-limited regime above ∼1100 °C and a reaction-controlled regime at lower temperatures. Together, these studies established silane as a kinetically tuneable and technologically viable precursor for thermal silicon CVD. Further experimental and kinetic studies have demonstrated that the initial step of silane pyrolysis is most likely the formation of highly reactive silylene species (:SiH2), accompanied by the release of molecular hydrogen as illustrated in Scheme 226. These intermediates readily insert into Si–H bonds of silanes, leading to the formation of higher polysilanes and initiating silicon hydride cluster growth. Subsequent dehydrogenation and rearrangement reactions generate unsaturated intermediates, including silenes and additional silylenes, which further propagate the reaction network. With increasing cluster size, intramolecular reactions can promote ring formation, yielding cyclic silicon hydrides that represent important intermediates in the early stages of silicon cluster and particle formation during silane pyrolysis. Depending on the reaction conditions, particularly temperature, pressure, and residence time, these species either contribute to heterogeneous film growth on heated surfaces or undergo homogeneous reactions leading to particle and powder formation in the gas phase. While the general mechanistic framework of silane pyrolysis is well established, the detailed reaction network remains highly complex and continues to be investigated through experimental and theoretical studies.327–329
 | ||
| Scheme 226 Simplified mechanism of silane pyrolysis illustrating the formation of higher silanes and cyclic silicon hydrides.327,328 | ||
Based on the operating pressure, TCVD can be further subdivided into atmospheric pressure CVD (APCVD), low-pressure CVD (LPCVD) and ultrahigh vacuum CVD (UHVCVD). In 1997, Sturm et al. demonstrated low-temperature LPCVD growth of β-SiC on Si using methylsilane as a single-source precursor, yielding polycrystalline, device-compatible SiC films.330 Two years later, Madaura et al. reported for the first time the APCVD of SiC films on a Si substrate using trimethylsilane as a single source precursor. Although trimethylsilane contains only a single Si–H bond and therefore falls outside the scope of classical hydrosilanes discussed in this review, this study is noteworthy as an early example of silicon deposition chemistry employing molecular silane precursors. The SEM microphotographs of the SiC/Si interface are shown in Fig. 20.331
 | ||
| Fig. 20 Microphotographs of the SiC/Si interface as seen by cross-sectional SEM adapted from Madapura et al.331 With permission from ECS – The Electrochemical Society, © 1999. | ||
Hazbun et al. investigated the use of tetrasilane (Si4H10) as a precursor for ultra-high-vacuum CVD of silicon epitaxial layers. They demonstrated that tetrasilane enables significantly higher growth rates than monosilane and allows crystalline silicon deposition at temperatures as low as 400 °C, making it suitable for low-temperature epitaxy.332 Furthermore, Byeon et al. investigated the epitaxial growth of Si and SiGe using high-order silanes such as disilane, trisilane, and tetrasilane under UHVCVD and LPCVD conditions without a carrier gas. They showed that higher-order silanes enable silicon epitaxy at reduced temperatures, with trisilane and tetrasilane providing significantly higher growth rates, while disilane yielded the highest crystal quality of the deposited films.333 Halogenated hydrosilanes were likewise employed as silicon precursors for both oxide and carbide thin films. In high-temperature thermal CVD, silicon tetrachloride in combination with propane enabled the growth of epitaxial SiC layers, where chlorine-containing species suppress gas-phase nucleation and improve stoichiometric control of Si:C ratios.334 In a related context, Sneh et al. reported atomic-layer controlled growth of SiO2 using a binary reaction sequence with SiCl4 and H2O.335 Nitrogen-functionalized hydrosilanes subsequently emerged as important precursors for the deposition of silicon nitride (Si3N4) thin films. Gumpher et al. demonstrated LPCVD of Si3N4 from di(t-butylamino)silane (BTBAS) and NH3 at 550–600 °C, producing high-quality nitride films suitable for microelectronic applications.336 The incorporation of preformed Si–N bonds in these precursors enhances surface reactivity while avoiding corrosive halide byproducts, thereby improving film purity and conformality in advanced deposition schemes.337
Silylphosphanes and related phosphinosilanes were explored primarily in patent literature as single-source Si–P precursors for in situ phosphorus doping during silicon CVD. Although conceptually attractive as potential alternatives to PH3, limited thermal robustness and the maturity of established PH3-based processes curtailed their broader implementation.338 Silylgermanes containing preformed Si–Ge bonds were systematically developed as molecular single-source precursors for SiGe thin-film growth. Kouvetakis and co-workers first established the complete hydride series (H3Ge)xSiH4−x (x = 1–4), including silylgermane H3GeSiH3 and tetrasilylgermane Ge(SiH3)4, and demonstrated their application in low-pressure deposition of epitaxial Si1−xGex layers at 300–450 °C. Thermal activation proceeds via Si–H and Ge–H bond cleavage with H2 elimination while preserving the intrinsic Si:Ge stoichiometry encoded in the precursor, enabling compositionally controlled alloy growth without separate silane and germane feeds. This molecular design strategy facilitates the preparation of a new class of Si-based semiconductors with Ge-rich stoichiometry. Fig. 21 shows a X-ray transmission electron microscopy (XTEM) image of a SiGe2 layer on a Si substrate.127
 | ||
| Fig. 21 XTEM micrograph of a SiGe2 layer grown on Si(100) adapted from Kouvetaksi et al.127 With permission from American Chemical Society, © 2005. | ||
More recently, Wagner and co-workers introduced mixed-substituted Si–Ge hydrides such as H3SiR2GeSiH3 (R = Ph, n-Bu), synthesized via Si2Cl6-mediated coupling and subsequent hydride formation, and demonstrated their conversion by low-pressure CVD into predominantly amorphous Si1−xGex coatings with predefined Si:Ge ratios and reduced pyrophoricity compared to purely hydridic systems.128
While thermal processes are widely used in industry, primarily due to the technical ease of implementation, they also exhibit clear disadvantages. In particular, temperature-sensitive substrates may be damaged by the high process temperature. For this reason, alternative energy sources have been developed over the decades of research.321,339,340
Plasma-enhanced chemical vapor deposition
PECVD, also known as glow-discharge chemical vapor deposition is a variation of CVD in which electrical energy is used to activate the chemical reaction. By applying a high frequency voltage between two electrodes at reduced pressure a glow discharge is developed in which a non-thermal plasma consisting of electrons, ions and electronically excited species is formed. Through electron impact within the plasma, the precursor gases are ionized and dissociated, thereby generating chemically active ions and radicals. As a result, the electron temperature can reach values of 2000 K or higher, while the gas temperature, depending on process conditions, remains near room temperature. The activated species can take part in heterogeneous reactions on or near the substrate surface, leading to the deposition of a thin film. Since the reactive species are predominantly formed in the gas phase, only minimal thermal activation is required. This enables PECVD processes to be carried out at significantly lower temperatures than TCVD, in many cases at temperatures below 200 °C. Consequently, depositions on temperature-sensitive substrates such as glass and polymeric materials becomes possible.321,339,341 The plasma-induced dissociation pathway of silane are summarized in the following Fig. 22. | ||
| Fig. 22 Schematic dissociation pathway of SiH4 and H2 in the plasma adapted from Kasap et al.313 With permission from Springer-Verlag US, © 2007. | ||
In 1965, Sterling and Swann342 were the first to report the deposition of an amorphous silicon layer from gaseous SiH4 using plasma-assisted CVD. In the early 1970s, a research group at Havard University demonstrated the incorporation of hydrogen into a-Si leading to the development of hydrogenated amorphous silicon (a-Si:H) with a significantly reduced defect density and improved electronical properties.343 Building on these findings, Spear and LeComber344 first demonstrated the controlled n- and p-doping of a-Si:H by adding phosphine and diborane to SiH4 in a reactor with a plasma at radio frequency voltages. This enabled precise control of the electrical conductivity over several orders of magnitude and paved the way for the realization of a-SiH-based devices, in particular thin-film solar cells and TFTs.345 As a result of these advances, PECVD became the standard procedure for the deposition of silicon thin films in the 1980s. In 1983, Matsuda et al. deposited amorphous a-SiC:H films from SiH4/CH4 mixtures at substrate temperatures of 300–400 °C, producing films suitable for microelectronic and optical applications.346 Pokhodnya et al. studied the intrinsic stress in a-Si:H films using cyclohexasilane (CHS) as a precursor.347 Lee et al. reported the deposition of trisilylamine using PECVD forming highly conformal SiCN films. In Fig. 23 a transmission electron microscopy (TEM) cross-sectional view of the substrate with the deposited layer is shown.348
 | ||
| Fig. 23 TEM cross-sectional view of the substrate with the deposited SiCN layer adapted from Lee et al.348 With permission from Elsevier B.V., © 2018. | ||
In the following decades, research efforts focused on optimizing the PECVD process with respect to material quality, increased deposition rates and long-term stability. New approaches such as very-high-frequency PECVD (VHF-PECVD),349 microwave-assisted CVD (MW-CVD)350 and “high power high pressure” regime RF-PECVD351 were developed. Today RF-PECVD at a excitation frequency of 13.56 MHz remains the dominating deposition technique, notwithstanding its relatively low deposition rate of 1–2 Å s−1.352
The following Fig. 24 shows a schematic illustration of a PECVD reactor.
 | ||
| Fig. 24 Schematic illustration of a plasma-enhanced chemical vapor deposition (PECVD) reactor. Graphic adapted from Verma et al.353 and altered with the help of AI. With permission from Springer Nature Singapore Pte Ltd., © 2023. | ||
Photo-assisted chemical vapor deposition
Photo-assisted chemical vapor deposition (PACVD), also known as photo-initiated CVD, is a variant of CVD in which photon energy, typically in the ultraviolet (UV) or vacuum-ultraviolet (VUV) range, is used to activate chemical reactions. Reaction initiation occurs via photolytic dissociation of the precursor molecules, leading to the formation of reactive radicals and electronically excited species, which subsequently participate in heterogeneous surface reactions resulting in thin film growth.220,354 In 1983, Migitaka et al. and Takahashi et al. reported the preparation of a-Si:H films from monosilane355 and disilane.356 A schematic illustration of a PACVD reactor can be found in the article of Dorval Dion et al.354A key advantage of PACVD is the absence of plasma, which eliminates ion-induced damage to both the substrate and the growing film. Furthermore, in contrast to TCVD, which relies on pyrolytic mechanisms and requires high substrate temperatures, PACVD enables deposition at significantly lower temperatures, in some cases at room temperature. As a result, the choice of substrate material is not restricted by thermal stability. PACVD also has limitations, including the requirement that precursor molecules exhibit sufficient absorption within the emission spectrum of the light source, as well as relatively low deposition rates.339,354
PACVD is often discussed together with laser-assisted CVD (LACVD), also known as laser chemical CVD, and the two processes are not always strictly distinguished terminologically in literature, as both rely on photonic activation. The crucial difference, however, lies in the method of energy input. In contrast to PACVD, LACVD uses coherent, high-intensity laser radiation. LACVD can generate very high local energy densities and induce both photolytic and locally pyrolytic processes, for example, through targeted heating of the substrate surface. Laser-CVD is therefore particularly suitable for locally confined deposition or structuring processes.220,339,357 A typical schematic of the LACVD process is shown in Fig. 25.
 | ||
| Fig. 25 Schematic illustration of a LACVD reactor. With permission from the Institute of Optics and Electronics, Chinese Academy of Science358 © 2022. | ||
Hot-wire chemical vapor deposition
Hot-wire chemical vapor deposition (HWCVD), also known as catalytic CVD (Cat-CVD), is a plasma-free CVD process in which the decomposition of precursor gases occurs on a highly heated metal filament. Tungsten or tantalum are typically used as the filament material, which can be heated to temperatures of approximately 1400 to 2100 °C. At the surface of the hot wire, gaseous silane can be efficiently dissociated, with an almost complete separation into its atomic components. The reactive species generated subsequently undergo further reactions with SiH4 in the gas phase, forming various secondary precursors that diffuse to the substrate surface and contribute to thin-film deposition.359,360 The following Fig. 26 gives a schematic view of a HWCVD reactor. | ||
| Fig. 26 Illustration of a schematic HWCVD reactor adapted from Schropp.360 With permission from ECS – The Electrochemical Society, © 2009. | ||
A characteristic feature of HWCVD is the spatial separation of precursor decomposition and layer growth. Since the generation of reactive species occurs exclusively on the filament, the process is often referred to as a remote decomposition method. The temperatures of the filament and substrate can be adjusted independently, enabling deposition at comparatively low substrate temperatures while simultaneously achieving high deposition rates.
The basic principle of HWCVD was first published and patented in 1979 by Wiesmann et al. under the name thermal vapor deposition.361 In the mid-1980s, Matsumura et al. first demonstrated the deposition of amorphous silicon using this method and introduced the term catalytic CVD.362 Independently, Doyle et al. described a comparable concept under the name evaporative surface decomposition (ESD).363
A significant milestone was achieved in 1991 with the work of Mahan et al., who demonstrated that amorphous silicon layers deposited using HWCVD can exhibit higher material quality than comparable films produced by PECVD.364 The high quality of the deposited layers is attributed to the efficient decomposition of the precursor gases at filament temperatures above approximately 1500 °C. Typical deposition rates for amorphous silicon range from 10 to 50 Å s−1, significantly exceeding those of conventional RF-PECVD processes.
Due to the absence of a plasma, ion-induced structural damage to the growing layer, that can be caused by ion bombardment or high-energy radiation, is largely avoided. The term Cat-CVD refers to the catalytic effect of the hot wire in the dissociation of the precursor gases, although the filament is not an ideal catalyst in the strict chemical sense, as it undergoes aging and erosion during the process.365 In the literature, the term HWCVD is predominantly used in physical and photovoltaic contexts, while Cat-CVD is particularly common in chemical research and Japanese literature. Current research is investigating, among other things, the application of HWCVD for the production of TFTs and thin-film solar cells.360
Atomic layer deposition
Atomic layer deposition is a thin film deposition technique, that can be classified as a special modification of CVD. In the literature, it is also referred to as atomic layer epitaxy or Atomic Layer Chemical Vapor Deposition (ALCVD). A key characteristic of ALD is the deposition of inorganic thin films with thicknesses down to a range of single monolayers, which is enabled by sequential, self-limiting surface reactions. As a result, highly homogeneous and conformal layers with excellent thickness control can be deposited.131,366In ALD, gaseous precursors are introduced into the reactor in a strictly sequential manner. Each precursor reacts exclusively with the available reactive sites on the substrate surface until a saturated adsorption level is reached. The reactor is purged with inert gas between the individual precursor pulses to remove excess precursor molecules and volatile byproducts. This procedure effectively suppresses gas-phase reactions, such that the film growth is governed almost entirely by surface reactions.133,367 The scheme of a sequential ALD cycle is shown in Fig. 27. Hirose et al. demonstrated the use of monosilane for room temperature ALD.368 Matsunami et al. established epitaxial growth of β-SiC (3C-SiC) on Si substrates by reacting SiH4 with hydrocarbons such as propane at temperatures above 1000 °C, enabling crystalline SiC layers for electronic applications and demonstrating in situ doping with nitrogen donors and aluminium or boron acceptors.369 Halogenated hydrosilanes were also employed for SiC epitaxy: Pedersen et al. employed methyltrichlorosilane in a hot-wall CVD reactor to obtain high-quality 4H-SiC layers.370 Additional chlorinated silanes, including trichlorosilane, dichlorosilane, and hexachlorodisilane (Si2Cl6), have likewise been utilized for high-quality SiC epitaxial growth.371
 | ||
| Fig. 27 Scheme of a sequential ALD cycle adapted from George.131 With permission from American Chemical Society, © 2009. | ||
Nitrogen-functionalized hydrosilanes became important precursors for the deposition of silicon dioxide (SiO2) thin films. Bis(dimethylamino)silane (BDMAS) was introduced for atomic layer deposition of SiO2 with H2O337 or O3372 as oxidants, where ligand-exchange reactions with surface OH groups followed by ozone or oxygen plasma oxidation yield SiO2.372 Furthermore Faraz et al. reported the deposition of silicon nitride layer through plasma-enhanced atomic layer deposition of di(sec-butylamino)silane (DSBAS) on planar and on high aspect ratio 3D trench nanostructures. Fig. 28 shows a TEM image of the layer as-deposited and post wet-etch with dilute hydrofluoric acid.373
 | ||
| Fig. 28 TEM image of the silicon nitride layer as-deposited (left) and post wet-etch with dilute hydrofluoric acid (right) adapted from Faraz et al.373 With permission from American Chemical Society, © 2017. | ||
In contrast to hydrosilanes, oxygen-functionalized silicon precursors such as tetraethyl orthosilicate (TEOS, Si(OEt)4) are widely used for the deposition of SiO2 thin films in LPCVD, PECVD, and sub-atmospheric CVD processes.374
Liquid phase deposition
A viable alternative to established vapour-based deposition techniques is solution-based processing of silicon, commonly referred to as liquid phase deposition. In contrast to CVD methods, which require pyrophoric precursor gases, elaborate vacuum or plasma infrastructure, and often elevated deposition temperatures, LPD can generally be carried out at lower temperatures and under atmospheric pressure. This reduces equipment complexity as well as operating costs. Furthermore, LPD is compatible with flexible, large-area, and temperature-sensitive substrates and can enable higher throughput, particularly when combined with coating or printing techniques. Such approaches also facilitate direct patterning of materials and may reduce the number of required processing steps. Consequently, LPD is considered a promising route for applications in printable electronics, including large-area flexible displays, thin-film solar cells, TFTs and silicon-based anode materials in lithium-ion batteries. The following section discusses the underlying chemical mechanisms of hydrosilane-based LPD and summarizes the key contributions in this field.19,375,376The conceptual foundations of hydrosilane-based liquid processing date back to the late 1980s. West and Wolff377 as well as Miller and Michl378 demonstrated that organopolysilanes can be deposited as thin films and subsequently converted into silicon-rich amorphous networks by thermal or photochemical treatment, thereby establishing the processing and photochemistry toolkit for later hydrosilanes routes. However, true hydrosilanes themselves were only mentioned in passing in these studies, and a dedicated liquid-phase deposition strategy based explicitly on hydrosilane precursors had not been established. A decisive step toward practical implementation was achieved by Shimoda et al. in 2006,19 who established cy-Si5H10 as a molecular precursor for solution-processed silicon films. Cy-Si5H10 was selected due to its favourable properties: it exhibits comparatively high stability and good availability making it suitable for handling and processing under inert conditions, and also exhibiting a high photoreactivity upon irradiation with ultraviolet light. In Shimodas LPD approach cy-Si5H10 is first irradiated with UV light with a wavelength of 365 nm leading to a ring-opening polymerization and the formation of polydihydrosilane. The resulting polyhydrosilane can be dissolved in organic solvents to yield a processable liquid silicon material, often referred to as “silicon ink”. The silicon ink is deposited by spin-coating on a substrate forming a uniform precursor. Subsequently thermal treatment induces the conversion of the polysilane network into a a-Si film. During initial heating, volatile components such as the solvent and cy-Si5H10 evaporate. At a temperature below approximately 280 °C cleavage of Si–Si bonds begins, leading to a release of SiH2 and SiH3 species. Above a temperature of around 300 °C the Si–H bonds break resulting in the formation of the amorphous silicon film.19,376,379–381 Fig. 29 illustrates the colour changes associated with variations in the chemical composition of the thin film at different annealing temperatures.
 | ||
| Fig. 29 Photographic image of the solution-processed polysilane films coated on quartz glass adapted from Trifunovic et al.381 With permission from American Institute of Physics, © 2015. | ||
The material can subsequently be further processed by additional thermal treatment or laser annealing to obtain polycrystalline silicon. Fig. 30 depicts a scheme for the solution-based formation of amorphous and polycrystalline silicon films.
 | ||
| Fig. 30 Scheme for the solution-based formation of amorphous and polycrystalline silicon films adapted from Gerwig et al.5 With permission from Chemistry – A European Journal published by Wiley-VCH GmbH, © 2024. | ||
In comparison to a-Si thin films formed through PECVD, the LPD produced films show a significantly lower mobility which is connected to a low concentration of hydrogen atoms. Films processed at temperatures below 300 °C exhibit charge carrier mobilities comparable to those obtained by PECVD. However, under these conditions the material can no longer be unambiguously classified as amorphous silicon and shows increased susceptibility to oxidation. Shimoda et al. therefore emphasizes the necessity of balancing low processing temperatures with sufficient structural stabilization and oxidation resistance in order to achieve device-relevant film quality.380
Beyond the preparation of intrinsic silicon inks, Shimoda and co-workers also developed n-type and p-type silicon inks by incorporating suitable dopant precursors, such as white phosphorus and decaborane, into the cy-Si5H10-derived system prior to film deposition. During subsequent thermal conversion, the dopant species are incorporated into the forming silicon network. These developments were later summarized under the term “liquid silicon family materials” (LSFMs), describing a materials platform in which cy-Si5H10 acts as a common precursor for different silicon-based functional layers. In this concept, intrinsic, n-type, and p-type silicon films as well as device structures such as TFTs and solar cells can be fabricated using entirely liquid-based processing routes.
Subsequent studies expanded the precursor chemistry, the available activation strategies as well as the deposition methods for hydrosilane-based LPD. Following Shimoda's initial work, cy-Si5H10 remained the dominant hydrosilane precursor, and numerous studies focused on refining its processing and activation. In contrast to the spin-coated polysilane ink approach described by Shimoda, Frey et al. employed an aerosol-assisted spray deposition technique using neat cy-Si5H10 as precursor. The cy-Si5H10 was atomized and deposited onto heated substrates under inert atmosphere, while ultraviolet irradiation was applied within the deposition chamber to promote precursor activation. Film formation proceeded primarily via thermally assisted decomposition of the liquid precursor during and after deposition, followed by high-temperature annealing (700–950 °C) to obtain polycrystalline silicon.382 Trifunovic et al. employed doctor-blade coating of cy-Si5H10, followed by ultraviolet curing at 365 nm and subsequent excimer laser at crystallization at 308 nm to obtain poly-Si at a maximum process temperature of 150 °C. In this approach, the solution-deposited precursor was directly transformed into crystalline silicon without requiring intermediate high-temperature annealing above 350 °C or a separate amorphous silicon conversion step. Therefore, this process is suitable for deposition on temperature-sensitive substrates like paper or polymer and facilitating low-cost electronics. Fig. 31 shows a poly Si film fabricated on paper.381
 | ||
| Fig. 31 Poly Si film fabricated on paper adapted from Trifunovic et al.381 With permission from American Institute of Physics, © 2015. | ||
In parallel, alternative activation methods for cy-Si5H10 were investigated. Cádiz Bedini et al. reported on ring-opening polymerisation induced by ultrasonic irradiation at a frequency of 26 kHz. The temperature was maintained below 75 °C, and it was confirmed, that no thermal activation of the precursor occurred under these conditions.
Masuda et al. demonstrated that cy-Si5H10-derived liquid silicon can be employed for solution-based silicon patterning using both inkjet printing and nanoimprinting strategies. In both approaches, cy-Si5H10 was first photochemically polymerised at 365 nm to yield a processable polysilane precursor. In the inkjet printing approach, the polysilane solution was deposited in a digitally defined, maskless manner onto the substrate. Subsequent thermal conversion at approximately 380–400 °C yielded hydrogenated amorphous silicon, while additional annealing at 1000 °C enabled crystallisation to polycrystalline silicon. The method further allowed the incorporation of p- and n-type dopants directly into the printed structures. Fig. 32 shows a printed silicon pattern on a glass substrate.383
 | ||
| Fig. 32 Inkjet printed silicon pattern on a glass substrate adapted from Masuda et al.383 With permission from Wiley-VCH GmbH, © 2020. | ||
In contrast, the nanoimprinting approach consisted of spin-coating the cy-Si5H10-derived Liq-Si precursor onto the substrate. The deposited film was subsequently pre-cured at 140–220 °C, which initiated partial crosslinking of the polysilane network. Successful pattern replication was achieved only within this temperature window under an applied pressure of 10 MPa. At lower temperatures, insufficient crosslinking prevented stable feature transfer, whereas at higher temperatures the film lost its deformability. Crosslinking proceeds via thermally activated 1,2-hydrogen shift reactions, accompanied by the release of SiH4 and H2, leading to progressive solidification of the film. After imprinting and controlled cooling, post-annealing at approximately 380 °C completed the conversion to amorphous silicon. Despite significant volumetric shrinkage during polymer-to-silicon transformation, nanoscale patterns were retained. Fig. 33 shows a nanoimprinted pattern of a-Si.384
 | ||
| Fig. 33 Nanoimprinted pattern of a-Si adapted from Masuda et al.384 With permission from American Chemical Society, © 2016. | ||
These studies demonstrate that cy-Si5H10-derived polysilanes are suitable precursors for lithography-free micro- and nanostructured silicon fabrication.
Gerwig et al. investigated the oligomerisation of cy-Si5H10 using electron spin resonance (ESR), NMR, and Raman spectroscopy supported by DFT and molecular dynamics simulations. Their results showed that cy-Si5H10 does not undergo a classical ring-opening polymerisation. Instead, short-lived silyl radicals are formed, which recombine to generate highly branched hydrooligosilanes. Silylene intermediates were found not to play a significant role. Competing decomposition and disproportionation reactions produce volatile fragments, reducing hydrogen content and overall yield. ESR measurements confirmed the presence of tertiary silyl radicals as reactive intermediates.5
The influence of solvent and processing parameters on film formation was further examined. Optimised spin-coating and UV irradiation conditions yielded homogeneous silicon films, which were subsequently passivated by hydrogen plasma. The resulting thin films and TFT devices exhibited performance comparable to PECVD-processed materials.
Shen et al. introduced a liquid-source vapor deposition (LVD), where cy-Si5H10 thermally decomposes between two parallel hot substrates at atmospheric pressure. In this configuration, a fraction of the precursor is directly converted into amorphous silicon on the substrate surface, while volatile silicon hydride radicals are simultaneously generated. These reactive intermediates diffuse within the confined deposition space and act as secondary growth species, thereby sustaining further film formation. This allows for the formation of denser amorphous silicon layers with reduced oxygen incorporation compared to conventional spin-coated films. The schematic set-up for LVD is shown in Fig. 34.385
 | ||
| Fig. 34 Schematic set-up of the LVD process with cy-Si5H10 as the precursor adapted from Shen et al.385 Published by Royal Society of Chemistry under CC-BY 4.0, © 2015. | ||
However, the demanding synthesis and purification of cy-Si5H10 highlight the need to consider alternative precursors such as cyclohexasilane (CHS) and NPS.5,376 Iyer et al. investigated CHS as an alternative cyclic hydrosilane precursor. The CHS solution was spin-coated onto the substrate under simultaneous UV irradiation to induce ring-opening polymerisation and network formation. Subsequent thermal annealing and excimer laser treatment enabled the transformation of the amorphous intermediate into crystalline silicon. Furthermore, hydrogen plasma exposure was applied to passivate residual defect states and improve the structural and electronic quality of the films.386 The following Fig. 35 shows SEM images of the surface of the thin film after different points in the formation process.
 | ||
| Fig. 35 SEM images of the thin film surface of (a) the amorphous silicon film, (b) the thermally treated silicon film, (c) the laser treated silicon film and (d) the laser treated silicon film at higher magnification adapted from Iyer et al.386 With permission from American Chemical Society, © 2012. | ||
Cadiz Bedini et al. demonstrated the preparation of silicon ink from trisilane using ultrasonic irradiation. After subsequent UV irradiation, they used the resulting oligomeric mixture deposit a a-Si–H thin film via spin coating. Furthermore, the authors also reported on the deposition of Si nanoparticles from the sonicated silicon ink.387
Bronger et al. were the first to introduce neopentasilane in solution-based silicon processing. Using spin-coating and slot-die coating as deposition techniques, they showed that the branched molecular structure and increased Si–Si bond content of NPS significantly influence the thin film formation. Compared to cy-Si5H10, NPS exhibits improved solubility, which results in enhanced film homogeneity and uniformity. In addition, they reported an improved p-type doping efficiency for NPS-derived films relative to cy-Si5H10. The electronic performance approached that of comparable PECVD-deposited materials.319 The working group of Haase executed further research to determine the usability of liquid silicon ink based on NPS for local n-and p-doping of solar cells.388
Further development explored hydrosilanes beyond simple cyclic or small branched molecules. Haas et al. introduced highly branched perhydrosilanes, namely 2,2,3,3-tetrasilyltetrasilane and 2,2,3,3,4,4-hexasilylpentasilane, which show a reduced pyrophoric character and a bathochromically shifted UV-absorption. After photolysis the obtained oligomeric polyhydrosilane mixture was spin-coated on glass substrates and thermally treated at 500 °C resulting in a homogeneous a-Si:H layer. Fig. 36 shows a photographic image and an optical micrograph of the solution processed a-Si:H film.21
 | ||
| Fig. 36 Photographic image (B) and optical micrograph (C) of the solution processed a-Si: film adapted from Haas et al.21 With permission from Wiley-VCH, © 2017. | ||
In this context, Christopoulos et al. reported the synthesis and structural characterisation of highly branched hydrogenated nonasilanes and decasilanes. Although no thin-film deposition experiments were performed, the study provided detailed information on molecular structure, bonding motifs, and stability of higher nuclearity hydrosilanes. Compared to smaller cyclic precursors, these compounds exhibit modified volatility and decomposition behaviour due to their increased silicon content and branching degree. The work therefore contributes to the understanding of structure–property relationships in hydrosilanes and identifies potential candidates for future liquid-phase silicon deposition studies.49
The increasing focus on precursor structure is also reflected in patent literature. Several patents disclose synthetic routes toward branched and doped hydridosilanes intended for thin-film deposition applications. These developments address precursor stability, controlled oligomerisation, and reduced pyrophoric character, highlighting the technological relevance of structure-tailored hydrosilanes for liquid-phase silicon processing.389 More recently, precursor-engineered strategies have focused on single-source hydrosilane-based precursors that contain one or more heteroatoms covalently linked to silicon to enable controlled Si–C network formation. Matsuda and co-workers developed a polymeric polydihydrosilane precursor with pendant hexyl groups (PSH), which was deposited by solution-based methods and subsequently pyrolyzed to form an a-SiC coating. Fig. 37 shows a photographic image of PSH films coated on glass substrates and pyrolyzed at different temperatures.390
 | ||
| Fig. 37 Photographic image of PSH films coated on glass substrates and pyrolyzed at different temperatures adapted from Masuda et al.390 With permission from Royal Society of Chemistry, © 2015. | ||
In a related molecular design approach, Haas and co-workers reported the systematic functionalization of higher silicon hydrides with carbon-containing substituents and demonstrated their application in LPD, where spin-coated precursor films were thermally converted into homogeneous Si/C thin films. Fig. 38 exemplary shows a picture and a light microscopy image of a thin layer formed from the precursor 1,1,1-trimethyl-2,2-disilyltrisilane.22
 | ||
| Fig. 38 A picture and a light microscopy image of a thin layer formed from the precursor 1,1,1-trimethyl-2,2-disilyltrisilane on glass adapted from Haas et al.22 Published by American Chemical Society under CC-BY 4.0, © 2023. | ||
Structure–property–application relationships
Across CVD and LPD, recent work clarifies how structure–property–application relationships are essential to fully exploit the potential of hydrosilanes in modern materials science. The molecular structure of hydrosilanes governs their activation routes, processing windows, and ultimately film properties. In CVD, high volatility and clean gas-phase decomposition are structure-dependent parameters. Small, volatile hydrides such as SiH4 or Si2H6 dissociate cleanly under plasma, hot-wire, or photon activation and remain advantageous for transport-limited growth formation of high-purity films. This includes hydrosilanes, where the incorporation of a Si–heteroatom bond can enhance surface reactivity, thereby improving the purity and conformality of a film. These attributes are particularly critical, as reduced impurity incorporation minimizes defect-mediated recombination, while improved conformality ensures uniform coverage and consistent electronic properties across complex device architectures. Consequently, both factors directly contribute to enhanced device performance and reliability in both microelectronics and thin-film solar cells.In LPD, the balance shifts toward solubility, manageable reactivity, and photochemical responsiveness: higher and especially branched hydrosilanes exhibit superior solubility, reduced pyrophoricity, and bathochromically shifted UV absorption, enabling efficient photochemical activation and yielding homogeneous a-Si:H films. These features promote homogeneous film formation and improved control over hydrogen incorporation and network structure in a-Si:H materials. For example, branched hydrosilanes support more uniform coating behavior and facilitate dopant incorporation, while cyclic precursors follow distinct radical-mediated pathways that yield highly branched intermediates, influencing film densification and defect evolution. Thus, precursor structure directly governs the resulting microstructure and optoelectronic properties of the deposited films.
Across both deposition regimes, molecular functionalization (e.g., preformed Si–N, Si–C, or Si–Ge bonds) provides a direct handle to encode composition and doping at the molecular level, linking precursor design directly to device-relevant properties. In the context of thin-film solar cells, such tailored precursors can influence key properties such as crystallinity, electronic structure, and impurity incorporation, thereby directly affecting device performance. These examples clearly demonstrate that the rational design of hydrosilane precursors-linking molecular structure to decomposition behavior and ultimately to material properties-represents a central strategy for advancing silicon-based technologies encoding film stoichiometry and network chemistry at the molecular level.
Conclusions
Over more than a century of research, the chemistry of hydrosilane has evolved from the early isolation of simple silanes to a sophisticated field spanning synthetic inorganic chemistry, materials science, and semiconductor technology. The development of reliable synthetic methodologies-from classical silicide hydrolysis to chlorosilane reduction, catalytic coupling reactions, and silanide-based strategies-has enabled access to a broad spectrum of linear, branched, cyclic, and cluster-type hydrosilanes. These advances have revealed the remarkable structural diversity of silicon hydrides and highlighted the different reactivity of Si–H and Si–Si bonds compared to their carbon analogues.A central theme emerging from recent research is the pivotal role of silanide intermediates and Si–H bond activation processes in hydrosilane functionalization. Alkali-metal silanides and related species provide powerful synthetic entry points to a wide range of silicon-element bonds, enabling the preparation of silyl derivatives with elements across the periodic table. In parallel, dehydrogenative coupling reactions have emerged as a particularly powerful methodology for the controlled construction of Si–Si bonds, allowing the transformation of simple monosilanes into higher oligohydrosilanes and polysilane frameworks through catalytic hydrogen elimination. Together with advances in silicon cluster chemistry, including the synthesis of silafullerenes and higher branched hydrosilanes, these developments have significantly expanded the conceptual and structural landscape of molecular silicon hydride chemistry.
Beyond their fundamental chemical interest, hydrosilanes have become indispensable precursors for the preparation of silicon-containing materials and thin films. Chemical vapour deposition represents the most widely employed industrial technique for silicon film formation, where simple hydrosilanes such as monosilane and disilane serve as key feedstocks for the growth of polycrystalline, amorphous, or epitaxial silicon layers used in semiconductor devices and photovoltaic technologies. The controlled thermal or plasma-assisted decomposition of these molecules enables the deposition of high-purity silicon films with precisely tuneable properties. Building on this established technology, recent research has increasingly explored molecularly defined higher hydrosilanes and functionalized derivatives as advanced precursors. In particular, solution-based approaches such as liquid-phase deposition has emerged as attractive complementary strategy that offers advantages in terms of processability, scalability, and compatibility with emerging device architectures.
Outlook and challenges
The chemistry of hydrosilanes has evolved from a largely academic field into a key enabling technology for semiconductor processing and silicon-based materials. As highlighted throughout this review, hydrosilanes represent highly versatile molecular precursors that bridge fundamental silicon chemistry with industrially relevant applications such as chemical vapor deposition (CVD) and liquid phase deposition (LPD).Building on this progress, future developments are expected to focus on improving synthetic accessibility, scalability, and the targeted functionalization of hydrosilane derivatives.
From an application perspective, hydrosilanes and especially higher oligomeric species offer significant potential as tailored single-source precursors for advanced materials. Their tunable reactivity and decomposition behavior enable precise control over film growth, composition, and doping in semiconductor fabrication. In particular, the development of functionalized hydrosilanes that incorporate heteroatoms directly into the molecular backbone represents a promising strategy toward cleaner and more efficient deposition processes, avoiding the need for co-feeding multiple gaseous precursors.
However, several key challenges currently limit the broader industrial implementation of these compounds. A major bottleneck is the economic feasibility, as the high cost of polysilanes has so far prevented their widespread use in large-scale applications such as solar cell manufacturing. Despite their attractive properties as solution-processable silicon precursors, their synthesis often involves multi-step procedures, low overall yields, and demanding purification protocols, all of which significantly increase production costs.
In addition, synthetic limitations remain a critical issue. For many technologically relevant doping elements, no scalable and robust synthetic routes to suitable hydrosilane-based precursors are currently available. While initial approaches toward Si–B, Si–P, or other heteroatom-functionalized systems have been reported, these methods often suffer from limited selectivity, stability issues, or poor scalability.
This lack of general and efficient synthetic strategies restricts the systematic exploration of doped silicon materials derived from hydrosilanes.
Another challenge lies in the control of reactivity and stability. The intrinsic reactivity of Si–H and Si–Si bonds, while advantageous for materials synthesis, can lead to undesired side reactions such as polymerization or decomposition, complicating handling and processing. This is particularly relevant for higher hydrosilanes, where competing reaction pathways often result in complex product mixtures and reduced selectivity.
Looking forward, addressing these challenges will require integrated efforts in synthesis, mechanistic understanding, and process development. Advances in catalytic methodologies, improved precursor design, and the development of scalable production routes will be essential to unlock the full potential of hydrosilanes in industrial applications. In this context, the continued interplay between fundamental research and applied process engineering will be crucial for translating laboratory-scale discoveries into viable technologies.
Author contributions
D. G., M. Hx., M. P. and P. V.: were responsible for visualization, data presentation and writing original draft (lead). M. H. was in charge for methodology and conceptualization, review and editing of the manuscript (lead), project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
This article is a review and does not report any original data. Therefore, no datasets were generated or analyzed during the current study.Acknowledgements
The financial support by the Austrian Federal Ministry of Labour and Economy, the National Foundation for Research, Technology and Development and the Christian Doppler Research Association is gratefully acknowledged. The authors acknowledge the use of artificial intelligence tools (OpenAi (GPT 5)) for language polishing of the manuscript.References
- (a) E. Hengge, H. Keller-Rudek, D. Koschel, U. Krüerke and P. Merlet, Gmelin Handbook of Inorganic Chemistry. Silicon, Supplement Vol. B1, Springer, Berlin, Heidelberg, 1982 Search PubMed; (b) J. H. Lorenz, Survey of the preparation, purity, and availability of silanes, Solar Energy Research Inst. (SERI), Golden, CO (United States) SERI/STR-211-2092, 1983 CrossRef PubMed.
- M. Akhtar, Preparation of Ultra-High Purity Higher Silanes and Germanes, Synth. React. Inorg. Met.-Org. Chem., 1986, 16, 729 CrossRef CAS.
- Encyclopedia of chemical technology. - 22: Silicon compounds to succinic acid and succinic anhydride: Silanes, ed. R. E. Kirk, D. F. Othmer, J. I. Kroschwitz, M. Howe-Grant and B. Arkles, Wiley, New York, N.Y., 1997 Search PubMed.
- J. Baumgartner and C. Grogger, in Comprehensive inorganic chemistry II. From elements to applications, ed. J. Reedijk, Elsevier, Amsterdam, 2nd edn, 2013, pp. 51–82 Search PubMed.
- M. Gerwig, U. Böhme and M. Friebel, Challenges in the Synthesis and Processing of Hydrosilanes as Precursors for Silicon Deposition, Chem. – Eur. J., 2024, 30, e202400013 CrossRef CAS PubMed.
- T. Wiesner and M. Haas, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2024 Search PubMed.
- E. Rivard, Group 14 inorganic hydrocarbon analogues, Chem. Soc. Rev., 2016, 45, 989 RSC.
- H. Buff and F. Wöhler, Ueber neue Verbindungen des Siliciums, Arch. Pharm., 1857, 142, 284 CrossRef.
- H. Moissan and S. Smiles, New Investigations of Liquid Silicon Hydride Si2H6, C. R. Chim., 1902, 569 Search PubMed.
- A. Stock and C. Somieski, Siliciumwasserstoffe VI.: Chlorierung und Methylierung des Monosilans, Ber. Dtsch. Chem. Ges. A/B, 1919, 52, 695 CrossRef.
- A. Stock and K. Somieski, Siliciumwasserstoffe, X.: Stickstoffhaltige Verbindungen, Ber. Dtsch. Chem. Ges. A/B, 1921, 54, 740 CrossRef.
- G. Fritz, Notizen: Neue Wasserstoffverbindungen des Siliciums und des Phosphors. Das Silylphosphin SiH3-PH2, Z. Naturforsch., B, 1953, 8, 776 CrossRef.
- B. J. Aylett, H. J. Emeléus and A. G. Maddock, Phosphine and arsine derivatives of monosilane, J. Inorg. Nucl. Chem., 1955, 1, 187 CrossRef CAS.
- M. Baudler, Franz Fehér (1903–1991), Eur. J. Inorg. Chem., 1998, 1998, 2089 CrossRef.
- M. A. Ring and D. M. Ritter, Preparation and Reactions of Potassium Silyl 1, J. Am. Chem. Soc., 1961, 83, 802 CrossRef CAS.
- E. Amberger and H. Boeters, The Preparation of Trisilylphosphine, Angew. Chem., Int. Ed. Engl., 1962, 1, 52 CrossRef.
- E. Hengge and G. Bauer, Cyclopentasilane, the First Unsubstituted Cyclic Silicon Hydride, Angew. Chem., Int. Ed. Engl., 1973, 12, 316 CrossRef.
- T. Lobreyer, H. Oberhammer and W. Sundermeyer, Synthesis and Structure of Tetrasilylgermane, Ge(SiH3)4 and Other Silylgermanes, Angew. Chem., Int. Ed. Engl., 1993, 32, 586 CrossRef.
- T. Shimoda, Y. Matsuki, M. Furusawa, T. Aoki, I. Yudasaka, H. Tanaka, H. Iwasawa, D. Wang, M. Miyasaka and Y. Takeuchi, Solution-processed silicon films and transistors, Nature, 2006, 440, 783 CrossRef CAS PubMed.
- J. Tillmann, J. H. Wender, U. Bahr, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, One-step synthesis of a 20silafullerane with an endohedral chloride ion, Angew. Chem., Int. Ed., 2015, 54, 5429 CrossRef CAS PubMed.
- M. Haas, V. Christopoulos, J. Radebner, M. Holthausen, T. Lainer, L. Schuh, H. Fitzek, G. Kothleitner, A. Torvisco, R. Fischer, O. Wunnicke and H. Stueger, Branched Hydrosilane Oligomers as Ideal Precursors for Liquid-Based Silicon-Film Deposition, Angew. Chem., Int. Ed., 2017, 56, 14071 CrossRef CAS PubMed.
- A. Sauermoser, T. Lainer, A. Knoechl, F. Goni, R. C. Fischer, H. Fitzek, M. Dienstleder, C. Prietl, A.-M. Kelterer, C. Bandl, G. Jakopic, G. Kothleitner and M. Haas, Fabrication of Amorphous Silicon-Carbon Hybrid Films Using Single-Source Precursors, Inorg. Chem., 2023, 62, 15490 CrossRef CAS PubMed.
- A. Stock and C. Somieski, Siliciumwasserstoffe. I. Die aus Magnesiumsilicid und Säuren entstehenden Siliciumwasserstoffe, Ber. Dtsch. Chem. Ges., 1916, 49, 111 CrossRef CAS.
- A. Stock, P. Stiebeler and F. Zeidler, Siliciumwasserstoffe, XVI.: Die höheren Siliciumhydride, Ber. Dtsch. Chem. Ges. A/B, 1923, 56, 1695 CrossRef.
- A. Stock, Siliciumwasserstoffe, Z. Elektrochem. Angew. Phys. Chem., 1926, 32, 341 CrossRef CAS.
- (a) F. Fehér, G. Kuhlbörsch and H. Luhleich, Beiträge zur Chemie des Siliciums und Germaniums. IV. Über die Drastellung des Rohsilas, Z. Anorg. Allg. Chem., 1960, 303, 283 CrossRef; (b) F. Fehér and H. Strack, Die gaschromatographische Trennung von Siliciumwasserstoffen an Squalan-Kieselgur-Trennsulen, Naturwissenschaften, 1963, 50, 570 CrossRef; (c) F. Fehér, D. Schinkitz and J. Schaaf, Ein Verfahren zur Darstellung höherer Silane, Z. Anorg. Allg. Chem., 1971, 383, 303 CrossRef; (d) F. Fehér, H. Baier, B. Enders, M. Krancher, J. Laakmann, F. J. Ocklenburg and D. Skrodski, Beiträge zur Chemie des Siliciums und des Germaniums. XXXVII. Weitere Untersuchungen zur Darstellung eines Silangemisches, Z. Anorg. Allg. Chem., 1985, 530, 191 CrossRef; (e) F. Fehér, P. Hädicke and H. Frings, Beiträge zur chemie des siliciums und germaniums, XXIII (1) physikalisch-chemische eigenschaften der silane von trisilan bis heptasilan, Inorg. Nucl. Chem. Lett., 1973, 9, 931 CrossRef; (f) F. Fehér and D. Skrodzki, Beiträge zur chemie des siliciums und germaniums, XXVI (1) ramanspektren des trisilans, der tetra-, penta-, hexasilane und des n-heptasilans, Inorg. Nucl. Chem. Lett., 1974, 10, 577 CrossRef.
- W. C. Johnson and S. Isenberg, Hydrogen Compounds of Silicon. I. The Preparation of Mono- and Disilane, J. Am. Chem. Soc., 1935, 57, 1349 CrossRef CAS.
- A. E. Finholt, A. C. Bond, K. E. Wilzbach and H. I. Schlesinger, The Preparation and Some Properties of Hydrides of Elements of the Fourth Group of the Periodic System and of their Organic Derivatives, J. Am. Chem. Soc., 1947, 69, 2692 CrossRef CAS.
- (a) F. Höfler and R. Jannach, Zur kenntnis des neopentasilans, Inorg. Nucl. Chem. Lett., 1973, 9, 723 CrossRef; (b) J. P. Cannady and X. Zhou, WO2008051328A1, 2008; (c) S. Wieber, M. Patz, M. Trocha, H. Rauleder, E. Müh, H. Stüger and C. Walkner, WO2011061088A1, 2011; (d) M.-Z. Oh, J. Haubrock, T. Schwärtzke, I. Moussallem and M. Trocha, WO2014095278A1, 2014.
- (a) L. M. Litz and S. A. Ring, US3163590, 1964; (b) W. Sundermeyer and L. M. Litz, Die elektrochemische Darstellung von Hydriden in geschmolzenen Salzen, Chem. Ing. Tech., 1965, 37, 14 CrossRef CAS.
- (a) E. Hengge and G. Bauer, Cyclopentasilan, das erste unsubstituierte cyclische Siliciumhydrid, Angew. Chem., 1973, 85, 304 Search PubMed; (b) E. Hengge and G. Bauer, Darstellung und Eigenschaften von Cyclopentasilan, Monatsh. Chem., 1975, 106, 503 CrossRef CAS; (c) E. Hengge and D. Kovar, Darstellung von Cyclohexasilan, Si6H12, Angew. Chem., 1977, 89, 417 CrossRef CAS.
- D. Schmidt, U. Böhme, J. Seidel and E. Kroke, Cyclopentasilane Si5H10: First single crystal X-ray structure of an oligosilane SixHy and thermal analysis with TG/MS, Inorg. Chem. Commun., 2013, 35, 92 CrossRef CAS.
- (a) P. Boudjouk, S. D. Kloos, B.-K. Kim, M. Page and D. Thweatt, An unexpected redistribution of trichlorosilane. Synthesis, structure and bonding of (N,N,N′,N′-tetraethylethylenediamine)-dichlorosilane, J. Chem. Soc., Dalton Trans., 1998, 877 RSC; (b) S. B. Choi, B. K. Kim, P. Boudjouk and D. G. Grier, Amine-promoted disproportionation and redistribution of trichlorosilane: formation of tetradecachlorocyclohexasilane dianion, J. Am. Chem. Soc., 2001, 123, 8117 CrossRef CAS PubMed; (c) A. Elangovan, K. Anderson, P. Boudjouk and D. L. Schulz, US8975429B2, 2015.
- J. Teichmann and M. Wagner, Silicon chemistry in zero to three dimensions: from dichlorosilylene to silafullerane, Chem. Commun., 2018, 54, 1397 RSC.
- H. Stüger, P. Lassacher and E. Hengge, Anorganische Bi(cyclopentasilanyle): Synthese und spektroskopische Charakterisierung, Z. Anorg. Allg. Chem., 1995, 621, 1517 CrossRef.
- (a) C. J. Bakay, US3968199A, 1976; (b) G. H. Wagner and C. E. Erickson, US2627451A, 1953.
- (a) K. Tachiki, S. Katakura and Y. Yamahita, JP37008374, 1962; (b) K. Tachiki and Y. Yamahita, JP36021507, 1961.
- J. Y. Corey, in Advances in Organometallic Chemistry Volume 51, ed. J. Y. Corey, Elsevier, 2004, vol. 51, pp. 1–52 Search PubMed.
- J. F. Harrod, in Inorganic and Organometallic Polymers, ed. M. Zeldin, K. J. Wynne and H. R. Allcock, American Chemical Society, Washington, DC, 1988, vol. 360, pp. 89–100 Search PubMed.
- (a) Y. Okumura, K. Takatsuna and J. Yagihashi, JP0218451, 1990; (b) H. Stüger, D. Wolf, B. Stützel, J. Sauer, Y. Onal, M. Trocha, G. Stochniol, A. Ebbers and N. Brausch, WO2010003729A1, 2010.
- G. Nikiforov and G. Itov, WO2020077182A1, 2020.
- A. D. Craig and A. G. MacDiarmid, Application of the Wurtz reaction to the synthesis of disilane and 1,2-dimethyldisilane, J. Inorg. Nucl. Chem., 1962, 24, 163 CrossRef.
- R. C. Kennedy, L. P. Freeman, A. P. Fox and M. A. Ring, Formation and cleavage of the silicon-silicon bond in disilane, J. Inorg. Nucl. Chem., 1966, 28, 1373 CrossRef CAS.
- W. R. Bornhorst and M. A. Ring, Formation and cleavage of the germanium-germanium bond in digermane, Inorg. Chem., 1968, 7, 1009 CrossRef CAS.
- F. Fehér and R. Freund, Contributions to the chemistry of silicon and germanium, XXII (1) new silanes, bromosilanes and phenylsilanes, Inorg. Nucl. Chem. Lett., 1973, 9, 937 CrossRef.
- T. Lobreyer, J. Oeler and W. Sundermeyer, Über die verbesserte Darstellung von Silyl– und Germylkalium sowie die Synthese von Silylgermanen, Chem. Ber., 1991, 124, 2405 CrossRef CAS.
- T. Lobreyer, W. Sundermeyer and H. Oberhammer, Über dehydrierende Aufbaureaktionen zu silylsubstituierten Alkalimetallgermaniden, –stanniden und –phosphiden; Molekülstruktur von Neopentasilan, Chem. Ber., 1994, 127, 2111 CrossRef CAS.
- H. Stueger, T. Mitterfellner, R. Fischer, C. Walkner, M. Patz and S. Wieber, Selective synthesis and derivatization of alkali metal silanides MSi(SiH3)3, Chem. – Eur. J., 2012, 18, 7662 CrossRef CAS PubMed.
- V. Christopoulos, M. Rotzinger, M. Gerwig, J. Seidel, E. Kroke, M. Holthausen, O. Wunnicke, A. Torvisco, R. Fischer, M. Haas and H. Stueger, Synthesis and Properties of Branched Hydrogenated Nonasilanes and Decasilanes, Inorg. Chem., 2019, 58, 8820 CrossRef CAS PubMed.
- X. Zhou, N. Chang, J. Young and X. Wang, Selective Synthesis of 2,2,4,4-Tetrasilylpentasilane, Inorg. Chem., 2019, 58, 12526 CrossRef CAS PubMed.
- H. Stüger, M. Haas, O. Wunnicke, M. D. Malsch and M. Holthausen, EP3590889A1, 2020.
- H. Gilman and R. L. Harrell, Hexakis(trimethylsilyl)disilane: A highly branched and symmetrical organopolysilane, J. Organomet. Chem., 1967, 9, 67 CrossRef CAS.
- G. Fritz, Bildung siliciumorganischer Verbindungen III. Mitt.: Zum thermischen Zerfall von SiH4, Z. Naturforsch., B, 1952, 507 CrossRef CAS.
- E. M. Tebben and M. A. Ring, Pyrolysis of disilane and trisilane, Inorg. Chem., 1969, 8, 1787 CrossRef CAS.
- (a) E. J. Spanier and A. G. MacDarmid, The Conversion of Silane to Higher Silanes in a Silent Electric Discharge, Inorg. Chem., 1962, 1, 432 CrossRef CAS; (b) S. D. Gokhale, J. E. Drake and W. L. Jolly, Synthesis of the higher silanes and germanes, J. Inorg. Nucl. Chem., 1965, 27, 1911 CrossRef CAS.
- H. Niki and G. J. Mains, The 3P1 Mercury-Photosensitized `Decomposition of Monosilane, J. Phys. Chem., 1964, 68, 304 CrossRef CAS.
- M. Bamberg, M. Bursch, A. Hansen, M. Brandl, G. Sentis, L. Kunze, M. Bolte, H.-W. Lerner, S. Grimme and M. Wagner, Cl@Si20H20-: Parent Siladodecahedrane with Endohedral Chloride Ion, J. Am. Chem. Soc., 2021, 143, 10865 CrossRef CAS PubMed.
- (a) J. Zhang, S. Yan, H. Qu, X. F. Yu and P. Peng, Alkali metal silanides α-MSiH3: A family of complex hydrides for solid-state hydrogen storage, Int. J. Hydrogen Energy, 2017, 42, 12405 CrossRef CAS; (b) A. Jain, H. Miyaoka, T. Ichikawa and Y. Kojima, Tailoring the absorption–desorption properties of KSiH3 compound using nano-metals (Ni, Co, Nb) as catalyst, J. Alloys Compd., 2015, 645, S144–S147 CrossRef CAS.
- (a) R. Janot, W. S. Tang, D. Clémençon and J.-N. Chotard, Catalyzed KSiH3 as a reversible hydrogen storage material, J. Mater. Chem. A, 2016, 4, 19045 RSC; (b) S. Sharma, F. Guo, T. Ichikawa, Y. Kojima, S. Agarwal and A. Jain, Iron based catalyst for the improvement of the sorption properties of KSiH3, Int. J. Hydrogen Energy, 2020, 45, 33681 CrossRef CAS.
- W. S. Tang, J.-N. Chotard, P. Raybaud and R. Janot, Enthalpy–Entropy Compensation Effect in Hydrogen Storage Materials: Striking Example of Alkali Silanides MSiH3 (M = K, Rb, Cs), J. Phys. Chem. C, 2014, 118, 3409 CrossRef CAS.
- P. Hagenmuller and M. Pouchard, Some reactions of monosilane with sodium metal, Bull. Soc. Chim. Fr., 1964, 1187 CAS.
- E. Amberger and R. Römer, Reaktionen der Hydrylanionen. I. Reaktionen des Silylanions SiH mit substituierten Boranen, Z. Anorg. Allg. Chem., 1966, 345, 1 CrossRef CAS.
- S. Cradock, G. A. Gibbon and C. H. van Dyke, Germyl chemistry. V. Hexamethylphosphoramide as a solvent for the preparation and reaction of alkali metal derivatives of silane and germane, Inorg. Chem., 1967, 6, 1751 CrossRef CAS.
- E. Amberger, R. Römer and A. Layer, Reaktionen der hydrylanionen V. Darstellung von SiH3Na, SiH3K. SiH3Rb, SiH3Cs und SnH3K und ihre Umsetzung mit CH3I, J. Organomet. Chem., 1968, 12, 417 CrossRef CAS.
- H. Bürger, R. Eujen and H. C. Marsmann, 1H- und 29Si-kernresonanzspektroskopische Charakterisierung der Anionen ⊖SiH3(n(SiH3)n, Z. Naturforsch., B, 1974, 29, 149 CrossRef.
- F. Fehér and R. Freund, Beiträge zur chemie des siliciums und germaniums, XXIV (1) darstellung und kernresonanz-spektroskopische untersuchung höherer silylanionen; Darstellung neuer phenyl- und alkylsilane, Inorg. Nucl. Chem. Lett., 1974, 10, 561 CrossRef.
- F. Fehér, G. Betzen and M. Krancher, Beiträge zur Chemie des Siliciums und Germaniums. XXXIII [1]. Zur Darstellung von Kaliumsilyl, Z. Anorg. Allg. Chem., 1981, 475, 81 CrossRef.
- F. Fehér and M. Krancher, Beiträge zur Chemie des Siliciums und Germaniums. XXXIV. Ein weiterer Beitrag zur Darstellung von Kaliumsilyl, Z. Anorg. Allg. Chem., 1984, 509, 95 CrossRef.
- F. Fehér and M. Krancher, Contributions to the Chemistry of Silicon and Germanium, XXXVIII. Systematic Investigations on the Reaction of Higher Silanes with Potassium Silyl, Z. Naturforsch., B, 1985, 40, 1010 CrossRef.
- B. F. Fieselmann and C. R. Dickson, Improved synthetic route to potassium silyl using crown ethers, potassium, and silane and its use to prepare methylsilane and disilylmethane, J. Organomet. Chem., 1989, 363, 1 CrossRef CAS.
- F. Fehér, M. Krancher and M. Fehér, Beiträge zur Chemie des Siliciums und Germaniums. XL. Die Bildung von Alkalimetallsilanylen, MSinH2n+1 (M = Na, K), – eine neue Methode zur Knüpfung von Silicium–Silicium–Bindungen ausgehend von SiH4 und Natrium oder Kalium, Z. Anorg. Allg. Chem., 1991, 606, 7 CrossRef.
- W. Sundermeyer, T. Lobreyer and O. Johannes, DE4139113A1, 1993.
- H. Stueger, V. Christopoulos, A. Temmel, M. Haas, R. Fischer, A. Torvisco, O. Wunnicke, S. Traut and S. Martens, Selective Synthesis and Derivatization of Germasilicon Hydrides, Inorg. Chem., 2016, 55, 4034 CrossRef CAS PubMed.
- T. Lainer, R. C. Fischer and M. Haas, The Synthesis of Tris(silyl)silanides Revisited. A Study of Reactivity and Stability, Z. Anorg. Allg. Chem., 2022, 648, e202200137 CrossRef CAS.
- V. Leich, T. P. Spaniol and J. Okuda, Formation of α-KSiH3 by hydrogenolysis of potassium triphenylsilyl, Chem. Commun., 2015, 51, 14772 RSC.
- D. Schuhknecht, V. Leich, T. P. Spaniol, I. Douair, L. Maron and J. Okuda, Alkali Metal Triphenyl- and Trihydridosilanides Stabilized by a Macrocyclic Polyamine Ligand, Chem. – Eur. J., 2020, 26, 2821 CrossRef CAS PubMed.
- S. M. Sze and K. K. Ng, Physics of Semiconductor Devices, Wiley, 2006 Search PubMed.
- S. Wolf and R. N. Tauber, Silicon processing for the VLSI era, Lattice Pr, Sunset Beach, Calif., 1986 Search PubMed.
- (a) A. J. Olivares, A. Zamchiy, V. S. Nguyen and P. Roca i Cabarrocas, Boron activation in silicon thin films grown by PECVD under epitaxial and microcrystalline conditions, Appl. Surf. Sci. Adv., 2023, 18, 100508 CrossRef; (b) Y. Kamochi, A. Motomiya, H. Habuka, Y. Ishida, S.-I. Ikeda and S. Hara, Boron-Silicon Thin Film Formation Using a Slim Vertical Chemical Vapor Deposition Reactor, Adv. Chem. Eng. Sci., 2023, 13, 7 CrossRef CAS.
- D. F. Gaines and T. V. Iorns, Group IV derivatives of pentaborane(9), J. Am. Chem. Soc., 1968, 90, 6617 CrossRef CAS.
- D. F. Gaines and T. V. Iorns, 1-Silyl derivatives of pentaborane(9), Inorg. Chem., 1971, 10, 1094 CrossRef CAS.
- T. C. Geisler and A. D. Norman, Preparation and properties of 2- and mu.-(halosilyl)pentaboranes(9), Inorg. Chem., 1970, 9, 2167 CrossRef CAS.
- T. C. Geisler, N. P. Soice and A. D. Norman, Relative stabilities of XSiH2B5H8 (X = H, SiH3, Cl) silylpentaborane(9)s: synthesis of chlorosilyl- and disilanyl-pentaboranes, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 132, 123 CrossRef CAS.
- T. Lainer, C. Walkner, N. M. Tasch, R. C. Fischer, A. Torvisco, H. Stueger and M. Haas, Synthesis and Characterization of Alkali Metal Hypersilyl Borates, Z. Anorg. Allg. Chem., 2024, 650, e202400054 CrossRef CAS.
- K. N. Semenenko and K. A. Taisumov, Preparation of monosilylalane, Russ. Chem. Bull., 1975, 24, 1578 CrossRef.
- E. Amberger and R. Römer, Notizen: Reaktionen der Hydrylanionen III. Synthese und Eigenschaften von Silyl-dibutylalan (n-C4H9)2AlSiH3, Z. Naturforsch., B, 1968, 23, 559 Search PubMed.
- J. M. Castillo, M. Klos, K. Jacobs, M. Horsch and H. Hasse, Characterization of Alkylsilane Self-Assembled Monolayers by Molecular Simulation, Langmuir, 2015, 31, 2630 CrossRef CAS PubMed.
- N. Aissaoui, L. Bergaoui, J. Landoulsi, J.-F. Lambert and S. Boujday, Silane layers on silicon surfaces: mechanism of interaction, stability, and influence on protein adsorption, Langmuir, 2012, 28, 656 CrossRef CAS PubMed.
- X. Jiang and S. F. Bent, Area-Selective ALD with Soft Lithographic Methods: Using Self-Assembled Monolayers to Direct Film Deposition, J. Phys. Chem. C, 2009, 113, 17613 CrossRef CAS.
- S. B. Khan, H. Wu, C. Pan and Z. Zhang, A Mini Review: Antireflective Coatings Processing Techniques, Applications and Future Perspective, Res. Rev. J Mat. Sci., 2017, 05, 36–54 Search PubMed.
- (a) S. Tannenbaum, S. Kaye and G. F. Lewenz, Synthesis and Properties of Some Alkylsilanes, J. Am. Chem. Soc., 1953, 75, 3753 CrossRef CAS; (b) M. E. Freeburger, B. M. Hughes, G. R. Buell, T. O. Tiernan and L. Spialter, Physical organosilicon chemistry. II. Mass spectral cracking patterns of phenylsilane and ortho-, meta- and para-substituted benzyl- and phenyltrimethylsilanes, J. Org. Chem., 1971, 36, 933 CrossRef CAS; (c) F. Mareš and V. Chvalovský, Organosilicon compounds XLV. Pyrolysis of trialkylsilanes, J. Organomet. Chem., 1966, 6, 327 CrossRef; (d) W. H. Nebergall, Some Reactions of Phenylsilane, J. Am. Chem. Soc., 1950, 72, 4702 CrossRef CAS.
- J. Zech and H. Schmidbaur, Synthetic pathways to disilylmethane, H3SiCH2SiH3, and methyldisilane, CH3SiH2SiH3, Chem. Ber., 1990, 123, 2087 CrossRef CAS.
- R. Hager, O. Steigelmann, G. Müller and H. Schmidbaur, A synthetic route to poly(silyl)methanes via poly(phenylsilyl)-methanes and poly(bromosily)methanes, Chem. Ber., 1989, 122, 2115 CrossRef CAS.
- R. Hager, O. Steigelmann, G. Müller, H. Schmidbaur, H. E. Robertson and D. W. H. Rankin, Tetrasilylmethane, C(SiH3)4, the Si/C-Inverse of Tetramethylsilane, Si(CH3)4, Angew. Chem., Int. Ed. Engl., 1990, 29, 201 CrossRef.
- V. N. Gevorgyan, L. M. Ignatovich and E. Lukevics, Reduction of alkoxysilanes, halo-silanes and -Germanes with lithium aluminium hydride under phase-transfer conditions, J. Organomet. Chem., 1985, 284, C31–C32 CrossRef CAS.
- H. Schmidbaur and J. Zech, An Improved Synthetic Pathway to Tetrasilylmethane and the Synthesis of 2,2-Disilylpropane, Eur. J. Solid State Inorg. Chem., 1992, 29, 5–21 CAS.
- D. V. Sidorov, P. A. Storozhenko, O. G. Shutova and B. E. Kozhevnikov, Synthesis of high-purity alkylsilanes, Theor. Found. Chem. Eng., 2007, 41, 668 CrossRef CAS.
- J. A. Morrison and J. M. Bellama, Synthesis and characterization of the (halosilyl)methylsilanes, J. Organomet. Chem., 1975, 92, 163 CrossRef CAS.
- I. N. Jung, S. H. Yeon and J. S. Han, Direct synthesis of bis(silyl)methanes containing silicon-hydrogen bonds, Organometallics, 1993, 12, 2360 CrossRef CAS.
- J.-H. Boo, K.-S. Yu, Y. Kim, S. H. Yeon and I. N. Jung, Growth of Cubic SiC Films Using 1,3-Disilabutane, Chem. Mater., 1995, 7, 694 CrossRef CAS.
- G. Fritz, St. Lauble, M. Breining, A. G. Beetz, A. M. Galminas, E. Matern and H. Goesmann, Bildung siliciumorganischer Verbindungen. 111. Die Hydrierung Si–chlorierter, C–spiroverbrückter 2,4−Disilacyclobutane mit LiAlH4 und i-Bu2AlH. Der Zugang zum Si8C3H20, Z. Anorg. Allg. Chem., 1994, 620, 127 CrossRef CAS.
- (a) G. Fritz and J. F. Willems, Fortschritte Der Chemischen Forschung, Springer-Verlag, Berlin/Heidelberg, 1964, p. 4/3 Search PubMed; (b) G. Fritz, H. J. Buhl and D. Kummer, Bildung Siliciumorganischer Verbindungen. XXII. Untersuchungen an Pyrolyseprodukten der Methylchlorsilane (SiH–haltige Silicium–methylene), Z. Anorg. Allg. Chem., 1964, 327, 165 CrossRef CAS.
- G. Fritz, Bildung siliciumorganischer Verbindungen. IV. Das thermische Verhalten von C2H5SiH3, (C2H5)2SiH2 und (C2H5)3SiH, Z. Anorg. Allg. Chem., 1953, 273, 275 CrossRef CAS.
- M. Bowrey and J. H. Purnell, Insertion reactions of SiH2 [silylene], J. Am. Chem. Soc., 1970, 92, 2594 CrossRef CAS.
- S. Bommers and H. Schmidbaur, Poly(trifluoromethane-sulfonatosilyl)methanes - Precursors to Polysilylmethanes, Z. Naturforsch., B, 1994, 49, 337 CrossRef CAS.
- H. Westermark, O. Theander and N. A. Sörensen, Preparation of Some Organic Silicon Hydrides, Acta Chem. Scand., 1954, 8, 1830 CrossRef CAS.
- U. Herzog and G. Roewer, Preparation of oligosilanes containing perhalogenated silyl groups and their hydrogenation by stannanes, J. Organomet. Chem., 1997, 544, 217 CrossRef CAS.
- (a) U. Herzog, E. Brendler and G. Roewer, Basekatalysierte hydrierung von methylchlortri- und -tetrasilanen mit trialkylstannanen zu methylchlorwasserstofftri- und -tetrasilanen, J. Organomet. Chem., 1996, 511, 85 CrossRef CAS; (b) U. Herzog and G. Roewer, Base catalysed hydrogenation of methylbromooligosilanes with trialkylstannanesidentification of the first methylbromohydrogenoligosilanes, J. Organomet. Chem., 1997, 527, 117 CrossRef CAS.
- U. Herzog, G. Roewer and U. Pätzold, Katalytische hydrierung chlorhaltiger disilane mit tributylstannan, J. Organomet. Chem., 1995, 494, 143 CrossRef CAS.
- (a) D. G. White and E. G. Rochow, Reactions of Silane with Unsaturated Hydrocarbons, J. Am. Chem. Soc., 1954, 76, 3897 CrossRef CAS; (b) R. N. Haszeldine, M. J. Newlands and J. B. Plumb, 375. Polyfluoroalkyl compounds of silicon. Part VI. Reaction of 3,3,3-trifluoropropene with silaneand the conversion of the products into silicones and polysiloxanes, J. Chem. Soc., 1965, 2101 RSC.
- A. J. Malcolm, C. R. Everly and G. E. Nelson, US4670574A, 1987.
- M. Kobayashi and M. Itoh, Hydrosilylation of Olefins with Monosilane in the Presence of Lithium Aluminum Hydride, Chem. Lett., 1996, 25, 1013 CrossRef.
- M. Itoh, K. Iwata and M. Kobayashi, Preparation of vinylsilane from monosilane and vinyl chloride, J. Organomet. Chem., 1999, 590, 36 CrossRef CAS.
- E. Amberger and E. Mühlhofer, Reaktionen der hydrylanionen II. Reaktionen des silylanions SiH3 und germylanions GeH3 mit monohalogenorganylverbindungen der 4. Hauptgruppe, J. Organomet. Chem., 1968, 12, 55 CrossRef CAS.
- T. Lobreyer, J. Oeler, W. Sundermeyer and H. Oberhammer, Über die–Synthese und Molekülstruktur von CH3M(SiH3)3 (M=Si, Ge), Chem. Ber., 1993, 126, 665 CrossRef CAS.
- T. Lainer, M. Leypold, C. Kugler, R. C. Fischer and M. Haas, Dodecamethoxyneopentasilane as a New Building Block for Defined Silicon Frameworks, Eur. J. Inorg. Chem., 2021, 2021, 529 CrossRef CAS.
- H. E. Opitz, J. S. Peake and W. H. Nebergall, The Preparation of Monobromosilane and Organic Silyl Derivatives, J. Am. Chem. Soc., 1956, 78, 292 CrossRef CAS.
- H. Schmidbaur and J. Ebenhöch, Synthetic Pathways to Simple Di- and Trisilylmethanes: Potential Starting Materials for the CVD Deposition of Amorphous Silicon a-SiC :H, Z. Naturforsch., B, 1986, 41, 1527 CrossRef.
- S. Xin, C. Aitken, J. F. Harrod, Y. Mu and E. Samuel, Redistribution reactions of alkoxy- and siloxysilanes, catalyzed by dimethyltitanocene, Can. J. Chem., 1990, 68, 471 CrossRef CAS.
- E. Belov, G. Dubrovskaya, N. Efimov, S. Kleshcevnikova, E. Korobkov and E. Lebedev, in Organosilicon Chemistry V: From Molecules to Materials, 2003, pp. 518–521 Search PubMed.
- M. Abedini and A. G. MacDiarmid, Synthesis of Methyldisilane by the Silent Electric Discharge of a Mixture of Silane and Dimethyl Ether, Inorg. Chem., 1966, 5, 2040 CrossRef CAS.
- W. J. Bolduc and M. A. Ring, Preparation of ethyldisilane, J. Organomet. Chem., 1966, 6, 202 CrossRef CAS.
- F. Fehér and R. Freund, Beiträge zur chemie des siliciums und germaniums, XXV (1) reaktionen von trisilan, n-tetrasilan und n-pentasilan mit n-butyllithium in n-hexan, Inorg. Nucl. Chem. Lett., 1974, 10, 569 CrossRef.
- (a) J. M. Hartmann, A. Abbadie, A. M. Papon, P. Holliger, G. Rolland, T. Billon, J. M. Fédéli, M. Rouvière, L. Vivien and S. Laval, Reduced pressure–chemical vapor deposition of Ge thick layers on Si(001) for 1.3–1.55-μm photodetection, J. Appl. Phys., 2004, 95, 5905 CrossRef CAS; (b) J. Aubin, J. M. Hartmann, M. Bauer and S. Moffatt, Very low temperature epitaxy of Ge and Ge rich SiGe alloys with Ge2H6 in a Reduced Pressure – Chemical Vapour Deposition tool, J. Cryst. Growth, 2016, 445, 65 CrossRef CAS; (c) T. I. Kamins, E. C. Carr, R. S. Williams and S. J. Rosner, Deposition of three-dimensional Ge islands on Si(001) by chemical vapor deposition at atmospheric and reduced pressures, J. Appl. Phys., 1997, 81, 211 CrossRef CAS; (d) M. Kummer, C. Rosenblad, A. Dommann, T. Hackbarth, G. Höck, M. Zeuner, E. Müller and H. von Känel, Low energy plasma enhanced chemical vapor deposition, Mater. Sci. Eng., B, 2002, 89, 288 CrossRef; (e) M. D. Stephens, M. Pikulin and C. Ritter, The Utility of Novel Single-Source Germyl Silanes, ECS Trans., 2006, 3, 183 CrossRef CAS; (f) J. C. Guillemin, L. Lassalle and T. Janati, Germane photochemistry. Photolysis of gas mixtures of planetary interest, Planet. Space Sci., 1995, 43, 75 CrossRef CAS PubMed; (g) J. Kouvetakis, J. Tolle, R. Roucka, V. R. D'Costa, Y. Fang, A. V. Chizmeshya and J. Menendez, Nanosynthesis of Si-Ge-Sn Semiconductors and Devices via Purpose-built Hydride Compounds, ECS Trans., 2008, 16, 807 CrossRef CAS; (h) J. Kouvetakis, J. Menendez and J. Tolle, Advanced Si-based Semiconductors for Energy and Photonic Applications, Solid State Phenom., 2009, 156–158, 77 Search PubMed; (i) S.-M. Jang, K. Liao and R. Reif, Chemical Vapor Deposition of Epitaxial Silicon–Germanium from Silane and Germane: II In Situ Boron, Arsenic, and Phosphorus Doping, J. Electrochem. Soc., 1995, 142, 3520 CrossRef CAS; (j) H. Oberhammer, T. Lobreyer and W. Sundermeyer, The Ge-Si bond in silylgermane. Discrepancy between experiment and theory, J. Mol. Struct., 1994, 323, 125 CrossRef CAS.
- R. Varma and A. P. Cox, Eine neue Synthese des Germylsilans, Angew. Chem., 1964, 76, 649 CrossRef CAS.
- P. L. Timms, C. C. Simpson and C. S. G. Phillips, 279. The silicon–germanium hydrides, J. Chem. Soc., 1964, 1467 RSC.
- J. Kouvetakis, C. J. Ritter, C. Hu, A. V. G. Chizmeshya, J. Tolle, D. Klewer and I. S. T. Tsong, Synthesis and fundamental studies of (H3Ge)xSiH4(x molecules: precursors to semiconductor hetero- and nanostructures on Si, J. Am. Chem. Soc., 2005, 127, 9855 CrossRef PubMed.
- B. Köstler, F. Jungwirth, L. Achenbach, M. Sistani, M. Bolte, H.-W. Lerner, P. Albert, M. Wagner and S. Barth, Mixed-Substituted Single-Source Precursors for Si1(xGex Thin Film Deposition, Inorg. Chem., 2022, 61, 17248 CrossRef PubMed.
- E. Wiberg, E. Amberger and H. Cambensi, Zur Kenntnis der Äthan–homologen Mischhydride H3Si–SnH3 und H3Ge–SnH3 sowie ihrer Phenyl–Acetato– und Chlorderivate, Z. Anorg. Allg. Chem., 1967, 351, 164 CrossRef CAS.
- B. Xu, L. Li, P. Shi, W. Yu, J. Zhao, X. Wang and L. Andrews, Matrix-Infrared Spectra and Structures of HM-SiH3 (M = Ge, Sn, Pb, Sb, Bi, Te Atoms), J. Phys. Chem. A, 2018, 122, 81 CrossRef CAS PubMed.
- S. M. George, Atomic layer deposition: an overview, Chem. Rev., 2010, 110, 111 CrossRef CAS PubMed.
- V. Miikkulainen, M. Leskelä, M. Ritala and R. L. Puurunen, Crystallinity of inorganic films grown by atomic layer deposition: Overview and general trends, J. Appl. Phys., 2013, 113, 21301 CrossRef.
- R. L. Puurunen, Surface chemistry of atomic layer deposition: A case study for the trimethylaluminum/water process, J. Appl. Phys., 2005, 97, 121301–121352 CrossRef.
- M. Leskelä and M. Ritala, Atomic layer deposition (ALD): from precursors to thin film structures, Thin Solid Films, 2002, 409, 138 CrossRef.
- B. D. Rekken and X. Zhou, WO2015184201A1, 2015.
- J. W. Kim, S. J. Jang, H. G. Kim, J. H. Ju, Y. H. Cho, J. D. Kim and M. W. Kim, KR101040325B1, 2011.
- H. Schuh, T. Schlosser, P. Bissinger and H. Schmidbaur, Disilanyl-amines—compounds comprising the structural unit Si-Si-N, as single-source precursors for plasma-enhanced chemical vapour deposition (PE-CVD) of silicon nitride, Z. Anorg. Allg. Chem., 1993, 619, 1347 CrossRef CAS.
- (a) W. Gollner, K. Renger and H. Stueger, Linear and Cyclic Polysilanes Containing the Bis(trimethylsilyl)amino Group: Synthesis, Reactions, and Spectroscopic Characterization, Inorg. Chem., 2003, 42, 4579 CrossRef CAS PubMed; (b) J. F. Lehmann and H. P. Withers Jr., US2012277457A1, 2012.
- H. Stüger, P. Lassacher and E. Hengge, Aminochlorodisilanes: Precursors to multifunctionalized disilane derivatives, J. Organomet. Chem., 1997, 547, 227 CrossRef.
- N. Chang, B. K. Hwang, B. D. Rekken and X. Zhou, WO2017200908A1, 2017.
- H. J. Emeléus and N. Miller, 175. Derivatives of monosilane. Part I. The reactions of chlorosilane with aliphatic amines, J. Chem. Soc., 1939, 819 RSC.
- D. G. Anderson and D. W. H. Rankin, Isopropyldisilylamine and disilyl-t-butylamine: preparation, spectroscopic properties, and molecular structure in the gas phase, determined by electron diffraction, J. Chem. Soc., Dalton Trans., 1989, 779 RSC.
- D. P. Spence, H. Chandra, B. Han, M. L. O'Neill, S. G. Mayorga and A. Mallikarjunan, EP2669248A1, 2013.
- A. Stock, Hydrides of Boron and Silicon, Cornell University Press; Oxford University Press, Ithaca, NY, London, 1933 Search PubMed.
- A. Stock and K. Somieski, Siliciumwasserstoffe, VIII: Halogen–Abkömmlinge des Disilans, Si2H6 und ihre Hydrolyse, Ber. Dtsch. Chem. Ges. A/B, 1920, 53, 759 CrossRef.
- B. J. Aylett and M. J. Hakim, The Preparation and Some Properties of Disilylamine, Inorg. Chem., 1966, 5, 167 CrossRef.
- A. B. Burg and E. S. Kuljian, Silyl-Amino Boron Compounds1, J. Am. Chem. Soc., 1950, 72, 3103 CrossRef CAS.
- R. L. Wells and R. Schaeffer, Studies of Silicon-Nitrogen Compounds. The Base-Catalyzed Elimination of Silane from Trisilylamine 1,2, J. Am. Chem. Soc., 1966, 88, 37 CrossRef CAS.
- M. Xiao, X. Lei, D. P. Spence, H. Chandra, B. Han, M. L. O'Neil, S. G. Mayorga and A. Mallikarjunan, US9337018B2, 2016.
- M. Abedini and A. G. MacDiarmid, The Preparation and Properties of Some New Nitrogen and Fluorine Derivatives of Disilane, Inorg. Chem., 1963, 2, 608 CrossRef CAS.
- B. J. Aylett and M. J. Hakim, Silicon–nitrogen compounds. Part VI. The preparation and properties of disilazane, J. Chem. Soc., A: Inorg. Phys. Theor., 1969, 639 RSC.
- B. J. Aylett, The silyl group as an electron acceptor, J. Inorg. Nucl. Chem., 1956, 2, 325 CrossRef CAS.
- L. Ward and A. G. MacDiarmid, The preparation and properties of bis-disilanyl sulphide and tris-disilanylamine, J. Inorg. Nucl. Chem., 1961, 21, 287 CrossRef CAS.
- S. G. Kim, J. J. Park, S. J. Jang, B. Yang, S. Lee, S. D. Lee, S. I. Lee and M. W. Kim, WO2018182305A1, 2018.
- S. W. Yoon, H. Y. Yang, H. N. Kim and G. H. Jo, US2023094481A1, 2023.
- M. Xiao, X. Lei and D. P. Spence, EP2818474A1, 2014.
- (a) Y. Jiang, X. Xu, W. Wang, Q. Liu and F. Tao, CN117510533A, 2024; (b) C. Tang, S. Zhu and J. Li, CN115677747A, 2023.
- M. Söldner, A. Schier and H. Schmidbaur, 1,2-Disilanediyl Bis(triflate), F3CSO3−SiH2SiH2−O3SCF3, as the Key Intermediate for a Facile Preparation of Open-Chain and Cyclic 1,1- and 1,2-Diaminodisilanes, Inorg. Chem., 1997, 36, 1758 CrossRef PubMed.
- B. M. Ketola, J. A. Maddock, B. D. Rekken, M. D. Telgenhoff and X. Zhou, WO2017106632A1, 2017.
- M. Xiao, M. R. MacDonald, R. Ho and X. Lei, EP2913334A1, 2015.
- A. Foss, J. Maddock, B. Rekken and M. Telgenhoff, WO2019226207A1, 2019.
- T. T. Nguyen, T. K. Mukhopadhyay, S. N. MacMillan, M. T. Janicke and R. J. Trovitch, Synthesis of Aminosilane Chemical Vapor Deposition Precursors and Polycarbosilazanes through Manganese-Catalyzed Si–N Dehydrocoupling, ACS Sustainable Chem. Eng., 2022, 10, 4218 CrossRef CAS.
- B. D. Rekken, US2021130374A1, 2021.
- A. Sanchez, G. Itov, P. Zhang and M. Stephens, WO2015048237A2, 2015.
- X. Zhou, WO2017106615A1, 2017.
- B. D. Rekken, X. Zhou, B. K. Hwang and B. Ketola, WO2017106587A1, 2017.
- H. J. Emeléus, A. G. Maddock and C. Reid, 68. Derivatives of monosilane. Part II. The iodo-compounds, J. Chem. Soc., 1941, 353 RSC.
- A. G. MacDiarmid, Pseudo-halogen derivatives of monosilane, J. Inorg. Nucl. Chem., 1956, 2, 88 CrossRef CAS.
- E. A. V. Ebsworth and J. C. Thompson, The preparation and properties of some silyl esters, J. Chem. Soc., A: Inorg. Phys. Theor., 1967, 69 RSC.
- C. Glidewell, Reactions of silylphosphine with amines and imines, Inorg. Nucl. Chem. Lett., 1974, 10, 39 CrossRef CAS.
- C. Glidewell, Some reactions of disilyl sulphide, J. Inorg. Nucl. Chem., 1969, 31, 1303 CrossRef CAS.
- E. A. V. Ebsworth and M. J. Mays, 945. The preparation and properties of silyl isocyanate, J. Chem. Soc., 1962, 4844 RSC.
- S. Sujishi and S. Witz, The Reaction of Silylamines with Boron Trifluoride. Methyl- and Silylaminoboron Difluorides, J. Am. Chem. Soc., 1957, 79, 2447 CrossRef CAS.
- W. M. Scantlin and A. D. Norman, Borane-catalyzed condensation of trisilazane and N-methyldisilazane, Inorg. Chem., 1972, 11, 3082 CrossRef CAS.
- E. A. V. Ebsworth and M. J. Mays, 653. The preparation and properties of silyl azide, J. Chem. Soc., 1964, 3450 RSC.
- G. Maier, J. Glatthaar and H. P. Reisenauer, Hetero–π–Systeme, 17: Aminosilylen (Aminosilandiyl), Chem. Ber., 1989, 122, 2403 CrossRef CAS.
- G. Fritz and H. O. Berkenhoff, Hydrolyse, Alkoholyse und Ammonolyse des Silylphosphins, Z. Anorg. Allg. Chem., 1957, 289, 250 CrossRef CAS.
- A. D. Norman and W. L. Jolly, Reaction of silylphosphine with ammonia, Inorg. Chem., 1979, 18, 1594 CrossRef CAS.
- E. A. V. Ebsworth and M. J. Mays, 959. The preparation and properties of disilylcyanamide, J. Chem. Soc., 1961, 4879 RSC.
- E. Ebsworth and M. J. Mays, The vibrational spectra and structure of (SiH3)2CN2, Spectrochim. Acta, 1963, 19, 1127 CrossRef CAS.
- C. Glidewell and A. G. Robiette, Gas phase electron diffraction study of SiH3N3 and (SiH3)2NCN, Chem. Phys. Lett., 1974, 28, 290 CrossRef CAS.
- H. Schmidbaur and H. Schuh, Die unterschiedliche Reaktivität von 1,4-Disilabutan und n-Tetrasilan gegenüber sekundären Aminen/Differences in Reactivity of 1,4-Disilabutane and n-Tetrasilane towards Secondary Amines, Z. Naturforsch., B, 1990, 45, 1679 CrossRef CAS.
- A. Sanchez, G. Itov, M. Khandelwal, C. Ritter, P. Zhang, J. M. Girard, Z. Wan, G. Kuchenbeiser, D. Orban, S. Kerrigan, R. Pesaresi, M. D. Stephens, Y. Wang and G. Husson, US2018072571A1, 2018.
- G. Fritz and P. Scheer, Silylphosphanes: Developments in Phosphorus Chemistry, Chem. Rev., 2000, 100, 3341 CrossRef CAS PubMed.
- S. M. Sze, Y. Li and K. K. Ng, Physics of semiconductor devices, Wiley, Hoboken, NJ, Chichester, West Sussex, 2021 Search PubMed.
- The Chemistry of Metal CVD, ed. T. T. Kodas and M. J. HampdenSmith, Wiley, Weinheim, New York, 1994 Search PubMed.
- Z. Rappoport and Y. Apeloig, The Chemistry of Organic Silicon Compounds, Wiley, 2001, vol. 3 Search PubMed.
- G. Fritz, Bildung, Eigenschaften und Reaktionen des Silylphosphins, Z. Anorg. Allg. Chem., 1955, 280, 332 CrossRef CAS.
- G. Fritz, G. Becker, G. Poppenburg, M. Rocholl and G. Trenczeck, Compounds of Phosphorus with Silicon and Aluminum, Angew. Chem., Int. Ed. Engl., 1966, 5, 53 CrossRef CAS.
- G. Fritz and H. O. Berkenhoff, Über ein Siliciumphosphid Si2P, Z. Anorg. Allg. Chem., 1959, 300, 205 CrossRef CAS.
- I. H. Sabherwal and A. B. Burg, A convenient synthesis of silylphosphine, Inorg. Nucl. Chem. Lett., 1972, 8, 27 CrossRef CAS.
- J. Blazejowski and F. W. Lampe, The IR multiphoton decomposition of PH3 SiH4 mixtures sensitized by SiF4, J. Photochem., 1984, 24, 235 CrossRef CAS.
- J. Blazejowski and F. W. Lampe, Monosilylphosphine formation by rapid silylene insertion in the IR photochemistry of SiH4-PH3 mixtures, J. Photochem., 1982, 20, 9 CrossRef CAS.
- J. Blazejowski and F. W. Lampe, The 147 nm photolysis of phosphine—silane mixtures, J. Photochem., 1981, 16, 105 CrossRef CAS.
- J. E. Drake and W. L. Jolly, Preparation of mixed hydrides of silicon, germanium, phosphorus, and arsenic, Chem. Ind., 1962, 1470 CAS.
- S. D. Gokhale and W. L. Jolly, The Synthesis of Disilanylphosphine and Disilylphosphine in a Silent Electric Discharge, Inorg. Chem., 1965, 4, 596 CrossRef CAS.
- S. D. Gokhale and W. L. Jolly, Disilanylphosphine and Disilylphosphine, Inorg. Chem., 1964, 3, 1141 CrossRef CAS.
- J. E. Drake, N. Goddard and J. Simpson, Some reactions of monohalogenated hydrides with monosilylphosphine and monosilylarsine, Inorg. Nucl. Chem. Lett., 1968, 4, 361 CrossRef CAS.
- J. E. Drake, N. Goddard and C. Riddle, Silicon–phosphorus hydrides. Part III. Exchange reactions of monosilylphosphine with chlorinated germanes and silanes, J. Chem. Soc., A: Inorg. Phys. Theor., 1969, 2704 RSC.
- E. Amberger and H. D. Boeters, Trisilylverbindungen, Chem. Ber., 1964, 97, 1999 CrossRef.
- A. J. Blake, E. A. V. Ebsworth and S. G. D. Henderson, Structure of trisilylphosphine, P(SiH3)3, at 100 K, Acta Crystallogr., Sect. C: Struct. Chem., 1991, 47, 486 CrossRef.
- C. Glidewell and G. M. Sheldrick, Preparation and properties of mono- and di-silylphosphine and mono-and disilyl-arsine, J. Chem. Soc., A: Inorg. Phys. Theor., 1969, 350 RSC.
- C. R. Russ and A. G. MacDiarmid, Cleavage and Addition Reactions of Silylphosphines and Silylarsines, Angew. Chem., Int. Ed. Engl., 1966, 5, 418 CrossRef CAS.
- D. E. Wingeleth and A. D. Norman, Redistribution of Primary Silyl- and Germylphosphines: Synthesis of Trisilyl- and Trigermylphosphines, Phosphorus Sulfur Relat. Elem., 1988, 39, 123 CrossRef CAS.
- J. E. Drake and N. Goddard, Silicon-phosphorus hydrides 2a comparative study by proton magnetic resonance spectroscopy of isomers disilanyl- and disilyl-phosphine, J. Chem. Soc., A: Inorg. Phys. Theor., 1969, 662–665 RSC.
- Hydrides of Groups IV and V, ed. W. L. Jolly, Interscience, New York, 1968, vol. 4 Search PubMed.
- J. B. Tice, A. V. G. Chizmeshya, J. Tolle, V. R. d’ Costa, J. Menendez and J. Kouvetakis, Practical routes to (SiH3)3P: applications in group IV semiconductor activation and in group III-V molecular synthesis, Dalton Trans., 2010, 39, 4551 RSC.
- F. Li, J. M. Girard and P. Zhang, WO2023121973A1, 2023.
- A. E. Finholt, C. Helling, V. Imhof, L. Nielsen and E. Jacobson, Complex Aluminum Hydrides Containing Nitrogen, Phosphorus, and Arsenic, Inorg. Chem., 1963, 2, 504 CrossRef CAS.
- A. D. Norman, A new general method for the synthesis of unsubstituted phosphino-silanes and -germanes, Chem. Commun., 1968, 812 RSC.
- G. Fritz and H. Schäfer, Bildung von NaAl(PH2)4, NaAl(HPCH3)4 und die präparative Darstellung von H3Si-PH2 und seiner PH-haltigen Derivate, Z. Anorg. Allg. Chem., 1971, 385, 243 CrossRef CAS.
- A. D. Norman and D. C. Wingeleth, Lithium tetraphosphinoaluminate phosphination of halosilanes and -germanes, Inorg. Chem., 1970, 9, 98 CrossRef CAS.
- A. D. Norman, Bis(phosphino)silane and tris(phosphino)silane, J. Am. Chem. Soc., 1968, 90, 6556 CrossRef CAS.
- G. Becker, B. Eschbach, D. Käshammer and O. Mundt, Metallderivate von Molekülverbindungen. VII. Bis[1,2-bis(dimethylamino)ethan-N,N′]lithium-disilylphosphanid Synthese und Struktur, Z. Anorg. Allg. Chem., 1994, 620, 29 CrossRef CAS.
- G. Fritz, H. Schäfer and W. Hölderich, Zur Metallierung der PH2−Gruppe in Silylphosphinen, Z. Anorg. Allg. Chem., 1974, 407, 266 CrossRef CAS.
- J. W. Anderson and J. E. Drake, Silicon–phosphorus hydrides. Part IV. The disilylphosphinoaluminate anion, J. Chem. Soc., A: Inorg. Phys. Theor., 1971, 2246 RSC.
- G. Fritz and G. Becker, Umsetzungen der Silylphosphine mit LiP(C2H5)2 und LiCH3, Z. Anorg. Allg. Chem., 1970, 372, 180 CrossRef CAS.
- S. Cradock, E. A. V. Ebsworth, H. Moretto, D. W. H. Rankin and W. J. Savage, Lithium-Derivate von Silanol und Verwandten Verbindungen, Angew. Chem., 1973, 85, 344 CrossRef CAS.
- S. Cradock, E. A. V. Ebsworth, D. W. H. Rankin and W. J. Savage, Preparation and spectroscopic studies of some reactions of lithium derivatives of silanol, disilylphosphine, and related compounds, J. Chem. Soc., Dalton Trans., 1976, 1661 RSC.
- H. O. Pierson, Handbook of chemical vapor deposition (CVD). Principles, technology, and applications, Noyes Publications, Park Ridge, N.J., U.S.A, 1992 Search PubMed.
- J. E. Drake and J. Simpson, Reactions of monosilylarsine with some boron Lewis acids and the reaction of monosilylphosphine with boron tribromide, J. Chem. Soc., A: Inorg. Phys. Theor., 1968, 1039 RSC.
- J. W. Anderson and J. E. Drake, Reactions of ‘LiAl(AsH2)4′, Inorg. Nucl. Chem. Lett., 1969, 5, 887 CrossRef CAS.
- J. W. Anderson and J. E. Drake, Preparation and properties of some primary silyl- and germyl-arsines, J. Chem. Soc., A: Inorg. Phys. Theor., 1970, 3131 RSC.
- A. Kashizadeh, R. Basnet, A. Liu, Z. Yang, L. Black, C. Sun, W. Han, Y. Wang and D. Macdonald, High Quality Antimony–Doped n–Type Silicon Wafers for Solar Cell Applications, Sol. RRL, 2025, 9, e2500335 CrossRef.
- E. Amberger and H. Boeters, Notizen: Darstellung und Eigenschaften von Trisilylstiban, Z. Naturforsch., B, 1963, 18, 157 CrossRef.
- M. Ohring, Materials Science of Thin Films, Elsevier, 2002 Search PubMed.
- C. M. Hessel, E. J. Henderson and J. G. C. Veinot, Hydrogen Silsesquioxane: A Molecular Precursor for Nanocrystalline Si−SiO2 Composites and Freestanding Hydride-Surface-Terminated Silicon Nanoparticles, Chem. Mater., 2006, 18, 6139 CrossRef CAS.
- A. Stock, C. Somieski and R. Wintgen, Siliciumwasserstoffe. III: Disiloxan, (SiH3)2O; zur Kenntnis des Tetrachlor monosilans, SiCl4, und des Hexachlor–disiloxans, (SiCl3)2O, Ber. Dtsch. Chem. Ges., 1917, 50, 1754 CrossRef CAS.
- H. J. Emeléus, A. G. MacDiarmid and A. G. Maddock, Sulphur and selenium derivatives of monosilane, J. Inorg. Nucl. Chem., 1955, 1, 194 CrossRef.
- W. A. Kriner, A. G. MacDiarmid and E. C. Evers, The Interaction of Disiloxane with Aluminum Halides 1, J. Am. Chem. Soc., 1958, 80, 1546 CrossRef CAS.
- L. G. L. Ward and A. G. MacDiarmid, The Preparation and Properties of Disilanyl Iodide and Bis-disilanyl Ether 1, J. Am. Chem. Soc., 1960, 82, 2151 CrossRef CAS.
- B. Sternbach and A. G. MacDiarmid, The Preparation and Properties of Silyl Methyl Ether 1, J. Am. Chem. Soc., 1961, 83, 3384 CrossRef CAS.
- M. Onyszchuk, The interaction of disiloxane with boron trifluoride and trichloride, Can. J. Chem., 1961, 39, 808 CrossRef CAS.
- C. H. van Dyke and A. G. MacDiarmid, The Reaction of 1,2-Disilyldisiloxane, 1-Silyldisiloxane, and 1,1,1-Trimethyldisiloxane with Boron Trichloride, Inorg. Chem., 1964, 3, 747 Search PubMed.
- C. H. van Dyke, The interaction of disiloxane and methoxysilane with phosphorus(III) halides, J. Inorg. Nucl. Chem., 1968, 30, 81 Search PubMed.
- C. Glidewell and D. W. H. Rankin, Germyl and silyl derivatives of phenol and thiophenol, J. Chem. Soc. A, 1969, 753 RSC.
- J. E. Drake, H. E. Henderson and R. T. Hemmings, Silyl and germyl derivatives of trifluoroacetic and trichloroacetic acid, Inorg. Chem., 1977, 16, 1682 CrossRef CAS.
- C. Glidewell, Reaction of silyl bromide with some group VI hydrido-anions, J. Chem. Soc. A, 1971, 823 RSC.
- F. Fehér, I. Fischer and D. Skrodzki, Beiträge zur Chemie des Siliciums und Germaniums. XXXI [1]. Die photochemische Darstellung neuer Isopropoxyderivate des Tri–, n–Tetra–, iso–Tetra– und n–Pentasilans und deren saure Hydrolyse zu Bis–Silanyl–Äthern, Z. Anorg. Allg. Chem., 1980, 466, 29 CrossRef.
- X. Li, W. Zhang, X. Guo, C. Lu, J. Wei and J. Fang, Constructing heterojunctions by surface sulfidation for efficient inverted perovskite solar cells, Science, 2022, 375, 434 CrossRef CAS PubMed.
- A. J. Downs and E. A. V. Ebsworth, 705. Silyl trifluoromethyl sulphide, J. Chem. Soc., 1960, 3516 RSC.
- B. Sternbach and A. G. MacDiarmid, The preparation and properties of silyl methyl sulphide, J. Inorg. Nucl. Chem., 1961, 23, 225 CrossRef CAS.
- M. Schmeißer and H. Frouzanfar, Darstellung von Mercaptosilanen, Z. Chem., 1968, 8, 254 CrossRef.
- E. A. V. Ebsworth, C. Glidewell and G. M. Sheldrick, Some reactions of trisilylphosphine, J. Chem. Soc. A, 1969, 352 RSC.
- J. E. Drake and C. Riddle, Electric discharge reactions of silane and germane with some volatile group VI species, J. Chem. Soc. A, 1970, 3134 RSC.
- J. W. Anderson and J. E. Drake, A. high yield synthesis of methylthio and methylseleno derivatives of silicon, germanium, and tin, Inorg. Nucl. Chem. Lett., 1971, 7, 1007 CrossRef CAS.
- S. Cradock, E. A. V. Ebsworth and H. F. Jessep, Ammonium salts of silanethiol and silaneselenol, J. Chem. Soc., Dalton Trans., 1972, 359 RSC.
- S. Cradock, E. A. V. Ebsworth, H. Moretto, D. W. H. Rankin and W. J. Savage, Lithium Derivatives of Silanol and Related Compounds, Angew. Chem., Int. Ed. Engl., 1973, 12, 317 CrossRef.
- M. A. Finch and C. H. van Dyke, Mixed silyl germyl Group VIA derivatives, Inorg. Chem., 1975, 14, 136 CrossRef CAS.
- A. Haas and M. Vongehr, Darstellung und Eigenschaften von Cyclotrisilathian sowie Trifluormethylchalkogenylsilanen (CF3X)nSiH4(n (X = S, Se und n = 1, 2, 3, 4), Z. Anorg. Allg. Chem., 1978, 447, 119 CrossRef CAS.
- A. Haas, R. Hitze, C. Krüger and K. Angermund, Darstellung und Charakterisierung von Silathia- und Silasela-Grundkörpern/Synthesis and Characterisation of Silathia and Silasela Basic Compounds, Z. Naturforsch., B, 1984, 39, 890 CrossRef.
- A. Haas, R. Süllentrup and C. Krüger, Synthese und Charakterisierung neuer Cyclischer und acyclischer Silachalkogenane mit Disilanyleinheiten, Z. Anorg. Allg. Chem., 1993, 619, 819 CrossRef CAS.
- (a) J. Kang, S. Tongay, J. Zhou, J. Li and J. Wu, Band offsets and heterostructures of two-dimensional semiconductors, Appl. Phys. Lett., 2013, 102, 012111 CrossRef; (b) M. Madhubalan, S. Sathish, M. Mahivardhan, P. Thandapani, K. K. Abimanyu, R. Kumar, J.-H. Seo, B.-W. Gu, S. M. Prabhu and B.-H. Jeon, Advances in Selenium–Based Materials for Supercapacitors: Chemistry, Interaction Mechanisms, and Practical Applications, Small Struct., 2025, 7, e202500715 CrossRef; (c) C. Wang, J. Guo, D. Liu, Z. Lin, S. Guo, S. Cai, J. Yan, B. He, Z. Zhang, M. Zhang and Y. Chai, Band-hybridized selenium contact for p-type semiconductors, Nat. Nanotechnol., 2026, 21, 207 CrossRef PubMed.
- E. A. V. Ebsworth, H. J. Emeléus and N. Welcman, 440. Perfluoroalkyl silyl selenides, J. Chem. Soc., 1962, 2290 RSC.
- S. Cradock, E. A. V. Ebsworth and D. W. H. Rankin, Studies in germyl chemistry. Part VIII. Digermyl selenide and digermyl telluride, J. Chem. Soc. A, 1969, 1628 RSC.
- G. K. Barker, J. E. Drake and R. T. Hemmings, Methylseleno-derivatives of Group IV. Part II. Hydrides, J. Chem. Soc., Dalton Trans., 1974, 450 RSC.
- J. E. Drake and R. T. Hemmings, Silyl and germyl derivatives of selenophenol and related species, J. Chem. Soc., Dalton Trans., 1976, 1730 RSC.
- J. E. Drake, B. M. Glavinčevski and R. T. Hemmings, Studies of silyl and germyl Group VI species. Part IV. Dimethyl- and tetramethyl-disilyl chalcogenides and related species, Can. J. Chem., 1980, 58, 2161 CrossRef CAS.
- A. Haas and R. Hitze, Preparation and Characterisation of Cyclotri(silaselane) (H2Si-Se)3, Z. Naturforsch., B, 1981, 36, 1069 CrossRef.
- (a) J. Zha, D. Dong, H. Huang, Y. Xia, J. Tong, H. Liu, H. P. Chan, J. C. Ho, C. Zhao, Y. Chai and C. Tan, Electronics and Optoelectronics Based on Tellurium, Adv. Mater., 2024, 36, e2408969 CrossRef PubMed; (b) S. Siddique, C. Chowde Gowda, S. Demiss, R. Tromer, S. Paul, K. K. Sadasivuni, E. F. Olu, A. Chandra, V. Kochat, D. S. Galvão, P. Kumbhakar, R. Mishra, P. M. Ajayan and C. S. Tiwary, Emerging two-dimensional tellurides, Mater. Today, 2021, 51, 402 CrossRef CAS; (c) J. Y. Kim, S. H. Park, H. J. Yang, M. W. Kim, D. H. Kim, S.-J. Choi and Y. J. Lee, Tellurium-based materials for nanoelectronics: applications, challenges, and outlooks, Microstructures, 2026, 6, 2026006 Search PubMed.
- H. Bürger and U. Goetze, Disilyltellurid, Inorg. Nucl. Chem. Lett., 1967, 3, 549 CrossRef.
- J. E. Drake and R. T. Hemmings, Studies of silyl and germyl Group 6 species. 5. Silyl and germyl derivatives of methane- and benzenetellurols, Inorg. Chem., 1980, 19, 1879 CrossRef CAS.
- (a) E. J. A. Pope and C. L. Hill, US8987402B2, 2015; (b) M. Bauer and S. G. Thomas, US8367528B2, 2013; (c) P. Tomasini and N. Cody, US7772097B2, 2010; (d) P. Tomasini, C. Arena, M. Bauer, C. Nyles, R. Betrram, W. Jianqing and M. D. Stephens, US2008026149A, 2008.
- (a) W. Kim, N. Hwang and Y. Lee, US2021222300A1, 2021; (b) T. Ying and Y. N. Sharad, WO2020123907A1, 2020.
- H. J. Emeléus and A. G. Maddock, 77. Derivatives of monosilane. Part III. The fluoromonosilanes, J. Chem. Soc., 1944, 293 RSC.
- D. Solan and A. B. Burg, Cocondensation reaction of difluorosilene with diborane. New polyfluoropolysilanes, Inorg. Chem., 1972, 11, 1253 CrossRef CAS.
- G. R. Langford, D. C. Moody and J. D. Odom, Reaction of silicon difluoride with phosphine, Inorg. Chem., 1975, 14, 134 CrossRef CAS.
- K. G. Sharp and J. F. Bald, Redistribution processes in mixed halodisilanes. Reaction of silicon difluoride with hydrogen bromide, Inorg. Chem., 1975, 14, 2553 CrossRef CAS.
- K. G. Sharp and J. L. Margrave, Silicon-fluorine chemistry. VII. Reaction of silicon difluoride with hydrogen sulfide, Inorg. Chem., 1969, 8, 2655 CrossRef CAS.
- J. J. D'Errico and K. G. Sharp, A new synthetic route to fluorodisilanes via selective reduction of halofluorodisilanes, Inorg. Chem., 1989, 28, 2886 CrossRef.
- J. E. Drake and N. Goddard, Halogenodisilanes, J. Chem. Soc. A, 1970, 2587 RSC.
- J. E. Drake, N. Goddard and N. P. C. Westwood, Halogenosilanes. Part III. Fluoro-, chloro-, and bromo-derivatives of trisilane, J. Chem. Soc. A, 1971, 3305 RSC.
- K. Hassler and W. Köll, Synthese und eigenschaften von 1,1,2,2-Tetrachlorund 1,1,2,2-Tetrafluordisilan, J. Organomet. Chem., 1995, 487, 223 CrossRef CAS.
- H. Stüger, Synthese 1,4-disubstituierter tetrasilane durch selektive spaltung von Si-phenyl-bindungen, J. Organomet. Chem., 1992, 433, 11 CrossRef.
- F. Höfler and R. Jannach, Zur darstellung fluorierter siliciumwasserstoffe, Inorg. Nucl. Chem. Lett., 1974, 10, 711 CrossRef.
- J. E. Drake and N. P. C. Westwood, Halogenosilanes. Part II. Electrical discharge production of halogenodisilanes, J. Chem. Soc. A, 1971, 3300 RSC.
- C. Ramírez-Márquez, Solar-Grade Silicon in the Energy Transition: A Strategic Commodity for the Global Photovoltaic Market, Commodities, 2025, 4, 18 CrossRef.
- (a) F. Chigondo, From Metallurgical-Grade to Solar-Grade Silicon: An Overview, Silicon, 2018, 10, 789 CrossRef CAS; (b) S. Yadav, K. Chattopadhyay and C. V. Singh, Solar grade silicon production: A review of kineticthermodynamic and fluid dynamics based continuum scale modeling, Renewable Sustainable Energy Rev., 2017, 78, 1288 CrossRef CAS.
- R. K. Agarwal, J. F. Lehmann, C. G. Coe and D. J. Tempel, US8206676B2, 2012.
- R. P. Hollandsworth, W. M. Ingle and M. A. Ring, Halogenation of silanes by silver chloride and silver bromide, Inorg. Chem., 1967, 6, 844 CrossRef CAS.
- G. Fritz and D. Kummer, Reaktionen der Phenylsilane mit Halogen und Halogenwasserstoffen, Z. Anorg. Allg. Chem., 1961, 308, 105 CrossRef CAS.
- C. H. van Dyke and A. G. MacDiarmid, Formation of diborane by the interaction of disilane with boron trichloride, J. Inorg. Nucl. Chem., 1963, 25, 1503 CrossRef CAS.
- J. E. Drake and N. Goddard, The formation and identification of chlorodisilanes and monochlorotrisilane, Inorg. Nucl. Chem. Lett., 1968, 4, 385 CrossRef CAS.
- R. P. Hollandsworth and M. A. Ring, Chlorodisilanes. Preparation and silicon-hydrogen stretching frequencies, Inorg. Chem., 1968, 7, 1635 CrossRef CAS.
- (a) R. P. Hollandsworth, W. M. Ingle and M. A. Ring, Halogenation of silanes by silver chloride and silver bromide, Inorg. Chem., 1967, 6, 844 CrossRef CAS; (b) A. J. Vanderwielen and M. A. Ring, Chlorination of silanes by silver chloride, Inorg. Chem., 1972, 11, 246 CrossRef CAS.
- M. Abedini, C. H. van Dyke and A. G. MacDiarmid, Preparation of disilanyl chloride and disilanyl bromide by the reaction of disilane with hydrogen halide, J. Inorg. Nucl. Chem., 1963, 25, 307 CrossRef CAS.
- F. Fehér and F. Ocklenburg, Beiträge zur Chemie des Siliciums und Germaniums. XXXV. Halogenierung von höheren Silanen mit Zinn(IV) – chlorid bzw. Quecksilber(II)–chlorid, Z. Anorg. Allg. Chem., 1984, 515, 36 CrossRef.
- F. Fehér, P. Plichta and R. Guillery, Chemistry of silicon and germanium. XII. Transformation of disilane, trisilane, and n-tetrasilane with elementary bromine and chlorine in Freon, Inorg. Chem., 1971, 10, 606 CrossRef.
- A. J. Vanderwielen and M. A. Ring, Chlorination of silanes by silver chloride, Inorg. Chem., 1972, 11, 246 CrossRef CAS.
- H. Stueger, T. Mitterfellner, R. Fischer, C. Walkner, M. Patz and S. Wieber, Partial halogenation of cyclic and branched perhydropentasilanes, Inorg. Chem., 2012, 51, 6173 CrossRef CAS PubMed.
- T. Lainer, R. Fischer, M. Leypold, M. Holthausen, O. Wunnicke, M. Haas and H. Stueger, Unusually selective synthesis of chlorohydrooligosilanes, Chem. Commun., 2020, 56, 13812 Search PubMed.
- F. Höfler, R. Jannach and W. Raml, Darstellung und Eigenschaften einiger Hochchlorierter Oligosilane, Z. Anorg. Allg. Chem., 1977, 428, 75 CrossRef.
- A. Gupper and K. Hassler, Synthesis and Properties of 1,2-Dichlorodisilane and 1,1,2-Trichlorodisilane, Eur. J. Inorg. Chem., 2001, 2001, 2007 CrossRef.
- K. Hassler and W. Köll, Chlor-, Brom- und Iodtrisilane: Synthesen und 29Si-Kernresonanzspektren, J. Organomet. Chem., 1997, 540, 113 CrossRef CAS.
- H. Söllradl and E. Hengge, Vergleichende 29Si-NMR-untersuchungen an verschiedenen disilanderivaten, J. Organomet. Chem., 1983, 243, 257 Search PubMed.
- W. Uhlig, Synthese funktionell substituierter Disilane auf der Basis von Triflatderivaten, Z. Anorg. Allg. Chem., 1993, 619, 1479 CrossRef CAS.
- (a) J. C. Schuhmacher, The production of solar-cell-grade silicon from bromosilanes, Washington, DC, 1979 Search PubMed; (b) J. C. Schuhmacher, EP0114876B1, 1990.
- N. S. Hosmane, Stannic bromide: A selective brominating agent for silanes, Inorg. Nucl. Chem. Lett., 1974, 10, 1077 CrossRef CAS.
- L. G. L. Ward, A. D. Norman, S. K. Gondal and A. G. MacDiarmid, in Inorganic Syntheses, ed. W. L. Jolly, Wiley, 1968, vol. 11, pp. 159–170 Search PubMed.
- T. C. Geisler, C. G. Cooper and A. D. Norman, Bromination of silanes and germane, Inorg. Chem., 1972, 11, 1710 CrossRef CAS.
- H. Stüger and P. Lassacher, Darstellung und spektroskopische charakterisierung verzweigter funktioneller hexasilangerüste, J. Organomet. Chem., 1993, 450, 79 CrossRef.
- (a) G. Tamizhmani, M. Cocivera, R. T. Oakley and P. Del Bel Belluz, Some physical properties of undoped amorphous silicon prepared by a new CVD process using iodosilanes, Chem. Mater., 1990, 2, 473 CrossRef CAS; (b) G. Kuchenbeiser and B. Lefevre, US9777373B2, 2017.
- A. G. Maddock, C. Reid and H. J. Emeléus, New Iodine and Fluorine Derivatives of Monosilane, Nature, 1939, 144, 328 CrossRef CAS.
- B. J. Aylett and I. A. Ellies, The preparation of iodosilanes from phenylchlorosilanes, J. Chem. Soc., 1960, 3415 CAS.
- F. Fehér, B. Mostert, A. G. Wronka and G. Betzen, Präprative Darstellung von Disilanyl- und Trisilanyljodiden, Monatsh. Chem., 1972, 103, 959 CrossRef.
- F. Fehér, P. Plichta and R. Guillery, Beiträge zur Chemie des Siliciums und Germaniums, XI. Darstellung und Untersuchung neuer Jodderivate des Disilans, Trisilans und n–Tetrasilans, Chem. Ber., 1970, 103, 3028 CrossRef.
- (a) K. Schenzel and K. Hassler, Bromdisilan: Verbesserte synthese, schwingungsspektren und normalkoordinatenanalyse, Spectrochim. Acta, Part A, 1994, 50, 139 CrossRef; (b) K. Hassler, W. Köll and K. Schenzel, Syntheses, infrared and Raman vibrational spectra, normal coordinate analyses and 29Si-NMR-Spectra of halogenated disilanes XnSi2H6(n (X = F, Cl, Br, I), J. Mol. Struct., 1995, 348, 353 CrossRef CAS; (c) K. Hassler and M. Pöschl, Synthese und Kernresonanzspektren von Bromdisilanen und Ioddisilanen, J. Organomet. Chem., 1990, 398, 225 CrossRef CAS.
- U. Pöschl and K. Hassler, Synthesis and Spectroscopy of Halogenated Cyclopentasilanes, Organometallics, 1996, 15, 3238 CrossRef.
- K. G. Sharp and J. F. Bald, Redistribution processes in mixed halodisilanes. Reaction of silicon difluoride with hydrogen bromide, Inorg. Chem., 1975, 14, 2553 CrossRef CAS.
- J. Emsley, Nature's building blocks: an A-Z guide to the elements, Oxford University Press, 2001 Search PubMed.
- R. A. Street, Hydrogenated Amorphous Silicon, Cambridge University Press, 2010 Search PubMed.
- J. A. Rossi and R. K. Willardson, Silicon Epitaxy, Elsevier textbooks, s.l., 1st edn, 2001, vol. 72 Search PubMed.
- Springer Handbook of Electronic and Photonic Materials, ed. S. Kasap and P. Capper, Springer US, Boston, MA, 2007 Search PubMed.
- P. Sutter, Introduction to semiconductors : from materials to devices, Springer, Cham, 2025 Search PubMed.
- Charge Transport in Disordered Solids with Applications in Electronics, ed. S. Baranovski, Wiley, Chichester, England, 2006 Search PubMed.
- (a) M. K. K. Takahashi, Amorphous silicon solar cells, Wiley, New York, 1986 Search PubMed; (b) J. Nelson, The Physics of Solar Cells, Published by Imperial College Press and distributed by World Scientific Publishing Co, 2003 CrossRef; (c) A. J. McEvoy, L. Castañer and T. Markvart, Solar cells : materials, manufacture and operation, ed. A. McEvoy, L. Castaner and T. Markvart, Elsevier, Waltham, MA, 2nd edn, 2013 Search PubMed; (d) S. K. Sharma, H. Im, D. Y. Kim and R. M. Mehra, Review on Se-and S-doped hydrogenated amorphous silicon thin films, Indian J. Pure Appl. Phys., 2014, 293 Search PubMed.
- A. Vora, PhD, Dissertation, Michigan Technological University, 2015.
- (a) Solution Processing of Inorganic Materials, ed. D. B. Mitzi, Wiley, Hoboken, New Jersey, 2008 Search PubMed; (b) M. Mews, C. Mader, S. Traut, T. Sontheimer, O. Wunnicke, L. Korte and B. Rech, Solution-processed amorphous silicon surface passivation layers, Appl. Phys. Lett., 2014, 105 Search PubMed.
- T. Bronger, P. H. Wöbkenberg, J. Wördenweber, S. Muthmann, U. W. Paetzold, V. Smirnov, S. Traut, Ü. Dagkaldiran, S. Wieber, M. Cölle, A. Prodi-Schwab, O. Wunnicke, M. Patz, M. Trocha, U. Rau and R. Carius, Solution–Based Silicon in Thin–Film Solar Cells, Adv. Energy Mater., 2014, 4, 1301871 CrossRef.
- M. Allendorf, From Bunsen to VLSI, Electrochem. Soc. Interface, 1998, 7, 36 CrossRef CAS.
- K. Choy, Chemical vapour deposition of coatings, Prog. Mater. Sci., 2003, 48, 57 CrossRef CAS.
- H. Katsui and T. Goto, in Multi-dimensional Additive Manufacturing, ed. S. Kirihara and K. Nakata, Springer Singapore, Singapore, 2021, pp. 75–95 Search PubMed.
- L. Sun, G. Yuan, L. Gao, J. Yang, M. Chhowalla, M. H. Gharahcheshmeh, K. K. Gleason, Y. S. Choi, B. H. Hong and Z. Liu, Chemical vapour deposition, Nat. Rev. Methods Primers, 2021, 1, 1–20 Search PubMed.
- E. C. Nnadozie, K. I. Ogunwa, V. I. Chukwuike, O. O. Nnadozie and C. Ehikhase, Synthesis and Characterization of Carbonaceous Materials for Medical Applications: A Comprehensive Review, BioMed, 2024, 4, 464 CrossRef.
- H. C. Theuerer, Epitaxial Silicon Films by the Hydrogen Reduction of SiCl4, J. Electrochem. Soc., 1961, 108, 649 CrossRef CAS.
- C. H. Lewis, H. C. Kelly, M. B. Giusto and S. Johnson, Preparation of High-Purity Silicon from Silane, J. Electrochem. Soc., 1961, 108, 1114 CrossRef CAS.
- W. O. Filtvedt, A. Holt, P. A. Ramachandran and M. C. Melaaen, Chemical vapor deposition of silicon from silane: Review of growth mechanisms and modeling/scaleup of fluidized bed reactors, Sol. Energy Mater. Sol. Cells, 2012, 107, 188 CrossRef CAS.
- K. Tonokura and M. Koshi, Reaction kinetics in silicon chemical vapor deposition, Curr. Opin. Solid State Mater. Sci., 2002, 6, 479 CrossRef CAS.
- (a) M. T. Swihart and S. L. Girshick, Thermochemistry and Kinetics of Silicon Hydride Cluster Formation during Thermal Decomposition of Silane, J. Phys. Chem. B, 1999, 103, 64 CrossRef CAS; (b) P. Zhang, J. Duan, G. Chen, J. Li and W. Wang, Production of polycrystalline silicon from silane pyrolysis: A review of fines formation, Sol. Energy, 2018, 175, 44 CrossRef CAS.
- C. W. Liu and J. C. Sturm, Low temperature chemical vapor deposition growth of β-SiC on (100) Si using methylsilane and device characteristics, J. Appl. Phys., 1997, 82, 4558 CrossRef CAS.
- S. Madapura, A. J. Steckl and M. Loboda, Heteroepitaxial Growth of SiC on Si(100) and (111) by Chemical Vapor Deposition Using Trimethylsilane, J. Electrochem. Soc., 1999, 146, 1197 CrossRef CAS.
- R. Hazbun, J. Hart, R. Hickey, A. Ghosh, N. Fernando, S. Zollner, T. N. Adam and J. Kolodzey, Silicon epitaxy using tetrasilane at low temperatures in ultra-high vacuum chemical vapor deposition, J. Cryst. Growth, 2016, 444, 21 CrossRef CAS.
- D.-S. Byeon, C. Cho, D. Yoon, Y. Choi, K. Lee, S. Baik and D.-H. Ko, Epitaxial Growth of Si and SiGe Using High-Order Silanes without a Carrier Gas at Low Temperatures via UHVCVD and LPCVD, Coatings, 2021, 11, 568 CrossRef CAS.
- G. Dhanaraj, Y. Chen, M. Dudley and H. Zhang, Growth and Surface Morphologies of 6H SiC Bulk and Epitaxial Crystals, Mater. Sci. Forum, 2006, 527–529, 67 CAS.
- O. Sneh, M. L. Wise, A. W. Ott, L. A. Okada and S. M. George, Atomic layer growth of SiO2 on Si(100) using SiCl4 and H2O in a binary reaction sequence, Surf. Sci., 1995, 334, 135 CrossRef CAS.
- J. Gumpher, W. Bather, N. Mehta and D. Wedel, Characterization of Low-Temperature Silicon Nitride LPCVD from Bis(tertiary-butylamino)silane and Ammonia, J. Electrochem. Soc., 2004, 151, G353 CrossRef CAS.
- B. B. Burton, S. W. Kang, S. W. Rhee and S. M. George, SiO 2 Atomic Layer Deposition Using Tris(dimethylamino)silane and Hydrogen Peroxide Studied by in Situ Transmission FTIR Spectroscopy, J. Phys. Chem. C, 2009, 113, 8249 CrossRef CAS.
- M. A. Todd, US2002173113A1, 2002.
- Chemical vapour deposition. Precursors, processes and applications, ed. A. C. Jones and M. L. Hitchman, Royal Society of Chemistry, Cambridge, UK, 2009 Search PubMed.
- F. Wang and J. Wu, in Modern Ion Plating Technology, Elsevier, 2023, pp. 247–285 Search PubMed.
- R. Murri, Silicon Based Thin Film Solar Cells, Bentham Science Publishers, 2013 Search PubMed.
- H. F. Sterling and R. Swann, Chemical vapour deposition promoted by r.f. discharge, Solid-State Electron., 1965, 8, 653 Search PubMed.
- M. H. Brodsky, Plasma preparations of amorphous silicon films, Thin Solid Films, 1978, 50, 57 CrossRef CAS.
- W. E. Spear and P. G. Le Comber, Substitutional doping of amorphous silicon, Solid State Commun., 1975, 17, 1193 CrossRef.
- P. G. Le Comber, W. E. Spear and A. Ghaith, Amorphous-silicon field-effect device and possible application, Electron. Lett., 1979, 15, 179 Search PubMed.
- A. Matsuda, Formation kinetics and control of microcrystallite in μc-Si:H from glow discharge plasma, J. Non-Cryst. Solids, 1983, 59–60, 767 CrossRef CAS.
- K. Pokhodnya, K. J. Anderson and P. R. Boudjouk, in 2014 IEEE 40th Photovoltaic Specialist Conference (PVSC), IEEE, 2014, 2014, pp. 3065–3067.
- W.-J. Lee and Y.-H. Choa, Highly conformal carbon-doped SiCN films by plasma-enhanced chemical vapor deposition with enhanced barrier properties, Thin Solid Films, 2018, 657, 32 CrossRef CAS.
- (a) H. Keppner, J. Meier, P. Torres, D. Fischer and A. Shah, Microcrystalline silicon and micromorph tandem solar cells, Appl. Phys. A, 1999, 69, 169 CrossRef CAS; (b) A. Shah, J. Meier, E. Vallat-Sauvain, N. Wyrsch, U. Kroll, C. Droz and U. Graf, Material and solar cell research in microcrystalline silicon, Sol. Energy Mater. Sol. Cells, 2003, 78, 469 CrossRef CAS.
- (a) W. J. Soppe, C. Devilee, M. Geusebroek, J. Löffler and H.-J. Muffler, The effect of argon dilution on deposition of microcrystalline silicon by microwave plasma enhanced chemical vapor deposition, Thin Solid Films, 2007, 515, 7490 CrossRef CAS; (b) W. J. Soppe, A. C. W. Biebericher, C. Devilee, H. Donker and H. Schlemm, High-rate growth of microcrystalline silicon by microwave-PECVD, Proc. 3rd World Conf. Photovolt. Energy Convers., 2003.
- (a) B. Rech, J. Müller, T. Repmann, O. Kluth, T. Roschek, J. Hüpkes, H. Stiebig and W. Appenzeller, Amorphous and Microcrystalline Silicon Based Solar Cells and Modules on Textured Zinc Oxide Coated Glass Substrates, MRS Proc., 2003, 762, 439–447 Search PubMed; (b) T. Roschek, T. Repmann, J. Müller, B. Rech and H. Wagner, Comprehensive study of microcrystalline silicon solar cells deposited at high rate using 13.56 MHz plasma-enhanced chemical vapor deposition, J. Vac. Sci. Technol., A, 2002, 20, 492 CrossRef CAS.
- (a) B. Rech and H. Wagner, Potential of amorphous silicon for solar cells, Appl. Phys. A, 1999, 69, 155 CrossRef CAS; (b) A. V. Shah, H. Schade, M. Vanecek, J. Meier, E. Vallat-Sauvain, N. Wyrsch, U. Kroll, C. Droz and J. Bailat, Thin–film silicon solar cell technology, Prog. Photovolt. Res. Appl., 2004, 12, 113 CrossRef CAS.
- Coating materials. Computational aspects, applications and challenges, ed. A. Verma, S. K. Sethi and S. Ogata, Springer, Singapore, 2023 Search PubMed.
- C. A. Dorval Dion and J. R. Tavares, Photo-initiated chemical vapor deposition as a scalable particle functionalization technology (a practical review), Powder Technol., 2013, 239, 484 CrossRef CAS.
- T. Saitoh, S. Muramatsu, T. Shimada and M. Migitaka, Optical and electrical properties of amorphous silicon films prepared by photochemical vapor deposition, Appl. Phys. Lett., 1983, 42, 678 CrossRef CAS.
- T. Inoue, M. Konagai and K. Takahashi, Photochemical vapor deposition of undoped and n -type amorphous silicon films produced from disilane, Appl. Phys. Lett., 1983, 43, 774 CrossRef CAS.
- T. F. Deutsch, D. J. Ehrlich and R. M. Osgood, Laser photodeposition of metal films with microscopic features, Appl. Phys. Lett., 1979, 35, 175 Search PubMed.
- Research progress of laser-assisted chemical vapor deposition, https://www.oejournal.org/oee/en/article/id/621efb7e99d88174c21c44a1, (accessed 2 March 2026).
- H. Matsumura, H. Umemoto and A. Masuda, Cat-CVD (hot-wire CVD): how different from PECVD in preparing amorphous silicon, J. Non-Cryst. Solids, 2004, 338–340, 19 CrossRef CAS.
- R. E. Schropp, Hot Wire Chemical Vapor Deposition: Recent Progress, Present State of the Art and Competitive Opportunities, ECS Trans., 2009, 25, 3 CrossRef CAS.
- (a) H. Wiesmann, A. K. Ghosh, T. McMahon and M. Strongin, a-Si : H produced by high-temperature thermal decomposition of silane, J. Appl. Phys., 1979, 50, 3752 CrossRef CAS; (b) M. Strongin, A. K. Ghosh, H. J. Wiesmann, E. B. Rock and H. A. Lutz III, US4237151A, 1980.
- (a) H. Matsumura, Catalytic Chemical Vapor Deposition (CTC–CVD) Method Producing High Quality Hydrogenated Amorphous Silicon, Jpn. J. Appl. Phys., 1986, 25, L949 CrossRef CAS; (b) H. Matsumura, Study on catalytic chemical vapor deposition method to prepare hydrogenated amorphous silicon, J. Appl. Phys., 1989, 65, 4396 CrossRef CAS.
- J. Doyle, R. Robertson, G. H. Lin, M. Z. He and A. Gallagher, Production of high-quality amorphous silicon films by evaporative silane surface decomposition, J. Appl. Phys., 1988, 64, 3215 CrossRef CAS.
- A. H. Mahan, J. Carapella, B. P. Nelson, R. S. Crandall and I. Balberg, Deposition of device quality, low H content amorphous silicon, J. Appl. Phys., 1991, 69, 6728 CrossRef CAS.
- R. E. I. Schropp and M. Zeman, Amorphous and Microcrystalline Silicon Solar cells: Modeling, Materials and Device Technology, Springer US, 1998 Search PubMed.
- T. Suntola, Atomic layer epitaxy, Thin Solid Films, 1992, 216, 84 CrossRef CAS.
- M. Leskelä and M. Ritala, Atomic layer deposition chemistry: recent developments and future challenges, Angew. Chem., Int. Ed., 2003, 42, 5548 CrossRef PubMed.
- F. Hirose, M. Suemitsu and N. Miyamoto, Silane gas-source atomic layer epitaxy, Appl. Surf. Sci., 1992, 60–61, 592 CrossRef CAS.
- H. Matsunami and T. Kimoto, Step-controlled epitaxial growth of SiC: High quality homoepitaxy, Mater. Sci. Eng., R, 1997, 20, 125 CrossRef.
- H. Pedersen, S. Leone, A. Henry, F. C. Beyer, V. Darakchieva and E. Janzén, Very high growth rate of 4H-SiC epilayers using the chlorinated precursor methyltrichlorosilane (MTS), J. Cryst. Growth, 2007, 307, 334 CrossRef CAS.
- (a) S. Nishino, T. Miyanagi and Y. Nishio, Epitaxial Growth of SiC on α-SiC Using Si2Cl6+C3H8+H2 System, Mater. Sci. Forum, 1998, 264–268, 139 CAS; (b) T. Rana, M. Chandrashekhar and T. S. Sudarshan, Vapor phase surface preparation (etching) of 4H–SiC substrates using tetrafluorosilane (SiF4) in a hydrogen ambient for SiC epitaxy, J. Cryst. Growth, 2013, 380, 61 CrossRef CAS.
- S. Kamiyama, T. Miura and Y. Nara, Comparison between SiO2 films deposited by atomic layer deposition with SiH2[N(CH3)2]2 and SiH[N(CH3)2]3 precursors, Thin Solid Films, 2006, 515, 1517 CrossRef CAS.
- T. Faraz, M. van Drunen, H. C. M. Knoops, A. Mallikarjunan, I. Buchanan, D. M. Hausmann, J. Henri and W. M. M. Kessels, Atomic Layer Deposition of Wet-Etch Resistant Silicon Nitride Using Di(sec-butylamino)silane and N2 Plasma on Planar and 3D Substrate Topographies, ACS Appl. Mater. Interfaces, 2017, 9, 1858 CrossRef CAS PubMed.
- (a) J. D. Ferguson, E. R. Smith, A. W. Weimer and S. M. George, ALD of SiO2 at Room Temperature Using TEOS and H2O with NH3 as the Catalyst, J. Electrochem. Soc., 2004, 151, G528 CrossRef CAS; (b) V. Y. Vasilyev, Evaluation of Low Temperature TEOS-Ozone Silicon Dioxide Thin Film CVD under Sub-Atmospheric Pressure Using Consecutively Pulsed Reactant Injection, ECS J. Solid State Sci. Technol., 2015, 4, N3164–N3167 CrossRef CAS.
- (a) D. B. Mitzi, Solution Processing of Chalcogenide Semiconductors via Dimensional Reduction, Adv. Mater., 2009, 21, 3141 CrossRef CAS; (b) D. B. Mitzi, L. L. Kosbar, C. E. Murray, M. Copel and A. Afzali, High-mobility ultrathin semiconducting films prepared by spin coating, Nature, 2004, 428, 299 CrossRef CAS PubMed.
- M. Gerwig, A. S. Ali, D. Neubert, S. Polster, U. Böhme, G. Franze, M. Rosenkranz, A. Popov, I. Ponomarev, M. P. M. Jank, C. Viehweger, E. Brendler, L. Frey, P. Kroll and E. Kroke, From Cyclopentasilane to Thin–Film Transistors, Adv. Electron. Mater., 2021, 7, 2000422 CrossRef CAS.
- A. R. Wolff and R. West, Photoinitiation of vinyl polymerization by polysilanes, Appl. Organomet. Chem., 1987, 1, 7 CrossRef CAS.
- R. D. Miller and J. Michl, Polysilane high polymers, Chem. Rev., 1989, 89, 1359 CrossRef CAS.
- (a) T. Shimoda, Nanoliquid Processes for Electronic Devices, Springer Singapore, Singapore, 2019 CrossRef; (b) T. Masuda, Y. Matsuki and T. Shimoda, Pyrolytic transformation from polydihydrosilane to hydrogenated amorphous silicon film, Thin Solid Films, 2012, 520, 6603 CrossRef CAS.
- T. Shimoda and T. Masuda, Liquid silicon and its application in electronics, Jpn. J. Appl. Phys., 2014, 53, 02BA01 CrossRef CAS.
- M. Trifunovic, T. Shimoda and R. Ishihara, Solution-processed polycrystalline silicon on paper, Appl. Phys. Lett., 2015, 106 Search PubMed.
- H. Frey, R. Lauth, H. Khan, N. Eisenreich and A. Koleczko, Deposition of Silicon Films from Liquid Cyclopentasilane Precursors Using High Pressure Spray System, Phys. Sci. Int. J., 2017, 14, 1 CrossRef.
- T. Masuda, M. Nakayama, K. Saito, H. Katayama and A. Terakawa, Inkjet Printing of Liquid Silicon, Macromol. Rapid Commun., 2020, 41, e2000362 CrossRef PubMed.
- T. Masuda, H. Takagishi, K. Yamazaki and T. Shimoda, Direct Imprinting of Liquid Silicon, ACS Appl. Mater. Interfaces, 2016, 8, 9969 CrossRef CAS PubMed.
- Z. Shen, T. Masuda, H. Takagishi, K. Ohdaira and T. Shimoda, Fabrication of high-quality amorphous silicon film from cyclopentasilane by vapor deposition between two parallel substrates, Chem. Commun., 2015, 51, 4417 RSC.
- G. R. S. Iyer, E. K. Hobbie, S. Guruvenket, J. M. Hoey, K. J. Anderson, J. Lovaasen, C. Gette, D. L. Schulz, O. F. Swenson, A. Elangovan and P. Boudjouk, Solution-based synthesis of crystalline silicon from liquid silane through laser and chemical annealing, ACS Appl. Mater. Interfaces, 2012, 4, 2680 CrossRef CAS PubMed.
- (a) A. P. Cádiz Bedini, B. Klingebiel, M. Luysberg and R. Carius, Sonochemical synthesis of hydrogenated amorphous silicon nanoparticles from liquid trisilane at ambient temperature and pressure, Ultrason. Sonochem., 2017, 39, 883 CrossRef PubMed; (b) A. P. Cádiz Bedini, S. Muthmann, J. Allgaier, K. Bittkau, F. Finger and R. Carius, Liquid hydridosilane precursor prepared from cyclopentasilane via sonication at low temperatures without the action of light, Ultrason. Sonochem., 2017, 34, 289 CrossRef PubMed; (c) TechConnect briefs 2017, ed. M. Laudon and B. Romanowicz, TechConnect, Danville, CA, U.S.A., 2017, vol. 1 Search PubMed.
- F. Haase, B. Lim, A. Merkle, T. Dullweber, R. Brendel, C. Günther, M. H. Holthausen, C. Mader, O. Wunnicke and R. Peibst, Printable liquid silicon for local doping of solar cells, Sol. Energy Mater. Sol. Cells, 2018, 179, 129 CrossRef CAS.
- (a) M. Holthausen, M. Roccaro, A. Pougin, D. Schmitt, J. Zoellner, C. Daeschlein and O. Wunnicke, EP3702397B1, 2021; (b) M. Holthausen, M. Roccaro, A. Pougin, D. Schmitt, J. Zöllner, C. Däschlein and O. Wunnicke, EP3702397A1, 2020; (c) H. Stüger, V. S. Christopoulos, M. Haas, O. Wunnicke, M. Roccaro and M. Holthausen, WO2022063680A1, 2022; (d) H. Stüger, M. Haas, O. Wunnicke, M. D. Malsch and M. Holthausen, EP3590889A1, 2020.
- T. Masuda, A. Iwasaka, H. Takagishi and T. Shimoda, Polymeric precursor for solution-processed amorphous silicon carbide, J. Mater. Chem. C, 2015, 3, 12212 RSC.
| This journal is © the Partner Organisations 2026 |