
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymers containing pendant lactone groups: synthesis and potential for sustainable materials
Azra Kocaarslan *a and
Baris Kumru*b
*a and
Baris Kumru*b
aInstitute of Functional Interfaces, Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen, Karlsruhe, Germany. E-mail: azra.kocaarslan@partner.kit.edu
bSchool of Energy Science and Engineering (ESE), Vidyasirimedhi Institute of Science and Technology (VISTEC), Wangchan Valley, 21210 Rayong, Thailand
First published on 28th May 2026
Abstract
The transition from fossil-based plastics to sustainable and circular polymer materials requires not only renewable feedstocks but also chemically diverse structural motifs. Lactones, cyclic esters present in natural products and pharmaceuticals, occupy a privileged position in polyester synthesis. While they are classically exploited as monomers for ring-opening polymerization to produce polyesters, vinyl-functionalized lactones and lactone-derived motifs offer a broader design space when incorporated as pendant groups into synthetic polymer backbones. In particular, the five- and six-membered ring structures of γ- and δ-lactones impart properties like those of their acrylate counterparts. Inspired by their natural occurrence and developments in biorefinery technologies, polymers bearing pendant lactone groups are emerging as attractive functional materials. In this minireview, we highlight polymers bearing pendant five- and six-membered lactone groups as an emerging platform for sustainable polymer materials.
1. Introduction
Synthetic polymers are found in almost every aspect of modern life, from packaging, coatings and textiles to electronics, energy technologies and aerospace.1,2 This versatility, however, comes at a cost. The overwhelming majority of today's polymers are derived from fossil resources, which translates into persistent greenhouse-gas emissions across the life cycle and a heavy reliance on non-renewable carbon.3 Moving towards sustainable polymers therefore requires more than just “drop-in” bio-based monomers: it calls for an integrated approach that couples renewable feedstocks with energy-efficient synthesis and compatibility with circular material flows.Nature offers a powerful source of inspiration for this transition.4 Natural macromolecules such as cellulose, starch, chitin, lignin, proteins and nucleic acids demonstrate how complex functions can be built from a limited set of simple, renewable building blocks under mild conditions. Cyclic motifs, including sugars, lactones and lactams, are particularly prominent and are used by biology to encode reactivity, recognition and dynamic behavior into macromolecular frameworks.5 The integration of vinyl polymerization with naturally derived monomers creates a powerful platform for designing sustainable, high-performance polymers. Towards this goal, lactone-derived vinyl monomers are particularly attractive because they combine a polymerizable carbon–carbon double bond with a cyclic ester functionality in a single molecular structure. In contrast to lactones used directly for ring-opening polymerization, the monomers considered in this review are vinyl-functionalized lactones in which chain-growth polymerization occurs primarily through the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. This distinction is important: polymerization of the double bond produces carbon–carbon polymer backbones analogous to those obtained from conventional acrylic and methacrylic monomers, while the lactone unit is retained as a pendant cyclic ester group. As a result, these monomers offer a route to acrylic-like materials with additional functionality, potential bio-based origin and opportunities for post-polymerization modification through the lactone ring. A representative example is α-methylene-γ-butyrolactone, which can be regarded as a cyclic analogue of methyl methacrylate (MMA). Polymerization through its exocyclic double bond affords polymers with high glass-transition temperatures (Tg around 195 °C), good durability and high refractive indices (1.540), while the pendant lactone ring provides a functional handle for further chemical transformation.6 Related vinyl lactone monomers have therefore attracted increasing interest as renewable alternatives or complements to petroleum-derived acrylates and methacrylates. Their reactivity, polymerization behavior, and structure–property relationships are strongly governed by the position and substitution pattern of the double bond relative to the lactone ring, distinguishing these monomers from both simple lactones and conventional vinyl monomers.
C bond. This distinction is important: polymerization of the double bond produces carbon–carbon polymer backbones analogous to those obtained from conventional acrylic and methacrylic monomers, while the lactone unit is retained as a pendant cyclic ester group. As a result, these monomers offer a route to acrylic-like materials with additional functionality, potential bio-based origin and opportunities for post-polymerization modification through the lactone ring. A representative example is α-methylene-γ-butyrolactone, which can be regarded as a cyclic analogue of methyl methacrylate (MMA). Polymerization through its exocyclic double bond affords polymers with high glass-transition temperatures (Tg around 195 °C), good durability and high refractive indices (1.540), while the pendant lactone ring provides a functional handle for further chemical transformation.6 Related vinyl lactone monomers have therefore attracted increasing interest as renewable alternatives or complements to petroleum-derived acrylates and methacrylates. Their reactivity, polymerization behavior, and structure–property relationships are strongly governed by the position and substitution pattern of the double bond relative to the lactone ring, distinguishing these monomers from both simple lactones and conventional vinyl monomers.
This review highlights the potential of lactone-pendant polymers and focuses on the synthesis, polymerization and functionalization of lactone-containing vinyl monomers. Particular emphasis is given on the reactivity of the polymerizable double bond and the properties of the resulting polymers bearing pendant lactone groups. Lactones that lack the vinyl functionality or are used solely as monomers for ring-opening polymerization are discussed briefly to not to lose sight of the strength of the respective monomers in ring-opening polymerization (ROP) and direct readers to potential post-polymerization modification strategies.
2. Synthesis of five- and six-membered lactones from bio-sources
Lactones are cyclic carboxylic esters that are typically formed by intramolecular esterification (“lactonization”) of hydroxycarboxylic acids, and they occur in ring sizes ranging from strained four-membered β-lactones to five- and six-membered γ- and δ-lactones and up to large macrocyclic systems.7 This simple motif appears widely in nature, where lactones serve as flavour and fragrance components (for example, many γ- and δ-lactones responsible for creamy, fruity aromas such as peach and coconut), pharmacophores in bioactive natural products such as macrolide antibiotics and sesquiterpene lactones,8 and key signals in chemical communication across insects, vertebrates, and bacteria. For key lactone motifs in natural signalling, we would like to guide readers to the review by Schulz and Hötling, which covers the topic from an organic chemistry perspective.9 Lactone properties are strongly shaped by ring size and substitution, such that ring strain and conjugation control the thermodynamics of ring-opening, while the presence of additional functional groups (e.g., aromatics, alkenes, halogens, or heteroatoms) modulates electrophilicity, hydrolytic stability, and volatility.10 From a synthetic standpoint, lactones can be accessed not only by classical acid- or base-catalysed lactonization of hydroxy acids, but also via modern methods such as the Baeyer–Villiger oxidation of cyclic ketones, transition-metal-catalysed oxidative cyclizations, and halolactonization of unsaturated acids, which together provide broad control over stereochemistry and substitution patterns.112.1 Ring-opening polymerization (ROP) of lactones
Beyond their roles as functional organic building blocks, lactones have emerged as a versatile class of monomers whose ROP provides access to polyesters with finely tunable thermal, mechanical, and degradation profiles.12 The thermodynamics of lactone ROP are governed primarily by ring strain and substitution patterns. Consequently, the chemical structure of the monomer, the reaction medium, and the nature of the catalyst collectively dictate the position of the polymer–monomer equilibrium and ultimately the attainable molar mass and conversion.13 In practice, a broad toolbox of catalysts is available, including metal-based coordination–insertion systems (e.g. Al, Sn, and Zn complexes), organic acids and bases, and bifunctional organocatalysts, enabling controlled ROP of diverse lactone families ranging from ε-caprolactone and δ-valerolactone to functionalized lactones and macrolactones, into well-defined polyesters.14,15 Complementary to these small-molecule approaches, enzymes such as lipases and cutinases can mediate lactone ROP under mild conditions, offering metal-free, often solvent-lean processes that are attractive from a green chemistry perspective; for a systematic overview of such biocatalytic systems, we refer readers to the review by Kara and co-workers.16 Another systematic investigation has been reported by the Zhao group, which examined the polymerization process at different temperatures catalyzed by different types and batches of lipases with varying water contents.17In fact, aliphatic polyesters obtained through ROP of moderately strained lactones represent an attractive platform for the development of closed-loop recyclable polymers. In a recent review, Li and co-workers summarized progress in polyesters derived from four-, five-, six-, and seven-membered lactones, with particular emphasis on the thermodynamic and kinetic factors that govern both polymerization and depolymerization. The review also discussed monomer design, polymerization methods, material properties, and strategies for chemical recycling. In this context, the design of lactone monomers is especially important. Suitable structural features must allow the monomer to polymerize efficiently while also enabling the resulting polyester to depolymerize back to the original monomer under appropriate conditions. This balance between polymerizability and depolymerizability is central to the development of closed-loop recyclable polyesters.18 Within this direction, for instance, Zhu and co-workers demonstrated a benzo-fusion strategy for the design of chemically recyclable semiaromatic polymers. By incorporating a fused aromatic ring into cyclic ester monomers, they showed that the monomer structure can be used to tune both the thermodynamics of ring-opening polymerization and the properties of the resulting polymers.19,20 These works clearly illustrate how rational monomer design can address the central challenge in closed-loop recyclable polymers by achieving efficient polymerization without sacrificing depolymerizability or material performance. The resulting polyesters can be designed to span from slowly degrading, high-Tg engineering materials to rapidly hydrolysable, bioresorbable matrices for biomedical and packaging applications, which reflect the structural and functional breadth of lactone-based platforms.18 In a closely related synthetic manifold, polythioesters with distinct degradation behavior and sulfur-containing functionalities are accessible via ROP of thiolactone monomers.21 Given the extensive number of studies and reviews dedicated to lactone ROP, readers are referred to the respective works for detailed discussions focusing specifically on ROP methodologies.22,23
In addition to larger lactone derivatives, five-membered ring lactones have also attracted interest as monomers for ROP. Their polymerization generally involves catalyst-mediated ring opening of the cyclic ester, followed by propagation to form the corresponding aliphatic polyester. Scheme 1 illustrates the ring-opening polymerization of α-methylene-γ-butyrolactone, yielding a polyester with pendant double bonds that can be further modified via PPM.
 | ||
| Scheme 1 Ring opening polymerization of α-methylene-γ-butyrolactone (MGBL) based on metal-based, organocatalytic and dual catalytic systems. Reproduced from ref. 24, published under a CC-BY 4.0 license.24 | ||
Beyond their role as monomers for ROP, lactones provide valuable functional groups for post-polymerization modification (PPM) reactions. Polymers bearing pendant lactone-derived cyclic ester groups, including γ-butyrolactone-, thiolactone-, and azlactone-based motifs, have been used as reactive platforms for PPM.24–26 Indeed, the PPM strategy provides access to polymer libraries with tunable chemical composition and material properties without requiring the preparation of a new monomer for each target structure. Therefore, compared with conventional polymers that lack reactive pendant groups, lactone-containing polymers provide an additional level of synthetic flexibility and represent attractive platforms for the development of functional and recyclable materials. Accordingly, ROP methodologies can be exploited as versatile further modification strategies.
3. Synthesis of five- and six-membered vinyl lactone monomers
Several vinyl lactones that are highly relevant to polymer chemistry originate directly from nature (Scheme 2). For example, α-methylene-γ-butyrolactone (MGBL), better known as tulipalin A, occurs in tulips and is formed enzymatically from its glucoside precursor, tuliposide, when bulb scales are damaged or incubated, where it functions as a biologically active defense compound.27 Another example is mevalonolactone, which is a six-membered δ-lactone in equilibrium with mevalonic acid, a central intermediate in isoprenoid biosynthesis pathways leading to squalene, cholesterol and a wide range of terpenes.28 | ||
| Scheme 2 The most commonly used vinyl lactone derivatives. This review focuses particularly on γ-valerolactone (GVL, green), γ-butyrolactone (GBL, blue), and mevalonolactone (MVL, red) derivatives. | ||
These natural examples clearly illustrate that lactone building blocks can be accessed through biological routes; however, their native abundance is far too limited to serve as realistic bulk feedstocks for polymer production. Instead, nature offers a conceptual blueprint: lactone-type structures can be obtained at scale via biorefining and biomass valorization.29 In modern biorefinery schemes, lignocellulosic carbohydrates and other renewable resources are converted into a small set of platform molecules, among which levulinic acid, γ-valerolactone (GVL), γ-butyrolactone (GBL) and itaconic acid are particularly attractive precursors for lactone polymer chemistry. These intermediates can be transformed into vinyl lactone monomers such as α-methylene-γ-valerolactone (MGVL) and α-methylene-γ-butyrolactone (MGBL), providing petroleum-free routes and building blocks suitable for both ROP and vinyl polymerization (Scheme 3).30–35
 | ||
| Scheme 3 Example of the synthesis of α-methylene-γ-valerolactone and α-methylene-γ-butyrolactone from levulinic acid and itaconic acid. | ||
The synthesis of MGBL has been demonstrated via enzyme engineering starting from isoprenyl acetate, showcasing how pathway design and biocatalysis can open direct selective routes to complex lactone monomers.36 In parallel, interest in mevalonolactone (MVL) and its parent mevalonic acid has expanded beyond classical isoprenoid biosynthesis: mevalonic acid is now recognized as a key intermediate for bio-based isoprene production, which can be upgraded to jet-fuel-range hydrocarbons.37 Industrial efforts mirror this scientific momentum: the US-based company Visolis produces mevalonic acid and isoprene from biomass at scale, and the FERMEVA project (funded by the Dutch Topsector Energie) brings together multiple industrial partners to establish mevalonic acid synthesis from renewable resources.38
Taken together, these developments highlight a clear convergence between biorefinery and polymer chemistry: on one side, intensified efforts to derive lactone-type monomers from renewable carbon, and on the other, growing interest in transforming these monomers into sustainable polymers. It is evident that the dynamic equilibrium between lactone formation and hydrolysis is extensively exploited in nature, and that vinyl-functionalized lactones either occur in biological systems or can simply be accessed via biomass-derived feedstocks.
In fact, considerable interest has been directed toward the development of renewable acrylic alternatives to petroleum-derived methyl methacrylate (MMA). Notably, biomass-derived cyclic analogues, including MGBL, β-Me-GBL, and MGVL, have emerged as promising candidates.6 For polymer chemists, this offers a clear design cue: lactones can be harnessed as nature-inspired functional pendant groups on robust polymer backbones. In this minireview, we adopt a distinct viewpoint on lactones as pendant motifs, surveying the synthetic literature and deciphering the potential of lactone-bearing, renewable polymers for next-generation sustainable and processable materials.
Table 1 lists the lactone-pendant polymers reported in the literature, including their polymerization strategies and key thermal properties, particularly glass-transition temperatures (Tg). The examples highlight the diversity of accessible architectures ranging from homopolymers to statistical and block copolymers, prepared by free-radical, coordination, living, and photo- or laser-initiated polymerizations, as well as the resulting Tg values and processing windows. Collectively, these studies illustrate how the incorporation of lactone side chains influences polymer structure–property relationships while underscoring the breadth of synthetic approaches available for constructing high performance bio-based functional materials.
| Lactone-pendant polymer structure | Notes | Ref. |
|---|---|---|
| Homopolymers | ||
 |
Polymerization methods: free radical, anionic and coordination, and controlled (RAFT) polymerization | 39, 41, 42, 44, 47, 54, 59–62 and 71 |
| Thermal properties: Tg = 195 °C for PMGBL and Tg = 225 °C for PMGVL54 | ||
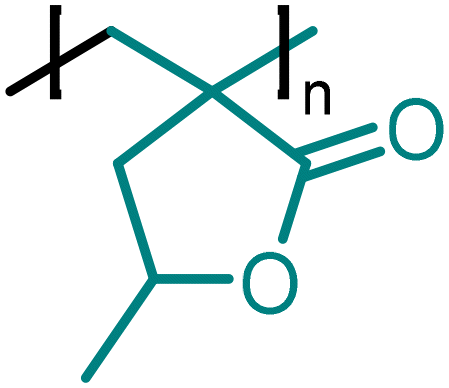 |
Polymerization methods: free radical, living and coordination polymerization | 44, 47, 54 and 59 |
| Thermal properties: Tg = 199 ± 27.5 °C | ||
 |
Polymerization methods: free radical and Lewis-pair mediated polymerization | 43 and 76–78 |
| Thermal properties: Tg between 206 °C and 220 °C, depending on molecular weight | ||
 |
Polymerization methods: NHC-catalyzed conjugate-addition polymerization | 84 |
| Thermal properties: Tg = 184 | ||
| Random copolymers | ||
 |
Polymerization methods: free radical, pulsed laser and spontaneous polymerization, and photo- (365 nm) and thermal initiation (Tpoly. = 50–70 °C reaction) | 50 |
 |
Polymerization methods: living polymerization | 49, 53, 54, 56 and 57 |
| Thermal properties: Tg = 213 °C | ||
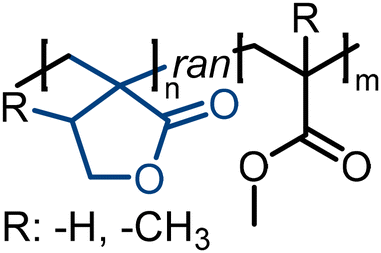 |
Polymerization methods: thermal or photoinitiated free radical polymerization and pulsed laser polymerization | 50, 51 and 62 |
| Thermal properties: Tg = 100–160 °C | ||
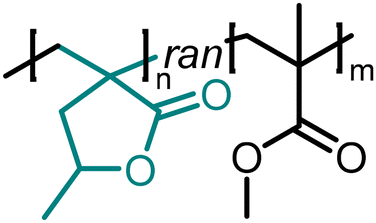 |
Polymerization methods: pulsed laser polymerization/size exclusion chromatography | 51 and 52 |
 |
Polymerization methods: thermally initiated free radical polymerization, | 47 |
| Thermal properties: Tg = 60–130 °C | ||
| Triblock copolymers | ||
 |
Polymerization methods: thermally induced atom transfer radical polymerization (ATRP) | 64 |
| Thermal properties: Tg = 60–130 °C | ||
 |
Polymerization methods: thermally initiated atom transfer radical polymerization, star-like polymer | 67 |
| Thermal properties: two distinct Tg values, corresponding to the PBA segment close to −50 °C and the PMGBL segment at 195 °C | ||
 |
Polymerization methods: ring-opening polymerization and thermally initiated atom transfer radical polymerization | 68–70 |
| Thermal properties: Tg = 70–190 °C | ||
3.1. Synthesis of five-membered lactone-pendant polymers
 | ||
| Scheme 4 (a) Free-radical and (b) anionic polymerization of MBL. | ||
A detailed kinetic study of FRP was conducted in 2015 by Lee and Pittman on β-methyl-α-methylene-γ-butyrolactone (β-Me-MGBL).43 The kinetic study of β-Me-MGBL showed that the reaction follows classical free-radical polymerization behavior, with the polymerization rate scaling half-order with respect to the initiator concentration and first-order with respect to the monomer concentration. The activation energy (∼87 kJ mol−1) exceeded that of MMA and related exocyclic methylene lactones, consistent with additional steric constraints during propagation. Although MMA and MGVL are often regarded as structural analogues, MGVL exhibits substantially higher reactivity due to the combined influence of ring strain and the exocyclic double bond. Mälmström and co-workers have reported a systematic comparison of MGBL- and MA-based acrylic polymers initiated by AIBN and lauroyl peroxide.44 MBL and MeMBL were successfully copolymerized with MMA and MA using AIBN or LP as the initiator. Monomer incorporation into the resulting copolymers was strongly governed by differences in monomer reactivity ratios, leading to compositional drift in certain systems. These variations in copolymer composition were reflected in the measured Tg. For instance, increasing the ratio of PMA in PMBL-co-PMA (3.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.2) shifted the Tg value from 190 °C to 138 °C. Notably, the incorporation of even low amounts of MBL or MGVL markedly enhanced the thermal stability of PMMA and eliminated the scission of head-to-head linkages.45 The choice of initiator had little effect on the polymerization kinetics or final polymer properties under the conditions studied, indicating that the monomer structure and reactivity dominate the polymerization outcome.
:6.2) shifted the Tg value from 190 °C to 138 °C. Notably, the incorporation of even low amounts of MBL or MGVL markedly enhanced the thermal stability of PMMA and eliminated the scission of head-to-head linkages.45 The choice of initiator had little effect on the polymerization kinetics or final polymer properties under the conditions studied, indicating that the monomer structure and reactivity dominate the polymerization outcome.
Tauer and Esposito substantially extended the structure–property relationship of MGVL-derived polymers by examining how different radical polymerization conditions, namely, thermal bulk, photoinitiated bulk, and photoinitiated heterophase polymerization, affect the resulting material properties.46,47 Their systematic study demonstrated that variations in initiation mode (thermal or photochemical) and reaction environment introduce significant and quantifiable changes in the molecular weight (aqueous heterophase polymerization), thermal degradation behaviour, and Tg, even in homopolymers. Thermally initiated bulk polymerization produced the most thermally robust materials across all monomers (MMA, MGVL and styrene (St)) investigated, with decomposition onset temperatures (Tdec,on) consistently higher than those of polymers formed under photoinitiated conditions. In contrast, heterogeneous photopolymerization yielded materials with substantially reduced thermal stability, attributable to structural irregularities and defect formation arising from the aqueous reaction environment. This effect is particularly pronounced for PMMA and PMGVL, which exhibit lower Tdec,on (∼80 °C) than their bulk-polymerized analogues (e.g., PMMA showing Tdec,on values near ∼150 °C and polystyrene (PS) ∼300 °C), whereas PS shows only modest sensitivity to the polymerization method. Differences are also reflected in the Tg: averaged across all polymerization routes, Tg values span 106 °C for PS, 116 °C for PMMA, and ∼200 °C for PMGVL, demonstrating the intrinsic rigidity imparted by the lactone ring in PMGVL as well as the variation introduced by polymerization history. Overall, these results reinforce a central principle in polymer science: the thermal properties of a polymer cannot be inferred from its nominal chemical structure alone but depend on the final polymer structure produced by a given polymerization route. Factors such as temperature, initiation chemistry, and reaction medium may influence thermal properties indirectly by altering the molecular weight, chain regularity, branching or crosslinking, end-group composition, and defect concentration.48
Copolymerization of MGVL and MGBL combines the exceptional rigidity and thermal stability imparted by the pendant lactone groups with the processability and well-established performance of conventional vinyl monomers. A range of polymerization strategies have been employed, including thermal and photochemical initiation, through controlled free-radical, Lewis-pair, and coordination polymerization methods.49 The first copolymerization study reported by Akkapeddi and co-workers involved free-radical copolymerization of MGBL with MMA, styrene (St), acrylamide (AA), acrylonitrile, and vinylene carbonate, respectively, using AIBN as the initiator at 60 °C.50 Later, D'Hooge and co-workers have studied the copolymerization of MGBL and MGVL with MMA, St and n-butyl acrylate (BA) using the pulsed laser polymerization/size exclusion chromatography technique.51 Under bulk conditions, copolymer composition data match the predictions of the thermal model, excluding MGVL, which shows preferential incorporation into copolymers and exhibits kinetic behaviour beyond the simple thermal model.52
Further collaboration between Herres-Pawlis and Henkel AG & Co. KGaA led to advances in the copolymerization of bio-derived lactones with established vinyl monomers, most notably acrylate derivatives (Scheme 5).53 The copolymerization behavior of MGBL and its higher homolog MGVL provides a representative case study demonstrating how subtle structural variations in renewable methylene lactones govern their reactivity, sequence distribution, and resulting material properties. In the Q–e framework, MGVL exhibits a significantly higher Q value (3.27) than MGBL (2.48) and a slightly lower polarity parameter (e = 0.61 vs. 0.83), indicating increased radical stabilization imparted by the α-methyl substituent of MGVL. This higher radical reactivity is consistent with the experimentally observed reactivity ratios r1 = 0.63 (MeGBL) and r2 = 1.51 (MGVL), which clearly reflect an alternating tendency between the two lactone monomers. As predicted, such complementary reactivity supports homogeneous copolymer formation with minimal compositional drift. These findings are corroborated by UV–vis turbidity screening at 450 nm, where poly(MGBL-co-MGVL) copolymers show extremely low absorbance (0.2–0.4 AU), confirming good miscibility and compatibility over a wide compositional range. This behavior stands in strong contrast to acrylate comonomers such as butyl acrylate (BA) or ethyl acrylate (EA), whose significant reactivity mismatches lead to pronounced turbidity and phase separation. The MGBL–MGVL pair, therefore, represents a model system in which structural similarity and matched reactivity profiles yield ideal, transparent copolymers.
 | ||
| Scheme 5 Schematic illustration of the free-radical copolymerization of vinyl lactones with conventional (meth)acrylate comonomers under thermal or photochemical initiation. Reproduced from ref. 53 with permission from the American Chemical Society,53 Copyright 2025. | ||
Apostolidis and colleagues focused on the polymerization behaviour of these monomers in renewable solvents using AIBN as a thermal initiator.54,55 Polymerizations carried out under an inert atmosphere in Cyrene®, GVL, and 2-methyltetrahydrofuran (2-MeTHF) proceeded via solution polymerization, and the polymer was isolated by precipitation into bio-based ethanol. Under these conditions, relatively modest number-average molar masses were obtained (Mn around 5500 g mol−1, Đ = 1.7 at 70% conversion after 5 h at 70 °C), resulting in a Tg value of 96.4 °C for PGVL. Inspired by the precipitation-based workup, they next explored polymerization directly in a series of bio-based alcohols (1-butanol, 2-phenylethanol, and 1,4-butanediol), which led to precipitation polymerization. In this case, the polymers exhibited significantly higher molar masses (Mn = 27–30 kg mol−1, Đ = 1.4–1.5 at 83–88% conversion after 5 h at 70 °C), while the Tg values remained relatively modest (94.7 °C for PGBL and 100 °C for PGVL), yet comparable to that of the commercial benchmark PMMA prepared by emulsion polymerization (103 °C). Films fabricated from the resulting polymer powders showed up to 98% transparency in the visible range (450–700 nm), again matching the optical performance of PMMA. In this setup, AIBN is the only non-renewable component; given its low loading, the overall process, from monomer synthesis to purified polymer powder, relies on more than 99% renewable inputs. Moreover, precipitation polymerization greatly simplifies polymer isolation and allows straightforward reuse of the reaction solvent as well as recycling of the bio-based ethanol used for washing.
C(OMe)OSi(iBu)3 (iBuSKA) as the initiator. The system also enabled the synthesis of well-defined block and statistical copolymers. Thermal analysis revealed exceptionally high Tg values characteristic of butyrolactone-derived polymers, with Tg = 194 °C for PMGBL and Tg = 225 °C for PMGVL, while the block copolymer PMGVL-co-PMGBL displays two distinct Tg values (199 °C and 211 °C) and random copolymers showed a single Tg value around 213 °C. Overall, the work represents a major advance in the controlled polymerization of bio-derived vinylidene monomers and provides a mechanistic foundation for engineering next-generation sustainable acrylic analogues.
 | ||
| Scheme 6 Living/controlled (meth)acrylate polymerization catalyzed by silyl ketene acetal. Reproduced from ref. 56, published under a CC-BY license. | ||
Later, the Chen group first attempted to provide a living character to the polymer chain via Lewis-pair polymerization using N-heterocyclic carbene/B(C6F5)3 initiators while avoiding chain termination.57,58 They achieved exceptionally rapid polymerization of the MGBL monomer, reaching 100% conversion within 1 minute and obtaining polymers with Mn = 33300 g mol−1. While the system demonstrated excellent polymerization activity for MGBL, it fell short of delivering true living polymerization characteristics.57 Subsequently, the Zhang group demonstrated living polymerization of MGBL with different Lewis pairs.59 The study reports the first successful living and controlled polymerization of MGBL and MGVL using an organic Lewis pair (LP) consisting of tris(pentafluorophenyl)borane (B(C6F5)3) and the strong organophosphorus superbase P(NIiPr)Ph2. This borane–phosphine Lewis pair enables exceptionally rapid polymerization, achieving 100% conversion in as little as 30 seconds, with a record turnover frequency (TOF) of 48000 h−1, the highest reported for living MGVL polymerization. The living nature was confirmed by successful multistep chain extension and the formation of well-defined diblock and random copolymers. The resulting polymer architectures translate directly into distinct structure–property relationships: random copolymers exhibit a single intermediate Tg (204 °C), while diblock copolymers display two separate Tg values corresponding to PGBL and PGVL domains, evidencing microphase-separated block structures. In a notable scope-extension study, Chen and co-workers demonstrated that a broad range of conjugated polar monomers, including methacrylates, MGBL, AA, and vinyl phosphonates, can be efficiently polymerized using classical and frustrated Lewis pairs based on the strong Lewis acid Al(C6F5)3 combined with diverse Lewis bases such as phosphines and N-heterocyclic carbenes, thereby establishing the versatility and mechanistic breadth of Lewis-pair-mediated polymerizations.60,61
Beyond metal-based and controlled radical strategies, N-heterocyclic carbene (NHC) organocatalysis has emerged as an effective metal-free approach for the controlled polymerization of methylene butyrolactones. Highly nucleophilic NHCs, particularly 1,3-di-tert-butylimidazolin-2-ylidene (ItBu), enable extremely rapid conjugate-addition polymerization of MGBL at room temperature, achieving quantitative conversion within minutes and producing polymers with Mn ≈ 70–85 kg mol−1 and dispersities around 1.5–2.0. Although initiation is slower than propagation and true living behavior is not maintained at high monomer-to-catalyst ratios due to internal chain transfer, predictable molecular weights, linear Mn-conversion relationships at early stages, and efficient chain reinitiation demonstrate a quasi-living, catalytic polymerization regime that is distinct from conventional radical processes.62
900 g mol−1) while maintaining control over polymerization (Đ = 1.14). The PMGBL-containing triblock copolymers exhibit exceptional thermal stability, with dual glass transitions at −50 °C and ∼195 °C and a rubbery plateau persisting to nearly 200–300 °C, far exceeding the thermal limits of conventional PMMA-based thermoplastic elastomers.65 Although PMGBL is often assumed to be a rigid polymer, primarily because of its high Tg (≈190 °C) and excellent mechanical hardness, Higaki and co-workers applied SEC-MALS and synchrotron SAXS to quantify the intrinsic chain stiffness of well-defined PMGBL, demonstrating that the polymer backbone behaves as a flexible worm-like chain in solution. PMGBL exhibits Kuhn lengths of 1.81 nm in GBL and 1.64 nm in DMF, only slightly larger than those of PMMA and comparable to PS, while its chain diameter (0.80–0.85 nm) also mirrors that of typical vinyl polymers.66
 | ||
| Scheme 7 Synthesis of the PMBL-b-PMMA-b-PMBL block copolymer through ATRP. Reproduced from ref. 64 with permission from the American Chemical Society,64 Copyright 2008. | ||
The proposed ATRP methodology was also used for creating star-like PBA-b-PMGBL block copolymers for high-temperature thermoplastic elastomer applications.67 Matyjaszewski and co-workers have synthesized the inner soft PBA and outer hard PMBL blocks using 10-arm and 20-arm PBA macroinitiators at 50 °C (Scheme 8). A halogen exchange strategy was employed during ATRP of PMBL to achieve cross-propagation rates comparable to propagation rates. Partial star coupling was observed during PMBL block extension from PBA arms, with coupling increasing as the number and length of arms increased. AFM and SAXS analyses revealed phase-separated morphologies, consisting of either hexagonally arranged cylindrical PMBL domains dispersed within a PBA matrix or lamellar structures in samples with higher PMBL content (21–27 wt%). Upon annealing at 230 °C, above the Tg of PMBL, a morphological transition was observed in all star-like copolymers, resulting in PMBL cylinders oriented perpendicular to the surface. The dynamic mechanical properties of multi-arm PBA–PMBL block copolymers were characterized by temperature-dependent measurements of the storage (G′) and loss (G″) moduli at a constant deformation frequency of 10 rad s−1. Two distinct Tg values, corresponding to the PBA segment close to −50 °C and the PMGBL segment at 195 °C, were observed for most copolymers, providing further evidence of microphase separation. The Tg of PMBL became less pronounced as the PMBL content decreased and was barely detectable at the lowest PMBL content. Dynamic mechanical analysis of multi-arm PBA–PMBL block copolymers revealed two distinct glass transition temperatures corresponding to PBA and PMBL, confirming microphase-separated structures. These findings suggest that PMBL-based copolymers could be exploited in specialized high-temperature applications.
 | ||
| Scheme 8 Synthesis of the multi-arm PBA-co-PMBL block copolymer via ATRP. Reproduced from ref. 67 with permission from Elsevier,67 Copyright 2010. | ||
The Hillmyer group synthesized another thermoplastic elastomer via sequential polymerization of the plant-based monomers menthide (M) and MBL (Scheme 9).68 The soft segment was synthesized through ring-opening transesterification polymerization of menthide (M) using diethylene glycol as the initiator, giving α,ω-dihydroxy poly(menthide) (HO-PM-OH, Tg = −22 °C), which was converted to α,ω-dibromo end-functionalized Br-PM-Br by esterification with excess 2-bromoisobutyryl bromide. The hard segment has been built on this macroinitiator through ATRP. The resulting triblock polymer, PMBL-b-PM-b-PMB, shows two distinct Tg values as expected, at −21 and 170–190 °C, consistent with a microphase-separated PM mid-block and PMBL end blocks. The triblock copolymer (5–100–5) maintained exceptional stretchability (>1300% strain) and meaningful mechanical strength even up to 100 °C, demonstrating superior high-temperature elastomeric performance compared with conventional styrenic triblock copolymers, whose elasticity typically deteriorates near their glass-transition temperature. The Hillmyer group applied this approach to prepare high-pressure-sensitive adhesives from PMBL-b-PM-b-PMBL renewable triblock copolymers with high Tg values (170–190 °C) via ATRP.69 Pressure-sensitive adhesives (PSAs) were formulated by blending PMeMBL-PM-PMeMBL triblock copolymers with rosin ester tackifiers (Sylvalite RE-85 and RE-10L). A representative formulation was solution-cast onto a PET film, producing a uniform adhesive coating. Adhesion testing showed high peel strength and strong loop tack, indicating excellent immediate and sustained adhesion. Most notably, the formulation exhibited an exceptionally high shear adhesion failure temperature (>150 °C), comparable to cross-linked PSA systems and significantly exceeding the performance of conventional non-cross-linked styrenic block copolymer PSAs.
 | ||
| Scheme 9 ABA-type triblock thermoplastic synthesis through subsequent ROP and ATRP from renewable sources. Reproduced from ref. 68 with permission from the American Chemical Society,68 Copyright 2012. | ||
Although ATRP enables a well-defined polymer architecture, its reliance on metal complexes can be a limitation. In contrast, nitroxide-mediated polymerization (NMP) offers a metal-free alternative for controlled radical processes. The first successful nitroxide-mediated polymerization (NMP) of MBL was achieved using the alkoxyamine initiator Dispolreg 007 at 80 °C in 50 wt% DMSO, targeting an Mn of 40 kg mol−1. The system exhibited hallmark features of reversible-deactivation radical polymerization (RDRP), including a linear increase of Mn with conversion, early-stage first-order kinetics, and moderate dispersity (Đ ≤ 1.45). High-end group fidelity was demonstrated through successful chain extension with both St and glycidyl methacrylate (GMA) at 120 °C, confirmed by clear GPC shifts to higher molecular weights.70 Overall, this NMP study highlights the potential of this methodology for creating thermoplastic elastomers, with glassy P(MGBL) end-blocks and a soft rubbery mid-block. Such systems offer a metal-free alternative to ATRP, eliminate catalyst-removal steps, avoid discoloration, and enable the design of polymers with high-Tg.
However, many attempts to polymerize MGBL and MGVL monomers still rely on using classical organic solvents, with the corresponding, generally high, environmental impact. Picchion and co-workers have demonstrated that the first RAFT polymerization of MGBL can be conducted in supercritical CO2 (ScCO2) using AIBN and 2-cyanoprop-2-yl dithiobenzoate (Scheme 10).71 Polymerizations were carried out in a high-pressure Parr batch reactor (max. 350 bar) under nitrogen at 80 °C for 24 h, yielding polymers with molecular weights in the range of 10–20 kg mol−1 and relatively low dispersities (Đ < 1.5). Materials produced in supercritical CO2 exhibited Tg values between 155 and 190 °C, consistent with the rigid lactone-based backbone. However, it should be noted that residual monomer entrapped within the polymer matrix depresses the measured Tg, reflecting plasticization effects associated with incomplete monomer removal. Later, the Hatton group investigated the first RAFT polymerization of MGVL at 80 °C in solvents (DMSO, cyrene, t-butanol, and methanol). Purified P(MGVL) homopolymers exhibited high Tg values (206–221 °C) and excellent thermal stability, where the onset of degradation was observed in the region of 345–366 °C.72
 | ||
| Scheme 10 RAFT polymerization of MGBL in supercritical CO2 at 300 bar. Reproduced from ref. 71 with permission from John Wiley & Sons,71 published under a CC BY-NC-ND license. | ||
Further studies from the Picchioni group have reported a two-step synthesis of linear PMBL-b-PHMA-b-PMBL copolymers comprising a soft/rubbery poly(n-hexylmethacrylate) (PHMA) inner block and two hard PMBL outer segments via metal-catalyst-free reversible addition–fragmentation chain-transfer (RAFT) polymerization.73 A two-step RAFT polymerization employing a bis-functional chain-transfer agent enabled precise control over the molecular weight (Mn = 25–35 kg mol−1), narrow dispersity (Đ = 1.1–1.3), and symmetric block growth. By varying the monomer feed during the chain-extension step, the hard/soft block ratio was systematically tuned, allowing direct control over morphology and mechanical properties. SAXS revealed that triblock copolymers form well-ordered microphase-separated morphologies, transitioning from spheres to cylinders and lamellae as the PMBL content increases, with domain spacings rising from ∼6 to ∼42 nm. Sharp higher-order reflections and dual Tg values (PHMA ∼ 5 °C; PMBL ∼ 160–195 °C) confirm strong phase separation and uniform block sizes. In contrast to SBS, whose polystyrene domains lose physical crosslinking near ∼100 °C, the high-Tg PMBL domains preserve the morphology and physical crosslinks at elevated temperatures, enabling superior thermal stability and reprocessability.
 | ||
| Scheme 11 Coordination polymerization of MGBL via metal catalysts (neutral lanthanide and cationic group 4 catalysts) at ambient temperature. Reproduced from ref. 75 with permission from the Royal Society of Chemistry.75 | ||
Further studies have shown that C2-symmetric zirconocenium (sandwich-type) catalysts and ansa-half-sandwich rare-earth-metal dialkyl and trialkyl complexes can precisely control the stereochemistry of MGBL and MGVL monomers, producing polymers ranging from stereo-random to perfectly isotactic, depending on the monomer structure.76–78 Two catalysts, rac-(EBI)Zr+ and rac-(EBDMI)Zr+, showed low activity and produced atactic PMGBL (mm ∼ 42%, Tg ∼ 195 °C, Mn ∼ 17–20 kg mol−1) and atactic PMGVL (mr ∼ 42%, Tg ∼ 227 °C, Mn ∼ 60–65 kg mol−1). In sharp contrast, polymerization of β-Me-MGBL yielded highly isotactic (mm = 95.2%) or perfectly isotactic (mm > 99%) polymers, with quantitative conversion, dramatically enhanced thermal properties (Tg up to 288 °C), and excellent solvent resistance. Computational and experimental data show that steric interactions between the β-methyl substituent, the growing chain, and the metallocene ligand during the initiation/propagation step enforce enantiofacial selectivity, directly translating catalyst structure into polymer stereoregularity and macroscopic performance.
While the use of renewable monomers aligns well with green chemistry principles, the polymerization process itself, as well as the associated separation, purification, and processing steps, play a decisive role in determining the overall environmental footprint of a polymer product. Several studies have therefore focused on tailoring synthetic conditions to maximize the “green” profile of lactone-functional polymer materials. Sebakhy and co-workers established a straightforward route to MGBL-based films via initiated chemical vapor deposition, enabling direct polymerization of the monomer in the gas phase onto target substrates using di-tert-butyl peroxide as a free-radical initiator.79 Experimentally, they correlated deposition kinetics, polymerization rate, and film thickness with thermodynamic parameters and optimized conditions to afford polymer films with an average molar mass of 9200 g mol−1 and a Tg value of 164 °C. The coatings exhibited good mechanical robustness, as evidenced by nanoindentation measurements, and high optical transparency, with film thicknesses of 2.6 µm and 4.2 µm demonstrated on a range of substrates. In short, this direct film fabrication strategy obviates the need for solution-based film-forming steps and represents a significant sustainability advancement for lactone-bearing polymer coatings.
Towards the goal of improving the polymerization process, Sebakhy and colleagues also reported a horseradish peroxidase-mediated, surfactant-free emulsion polymerization of MGBL in a water/2,4-pentanedione mixture at room temperature (Fig. 1).80 Polymerization conducted in air for 3 h resulted in a 98% solid yield and an average molar mass of 73400 g mol−1, with a relatively high dispersity (Đ = 2.4) for an emulsion system. The presence of air in the reaction medium accelerated the polymerization process as oxygen plays a key role in active radical generation, whereas polymerization under an inert atmosphere took more than 24 hours to complete. The stable latex displayed an average particle diameter of 131 nm and a high Tg of 200 °C. This approach relies on a simple experimental setup and eliminates the need for expensive inert gas streams that are typically required in free-radical polymerizations. Rapid polymerization in air thus provides a direct and operationally simple route to green MGBL-based polymer latexes.
 | ||
| Fig. 1 Proposed mechanism of surfactant-free horseradish peroxidase-mediated radical polymerization of MGBL in water. Reproduced from ref. 80, published under a CC-BY 4.0 license. | ||
3.2 Synthesis of six-membered lactone-pendant polymers
Only a few studies have reported the ROP of mevalonolactone (MVL); in contrast, its methacrylate derivative offers substantial potential as a radically polymerizable monomer to introduce mevalonolactone pendant units into polymer backbones. The monomer is obtained by treating mevalonolactone with methacryloyl chloride in the presence of a base, thereby installing a methacrylate functionality on the hydroxyl group of mevalonolactone. Surprisingly, despite this straightforward synthesis and attractive structure, mevalonolactone methacrylate (MVLMA) has not yet been widely explored by the polymer community, and to date, only three publications describe its polymerization and potential applications. | ||
| Fig. 2 (A) Structure of the MVLMA copolymer prepared by free radical copolymerization for photoresist applications,84 (B) digital image of the MVLMA homopolymer in water and its treatment with hydrazine leading to the formation of a water soluble polymer within seconds. Reproduced from ref. 85, published under a CC-BY license. | ||
Subsequently, a similar formulation strategy was explored by Fujitsu.84 Copolymers with varying MVLMA/2-MAdMA (2-methyl-2-adamantane methacrylate, 2-MAdMA) molar ratios were synthesized in γ-butyrolactone using 15 mol% AIBN at 70 °C for 8 h. Polymer yields comparable to those in the AZ Electronics study were obtained but significantly lower Tg values ranging from 92–105 °C were reported, possibly due to the high initiator loading leading to reduced molar mass. These copolymers were fully compatible with industrial lithographic processes and enabled high-resolution patterning below 100 nm using an ArF excimer laser (193 nm). Collectively, these industry-driven studies show the promise of polymers bearing pendant mevalonolactone units as high-performance materials for advanced microelectronics and semiconductor lithography.
In 2021, our group adopted an empirical approach to investigate light-induced free-radical and controlled radical homopolymerization of MVLMA, providing the only detailed study on its homopolymer synthesis and properties.85 We employed the commercial photoinitiator 2,4,6-trimethylbenzoyl diphenylphosphine oxide (TPO), the RAFT agent ethyl 2-(phenylcarbonothioylthio)-2-phenylacetate (dithiobenzoate), and a UV light source (30 W). Conventional free-radical homopolymerization of MVLMA in toluene for 16 h under UV irradiation afforded a white polymer powder in 57% isolated yield after purification, and the polymer structure was confirmed by NMR spectroscopy. Size-exclusion chromatography (SEC) revealed a number-average molar mass of 29 kg mol−1 with a Đ value of 2.2, and the homopolymer exhibited a high Tg value of 154 °C. The material showed high hydrolytic stability with no detectable structural changes after 2 days in D2O. To our surprise, rapid nucleophile-induced ring-opening was observed within seconds, accompanied by a notable increase in polymer hydrophilicity. To harness this reactivity, thin films of the homopolymer were spin-coated onto glass substrates; subsequent treatment with hydrazine or thioglycerol led to instantaneous lactone ring-opening and conversion to a hydrophilic surface, clearly demonstrating the responsive character of MVLMA-based polymers. In parallel, we explored photoinduced RAFT homopolymerization, which yielded a polymer in 20% isolated yield with a molar mass of 7.5 kg mol−1 and Đ = 1.5. The living character of this system was confirmed by successful chain extension with styrene under light irradiation, although the resulting block copolymer was obtained in only 14% yield. All polymer structures were analyzed and confirmed by NMR spectroscopy, and the (co)polymers were found to be soluble in DMSO, NMP, CHCl3, and DMF. Based on the results, MVLMA is a robust yet underexploited platform for designing nucleophile-responsive, high-Tg materials with significant potential in advanced coatings and functional polymer architectures.
Very recently, Chen and co-workers demonstrated that bio-based α-methylene-δ-valerolactone (MVL) and α-methylene-δ-decalactone (MDL) provide lactone-containing acrylic polymers with both performance and recyclability advantages (Scheme 12).86 MVL underwent rapid NHC-catalyzed conjugate-addition polymerization under solvent-free ambient conditions, reaching essentially quantitative conversion within seconds and affording PMVL with Mn up to 2.0 × 105 g mol−1. PMVL could also be obtained by AIBN-initiated bulk radical polymerization at 60 °C for 1 h. The resulting PMVL displayed a high Tg value of 184 °C, compared with ca. 110 °C for atactic PMMA, while PMDL showed a Tg value of 122 °C. Despite their high thermal performance, PMVL and PMDL could be selectively depolymerized in bulk under vacuum for 1 h at 220 and 210 °C, respectively, affording MVL and MDL in 99.5% and 90% isolated yields. In comparison, PMMA required 400 °C and gave only 53% monomer recovery under the reported conditions. The pendant lactone ring also enabled post-polymerization modification, as partial NaOH-mediated ring opening of PMVL at 80 °C generated water-soluble ionomeric materials. These results highlight the dual role of the lactone unit in vinyl lactone polymers: it enhances thermal and solvent-resistance properties while also serving as a reactive handle for depolymerization, copolymer design, and post-polymerization functionalization.

4. Recycling potential
ROP of lactones typically yields aliphatic polyesters in which ester linkages are incorporated into the polymer backbone, thereby providing potential sites for hydrolytic degradation or chemical recycling. In contrast, vinyl polymers bearing pendant lactone groups contain the lactone moieties as side-chain functionalities rather than backbone-cleavable units. Consequently, the presence of lactone groups alone does not necessarily impart polyester-like degradability or straightforward whole-polymer recyclability.In order to assess the chemical recyclability of lactone-bearing polymers, PMMA was benchmarked against PMGBL and PMGVL under identical thermolysis conditions (400 °C, 50 mTorr, 3 h) in simple glassware.87 Under these conditions, PMMA afforded only ∼53% recovery of methyl methacrylate (MMA), with the remaining ∼47% of the material converting into intractable carbonized char, whereas the recovered MMA required further purification due to detectable side products. In sharp contrast, PMGBL and PMGVL displayed markedly enhanced depolymerization selectivity: PMGBL delivered ∼65% monomer (MGBL) with the entire mass balance accounted for as a volatile monomer plus a non-carbonized oligomeric residue (Mn ∼ 1.5 kg mol−1), while PMGVL reached 76 ± 1% monomer recovery of MGVL, again with the remaining ∼24% as an oligomer and no char formation (Scheme 13). Notably, MGVL was recovered as a pure monomer. To rationalize this unexpectedly high recyclability, DFT calculations showed that the estimated bulk ceiling temperatures (Tc) of PGBL (500 °C) and polyMGVL (405 °C at 9.36 M) are in fact higher than that of PMMA (∼300 °C), ruling out the low-Tc hypothesis. The tethered five-membered lactone ring stabilizes highly reactive primary macroradicals and suppresses unproductive fragmentation pathways that in PMMA lead to CO/CO2 evolution, char formation, and loss of monomer yield. In short, beyond their renewable feedstock origin, bio-based lactone monomers offer attractive process tunability and outstanding chemical recyclability for truly sustainable polymer manufacturing.
 | ||
| Scheme 13 The proposed tethering effect of the lactone during the thermally induced depolymerization of PMGVL. Reproduced from ref. 87, published under a CC-BY license. | ||
The study was supported by DFT calculations, which estimated Tc values of MGBL > MGVL > MMA, consistent with the observed reactivity and polymerizability trends. Depolymerization of PMGBL demonstrates improved monomer recovery and reduced carbonization compared to PMMA, although the recovered monomer is not fully spectroscopically pure. The presence of pendant lactone groups appears to suppress side reactions and favor productive unzipping pathways. In contrast, PGVL exhibits higher monomer recovery and purity, which has been attributed to additional γ-methyl substitution that enhances radical stabilization via hyperconjugation. Notably, the residual material from PMGBL depolymerization consists mainly of oligomeric species rather than intractable char, highlighting the potential of lactone-functional vinyl polymers for chemical recycling, while also underscoring the need for further optimization to achieve quantitative monomer recovery.
Another recycling study has been reported by the Hong group, which stressed not only the post-polymerization modification possibility of lactone monomers, but also demonstrated that the thermal depolymerization profile of these polymers can be achieved with pure monomer recovery.88 In their study, the thermal recyclability of PMGBL was demonstrated using bulk thermolysis under reduced pressure (100 mTorr) in a temperature-controlled furnace equipped with a cold trap to collect volatile products. Depolymerization efficiency strongly depended on temperature; at 310 °C (slightly above Td), only limited monomer recovery (17.4%) was observed. Increasing the temperature to 330 °C improved recovery to 34.0%, while heating at 370 °C enabled quantitative monomer recovery with high purity (GC-MS purity up to 95.7%). The volatile monomer was distilled under vacuum and condensed in the cold trap, confirming complete thermal recyclability under optimized conditions.
To date, reported recycling strategies for such materials have focused primarily on exploiting thermal ceiling temperatures, which highlights both the promise and the current limitations of this design approach. However, emerging depolymerization concepts developed in controlled polymerization for PMMA and reversible covalent chemistry may provide promising routes for the upcycling or recycling of lactone-functional polymer systems.89,90
5. Summary and outlook
Lactones are functional cyclic esters that are present in many key biological systems and pharmaceuticals. Functionalized lactones can be obtained from biomass refining to increase their availability for further use. From a synthetic polymer chemistry perspective, lactones are primarily studied as monomers for ROP to afford functional polyesters. However, as explained in detail in this manuscript, their role can be extended beyond ROP when a methylene group is introduced into the lactone building block. To date, lactone-pendant functional polymers have been accessed via a broad range of synthetic methodologies, spanning radical and ionic to coordination polymerization. These studies explore the polymerization behavior of lactone-bearing monomers, evaluate their homo- and copolymerization reactivities, and assess the properties of the resulting materials. Their application potential is often demonstrated through favorable thermal (notably glass transition temperatures, Tg) and optical (transparency) properties, positioning lactone-pendant polymers as promising alternatives to poly(methyl methacrylate). In addition, MVL-bearing polymers have been studied for their exceptionally high reactivity towards nucleophiles and for their use as film resists in optical lithography and patterning.Lactone-functional polymers share an intriguing commonality in that the earliest reports of their synthesis originate from industry-affiliated research. Although these studies have not (to the best of our knowledge) yet been translated into commercial lactone-pendant materials, they have clearly opened an inspirational pathway for academic research. Despite their frequent association with sustainable polymer design, it is important to recognize that the sustainability advantages of lactone-pendant polymers have not yet been fully realized. In many cases, the employed monomers are still derived from petrochemical sources, and reported recycling strategies often rely on energy-intensive thermal depolymerization processes. As such, the current state of the art should be viewed as a promising platform rather than a fully established sustainable solution.
Looking forward, several key directions can be identified to enhance the sustainability profile of these materials. First, the development of scalable and economically viable routes to bio-based lactone monomers is essential. Although certain monomers such as MGBL (tulipalin A) are naturally occurring, their large-scale production from renewable feedstocks remains limited, and many studies continue to rely on petrochemical synthesis. Advancing biorefinery technologies and integrating partially bio-based carbon sources may provide realistic near-term solutions. Another key prerequisite is transparent reporting on monomer origin. For example, although MGBL is naturally occurring, most studies still employ petrochemically derived MGBL and should therefore not be labelled as “renewable”. Similar considerations apply to other lactone monomers as well. Polymer chemists need to articulate the property and sustainability advantages of these structures clearly enough to justify investment from the biorefinery community in scalable routes, even if only partially bio-based carbon solutions are realistic in the short term.
Second, as also supported by research studies, rational polymer and monomer design will play a central role in enabling circularity. Tuning ceiling temperatures, incorporating cleavable linkages, or designing systems that respond to orthogonal stimuli could allow for more efficient and selective depolymerization.
Third, improvements in chemical recycling strategies are required. While thermally induced depolymerization based on ceiling temperature concepts has demonstrated the feasibility of monomer recovery, future efforts should focus on reducing energy input and increasing selectivity inspired by current acrylic polymer recycling strategies. The development of catalytic or chemically triggered depolymerization pathways that operate under milder conditions would represent a significant step toward practical closed-loop recycling. In this context, emerging concepts from reversible-deactivation radical polymerization and dynamic covalent chemistry may offer valuable design principles.
Beyond recycling, the intrinsic functionality of lactone units provides opportunities for post-polymerization modification, enabling the design of advanced materials such as drug conjugates, self-assembled systems, and stimuli-responsive architectures. Recent work by Du Prez and co-workers highlights the potential of pendant lactones in dynamic polymer networks, where reversible lactone ring opening has been used to construct vitrimer systems.91 Such approaches illustrate how the lactone functionality can be leveraged not only for recyclability but also for adaptive and reprocessable materials.
Finally, to substantiate sustainability claims, future studies should incorporate quantitative metrics such as life-cycle assessment (LCA) to benchmark lactone-pendant polymers against established materials. Chen and co-workers reported a techno-economic analysis and life-cycle assessment for the production of PMVL from bio-based sources.86 Their analysis suggested that bio-based PMVL could become economically competitive with PMMA. The study also indicated that PMVL may offer environmental advantages over PMMA under optimized production and recycling conditions. These benefits were most apparent under ambitious recycling scenarios that exploit the inherent chemical recyclability of PMVL. This example highlights the importance of combining monomer sourcing, recycling efficiency, and LCA when evaluating the sustainability of lactone-based polymers.
In summary, lactone-pendant polymers represent a versatile and conceptually powerful platform at the interface of functional and potentially sustainable polymer design. While significant challenges remain, particularly in monomer sourcing, energy-efficient recycling, and process scalability, continued advances in polymer chemistry and materials engineering are expected to unlock their full potential as components of next-generation circular polymer systems.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review. All the literature findings can be found in the reference section. The DOI hyperlinks of research and review articles are also included in the reference section.Acknowledgements
A. K. is thankful for the Liebig Fellowship supported by the Verband der Chemischen Industrie e.V. (VCI). B. K. acknowledges continuous support from VISTEC and colleagues.During the preparation of this work, the authors used a GPT LLM to enhance language and readability. After using this tool, the authors reviewed and edited the content as needed and took full responsibility for the content of the published article.
References
- V. Yaghoubi and B. Kumru, Retrosynthetic Life Cycle Assessment: A Short Perspective on the Sustainability of Integrating Thermoplastics and Artificial Intelligence Into Composite Systems, Adv. Sustainable Syst., 2024, 8, 2300543, DOI:10.1002/adsu.202300543.
- T. Luo, Y. Hu, M. Zhang, P. Jia and Y. Zhou, Recent advances of sustainable and recyclable polymer materials from renewable resources, Resour. Chem. Mater., 2025, 4, 100085, DOI:10.1016/j.recm.2024.10.004.
- K. Houssini, J. Li and Q. Tan, Complexities of the global plastics supply chain revealed in a trade-linked material flow analysis, Commun. Earth Environ., 2025, 6, 257, DOI:10.1038/s43247-025-02169-5.
- H. Fouilloux and C. M. Thomas, Production and Polymerization of Biobased Acrylates and Analogs, Macromol. Rapid Commun., 2021, 42, 2000530, DOI:10.1002/marc.202000530.
- E. Caselli, R. A. Powers, L. C. Blasczcak, C. Y. E. Wu, F. Prati and B. K. Shoichet, Energetic, structural, and antimicrobial analyses of β-lactam side chain recognition by β-lactamases, Chem. Biol., 2001, 8, 17, DOI:10.1016/S1074-5521(00)00052-1.
- S. Agarwal, Q. Jin and S. Maji, Biobased polymers from plant-derived tulipalin A, in Biobased monomers, polymers, and materials, ACS Publications, 2012, p. 197 Search PubMed.
- C. Tănase, L. Pintilie and R. E. Tănase, Tănase, Lactones in the synthesis of prostaglandins and prostaglandin analogs, Int. J. Mol. Sci., 2021, 22, 1572, DOI:10.3390/ijms22041572.
- Y. Shi, T. Dong, B. Zeng, M. Yao, Y. Wang, Z. Xie, W. Xiao and Y. Yuan, Production of plant sesquiterpene lactone parthenolide in the yeast cell factory, ACS Synth. Biol., 2022, 11, 2473, DOI:10.1021/acssynbio.2c00132.
- S. Schulz and S. Hötling, The use of the lactone motif in chemical communication, Nat. Prod. Rep., 2015, 32, 1042, 10.1039/C5NP00006H.
- R. R. Kitson, A. Millemaggi and R. J. Taylor, The renaissance of α–methylene–γ–butyrolactones: new synthetic approaches, Angew. Chem., Int. Ed., 2009, 48, 9426, DOI:10.1002/anie.200903108Digital.
- H. Hoffmann and J. Rabe, Synthesis and biological activity of α–methylene–γ–butyrolactones, Angew. Chem., Int. Ed., 1985, 24, 94, DOI:10.1002/anie.198500941.
- Q. Song, C. Pascouau, J. Zhao, G. Zhang, F. Peruch and S. Carlotti, Ring-opening polymerization of γ-lactones and copolymerization with other cyclic monomers, Prog. Polym. Sci., 2020, 110, 101309, DOI:10.1016/j.progpolymsci.2020.101309.
- P. Olsén, K. Odelius and A.-C. Albertsson, Thermodynamic presynthetic considerations for ring-opening polymerization, Biomacromolecules, 2016, 17, 699, DOI:10.1021/acs.biomac.5b01698.
- P. McMichael, X. Schultze, H. Cramail and F. Peruch, Sourcing, thermodynamics, and ring-opening (co) polymerization of substituted δ-lactones: a review, Polym. Chem., 2023, 14, 3783, 10.1039/D3PY00657C.
- J. Zhang, L. Jiang, S. Liu, J. Shen, P. Braunstein, Y. Shen, X. Kang and Z. Li, Bifunctional and recyclable polyesters by chemoselective ring-opening polymerization of a δ-lactone derived from CO2 and butadiene, Nat. Commun., 2024, 15, 8698, DOI:10.1038/s41467-024-52090-2.
- J. Engel, A. Cordellier, L. Huang and S. Kara, Enzymatic ring–opening polymerization of lactones: Traditional approaches and alternative strategies, ChemCatChem, 2019, 11, 4983, DOI:10.1002/cctc.201900976.
- H. Zhao, G. A. Nathaniel and P. C. Merenini, Enzymatic ring-opening polymerization (ROP) of lactides and lactone in ionic liquids and organic solvents: digging the controlling factors, RSC Adv., 2017, 7, 48639, 10.1039/C7RA09038B.
- Z. Li, Y. Shen and Z. Li, Ring-opening polymerization of lactones to prepare closed-loop recyclable polyesters, Macromolecules, 2024, 57, 1919, DOI:10.1021/acs.macromol.3c01912.
- Z. Cai, Y.-M. Tu, H.-Z. Fan and J.-B. Zhu, Benzo-Fused Monomer Design toward Semiaromatic Polymers for a Circular Plastic Economy, Acc. Chem. Res., 2026, 20591, DOI:10.1021/acs.accounts.5c00743.
- Y.-C. Ye, Y.-H. Luo, M. Xie, Q. Cao, Z. Cai and J.-B. Zhu, Selectively controlled ring-opening copolymerization to chemically recyclable thermoplastic elastomers, Polym. Chem., 2026, 17, 1377, 10.1039/D6PY00127K.
- A. W. Woodhouse, A. Kocaarslan, J. A. Garden and H. Mutlu, Unlocking the potential of polythioesters, Macromol. Rapid Commun., 2024, 45, 2400260, DOI:10.1002/marc.202400260.
- R. J. Anderson, T. Akiyama and I. A. Tonks, Synthesis and ring-opening (co) polymerization of lactones derived from the cotelomerization of isoprene, butadiene, and CO 2, Faraday Discuss., 2026, 262, 94, 10.1039/D5FD00034C.
- A.-C. Albertsson and I. K. Varma, Recent developments in ring opening polymerization of lactones for biomedical applications, Biomacromolecules, 2003, 4, 1466, DOI:10.1021/bm034247a.
- J. Kollár, M. Danko, F. Pippig and J. Mosnáček, Functional polymers and polymeric materials from renewable alpha-unsaturated gamma-butyrolactones, Front. Chem., 2019, 7, 845, DOI:10.3389/fchem.2019.00845.
- M. W. Jones, S.-J. Richards, D. M. Haddleton and M. I. Gibson, Poly (azlactone) s: versatile scaffolds for tandem post-polymerisation modification and glycopolymer synthesis, Polym. Chem., 2013, 4, 717, 10.1039/C2PY20757E.
- P. Espeel and F. E. Du Prez, One-pot multi-step reactions based on thiolactone chemistry: A powerful synthetic tool in polymer science, Eur. Polym. J., 2015, 62, 247, DOI:10.1016/j.eurpolymj.2014.07.008.
- M. W. van Rossum, M. Alberda and L. H. van der Plas, Tulipaline and tuliposide in cultured explants of tulip bulb scales, Phytochemistry, 1998, 49, 723, DOI:10.1016/S0031-9422(98)00199-X.
- J. W. Doyle and A. A. Kandutsch, Requirement for mevalonate in cycling cells: quantitative and temporal aspects, J. Cell. Physiol., 1988, 137, 133, DOI:10.1002/jcp.1041370116.
- D. Kiani, R. Eaglesfield, J. H. May, A. Z. Werner, E. Y.-X. Chen, Y. Román-Leshkov, Y. J. Pagán-Torres and G. T. Beckham, Production of bio-based lactones as monomers for a circular polymer economy, Nat. Rev. Chem., 2025, 1, DOI:10.1038/s41570-025-00765-9.
- L. E. Manzer, Catalytic synthesis of α-methylene-γ-valerolactone: a biomass-derived acrylic monomer, Appl. Catal., A, 2004, 272, 249, DOI:10.1016/j.apcata.2004.05.048.
- M. Al-Naji, B. Puértolas, B. Kumru, D. Cruz, M. Bäumel, B. V. Schmidt, N. V. Tarakina and J. Pérez-Ramírez, Sustainable continuous flow valorization of γ–valerolactone with trioxane to α–Methylene–γ–Valerolactone over basic beta zeolites, ChemSusChem, 2019, 12, 2628, DOI:10.1002/cssc.201900418.
- M. Bilal and M. Al-Naji, Recent developments of the biorefinery of γ-valerolactone, Discover Catalysis, 2025, 2, 10, DOI:10.1007/s44344-025-00016-5.
- R. R. Gowda and E. Y.-X. Chen, Synthesis of β-methyl-α-methylene-γ-butyrolactone from biorenewable itaconic acid, Org. Chem. Front., 2014, 1, 230, 10.1039/C3QO00089C.
- X. Tang, M. Hong, L. Falivene, L. Caporaso, L. Cavallo and E. Y. X. Chen, The Quest for Converting Biorenewable Bifunctional α-Methylene-γ-butyrolactone into Degradable and Recyclable Polyester: Controlling Vinyl-Addition/Ring-Opening/Cross-Linking Pathways, J. Am. Chem. Soc., 2016, 138, 14326, DOI:10.1021/jacs.6b07974.
- J. T. Trotta, M. Jin, K. J. Stawiasz, Q. Michaudel, W.-L. Chen and B. P. Fors, Synthesis of methylene butyrolactone polymers from itaconic acid, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2730, DOI:10.1002/pola.28654.
- A. Nigl, V. Delsoglio, L. Sovic, M. Grgić, L. Malihan-Yap, K. Myrtollari, J. Spasic, M. Winkler, G. Oberdorfer, A. Taden, I. Anić and R. Kourist, Engineering of Transmembrane Alkane Monooxygenases to Improve a Key Reaction Step in the Synthesis of Polymer Precursor Tulipalin A, Angew. Chem., Int. Ed., 2025, 64, e202503464, DOI:10.1002/anie.202503464.
- N. R. Baral, M. Yang, B. G. Harvey, B. A. Simmons, A. Mukhopadhyay, T. S. Lee and C. D. Scown, Production Cost and Carbon Footprint of Biomass-Derived Dimethylcyclooctane as a High-Performance Jet Fuel Blendstock, ACS Sustainable Chem. Eng., 2021, 9, 11872, DOI:10.1021/acssuschemeng.1c03772.
- ] Visolis Inc., inv. a. B. N. Deepak Dugar, 2015.
- M. K. Akkapeddi, Poly(α-methylene-γ-butyrolactone) Synthesis, Configurational Structure, and Properties, Macromolecules, 1979, 12, 546, DOI:10.1021/ma60070a002.
- H. Elhaddad and D. W. Yee, Overview of Tacticity Control in Radical Polymerization, Chimia, 2024, 78, 831, DOI:10.2533/chimia.2024.831.
- Y. Hu, L. O. Gustafson, H. Zhu and E. Y.-X. Chen, Anionic polymerization of MMA and renewable methylene butyrolactones by resorbable potassium salts, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2008, DOI:10.1002/pola.24628.
- J. Suenaga, D. M. Sutherlin and J. K. Stille, Polymerization of (RS)- and (R)-α-methylene-γ-methyl-γ-butyrolactone, Macromolecules, 1984, 17, 2913, DOI:10.1021/ma00142a080.
- C. U. Pittman Jr. and H. Lee, Radical-initiated polymerization of β-methyl-α-methylene-γ-butyrolactone, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1759, DOI:10.1002/pola.10719.
- M. Mousa, H. Bergenudd, A. L. Kron and E. Mälmström, Biobased Lactones—Exploring Their Free-Radical Polymerization and Polymer Properties, Macromolecules, 2021, 54, 6127, DOI:10.1021/acs.macromol.1c00543.
- M. Ferriol, A. Gentilhomme, M. Cochez, N. Oget and J. Mieloszynski, Thermal degradation of poly (methyl methacrylate)(PMMA): modelling of DTG and TG curves, Polym. Degrad. Stab., 2003, 79, 271, DOI:10.1016/S0141-3910(02)00291-4.
- C. Wei, D. Esposito and K. Tauer, Thermal properties of thermoplastic polymers: Influence of polymer structure and procedure of radical polymerization, Polym. Degrad. Stab., 2016, 131, 157, DOI:10.1016/j.polymdegradstab.2016.07.016.
- Z. Vobecka, C. Wei, K. Tauer and D. Esposito, Poly(α-methylene-γ-valerolactone) 1. Sustainable monomer synthesis and radical polymerization studies, Polymer, 2015, 74, 262, DOI:10.1016/j.polymer.2015.04.074.
- E. Wojtczak, P. Kubisa and M. Bednarek, Thermal stability of polylactide with different end-groups depending on the catalyst used for the polymerization, Polym. Degrad. Stab., 2018, 151, 100, DOI:10.1016/j.polymdegradstab.2018.03.003.
- C. Lee and H. Hall Jr, Photocopolymerizations of electron-rich olefins with electron-poor olefins by irradiation of their EDA complexes, Macromolecules, 1989, 22, 21, DOI:10.1021/ma00191a004.
- M. K. Akkapeddi, The free radical copolymerization characteristics of α-methylene γ-butyrolactone, Polymer, 1979, 20, 1215, DOI:10.1016/0032-3861(79)90145-9.
- Y. W. Marien, M. Edeleva, P. H. M. Van Steenberge and D. R. D'Hooge, Exploiting the pulsed laser polymerization-size exclusion chromatography technique to retrieve kinetic parameters in radical polymerization: State-of-the-art and future challenges, in Adv. Chem. Eng, ed. D. Moscatelli and M. Sponchioni, Academic Press, 2020, ch. 3, p. 59 Search PubMed.
- R. A. Cockburn, R. Siegmann, K. A. Payne, S. Beuermann, T. F. L. McKenna and R. A. Hutchinson, Free Radical Copolymerization Kinetics of γ-Methyl-α-methylene-γ-butyrolactone (MeMBL), Biomacromolecules, 2011, 12, 2319, DOI:10.1021/bm200400s.
- K. Mohammad, H. Luetzen, P. Sielaff, B. Spiegelberg, A. Taden and S. Herres-Pawlis, Free-Radical (Co)polymerization of Tulipalin A—Analyzing Copolymerization Behavior, Macromolecules, 2025, 58, 9607, DOI:10.1021/acs.macromol.5c00640.
- D. Apostolidis, W. E. Dyer, C. A. Dransfeld and B. Kumru, Solution and precipitation based radical polymerization of renewable vinyl lactones in renewable solvents, RSC Adv., 2025, 15, 21659, 10.1039/D5RA02151K.
- Netherlands, inv. K. B. a. A. D. Clemens, 2022.
- G. M. Miyake, Y. Zhang and E. Y. X. Chen, Living Polymerization of Naturally Renewable Butyrolactone-Based Vinylidene Monomers by Ambiphilic Silicon Propagators, Macromolecules, 2010, 43, 4902, DOI:10.1021/ma100615t.
- J. Chen and E. Y.-X. Chen, Lewis Pair Polymerization of Acrylic Monomers by N-Heterocyclic Carbenes and B(C6F5)3, Isr. J. Chem., 2015, 55, 216, DOI:10.1002/ijch.201400136.
- Y. Zhang, G. M. Miyake and E. Y.-X. Chen, Alane-Based Classical and Frustrated Lewis Pairs in Polymer Synthesis: Rapid Polymerization of MMA and Naturally Renewable Methylene Butyrolactones into High-Molecular-Weight Polymers, Angew. Chem., Int. Ed., 2010, 49, 10158, DOI:10.1002/anie.201005534.
- Y. Bai, L. Liao, S. Liu, Y. Li, J. He and Y. Zhang, Rapid and living polymerization of renewable methylene butyrolactones via borane/phosphine Lewis pairs, Polym. Chem., 2025, 16, 4804, 10.1039/D5PY00789E.
- Y. Zhang, G. M. Miyake, M. G. John, L. Falivene, L. Caporaso, L. Cavallo and E. Y. X. Chen, Lewis pair polymerization by classical and frustrated Lewis pairs: acid, base and monomer scope and polymerization mechanism, Dalton Trans., 2012, 41, 9119, 10.1039/C2DT30427A.
- Y. Bai, H. Wang, J. He, Y. Zhang and E. Y.-X. Chen, Dual-initiating and living frustrated Lewis pairs: expeditious synthesis of biobased thermoplastic elastomers, Nat. Commun., 2021, 12, 4874, DOI:10.1038/s41467-021-25069-6.
- Y. Zhang, M. Schmitt, L. Falivene, L. Caporaso, L. Cavallo and E. Y. X. Chen, Organocatalytic Conjugate-Addition Polymerization of Linear and Cyclic Acrylic Monomers by N-Heterocyclic Carbenes: Mechanisms of Chain Initiation, Propagation, and Termination, J. Am. Chem. Soc., 2013, 135, 17925, DOI:10.1021/ja4088677.
- P. B. V. Scholten, D. Moatsou, C. Detrembleur and M. A. R. Meier, Progress Toward Sustainable Reversible Deactivation Radical Polymerization, Macromol. Rapid Commun., 2020, 41, 2000266, DOI:10.1002/marc.202000266.
- J. Mosnáček and K. Matyjaszewski, Atom Transfer Radical Polymerization of Tulipalin A: A Naturally Renewable Monomer, Macromolecules, 2008, 41, 5509, DOI:10.1021/ma8010813.
- J. Mosnáček, J. A. Yoon, A. Juhari, K. Koynov and K. Matyjaszewski, Synthesis, morphology and mechanical properties of linear triblock copolymers based on poly(α-methylene-γ-butyrolactone), Polymer, 2009, 50, 2087, DOI:10.1016/j.polymer.2009.02.037.
- Y. Higaki, R. Okazaki, T. Ishikawa, M. Kikuchi, N. Ohta and A. Takahara, Chain stiffness and chain conformation of poly(α-methylene-γ-butyrolactone) in dilute solutions, Polymer, 2014, 55, 6539, DOI:10.1016/j.polymer.2014.10.026.
- A. Juhari, J. Mosnáček, J. A. Yoon, A. Nese, K. Koynov, T. Kowalewski and K. Matyjaszewski, Star-like poly (n-butyl acrylate)-b-poly (α-methylene-γ-butyrolactone) block copolymers for high temperature thermoplastic elastomers applications, Polymer, 2010, 51, 4806, DOI:10.1016/j.polymer.2010.08.017.
- J. Shin, Y. Lee, W. B. Tolman and M. A. Hillmyer, Thermoplastic Elastomers Derived from Menthide and Tulipalin A, Biomacromolecules, 2012, 13, 3833, DOI:10.1021/bm3012852.
- K. Ding, A. John, J. Shin, Y. Lee, T. Quinn, W. B. Tolman and M. A. Hillmyer, High-Performance Pressure-Sensitive Adhesives from Renewable Triblock Copolymers, Biomacromolecules, 2015, 16, 2537, DOI:10.1021/acs.biomac.5b00754.
- A. Métafiot and M. Marić, Nitroxide mediated controlled radical polymerization of α-Methylene-γ-Butyrolactone, J. Polym. Sci., 2024, 62, 3399, DOI:10.1002/pol.20240115.
- F. G. Versteeg, N. C. Hegeman, K. O. Sebakhy and F. Picchioni, RAFT Polymerization of a Biorenewable/Sustainable Monomer via a Green Process, Macromol. Rapid Commun., 2022, 43, 2200045, DOI:10.1002/marc.202200045.
- O. J. Harris, R. R. Larder, B. Jordan, I. Prior, R. El-Khoury, K. O. Sebakhy and F. L. Hatton, RAFT solution polymerisation of bio-based γ-methyl-α-methylene-γ-butyrolactone monomer in DMSO and Cyrene, Chem. Commun., 2024, 60, 14399, 10.1039/D4CC04571H.
- F. G. Versteeg, T. Pelras, G. Portale and F. Picchioni, Synthesis of a thermoplastic elastomer from α-methylene-γ-butyrolactone for high temperature applications, Mater. Adv., 2024, 5, 7446, 10.1039/D4MA00583J.
- Y. Hu, X. Xu, Y. Zhang, Y. Chen and E. Y. X. Chen, Polymerization of Naturally Renewable Methylene Butyrolactones by Half-Sandwich Indenyl Rare Earth Metal Dialkyls with Exceptional Activity, Macromolecules, 2010, 43, 9328, DOI:10.1021/ma101901y.
- G. M. Miyake, S. E. Newton, W. R. Mariott and E. Y. X. Chen, Coordination polymerization of renewable butyrolactone-based vinyl monomers by lanthanide and early metal catalysts, Dalton Trans., 2010, 39, 6710, 10.1039/C001909G.
- X. Chen, L. Caporaso, L. Cavallo and E. Y. X. Chen, Stereoselectivity in Metallocene-Catalyzed Coordination Polymerization of Renewable Methylene Butyrolactones: From Stereo-random to Stereo-perfect Polymers, J. Am. Chem. Soc., 2012, 134, 7278, DOI:10.1021/ja301811s.
- Y. Hu, G. M. Miyake, B. Wang, D. Cui and E. Y.-X. Chen, ansa-Rare-Earth-Metal Catalysts for Rapid and Stereoselective Polymerization of Renewable Methylene Methylbutyrolactones, Chem. – Eur. J., 2012, 18, 3345, DOI:10.1002/chem.201102677.
- Y. Hu, X. Wang, Y. Chen, L. Caporaso, L. Cavallo and E. Y. X. Chen, Rare-Earth Half-Sandwich Dialkyl and Homoleptic Trialkyl Complexes for Rapid and Stereoselective Polymerization of a Conjugated Polar Olefin, Organometallics, 2013, 32, 1459, DOI:10.1021/om301128g.
- V. Graur, A. Mukherjee, K. O. Sebakhy and R. K. Bose, Initiated Chemical Vapor Deposition (iCVD) of Bio-Based Poly(tulipalin A) Coatings: Structure and Material Properties, Polymers, 2022, 14, 3993, DOI:10.3390/polym14193993.
- A. Elshewy, M. E. Hariri El Nokab, J. E. Sayed, Y. A. Alassmy, M. M. Abduljawad, D. R. D′hooge, P. H. M. Van Steenberge, M. H. Habib and K. O. Sebakhy, Surfactant-Free Peroxidase-Mediated Enzymatic Polymerization of a Biorenewable Butyrolactone Monomer via a Green Approach: Synthesis of Sustainable Biobased Latexes, ACS Appl. Polym. Mater., 2024, 6, 115, DOI:10.1021/acsapm.3c01740.
- R. Dammel, M. Cook, A. Klauck-Jacobs, T. Kudo, S. Mehtsun, J. Oberlander, M. Padmanaban and M. Dalil Rahman, J. Photopolym. Sci. Technol., 1999, 12, 433, DOI:10.2494/photopolymer.12.433.
- K. Nozaki and E. Yano, High-performance resist materials for ArF excimer laser and electron beam lithography, Fujitsu Sci. Tech. J., 2002, 38, 3 CAS.
- R. Dammel, M. Cook, A. Klauck-Jacobs, T. Kudo, S. Mehtsun, J. Oberlander, M. Padmanaban and D. L. Rahman, 193 nm Resists for Deep Sub-Wavelength Applications, J. Photopolym. Sci. Technol., 1999, 12, 433, DOI:10.2494/photopolymer.12.433.
- K. N. a. E. Yano, High-Performance Resist Materials for ArF Excimer Laser and Electron Beam Lithography, Fujitsu Sci. Tech. J., 2001, 38 Search PubMed.
- C. Esen, M. Antonietti and B. Kumru, On the photopolymerization of mevalonic lactone methacrylate: exposing the potential of an overlooked monomer, Polym. Chem., 2022, 13, 139, 10.1039/D1PY01497H.
- R. A. Gilsdorf, E. R. Chokkapu, A. Athaley, T. Uekert, R. R. Gowda, A. Singh, J. S. DesVeaux, G. T. Beckham and E. Y.-X. Chen, Bio-based lactone acrylic plastics with performance and recyclability advantages, Cell Rep. Phys. Sci., 2024, 5, DOI:10.1016/j.xcrp.2024.101938.
- R. A. Gilsdorf, M. A. Nicki and E. Y. X. Chen, High chemical recyclability of vinyl lactone acrylic bioplastics, Polym. Chem., 2020, 11, 4942, 10.1039/D0PY00786B.
- Z.-H. Zhang, X. Wang, B. Weng, Y. Zhang, G. Zhang and M. Hong, Zinc-mediated allylation-lactonization one-pot reaction to methylene butyrolactones: Renewable monomers for sustainable acrylic polymers with closed-loop recyclability, ACS Polym. Au, 2022, 2, 266, DOI:10.1021/acspolymersau.2c00001.
- G. C. Gilchrist, R. W. Hughes, T. Maity, S. R. Gitter, N. Izadi, J. D. Marquez, J. B. Young, A. M. Evans and B. S. Sumerlin, Electrochemically Initiated Depolymerization of Poly(Methyl Methacrylate), J. Am. Chem. Soc., 2026, 148, 3336, DOI:10.1021/jacs.5c17930.
- T. Maity, D. X. Bones, R. W. Hughes, K. C. Stevens, W. M. Gramlich and B. S. Sumerlin, Depolymerization of Vinyl Polymers, ACS Macro Lett., 2026, 15, 17–32, DOI:10.1021/acsmacrolett.5c00740.
- B. Daelman, J. Debuyck, V. Scholiers, J. M. Winne and F. E. Du Prez, Creep Resistant and Reprocessable Polyamide Networks Based on Reversible Lactone Ring-Opening, Angew. Chem., Int. Ed., 2026, 65, e19828, DOI:10.1002/anie.202519828.
| This journal is © The Royal Society of Chemistry 2026 |