
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development and modification of porous polymer structures in the vicinity of cellulose fibers
Sebastian
Pusse
 a,
Sebastian
Heinz
a,
Waranya
Limprasart
b,
Lea
Gemmer
a,
Suteera
Witayakran
ad,
Samuel
Schabel
c,
Volker
Presser
efg,
Torsten
Gutmann
*bh and
Markus
Gallei
*ag
a,
Sebastian
Heinz
a,
Waranya
Limprasart
b,
Lea
Gemmer
a,
Suteera
Witayakran
ad,
Samuel
Schabel
c,
Volker
Presser
efg,
Torsten
Gutmann
*bh and
Markus
Gallei
*ag
aPolymer Chemistry, Saarland University, Campus C4 2, 66123 Saarbrücken, Germany. E-mail: markus.gallei@uni-saarland.de
bEduard-Zintl-Institute for Inorg. and Phys. Chemistry, Technical University Darmstadt, Peter-Grünberg-Straße 8, 64287 Darmstadt, Germany. E-mail: gutmann@chemie.tu-darmstadt.de
cMechanical Engineering, Chair of Paper Technology and Mechanical Process Engineering, Technical University of Darmstadt, Alexanderstraße 8, Darmstadt, 64283, Germany
dMax Planck Institute for Informatics, Saarland Informatics Campus, E1 4, 66123 Saarbrücken, Germany
eINM - Leibniz Institute for New Materials, Saarbrücken 66123, Germany
fDepartment of Materials Science and Engineering, Saarland University, Campus D2 2, Saarbrücken 66123, Germany
gSaarene, Saarland Center for Energy Materials and Sustainability, Saarland University, Campus C4 2, 66123 Saarbrücken, Germany
hDepartment of Chemistry, Physical Chemistry, University of Paderborn, Warburger Straße 100, 33098 Paderborn, Germany
First published on 23rd March 2026
Abstract
In this work, hierarchically porous materials have been prepared by the self-assembly and pore formation of different amphiphilic block copolymers (BCPs) in the vicinity of cellulose paper sheets. For this, polystyrene-block-polysolketal methacrylate (PS-b-SMA) as a linear BCP and polymethyl methacrylate-block-polysolketal methacrylate (PMMA-b-SMA)n as a star-shaped BCP were prepared using living anionic polymerization. Under mild acidic conditions, the amphiphilic properties were revealed by converting the PSMA block segment to poly(dihydroxypropyl methacrylate) (PDHPMA). The BCPs were incorporated onto cellulose linters fiber-based sheets by a self-assembly and nonsolvent-induced phase separation (SNIPS) process. The resulting porous materials have been further modified with 3-aminopropyltriethoxysilane (APTES) and 3,3,3-trifluoropropyl dimethyl chlorosilane (TFPCS) using a vapor-phase modification approach. This strategy enabled further tuning of the surface properties of the resulting porous structures to adjust surface polarity. The characteristics of the modified porous materials were confirmed at the microscopic scale by solid-state nuclear magnetic resonance (NMR) combined with selectively enhanced dynamic nuclear polarization (DNP) and Fourier transform infrared (FTIR) spectroscopy. The influence of APTES and TFPCS was further analyzed at the macroscopic level using water contact angle (WCA) measurements and water permeance testing, where changes were observed for both modifiers. Using this convenient strategy, the fabrication of functional porous cellulose composite materials is demonstrated, paving the way for a new family of cellulose-based porous materials.
Introduction
The availability of freshwater is becoming a global challenge for humankind, with up to 87 out of 180 countries expected to face water scarcity by 2050. Yet, the small amount of fresh drinking water is further reduced by human-made contamination. It is worth mentioning two very recent examples that have only gained significant attention in recent years: one is the pollution of our environment by microplastics and the health concerns it raises.1–4 The second example is per- and polyfluoroalkyl substances (PFASs) due to their high persistence in the environment and the human body.5–7 To fight against such health hazards, an ever-increasing range of tools is required now and in the future. One approach to addressing environmental pollution is the use of porous functional materials.8–10 Such porous materials are used in filtration and adsorption systems and are often composed of polyvinylidene fluoride (PVDF), polyacrylonitrile (PAN), polyether sulfones (PESs), polyamide (PA), or cellulose.11–13 The last example is considered more sustainable, as it is derived from natural and renewable resources such as wood, cotton, hemp, algae, and bacteria, rather than petroleum-based products.Cellulose, the most abundant renewable biopolymer on Earth, is a linear polysaccharide composed of β (1 → 4)-linked D-glucose units biosynthesized by plants, various types of algae, and bacteria.14 Due to its renewability, biodegradability, and beneficial mechanical properties, cellulose has become a sustainable alternative to petroleum-based polymers in materials science and engineering, helping reduce environmental impacts and decrease reliance on non-renewable resources.15 Because cellulose fibers are lightweight and exhibit high tensile strength and good thermal stability, these qualities make them valuable for manufacturing paper and packaging, textiles, films, membranes, biocomposites and emerging technologies such as biomedical devices and smart electronics.16,17 However, the intrinsic hydrophilicity of cellulose, caused by the abundance of surface hydroxy moieties in its backbone, limits its compatibility with hydrophobic matrices and hampers performance under humid conditions. To overcome these challenges, surface modification has been widely studied to improve or tailor interfacial and chemical wettability and barrier properties to meet the desired requirements of advanced materials. Various surface modification techniques have been developed, including chemical grafting, silanization, polymer grafting, plasma or vapor deposition and surface coating.14,18,19 Among these, polymer coating has gained significant attention due to its simplicity, scalability and ability to preserve the native structure of the fiber substrate.20,21 Applying functional polymer layers onto cellulose fibers or paper allows for adjustments in surface wettability, barrier properties and mechanical strength, making it suitable for applications such as sustainable packaging,22 filtration membranes,23 microfluidics,24 sensors,25,26 and advanced functional composites.27,28 Besides improving the intrinsic properties of cellulose fibers or paper, the coating process can also benefit from the presence of the cellulose substrate itself. Recent studies have shown that the cellulose substrate has a potential impact on molecular alignment or enhances the structural organization to produce highly ordered microstructures of amphiphilic block copolymers (BCPs), such as polystyrene-block-poly(2-hydroxyethyl methacrylate) (PS-b-PHEMA)29 and polyisoprene-block-polyvinyl pyridine (PI-b-P2VP),30 in direct contact with cellulose fibers. The self-assembly of BCPs, such as PS-b-PHEMA or PI-b-P2VP, is strongly influenced by the presence of cellulose fibers, thereby affecting domain orientation and pore-forming capabilities. By combining a wide range of BCPs with specific functionalities, the applicability of porous cellulose materials can be significantly enhanced. For example, cellulose can be equipped with BCPs, which not only provide a hierarchical porous structure but also feature stimuli responsiveness. Temperature as a trigger has been shown to alter pore size and polarity in so-called smart membranes.31,32 A change in membrane characteristics can be obtained as a response to changes in the pH of a filtrate,33–35 or the presence of sugars.36 BCPs can be used not only for the fabrication of stimuli-responsive membranes but also for targeting specific pollutants, such as heavy metal ions or perfluorinated pollutants.37 For each example presented, the functionality is already implemented into the polymer before membrane fabrication. For functionalities that interfere with the membrane-forming process or cannot be polymerized and integrated into BCP segments, an alternative approach for application-specific membranes is the post-modification of already prepared BCP membranes. When direct synthesis or membrane preparation is not suitable, a BCP with an addressable functional group is synthesized, converted into a membrane, and then post-modified. Membranes derived from the self-assembly of polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP) represent a feasible polymer system for post-modification strategies. Here, the P4VP block segment can be modified to form a polyelectrolyte brush, enhancing anti-fouling properties.38 The already mentioned PS-b-PHEMA represents another family of BCP motifs. PHEMA provides hydroxy groups as addressable functional groups. These groups already impart anti-fouling properties39,40 but also allow further modification with amines, peptides41 or esters42 for protein-selective separation strategies. In all the examples presented above, the membranes originate from a special procedure, namely the self-assembly and non-solvent induced phase separation (SNIPS) process.43,44 The SNIPS process represents a very feasible tool for the fabrication of such high-performance membranes, where the pore size can be defined by the material used during membrane preparation.45 Ultrafiltration membranes with a uniform pore-size distribution, combined with high pore order and pore density, provide a sharp cutoff for filtrates, forming the foundation here. The BCP is a central element in this fabrication protocol. To produce such uniform membranes, tailored BCPs serve as the basis for their composition and architecture. To approach such materials, controlled radical polymerizations,46–50 as well as living anionic polymerization,37,41,51–53 play a crucial role in material preparation. These controlled polymerization techniques can not only be used for the synthesis of linear block copolymers but also allow a wide variety of block copolymer architectures. Here, the architectures of star-shaped block copolymers in the form of (AB)n, with n indicating the number of arms, are of special interest. Influenced by BCP architecture, the gyroidal morphology becomes more stable over a wider range of BCP compositions54–57 compared to a rather narrow section of the mean-field diagram for a common linear block copolymer.58 Based on that observation, the application of star-shaped BCPs can lead to an alternative structure of the surface layer of a SNIPS membrane. Instead of dense, hexagonally packed cylindrical pores in the top layer,43 which must be trapped kinetically,59 interconnected porous structures with an isotropic orientation can be introduced using star-shaped block copolymers. Such structures tend to allow a higher water flux.60,61
In this work, we prepared two types of amphiphilic block copolymers via living anionic polymerization. First, a polystyrene-block-poly(2,3-dihydroxypropyl methacrylate) (PS-b-PDHPMA) BCP was prepared using solketal methacrylate (SMA) as a precursor for DHPMA. The second developed polymer was an amphiphilic star-shaped BCP prepared via a convergent arm-first route. For the more hydrophobic major block, methyl methacrylate (MMA) was used. The second, minor block is based on SMA. The more hydrophilic character is revealed during the deprotection step of the PMMA-b-PSMA block copolymer, converting it into PMMA-b-PDHPMA. Both polymers were fabricated into asymmetric porous structures using the SNIPS process and modified further with (3-aminopropyl) triethoxysilane (APTES) and 3,3,3-trifluoropropyl dimethyl chlorosilane (TFPCS) via vapor phase reactions. Both membranes were characterized using FTIR spectroscopy and water contact angle measurements. These were further complemented by selectively enhanced dynamic nuclear polarization (DNP) solid-state NMR spectroscopy, which has been established as a powerful tool to specifically address structural moieties at the molecular level in highly complex paper/polymer hybrid materials.62,63 In the final step, the effect of surface modification on the membrane's water permeance was investigated.
Experimental section
Materials
All chemicals and solvents were purchased from TCI Chemicals, Sigma-Aldrich, Fisher Scientific, and BLD Pharma. All chemicals and solvents were used as received unless otherwise stated. All monomers were prepared according to standard procedures typical of anionic polymerization. The monomers were dried over CaH2 and distilled prior to use. Tetrahydrofuran (THF) was dried with diphenylhexyllithium (DPHLi) and transferred freshly into a reaction ampulla using cryotransfer. MeOH used for quenching of active chain ends was dried over molecular sieves (3 Å) before cryotransfer and storage in a glovebox. The monomer SMA was synthesized as described in the literature, using toluene as a solvent instead of benzene.64 Linters paper sheets with a weight of 50.0 ± 1.6 g m−2 were prepared by the Chair of Paper Technology and Mechanical Process Engineering (PMV), following the Rapid-Köthen paper sheet formation procedure.Polymer synthesis
Membrane formation via the SNIPS protocol
In the first step of the membrane preparation process, a solvent mixture was prepared as a stock solution, containing THF, dimethylformamide (DMF), and 1,4-dioxane (DOX) in a weight ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. For each polymer, a casting solution was prepared by combining the polymer, the solvent mixture, and additives, as presented in Tables S1, S3 and S5. To ensure complete polymer dissolution, the mixture was slowly shaken for up to 48 hours at ambient temperature. The paper supports were pre-conditioned using THF. The highly viscous casting solution was spread using a doctor blade with a 200 µm gap size. After a certain evaporation time, the evenly coated paper was transferred into a precipitation bath containing deionized water. After 10 minutes in the precipitation bath, the membranes were first dried under ambient conditions and then under reduced pressure at elevated temperatures.
:1:1. For each polymer, a casting solution was prepared by combining the polymer, the solvent mixture, and additives, as presented in Tables S1, S3 and S5. To ensure complete polymer dissolution, the mixture was slowly shaken for up to 48 hours at ambient temperature. The paper supports were pre-conditioned using THF. The highly viscous casting solution was spread using a doctor blade with a 200 µm gap size. After a certain evaporation time, the evenly coated paper was transferred into a precipitation bath containing deionized water. After 10 minutes in the precipitation bath, the membranes were first dried under ambient conditions and then under reduced pressure at elevated temperatures.
Vapor phase functionalization with (3-aminopropyltriethoxysilane) (APTES) and (3,3,3-trifluoropropyl) dimethyl chlorosilane (TFPCS)
2 mL of APTES or TFPCS was placed in an evaporating dish inside a desiccator. The membrane was positioned face up directly above the liquid APTES or TFPCS, and the desiccator was gradually evacuated to 0.02 mbar or 0.15 mbar to promote vapor-phase reactions of APTES and TFPCS, respectively. After 72 h under these conditions, the functionalized membrane was removed from the desiccator and allowed to dry at room temperature.Material characterization
Standard size exclusion chromatography (SEC) was performed with a 1260 Infinity II (Agilent Technologies) system. In this study, THF was used as the mobile phase (HPLC grade, flow rate: 1 mL min−1) on an SDV column set from Polymer Standard Service (PSS) (SDV 103 Å, SDV 105 Å, SDV 106 Å, 5 µm) with a PSS SECurity2 RI/UV detector. Calibration was carried out using polystyrene (PS) standards from PSS. PSS WinGPC® UniChrom V 8.31 was used for data acquisition and measurement evaluation.With dimethylformamide (DMF) as the mobile phase (flow rate 1 mL min−1, containing 1 g L−1 LiBr) a PSS GRAM analytical column set from PSS (30 Å, 103 Å, 103 Å) was used at a temperature of 50 °C. The calibration for this set-up is based on poly(methyl methacrylate) (PMMA) standards from PSS.
Fourier-transform infrared spectroscopy (FTIR) spectra were recorded using an attenuated total reflectance Fourier transform infrared (ATR-FTIR, diamond ATR) instrument (Bruker Alpha II) in the 400–4000 cm−1 range with a resolution of 4 cm−1.
Scanning electron microscopy (SEM) was performed with a ZEISS GEMINI 500 microscope using SmartSEM Version 6.07. The samples were mounted on an aluminum stud using adhesive copper tape and sputter-coated with approximately 6 nm platinum using a PLASMATOOL 125 SIN 2020_131 automatic turbo coater from Ingenieurbüro Peter Liebscher.
Static water contact angle (WCA) measurements were performed using a Hamilton syringe (100 µL), a syringe pump (KD Scientific, adjusted to 10 µL), and a custom-made xyz-positioning table. In standard measurements, deionized water was used. Photographs were collected using a Nikon D54000 and digiCamControl 2.1.2.0, and OpenDrop 3.3.165 was used for evaluation. The WCA was measured from three different spots on the sample. The absorption time is the total duration from the initial contact of the water droplet until it visibly disappears. Data are presented as mean ± standard deviation (SD).
Nuclear magnetic resonance (NMR) spectra for the characterization of the different polymers in solution were recorded using a Bruker Avance II 400 spectrometer with a 9.4 T Ultrashield Plus Magnet and a BBFO probe and were referenced to the solvent signals.66 For processing and evaluation of the spectra, MestReNova 14.2.0 was used.
DNP-enhanced solid-state NMR experiments were performed on samples prepared according to the literature.63,67 Typically, 10 mg of each material was mixed with ca. 10 μL of a 15 mM AMUPol D2O/H2O (9:1) (AMU) or a 15 mM TEKPol in TCE (TEK) solution. Samples were packed into 3.2 mm sapphire rotors and sealed with a Teflon plug and a ZrO2 driving cap.
All DNP-enhanced solid-state NMR spectra were recorded using a Bruker Avance III 400 MHz DNP spectrometer operating at 9.4 T according to frequencies of 401.63 MHz, 100.99 MHz, and 79.79 MHz for 1H, 13C, and 29Si, respectively. This spectrometer was equipped with a 9.7 T Bruker gyrotron system generating microwaves with a frequency of 263 GHz. The spectra were recorded at nominally 120 K with a 3.2 mm low temperature probe operated in 1H/X double mode.
1H → 13C CP MAS DNP spectra were recorded at a spinning rate of 8 kHz. Spectra were acquired with a contact time of 2 ms. The acquisition time was set to 30 ms, and the excitation length of the 90° pulse on the 1H channel was set to 3 µs. The recycle delay was varied depending on the sample and radical matrix, as summarized in Tables S8 and S9. Each spectrum was recorded, accumulating 512 scans. Heteronuclear decoupling was performed during data acquisition employing a tppm15 decoupling sequence.68 Referencing was done with respect to TMS (0 ppm) according to the literature.69
1H → 29Si CP MAS DNP spectra were recorded at a spinning rate of 8 kHz. The spectra were acquired with a contact time of 0.5 ms. The acquisition time was set to 25 ms and the excitation pulse length of the 90° pulse on the 1H channel to 3 µs. The recycle delay was varied depending on the sample and radical matrix, as summarized in Tables S8 and S9. Each spectrum was recorded, accumulating 5120 scans. Heteronuclear decoupling was performed during data acquisition employing the tppm15 decoupling sequence.68 Referencing was done with respect to TSP (0 ppm).
Permeance measurements were carried out using a measuring cell as a dead-end setup with a total volume of 400 mL. For all measurements, an external pressure of 0.2 bar was applied using N2. Membranes were measured over a time of up to three hours in intervals of approximately 10 to 11 min. For all measurements, circular samples with a diameter of 10 mm were used. All samples were preconditioned by immersing them for 30 min in deionized water (MilliQ).
Results and discussion
Synthesis of linear and star-shaped block copolymers
The linear amphiphilic AB BCP was synthesized using a sequential approach via living anionic polymerization (Scheme 1a). First, the more hydrophobic block segment was prepared from styrene using s-BuLi as the initiator. The living chain ends were modified using DPE to ensure the polymerization of the following methacrylate-based monomer.51,52,70,71 SMA was used to incorporate a more hydrophilic second block segment. The living chain ends were quenched after the second polymerization step using MeOH as a proton source. A corresponding 1H NMR spectrum of the BCP is displayed in Fig. 1a. Characteristic signals were observed for each block, as assigned in the spectrum. To reveal the amphiphilic character of the BCP, the polymer was treated with HCl in an aqueous environment to cleave off acetone as the protecting group, thereby yielding two hydroxy moieties and converting the PSMA block into a poly(2,3-dihydroxypropyl methacrylate) (PDHPMA) block segment. The corresponding 1H NMR spectrum for PS-b-PDHPMA is shown in Fig. 1c. The SEC measurements for each synthesis step are displayed in Fig. 2. Fig. 2a shows the linear BCP with the more hydrophobic block segment (blue) and the BCP (red), and after deprotection in Fig. 2b. As expected for this polymerization technique, the BCP shows a narrow molar mass distribution, confirming good control over the polymerization. Compared to the SEC trace for its first block (Fig. 2a), made from styrene, no broadening of the molecular weight distribution was observed. Only an increase in molecular weight was observed. The dispersity of Đ = 1.07 (Table 1) indicated good control over the polymerization without noticeable termination reactions during the initiation of the second monomer. For the deprotected PDHPMA, a small increase in molecular weight is observed (Fig. 2b) compared to the protected PSMA form (Fig. 2a), likely due to interactions between the PDHPMA hydroxy moieties. The polymer also showed an increased dispersity.52 For the preparation of the star-shaped (AB)n BCP and for gaining access to additional morphologies,72,73 another suitable synthesis pathway was used (Scheme 1b). The star-shaped BCP was prepared in a convergent arm-first approach using living anionic polymerization. First, to enable an architecture in which the more hydrophilic block is on the outside of the star structure, the more hydrophilic block (based on SMA) was polymerized first using DPHLi. This time, MMA was used for the second, more hydrophobic block of the BCP because of its greater reactivity in living anionic polymerization than styrene. To branch the linear BCPs and thus generate star-shaped (AB)n BCPs, 1,4-butanediol dimethacrylate (BDDMA) was added in a third step and stirred for several hours. To unveil the hydroxy moieties, the received polymerization product was treated in a manner similar to that of the linear PS-b-PSMA (Scheme 1a). A 1H NMR spectrum is presented in Fig. 1b, revealing characteristic signals for both PMMA and PSMA. For BDDMA, no characteristic signals are observed due to its low number of repeating units. SEC traces of (PSMA-b-PMMA)n are shown in Fig. 2c. Fig. 1d shows the NMR spectrum of its deprotected form PDHPMA-b-PMMA recorded in pyridine-d5, again displaying three signals at 4.63 ppm, 4.51 ppm, and 4.11 ppm, assigned to the side chain of PDHPMA. In the SEC measurement for the crosslinking product, a bimodal distribution is observable (Fig. 2d). This distribution indicated a mixture of two types of star-shaped BCPs. An assumption based on the trace maxima is presented in Fig. S1. It indicates a combination of a linear ABBA BCP and a four-arm BCP by comparison of the molecular weight of both maxima and the molecular weight of the linear precursor listed in Table 1. The SEC measurement of PSMA-b-PMMA unveils two more species at lower molecular weights (Fig. 2c). These species can be connected to unreacted PSMA and unreacted PSMA-b-PMMA. Similar to a linear BCP, the molar fraction of the second block (Table 1) is calculated from the 1H NMR spectrum of the PSMA-b-PMMA displayed in Fig. 1c, following eqn (S1). Like linear PS-b-PDHPMA, the molecular weight distribution of the star-shaped PMMA-b-PDHPMA has a broader molar mass distribution (Fig. 2d), featuring a dispersity of Đ = 1.52 compared to ĐStar BCP = 1.13. A significant increase in the apparent molecular weight was also observed. For a better comparison, the linear BCP and the star-shaped BCP were protected using benzoic anhydride in pyridine to protect the hydroxy moieties of the PDHPMA block.52 Both BCPs were then analyzed in SEC using THF and DMF as solvents. The results are given in Fig. S2 and Table S1. Each Bz-protected species appears with a slightly increased molecular weight compared to the PSMA species in THF. The received molecular weights for the Bz-protected species in both samples in DMF are lower than those in THF. This could be given due to different setups for both solvents. Since both samples follow the same trend for the Bz-protected species, the contradictory observations could be based on the interactions between the hydroxy moieties of the more hydrophilic block. Furthermore, the isolation of the star-shaped BCP via inverse precipitation was not successful. Because of this, the polymer was used further as a blend for the membrane preparation. | ||
| Scheme 1 Synthesis route to the linear BCP (a) and one possible star-shaped BCP (b), including the deprotection step of PSMA (red) to unveil the more hydrophilic PDHPMA block (red). | ||
 | ||
| Fig. 1 1H NMR spectra of the linear BCP before (a) and after deprotection (c), as well as the star-shaped BCP before (b) and after deprotection (d), with the assigned hydrogens in blue. The NMR spectra of the protected form were recorded in CDCl3. For the deprotected polymer, pyridine-d5 was used as a solvent. | ||
 | ||
| Fig. 2 Compilation of SEC measurements for each step in polymer synthesis for the linear BCP (a) in THF with PS calibration and deprotected (b) in DMF vs. PMMA calibration. The star-shaped BCP in THF vs. PS calibration is shown in (c) with its deprotected form in DMF vs. PMMA calibration (d). | ||
| M n [g mol−1] (Đ) |
x
hydrophilic
|
|||||
|---|---|---|---|---|---|---|
| Hydrophobic block | PSMA block | AB BCP | (AB)n BCP | Deprotectedc | ||
| a SEC measurement in THF against PS calibration. b Calculated from NMR before deprotection. c SEC measurement in DMF against PMMA calibration. | ||||||
| Linear BCP | 122000a (1.07) |
33600b |
138200a (1.07) |
— | 143740 (1.19) |
0.14 |
| Star BCP | 90700b |
25400a (1.05) |
163500a (1.03) |
538000a (1.13) |
439830 (1.52) |
0.12 |
Membrane preparation and post-modification
For membrane preparation, substrates consisting of neat cellulose fibers were used to maximize the effect of the polar BCP segment on the cellulose fiber surface. These cellulose sheets originate from cotton linters fibers and are therefore referred to as linters paper in the following text. The membranes were prepared according to a SNIPS protocol, and different batches were prepared. To estimate the evaporation time, a crucial parameter for the pore-formation process, a first batch comprising three membranes, M1a–M1c (Fig. S3), was prepared. The composition of the highly viscous polymer solution is listed in Table S2. For all batches, a solvent mixture containing THF, DMF, and DOX at a weight ratio of 2:1:1 was used. Additionally, CuCl2 was used as an additive to increase the solution viscosity and thus enhance pore order.52,74 THF was used for the major, more hydrophobic block, while DMF was used for the minor, more hydrophilic segment.52 DOX was used for the modification of the solvent blend and thus enhanced pore formation.75 Each membrane of the first batch differs in evaporation time: 10, 22, and 16 seconds (Table S3). Other conditions to be considered are listed in detail there. Based on the surface structure and the uniformity of the porous structure, an evaporation time of 16 seconds was chosen for further membrane fabrication. As presented exemplarily in Fig. S4, M1a (left) exhibited a sponge-like substructure beneath the selective layer. For M1c, a surface defect is displayed in Fig. S4 (right). This surface defect unveils a porous substructure for this sample as well. To supply larger membranes for the subsequent post-modification step, a second batch was produced under the same conditions. By maintaining the relative humidity and temperature during membrane preparation for the second batch (Table S4), as in the first batch, the membranes exhibited average pore sizes of 49.9 ± 14.9 nm for M2a and 42.3 ± 9.9 nm for M2b. A compilation for both membranes is given in Table S5. Both membranes are presented in Fig. 3a and c. Close-ups of both samples are also presented in Fig. 3b and d. These images were also used to determine the average pore diameter. An example of such a measurement is shown in Fig. S5, where each diameter taken was highlighted in yellow. Furthermore, a histogram for each pore diameter determination is given in Fig. S6. Compared to the first batch, the pores appear approximately 13 nm larger for M2a (Table 2 and Table S3). As displayed in Fig. 2a and 3a, the surface of M2a appeared more ruptured and thus less uniform in pore structure. This could result in a larger average pore diameter, which is consistent with the larger standard deviation for M2a shown in Table 2. M2b (Fig. 3b), on the other hand, exhibited a surface structure like that of M1c presented in Fig. S3. The pore-size distribution for M2b was similar to that of M1c. Only the pores are about 7.5 nm larger in diameter. For the membrane (M3, Tables S6 and S7), originating from a star-shaped BCP, porous structures were obtained (Fig. 3c and d), with an average pore diameter of 35.2 ± 6.2 nm (Table 2). The pores appear smaller than those in the second batch based on the linear BCP while maintaining a similar block distribution. For investigations of branched BCPs in the bulk state, observations were made where certain domains appeared narrower compared to microphase separations of similar BCPs in a linear configuration.73 Based on the behavior in the bulk state, branched systems such as these star-shaped BCPs could thus lead to a smaller pore diameter on average in such porous materials. Similar behavior was also observed in blend systems of linear block copolymers.76
 | ||
| Fig. 3 The prepared membrane M2a is shown as an overview (a) and with magnification (b). Membrane M2b is displayed as an overview (c) and with magnification (d). The received membrane M3 is displayed as an overview (e) and with magnification (f). | ||
| Membrane | Type of polymer used | Modification | Pore size (nm) |
|---|---|---|---|
| M2a | Linear BCP | Pristine | 49.9 ± 14.9 |
| APTES | 37.6 ± 9.4 | ||
| M2b | Linear BCP | Pristine | 42.3 ± 9.9 |
| TFPCS | 38.4 ± 9.6 | ||
| M3 | Star-shaped BCP | Pristine | 35.2 ± 6.2 |
| APTES | 26.7 ± 6.3 | ||
| TFPCS | — |
Qualitative characterization of modified porous structures using FTIR and solid-state NMR spectroscopy
The cellulose linters fiber-based hybrid membranes were further modified with TFPCS or APTES to tune the surface properties of the porous materials, as shown in Scheme 2, with APTES in orange and TFPCS in yellow. In this section, molecular analyses following the post-modification protocols were performed to confirm the successful vapour-phase reaction steps and cellulose modification. By using the surface-exposed hydroxy moieties derived from the PDHPMA block of the BCP (red), new functional groups can be introduced via substitution reactions at silicon units bearing functional groups. The post-modification of the membranes was carried out using a vapor phase modification method.77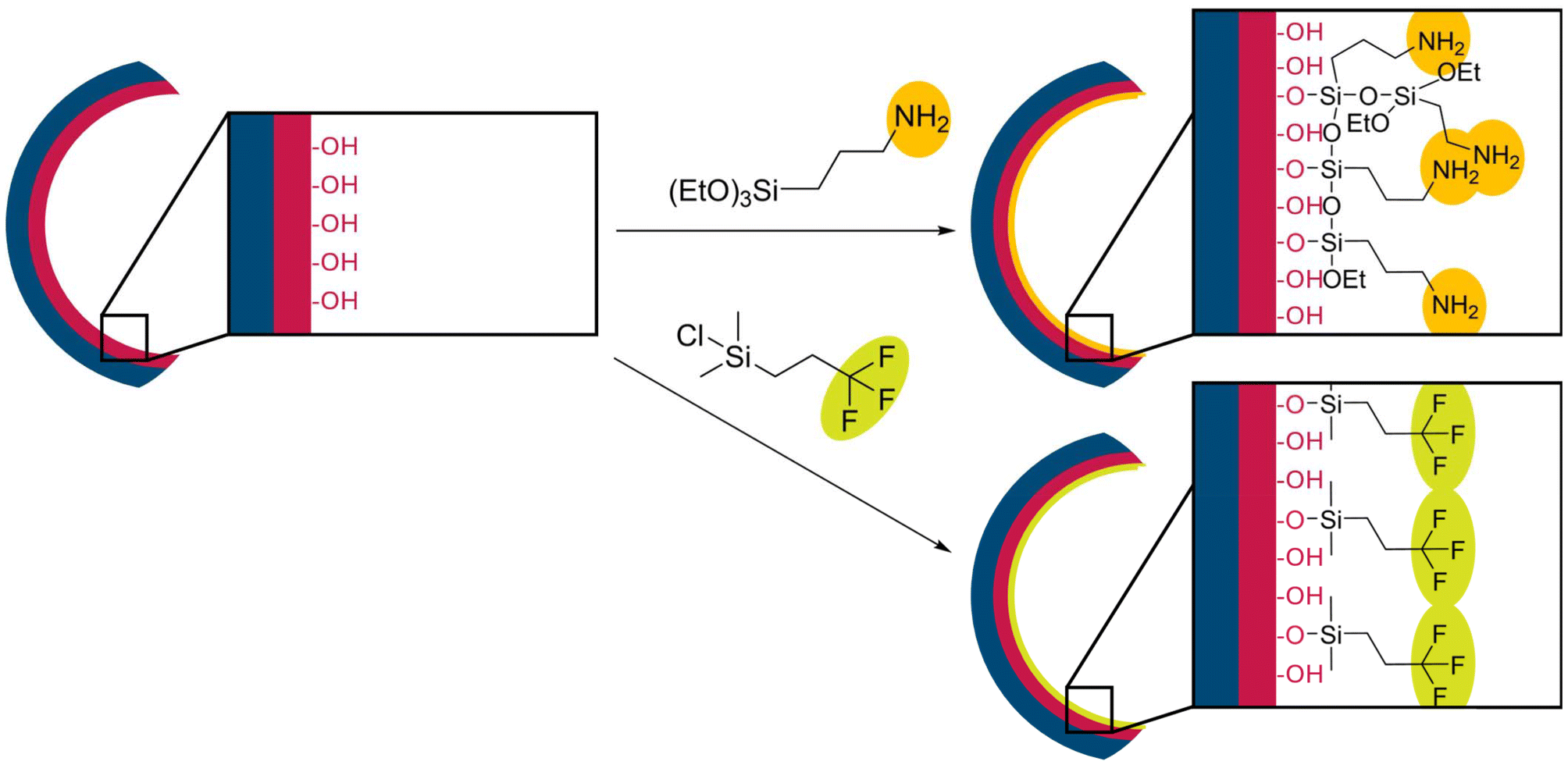 | ||
| Scheme 2 Post-modification process of a pore of APTES with possible multilayer formation (top) and TFPCS as a monolayer (bottom). | ||
The characterization of the membranes M2a and M2b was done by FTIR spectroscopy, as shown in Fig. 4. For reference, the spectrum of a pristine membrane sample is displayed in black (Fig. 4, top), an APTES-modified membrane is shown in blue (Fig. 4, middle), and a membrane after TFPCS is presented in red (Fig. 4, bottom). The pristine membrane spectrum displayed characteristic absorption bands of the coated linear BCP, including a broad OH stretching band at 3600–3100 cm−1, C–H aromatic stretching at 3080–3027 cm−1, C–H aliphatic stretching at 2924–2850 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1724 cm−1, aromatic ring CC stretching or ring vibrations at 1603–1452 cm−1, and C–O stretching at 1158 cm−1. After modification, the APTES-modified spectrum showed a clear band at 1550 cm−1, indicating N–H bending, and an increased intensity in the Si–O–Si/Si–O–C stretching region between 1000 and 1100 cm−1, confirming the presence of amine and siloxane groups from APTES.78 Similarly, the TFPCS-modified spectrum revealed the appearance of specific peaks, including the C–F stretching of the CF3 group at 1263 cm−1 and the Si–C stretching at 842 cm−1, providing strong evidence for the covalent attachment of the fluorinated silane (TFPCS).79 Both modifications in the gas phase appear to affect the membrane structure, with the pore size decreasing during the modifications. Surfaces and pore diameters are compiled in Fig. 4 and Table 2. At higher magnification, the surface structure of M2a before (Fig. 5a) and after modification (Fig. 5b) was observed. Changes to the TFPCS approach can also be observed in Fig. 5c and d. The change in surface structure resulted in a smaller pore size for both membranes, as listed in Table 2. For APTES modification, a decrease in pore diameter of approximately 12 nm was observed. For TFPCS modification, a decrease of approximately 4 nm was observed. It could be correlated with the type of anchoring unit. A trifunctional anchoring unit can lead to a multilayer coating and, consequently, a thicker layer, as indicated in Scheme 2. For M3, the post-modification was performed using the same protocol as that for M2a and M2b. For sample M3, post-modification with APTES resulted in a decrease in pore diameter of approximately 9 nm while retaining the surface appearance (Fig. 5e and f). This matches the observations made before. Surprisingly, for the sample reacted with TFPCS, the porous structure vanished completely, as shown in Fig. 5g. APTES with its three ethoxy moieties is able to generate a multi-layer structure at the pore surface. Such a multi-layer could be capable of stabilizing the structure during the post-modification process.62,80 In contrast, the anchoring unit, which is used in TFPCS, is only capable of forming a monolayer. Based on that, a loss of structure on the surface could be attributed to the ability of the post-modification reagent to build stabilizing multi-layer structures.
O stretching at 1724 cm−1, aromatic ring CC stretching or ring vibrations at 1603–1452 cm−1, and C–O stretching at 1158 cm−1. After modification, the APTES-modified spectrum showed a clear band at 1550 cm−1, indicating N–H bending, and an increased intensity in the Si–O–Si/Si–O–C stretching region between 1000 and 1100 cm−1, confirming the presence of amine and siloxane groups from APTES.78 Similarly, the TFPCS-modified spectrum revealed the appearance of specific peaks, including the C–F stretching of the CF3 group at 1263 cm−1 and the Si–C stretching at 842 cm−1, providing strong evidence for the covalent attachment of the fluorinated silane (TFPCS).79 Both modifications in the gas phase appear to affect the membrane structure, with the pore size decreasing during the modifications. Surfaces and pore diameters are compiled in Fig. 4 and Table 2. At higher magnification, the surface structure of M2a before (Fig. 5a) and after modification (Fig. 5b) was observed. Changes to the TFPCS approach can also be observed in Fig. 5c and d. The change in surface structure resulted in a smaller pore size for both membranes, as listed in Table 2. For APTES modification, a decrease in pore diameter of approximately 12 nm was observed. For TFPCS modification, a decrease of approximately 4 nm was observed. It could be correlated with the type of anchoring unit. A trifunctional anchoring unit can lead to a multilayer coating and, consequently, a thicker layer, as indicated in Scheme 2. For M3, the post-modification was performed using the same protocol as that for M2a and M2b. For sample M3, post-modification with APTES resulted in a decrease in pore diameter of approximately 9 nm while retaining the surface appearance (Fig. 5e and f). This matches the observations made before. Surprisingly, for the sample reacted with TFPCS, the porous structure vanished completely, as shown in Fig. 5g. APTES with its three ethoxy moieties is able to generate a multi-layer structure at the pore surface. Such a multi-layer could be capable of stabilizing the structure during the post-modification process.62,80 In contrast, the anchoring unit, which is used in TFPCS, is only capable of forming a monolayer. Based on that, a loss of structure on the surface could be attributed to the ability of the post-modification reagent to build stabilizing multi-layer structures.
 | ||
| Fig. 4 FTIR spectra of the cellulose-based membrane coated with a porous structure based on PS-b-PDHPMA before post-modification (black) and after modification with APTES (blue) and TFPCS (red). On the left side, an overview is given, while on the right, a detailed presentation is given. | ||
 | ||
| Fig. 5 Membranes based on the linear BCP are shown before post-modification (a, c) and after post-modification with APTES (b) and TFPCS (d). Membranes based on star-shaped BCPs (M3) before post-modification (e) and after post-modification with APTES (f) and TFPCS (g). | ||
Comparison of the FTIR spectra confirmed the successful surface modification of PS-b-PDHPMA membranes using linters paper. By varying functional groups, e.g., fluorinated functional groups in the case of TFPCS or amines for APTES, surface properties like polarity can be tuned. The consequences of both surface modifications are discussed in the following in the context of water contact angle measurements and water permeance.
To analyze the detailed structure of the distorted, highly complex membrane and modified membrane systems, as a complementary analytical approach, selectively enhanced DNP solid-state NMR was applied. With this technique, it is feasible to easily distinguish between structure moieties in more hydrophobic or more hydrophilic environments. When comparing the DNP-enhanced 1H → 13C CP MAS spectra (Fig. 6a) of the linters paper, modified with PS-b-PDHPMA (M2) prepared with TEK as a radical–solvent system, no pronounced differences were observed among the three membranes. This indicates that the environments of the carbon nuclei of the cellulose and polymer matrix remain essentially unchanged after surface modification. In contrast, the DNP-enhanced 1H → 13C CP MAS spectra of the linters paper modified with PMMA-b-PDHPMA (M3) (Fig. 6b) show noticeable changes only for the TFPCS-modified membrane. The characteristic peaks of the TFPCS moiety appear at 2, 10, and 29 ppm, whereas no new signals are observed for the APTES-modified membrane. To confirm that the newly observed peaks indeed originate from the TFPCS functionality, the spectrum was compared with that of TFPCS-modified SBA-15 (Fig. 6c). The identical peak positions in both materials strongly indicate the successful incorporation of TFPCS on the membrane surface. Then, DNP-enhanced 1H → 29Si CP MAS spectra (Fig. 6d) were analyzed to probe silicon-containing functional groups introduced during the modification and to distinguish between APTES- and TFPCS-functionalized surfaces clearly. The DNP-enhanced 1H → 29Si CP MAS spectrum of the APTES-modified membrane shows a broad asymmetric signal at −68 ppm, which is assigned to Tn groups [CH3(OSi)3] originating from the covalent attachment of the APTES linker to the membrane surface. In contrast, the TFPCS-modified membrane shows a signal around +7 ppm, corresponding to M groups [(CH3)3Si], indicative of trimethylsilyl groups. The presence of these distinct signals clearly underlines the successful surface modification by APTES and TFPCS, respectively. The TFPCS-modified M3 membrane displays a significantly more intense M-group signal compared to the TFPCS-modified M2 membrane, suggesting that TFPCS couples more effectively or in higher amounts to the M3 membrane. In contrast, the APTES-modified membranes show comparable Tn signal intensities for both M2 and M3, indicating that the extent of APTES functionalization is similar across both membrane systems.
 | ||
| Fig. 6 (a) DNP-enhanced 1H → 13C CP MAS spectra of the unmodified membrane (linters paper + PS-b-PDHPMA membrane (M2)) and the APTES- and TFPCS-modified membranes. (b) DNP-enhanced 1H → 13C CP MAS spectra of the unmodified membrane (linters paper + PMMA-b-PDHPMA membrane (M3)) and the APTES- and TFPCS-modified membranes. (c) DNP-enhanced 1H → 13C CP MAS spectra of TFPCS-modified SBA-15 and TFPCS-modified membranes. (d) DNP-enhanced 1H → 29Si CP MAS spectra of APTES- and TFPCS-modified membranes. Note: a 15 mM TEKPol in TCE (TEK) radical–solvent system was used for sample preparations. All spectra were recorded at a spinning rate of 8 kHz. Spinning sidebands are marked with an asterisk. Signals referring to the polymer are marked with “P”. | ||
Macroscopic effects of surface modification on the water contact angle and permeance of the substrate
To further evaluate the influence of post-modification, the materials were studied via WCA measurements in different stages before and after application of APTES or TFPCS. For reference, pure linters paper and linters paper with APTES-modified and TFPCS-modified surfaces were tested. The results are presented comparatively in Table 3 and discussed in the following text. During WCA measurement, the water droplet slowly spread and then disappeared on the porous substrates. Therefore, the static contact angle was measured at 3 s after a drop was deposited on the surface, and the absorption time was also measured for comparison. All WCA and absorption times discussed in the following are displayed in Table 3. The WCA and absorption time results clearly show that the unmodified cellulose linters substrate, which is hydrophilic and porous, exhibited complete and rapid wetting (absorption time < 1 s and WCA = 0). After functionalization of the paper with APTES and TFPCS, the WCAs of these modified linters paper samples were difficult to determine. The water droplet spread out and vanished completely in 12 seconds for APTES-modified linters paper and in less than 1 s for TFPCS-modified linters paper. This may be due to the modified cellulose fiber substrate remaining highly porous, since only a thin monolayer of APTES or TFPCS molecules was deposited on the paper surface through vapor deposition. However, APTES-modified paper exhibited a slight increase in wetting resistance, with an absorption time of 12 ± 1 s, compared to unmodified paper. In contrast, the application of the linear BCP resulted in a significant increase in the absorption time (29 ± 10 s). A water contact angle of 25° ± 2° was obtained for the cellulose polymer hybrid M2. After functionalization, both functionalized M2 substrates showed a clear shift toward increased hydrophobicity. The APTES modification caused the most notable change, increasing the WCA to 39° ± 5° and significantly increasing the absorption time to 106 ± 38 s. Similarly, TFPCS modification increased the WCA to 35° ± 5° and the absorption time to 48 ± 6 s for M2. This boost in hydrophobicity additionally confirmed that the amine- and fluorinated-silane groups were successfully attached to the M2 membrane surface, consistent with the expected decrease in surface energy.| Sample | Absorption time [s] | WCA [°] |
|---|---|---|
| a The droplet was continuously spreading, making it difficult to obtain an accurate static contact angle. | ||
| Linters paper | <1 | 0 |
| Linters paper APTES-modified | 12 ± 1 | N/Aa |
| Linters paper TFPCS-modified | <1 | 0 |
| M2b | 29 ± 10 | 25 ± 2 |
| M2 APTES-modified | 106 ± 38 | 39 ± 5 |
| M2 TFPCS-modified | 48 ± 6 | 35 ± 5 |
In the last step, both modified membranes were tested for their water permeance at a slightly increased water pressure of 0.2 bar. The performance was then compared with unmodified M2a and M2b samples. The development of permeance for each sample over time is presented in Fig. 7 at the top. On the left side, M2a is shown without (black) and with APTES modification (blue), while the image on the right side represents the development of M2b without modification (black) and with TFPCS modification (red). Membrane M2a shows higher overall water permeance than M2b. This correlates with the larger pore diameter displayed in Table 2. For both membranes, a strong decrease in permeance is observed for the first 60 to 75 minutes. This was observed earlier as well and can be attributed to the swelling of the more hydrophilic block coating the inner pore surface.46,49 After 75 minutes, only a small decrease is observed over up to 125 minutes for M2a and 180 minutes for M2b with a permeance of approximately 120 L bar−1 m−2 h−1 for M2a and 212 L bar−1 m−2 h−1 for M2b. For both samples, after modification with APTES or TFPCS, an increase in water permeance is observed for the whole test time. At 82 minutes, the permeance is three times that of the APTES-modified sample with 1885 L bar−1 m−2 h−1. The permeance for M2b is doubled due to TFPCS modification with 454 L bar−1 m−2 h−1. Such observations contrast with the results for the membranes, particularly for the modified membranes based on the star-shaped BCP M3. The overall development of permeance over the entire test period matches the observations from the linear BCP presented earlier. For the pristine sample (Fig. 7, black), the permeance is approximately 100 L bar−1 m−2 h−1, in a similar order of magnitude to M2b. In contrast to M2a and M2b, no significant influence of the APTES modification for M3 was observed. While the permeance of the APTES-modified membrane was quite similar, the TFPCS modification reduced the membrane's permeance compared to the pristine M3 sample. This observation aligns with SEM imaging results and is linked to the loss of pores on the material surface during the post-modification process. Overall, the membranes shown here operate without but also with post-modification in the range of 100–1900 L bar−1 m−2 h−1, which is typical of amphiphilic SNIPS-like membranes shown in the literature before.41,46,48,81 With a focus on the post-modification of porous structures, the implemented structures during membrane casting could be preserved for APTES modification. Only during TFPCS modification of M3, a loss of structure was observed.
 | ||
| Fig. 7 Water permeance measurements of membranes M2a (top left) and M2b (top right) before and after surface modification with APTES (left) and TFPCS (right). A compilation of M3-based membranes is shown at the bottom. | ||
Conclusion
A sequential procedure for the synthesis of an amphiphilic block copolymer based on solketal methacrylate SMA was established using living anionic polymerization. A defined polystyrene-block-polysolketal methacrylate (PS-b-PSMA) BCP with a molecular weight of Mn = 138200 g mol−1 and a dispersity of Đ = 1.07 was achieved and converted into polystyrene-block-poly(dihydroxypropyl methacrylate) PS-b-PDHPMA through an acid-mediated deprotection step under mild conditions. The protocol was further developed for the preparation of a poly(dihydroxypropyl methacrylate)-block-polymethyl methacrylate (PDHPMA-b-PMMA)n star-BCP using a convergent arm-first strategy with 1,4-butanediol dimethacrylate (BDDMA) as a linking unit. Here, a molecular weight of Mn = 538000 gmol−1 and a dispersity of Đ = 1.13 were obtained before conversion of the PSMA block. Both polymers were converted into porous materials in the vicinity of cellulose paper sheets following an SNIPS protocol. For both polymers, porous structures were obtained, while the average pore diameter for the linear BCP tends to be larger compared to the star-shaped BCP. Pore diameters ranging from 25 to 50 nm were achieved. Using surface-exposed hydroxy moieties, amines, and fluorinated functionalities, the surface properties of the porous materials were manipulated. Using a vapor-phase approach, the surfaces of the resulting porous materials were modified with 3-aminopropyltriethoxysilane (APTES) and 3,3,3-trifluoropropyl dimethyl chlorosilane (TFPCS), respectively. SEM imaging indicates a decrease in pore diameter of approximately 5 nm. The influence of the post-modification step was then investigated on a molecular scale using FTIR and DNP-enhanced solid-state NMR, revealing that TFPCS modification is much more efficient for the PMMA-b-PDHPMA membrane (M3) than for the PS-b-PDHPMA membrane (M2), whereas APTES modification shows similar efficiency for both membranes. The influence of post-modification on a macroscopic scale was examined using water contact angle measurements. Here, for both reagents, a significant increase in contact angle and absorption time was observed. Finally, porous materials with and without post-modification were tested as candidates for cellulose-based membrane materials by investigating the development of water permeance over time at slightly elevated pressure. In summary, this work presents promising prospects for producing tailor-made porous cellulose-based materials with precise control over their properties for specific applications.
Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data that support the findings of this study are available from the corresponding author upon request.The data supporting this publication is included in the supplementary information (SI): composition of membrane solutions, further details for the characterization of polymers and porous structures and more details regarding solid-state NMR. See DOI: https://doi.org/10.1039/d5py01203a.
Acknowledgements
T. G. gratefully acknowledges the Deutsche Forschungsgemeinschaft (DFG) under grant GU-1650/3-2. W. L. thanks the Development and Promotion of Science and Technology Talents Project (DPST), the Royal Government of Thailand Scholarship, and the Institute for the Promotion of Teaching Science and Technology (IPST) for financial support. This study was performed within the project “Switchable Membrane Systems – SWIMEMSYS”, which is financed by the European Regional Development Fund and Saarland funds. M. G., S. P., and S. H. would like to thank the European Union and the Ministry of Economics, Innovation, Digitalisation, and Energy for financial support.References
- H. Park and B. Park, Review of Microplastic Distribution, Toxicity, Analysis Methods, and Removal Technologies, Water, 2021, 13(19), 2736, DOI:10.3390/w13192736.
- M. A. Dar, P. Palsania, S. Satya, M. Dashora, O. A. Bhat, S. Parveen, S. K. Patidar and G. Kaushik, Microplastic pollution: A global perspective in surface waters, microbial degradation, and corresponding mechanism, Mar. Pollut. Bull., 2025, 210, 117344, DOI:10.1016/j.marpolbul.2024.117344.
- M. K. Nguyen, C. Lin and D. D. Nguyen, Microplastic pollution associated with probabilistic human health risks: Potential hazards, critical factors, challenges, and limitations, Mar. Pollut. Bull., 2025, 221, 118528, DOI:10.1016/j.marpolbul.2025.118528.
- P. Ahmadi, D. Doyle, N. Mojarad, S. Taherkhani, A. Janzadeh, M. Honardoost and M. Gholami, Effects of Micro- and Nanoplastic Exposure on Macrophages: A Review of Molecular and Cellular Mechanisms, Toxicol. Mech. Methods, 2025, 1–40, DOI:10.1080/15376516.2025.2500546.
- J. Gluge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, An overview of the uses of per- and polyfluoroalkyl substances (PFAS), Environ. Sci.: Processes Impacts, 2020, 22(12), 2345–2373, 10.1039/d0em00291g.
- M. Bonato, F. Corra, M. Bellio, L. Guidolin, L. Tallandini, P. Irato and G. Santovito, PFAS Environmental Pollution and Antioxidant Responses: An Overview of the Impact on Human Field, Int. J. Environ. Res. Public Health, 2020, 17(21), 8020, DOI:10.3390/ijerph17218020.
- H. Rekik, H. Arab, L. Pichon, M. A. El Khakani and P. Drogui, Per-and polyfluoroalkyl (PFAS) eternal pollutants: Sources, environmental impacts and treatment processes, Chemosphere, 2024, 358, 142044, DOI:10.1016/j.chemosphere.2024.142044.
- Y. Song, J. Phipps, C. Zhu and S. Ma, Porous Materials for Water Purification, Angew. Chem., Int. Ed., 2023, 62(11), e202216724, DOI:10.1002/anie.202216724.
- S. M. Al-Jubouri, S. Al-Batty, R. K. S. Al-Hamd, R. Sims, M. W. Hakami and M. H. Sk, Sustainable environment through using porous materials: A review on wastewater treatment, Asia–Pac. J. Chem. Eng., 2023, 18(4), e2941, DOI:10.1002/apj.2941.
- K. C. Khulbe and T. Matsuura, Removal of heavy metals and pollutants by membrane adsorption techniques, Appl. Water Sci., 2018, 8(1), 19, DOI:10.1007/s13201-018-0661-6.
- C. Regula, E. Carretier, Y. Wyart, G. Gesan-Guiziou, A. Vincent, D. Boudot and P. Moulin, Chemical cleaning/disinfection and ageing of organic UF membranes: a review, Water Res., 2014, 56, 325–365, DOI:10.1016/j.watres.2014.02.050.
- A. V. Bildyukevich, T. V. Plisko, F. Lipnizki and S. A. Pratsenko, Correlation between membrane surface properties, polymer nature and fouling in skim milk ultrafiltration, Colloids Surf., A, 2020, 605, 125387, DOI:10.1016/j.colsurfa.2020.125387.
- M. T. Aminzai and E. Yabalak, Advanced polymeric membranes for environmental remediation: emerging roles of hydrochar and biochar composites, J. Mater. Sci., 2025, 60(40), 18710–18733, DOI:10.1007/s10853-025-11508-y.
- M. Madhushree, P. Vairavel, G. T. Mahesha and K. S. Bhat, A Comprehensive Review of Cellulose and Cellulose-Based Materials: Extraction, Modification, and Sustainable Applications, J. Nat. Fibers, 2024, 21(1), 2418357, DOI:10.1080/15440478.2024.2418357.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Cellulose: Fascinating biopolymer and sustainable raw material, Angew. Chem., Int. Ed., 2005, 44(22), 3358–3393, DOI:10.1002/anie.200460587.
- T. Aziz, A. Farid, F. Haq, M. Kiran, A. Ullah, K. C. Zhang, C. Li, S. Ghazanfar, H. Y. Sun and R. Ullah, et al., A Review on the Modification of Cellulose and Its Applications, Polymers, 2022, 14(15), 3206, DOI:10.3390/polym14153206.
- B. Thomas, M. C. Raj, K. B. Athira, M. H. Rubiyah, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Nanocellulose, a Versatile Green Platform: From Biosources to Materials and Their Applications, Chem. Rev., 2018, 118(24), 11575–11625, DOI:10.1021/acs.chemrev.7b00627.
- F. Zhang, K. Xu, Y. A. Bai and P. X. Wang, Multifunctional cellulose paper-based materials, Cellulose, 2023, 30(14), 8539–8569, DOI:10.1007/s10570-023-05426-y.
- S. Liyanage, S. Acharya, P. Parajuli, J. L. Shamshina and N. Abidi, Production and Surface Modification of Cellulose Bioproducts, Polymers, 2021, 13(19), 3433, DOI:10.3390/polym13193433.
- L. Yang, Q. Y. Yuan, C. W. Lou, J. H. Lin and T. T. Li, Recent Research Progress of Polysaccharide Polymer Coatings for Improving Properties of Paper-Based Packaging Materials, Coatings, 2025, 15(3), 326, DOI:10.3390/coatings15030326.
- A. Solberg, J. Zehner, F. Somorowsky, K. Rose, A. Korpela and K. Syverud, Material properties and water resistance of inorganic-organic polymer coated cellulose paper and nanopaper, Cellulose, 2023, 30(2), 1205–1223, DOI:10.1007/s10570-022-04925-8.
- M. Mujtaba, J. Lipponen, M. Ojanen, S. Puttonen and H. Vaittinen, Trends and challenges in the development of bio-based barrier coating materials for paper/cardboard food packaging; a, Sci. Total Environ., 2022, 851, 158328, DOI:10.1016/j.scitotenv.2022.158328.
- M. D. Islam, F. J. Uddin, T. U. Rashid and M. Shahruzzaman, Cellulose acetate-based membrane for wastewater treatment-A state-of-the-art review, Mater. Adv., 2023, 4(18), 4054–4102, 10.1039/d3ma00255a.
- J. Lee, H. Luo, Y. Y. Chen, K. Ilestad, D. Yu, M. Ybarra and C. Ma, Paper-Based Microfluidics for Tissue Engineering and Regenerative Medicine, Cells Tissues Organs, 2025, 214(6), 512–528, DOI:10.1159/000545248.
- M. Sheraz, X. F. Sun, A. Siddiqui, Y. K. Wang, S. H. Hu and R. Sun, Cellulose-Based Electrochemical Sensors, Sensors, 2025, 25(3), 645, DOI:10.3390/s25030645.
- C. M. Lin, Z. H. Pei, J. Chen, J. X. Lan, H. Huang, X. J. Ma and S. L. Cao, An Eco-friendly Cellulose Paper-Based Tactile Sensor Driven by the Triboelectric Effect, ACS Appl. Polym. Mater., 2025, 7(3), 1225–1235, DOI:10.1021/acsapm.4c02682.
- M. L. Huang, Y. J. Tang, X. Y. Wang, P. Zhu, T. Y. Chen and Y. M. Zhou, Preparation of polyaniline/cellulose nanocrystal composite and its application in surface coating of cellulosic paper, Prog. Org. Coat., 2021, 159, 106452, DOI:10.1016/j.porgcoat.2021.106452.
- L. Wang, P. V. Kelly, N. Ozveren, X. F. Zhang, M. Korey, C. Chen, K. Li, S. Bhandari, H. Tekinalp and X. H. Zhao, et al. Multifunctional polymer composite coatings and adhesives by incorporating cellulose nanomaterials, Matter, 2023, 6(2), 344–372, DOI:10.1016/j.matt.2022.11.024.
- M. Plank, F. Hartmann, B. Kuttich, T. Kraus and M. Gallei, Self-assembly of amphiphilic poly(2-hydroxyethyl methacrylate)-containing block copolymers in the vicinity of cellulose fibres, Eur. Polym. J., 2020, 141, 110059, DOI:10.1016/j.eurpolymj.2020.110059.
- L. Gemmer, Q. W. Hu, B. J. Niebuur, T. Kraus, B. N. Balzer and M. Gallei, A block copolymer templated approach for the preparation of nanoporous polymer structures and cellulose fiber hybrids by ozone treatment, Polym. Chem., 2022, 13(27), 4028–4046, 10.1039/d2py00562j.
- F. V. Frieß, F. Hartmann, L. Gemmer, J. Pieschel, B. J. Niebuur, M. Faust, T. Kraus, V. Presser and M. Gallei, Thermo–Responsive Ultrafiltration Block Copolymer Membranes Based on Polystyrene–block–Poly(diethyl acrylamide), Macromol. Mater. Eng., 2023, 2300113, DOI:10.1002/mame.202300113.
- J. I. Clodt, V. Filiz, S. Rangou, K. Buhr, C. Abetz, D. Höche, J. Hahn, A. Jung and V. Abetz, Double Stimuli–Responsive Isoporous Membranes via Post–Modification of pH–Sensitive Self–Assembled Diblock Copolymer Membranes, Adv. Funct. Mater., 2012, 23(6), 731–738, DOI:10.1002/adfm.201202015.
- L. Hub, J. Koll, K. Buhr, M. Radjabian and V. Abetz, pH-responsive size- and charge-selective block copolymer membrane for the separation of small proteins, J. Membr. Sci., 2024, 708, 123021, DOI:10.1016/j.memsci.2024.123021.
- X. Luo, Z. Q. Wang, M. X. Yan, J. H. Xin and Y. Q. Yang, One-pot three-step synthesis of PDMAEMA-PMMA-PNIPAM triblock block copolymer for preparing pH and thermal dual-responsive PVDF blend membranes, Polym. Eng. Sci., 2023, 63(10), 3343–3352, DOI:10.1002/pen.26448.
- S. P. Nunes, A. R. Behzad, B. Hooghan, R. Sougrat, M. Karunakaran, N. Pradeep, U. Vainio and K. V. Peinemann, Switchable pH-responsive polymeric membranes prepared via block copolymer micelle assembly, ACS Nano, 2011, 5(5), 3516–3522, DOI:10.1021/nn200484v.
- Z. Shu, H. Z. Li, Y. Shi, D. Y. Zuo, Z. Yi and C. J. Gao, Dual sugar and temperature responsive isoporous membranes for protein sieving with improved separation coefficient and decreased denaturation, J. Membr. Sci., 2023, 672, 121450, DOI:10.1016/j.memsci.2023.121450.
- T. Rittner, S. Pusse, B. Bossmann, K. Staudt, A. Haben, R. Kautenburger, H. P. Beck and M. Gallei, Metallopolymer-based block copolymers for perfluorinated substances (PFAS) and ion removal, J. Mater. Chem. C, 2024, 12(47), 19116–19129, 10.1039/d4tc03546a.
- Z. Z. Zhang, M. M. Rahman, B. Bajer, N. Scharnagl and V. Abetz, Highly selective isoporous block copolymer membranes with tunable polyelectrolyte brushes in soft nanochannels, J. Membr. Sci., 2022, 646, 120266, DOI:10.1016/j.memsci.2022.120266.
- Y. Miyoshi, Y. Nakata, T. Kitagawa, H. Matsuyama, T. Yoshioka and K. Nakagawa, Effect of Surface Modification of Polyamide-Based Reverse Osmosis Membranes by Glycerol Monoacrylate-Butyl Acrylate Copolymers on Antifouling, Ind. Eng. Chem. Res., 2024, 63(9), 4124–4133, DOI:10.1021/acs.iecr.4c00153.
- C. Zhao, L. Li, Q. Wang, Q. Yu and J. Zheng, Effect of film thickness on the antifouling performance of poly(hydroxy-functional methacrylates) grafted surfaces, Langmuir, 2011, 27(8), 4906–4913, DOI:10.1021/la200061h.
- S. Schöttner, M. Brodrecht, E. Uhlein, C. Dietz, H. Breitzke, A. A. Tietze, G. Buntkowsky and M. Gallei, Amine-Containing Block Copolymers for the Bottom-Up Preparation of Functional Porous Membranes, Macromolecules, 2019, 52(7), 2631–2641, DOI:10.1021/acs.macromol.8b02758.
- J. L. Wang, M. M. Rahman, C. Abetz and V. Abetz, Bovine serum albumin selective integral asymmetric isoporous membrane, J. Membr. Sci., 2020, 604, 118074, DOI:10.1016/j.memsci.2020.118074.
- K. V. Peinemann, V. Abetz and P. F. Simon, Asymmetric superstructure formed in a block copolymer via phase separation, Nat. Mater., 2007, 6(12), 992–996, DOI:10.1038/nmat2038.
- L. Tsaur and U. B. Wiesner, Non-Equilibrium Block Copolymer Self-Assembly Based Porous Membrane Formation Processes Employing Multicomponent Systems, Polymers, 2023, 15(9) DOI:10.3390/polym15092020.
- M. Gallei, S. Rangou, V. Filiz, K. Buhr, S. Bolmer, C. Abetz and V. Abetz, The Influence of Magnesium Acetate on the Structure Formation of Polystyrene-block-poly(4-vinylpyridine)-Based Integral-Asymmetric Membranes, Macromol. Chem. Phys., 2013, 214(9), 1037–1046, DOI:10.1002/macp.201200708.
- F. V. Frieß, Q. Hu, J. Mayer, L. Gemmer, V. Presser, B. N. Balzer and M. Gallei, Nanoporous Block Copolymer Membranes with Enhanced Solvent Resistance Via UV-Mediated Cross-Linking Strategies, Macromol. Rapid Commun., 2022, 43(3), e2100632, DOI:10.1002/marc.202100632.
- L. Gemmer, B.-J. Niebuur, C. Dietz, D. Rauber, M. Plank, F. V. Frieß, V. Presser, R. W. Stark, T. Kraus and M. Gallei, Polyacrylonitrile-containing amphiphilic block copolymers: self-assembly and porous membrane formation, Polym. Chem., 2023, 14(42), 4825–4837, 10.1039/d3py00836c.
- M. Plank, F. V. Frieß, C. V. Bitsch, J. Pieschel, J. Reitenbach and M. Gallei, Modular Synthesis of Functional Block Copolymers by Thiol–Maleimide “Click” Chemistry for Porous Membrane Formation, Macromolecules, 2023, 56(4), 1674–1687, DOI:10.1021/acs.macromol.2c02255.
- S. Pusse, B. J. Niebuur, T. Kraus, V. Presser, B. N. Balzer and M. Gallei, Synthesis and Self-Assembly of Pore-Forming Three-Arm Amphiphilic Block Copolymers, Macromol. Rapid Commun., 2025, e2500077, DOI:10.1002/marc.202500077.
- S. Heinz, L. Gemmer, O. Janka and M. Gallei, Ferrocene-Modified Polyacrylonitrile-Containing Block Copolymers as Preceramic Materials, Polymers, 2024, 16(15), 2142, DOI:10.3390/polym16152142.
- S. Schöttner, R. Hossain, C. Ruttiger and M. Gallei, Ferrocene-Modified Block Copolymers for the Preparation of Smart Porous Membranes, Polymers, 2017, 9(10), 491, DOI:10.3390/polym9100491.
- S. Schöttner, H.-J. Schaffrath and M. Gallei, Poly(2-hydroxyethyl methacrylate)-Based Amphiphilic Block Copolymers for High Water Flux Membranes and Ceramic Templates, Macromolecules, 2016, 49(19), 7286–7295, DOI:10.1021/acs.macromol.6b01803.
- S. Saleem, S. Rangou, C. Abetz, V. Filiz and V. Abetz, Isoporous Membranes from Novel Polystyrene-b-poly(4-vinylpyridine)-b-poly(solketal methacrylate) (PS-b-P4VP-b-PSMA) Triblock Terpolymers and Their Post-Modification, Polymers, 2019, 12(1), 41, DOI:10.3390/polym12010041.
- D. B. Alward, D. J. Kinning, E. L. Thomas and L. J. Fetters, Effect of Arm Number and Arm Molecular-Weight on the Solid-State Morphology of Poly(Styrene-Isoprene) Star Block Copolymers, Macromolecules, 1986, 19(1), 215–224, DOI:10.1021/ma00155a034.
- D. J. Kinning, E. L. Thomas, D. B. Alward, L. J. Fetters and D. L. Handlin, Sharpness of the functionality-induced structural transition in poly(styrene-isoprene) star block copolymers, Macromolecules, 2002, 19(4), 1288–1290, DOI:10.1021/ma00158a067.
- E. L. Thomas, D. B. Alward, D. J. Kinning, D. C. Martin, D. L. Handlin and L. J. Fetters, Ordered Bicontinuous Double-Diamond Structure of Star Block Copolymers - a New Equilibrium Microdomain Morphology, Macromolecules, 1986, 19(8), 2197–2202, DOI:10.1021/ma00162a016.
- M. W. Matsen, Effect of Architecture on the Phase Behavior of AB-Type Block Copolymer Melts, Macromolecules, 2012, 45(4), 2161–2165, DOI:10.1021/ma202782s.
- L. Leibler, Theory of Microphase Separation in Block Copolymers, Macromolecules, 1980, 13(6), 1602–1617, DOI:10.1021/ma60078a047.
- L. Upadhyaya, M. Semsarilar, S. Nehache, A. Deratani and D. Quemener, Filtration membranes from self-assembled block copolymers - a review on recent progress, Eur. Phys. J.: Spec. Top., 2015, 224(9), 1883–1897, DOI:10.1140/epjst/e2015-02507-7.
- L. Li, P. Szewczykowski, L. D. Clausen, K. M. Hansen, G. E. Jonsson and S. Ndoni, Ultrafiltration by gyroid nanoporous polymer membranes, J. Membr. Sci., 2011, 384(1–2), 126–135, DOI:10.1016/j.memsci.2011.09.012.
- S. Park, Y. Kim, H. Ahn, J. H. Kim, P. J. Yoo and D. Y. Ryu, Giant Gyroid and Templates from High-Molecular-Weight Block Copolymer Self-assembly, Sci. Rep., 2016, 6, 36326, DOI:10.1038/srep36326.
- T. Gutmann, B. Kumari, L. Zhao, H. Breitzke, S. Schöttner, C. Rüttiger and M. Gallei, Dynamic Nuclear Polarization Signal Amplification as a Sensitive Probe for Specific Functionalization of Complex Paper Substrates, J. Phys. Chem. C, 2017, 121(7), 3896–3903, DOI:10.1021/acs.jpcc.6b11751.
- M. V. Höfler, J. Lins, D. Seelinger, L. Pachernegg, T. Schäfer, S. Spirk, M. Biesalski and T. Gutmann, DNP enhanced solid-state NMR – A powerful tool to address the surface functionalization of cellulose/paper derived materials, J. Magn. Reson. Open, 2024, 21, 100163, DOI:10.1016/j.jmro.2024.100163.
- S. O. Kyeremateng, E. Amado and J. Kressler, Synthesis and characterization of random copolymers of (2,2-dimethyl-1,3-dioxolan-4-yl)methyl methacrylate and 2,3-dihydroxypropyl methacrylate, Eur. Polym. J., 2007, 43(8), 3380–3391, DOI:10.1016/j.eurpolymj.2007.04.048.
- E. Huang, A. Skoufis, T. Denning, J. Qi, R. Dagastine, R. Tabor and J. Berry, OpenDrop: Open-source software for pendant drop tensiometry contact angle measurements, J. Open Source Software, 2021, 6(58), 2604, DOI:10.21105/joss.02604.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist, Organometallics, 2010, 29(9), 2176–2179, DOI:10.1021/om100106e.
- W. Limprasart, M. V. Höfler, N. Kunzmann, L. Rösler, K. Herr, H. Breitzke and T. Gutmann, Peptides as Model Systems for Biofunctionalizations of Cellulose–Synthesis and Structural Characterization by Advanced Solid-State Nuclear Magnetic Resonance Techniques, J. Phys. Chem. C, 2023, 127(45), 22129–22138, DOI:10.1021/acs.jpcc.3c05068.
- A. E. Bennett, C. M. Rienstra, M. Auger, K. V. Lakshmi and R. G. Griffin, Heteronuclear decoupling in rotating solids, J. Chem. Phys., 1995, 103(16), 6951–6958, DOI:10.1063/1.470372.
- M. Brodrecht, B. Kumari, A. Thankamony, H. Breitzke, T. Gutmann and G. Buntkowsky, Structural Insights into Peptides Bound to the Surface of Silica Nanopores, Chemistry, 2019, 25(20), 5214–5221, DOI:10.1002/chem.201805480.
- M. Gallei, R. Klein and M. Rehahn, Silacyclobutane-Mediated Re-Activation of “Sleeping” Polyvinylferrocene Macro-Anions: A Powerful Access to Novel Metalloblock Copolymers, Macromolecules, 2010, 43(4), 1844–1854, DOI:10.1021/ma902092j.
- D. Baskaran, Strategic developments in living anionic polymerization of alkyl (meth)acrylates, Prog. Polym. Sci., 2003, 28(4), 521–581, DOI:10.1016/s0079-6700(02)00083-7.
- F. Hartmann, R. Dockhorn, S. Pusse, B. J. Niebuur, M. Koch, T. Kraus, A. Schiesser, B. N. Balzer and M. Gallei, Design and Self-Assembly of Second-Generation Dendrimer-like Block Copolymers, Macromolecules, 2024, 57(15), 7098–7111, DOI:10.1021/acs.macromol.4c00944.
- F. Hartmann, B. J. Niebuur, M. Koch, T. Kraus and M. Gallei, Synthesis and microphase separation of dendrimer-like block copolymers by anionic polymerization strategies, Eur. Polym. J., 2023, 187, 111894, DOI:10.1016/j.eurpolymj.2023.111894.
- L. F. Villalobos, M. Karunakaran and K. V. Peinemann, Complexation-induced phase separation: preparation of composite membranes with a nanometer-thin dense skin loaded with metal ions, Nano Lett., 2015, 15(5), 3166–3171, DOI:10.1021/acs.nanolett.5b00275.
- S. P. Nunes, M. Karunakaran, N. Pradeep, A. R. Behzad, B. Hooghan, R. Sougrat, H. He and K. V. Peinemann, From micelle supramolecular assemblies in selective solvents to isoporous membranes, Langmuir, 2011, 27(16), 10184–10190, DOI:10.1021/la201439p.
- M. Steube, M. Plank, M. Gallei, H. Frey and G. Floudas, Building Bridges by Blending: Morphology and Mechanical Properties of Binary Tapered Diblock/Multiblock Copolymer Blends, Macromol. Chem. Phys., 2021, 222(4), 2000373, DOI:10.1002/macp.202000373.
- K. Schellnhuber, J. Blass, H. Hubner, M. Gallei and R. Bennewitz, Single-Polymer Friction Force Microscopy of dsDNA Interacting with a Nanoporous Membrane, Langmuir, 2024, 40(1), 968–974, DOI:10.1021/acs.langmuir.3c03190.
- N. S. K. Gunda, M. Singh, L. Norman, K. Kaur and S. K. Mitra, Optimization and characterization of biomolecule immobilization on silicon substrates using (3-aminopropyl)triethoxysilane (APTES) and glutaraldehyde linker, Appl. Surf. Sci., 2014, 305, 522–530, DOI:10.1016/j.apsusc.2014.03.130.
- Y. S. Li, N. E. Vecchio and W. Lu, Infrared and Raman spectra of (3,3,3-trifluoropropyl)trimethoxysilane, its sol and xerogel, Spectrochim. Acta, Part A, 2013, 105, 213–217, DOI:10.1016/j.saa.2012.12.022.
- J. Elbert, M. Gallei, C. Rüttiger, A. Brunsen, H. Didzoleit, B. Stühn and M. Rehahn, Ferrocene Polymers for Switchable Surface Wettability, Organometallics, 2013, 32(20), 5873–5878, DOI:10.1021/om400468p.
- K. Foroutani, S. M. Ghasemi and B. Pourabbas, Ordered isoporous membranes from ionic diblock copolymers via SNIPS: Optimizing effective factors with a structural survey, Prog. Org. Coat., 2021, 161, 106554, DOI:10.1016/j.porgcoat.2021.106554.
| This journal is © The Royal Society of Chemistry 2026 |