
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2/epoxides ring-opening copolymerization towards hydroxy-functionalized polycarbonates
Nishant
Chaudhary
ab,
A. Stephen K.
Hashmi
 b,
Jean-François
Carpentier
*a and
Sophie M.
Guillaume
*a
b,
Jean-François
Carpentier
*a and
Sophie M.
Guillaume
*a
aUniv. Rennes, CNRS, Institut des Sciences Chimiques de Rennes, UMR 6226, F-35042 Rennes, France. E-mail: sophie.guillaume@univ-rennes.fr; jean-francois.carpentier@univ-rennes.fr
bOrganisch-Chemisches Institut, Universität Heidelberg, Catalysis Research Laboratory (CaRLa), Im Neuenheimer Feld 271, 69120, Heidelberg, Germany. E-mail: hashmi@hashmi.de
First published on 4th February 2026
Abstract
Ring-opening copolymerization (ROCOP) of CO2 and epoxides witnesses continued interest to access sustainable polycarbonates. Introduction of an exocyclic functional group onto the epoxides enables to tune and diversify the properties of the resulting CO2-based polycarbonates. Herein, the CO2/benzyl glycidyl ether (BnGE) or CO2/cyclohexene oxide (CHO) ROCOP has been performed, using a bicomponent catalyst system composed of either a {diamino-bisphenolate}MCl (Al, Fe) or {Salphen}CoCl complex or triethylborane (BEt3) as catalyst, combined with bis(triphenylphosphoranylidene)ammonium chloride (PPNCl) as initiator. While the Al/Fe-based catalyst systems selectively returned the corresponding benzyloxymethylene five-membered cyclic carbonate (5CCOBn) with poor activity in the copolymerization of CO2/BnGE, the {Salphen}CoCl/PPNCl and BEt3/PPNCl systems produced poly(benzyl glycidyl ether carbonate) (PBnGEC) with high chemoselectivity (∼80% and >98%) and regioselectivity (>99% and ∼84%), featuring >99% and ∼85% of carbonate linkages, respectively. Investigation of the {Salphen}CoCl/PPNCl and BEt3/PPNCl catalytic systems in the ROCOP of CO2/BnGE/CHO with different comonomers loadings, enabled to prepare a series of tunable P(BnGEC-co-CHC) terpolymers with similar selectivities. Subsequent hydrogenolysis of these hydrophobic polymers using Pd/C resulted in the deprotection of the side-chain benzyloxy moieties, affording the corresponding hydrophilic P(GC-co-CHC) polymers featuring hydroxyl pendant groups; yet, significant degradation of the polycarbonate main chain was observed for hydroxyl contents >15 mol%. Depolymerization of PBnGEC, PCHC and P(BnGEC-co-CHC) using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as catalyst quantitatively returned both corresponding 5CCs. In-depth characterizations by NMR spectroscopy, mass spectrometry, SEC, TGA, and DSC analyses supported well-defined protected and deprotected terpolymers with tunable chemical and thermal properties, providing opportunities for biomedical and/or industrial outcomes.
Introduction
The utilization of carbon dioxide has attracted considerable attention over the past few decades driven by the quest of a more sustainable future. The development of CO2-derived polymers represents a promising strategy for converting this abundant greenhouse gas into value-added materials. These polymers offer numerous advantages, typically including a wide range of applications, recyclability, degradability, and a decreased dependence on fossil fuel resources. Among them, aliphatic polycarbonates synthesized by ring-opening copolymerization (ROCOP) of CO2 and epoxides have emerged as key alternatives to conventional petrochemical-based polymers.1–5 Since the pioneering work of Inoue et al. in 1969, using ZnEt2 and water as a catalyst system for such copolymerizations,6,7 significant advances have been made in the design and optimization of metal-based catalysts. Despite these achievements, the field remains open, demanding continued research efforts towards more effective catalyst systems and a broader diversity of polycarbonate materials.8,9A wide range of catalyst systems have been explored for the ROCOP of epoxides with CO2. Transition/main group (Zn, Co, Cr, Mg, Al, etc.) metal-based catalysts initially dominated the field due to their efficiency in mediating controlled polymerization, as illustrated by seminal advanced contributions from the research groups of Darensbourg,10–12 Coates,13 Williams,14,15 among others.6,7,16 Originally focusing on mononuclear metal complexes, progress has evolved towards bifunctional and, more recently, di- or multi-nuclear metal catalysts, reflecting a diversification in the catalyst architecture.17,18 Despite their efficiency, metal-based catalytic systems can suffer from certain limitations, especially ligand multistep synthesis, potential issues related to metal/ligand toxicity, and possible coloration of the resulting polymers due to residual catalyst contamination.19,20
In 2016, a breakthrough in the ROCOP of CO2 and epoxide was achieved by Gnanou, Feng, and co-workers who reported systems based on a simple Lewis acid catalyst, namely BEt3, combined to an onium halide (e.g., bis(triphenylphosphoranylidene)ammonium chloride (PPNCl)) or an alkoxide acting as initiator. The efficient anionic copolymerization of CO2 with common epoxides such as propylene oxide (PO) or cyclohexene oxide (CHO) thus returned colorless polycarbonates with high selectivity (94% and >99%, respectively), significant activity (turnover frequency (TOF), calculated on the basis of the default initiator of 50 h−1 and 600 h−1, respectively), and dispersity value typically in the range ĐM = 1.1–1.9.21 Density Functional Theory (DFT) calculations suggested that the role of BEt3 extended beyond epoxide activation, specifically stabilizing the nucleophilicity of the propagating anionic chain, thereby mitigating undesirable backbiting reactions (and eventually increasing the selectivity towards polymer vs. cyclic carbonate formation).22 These pioneering findings established the viability of organoborane catalysts for the production of CO2-derived polycarbonates and carbonyl sulfide (COS) derived-polythiocarbonate.23–25 Subsequent investigations by Kerton and coworkers explored a series of alkyl- and aryl-substituted boranes with variable Lewis acidities.26 Their studies revealed that stronger Lewis acids such as BPh3 or B(C6F5)3, when paired with PPNCl under the optimized conditions previously established with BEt3/PPNCl system, were inactive in the ROCOP of CO2/PO, yielding instead the five-membered cyclic carbonate (5CC) at 100 °C, and only moderately active for CO2/CHO copolymerization at 60 °C. Further developments in CO2/epoxide ROCOP catalysis mediated by organoboron derivatives involved binary frustrated Lewis pair (FLP) systems.27,28 For instance, BEt3 associated with NEt3 generates a zwitterionic species which initiates ROCOP. Screening various FLP-based systems combining trialkylborane and tertiary phosphines revealed that steric and electronic effects significantly impact the catalytic activity and the control of the ROCOP. Notably, the PtBu3/BnBu3 system (featuring a bulky, strong nucleophilic phosphine, and a bulky, mild Lewis acidic borane) showcased TOF up to 447 mol(polymer) mol(PtBu3)−1 h−1 at 80 °C, with up to 96% carbonate linkages, and a dispersity ĐM value of 1.07. Overall, these studies highlighted the critical synergistic roles of the Lewis acidity, steric hindrance and size of the cation in the organic salt, as well as its ability to interact with CO2.22 Feng and coworkers further explored BEt3-based catalytic systems to synthesize ABA-type triblock copolymers incorporating structurally distinct epoxides, thereby expanding the functionality and application scope of these materials.29 Moreover, Wu and coworkers reported bifunctional organoborane catalysts combining both the Lewis acidic boron center and the nucleophilic quaternary ammonium/phosphonium halide salts for CO2/epoxides ROCOP that outperform the binary catalytic systems, attributing the enhanced activity and efficiency to intramolecular synergistic effects.30 Subsequent research introduced a variety of mono-, di-, and multinuclear organoborane catalysts through diverse strategies towards the synthesis of block copolymers.31,32
In the present study, catalytic systems based on aluminum (1–2), iron (3), cobalt (4) complexes and BEt3 compounds were explored for the ROCOP of CO2 with benzyl glycidyl ether (BnGE), a functional monomer featuring an exocyclic benzyloxymethyl moiety (–CH2OCH2Ph), to synthesize copolymers and terpolymers bearing functional pendant groups along the polymer backbone (Scheme 1). The latter most performant {Salphen}CoCl (4)/PPNCl and BEt3/PPNCl systems were next comparatively investigated in the ROCOP of CO2/BnGE/CHO, resulting in PBnGEC-based terpolymers with tunable BnGE/CHO carbonate contents and adjustable thermal properties (Scheme 2). Although prior studies from the groups of Frey and Grinstaff have reported the use of zinc33,34 and cobalt35,36 catalysts, respectively, for the CO2/BnGE ROCOP to prepare PBnGEC, to our knowledge, this is the first report involving an organoborane catalyst system for this ROCOP. The benzyloxymethyl pendant groups were subsequently transformed via hydrogenolysis into hydroxymethyl side-chain moieties. Finally, depolymerization of the PBnGEC, PCHC copolymers and P(BnGEC-co-CHC) terpolymers was explored with different catalytic systems to assess their susceptibility to chemical breakdown (Scheme 3). Comprehensive structural and thermal characterizations of the recovered copolymers and terpolymers using NMR spectroscopy, size-exclusion chromatography (SEC), matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF MS), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC), provided insights into structure–property relationships.
 | ||
| Scheme 1 ROCOP of CO2/BnGE catalyzed by (a) {ONNOR2}MCl/PPNCl (R = tBu, Me, Cl, M = Al, Fe) complexes resulting in the corresponding five-membered ring benzyloxymethylene cyclic carbonate (5CCOBn), and (b) {Salphen}CoCl/PPNCl or BEt3/PPNCl system, resulting in poly(benzyl glycidyl ether carbonate) (PBnGEC), poly(benzyl glycidyl ether) (PBnGEE) and cyclic carbonate (5CCOBn). | ||
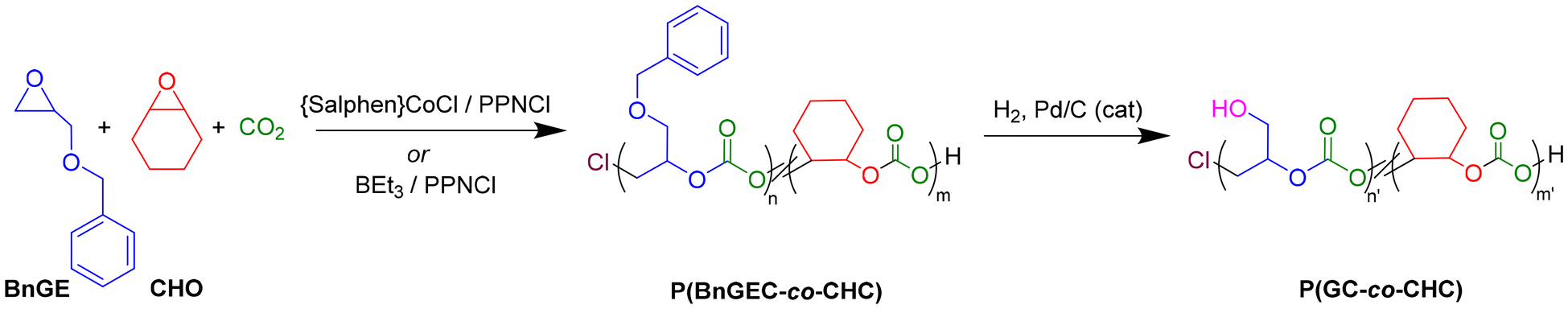 | ||
| Scheme 2 Terpolymerization of CO2/BnGE/CHO into P(BnGEC-co-CHC) catalyzed by the {Salphen}CoCl/PPNCl or BEt3/PPNCl system, followed by Pd-catalyzed debenzylation resulting into P(GC-co-CHC) terpolymers. | ||
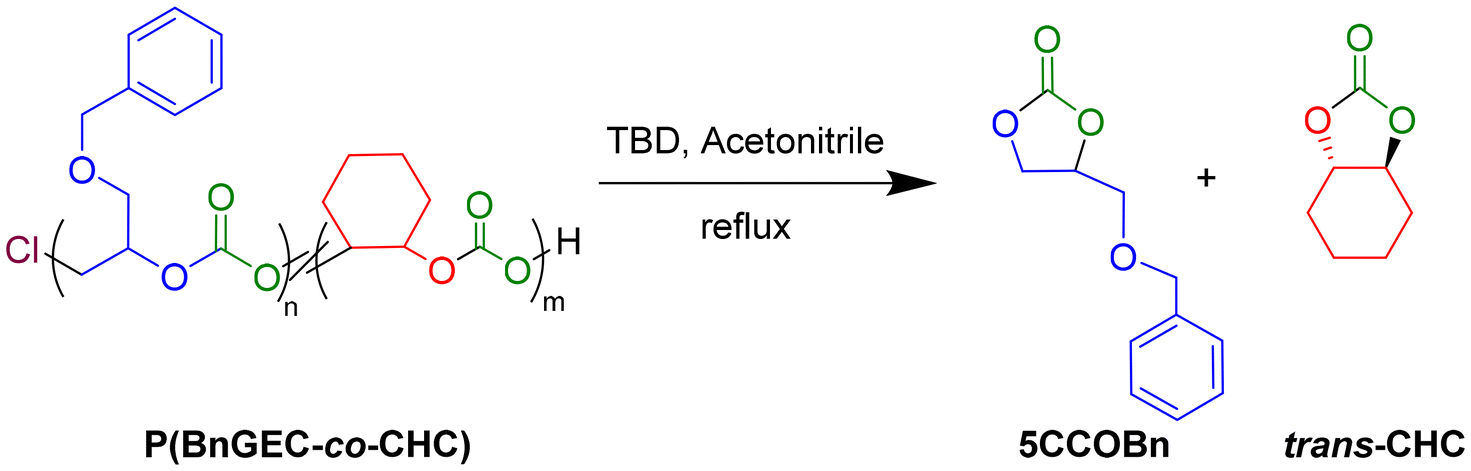 | ||
| Scheme 3 Depolymerization of P(BnGEC-co-CHC) terpolymer using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). | ||
Experimental section
Materials and methods
All reactions were performed under inert atmosphere of argon using Schlenk-line and glovebox techniques. Benzyl glycidyl ether (BnGE, BLDpharm, 98%) and cyclohexene oxide (CHO, TCI Europe, 98%) were dried over CaH2 for 3–4 days, distilled and stored under argon. Bis(triphenylphosphoranylidene)ammonium chloride (PPNCl) (Sigma-Aldrich, 97%) was heated under vacuum at 140 °C for 24 h and stored in the glovebox at room temperature. THF was dried over Na/benzophenone under argon and freshly distilled before use. BEt3 (1.0 M solution in hexanes, Sigma-Aldrich) and CO2 (Air Liquide, H2O < 7 ppm) were used as received. (Diamino-bisphenolate)-aluminum and iron chloride complexes {ONNOR2}MCl (M = Al, R = tBu (1) or Me (2); M = Fe, R = Cl (3)) were synthesized according to the literature procedure.37–39 Salphen-cobalt-chloride complex 4 was synthesized using the literature procedure.40 All other solvents and reagents were purchased from commercial suppliers and used without purification, unless otherwise mentioned. Hydrogenolysis of the terpolymers was performed as previously reported using Pd/C (10 wt%, Sigma-Aldrich).33–36
1H and 13C NMR spectra were recorded on Bruker Avance AM 500 or Ascend 400 spectrometers at 25 °C and were processed with Mestrenova software. Chemical shifts (δ) are reported in ppm and were referenced internally relative to SiMe4 (δ = 0 ppm) using the residual solvent resonances. The number- and weight-average molar mass (Mn,SEC and Mw,SEC, respectively) and dispersity (ĐM = Mw,SEC/Mn,SEC) values of polymer samples were determined by size-exclusion chromatography (SEC) in THF (HPLC-grade) at 30 °C (flow rate = 1.0 mL min−1) on a Agilent Technologies 1260 Infinity II instrument equipped with a RI detector. The polymer samples were dissolved in THF (1.0 mg mL−1) and were filtered through an Agilent PTFE 0.2 µm PTFE filter. The SEC instrument was calibrated using narrow-Mn polystyrene standards of molar mass in the range 3300–70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1.
500 g mol−1.
After purification of the copolymers and terpolymers isolated from the crude reaction mixture (vide infra), the composition in carbonate and ether segments was determined by analysis of the aliphatic region of the 1H NMR spectra. The characteristic resonances associated with the carbonate repeating units were observed at δ = 3.47–3.88 ppm (2H, side-chain methylene, –CH2CH(CH2OBn)OCOO–), δ = 4.16–4.67 ppm (4H, in-chain methylene and side-chain benzyl methylene, –CH2CH(CH2OCH2Ph)OCOO–), and δ = 4.97–5.17 ppm (1H, in-chain methine, –CH2CH(CH2OBn)OCOO–). The ether repeating units were typically observed at δ = 3.41–3.80 ppm (5H, in-chain methylene and methine, and side-chain methylene, –CH2CH(CH2OBn)O–) and δ = 4.39–4.52 ppm (2H, side-chain benzyl methylene, –CH2CH(CH2OCH2Ph)O–).
In copolymers containing both carbonate and ether linkages, signals do overlap within the δ = 3.47–3.88 ppm and δ = 4.16–4.67 ppm regions. The ether segment content (y) was determined, taking into account these overlapping regions using: y = [{(I3.47–3.88) − 2}/5 + {(I4.16–4.67) − 4}/2]/2, where I is the integral value over the specified chemical shift range. The relative contents in carbonate and ether linkages in the copolymer was then calculated according to: carbonate content (%) = {x/(x + y)} × 100, and ether content (%) = {y/(x + y)} × 100 or 100 – carbonate linkage, where x is the carbonate content and is set to 1. This method thus enabled a fair quantitative estimation of the composition of the copolymers and terpolymers, in spite of overlapping signals in the aliphatic region.
Thermogravimetric analyses (TGA) were performed on a Mettler Toledo TGA/DSC1 instrument by heating the polymer samples at a rate of 10 °C min−1 from +25 to +500 °C under a dynamic nitrogen atmosphere (flow rate = 10 mL min−1). The onset decomposition temperature (Td) was defined as the temperature for 5% weight loss.
Differential scanning calorimetry (DSC) analyses were performed on a DSC 2500 TA instrument calibrated with indium using aluminum capsules (40 μL). All samples (2–10 mg) were analyzed over three successive heating and cooling cycles over a temperature range of −80 to +220 °C at a rate of 10 °C min−1. The second cycle was used to determine the Tg value.
:1 v/v), and palladium on activated charcoal (Pd/C, 10 wt%; 18 mg, 10 mol% Pd vs. OBn). The autoclave was sealed and pressurized with hydrogen (40 bar), heated at 40 °C in an oil bath, with continuous stirring using a magnetic stir bar. After 24 h, the reactor was cooled down to room temperature, H2 was vented and the solution was filtered through a Celite pad along with washing with a solvent mixture of ethyl acetate and methanol (1:1 v/v). The resulting filtrate was collected, the solvent was evaporated under vacuum, and the recovered white solid polymer was dried under vacuum for 24 h.
Results and discussion
Metal-catalyzed CO2/epoxide ROCOP
The ROCOP of CO2 and BnGE was first investigated using some metal-based catalysts. As previously reported by Kozak, Kerton and coworkers for the successful CO2/CHO copolymerization into poly(cyclohexene carbonate) (PCHC), the {diamino-bisphenolate}-aluminum(III) or -iron(III) chloride complexes {ONNOR2}MCl (Scheme 1, M = Al, R = tBu (1), Me (2); M = Fe, R = Cl (3)) were assessed under the typical optimal conditions therein established, namely low temperature (40 °C) and moderate CO2 pressure (30 bar).37,39 All three complexes 1–3 revealed active, yet selectively producing the corresponding –CH2OBn substituted five-membered cyclic carbonate (5CCOBn) (Table S1). In order to mitigate the 5CCOBn formation, we next explored the one-pot terpolymerization of CO2 with BnGE and CHO, using iron complex 3, under the aforementioned conditions reported for CHO.39 The hypothesis was that the active growing oxianionic chain of CHO could effectively incorporate BnGE units, thus suppressing back-biting side-reactions and eventually promoting the effective terpolymer formation. Yet, although a small amount of terpolymer containing ∼5 mol% of BnGE was thus obtained, the catalytic activity and productivity remained poor (Table S2). Hence, overall, these Al/Fe-catalyzed CO2/BnGE ROCOP did not enable the selective preparation of polycarbonates.On the other hand, the well-established {Salcy}CoX/PPNY catalyst system was previously reported to be effective in the copolymerization and terpolymerization of BnGE (up to 99% carbonate linkages and Mn up to 41.0 kg mol−1).35,36 Similarly, we investigated the {Salphen}CoCl/PPNCl catalyst system in the copolymerization of BnGE/CO2, and CHO/CO2 (Scheme 1 and Table 1). The ROCOP of BnGE and CO2, performed at 0.1 mol% catalyst loading under the previously optimized conditions of 25 °C and 15 bar of CO2, yielded PBnGEC with up to ∼80% chemoselectivity, >99% selective towards head-to-tail (HT) regiosequence, and >99% carbonate linkages (Table 1, entries 1–3; and Fig. S2, S3;). The ROCOP of CHO and CO2 was found to proceed sluggishly under the reaction conditions optimized for the BnGE/CO2 system, possibly owing to its rigid monomer structure (Table 1, entry 9). On the other hand, the terpolymerization of BnGE, CHO, and CO2 – provided BnGE is introduced in equal or higher molar loading than CHO – proceeded smoothly, with TOF up to 15 mol(polym) mol(PPNCl)−1 h−1 (Scheme 2 and Table 1, entry 4), and CHO was effectively incorporated within the terpolymer which was formed in ∼90% selectivity (vs. 5CCs) (Table 1, entries 4–7). The terpolymer composition as determined by 1H NMR closely reflected the initial monomers feed ratio, with 99% carbonate linkages. When CHO was introduced in excess relative to BnGE, the polymerization rate decreased significantly, and the resulting terpolymer composition deviated from the feed ratio, consistent with the lower reactivity of the cobalt catalyst toward the CHO/CO2 ROCOP (Table 1, entry 8). SEC-determined number-averaged molar mass values (Mn, up to 20.9 kg mol−1) over the bimodal trace (ĐM = 1.15–1.22) of the copolymers and terpolymers were notably lower than the predicted ones; we assume this is likely due to the presence of adventitious water traces in the system (CO2), despite the care taken in the purity/purification of reagents and solvent (vide infra). Residual water may have induced side reactions such as chain-transfer processes and epoxide hydrolysis, generating multiple initiation sites and consequently the lowering of the molar mass.41,42
| Entry |
f
BnGEb |
Time (h) | Conv.c (%) | Selectivity BnGEc (polymer vs. CC) | P(BnGEC-co-CHC)dfBnGE |
M
n,theo
(kg mol−1) |
M
n,SEC,THF
(kg mol−1) |
Đ
M
|
T
g,DSCh (°C) |
Yieldi (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BnGE | CHO | ||||||||||
|
a All reactions were performed with [monomer]0/[catalyst]0/[cocatalyst]0 = 1000:1:1, where PPNCl was used as a cocatalyst, in neat BnGE/CHO at 25 °C under 15 bar of CO2. The isolated copolymers and terpolymers contained >99% carbonate linkages and >99% polymer selectivity for CHO, as determined by 1H NMR analysis.
b
f
BnGE = molar fraction of BnGE vs. CHO in substrate.
c % Monomer conversion and CO2 selectivity determined by 1H NMR analysis of the crude reaction mixture.
d BnGE molar fraction determined by 1H NMR analysis of the isolated polymer.
e Calculated molar mass of P(BnGEC-co-CHC) according to Mn,theo = [(BnGE carbonate unit × 208) + (BnGE ether unit × 164)] + [(CHO carbonate unit × 142) + (CHO ether unit × 98)] + mass of end-groups (–Cl, –H).
f Experimental molar mass of polymer determined by SEC in THF with a calibration using polystyrene standards; the molar mass value is the average of the bimodal trace.
g Dispersity value determined by SEC in THF.
h
T
g, glass transition temperature measured by DSC.
i Yield (%) = [Wt. of isolated polymer obtained(g)/(monomer used + CO2 consumed) (g)] × 100.
j 0.4 mol% of the catalyst was used.
k THF was used as a solvent for homogeneity.
|
|||||||||||
| 1 | 1.00 | 4 | 19 | — | 68 | 1.00 | 26.9 | 5.4 | 1.15 | — | |
| 2 | 1.00 | 18 | 42 | — | 86 | 1.00 | 75.1 | 8.6 | 1.19 | 13 | 20 |
| 3 | 1.00 | 48 | 56 | — | 68 | 1.00 | 79.2 | 13.8 | 1.24 | 9 | 23 |
| 4 | 0.90 | 48 | 77 | 64 | 88 | 0.87 | 135.9 | 17.1 | 1.22 | 16 | 61 |
| 5 | 0.80 | 48 | 79 | 63 | 93 | 0.80 | 140.0 | 18.3 | 1.22 | 25 | 67 |
| 6j,k | 0.50 | 24 | 98 | 92 | 92 | 0.54 | 39.6 | 11.3 | 1.18 | 46 | 46 |
| 7 | 0.50 | 48 | 71 | 50 | 95 | 0.52 | 105.3 | 20.9 | 1.16 | 45 | 46 |
| 8k | 0.10 | 68 | 36 | 25 | 85 | 0.52 | 38.0 | 9.1 | 1.16 | 40 | — |
| 9k | 0.00 | 68 | — | 6 | — | — | — | — | — | ||
BEt3/PPNCl-catalyzed CO2/epoxide ROCOP
The copolymerization of CO2 and BnGE was next investigated using the Lewis acid–base pair catalytic system comprising BEt3 as activator and PPNCl as initiator, at 40 °C in THF under 30 bar of CO2 (Scheme 1 and Table 2). 1H NMR monitoring of the reaction showed the formation of the awaited polymer with PBnGEC polycarbonate and PBnGEE polyether units, alongside formation of the cyclic carbonate (5CCOBn) and some unreacted monomer (Fig. S1). The recovered sample was next repeatedly precipitated in CH2Cl2/methanol, until complete disappearance of 5CCOBn NMR signals (δ = 4.73–4.81 ppm). In order to carefully assess the chemical composition of the macromolecules formed, the corresponding pristine polyether was synthesized from the ROP of BnGE performed in THF at 40 °C, to serve as a reference for quantifying ether linkages (typical signals: δ = 3.41–3.84 ppm (5H, in-chain methylene and methine, and side-chain methylene, –CH2CH(CH2OBn)O–), 4.46 ppm (2H = side-chain benzyl methylene –CH2CH(CH2OCH2Ph)O–)) within the copolymer (Fig. 1). The presence of both carbonate and ether units in the isolated polymer sample was further corroborated by 13C NMR spectroscopy, showing two distinct methine resonances (δcarbonate-C3 = 74.5 ppm and δether-C3′ = 78.9 ppm; Fig. S7). | ||
| Fig. 1 Representative 1H NMR (400 MHz, CDCl3, 23 °C) spectra of PBnGEC with minor ether linkages (a)-bottom, and pristine PBnGEE (b)-top prepared with the BEt3/PPNCl catalyst system. | ||
| Entry | [M]0/[A]0/[I]0 (T, PCO2) | Time (h) | Conv.a (%) | TOFb (h−1) | Selectivity of the crude reaction mixturea | Isolated polymer | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Polym. | 5CC | Polyether | Carbonate vs. ether linkagesc1H (13C) |
M
n,theod (kg mol−1) |
M
n,SECe (kg mol−1) |
Đ
Mf |
Yieldg (%) | |||||
| Monomer (BnGE), activator (BEt3), initiator (PPNCl). THF was used as solvent, introduced in a volume equal to that of the monomer.a % Monomer conversion and CO2 selectivity determined by 1H NMR analysis of the crude reaction mixture.b Turnover frequency (TOF) = mol(polymer) mol(PPNCl) (h−1).c Determined by 1H NMR analysis of the isolated PBnGEC.d Mn,theo = (no. of carbonate units × 208) + (no. of ether units × 164) + mass of end-groups.e Experimental molar mass of PBnGEC determined by SEC in THF with a calibration using polystyrene standards; the molar mass value is the average of the bimodal trace.f Dispersity value determined by SEC in THF.g Yield (%) = [Wt. of isolated polymer obtained(g)/(monomer used + CO2 consumed) (g)] × 100. | ||||||||||||
| 1 | 100:1.5:1 (40,15) |
15 | 79 | 5 | — | 99 | — | — | — | — | — | — |
| 2 | 100:2:1 (40,15) |
15 | 87 | 6 | 97 | 0 | 3 | 83:17 |
16.9 | 8.7 | 1.13 | 65 |
| 3 | 100:2:1 (40,15) |
15 | 89 | 6 | 93 | 0 | 6 | 54:46 |
15.6 | 9.3 | 1.11 | 46 |
| 4 | 100:2:1 (40,15) |
15 | 77 | 5 | 87 | 3 | 10 | 71:28 |
13.2 | 9.1 | 1.12 | 47 |
| 5 | 100:4:1 (40,20) |
15 | 80 | 5 | 97 | 0 | 2 | 54:46 |
14.6 | 6.3 | 1.17 | 17 |
| 6 | 100:4:1 (40,30) |
15 | 78 | 5 | 98 | 1 | 1 | 68:32 |
14.8 | 7.3 | 1.14 | 48 |
| 7 | 100:6:1 (40,30) |
15 | 89 | 6 | 97 | 2 | 0 | 82:18(92:8) |
17.3 | 5.4 | 1.16 | 65 |
| 8 | 100:6:1 (40,30) |
15 | 82 | 5 | 98 | 2 | 0 | 84:16 |
16.1 | 7.9 | 1.16 | 67 |
| 9 | 100:6:1 (60,10) |
5 | 95 | 19 | 76 | 13 | 11 | 75:25 |
14.4 | 8.9 | 1.15 | 38 |
| 10 | 100:2:1 (60,30) |
5 | 91 | 18 | 98 | 2 | 0 | 77:23 |
17.6 | 9.2 | 1.12 | 72 |
| 11 | 100:6:1 (40,40) |
16 | 73 | 5 | 83 | 2 | 15 | 80:20 |
12.2 | 4.5 | 1.16 | 26 |
| 12 | 100:8:1 (40,30) |
16 | 90 | 6 | 95 | 2 | 3 | 83:17 |
13.8 | 4.9 | 1.13 | 64 |
| 13 | 100:10:1 (40,30) |
16 | 79 | 5 | 87 | 2 | 11 | 81:19 |
13.7 | 6.5 | 1.16 | 62 |
| 14 | 100:12:1 (40,30) |
16 | 95 | 6 | 82 | 0 | 18 | 83:17 |
15.6 | 7.5 | 1.13 | 60 |
The main reaction parameters (temperature, pressure, BEt3/PPNCl ratio) were screened to establish the optimized operating conditions (Fig. 2 and Table 2). Under the standard conditions previously established for optimal efficiency of BEt3/PPNCl in CO2/PO ROCOP, namely with an activator/initiator loading of 2:1,21 the CO2/BnGE ROCOP performed at 40 °C and PCO2 = 15 bar produced PBnGEC, yet with variable carbonate and ether linkage contents, possibly due to uncontrolled copolymerization (Fig. S10). Using a BEt3/PPNCl loading reduced to 1.5:1 selectively returned 5CCOBn, most likely owing to the highly nucleophilic propagating anionic chain.21 Conversely, raising the BEt3/PPNCl molar ratio to 4:1 resulted in effective, yet still incompletely controlled ROCOP, and in a polycarbonate contaminated with ether linkages (Fig. S10).
 | ||
| Fig. 2 Effects of the BEt3/PPNCl ratio, reaction temperature and CO2 pressure on the selectivity of the CO2/BnGE ROCOP (Table 2). | ||
Zhang and coworkers previously reported the BEt3/PPNCl catalyzed ROCOP of CO2/phenyl glycidyl ether (PGE) − an epoxide analogous to BnGE, yet with slightly differentiated electronic features (phenyl vs. benzyl) −, which proceeded to give up to 99% of carbonate content in the resulting copolymer.43 Their findings indicated that this epoxy substrate required a larger excess of BEt3 (6:1 ratio) for a fully effective copolymerization. Applying this rationale to our system upon increasing the BEt3/PPNCl ratio to 6:1 (at 40 °C and PCO2 of 30 bar) actually enabled to improve the carbonate content to 85% with a TOF of 6 mol(polym) mol(PPNCl)−1 h−1 and an isolated polymer yield of 65% with a high head-to-tail regioselectivity of 84% (Fig. 2 and Table 2). Increasing further the BEt3 charge to a ratio of 8:1, 10:1, or 12:1 (under the same operating conditions) did not really alter the copolymer composition, but the fraction of polyether oligomers increased, thereby reducing the chemoselectivity. No matter of the slightly differentiated electronic features between the two monomers (phenyl in PGE vs. benzyl in BnGE), we assume that this [BEt3/PPNCl]-catalyzed copolymerization is more an equilibrium-driven process where excess BEt3 (i.e. 6 equiv.) is required to push the reaction forward.
A higher reaction temperature (60 °C − as optimized with PGE) enhanced the activity (TOF = 18 h−1vs. 6 h−1 at 40 °C) while reducing the carbonate/ether linkage selectivity (from 85:15 down to 75:25 at 40 °C), as anticipated from the thermodynamics of ROCOP.44 Additionally, increasing the monomer loading above 100 equiv. of BnGE adversely affected the selectivity, shifting the reaction towards 5CCOBn formation. This may be attributed to a dilution effect, wherein lower catalyst concentration hindered the cooperative action of the Lewis pair required for an effective ROCOP (Fig. 3).45 This contrasts with the {SalPhen}CoCl/PPNCl system which readily withstands [monomer]0/[catalyst]0/[cocatalyst]0 ratios of 1000:1:1 (Table 1). To explore potential intramolecular synergistic interactions,46 a bifunctional organoborane catalyst, namely [Et3N-C5-BBN][Cl], was also examined, in place of the binary catalyst system for the ROCOP of BnGE/CO2, under varied conditions; however, the reaction exclusively returned the cyclic carbonate.
 | ||
| Fig. 3 Effect of the BEt3/PPNCl catalyst loading on the selectivity of the CO2/BnGE ROCOP (Table S3). Under the optimized conditions thus established for the CO2/BnGE ROCOP (BEt3/PPNCl molar ratio = 6:1, 40 °C, PCO2 = 30 bar), the alike ROCOP of CO2 with CHO selectively provided PCHC with a carbonate content >99%, as no NMR signal was observed for the ether linkages (δ = 3.4–3.5 ppm) (Fig. 4, fBnGE = 0). | ||
SEC traces of all CO2/BnGE and CO2/CHO copolymers revealed a bimodal distribution, with a minor population at lower molar masses (Fig. S15 and 16). As for Co catalysis (vide supra), this suggests concomitant initiation by adventitious water, in agreement with prior literature where bimodal distributions are quite commonly observed and unambiguously assigned to water co-initiation.41,42 The Mn values over the bimodal trace ranges from 5–10 kg mol−1 with ĐM ≤ 1.2. MALDI-ToF mass spectrometry analysis of the copolymers supported the alternating CO2/BnGE structure with minor ether incorporation (Fig. S26). The major population observed in MALDI-ToF MS was attributed to α,ω-dihydroxy telechelic PBnGEC with some ether insertions (three intense series observed with, respectively, one, two and three ether unit(s) incorporated). As mentioned above, such dihydroxy end-capped PBnGEC most likely resulted from concomitant initiation and/or chain transfer reactions mediated by adventitious water. The minor population (also with three series) observed in MALDI-ToF-MS was assigned to the α-chlorine, ω-hydroxy end-capped PBnGEC, arising from PPNCl initiation.
BEt3/PPNCl-catalyzed CO2/BnGE/CHO terpolymerization
A series of P(BnGEC-co-CHC) terpolymers was synthesized from various feed ratios of BnGE and CHO, under the optimized conditions ([monomer]0/[BEt3]0/[PPNCl]0 = 100:6:1, 40 °C, PCO2 = 30 bar, 15 h; Scheme 2 and Table 3). The BnGE-to-CHO molar fraction was varied in the range 0.90:0.10 to 0.02:0.98. 1H NMR analysis of the recovered polymer samples unambiguously showed distinguishable signals for each carbonate unit methine hydrogens (δCH,PBnGEC = 5.04 ppm, 1H; δCH,PCHC = 2.09 ppm, 2H), which served to determine the polymer composition (Fig. 4). Regardless of the monomers loading, all terpolymerizations exhibited good reactivity, with an average isolated yield of 75%, and high selectivity (∼98%) towards polymer formation. The carbonate-to-ether linkage ratios, ca. 85:15 for BnGE and 99:1 for CHO, were consistent with those obtained above from their individual ROCOP with CO2, respectively (Table S3). The experimentally determined molar fraction of PBnGEC within the polymer backbone (fPBnGEC) matched well the initial monomer feed ratio, with a maximum integrated fraction of 0.78 from a loading of 0.90 (Table 3, entry 1). The incorporation of PBnGEC and PCHC units in the terpolymer varied monotonously with their respective loading ratio (Fig. 4).
 | ||
| Fig. 4 1H NMR spectra (400 MHz, CDCl3, 23 °C) of P(BnGEC-co-CHC) terpolymers synthesized with the BEt3/PPNCl system at various BnGE/CHO loadings (Table 3). | ||
| Entry |
f
BnGE
|
BnGE/CHO Conv.c (%) | P(BnGEC-co-CHC) | P(GC-co-CHC) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
f
PBnGEC,theod |
f
PBnGEC,expe |
M
n,theof (kg mol−1) |
M
n,SECg (kg mol−1) |
Đ
Mh |
T
d,TGAi (°C) (5%) |
T
g,theoj (°C) |
T
g,DSCk (°C) |
f
Glycidoll |
M
n,theom (kg mol−1) |
M
n,SEC,THFn (kg mol−1)/ĐMh |
M
n,SEC,DMFo (kg mol−1)/ĐMp |
T
d,TGAi (°C) (5%) |
T
g,DSCk (°C) |
|||
|
a All reactions were performed with a [monomer]0/[activator]0/[initiator]0 of 100:6:1 molar ratio, at 40 °C, PCO2 = 30 bar, for 15 h. THF was used as solvent, introduced in a volume equal to that of the monomer.
b
f
BnGE = Molar fraction of BnGE in substrate.
c Monomers conversion determined by 1H NMR analysis of the crude reaction mixture.
d Calculated PBnGEC molar fraction determined by 1H NMR analysis of the crude reaction mixture.
e Experimental PBnGEC molar fraction determined by 1H NMR analysis of the isolated P(BnGEC-co-CHC).
f Calculated molar mass of P(BnGEC-co-CHC) according to Mn,theo = [(BnGE carbonate unit × 208) + (BnGE ether unit × 164)] + [(CHO carbonate unit × 142) + (CHO ether unit × 98)] + mass of end-groups (–Cl, –H).
g Experimental molar mass of P(BnGEC-co-CHC) determined by SEC in THF with a calibration using polystyrene standards; the molar mass value is the average of the bimodal trace and is uncorrected for possible hydrodynamic volume differences.
h Dispersity value determined by SEC in THF.
i Decomposition temperature at 5 wt% loss determined by TGA.
j Calculated glass transition temperature of P(BnGEC-co-CHC) determined using Fox eqn (1/Tg,terpolymer = (XPBnGEC/Tg,PBnGEC) + (XPCHC/Tg,PCHC) with X = molar fraction of PBnGEC/PCHC and Tg expressed in °K).
k Experimental glass transition temperature of P(BnGEC-co-CHC) and P(GC-co-CHC) measured by DSC.
l Calculated glycidol molar fraction determined by 1H NMR analysis of the isolated P(GC-co-CHC).
m Calculated molar mass of P(GC-co-CHC) according to Mn,theo. = Mn,SEC × [1-(fGlycidol × 74/164)].
n Experimental molar mass of P(GC-co-CHC) determined by SEC in THF with a calibration using polystyrene standards; the molar mass value is the average of the bimodal trace and is uncorrected for possible hydrodynamic volume differences.
o Experimental molar mass of P(GC-co-CHC) determined by SEC in DMF with a calibration using polymethymetacrylate standards; the molar mass value is the average of the bimodal trace and is uncorrected for possible hydrodynamic volume differences.
p Dispersity value determined by SEC in DMF.
|
||||||||||||||||
| 1 | 0.90 | 88/98 | 0.89 | 0.78 | 17.1 | 6.2 | 1.18 | 207 | 16 | 8 | 0.77 | 4.0 | 0.6/1.18 | 1.0/1.15 | ||
| 2 | 0.80 | 89/99 | 0.78 | 0.70 | 16.9 | 5.7 | 1.17 | 26 | 13 | 0.64 | 4.0 | — | 1.1/1.27 | |||
| 3 | 0.50 | 93/99 | 0.48 | 0.43 | 16.1 | 6.4 | 1.18 | 215 | 54 | 37 | 0.43 | 5.1 | 1.2/1.68 | 1.4/1.32 | ||
| 4 | 0.30 | 96/94 | 0.30 | 0.26 | 14.8 | 7.0 | 1.24 | 76 | 57 | 0.23 | 6.2 | 3.7/1.47 | 2.6/1.61 | 23 | ||
| 5 | 0.10 | 99/93 | 0.10 | 0.12 | 13.8 | 7.3 | 1.25 | 96 | 85 | 0.11 | 6.9 | 7.0/1.13 | 4.8/1.48 | 175 | 59 | |
| 6 | 0.05 | 99/99 | 0.04 | 0.08 | 14.7 | 8.8 | 1.19 | 201 | 102 | 84 | 0.07 | 8.5 | 6.3/1.28 | 7.1/1.31 | 68 | |
| 7 | 0.02 | 99/99 | 0.02 | 0.05 | 14.5 | 8.5 | 1.19 | 107 | 91 | — | — | — | — | |||
The microstructure of carbonate units within the P(BnGEC-co-CHC) terpolymers was assessed from 13C{1H} NMR spectra, by comparison with those of PBnGEC copolymers (Fig. 5). Notable splitting of the C1 and C2 resonances (corresponding to in-chain methylene and methine carbons, respectively) in the PBnGEC copolymer into distinct C1′ and C2′ resonances, respectively, in the P(BnGEC-co-CHC) terpolymer, indicates a distinct chemical environment. This observation supports the formation of terpolymers composed of randomly distributed BnGE and CHO carbonate units rather than block sequenced materials (i.e., P(BnGEC-b-CHC)).35 Again, the SEC traces of all terpolymers were bimodal, with molar mass values ranging from 5700 to 8800 g mol−1 along with rather narrow dispersity values (1.17 < ĐM < 1.25; Fig. S21).
 | ||
| Fig. 5 Representative 13C{1H} NMR spectra (100 MHz, CDCl3, 23 °C) of a PBnGEC copolymer (a-bottom, Table 2, entry 7) and a P(BnGEC-co-CHC) terpolymer (b-top, Table 3, entry 3) synthesized with BEt3/PPNCl catalyst system, showing the notable splitting of the backbone methylene (C1/C1′) and methine (C2/C2′) resonances in the terpolymer in comparison to its copolymer analogue. | ||
Benzyl deprotection of P(BnGEC-co-CHC) terpolymers into hydroxy-functional P(GC-co-CHC) polycarbonates
The benzyloxy groups of P(BnGEC-co-CHC) terpolymers were quantitatively deprotected upon catalytic hydrogenolysis with Pd/C (10% Pd vs. OBn, 40 °C, ethyl acetate/methanol (1:1 v/v), 24–48 h, 40 bar H2) affording the corresponding poly((1,2-glycerol carbonate)-co-CHC) (P(GC-co-CHC)) (Scheme 2). The deprotected polymers were isolated as white solid materials, partially soluble in THF and CDCl3, yet readily soluble in DMSO at room temperature. Expectedly, this latter behavior hints at more hydrophilic materials than the BnO-protected parent polymers. Completion of the reaction was confirmed by 1H NMR analysis in DMSO-d6, evidencing complete disappearance of the phenyl hydrogens signal (δ = 7.30 ppm, m, 5H) concomitant with the growing of a broad hydroxy signal (δ = 5.04 ppm, 1H) (Fig. S15). Notably, efficient deprotection of terpolymers with a low benzyl content (fBnGE = 0.08) required increased catalyst loading (50 mol%) and prolonged reaction time (48 h). This behavior can be tentatively rationalized by the limited accessibility of benzyl moieties at low concentrations, reducing the probability of catalytic activation. SEC analysis of the deprotected P(GC-co-CHC) terpolymers in DMF (vs. PMMA standards) revealed that hydrogenolysis of terpolymers with a higher benzyloxy content (>15 mol%) induced significant concomitant polymer main-chain cleavage. Repeated experiments under different deprotection conditions (solvent, temperature, concentration, reaction time) systematically returned the same results, in contrast to previous reports in the literature.33–36 Kinetic monitoring by SEC showed that backbone cleavage does take place concomitantly with OBn hydrogenolysis. We assume that this apparent cleavage of the polymer backbone is likely resulting from nucleophilic attack of the main-chain carbonate moieties by (abundant) pendant hydroxyl groups. In contrast and in line with the above hypothesis, terpolymers with a lower benzyloxy content (<15 mol%) largely retained the integrity of the polymer backbone. For terpolymers enriched in PCHC linkages and featuring low hydroxyl content, the molar mass values determined by SEC were in good correlation with the molar mass calculated based on each monomer consumption (Table 3, entries 5 and 6). Given their degradability and versatility based on their functionalization opportunities, such hydroxylated polycarbonates provide valuable candidates for biomedical applications such as drug delivery or scaffold materials for tissue engineering.47–49
Thermal features of P(BnGEC-co-CHC) terpolymers
The thermal properties of the synthesized terpolymers – both BnO-protected ones obtained from BEt3/PPNCl and 4/PPNCl catalyst systems, and deprotected ones – were examined by thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC). All P(BnGEC-co-CHC) samples synthesized from BEt3/PPNCl were recovered as colorless (in contrast, the polymers synthesized using cobalt catalyst featured a pale orange/brown color; no specific treatment was attempted to remove metal catalyst residues from the synthesized polymers), either sticky soft solids for PBnGEC-enriched terpolymers or powdery solid materials for PCHC-rich terpolymers (Fig. 6a). | ||
| Fig. 6 Illustration of (a) the powdery-to-soft/sticky nature of P(BnGEC-co-CHC) terpolymers upon increasing the PBnGEC content (from left to right), and (b) correlation of the glass transition temperature (Tg) values of these same terpolymers as determined by DSC analysis (Tg,exp) and compared to the calculated values from Fox equation (Tg,theo). | ||
The decomposition temperature at 5 wt% loss (T5%d) for PBnGEC and PCHC samples synthesized by the BEt3/PPNCl system was measured at 225 and 237 °C, respectively (Fig. S27 and 28), while the T5%d for P(BnGEC-co-CHC) terpolymers was recorded in the range of 200–215 °C (Table 3). DSC analysis of the homopolymers revealed a glass transition temperature (Tg) of 113 °C for PCHC (Table S3, entry 10), while in the case of PBnGEC, the first heating cycle displayed an exothermic peak around 164 °C (Fig. S31). Subsequent 1H NMR analysis of this latter heated sample (after three heating–cooling cycles) indicated the selective formation of cyclic carbonate 5CCOBn (Fig. S45); this is attributed to thermally-induced depolymerization and/or intramolecular cyclization facilitated by backbiting, accompanied by heat release. A similar exothermic behavior was observed in terpolymers with a high BnGE content (fBnGE = 0.78 and 0.70; Table 3, entries 1 and 2), while PCHC-rich terpolymers did not exhibit such a transition; we assume this is possibly due to the increased chain rigidity imparted by CHO monomer incorporation, as previously observed for poly(propylene carbonate).50 Upon lowering the upper temperature limit to 140 °C, the Tg values measured for all P(BnGEC-co-CHC) terpolymers were within the range defined by the Tg values of PBnGEC (−1 °C) (Fig. S32) and PCHC (113 °C) (Fig. S34), aligning well with predicted values derived from the Fox equation (Fig. 6b). Also, the Tg value of PBnGEC synthesized using cobalt catalyst 4 containing 99% carbonate linkages was measured at 9 °C (Fig. S33), consistent with previously reported values,35 whereas the polymer prepared using the BEt3/PPNCl catalytic system exhibited a lower Tg of −1 °C, attributed to the presence of ether linkages (∼15%) in the macromolecular chain. Similarly, for the terpolymers synthesized using the BEt3/PPNCl system, the Tg values measured were again lower than those of terpolymers prepared with cobalt catalyst 4, similarly arising from the presence of ether linkages.
Following benzyl deprotection, the T5%d value of the resulting P(GC-co-CHC) terpolymer with fGC ≈ 0.10 showed a decrease of ∼25 °C in comparison to its protected parent terpolymer. DSC analysis of the hydroxy-functionalized terpolymers enriched in PCHC linkages resulted in a lowering of Tg values (∼20 °C) relative to their protected counterparts, in line with the observed reduction in molar mass due to chain scission, as indicated by SEC(Table 3, entries 5 and 6).
Depolymerization of PBnGEC, PCHC and P(BnGEC-co-CHC) copolymers and terpolymers
Investigation of the depolymerization ability of the copolymers and terpolymers via organocatalysis enabled to assess their potential for chemical recycling.51 Following the procedure previously reported by Kleij and coworkers on the depolymerization of biohybrid polycarbonates derived from terpenes, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were investigated as potential organocatalysts (Fig. S46).52 Depolymerization of the PBnGEC, PCHC and terpolymers, using 1 mol% TBD under reflux in acetonitrile or toluene within 2 h, returned the corresponding cyclic carbonate in moderate yield, i.e., BnGE five-membered ring cyclic carbonate (5CCOBn, 23%) and trans-cyclohexene carbonate (t-CHC, 18%), respectively (Table S4, entries 1 and 15). Increasing the catalyst loading to 5 mol% enhanced the depolymerization efficiency with quantitative yields of cyclic carbonates (Table S4, entries 2 and 16). The rate of depolymerization to 5CCOBn was comparatively faster than of t-CHC, as observed in their individual copolymer depolymerization (Table S4, entries 1 vs. 15, entries 2 vs. 16). Depolymerization of a terpolymer comprising nearly equal amounts of BnGE and CHO carbonate units released BnGE (99%) faster than CHO (73%) (Table S4, entry 5).Higher catalyst inputs resulted in complete and faster conversion to cyclic carbonates without any detectable epoxide residues, indicating high activity and high catalytic selectivity (Table S4, entries 7, 8 and 10). Terpolymers with a 0.80 molar fraction of BnGE carbonate unit underwent efficient depolymerization regardless of their molar mass (Mn = 6.6–18.3 kg mol−1), affording both cyclic carbonates accordingly to their original composition ratio (Table S4, entries 3 and 4). Switching from acetonitrile to toluene (111 °C) significantly decreased the depolymerization rate, while deleteriously impacting the formation of t-CHC (Table S4, entries 7, 8, 11 and 12). Screening the DBU-catalyzed depolymerization, even at higher organobase loadings (25 mol%), revealed a lower activity as compared to TBD (Table S4, entries 12 and 13). Overall, regardless of the terpolymer initial composition in BnGEC/CHC, TBD outperformed DBU. We assume this most likely arose from the presence of hydrogen-bonding interactions which stabilize the transition state, as evidenced by Kleij et al. using detailed DFT studies,53 thereby exhibiting superior catalytic efficiency and especially selectivity towards the 5CCs, with acetonitrile proving to be the most effective solvent.
Conclusion
The {Salphen}cobalt (4)/PPNCl and borane/PPNCl catalytic systems, successfully herein applied for the first time to the preparation of PBnGEC by ROCOP involving CO2 and BnGE, outperformed the discrete aluminum and iron metal-based PPNCl alike catalytic systems. Indeed, the latter ones exclusively produced the corresponding 5CCOBn cyclic carbonate, despite these same catalysts previously proved quite effective for other related ROCOP reactions. Optimization of the reaction conditions ([monomer]0/[activator]0/[initiator]0 = 100:6:1, 40 °C, PCO2 = 30 bar, 15 h) enabled to reproducibly synthesize CO2/BnGE copolymers with high contents of carbonate linkage and highlighted the significant role of dilution in governing selectivity at higher monomer loading (the latter condition returning 5CCOBn). The {Salphen}CoCl/PPNCl catalyst system also selectively afforded PBnGEC polycarbonates from the ROCOP of CO2 and BnGE. While this cobalt catalyst endured large amounts of monomer (1000 equiv.) and returned high molar mass (∼20 kg mol−1) PBnGEC with high chemoselectivity (80%), regioselectivity (>99%), and carbonate content (∼85%), the borane-based catalyst required lower monomer amounts (<100–250 equiv.) to achieve optimal performance, that is higher chemoselectivity (>98%) but relatively lower regioselectivity (∼84%) with moderate carbonate content (∼85%) and molar mass (∼9 kg mol−1).
In the terpolymerization of CO2/BnGE/CHO, the {Salphen}CoCl/PPNCl system was found to be also effective at low (0.1 mol%) catalyst loading, provided BnGE was loaded in equal or higher amounts than CHO, returning terpolymers in high yields and selectivity. However, it proved ineffective for the terpolymerization in the presence of higher equivalents of CHO against BnGE. Notably, the CO2/BnGE/CHO ROCOP catalyzed by BEt3/PPNCl successfully enabled to prepare the corresponding terpolymers with quantitative monomers consumption, yet with a large (6 mol%) BEt3 loading. The recovered P(BnGEC-co-CHC) terpolymers displayed a composition in PBnGEC and PCHC units closely matching the monomers initial loadings with fBnGE = 0.05–0.78.
Subsequent hydrogenolysis of the polycarbonates using Pd/C returned the corresponding terpolymers with hydroxyl pendant functionalities. Benzyl deprotection efficiency and preservation of polymer architecture were evidenced to be strongly dependent on the benzyloxy content. The observed lowering of molar masses for copolymers containing more than 15 mol% of benzyloxy/hydroxy pendant groups indicates a self-immolative materials-like behavior; this contrasts with previous literature reports claiming integrity of the polymer main-chain/backbone under similar hydrogenolysis conditions.
The P(BnGEC-co-CHC) colorless samples revealed either powdery or sticky soft solids depending on a low or high BnGE-to-CHO ratio, respectively. Accordingly, thermal analyses highlighted the possibility to tune the glass transition temperature according to the comonomers relative feeding ratio (Tg = 8–91 °C) thereby paving the way to various applications of such thermoplastic polymer materials. The thermal investigations further underlined the ability of polycarbonates to undergo backbiting at elevated temperatures. Benzyloxy deprotection of the copolymers significantly lowered the Tg values (by ∼20 °C) and generated less thermally stable alike copolymers (Td decreased by ∼25 °C).
Depolymerization of the P(BnGEC-co-CHC) catalyzed by the commercial organobase TBD rewardingly returned the two cyclic carbonates, 5CCOBn and t-CHC, highlighting the chemical recycling potential of the polymer materials into molecular resources/monomers retaining CO2. These findings provide strategies for the design, post-functionalization, and depolymerization of polycarbonates with tailored thermal and mechanical properties.
Author contributions
Nishant Chaudhary: investigation, formal analysis, data curation, writing original draft. A. Stephen K. Hashmi: funding acquisition, conceptualization. Jean-François Carpentier and Sophie Guillaume: funding acquisition, conceptualization, supervision, formal analysis, writing final draft.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: complementing data on the synthesis and depolymerization of the polymers, NMR, SEC, MALDI-ToF MS, DSC, TGA characterizations of polymers. See DOI: https://doi.org/10.1039/d5py01191d.Data will be made available upon reasonable request to the corresponding author.
Acknowledgements
This work was performed with the contribution of European Union Grant MSCA-DN-DCarbonize 101073223 (Ph.D. grant to N.C.), Université de Rennes and CNRS.References
- F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Designing a Circular Carbon and Plastics Economy for a Sustainable Future, Nature, 2024, 626, 45–57 CrossRef CAS PubMed.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, The Technological and Economic Prospects for CO2 Utilization and Removal, Nature, 2019, 575, 87–97 Search PubMed.
- G.-W. Yang, R. Xie, Y.-Y. Zhang, C.-K. Xu and G.-P. Wu, Evolution of Copolymers of Epoxides and CO2: Catalysts, Monomers, Architectures, and Applications, Chem. Rev., 2024, 124, 12305–12380 CrossRef CAS PubMed.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Advances in the Use of CO2 as a Renewable Feedstock for the Synthesis of Polymers, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- C. Zhang, X. Geng, X. Zhang, Y. Gnanou and X. Feng, Alkyl Borane-Mediated Metal-Free Ring-Opening (Co)Polymerizations of Oxygenated Monomers, Prog. Polym. Sci., 2023, 136, 101644 Search PubMed.
- S. Inoue, H. Koinuma and T. Tsuruta, Copolymerization of Carbon Dioxide and Epoxide, J. Polym. Sci., B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- S. Inoue, H. Koinuma and T. Tsuruta, Copolymerization of Carbon Dioxide and Epoxide with Organometallic Compounds, Makromol. Chem., 1969, 130, 210–220 CrossRef CAS.
- H. Wang, F. Xu, Z. Zhang, M. Feng, M. Jiang and S. Zhang, Bio-Based Polycarbonates: Progress and Prospects, RSC Sustainability, 2023, 1, 2162–2179 Search PubMed.
- G. Rosetto, F. Vidal, T. M. McGuire, R. W. F. Kerr and C. K. Williams, High Molar Mass Polycarbonates as Closed-Loop Recyclable Thermoplastics, J. Am. Chem. Soc., 2024, 146, 8381–8393 CrossRef CAS PubMed.
- D. J. Darensbourg and M. W. Holtcamp, Catalytic Activity of Zinc(II) Phenoxides Which Possess Readily Accessible Coordination Sites. Copolymerization and Terpolymerization of Epoxides and Carbon Dioxide, Macromolecules, 1995, 28, 7577–7579 CrossRef CAS.
- D. J. Darensbourg, Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- Y. Wang and D. J. Darensbourg, Carbon Dioxide-Based Functional Polycarbonates: Metal Catalyzed Copolymerization of CO2 and Epoxides, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, Single-Site β-Diiminate Zinc Catalysts for the Alternating Copolymerization of CO2 and Epoxides: Catalyst Synthesis and Unprecedented Polymerization Activity, J. Am. Chem. Soc., 2001, 123, 8738–8749 CrossRef CAS PubMed.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Highly Active Dizinc Catalyst for the Copolymerization of Carbon Dioxide and Cyclohexene Oxide at One Atmosphere Pressure, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed.
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, Co(III)/Alkali-Metal(I) Heterodinuclear Catalysts for the Ring-Opening Copolymerization of CO2 and Propylene Oxide, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed.
- H. Cao, S. Liu and X. Wang, Environmentally Benign Metal Catalyst for the Ring-Opening Copolymerization of Epoxide and CO2: State-of-the-Art, Opportunities, and Challenges, Green Chem. Eng., 2022, 3, 111–124 Search PubMed.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, Multifunctional Catalysts for Ring-Opening Copolymerizations, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- W. T. Diment, W. Lindeboom, F. Fiorentini, A. C. Deacy and C. K. Williams, Synergic Heterodinuclear Catalysts for the Ring-Opening Copolymerization (ROCOP) of Epoxides, Carbon Dioxide, and Anhydrides, Acc. Chem. Res., 2022, 55, 1997–2010 CrossRef CAS PubMed.
- S. S, J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, A Highly Active and Recyclable Catalytic System for CO2/Propylene Oxide Copolymerization, Angew. Chem., Int. Ed., 2008, 47, 7306–7309 CrossRef PubMed.
- B. Bahramian, Y. Ma, R. Rohanizadeh, W. Chrzanowski and F. Dehghani, A New Solution for Removing Metal-Based Catalyst Residues from a Biodegradable Polymer, Green Chem., 2016, 18, 3740–3748 Search PubMed.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Metal-Free Alternating Copolymerization of CO2 with Epoxides: Fulfilling “Green”, Synthesis and Activity, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- D.-D. Zhang, X. Feng, Y. Gnanou and K.-W. Huang, Theoretical Mechanistic Investigation into Metal-Free Alternating Copolymerization of CO2 and Epoxides: The Key Role of Triethylborane, Macromolecules, 2018, 51, 5600–5607 CrossRef CAS.
- D. Zhang, H. Zhang, N. Hadjichristidis, Y. Gnanou and X. Feng, Lithium-Assisted Copolymerization of CO2/Cyclohexene Oxide: A Novel and Straightforward Route to Polycarbonates and Related Block Copolymers, Macromolecules, 2016, 49, 2484–2492 CrossRef CAS.
- N. G. Patil, S. K. Boopathi, P. Alagi, N. Hadjichristidis, Y. Gnanou and X. Feng, Carboxylate Salts as Ideal Initiators for the Metal-Free Copolymerization of CO2 with Epoxides: Synthesis of Well-Defined Polycarbonates Diols and Polyols, Macromolecules, 2019, 52, 2431–2438 Search PubMed.
- J. L. Yang, H. L. Wu, Y. Li, X. H. Zhang and D. J. Darensbourg, Perfectly alternating and regioselective copolymerization of carbonyl sulfide and epoxides by metal–free Lewis pairs, Angew. Chem., Int. Ed., 2017, 56, 5774–5779 CrossRef CAS PubMed.
- K. A. Andrea and F. M. Kerton, Triarylborane-Catalyzed Formation of Cyclic Organic Carbonates and Polycarbonates, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS.
- Y. Wang, J.-Y. Zhang, J.-L. Yang, H.-K. Zhang, J. Kiriratnikom, C.-J. Zhang, K.-L. Chen, X.-H. Cao, L.-F. Hu, X.-H. Zhang and B. Z. Tang, Highly Selective and Productive Synthesis of a Carbon Dioxide-Based Copolymer upon Zwitterionic Growth, Macromolecules, 2021, 54, 2178–2186 CrossRef CAS.
- Y. Wang, Z. Liu, W. Guo, C. Zhang and X. Zhang, Phosphine-Borane Frustrated Lewis Pairs for Metal-Free CO2/Epoxide Copolymerization, Macromolecules, 2023, 56, 4901–4909 CrossRef CAS.
- M. Jia, D. Zhang, G. W. de Kort, C. H. R. M. Wilsens, S. Rastogi, N. Hadjichristidis, Y. Gnanou and X. Feng, All-Polycarbonate Thermoplastic Elastomers Based on Triblock Copolymers Derived from Triethylborane-Mediated Sequential Copolymerization of CO2 with Various Epoxides, Macromolecules, 2020, 53, 5297–5307 CrossRef CAS PubMed.
- G.-W. Yang, Y.-Y. Zhang, R. Xie and G.-P. Wu, Scalable Bifunctional Organoboron Catalysts for Copolymerization of CO2 and Epoxides with Unprecedented Efficiency, J. Am. Chem. Soc., 2020, 142, 12245–12255 CrossRef CAS PubMed.
- Y.-Y. Zhang, G.-W. Yang, R. Xie, X.-F. Zhu and G.-P. Wu, Sequence-Reversible Construction of Oxygen-Rich Block Copolymers from Epoxide Mixtures by Organoboron Catalysts, J. Am. Chem. Soc., 2022, 144, 19896–19909 Search PubMed.
- G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, X.-F. Zhu and G.-P. Wu, Pinwheel-Shaped Tetranuclear Organoboron Catalysts for Perfectly Alternating Copolymerization of CO2 and Epichlorohydrin, J. Am. Chem. Soc., 2021, 143, 3455–3465 Search PubMed.
- J. Geschwind and H. Frey, Poly(1,2-Glycerol Carbonate): A Fundamental Polymer Structure Synthesized from CO2 and Glycidyl Ethers, Macromolecules, 2013, 46, 3280–3287 CrossRef CAS.
- J. Hilf, A. Phillips and H. Frey, Poly(Carbonate) Copolymers with a Tailored Number of Hydroxyl Groups from Glycidyl Ethers and CO2, Polym. Chem., 2014, 5, 814–818 Search PubMed.
- H. Zhang and M. W. Grinstaff, Terpolymerization of Benzyl Glycidyl Ether, Propylene Oxide, and CO2 Using Binary and Bifunctional [Rac–SalcyCoIII X] Complexes and the Thermal and Mechanical Properties of the Resultant Poly(Benzyl 1,2−glycerol–co–propylene Carbonate)s and Poly(1,2−glycerol–co–propylene Carbonate)s, J. Appl. Polym. Sci., 2014, 131, 39893 Search PubMed.
- H. Zhang and M. W. Grinstaff, Synthesis of Atactic and Isotactic Poly(1,2-Glycerol Carbonate)s: Degradable Polymers for Biomedical and Pharmaceutical Applications, J. Am. Chem. Soc., 2013, 135, 6806–6809 Search PubMed.
- H. Plommer, L. Stein, J. N. Murphy, N. Ikpo, N. Mora-Diez and F. M. Kerton, Copolymerization of CHO/CO2 Catalyzed by a Series of Aluminum Amino-Phenolate Complexes and Insights into Structure–Activity Relationships, Dalton Trans., 2020, 49, 6884–6895 Search PubMed.
- A. M. Reckling, D. Martin, L. N. Dawe, A. Decken and C. M. Kozak, Structure and C–C Cross-Coupling Reactivity of Iron(III) Complexes of Halogenated Amine-Bis(Phenolate), Ligands, J. Organomet. Chem., 2011, 696, 787–794 Search PubMed.
- K. A. Andrea, E. D. Butler, T. R. Brown, T. S. Anderson, D. Jagota, C. Rose, E. M. Lee, S. D. Goulding, J. N. Murphy, F. M. Kerton and C. M. Kozak, Iron Complexes for Cyclic Carbonate and Polycarbonate Formation: Selectivity Control from Ligand Design and Metal-Center Geometry, Inorg. Chem., 2019, 58, 11231–11240 Search PubMed.
- W.-Z. Wang, K.-Y. Zhang, X.-G. Jia, L. Wang, L.-L. Li, W. Fan and L. Xia, A New Dinuclear Cobalt Complex for Copolymerization of CO2 and Propylene Oxide: High Activity and Selectivity, Molecules, 2020, 25, 4095 CrossRef CAS PubMed.
- M. Jia, N. Hadjichristidis, Y. Gnanou and X. Feng, Monomodal Ultrahigh-Molar-Mass Polycarbonate Homopolymers and Diblock Copolymers by Anionic Copolymerization of Epoxides with CO2, ACS Macro. Lett., 2019, 8, 1594–1598 Search PubMed.
- G.-P. Wu and D. J. Darensbourg, Mechanistic Insights into Water-Mediated Tandem Catalysis of Metal-Coordination CO2/Epoxide Copolymerization and Organocatalytic Ring-Opening Polymerization: One-Pot, Two Steps, and Three Catalysis Cycles for Triblock Copolymers Synthesis, Macromolecules, 2016, 49, 807–814 Search PubMed.
- Z. Chen, J.-L. Yang, X.-Y. Lu, L.-F. Hu, X.-H. Cao, G.-P. Wu and X.-H. Zhang, Triethyl Borane-Regulated Selective Production of Polycarbonates and Cyclic Carbonates for the Coupling Reaction of CO2 with Epoxides, Polym. Chem., 2019, 10, 3621–3628 Search PubMed.
- M. Taherimehr and P. P. Pescarmona, Green Polycarbonates Prepared by the Copolymerization of CO2 with Epoxides, J. Appl. Polym. Sci., 2014, 131, 41141 Search PubMed.
- S. Naumann, Borane Catalysis for Epoxide (Co)Polymerization, Polym. Chem., 2023, 14, 1834–1862 Search PubMed.
- C.-K. Xu, C. Lu, S. Zhao, G.-W. Yang, W. Li, J. Wang and G.-P. Wu, Rational optimization of ammonium and phosphonium cations of Bifunctional organoborane catalysts for copolymerization of propylene oxide with CO2 to afford poly(propylene carbonate), Macromolecules, 2024, 57, 9076–9087 Search PubMed.
- P. Verkoyen and H. Frey, Amino-Functional Polyethers: Versatile, Stimuli-Responsive Polymers, Polym. Chem., 2020, 11, 3940–3950 Search PubMed.
- R. Matthes and H. Frey, Polyethers Based on Short-Chain Alkyl Glycidyl Ethers: Thermoresponsive and Highly Biocompatible Materials, Biomacromolecules, 2022, 23, 2219–2235 CrossRef CAS PubMed.
- M. Scharfenberg, J. Hilf and H. Frey, Functional Polycarbonates from Carbon Dioxide and Tailored Epoxide Monomers: Degradable Materials and Their Application Potential, Adv. Funct. Mater., 2018, 28, 1704302 Search PubMed.
- G. A. Luinstra and E. Borchardt, Material Properties of Poly(Propylene Carbonates), in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 29–48 Search PubMed.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Organocatalysis for depolymerisation, Polym. Chem., 2019, 10, 172–186 Search PubMed.
- T. Senthamarai, E. Lanaro, J. Tinker, A. Buchard and A. W. Kleij, Synthesis and depolymerization studies of biohybrid polycarbonates derived from terpenes, Polym. Chem., 2025, 16, 2784–2790 Search PubMed.
- D. H. Lamparelli, A. Villar-Yanez, L. Dittrich, J. Rintjema, F. Bravo, C. Bo and A. W. Kleij, Bicyclic guanidine promoted mechanistically divergent depolymerization and recycling of a biobased polycarbonate, Angew. Chem., Int. Ed., 2023, 62, e202314659 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |