
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of biobased (co)polyesters derived from cyclic monomers: camphoric acid and 1,4-cyclohexanedimethanol
Syaiful
Ahsan
 ab,
Fitrilia
Silvianti
c,
Cornelis
Post
ad,
Vincent S. D.
Voet
d,
Rudy
Folkersma
d,
Jeffy
Joji
e,
Louis M.
Pitet
e,
Subin
Damodaran
f,
Katja
Loos
a and
Dina
Maniar
*a
ab,
Fitrilia
Silvianti
c,
Cornelis
Post
ad,
Vincent S. D.
Voet
d,
Rudy
Folkersma
d,
Jeffy
Joji
e,
Louis M.
Pitet
e,
Subin
Damodaran
f,
Katja
Loos
a and
Dina
Maniar
*a
aMacromolecular Chemistry & New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 3, 9747 AG, Groningen, the Netherlands. E-mail: d.maniar@rug.nl
bPoliteknik STMI Jakarta, the Ministry of Industry of the Republic of Indonesia, Jl. Letjen Suprapto 26, Jakarta, Indonesia
cLife Science Department, Academy Tech & Design, NHL Stenden University of Applied Sciences, Van Schaikweg 94, 7811 KL, Emmen, the Netherlands
dCircular Plastics, Academy Tech & Design, NHL Stenden University of Applied Sciences, Van Schaikweg 94, 7811 KL, Emmen, the Netherlands
eAdvanced Functional Polymers (AFP) Laboratory, Institute for Materials Research (imo-imomec), Hasselt University, Martelarenlaan 42, 3500 Hasselt, Belgium
fTosoh Bioscience GmbH, Im Leuschnerpark 4, 64347 Griesheim, Germany
First published on 28th January 2026
Abstract
Annual plastic production volumes are more than 400 million tons and are anticipated to continue increasing over the next decade. The majority of plastics originate from fossil resources. Limited raw material reserves and ongoing utilization of plastics contribute to elevated CO2 emissions, ultimately contributing to climate change. Development of green polymers (i.e., biobased) is one way to reduce our environmental impact. Using renewable resources as raw materials for polymer synthesis reduces the reliance on petroleum and in some cases enables recycling and/or biodegradation. Various aliphatic biobased polyesters have been studied; however, they typically have low glass transition temperatures (Tg) and poor thermomechanical performance, which may limit their applications. In this work, we investigate the synthesis route and structure–property relationships of (co)polyesters from cyclic biobased monomers, camphoric acid and 1,4-cyclohexanedimethanol (CHDM). We observed that increasing the reaction temperature and extending the reaction time led to increased molecular weight and yield of poly(cyclohexanedimethylene camphorate) (PCHC). Conversely, substituting p-toluenesulfonic acid (p-TSA) with a titanium(IV) isopropoxide (TTIP) catalyst led to reductions in both the molecular weight and yield. Furthermore, (co)polyesters with Tg values ranging from –29 to +56 °C were successfully synthesized. DSC and WAXD analyses suggest that the polyesters derived from camphoric acid and the linear diols were amorphous, whereas those based on CHDM were semicrystalline. This work helps address existing knowledge gaps in biobased polymer development by introducing cyclic biobased monomers that expand the current library of renewable materials, thereby broadening opportunities for advanced applications such as coating and packaging materials.
Introduction
The predominant monomers utilized in plastic production come from fossil sources, with an increase of global plastic production from 371 million tons (Mt) in 2018 to 414 Mt in 2023, contributing to an increase in CO2 emissions with its ongoing use.1,2 Currently, one approach to address this issue is the development of green polymers.3,4 Green polymer chemistry is characterized by 12 principles, including two primary features: the utilization of renewable resources as raw materials or building blocks for polymer synthesis and the adoption of an environmentally friendly approach for synthesis.5 Recently, significant progress has been made in the development of biobased polymers derived from sustainable sources, with increasing interest focused on linear aliphatic polyesters for their biodegradability and on semiaromatic polyesters for their enhanced thermal and mechanical properties. Numerous studies have explored diverse aliphatic biobased monomers and polymerization techniques to optimize their mechanical properties, thermal stability, and degradation behavior, as well as improve the sustainability of the synthetic route.6–9 However, these aliphatic polyesters are typically semicrystalline with low glass transition temperatures (Tg) and limited thermomechanical performance, restricting their utilization for applications demanding high optical clarity, thermal stability, and impact resistance. As a result, designing and synthesizing rigid and amorphous polymers represents a promising strategy for developing biobased materials with enhanced properties.10The incorporation of rigid cyclic monomers into polymer backbones is a promising approach to improve the polymer chain stiffness and thermomechanical properties of amorphous polymers. In this context, biobased cyclic monomers have received increasing attention, with camphoric acid from camphor gaining significant interest as a renewable building block for the synthesis of biobased polyesters. Camphor is one of the most widely used commercial aroma chemicals and can be obtained by distilling wood from the camphor laurel tree (Cinnamomum camphora).11 It serves as a valuable starting material in the synthesis of a variety of useful chemicals.12–14 However, only a limited number of studies have explored the polymerization of camphor-derived monomers.15–20 The oxidation of the bicyclic terpene (1R)-(+)-camphor results in the production of biobased rigid, five-membered ring diacids known as (1R,3S)-(+)-camphoric acid.15–19 The bioavailability, scalability, and nontoxic nature of camphoric acid make it an appealing platform for sustainable polymer design.21 Its rigid structure facilitates the production of polymers with enhanced thermal and mechanical performance, as demonstrated by Ouhichi et al., who developed UV-curable polyester resins from camphoric acid, itaconic acid, and 1,6-hexanediol for coating applications.16 Guidotti et al. synthesized polyester from camphoric acid, butanediol, and another rigid diacid, achieving high barrier properties and mechanical resistance up to 31 MPa, which is suitable for food packaging.17,18 Additionally, Hu et al. reported a novel biobased epoxy resin from camphoric acid with superior ultraviolet resistance, impact strength, and toughness, offering a sustainable alternative to commercial bisphenol A-based epoxy resins.20 Moreover, Nsengiyumva and Miller demonstrated the degradability of polyesters derived from camphoric acid through water-degradation studies, further emphasizing their potential for sustainable polymer applications.15
Another cyclic monomer, 1,4-cyclohexanedimethanol (CHDM), has attracted our interest as a valuable cyclic diol for the synthesis of high-performance polyesters. It is traditionally synthesized through a two-step industrial process, wherein dimethyl terephthalate (DMT) is first converted to dimethyl 1,4-cyclohexanedicarboxylate (DMCD), which is subsequently hydrogenated to yield CHDM.22 Alternatively, one-pot direct conversion of DMT to CHDM can be employed.23 It can also be synthesized from the upcycling of poly(ethylene terephthalate) (PET) waste through tandem reactions utilizing metal catalysts.24,25 In an effort to explore possible renewable routes to CHDM, the monomer was synthesized from plant-based acrylate and acetaldehyde, which are alicyclic glycol groups.26 It is commonly used to alter the physical characteristics of PET.27 CHDM was also utilized to increase the melting temperature and glass transition temperature of poly(butylene 2,5-furandicarboxylate) (PBF), a recognized semicrystalline furanic polyester, which is considered a promising renewable alternative to poly(butylene terephthalate) (PBT) for producing food packaging materials.28 Similar to that of camphoric acid, the rigid structure of CHDM has been reported to promote the production of polyesters, which exhibit high thermomechanical properties and excellent barrier properties.29,30 Interestingly, Tsai et al. investigated the enzymatic hydrolysis of CHDM-based copolyesters and reported that these polymers undergo surface erosion through random endo-type scission, with the rate of degradation being influenced primarily by surface hydrophilicity rather than crystallinity.31 Finally, although polymers based on camphoric acid and CHDM have been studied individually, to our knowledge, no investigations have explored polymers derived from their combination. Moreover, a comprehensive understanding of their reaction kinetics, thermomechanical behavior, and biodegradability remains limited. Thus, camphoric acid and CHDM represent promising biobased precursors for synthesizing rigid and amorphous polyesters that combine excellent thermomechanical performance with potential biodegradability.
In this work, we report the synthesis and characterization of biobased (co)polyesters derived from camphoric acid and CHDM. Two aliphatic diols, ethylene glycol (EG) and 1,8-octanediol (OD), as well as cyclic diol CHDM, were used to produce the corresponding polyesters poly(ethylene camphorate) (PEC), poly(octamethylene camphorate) (POC), and poly(cyclohexanedimethylene camphorate) (PCHC), along with two copolyesters: poly(cyclohexanedimethylene-co-ethylene camphorate) (P(CHC-co-EC)) and poly(cyclohexanedimethylene-co-octamethylene camphorate) (P(CHC-co-OC)). The variations in these (co)polyesters were explored to understand how the presence of cyclic-aliphatic and cyclic-cyclic polyester structures, as well as their counterpart copolyester polymer chains, influences their properties. The polymerization method was thoroughly investigated, including a kinetic study to examine the distinct catalytic activities and the effects of catalyst variations. To explore potential applications, the correlation between the structure and properties of the obtained polymers was also investigated.
Results and discussion
Synthesis of camphoric acid-based polyesters
Camphoric acid-based polyesters were synthesized via a two-step melt polycondensation process—a bulk polymerization method widely favored in industry due to its operational simplicity and efficiency.32 Unlike solvent-based polymerizations, which typically require multiple washing steps and are associated with environmental and economic concerns,33 the solvent-free approach minimizes such drawbacks. Additionally, reducing or eliminating the solvent content can suppress undesired hydrolysis.34 The synthesis was based on a modified version of the method reported by Nsengiyumva and Miller for PEC,15 building upon their approach to expand the scope of camphoric acid–based polyesters. Here, the method was adapted to produce two additional novel polyesters, including one incorporating the cyclic diol CHDM (Table 1). All three camphoric acid-based polyesters were synthesized using a camphoric acid/diol molar ratio of 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 (Scheme 1). This ratio was used to compensate for the tendency of diols to evaporate under high-temperature and low-pressure conditions. Polymerization was carried out for 3 or 24 hours at 180 °C in a nitrogen atmosphere, followed by 19 or 7 hours at 2 mmHg when the temperature was increased to 200 or 230 °C. High temperature and vacuum removed water and residual diol, thereby increasing the molecular weight. The products comprised two dark brown sticky polymers (PEC and POC) and one light brown solid polymer (PCHC) (Fig. S12). A summary of the synthesized polyesters is presented in Table 1.
:1.2 (Scheme 1). This ratio was used to compensate for the tendency of diols to evaporate under high-temperature and low-pressure conditions. Polymerization was carried out for 3 or 24 hours at 180 °C in a nitrogen atmosphere, followed by 19 or 7 hours at 2 mmHg when the temperature was increased to 200 or 230 °C. High temperature and vacuum removed water and residual diol, thereby increasing the molecular weight. The products comprised two dark brown sticky polymers (PEC and POC) and one light brown solid polymer (PCHC) (Fig. S12). A summary of the synthesized polyesters is presented in Table 1.
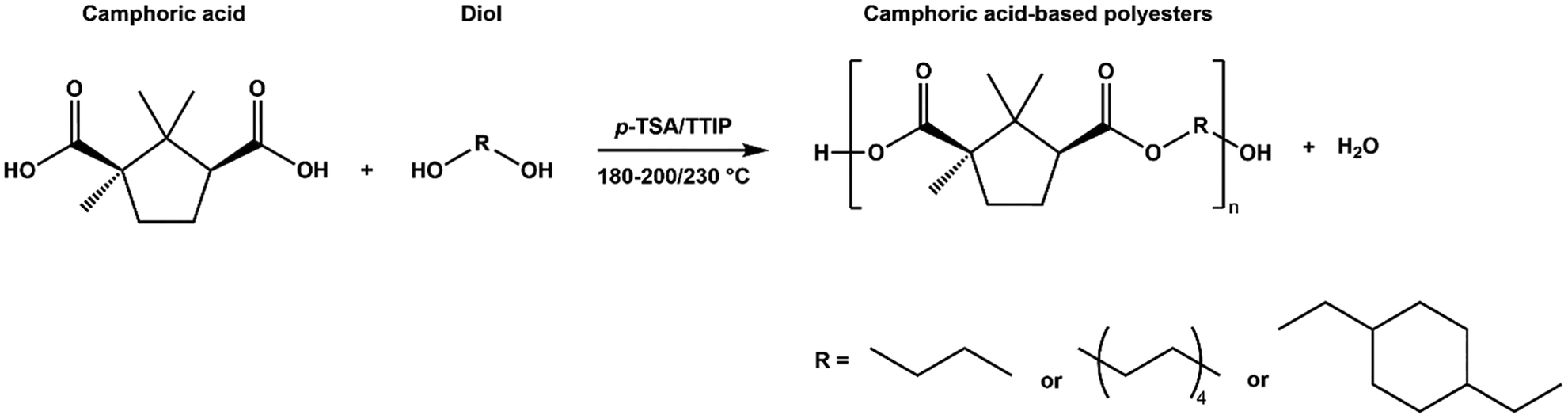 | ||
| Scheme 1 Bulk polymerization of camphoric acid and various diols using either p-TSA or TTIP as catalysts. | ||
| Abbreviation | Structure | Reaction conditionsa |
|
|
Đ | y (%) |
|---|---|---|---|---|---|---|
| a Catalyst type, esterification and polycondensation temperature range, and total reaction time. b Molecular weights were determined by SEC in chloroform. c Isolated yield. | ||||||
| PEC |
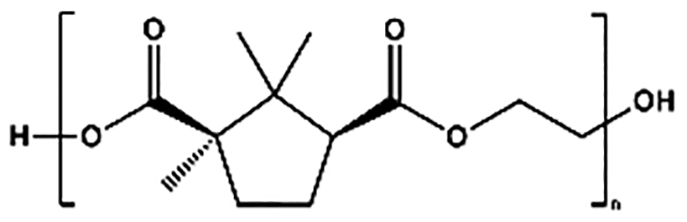
|
p-TSA, 180–200 °C, 22 h | 14000 |
23200 |
1.7 | 43 |
| POC |
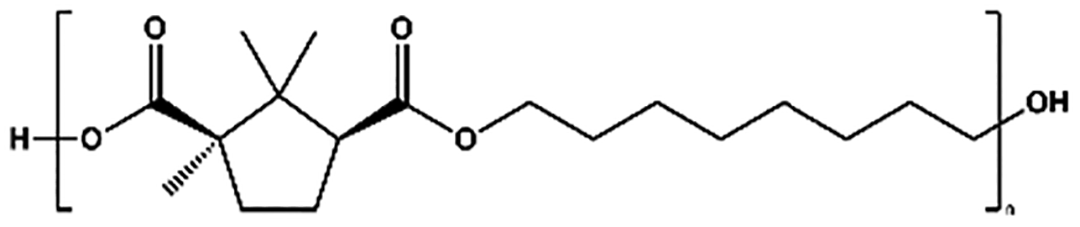
|
p-TSA, 180–200 °C, 22 h | 14700 |
49500 |
3.4 | 63 |
| PCHC |
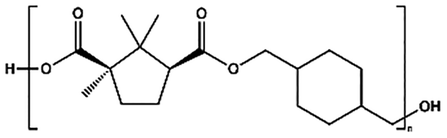
|
p-TSA, 180–200 °C, 22 h | 4500 |
8600 |
1.9 | 65 |
| PCHC |
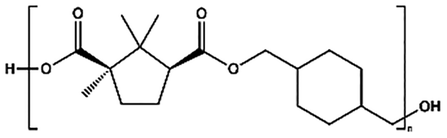
|
p-TSA, 180–230 °C, 31 h | 7800 |
18700 |
2.4 | 79 |
| PCHC |
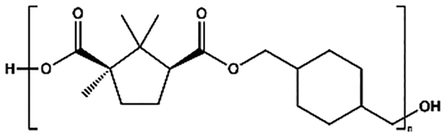
|
TTIP, 180–230 °C, 31 h | 5800 |
13500 |
2.3 | 48 |


The chemical structures of the obtained polyesters were studied via ATR-FTIR and 1H-NMR spectroscopy. The detailed NMR and IR peak assignments are available in the Experimental section. Fig. 1a shows the ATR-FTIR spectra of the obtained polyesters, confirming the formation of ester linkages through the appearance of an absorption band at 1720–1722 cm−1, which corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of the ester group. This band represents a shift from the CO stretching vibration of the carboxylic acid group in camphoric acid (CA), which was originally observed at 1690 cm−1, indicating successful esterification. Additionally, strong absorption bands in the range of 991–1257 cm−1 are observed in all the polyester spectra, corresponding to the asymmetric and symmetric stretching vibrations of C–O bonds. The FTIR spectrum of camphoric acid shows a broad absorption band between 2400–3250 cm−1, attributed to O–H stretching vibrations of hydroxyl groups. This broad band is absent in the spectra of the polyesters, which only show C–H stretching vibrations at 2892–2970 cm−1, further supporting the complete conversion of hydroxyl and carboxylic acid groups into ester functionalities.
O vibration of the ester group. This band represents a shift from the CO stretching vibration of the carboxylic acid group in camphoric acid (CA), which was originally observed at 1690 cm−1, indicating successful esterification. Additionally, strong absorption bands in the range of 991–1257 cm−1 are observed in all the polyester spectra, corresponding to the asymmetric and symmetric stretching vibrations of C–O bonds. The FTIR spectrum of camphoric acid shows a broad absorption band between 2400–3250 cm−1, attributed to O–H stretching vibrations of hydroxyl groups. This broad band is absent in the spectra of the polyesters, which only show C–H stretching vibrations at 2892–2970 cm−1, further supporting the complete conversion of hydroxyl and carboxylic acid groups into ester functionalities.
 | ||
| Fig. 1 (a) ATR-FTIR specctra of camphoric acid (CA) and the PEC, POC, and PCHC. (1) O–H stretching, (2) CO stretching, and (3) C–O stretching. (b) 1H-NMR spectrum of PCHC in CDCl3 as the solvent. | ||
The polymeric structure was further confirmed by 1H-NMR spectroscopy. A representative spectrum of PCHC is shown in Fig. 1b, and those of PEC and POC are provided in the SI (Fig. S1 and S2, respectively). The successful formation of ester linkages is evidenced by the appearance of signals at 4.00 ppm (g – CHDM, CH2O, cis) and 3.91 ppm (g – CHDM, CH2O, trans). These peaks shift downfield from the CH2OH proton signals of the CHDM monomer, which were originally observed at 3.51 ppm (cis-1,4-CHDM) and 3.43 ppm (trans-1,4-CHDM) (Fig. S5), consistent with the conversion of the hydroxyl groups into ester functionalities. The residual signals at 3.53 ppm (CH2OH, cis) and 3.46 ppm (CH2OH, trans) are assigned to the end groups (g′), suggesting the presence of hydroxyl functionalities at the polymer chain ends, as previously reported.30
Table 1 lists the abbreviations, polymer structures, molecular weights, dispersities (Đ), and yields of the synthesized polyesters. For polyesters based on linear diol comonomers, PEC and POC, comparable number-average molecular weights  of 14000 and 14700 g mol−1, respectively, were obtained. However, POC exhibited a significantly higher weight-average molecular weight
of 14000 and 14700 g mol−1, respectively, were obtained. However, POC exhibited a significantly higher weight-average molecular weight 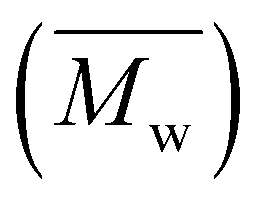 , leading to a high dispersity of 3.4, suggesting a broader molecular weight distribution, likely caused by side reactions or non-uniform mixing as the viscosity increased during polymerization. In contrast, the polyester obtained from the polymerization of camphoric acid with the cyclic diol comonomer CHDM (PCHC) had a considerably lower
, leading to a high dispersity of 3.4, suggesting a broader molecular weight distribution, likely caused by side reactions or non-uniform mixing as the viscosity increased during polymerization. In contrast, the polyester obtained from the polymerization of camphoric acid with the cyclic diol comonomer CHDM (PCHC) had a considerably lower  value of 4500 g mol−1. The difficulty in removing excess CHDM during condensation, compared to EG or OD, might be a contributing factor. In addition to that, it could also indicate that the bulky structure of cyclic diols (i.e., CHDM) decreases the reaction rate.35 Compared with the work of Nsengiyumva and Miller, who also reported that PEC and related camphoric acid–based polyesters from C2–C6 diols with
value of 4500 g mol−1. The difficulty in removing excess CHDM during condensation, compared to EG or OD, might be a contributing factor. In addition to that, it could also indicate that the bulky structure of cyclic diols (i.e., CHDM) decreases the reaction rate.35 Compared with the work of Nsengiyumva and Miller, who also reported that PEC and related camphoric acid–based polyesters from C2–C6 diols with 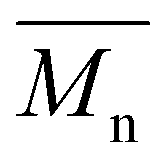 values of up to 20200 g mol−1 and Đ ranging from 2.7–4.0,15 our synthesis employed milder conditions; shorter reaction times and lower temperatures; to reduce energy demand and improve sustainability. Under these milder conditions, PEC showed a lower
values of up to 20200 g mol−1 and Đ ranging from 2.7–4.0,15 our synthesis employed milder conditions; shorter reaction times and lower temperatures; to reduce energy demand and improve sustainability. Under these milder conditions, PEC showed a lower 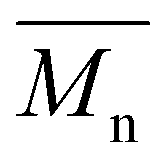 but narrower dispersity of 1.7. Nevertheless, this approach enables the synthesis of novel POC (C8) and PCHC, expanding the range of accessible camphoric acid–based polyesters.
but narrower dispersity of 1.7. Nevertheless, this approach enables the synthesis of novel POC (C8) and PCHC, expanding the range of accessible camphoric acid–based polyesters.
The use of different catalysts and reaction conditions was further investigated to optimize PCHC synthesis. Two catalysts, the organocatalyst p-TSA and the metal-based catalyst TTIP, were used and evaluated. TTIP has shown exceptional catalytic activity for polyester synthesis, especially in the production of moderate to high-molecular-weight polyesters, making it a superior choice compared with other metal catalysts.36–38 TTIP offers the advantage of high efficiency while maintaining the desired polymer properties.39 By comparison, p-TSA has proven to be highly effective in synthesizing a wide range of polyesters,40–42 including moderate to high-molecular-weight polyesters derived from camphoric acid.15 It is considered a green, efficient, and reusable catalyst that is also used to catalyze the synthesis of jasminaldehyde.43 We observed that for PCHC synthesis, in comparison with p-TSA, the use of TTIP as a catalyst resulted in a lower molecular weight and yield (Table 1). Although TTIP is commonly used as a catalyst for camphoric acid-based polyesters,17,18 the presence of water as a byproduct degrades TTIP to generate TiO2, which results in decreased catalytic activity.44 However, we observed that products with a brighter color were obtained from reactions catalyzed by TTIP, since strongly acidic catalysts (e.g., p-TSA) used to catalyze condensation reactions tend to promote discoloration (and hydrolysis of the reaction products can occur) if the catalyst is not neutralized and separated from the product.34 Furthermore, extended reaction times and elevated temperatures led to a significant increase in both 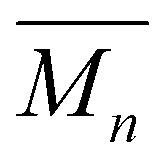 and yield, from 4500 to 7800 g mol−1 and from 65% to 79%, respectively.
and yield, from 4500 to 7800 g mol−1 and from 65% to 79%, respectively.
To gain deeper insights into the polymerization conditions of PCHC synthesis, the polymerization kinetics were studied by 1H-NMR and SEC. These investigations are important for understanding the growth dynamics of the polymer chain over time and can therefore provide fundamental information for optimizing the reaction conditions. The two-stage method was applied with the following reaction conditions: the reaction was first conducted for 3 hours in a nitrogen atmosphere, followed by 19 hours under vacuum at elevated temperatures of 190–200 °C with p-TSA as the catalyst. At selected time intervals, small amounts of the reaction mixture were withdrawn for 1H-NMR analysis. The 1H-NMR spectra from the kinetic studies are shown in Fig. 2. We observed that ester bonds were already produced within 30 minutes of reaction, as evidenced by the emergence of a new signal at 4.00 ppm (cis) and 3.91 ppm (trans) (g, highlighted in light green), corresponding to the methylene group next to the ester bond (COOCH2). Furthermore, the proton signal attributed to the methylene protons of the hydroxymethyl group (CH2OH) in the monomer CHDM exhibited a decrease in intensity and a shift from 3.55 ppm (cis) and 3.47 ppm (trans) to 3.54 ppm (cis) and 3.46 ppm (trans) (highlighted in light blue), indicating the conversion of the CHDM monomer into oligoesters. This monomer peak disappeared completely after 6 h (3 h vacuum), resulting in the residual signals of the end groups (g′). Similarly, protons b and c in the methyl groups shifted from 0.86 ppm to 0.78 ppm and from 1.25 ppm to 1.21 ppm, respectively. These upfield shifts suggest increased shielding of the protons, likely due to the consumption of electron-withdrawing end groups and their repositioning within the more electron-rich polymer backbone during the monomer-to-polymer conversion. We also observed an evolution in the ratio between these peaks throughout the reaction time, with the polymer-associated peaks increasing in intensity relative to the monomer peaks, indicating the progressive conversion of monomers into the corresponding polymer. However, when the polymerization was extended from 22 to 26 h, no significant change was observed in the peak intensities, indicating that the reaction was essentially complete. SEC analysis (Table S1 and Fig. S6) corroborated these findings, revealing a gradual increase in molecular weight during the initial 3 h, followed by significant growth after the pressure decreased at 6, 9, and 22 h. Collectively, the 1H-NMR and SEC results indicate that there is no significant difference between 22, 24, and 26 h, suggesting that a 22 h reaction at 200 °C is sufficient for complete polymerization.
 | ||
| Fig. 2 1H-NMR spectra of the mixture from the p-TSA-catalyzed polycondensations of camphoric acid and CHDM in bulk, carried out at 180 °C for the first 3 h under nitrogen and increased gradually to 200 °C under dynamic vacuum until 2 mmHg was reached. | ||
Thermal properties, crystallinity, and surface wettability of the camphoric acid-based polyesters
Table 2 summarizes the thermal properties and crystallinity of the synthesized camphoric acid-based polyesters, as determined by TGA, DSC, and WAXD. For polyesters containing linear diol comonomers, glass transition temperatures (Tg) of +11 °C and −29 °C were observed in the second DSC heating curves for PEC and POC, respectively (Fig. 3a). The Tg of PEC in this work was significantly lower than previously reported values (44 °C),15 which can be attributed to the substantially lower molecular weight obtained here. Furthermore, the Tg decreased progressively as the alkylene spacer length (n) in the linear diol comonomers increased from 2 to 8, which is consistent with observations by Nsengiyumva and Miller and other studies.15 This trend is due to the increased chain flexibility and mobility associated with longer alkylene segments. Both the DSC and WAXD (Fig. 5) results indicate that PEC and POC are amorphous, as no melting transitions or sharp diffraction peaks are detected. The asymmetrical structure of camphoric acid results in a random regiochemistry, which presumably prevents regular chain packing. Consequently, these polymers lack the long-range stereochemical and conformational order required for crystalline structures. Similarly, other studies have reported a decrease in crystallinity with increasing amounts of camphoric moieties.17,18 | ||
| Fig. 3 DSC thermograms of (a) the second heating curve of the PEC, POC, and PCHC and (b) the first heating, cooling, and second heating curves of the PCHC. | ||
| Polyesters | DSCa | TGAb | WAXDc | |||
|---|---|---|---|---|---|---|
| T m | T g | T d5% | T dmax | 2θ | χ c | |
| (°C) | (°C) | (°C) | (°C) | (°) | (%) | |
| a T m (melting temperature) was obtained from the first DSC heating scan; Tg (glass transition temperature) was obtained from the second DSC heating scan. b T d5% (decomposition temperature at 5% weight loss) and Tdmax (temperature at maximum rate of decomposition) were determined via TGA. c 2θ (diffraction peak) and χc (degree of crystallinity) were determined via WAXD. | ||||||
| PEC | — | 11 | 361 | 439 | — | 0 |
| POC | — | −29 | 271 | 409 | — | 0 |
| PCHC | 99/162 | 56 | 354 | 427 | 14.5/16.9 | 46 |
Interestingly, the polymerization of camphoric acid with a rigid cyclic diol, CHDM, appears to override this crystallinity disruption, as suggested by the DSC results. Two melting transitions were observed at 99 and 162 °C in the first DSC heating curves of PCHC (Fig. 3b). This finding is further supported by the WAXD results, which show two distinct diffraction peaks at 14.5° and 16.9° (Fig. 4a), with a crystallinity of 46%, as determined via peak deconvolution (Fig. S11). The bulky, symmetric, and conformationally constrained CHDM units can promote more regular chain alignment, partially compensating for the regiochemical disorder of camphoric acid. This enhanced packing efficiency, combined with the restricted rotational freedom of the rigid cyclic structure, enables the formation of ordered domains, leading to semicrystalline polyesters despite the presence of camphoric acid. Additionally, this result is consistent with the study by Wang et al., who reported that the increased symmetry and rigidity of trans-CHDM increase chain regularity and thereby improve the crystallization capacity of poly(ethylene-co-1,4-cyclohexanedimethylene 2,5-furandicarboxylate) (PECF).45 Hence, the interaction between trans-CHDM units is expected to be favorable over the disordering effect of the camphoric acid units, thus allowing crystallization. The polymerization of camphoric acid with the cyclic monomer CHDM not only results in a semicrystalline structure but also leads to an increased glass transition temperature (Tg), as anticipated. This increase in the Tg is consistent with previous reports, in which the incorporation of rigid cyclic units was shown to increase the glass transition temperature of polyesters.15,19,46
 | ||
| Fig. 4 WAXD spectra of (a) poly(ethylene camphorate) (PEC), poly(octamethylene camphorate) (POC) and poly(cyclohexanedimethylene camphorate) (PCHC), and (b) PCHC before and after reheating. | ||
No crystallization was observed in the DSC cooling scan of the PCHC, and no melting peak appeared in the second DSC heating scan. This indicated that the crystallization rate of this polyester is relatively slow. PCHC demonstrated two different broad melting peaks, which could be associated with polymorphisms, the melting-recrystallization mechanism, the double lamellar thickness, physical ageing, or different molecular weight species.47–53 To further explain the double melting behavior, an additional melting observation via DSC revealed that the PCHC sample heated until the first melting point and then cooled to room temperature exhibited the same WAXD pattern as the pristine PCHC sample did (Fig. 4b). This indicates that there is no difference in the crystalline structure between the heated PCHC and the pristine sample, eliminating the possibility of polymorphism. Nonetheless, we note that the current data cannot unequivocally distinguish among other factors, and additional characterization, such as small-angle X-ray scattering (SAXS), is required to confirm the origin of the double melting behavior.
The thermal stability of the obtained camphoric-acid based polyesters was evaluated via TGA under an inert (N2) atmosphere and quantified by determining the Td5% and Tdmax values, which are the temperatures at which 5% and maximum weight loss, respectively, are reached. The weight loss versus temperature profiles and their derivatives are shown in Fig. 5a and b, and the corresponding degradation temperatures are summarized in Table 2. The thermal degradation of PEC and PCHC already begins at approximately 350 °C (Td5%), whereas that of POC starts at 271 °C (Td5%). All synthesized biobased polyesters showed TGA traces with a single-step degradation profile and displayed maximum decomposition temperatures (Tdmax) exceeding 400 °C. Although longer alkylene spacers in the polymer backbone typically lead to higher decomposition temperatures, in our case, POC shows lower thermal stability than PEC does. This deviation can be attributed to the broad dispersity of POC, where the presence of low-molecular-weight chain fractions increases the proportion of thermally labile chain ends, which may degrade at lower temperatures. In addition, all the polymers exhibited minimal char yields of less than 0.62% at 700 °C.
 | ||
| Fig. 5 (a) TGA results of the biobased polyesters: poly(ethylene camphorate) (PEC), poly(octamethylene camphorate) (POC) and poly(cyclohexanedimethylene camphorate) (PCHC). (b) Derivative weight loss curves versus temperature of PEC, POC, and PCHC. | ||
Wettability reflects how strongly a liquid is attracted to a solid surface. The shape of the liquid droplet is determined by the balance between cohesive forces within the liquid and adhesive forces between the liquid and the solid surface.54 According to the standard criteria for wettability, systems with a static water contact angle of less than 90° are considered hydrophilic, whereas systems with a contact angle larger than 90° are considered hydrophobic.55,56 In this study, static contact angle measurements were used to evaluate the surface wettability of the synthesized polyester. The contact angles were then determined via the drop snake algorithm.57Fig. 6 shows the water contact angles (WCAs) of the CA-derived polyesters. All the measured values are less than 90°, indicating that the polyesters are relatively hydrophilic. PEC and PCHC have similar water contact angle (WCA) values of 82.3 and 87.8°, respectively, whereas POC has a significantly lower WCA of 55.5°. This difference may be attributed to the broader molecular weight distribution of POC, as the greater diversity of chain lengths can expose more hydrophilic chain ends, such as hydroxyl (–OH) and carboxylic acid (–COOH) groups. Additionally, surface roughness, which is known to influence water contact angles, should be considered when these results are interpreted.57 The incorporation of hydrophilic groups into polymer backbones allows fine-tuning of their wettability, which can, in turn, influence (bio)degradation.58 The observed hydrophilicity of PEC, POC, and PCHC suggests their potential for degradation in aqueous environments, which is consistent with preliminary reports showing that polyethylene camphorate (PEC) degrades under both acidic and neutral conditions.15
 | ||
| Fig. 6 Drop profile and water contact angle value of (a) PEC, (b) POC, and (c) PCHC. | ||
Camphoric acid and CHDM-based copolyesters
To further augment the thermomechanical properties of PCHC polyesters, two different series of copolyesters, namely, P(CHC-co-EC) and P(CHC-co-OC), were synthesized (Table 3). Two different diols, ethylene glycol (EG) or 1,8-octanediol (OD), and CHDM were polymerized with camphoric acid, with varying feed fractions, as listed in Table 4. The polymerization was carried out via two-step melt polycondensations for 22 hours at 180–200 °C (Scheme S1). The chemical structures and compositions of both copolyesters were confirmed via1H-NMR spectroscopy (SI Fig. S3 and S4). The detailed 1H-NMR peak assignments are available in the Experimental section.| Abbreviation | Copolyesters | Structure |
|---|---|---|
| P(CHC-co-EC) | Poly(cyclohexanedimethylene-co-ethylene camphorate) |
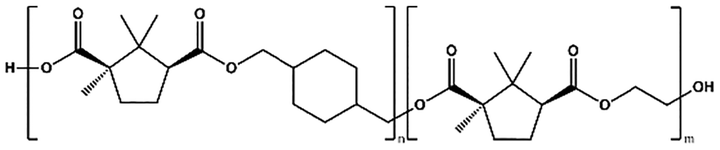
|
| P(CHC-co-OC) | Poly(cyclohexanedimethylene-co-octamethylene camphorate) |
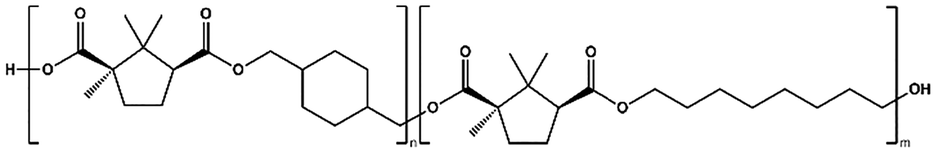
|


| F CHC | F OC | X CHC | X OC | |||||
|---|---|---|---|---|---|---|---|---|
| a F CHC, FEC, and FOC represent the molar feed ratios of PCHC, PEC, and POC, respectively. b X CHC, XEC, and XOC represent the molar fractions of the PCHC, PEC, and POC segments in the obtained copolyesters, respectively, as determined by 1H-NMR, with calculation details provided in the SI. c Molecular weights were determined by SEC in chloroform. d Isolated yield. | ||||||||
| P(CHC-co-OC)25 | 25 | 75 | 28 | 72 | 11100 |
28400 |
2.6 | 82 |
| P(CHC-co-OC)50 | 50 | 50 | 49 | 51 | 5700 |
19600 |
3.4 | 69 |
| P(CHC-co-OC)75 | 75 | 25 | 72 | 28 | 8600 |
19800 |
2.3 | 81 |
A series of each copolymer was synthesized with three different monomer feed ratios: 25/75, 50/50, and 75/25, corresponding to low, equimolar, and high incorporation of the CHDM monomer, respectively. The resulting copolyesters presented 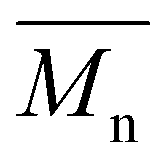 values ranging from 5600 to 11100 g mol−1, with satisfactory yields ranging from 69 to 82% (Table 4). A key objective of this study was to elucidate the monomer incorporation mechanism during copolymerization, which provides guidance for tuning the thermomechanical properties of the resulting polyesters. We observed that increasing the PCHC molar feed fraction to 50% led to a significant reduction in both the
values ranging from 5600 to 11100 g mol−1, with satisfactory yields ranging from 69 to 82% (Table 4). A key objective of this study was to elucidate the monomer incorporation mechanism during copolymerization, which provides guidance for tuning the thermomechanical properties of the resulting polyesters. We observed that increasing the PCHC molar feed fraction to 50% led to a significant reduction in both the 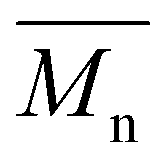 and
and 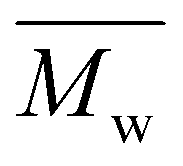 values of P(CHC-co-EC)s and P(CHC-co-OC)s. Further increasing the CHDM content resulted in a subsequent increase in the molecular weight. Although monomer reactivity often influences polycondensations, the close match between the initial feed ratios (FCHC, FEC and FOC) and the resulting copolymer compositions (XCHC, XEC, and XOC) indicates that differences in monomer reactivity do not explain the observed decrease in molecular weight at intermediate CHDM contents. This consistency suggests that all three diol monomers have relatively similar reactivities. At intermediate compositions, the randomness in the copolymer chain sequence often increases, which may reduce the polymerization efficiency and lead to a lower molecular weight and broader dispersity, as evidenced by prior studies.30,59,60 This finding is also consistent with our observation that P(CHC-co-OC)50 has a broad molecular weight distribution of 3.4. Ultimately, the differences in molar mass are relatively small, considering that small differences in conversion in a step-growth polymerization can lead to rather significant differences in molar mass. In this case, a difference of conversion of less than 2% would account for the discrepancy in molar mass observed by SEC measurements.
values of P(CHC-co-EC)s and P(CHC-co-OC)s. Further increasing the CHDM content resulted in a subsequent increase in the molecular weight. Although monomer reactivity often influences polycondensations, the close match between the initial feed ratios (FCHC, FEC and FOC) and the resulting copolymer compositions (XCHC, XEC, and XOC) indicates that differences in monomer reactivity do not explain the observed decrease in molecular weight at intermediate CHDM contents. This consistency suggests that all three diol monomers have relatively similar reactivities. At intermediate compositions, the randomness in the copolymer chain sequence often increases, which may reduce the polymerization efficiency and lead to a lower molecular weight and broader dispersity, as evidenced by prior studies.30,59,60 This finding is also consistent with our observation that P(CHC-co-OC)50 has a broad molecular weight distribution of 3.4. Ultimately, the differences in molar mass are relatively small, considering that small differences in conversion in a step-growth polymerization can lead to rather significant differences in molar mass. In this case, a difference of conversion of less than 2% would account for the discrepancy in molar mass observed by SEC measurements.
Effect of the monomer feed composition on the thermal properties and crystallinity of the copolyesters
The thermal stability of the synthesized copolyesters was evaluated by TGA following the same procedure described above for the camphoric acid–based polyesters. The results are summarized in Table 5 and depicted in Fig. S15. All the copolyesters exhibited high thermal stability, with Td5% values ranging from 308 to 385 °C and Tdmax values between 409 and 437 °C. Consistent with their corresponding homopolyesters, all Tdmax values exceeded 400 °C, whereas the char yields remained below 0.62% at 700 °C. The P(CHC-co-EC) and P(CHC-co-OC) series showed comparable thermal stabilities, with no significant differences between them. This suggests that the incorporation of either C2 or C8 aliphatic units has a similar effect on the overall thermal stability of the copolyesters, indicating that the PCHC segments play a dominant role in governing their decomposition behavior. Furthermore, the decomposition temperature (Td5% and Tdmax) did not vary significantly as the PCHC fraction increased from 25 to 75 mol%. This is consistent with previous reports on CHDM-based copolyesters, where the decomposition temperature is largely independent of the monomer feed ratio.29 These results indicate that decomposition is triggered by bond scission, which is not strongly affected by this compositional change.| Copolyesters (°C) | DSCa | TGAb | WAXDc | WCAd | |||
|---|---|---|---|---|---|---|---|
| T m (°C) | T g (°C) | T d5% (°C) | T dmax | 2θ (°) | χ c (%) | θ (°) | |
| a T m (melting temperature) was obtained from the first DSC heating scan; Tg (glass transition temperature) was obtained from the second DSC heating scan. b T d5% (decomposition temperature at 5% weight loss) and Tdmax (temperature at maximum rate of decomposition) were determined via TGA. c 2θ (diffraction peak) and χc (degree of crystallinity) were determined via WAXD. d Water contact angle. | |||||||
| P(CHC-co-EC)25 | — | 17 | 308 | 437 | — | 0 | 83.3 ± 0.3 |
| P(CHC-co-EC)50 | — | 29 | 320 | 433 | — | 0 | 94.1 ± 1.5 |
| P(CHC-co-EC)75 | 95/106 | 48 | 385 | 426 | 14.4/16.8 | 25 | 101.5 ± 0.4 |
| P(CHC-co-OC)25 | — | −18 | 368 | 409 | — | 0 | 91.2 ± 0.8 |
| P(CHC-co-OC)50 | 82 | 2 | 365 | 415 | 14.5/17.1 | 8 | 87.4 ± 0.5 |
| P(CHC-co-OC)75 | 85/109 | 33 | 377 | 420 | 14.4/16.9 | 29 | 105.2 ± 0.3 |
An increase in the CHDM feed increased the water contact angle from 83.3° to 101.5° for the P(CHC-co-EC) series and from 87.4° to 105.2° for the P(CHC-co-OC) series. This suggests that the cyclohexanedimethylene-camphorate moiety (the more hydrophobic segment) with lower surface energy, compared with both the ethylene-camphorate and octamethylene-camphorate moieties, segregated to the surface during spin-coating.61,62 Moreover, higher crystallinity increases the molecular mobility of the low surface energy component.61,63 However, in the latter series, P(CHC-co-OC)50 decreased (87.4°), which can be explained by its broad dispersity (3.4), which is equal to that of POC. The greater variety in the length of the chains would expose their hydrophilic chain ends. This results in the hydrophobicity of polymers that contain a minimum of 50% CHDM (except for P(CHC-co-OC)50), perhaps slightly reducing their potential susceptibility to hydrolytic degradation. This remains to be seen in a future extension of this work.
The thermophysical behavior of the copolyester series was analyzed via DSC, and the Tg and Tm values are summarized in Table 5. For the P(CHC-co-EC) series, the Tg ranged from +17 to +48 °C, whereas for the P(CHC-co-OC) series, they ranged from −18 to +33 °C. In both series, the Tg values increased with increasing CHDM content, indicating increased chain rigidity and reduced mobility due to the presence of bulky, rigid PCHC segments. These results are consistent with prior research on CHDM-containing copolyesters.31,64,65 For example, Diao et al. reported that by utilizing CHDM, 1,4-butanediol and furandicarboxylic acid, the Tg of polyesters increased from +40 to +87 °C with increasing CHDM feed from 0 to 100%.
All the copolyesters had only one Tg value, which suggests a uniform amorphous phase. The experimentally obtained Tg values were compared with theoretically derived Tg values on the basis of the Tg values of the homopolyesters PEC, POC, and PCHC (Table 2). The theoretical values of the glass transition temperatures of the copolyesters were calculated via the Fox equation, as described in the Experimental section.31,65–70 A comparison between the theoretical and experimental values is shown in Fig. 7. We observed that there was no significant difference between the theoretical values and the experimental data regarding the Tg values of the copolyesters, suggesting that the copolyesters are random copolymers with good thermodynamic compatibility.69,70
 | ||
| Fig. 7 Comparison of theoretical and experimental glass transition temperatures for (a) P(CHC-co-EC) and (b) P(CHC-co-OC) copolyesters. | ||
T m is a first-order transition associated with the crystalline domains of the polymer and was observed only in P(CHC-co-EC)75, P(CHC-co-OC)50, and P(CHC-co-OC)75 (Table 5 and Fig. S14). WAXD results further confirmed the semicrystallinity of these copolyesters, showing sharp peaks at 2θ ≈ 14° and 17°, corresponding to their crystalline domains (Fig. S10). In the P(CHC-co-EC)75 sample, two melting endotherms were detected at 95 °C and 106 °C, indicating that semicrystallinity develops only when the PCHC segment content reaches 75 mol%. In contrast, in the P(CHC-co-OC) series, P(CHC-co-OC)50 already exhibited melting transitions at 82 °C, and P(CHC-co-OC)75 did so at 85 and 109 °C, indicating that a lower PCHC fraction (50 mol%) is sufficient to induce crystallinity in this series. This difference suggests that longer octamethylene segments facilitate more effective packing of CHDM-rich regions, enabling crystallization at lower PCHC contents than shorter ethylene segments do. Both crystallinity and surface hydrophilicity are significant factors influencing polymer biodegradation and should be considered, among other factors.71
Conclusions
In summary, a series of novel (co)polyesters were synthesized in bulk from the rigid, cyclic, biobased monomers camphoric acid and CHDM. Copolymerization of camphoric acid with aliphatic diols and/or CHDM yielded copolyesters with molecular weights up to 14700 g mol−1. The polycondensations of camphoric acid and CHDM were investigated under various conditions, including different reaction times, temperatures, and the use of two catalysts. The results showed that using p-TSA with a 22 h reaction at 200 °C was sufficient to achieve complete polycondensations of camphoric acid and CHDM. The rigid structure of CHDM resulted in an increase in the Tg from 29 °C for POC to +56 °C for PCHC, demonstrating that its incorporation as a cyclic comonomer enhances the thermal properties of the polyesters. This trend was further corroborated in the copolyester series: as the PCHC content increased, the Tg values shifted consistently higher, reflecting the restricted chain mobility imparted by the cyclic comonomer. These results demonstrate that Tg can be effectively tuned by adjusting the CHDM incorporation ratio. Moreover, the observed Tg values are in good agreement with theoretical predictions based on the Fox equation.
DSC and WAXD analyses revealed that the incorporation of CHDM promoted semicrystalline behavior in the camphoric acid-based polyesters. For example, PCHC exhibited melting peaks at 99 °C and 162 °C and a degree of crystallinity of 46%. In the case of the copolyesters, the effect of CHDM content on crystallization is also evident: P(CHC-co-EC)75 shows two melting endotherms at 95 °C and 106 °C, whereas P(CHC-co-OC)50 and P(CHC-co-OC)75 display melting peaks at 82–85 °C and 109 °C, reflecting increasing crystallinity with increasing CHDM content. These results demonstrate that higher CHDM incorporation enhances chain regularity, promoting semicrystallinity and improving thermal stability, whereas camphoric acid alone predominantly forms amorphous polymers.
All the homopolyesters and the two copolyesters are hydrophilic, with water contact angles ranging from 55.5 to 87.8°, indicating favorable interactions with aqueous environments and potential susceptibility to hydrolytic degradation. All the obtained polyesters exhibited high thermal stability, with Tdmax values exceeding 400 °C and minimal char formation (<0.6% at 700 °C), demonstrating robust polymer backbones capable of withstanding elevated temperatures. These results suggest that the structural durability and environmental responsiveness of polyesters derived from camphoric acid and CHDM are balanced, although further investigations into their degradability and their long-term performance are still necessary. Finally, these findings demonstrate that biobased rigid monomers such as camphoric acid and CHDM can be effectively utilized to synthesize (co)polyesters with tunable thermal and physicochemical properties, making them promising candidates for applications such as coatings, packaging, and other sustainable materials.
Experimental section
Materials
(1R,3S)-(+)-Camphoric acid (CA, 99%), ethylene glycol (EG, EMPLURA®), 1,8-octanediol (OD, 98%), 1,4-cyclohexane dimethanol (CHDM, mixture of cis and trans, 99%), p-toluenesulfonic acid (p-TSA, monohydrate, ≥98%), titanium(IV) isopropoxide (TTIP, 97%), chloroform (CHCl3, amylene stabilized, HPLC grade, >99.8%), and deuterated chloroform (CDCl3, 99.8 atom% D) were purchased from Sigma–Aldrich. Chloroform (CHCl3, ChromAR HPLC, ethanol stabilized) and methanol (MeOH, AR®, anhydrous) were purchased from Macron Fine Chemicals. All chemicals were used as received.General synthetic procedure for the synthesis of camphoric acid-derived biobased (co)polyesters
The synthesis route for camphoric acid and CHDM-based (co)polyesters was modified on the basis of the work of Nsengiyumva and Miller.15 A two-stage melt polycondensation method was applied. The typical reaction setup is carried out in a 50 ml round-bottom flask equipped with a magnetic stirring egg and connected to a Schlenk line to allow switching between an inert argon flow and high vacuum (2 mmHg). The molar ratio of CA to diol (EG, OD, and CHDM) in the reaction mixture was maintained at 1.0/1.2. The total mass of the monomers was 2.0 g, with an additional 50.0 mg of p-TSA used as the catalyst. As an example, the synthesis of poly(ethylene camphorate) (PEC) is as follows: 1.458 g CA (7.282 mmol), 0.542 g CHDM (8.732 mmol), and 50.0 mg p-TSA were added to the flask. During the initial 3 h period, the temperature was adjusted to 180 °C while nitrogen was used to create an inert atmosphere. In the second stage of the experiment, the temperature was increased to 190 °C, and a dynamic vacuum was employed until it reached a pressure of 2 mmHg for 19 hours while the temperature was increased to 200 °C.The synthesis of poly(cyclohexanedimethylene camphorate) (PCHC) required two additional extended procedures, which were adjusted from the previous procedure. In one case, a nitrogen atmosphere was applied for 24 h at 180 °C, followed by 200–230 °C for 7 h in dynamic vacuum until 2 mmHg was reached. In another case, when a similar procedure was used, the catalyst was changed to TTIP.
The polyester products were dissolved in chloroform and filtered, and then the polyesters were precipitated in cold methanol. The precipitation products were centrifuged and decanted, and the obtained polyesters were dried in a vacuum oven at 40 °C for 72 hours and characterized by 1H-NMR, FTIR, SEC, DSC, TGA, WAXD, and WCA.
Camphoric acid and camphoric acid-derived biobased (co)polyesters
ATR-FTIR (ν, cm−1): camphoric acid. 2400–3250 (O–H stretching vibration); 2892–2970 (C–H stretching vibrations); 1690 (CO stretching vibrations); 1411–1458 (C–H bending vibration); 1120–1280 (C–O stretching vibrations); 933 (O–H bending vibrations). Camphoric acid-based (co)polyesters. 2854–2966 (C–H stretching vibrations); 1720–1722 (CO stretching vibrations); 1353–1460 (C–H bending and stretching vibrations); 991–1257 (C–O stretching vibrations); 754 (C–H bending (rocking) vibrations in POC which possesses more than four CH2 groups in an aliphatic chain).
Kinetics study: polycondensations of camphoric acid (CA) and 1,4-cyclohexane dimethanol (CHDM)
Instrumental methods
Proton nuclear magnetic resonance (1H-NMR) measurements were conducted with a Bruker Ascend™ NMR600 (1H: 600 MHz) using CDCl3 as the solvent. The number of scans was 64, and the chemical shifts are reported in parts per million.Fourier transform infrared (FTIR) spectroscopy was conducted with a Bruker VERTEX 70 spectrometer equipped with an attenuated total reflectance (ATR) unit. The spectra were collected in the range of 4000–400 cm−1 (16 scans, 4 cm−1 resolution) at room temperature. Atmospheric compensation and baseline correction were applied to the collected spectra.
Size-exclusion chromatography (SEC) was employed to investigate the molecular weights of the (co)polyesters. The samples were dissolved in purified chloroform (CHCl3) with 1000 ppm toluene as an internal standard at a concentration of 5.0 ± 0.1 mg mL−1 and subsequently filtered through a 0.45 PTFE syringe filter. A volume of 100 µL was introduced into an SEC system with an integrated dual flow refractive index detector (HLC-8420 EcoSEC Elite, Tosoh Bioscience) operating at a flow rate of 1 mL min−1. The column setup included a PSS SDV precolumn (5 µm particle size, 50 × 8 mm) followed by two PSS SDV analytical linear M columns (300 × 8 mm). The acquisition and processing of data were conducted via SECView software from Tosoh Bioscience. The weight-average molecular weight  and dispersity (Đ) of all samples were determined exclusively from the RI signal via calibration with polystyrene standards (Polymer Standards Service; PSS),
and dispersity (Đ) of all samples were determined exclusively from the RI signal via calibration with polystyrene standards (Polymer Standards Service; PSS),  = 400–1000000 g·mol−1.
= 400–1000000 g·mol−1.
Differential scanning calorimetry (DSC) measurements were conducted to analyze the thermal transitions of the rendered (co)polyesters. The measurements were performed on a TA-Instrument Q1000 DSC by heating–cooling-heating scans in the range of −50–200 °C with heating and cooling rates of 10 °C min−1.
The Tg values of the copolyesters were calculated via the Fox equation:
 | (1) |
T g1 is the glass transition temperature of PCHC, while Tg2 is the glass transition temperature of either PEC or POC. W1 is the molar fraction of the CHDM content in the copolyesters (P(CHC-co-EC) or P(CHC-co-OC)), whereas W2 is the molar fraction of either the ethylene glycol or 1,8-octanediol content in the copolyesters.
Thermogravimetric analysis (TGA) was performed on a TA Instruments Discovery TGA 5500 instrument by heating the sample in the range of 25–700 °C at a 10 °C min−1 scan rate in a nitrogen environment via platinum pans. Prior to measurement, isothermal preheating for 30 minutes was performed at 100 °C to remove moisture.
Wide-angle X-ray diffraction (WAXD) patterns were recorded on a Bruker D8 Endeavor diffractometer with Cu Kα radiation in the angular range of 4–50° (2θ) at room temperature. WAXD analysis was performed on the polyester powders, and the obtained patterns were analyzed with the software OriginPro 9.0.0 to determine the degree of crystallinity via peak deconvolution.
Water contact angles (WCAs) were measured via a VCA-2500XE (AST) system. The flat films of the (co)polyesters were produced via a Headway Research spin coater model PWM32. A drop of 4 μL of Milli-Q water was dispersed in the flat film sample area via a syringe. The water drop image was stored by a video camera, and the contact angle (θ) was calculated via ImageJ software. Drop Analysis – DropSnake plugins were utilized to analyze the shape of the drop. The contact angles were measured under ambient conditions. Each polymer was measured in triplicate and averaged.
Author contributions
S. Ahsan: investigation, conceptualization, methodology, formal analysis, data curation, visualization, writing – original draft. F. Silvianti: methodology, writing – review & editing. C. Post: methodology, formal analysis, writing – review & editing. V. S. D. Voet, R. Folkersma: project administration, writing – review & editing. J. Joji, L. M. Pitet, S. Damodaran: methodology, formal analysis, writing – review & editing. K. Loos: supervision, resources, project administration, funding acquisition, writing – review & editing. D. Maniar: conceptualization, methodology, formal analysis, supervision, resources, project administration, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: spectra of PEC, POC, PCHC, P(CHC-co-EC), P(CHC-co-OC), and CHDM in CDCl3 (Fig. S1–S5). Molecular weights and SEC chromatograms of the mixture from the p-TSA-catalyzed polycondensations of camphoric acid and CHDM in bulk (Table S1 and Fig. S6). SEC chromatograms of PEC, POC, and PCHC (Fig. S7). SEC chromatograms of PCHC from variation of polymerization conditions (Fig. S8). SEC chromatograms of P(CHC-co-EC) and P(CHC-co-OC) (Fig. S9). WAXD patterns of P(CHC-co-EC) and P(CHC-co-OC) (Fig. S10). Multi-peak fitting of WAXD spectra of PCHC, P(CHC-co-EC)75, P(CHC-co-OC)50, and P(CHC-co-OC)75 (Fig. S11). Product appearance of PEC, POC, and PCHC (Fig. S12). Product appearances of P(CHC-co-EC)25, P(CHC-co-EC)50, P(CHC-co-EC)75, P(CHC-co-OC)25, P(CHC-co-OC)50, and P(CHC-co-OC)75 (Fig. S13). DSC thermograms of P(CHC-co-EC)25, P(CHC-co-EC)50, P(CHC-co-EC)75, P(CHC-co-OC)25, P(CHC-co-OC)50, and P(CHC-co-OC)75 (Fig. S14). TGA results of P(CHC-co-EC)s and P(CHC-co-OC)s (Fig. S15). Drop profile and water contact angle value of P(CHC-co-EC)25, P(CHC-co-EC)50, P(CHC-co-EC)75, P(CHC-co-OC)25, P(CHC-co-OC)50, and P(CHC-co-OC)75 (Fig. S16). See DOI: https://doi.org/10.1039/d5py01182e.Acknowledgements
Jur van Dijken is gratefully acknowledged for his assistance with the TGA measurements. The first author (S. Ahsan) expresses gratitude for the financial assistance provided by the Indonesia Endowment Funds for Education of the Ministry of Finance (LPDP Kemenkeu) of the Republic of Indonesia.References
- M. Zhu and Q. Huang, Front. Environ. Sci., 2025, 13, 1603040 CrossRef.
- B. M. Stadler, A. Brandt, A. Kux, H. Beck and J. G. de Vries, ChemSusChem, 2020, 13, 556–563 CrossRef CAS PubMed.
- R. Mülhaupt, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef.
- S. Nandy, E. Fortunato and R. Martins, Prog. Nat. Sci.:Mater. Int., 2022, 32, 1–9 CrossRef CAS.
- S. Kobayashi, Struct. Chem., 2017, 28, 461–474 CrossRef CAS.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein and K. Loos, Biomolecules, 2013, 3, 461–480 CrossRef PubMed.
- Y. Jiang, G. O. R. A. Van Ekenstein, A. J. J. Woortman and K. Loos, Macromol. Chem. Phys., 2014, 215, 2185–2197 Search PubMed.
- D. Maniar, C. Fodor, I. K. Adi, A. J. J. Woortman, J. van Dijken and K. Loos, Polym. Int., 2021, 70, 555–563 CrossRef CAS.
- P. Shan, X. Lian, W. Lu, X. Yin, Y. Lu, M. Zhang, X. Wen, G. Xin, Z. Li and Z. Li, Biomacromolecules, 2023, 24, 2563–2574 CrossRef CAS PubMed.
- N. G. Valsange, M. N. Garcia Gonzalez, N. Warlin, S. V. Mankar, N. Rehnberg, S. Lundmark, B. Zhang and P. Jannasch, Green Chem., 2021, 23, 5706–5723 Search PubMed.
- S. Guo, Z. Geng, W. Zhang, J. Liang, C. Wang, Z. Deng and S. Du, Int. J. Mol. Sci., 2016, 17, 1836 CrossRef PubMed.
- Y. Zhang, P. Zhao, S. Sun, Q. Wu, E. Shi and J. Xiao, Commun. Chem., 2023, 6, 133 CrossRef CAS PubMed.
- Z. Rafiński and A. Kozakiewicz, J. Org. Chem., 2015, 80, 7468–7476 CrossRef PubMed.
- S. T. Duong and M. Fujiki, Polym. Chem., 2017, 8, 4673–4679 Search PubMed.
- O. Nsengiyumva and S. A. Miller, Green Chem., 2019, 21, 973–978 Search PubMed.
- R. Ouhichi, A. Bougarech, M. Kluge, S. Pérocheau Arnaud, S. Abid, M. Abid and T. Robert, Eur. Polym. J., 2021, 151, 110423 Search PubMed.
- G. Guidotti, M. Soccio, M. Gazzano, V. Siracusa and N. Lotti, Molecules, 2023, 28, 4056 Search PubMed.
- G. Guidotti, G. Burzotta, M. Soccio, M. Gazzano, V. Siracusa, A. Munari and N. Lotti, Polymers, 2021, 13, 2707 Search PubMed.
- C. Pang, X. Jiang, Y. Yu, L. Chen, J. Ma and H. Gao, ACS Macro Lett., 2019, 8, 1442–1448 CrossRef CAS PubMed.
- J. Hu, C. Wang, J. Dai, N. Teng, S. Wang, L. Zhang, Y. Jiang and X. Liu, Polym. Adv. Technol., 2021, 32, 3701–3713 CrossRef CAS.
- C. E. Carraher, M. R. Roner, A. G. Campbell, A. Moric-Johnson, L. Miller, P. Slawek, F. Mosca, J. D. Einkauf, J. E. Haky and R. Crichton, J. Inorg. Organomet. Polym. Mater., 2018, 28, 481–491 CrossRef CAS.
- S. R. Turner, J. Polym. Sci., Part A:Polym. Chem., 2004, 42, 5847–5852 Search PubMed.
- X. Xiao, H. Xin, Y. Qi, C. Zhao, P. Wu and X. Li, Appl. Catal., A, 2022, 632, 118510 Search PubMed.
- W. Ren, C. Gang, C. Zhao, P. Wu and X. Li, Appl. Catal., A, 2025, 698, 120233 CrossRef CAS.
- S. Mo, J. Kou, J. Zeng, K. Song, Y. Zhang, S. He, Y. Hu, Y. Guo, X. Liu, X. Chen and Y. Wang, Chem. Eng. J., 2024, 500, 157249 CrossRef CAS.
- L. Yuan, Y. Hu, Z. Zhao, G. Li, A. Wang, Y. Cong, F. Wang, T. Zhang and N. Li, Angew. Chem., Int. Ed., 2022, 61, e202113471 Search PubMed.
- H. J. Bang, H. Y. Kim, F. L. Jin and S. J. Park, J. Ind. Eng. Chem., 2011, 17, 805–810 Search PubMed.
- L. Diao, K. Su, Z. Li and C. Ding, RSC Adv., 2016, 6, 27632–27639 RSC.
- J. Wang, X. Liu, Y. Zhang, F. Liu and J. Zhu, Polymer (Guildf.), 2016, 103, 1–8 CrossRef CAS.
- L. Urpí, A. Alla and A. Martínez de Ilarduya, Polymer (Guildf.), 2023, 266, 125624 CrossRef.
- Y. Tsai, L. C. Jheng and C. Y. Hung, Polym. Degrad. Stab., 2010, 95, 72–78 Search PubMed.
- C. Post, J. van der Vlist, J. A. Jongstra, R. Folkersma, V. S. D. Voet and K. Loos, Eur. Polym. J., 2025, 222, 113594 CrossRef CAS.
- I. K. Park, H. Sun, S. H. Kim, Y. Kim, G. E. Kim, Y. Lee, T. Kim, H. R. Choi, J. Suhr and J. Do Nam, Sci. Rep., 2019, 9, 7033 Search PubMed.
- A. K. Chaudhary, J. Lopez, E. J. Beckman and A. J. Russell, Biotechnol. Prog., 1997, 13, 318–325 Search PubMed.
- W. J. Yoon, S. Y. Hwang, J. M. Koo, Y. J. Lee, S. U. Lee and S. S. Im, Macromolecules, 2013, 46, 7219–7231 Search PubMed.
- Z. Terzopoulou, E. Karakatsianopoulou, N. Kasmi, M. Majdoub, G. Z. Papageorgiou and D. N. Bikiaris, J. Anal. Appl. Pyrolysis, 2017, 126, 357–370 Search PubMed.
- Z. Terzopoulou, E. Karakatsianopoulou, N. Kasmi, V. Tsanaktsis, N. Nikolaidis, M. Kostoglou, G. Z. Papageorgiou, D. A. Lambropoulou and D. N. Bikiaris, Polym. Chem., 2017, 8, 6895–6908 Search PubMed.
- J. Zhu, J. Cai, W. Xie, P. H. Chen, M. Gazzano, M. Scandola and R. A. Gross, Macromolecules, 2013, 46, 796–804 CrossRef CAS.
- A. G. Amador, A. Watts, A. E. Neitzel and M. A. Hillmyer, Macromolecules, 2019, 52, 2371–2383 Search PubMed.
- M. S. Mohammadnia, P. Salaryan, Z. K. Azimi and F. T. Seyidov, Int. J. Chem. Biochem. Sci., 2012, 2, 36–41 Search PubMed.
- C. Q. Huang, S. Y. Luo, S. Y. Xu, J. B. Zhao, S. L. Jiang and W. T. Yang, J. Appl. Polym. Sci., 2010, 115, 1555–1565 CrossRef CAS.
- T. Zhang, B. A. Howell, P. K. Martin, S. J. Martin and P. B. Smith, Green Polym. Chem., 2013, 1144, 281–290 Search PubMed.
- V. S. R. Ganga, S. H. R. Abdi, R. I. Kureshy, N. U. H. Khan and H. C. Bajaj, J. Mol. Catal. A:Chem., 2016, 420, 264–271 CrossRef CAS.
- K. S. Naidu, A. S. Kate, V. Kshirsagar, R. Ganeshan, T. Gunale, B. Zhou, S. Anapat, Y. Sulub, A. Kumar and N. Rao, Polym. Test., 2020, 86, 106446 CrossRef.
- J. Wang, X. Liu, Z. Jia, L. Sun, Y. Zhang and J. Zhu, Polymer (Guildf.), 2018, 137, 173–185 CrossRef CAS.
- S. Curia, A. Biundo, I. Fischer, V. Braunschmid, G. M. Gübitz and J. F. Stanzione, ChemSusChem, 2018, 11, 2529–2539 CrossRef CAS PubMed.
- P. Pan and Y. Inoue, Prog. Polym. Sci., 2009, 34, 605–640 CrossRef CAS.
- M. C. Righetti, P. Marchese, M. Vannini, A. Celli, C. Lorenzetti, D. Cavallo, C. Ocando, A. J. Müller and R. Androsch, Biomacromolecules, 2020, 21, 2622–2634 Search PubMed.
- Y. T. Shieh and G. L. Liu, J. Polym. Sci., Part B:Polym. Phys., 2007, 45, 466–474 Search PubMed.
- G. Stoclet, R. Seguela and J. M. Lefebvre, Polymer (Guildf.), 2011, 52, 1417–1425 Search PubMed.
- E. Núñez, C. Ferrando, E. Malmström, H. Claesson, P. E. Werner and U. W. Gedde, Polymer (Guildf.), 2004, 45, 5251–5263 CrossRef.
- D. C. Bassett, R. H. Olley, I. A. M. Ai and R. J. J. Thomson, Polymer (Guildf.), 1988, 29, 1745–1754 Search PubMed.
- G. F. Shan, W. Yang, X. G. Tang, M. B. Yang, B. H. Xie, Q. Fu and Y. W. Mai, Polym. Test., 2010, 29, 273–280 Search PubMed.
- C. H. Kung, P. K. Sow, B. Zahiri and W. Mérida, Adv. Mater. Interfaces, 2019, 6, 1900839 CrossRef CAS.
- N. Verplanck, Y. Coffinier, V. Thomy and R. Boukherroub, Nanoscale Res. Lett., 2007, 2, 577–596 CrossRef CAS.
- F. Zhang and H. Y. Low, Langmuir, 2007, 23, 7793–7798 CrossRef CAS PubMed.
- H. Gu, C. Wang, S. Gong, Y. Mei, H. Li and W. Ma, Surf. Coat. Technol., 2016, 292, 72–77 CrossRef CAS.
- R. Belibel, T. Avramoglou, A. Garcia, C. Barbaud and L. Mora, Mater. Sci. Eng. Carbon, 2016, 59, 998–1006 Search PubMed.
- M. Safari, A. Mugica, M. Zubitur, A. M. de Ilarduya, S. Muñoz-Guerra and A. J. Müller, Polymers, 2020, 12, 17 CrossRef CAS PubMed.
- M. Safari, I. Otaegi, N. Aramburu, G. Guerrica-Echevarria, A. M. De Ilarduya, H. Sardon and A. J. Müller, Polymers, 2021, 13, 2263 CrossRef CAS PubMed.
- C. M. Chan and L. T. Weng, Materials, 2016, 9, 655 Search PubMed.
- K. L. A. Cimatu, U. I. Premadasa, T. D. Ambagaspitiya, N. M. Adhikari and J. H. Jang, J. Colloid Interface Sci., 2020, 580, 645–659 CrossRef CAS PubMed.
- G. Zorn, F. I. Simonovsky, B. D. Ratner and D. G. Castner, Adv. Healthcare Mater., 2022, 11, 2100894 CrossRef CAS PubMed.
- Y. Liu, E. Ranucci, M. S. Lindblad and A. C. Albertsson, J. Polym. Sci., Part A:Polym. Chem., 2001, 39, 2508–2519 CrossRef CAS.
- L. Diao, K. Su, Z. Li and C. Ding, RSC Adv., 2016, 6, 27632–27639 RSC.
- Y. Tsai, C. H. Fan, C. Y. Hung and F. J. Tsai, Eur. Polym. J., 2009, 45, 115–122 Search PubMed.
- Y. L. Li, X. M. Jia, X. Z. Zhang, Z. Y. Lu and H. J. Qian, Soft Matter, 2022, 19, 128–136 Search PubMed.
- S. Feng, H. Yang and Z. Qiu, Polym. Chem., 2025, 16, 2133–2142 Search PubMed.
- Y. Zhuang, X. Liu and Y. Gu, Polym. Chem., 2012, 3, 1517–1525 Search PubMed.
- H. Hu, R. Zhang, W. Bin Ying, L. Shi, C. Yao, Z. Kong, K. Wang, J. Wang and J. Zhu, Polym. Chem., 2019, 10, 1812–1822 Search PubMed.
- K. S. Savitha, B. R. Paghadar, M. Senthil Kumar and R. L. Jagadish, Polym. Chem., 2022, 13, 3562–3612 RSC.
| This journal is © The Royal Society of Chemistry 2026 |