
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvercoming stromal barriers in pancreatic cancer via size-engineered carrier-free nano-prodrugs
Mengheng
Yang
 a,
Ryuju
Suzuki
b,
Yoshitaka
Koseki
c,
Shuto
Kodera
d,
Ken
Saijo
d,
Hisato
Kawakami
d,
Keita
Tanita
a,
Sanjay
Kumar
a,
Kouki
Oka
a and
Hitoshi
Kasai
*a
a,
Ryuju
Suzuki
b,
Yoshitaka
Koseki
c,
Shuto
Kodera
d,
Ken
Saijo
d,
Hisato
Kawakami
d,
Keita
Tanita
a,
Sanjay
Kumar
a,
Kouki
Oka
a and
Hitoshi
Kasai
*a
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Japan. E-mail: kasai@tohoku.ac.jp
bNational Institute of Technology, Sendai College, Sendai, Japan
cDepartment of Life and Environmental Sciences, Faculty of Bioresource Sciences, Prefectural University of Hiroshima, Hiroshima, Japan
dDepartment of Clinical Oncology, Tohoku University Graduate School of Medicine, Sendai, Japan
First published on 30th January 2026
Abstract
Pancreatic cancer remains a major therapeutic challenge due to its dense desmoplastic stroma, which limits drug penetration and reduces chemotherapy efficacy. Here, we report a carrier-free nanoprodrug (CFNPG) based on SNC4DC, a homodimeric prodrug of SN-38, enabling controlled SN-38 release in pancreatic cancer cells. Using a precisely controlled reprecipitation method, we generated stable CFNPGs with tunable particle sizes down to ∼30 nm, resulting in enhanced tissue penetration. These nanoparticles exhibited high drug loading, absence of carrier-associated toxicity, potent antitumor activity, and minimal systemic side effects in an orthotopic pancreatic cancer model, providing a physiologically relevant assessment of drug delivery. Our findings demonstrate that precise size engineering of carrier-free nanoprodrugs can significantly improve tissue penetration and therapeutic efficacy, providing a clinically translatable strategy for pancreatic cancer therapy.
Introduction
Targeted cancer therapy exploiting the enhanced permeability and retention (EPR) effect represents a promising strategy for systemic treatment.1–8 Nanomedicines preferentially accumulate in tumors due to high vascular permeability and impaired lymphatic drainage, achieving higher intratumoral concentrations than small-molecule drugs.9–15Particle size plays a pivotal role in determining tumor targeting efficiency. Nanoparticles larger than 100 nm exhibit limited penetration in poorly permeable tumors such as pancreatic cancer, whereas those smaller than 100 nm show improved tissue penetration. In particular, particles below 50 nm are more likely to penetrate the dense stromal matrix characteristic of pancreatic cancer (Scheme 1).16–19 Studies on poly(ethylene glycol) (PEG)-based nanomedicines have corroborated this size-dependent behavior: sub-100 nm PEG nanoparticles readily extravasate in highly vascularized tumors, whereas only particles below 50 nm are effective in poorly permeable tumors.4,20 However, these evaluation systems use xenograft models, which hinder the analysis of essential tumor formation in the organs where the tumor originates. Even with precise size control, the use of conventional nanocarriers is often associated with systemic toxicity and off-target effects.
 | ||
| Scheme 1 Illustration of EPR effect (nanoparticles with sizes ranging from 10 to 200 nm tend to extravasate through leaky tumor vasculature and accumulate at tumor sites), pancreatic tumor microenvironment, and mechanism of action of nanoparticles <30 nm. Small nanoparticles penetrate dense stroma and release SN-38 intracellularly via GSH-mediated metabolism. | ||
Carrier-free nanomedicines have recently emerged as a promising alternative to mitigate carrier-related toxicity.9,21–24 For instance, SNC4DC, a homodimeric prodrug of the anticancer agent SN-38,21,25 can be processed into stable nanosuspensions via the reprecipitation method, typically yielding particles with a diameter of ∼75 nm. This technique relies on a rapid solubility change between solvent and antisolvent phases to induce nanoparticle formation, making drug solubility a key determinant of the particle size.26 Previous studies have demonstrated the promising antitumor properties of SNC4DC and the potential of further reducing the particle size in the context of enhancing tumor penetration and therapeutic efficacy.
The in vivo evaluation of the nanoparticle performance strongly depends on the choice of the tumor model. Most existing studies employ subcutaneous implantation, which inadequately reproduces the complex tumor microenvironment of refractory cancers such as pancreatic cancer. Meanwhile, orthotopic xenograft models provide a more physiologically relevant alternative. In fact, a previous study has shown that nanoparticles can be selectively delivered and exert their therapeutic effects in orthotopic pancreatic-cancer models.27
In the present study, aiming to decrease the size of SNC4DC nanoparticles and elucidate the size–efficacy relationship, we optimized the reprecipitation parameters to prepare SNC4DC nanosuspensions with particle sizes ranging from 30 to 400 nm and systematically evaluated their antitumor efficacy in vitro using pancreatic-cancer-cell models. For the in vivo validation, we employed an orthotopic pancreatic-cancer model incorporating bioluminescence imaging for non-invasive tumor monitoring.
Experimental
General preparation of nanoparticle dispersions
The nanoparticle dispersions were prepared using the reprecipitation method.28 The SNC4DC prodrug was transformed into a nanoparticle dispersion by inducing a rapid change in solubility using a 10 mM solution of SNC4DC in a good solvent (THF; 100 μL) and distilled water as the poor solvent. Using a 250 μL microsyringe, the SNC4DC solution was rapidly injected into water while stirring at a speed of 1500 rpm, followed by the cessation of stirring shortly after. The final concentration of the dispersion after reprecipitation was 0.1 mM (equal to that of SN-38).The prepared nanoparticles were evaluated using DLS (Zetasizer Nano series Nano-ZS; Malvern Instruments, Ltd) and SEM (S-4800; Hitachi). After performing the DLS and SEM measurements, the samples were stored in a refrigerator at 4 °C for four weeks, before the measurements were repeated.
Drug-release behavior
In centrifuge tubes, 500 μL of 100 μM (equal to that of SN-38) dispersions of 30 nm and 200 nm nanoparticles were prepared. To each tube, 500 μL of a 10 mM GSH solution (PBS/EtOH = 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, pH adjusted to 7.4) was added. The samples were incubated at 37 °C, and aliquots were collected after 1, 3, 6, and 24 h. At each time point, 12.5 μL of the sample was mixed with 987.5 μL of acetonitrile (MeCN) containing 3% acetic acid (AcOH). The concentrations of SN-38 and SNC4DC in the samples were quantified via LC-MS/MS based on standard calibration curves, using a Shimadzu LC-MS/MS instrument and a 3 μm C8 column (Imtakt Unison UK-C8, UK824, 2 mm ID × 100 mm length, 3 μm, C8, end-capped). The column temperature was maintained at 40 °C, and the flow rate was set to 0.3 mL min−1. The mobile phase consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in MeCN).
:3, pH adjusted to 7.4) was added. The samples were incubated at 37 °C, and aliquots were collected after 1, 3, 6, and 24 h. At each time point, 12.5 μL of the sample was mixed with 987.5 μL of acetonitrile (MeCN) containing 3% acetic acid (AcOH). The concentrations of SN-38 and SNC4DC in the samples were quantified via LC-MS/MS based on standard calibration curves, using a Shimadzu LC-MS/MS instrument and a 3 μm C8 column (Imtakt Unison UK-C8, UK824, 2 mm ID × 100 mm length, 3 μm, C8, end-capped). The column temperature was maintained at 40 °C, and the flow rate was set to 0.3 mL min−1. The mobile phase consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in MeCN).
Cellular uptake
The Förster-resonance-energy-transfer (FRET) principle was used to investigate the intracellular dynamics, and FRET-SNC4DC nanoparticles composed of SNC4DC (FRET donor) and the dye bodipy FL (BPFL)-cholesterol (FRET acceptor) were fabricated according to our previous study.21In the nanoparticle state, BPFL-cholesterol is very close to SNC4DC, enabling FRET to occur. In contrast, the dissolution of SNC4DC or BPFL-cholesterol molecules increases the distance between the FRET donor and the acceptor, thereby quenching the FRET signal. The fabricated FRET-SNC4DC nanoparticles were added to a culture dish seeded with BxPC-3 cells, and the intracellular dynamics of the FRET-SNC4DC nanoparticles were observed using CLSM.
In a six-well plate, 1 mL of the BxPC-3 cell suspension at a density of 2 × 105 cells per mL was added to each well. After 24 h, 1 mL of 0.1 mM SNC4DC nanoparticle dispersion was added to the wells. The cells were incubated at 37 °C for 1, 3, and 6 h, before the supernatant was collected. Subsequently, 200 μL of EDTA-containing trypsin solution was added to the wells, and the cells were incubated at 37 °C for 5 min before being collected via centrifugation (1000 rpm for 5 minutes). From the collected supernatant, 20 μL was taken and mixed with 980 μL of MeCN containing 3% AcOH. Meanwhile, 1000 μL of dichloromethane/MeOH (1:1) was added to the collected cells. From the resulting solution, 70 μL of the supernatant was collected and mixed with 630 μL of MeCN containing 3% AcOH. The concentrations were then measured using LC-MS/MS.
Cytotoxicity studies
BxPC-3 cells were cultured in Dulbecco's Modified Eagle Medium containing 10% fetal bovine serum under 5% CO2 at 37 °C. The cultured cells were seeded in 96-well plates (2 × 105 cells per well) and incubated for 24 h. Then, the culture medium was removed, and 100 μL of medium containing the nano-prodrug was added. After 48 h, the number of viable cells was evaluated using a cell-counting kit-8 (DOJINDO) and a microplate reader (iMark; Bio-Rad Laboratories, Inc.). Cell viabilities were normalized to the difference of the optical densities at 450 and 620 nm (OD450 − OD620) for the untreated cells. Assays were performed in triplicate.Animal experiments
The Ethical Committee of the Graduate School of Medicine, Tohoku University, approved the protocol under no. 2023MdA-077. All experiments were conducted in accordance with the approved guidelines.Pancreatic cancer was orthotopically implanted in the pancreas of C57BL/6 mice using KPC-LUC cells.29–31 The orthotopic pancreatic-cancer model was established as follows: KPCC57LUC cells were suspended in PBS. Female C57BL/6 mice, five weeks of age, were anesthetized, and a laparotomy was performed to expose the abdominal cavity. The pancreas, attached to the spleen, was gently exteriorized. A total of 50 μL of the cell suspension containing 5 × 105 KPCC57LUC cells was carefully injected into the tail of the pancreas. After injection, the pancreas and spleen were returned to the peritoneal cavity, and the abdominal wall and skin were closed with surgical sutures.
Seven days after implantation, D-luciferin was administered via intraperitoneal injection, and tumor growth was monitored using in vivo bioluminescence imaging with an IVIS system. Mice were then randomized into experimental groups such that the average tumor burden was equivalent across all groups.
Following randomization, treatment was initiated. Mice received intravenous injections via the tail vein every other day for a total of five doses over 10 days. The control group received 100 μL of physiological saline, while the experimental groups were administered SNC4DC nanodispersions with an average nanoparticle size of 30, 100, or 200 nm, each at a concentration of 2.56 mM. Tumor progression was monitored via bioluminescence imaging using intraperitoneal injection of D-luciferin, and the signal intensities were quantified using the Living Image software suite (Xenogen). The body weight was recorded throughout the experimental period. One week after the final injection, mice were euthanized and tumors were harvested and weighed.
Results and discussion
Preparation of 30 nm nanoparticles
Reprecipitation was performed by modulating the temperatures of both the good and poor solvent. The good solvent was heated in an oven (50 °C; 10 min) and the poor solvent was heated to 50 °C using a hot plate. Cooling was accomplished by immersing the solvents in an ice/water bath.Preparation of 100 nm, 200 nm and 400 nm nanoparticles
Keeping the volume of the poor solvent constant, THF was added to it. Reprecipitation was conducted by varying the proportion of the poor solvent mixed with THF using the same procedure. Subsequently, THF was removed using a rotary evaporator, and pure water was added to dilute the solution to a final concentration of 0.1 mM (equal to that of SN-38).Tuning the nanoparticle size
By modulating the temperatures of the good solvent (THF, T1) and the poor solvent (water, T2), as well as the solvent composition during reprecipitation, the nanoparticle size of SNC4DC could be precisely tuned from 30 nm to 400 nm. Optimal conditions (T1 = 50 °C, T2 = 0 °C) yielded ∼30 nm nanoparticles with excellent dispersion stability over four weeks at 4 °C (Fig. 1a). By varying the temperature, the nanoparticle size was tuned within the range of 30–75 nm (Fig. 1b). The size variation was primarily governed by supersaturation (Scheme 2): lowering T2 reduced the solubility of SNC4DC in water and increased supersaturation, promoting the formation of a larger number of nuclei and thus smaller nanoparticles.26,32 In contrast, increasing the THF ratio in the poor solvent decreased supersaturation and favored nanoparticle growth, producing particles up to 400 nm (Fig. 1c–e). Both 30 nm and 400 nm formulations remained stable without aggregation or sedimentation during long-term storage. Further mechanistic insights—such as the influence of hydrogen bonding at low T2, the role of THF–water interactions, and the impact of evaporation dynamics on nucleation and growth—are discussed in the SI.33–36 | ||
| Fig. 1 (a) SEM image of 30 nm SNC4DC nanoparticles; (b) size variation under different temperature conditions; (c) SEM image of 100 nm nanoparticles; (d) SEM image of 200 nm nanoparticles; (e) size as a function of THF ratio in poor solvent; (f) drug-release profiles of 30, 100, 200 nm nanoparticles with 10 mM GSH. | ||
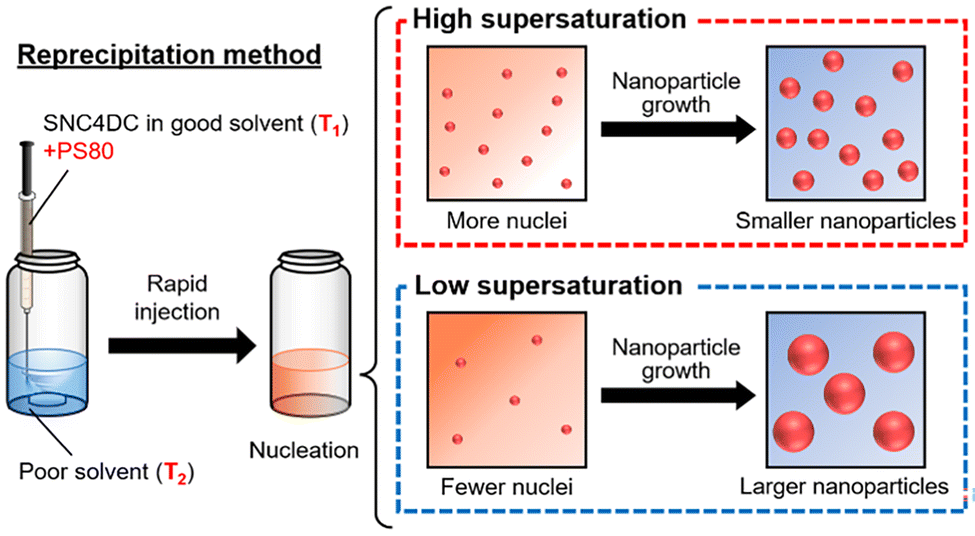 | ||
| Scheme 2 Effect of supersaturation on nanoparticle size during reprecipitation. | ||
Collectively, these results demonstrate that precise modulation of solvent conditions enables controlled tuning of SNC4DC nanoparticle size while maintaining high colloidal stability.
Size-dependent drug release and cellular uptake
SNC4DC releases SN-38 through a two-step metabolism catalyzed by glutathione (GSH) and hydrolases.21 Nanoparticles with average diameters of 30 and 200 nm exhibited different drug release rates at the same concentration of GSH. The release rate of the 200 nm nanoparticles was clearly slower, with half of the drug still remaining after 24 h. Conversely, the 30 nm nanoparticles released the drug more rapidly, with almost no residual drug remaining after 24 h (Fig. 1f). The incomplete release of SN-38 may be attributed to partial hydrolysis of ester or disulfide bonds during linker degradation, resulting in partially cleaved intermediates. The difference in drug release rates can be ascribed to the variation in specific surface area among nanoparticles of different sizes. Smaller nanoparticles possess a larger specific surface area, allowing more SNC4DC molecules to directly interact with GSH and thereby accelerating the release rate. In contrast, larger nanoparticles have a smaller contact area with GSH, reducing the number of accessible disulfide bonds and consequently requiring a longer time for complete SN-38 release.Compared to commercial irinotecan, SNC4DC exhibited stronger cytotoxicity against human pancreatic-cancer cell line BxPC-3. In addition, SNC4DC with different nanoparticle sizes showed varying levels of cytotoxicity, whereby smaller nanoparticles demonstrated stronger cytotoxicity (Fig. 2a).
 | ||
| Fig. 2 (a) Cytotoxic effects on BxPC-3 cells; (b) intracellular and total SN-38 levels at 1, 3, 6 h; (c) CLSM images: green = molecular SNC4DC, red = nanoparticle, white = colocalization. | ||
A quantitative analysis revealed that the amount of SN-38, both within the cells and in the culture medium, was substantially higher in the 30 nm group than in the 200 nm group (Fig. 2b). Consistently, uptake assays performed after 1, 3, and 6 h, with cell viability maintained >95%, showed that cells treated with 30 nm nanoparticles exhibited lower residual levels of SNC4DC in both molecular and nanoparticle form in the cellular fraction and the supernatant. These results indicate that a greater proportion of internalized SNC4DC was converted into SN-38 in the 30 nm group.
Furthermore, the time-dependent cellular uptake of nanoparticles with different sizes was visualized using confocal laser scanning microscopy (CLSM). Consistent with prior findings,37,38 nanoparticles were internalized intact, subsequently dissolving within the cells to exert their pharmacological activity. In the CLSM images (Fig. 2c), green fluorescence represents the molecular (dissolved) state of SNC4DC, red fluorescence indicates the nanoparticle form of SNC4DC, and white regions in the merged channels denote areas of colocalization (overlap). The image analysis demonstrated significantly higher uptake and dissolution of 30 nm nanoparticles after 3 h, whereas their intracellular presence decreased after 6 h. In contrast, the 200 nm nanoparticles exhibited a sustained uptake, remaining detectable within the cells for 6 h. These observations indicate that the 30 nm nanoparticles are more rapidly internalized and dissolved within BxPC-3 cells.
Antitumor effect in the orthotopic pancreatic-cancer model
To elucidate the influence of nanoparticle size on therapeutic performance, an orthotopic pancreatic cancer model was established in mice and treated with SNC4DC nano-prodrugs of distinct sizes (30, 100, and 200 nm).21,29–31 The experimental schedule is shown in Fig. 3a. Tumor progression was longitudinally monitored by IVIS bioluminescence imaging (Fig. 3b). A clear size-dependent therapeutic trend was observed: treatment with 30 nm SNC4DC markedly suppressed tumor growth, reducing final tumor volumes to approximately one third of those in saline-treated controls, whereas the 200 nm formulation exhibited negligible inhibition (Fig. 3c and d). The 100 nm formulation demonstrated intermediate efficacy between the 30 nm and 200 nm counterparts. This observation aligns with previous findings that only nanoparticles smaller than 50 nm can effectively penetrate poorly vascularized tumors.10 | ||
| Fig. 3 (a) Experimental workflow; (b) IVIS images and bioluminescence quantification; (c) final tumor weights (N = 5, SD); (d) representative excised tumors; (e) body-weight changes of mice during the treatment period. | ||
Importantly, administration of 30 nm SNC4DC did not cause significant body-weight loss (Fig. 3e), indicating minimal systemic toxicity. The superior efficacy of the smaller nanoparticles may be attributed to their enhanced penetration through the dense stromal matrix of pancreatic tumors, improved cellular uptake, and faster intracellular drug release, as supported by in vitro release studies. Collectively, these results highlight the favorable safety profile and potent antitumor activity of the 30 nm SNC4DC nano-prodrug, underscoring the crucial role of precise size control in optimizing therapeutic performance in solid tumors.
Conclusions
High drug loading and the elimination of carrier-associated toxicity enabled potent antitumor efficacy in physiologically relevant models.Building on these advantages, SNC4DC nanoparticles precisely engineered to 30 nm exhibited rapid drug release, efficient cellular uptake, and strong antitumor activity in orthotopic pancreatic cancer models, while minimizing systemic toxicity.
This carrier-free system also allows further fine-tuning of nanoparticle size, offering opportunities to optimize pharmacological activity below 30 nm.
Collectively, the combination of high drug loading, carrier-free design, and precise size control provides a clinically translatable strategy to enhance drug delivery and overcome stromal barriers in pancreatic cancer therapy.
Author contributions
M. Y.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, validation, visualization, writing – original draft, R. S.: conceptualization, writing – review & editing, Y. K.: conceptualization, data curation, funding acquisition, writing – review & editing, S. K.: data curation, methodology, writing – review & editing, K. S.: resources, supervision, writing – review and editing, H. K.: resources, writing – review and editing, K. T.: data curation, writing – review and editing, K. O.: supervision, writing – review and editing, H. K.: conceptualization, funding acquisition, project administration, resources, supervision, visualization, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). The supplementary information includes IC50 data of SNC4DC nanoparticles in BxPC-3 cells, cytotoxicity results in normal cells, and additional characterization of NPDs, including photographs and stability data. See DOI: https://doi.org/10.1039/d5pm00364d.Further data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgements
This work was supported by JST SPRING (grant number JPMJSP2114) and JSPS Grants-in-Aid for Scientific Research (22H00328 and 25K09889). The authors wish to express their gratitude to Ms. Yuriko Fujinoya for invaluable experimental assistance.References
- M. Ikeda-Imafuku, L. L.-W. Wang, D. Rodrigues, S. Shaha, Z. Zhao and S. Mitragotri, J. Controlled Release, 2022, 345, 512 CrossRef CAS PubMed.
- F. Alexis, E. M. Pridgen, R. Langer and O. C. Farokhzad, Drug Delivery, 2009, 55 Search PubMed.
- C. Wong, T. Stylianopoulos, J. Cui, J. Martin, V. P. Chauhan, W. Jiang, Z. Popovic, R. K. Jain, M. G. Bawendi and D. Fukumura, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2426 CrossRef CAS PubMed.
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. W. Chan, Nano Lett., 2009, 9, 1909 CrossRef CAS PubMed.
- W. J. Gradishar, S. Tjulandin, N. Davidson, H. Shaw, N. Desai, P. Bhar, M. Hawkins and J. O'Shaughnessy, J. Clin. Oncol., 2005, 23, 7794 CrossRef CAS PubMed.
- S. Mansoor, P. P. D. Kondiah, Y. E. Choonara and V. Pillay, Polymers, 2019, 11, 1380 Search PubMed.
- Y. Bao, M. Maeki, A. Ishida, H. Tani and M. Tokeshi, PLoS One, 2022, 17, e0271050 CrossRef CAS PubMed.
- X. Gu and T. Minko, Cancers, 2024, 16, 1589 CrossRef CAS PubMed.
- H. Kasai, T. Murakami, Y. Ikuta, Y. Koseki, K. Baba, H. Oikawa, H. Nakanishi, M. Okada, M. Shoji, M. Ueda, H. Imahori and M. Hashida, Angew Chem., Int. Ed., 2012, 51, 10315 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815 Search PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387 CAS.
- X. Liu, L. Y. Lin, F. Y. Tseng, Y. C. Tan, J. Li, L. Feng, L. Song, C. F. Lai, X. Li, J. H. He, R. Sakthivel and R. J. Chung, Analyst, 2021, 146, 4066 Search PubMed.
- B. Feng, F. Zhou, B. Hou, D. Wang, T. Wang, Y. Fu, Y. Ma, H. Yu and Y. Li, Adv. Mater., 2018, 30, 1803001 CrossRef PubMed.
- T.-L.-H. Ngo, K.-L. Wang, W.-Y. Pan, T. Ruan and Y.-J. Lin, ACS Nano, 2024, 18, 5632 CrossRef CAS PubMed.
- J. Dong, J. Ding, S. Luo, R. Li, Y. Wang, B. Xiao, Y. Pei, X. Chen, W. Sun and Z. Pei, J. Nanobiotechnol., 2025, 23, 123 CrossRef CAS PubMed.
- P. S. Uster, P. K. Working and J. Vaage, Int. J. Pharm., 1998, 162, 77 CrossRef CAS.
- S. Unezaki, K. Maruyama, J.-I. Hosoda, I. Nagae, Y. Koyanagi, M. Nakata, O. Ishida, M. Iwatsuru and S. Tsuchiya, Int. J. Pharm., 1996, 144, 11 Search PubMed.
- M. R. Kano, Y. Bae, C. Iwata, Y. Morishita, M. Yashiro, M. Oka, T. Fujii, A. Komuro, K. Kiyono and M. Kaminishi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3460 Search PubMed.
- M. Yang, K. Yang, B. Gao, P. Wang, T. Li, Y. Zheng, Y. Pei, Z. Pei and Y. Lv, Chem. Commun., 2022, 58, 11147 RSC.
- M. R. Dreher, W. Liu, C. R. Michelich, M. W. Dewhirst, F. Yuan and A. Chilkoti, J. Natl. Cancer Inst., 2006, 98, 335 CrossRef CAS PubMed.
- K. Tanita, Y. Koseki, S. Kumar, F. Taemaitree, A. Mizutani, H. Nakatsuji, R. Suzuki, A. T. N. Dao, F. Fujishima, H. Tada, T. Ishida, K. Saijo, C. Ishioka and H. Kasai, Nanoscale, 2024, 16, 15256 Search PubMed.
- A. Shibata, Y. Koseki, K. Tanita, S. Kitajima, K. Oka, K. Maruoka, R. Suzuki, A. T. N. Dao and H. Kasai, Chem. Commun., 2024, 60, 6427 RSC.
- Y. Koseki, Y. Ikuta, L. Cong, M. Takano-Kasuya, H. Tada, M. Watanabe, K. Gonda, T. Ishida, N. Ohuchi, K. Tanita, F. Taemaitree, A. T. N. Dao, T. Onodera, H. Oikawa and H. Kasai, Bull. Chem. Soc. Jpn., 2019, 92, 1305 CrossRef CAS.
- M. Yang, A. T. Dao, H. Nakatani, Y. Eguchi, R. Suzuki, Y. Koseki, K. Oka and H. Kasai, MRS Commun., 2025, 1 Search PubMed.
- W. Jin, Z. Chen, S. Yang, Y. Qu, Y. Pei and Z. Pei, Chem. Commun., 2022, 58, 12584 RSC.
- J. Mori, Y. Miyashita, D. Oliveira, H. Kasai, H. Oikawa and H. Nakanishi, J. Cryst. Growth, 2009, 311, 553 CrossRef CAS.
- C. R. Patra, R. Bhattacharya, E. Wang, A. Katarya, J. S. Lau, S. Dutta, M. Muders, S. Wang, S. A. Buhrow, S. L. Safgren, M. J. Yaszemski, J. M. Reid, M. M. Ames, P. Mukherjee and D. Mukhopadhyay, Cancer Res., 2008, 68, 1970 Search PubMed.
- H. Kasai, H. S. Nalwa, H. Oikawa, S. Okada, H. Matsuda, N. Minami, A. Kakuta, K. Ono, A. Mukoh and H. Nakanishi, Jpn. J. Appl. Phys., 1992, 31, L1132 CrossRef CAS.
- E. S. Lee, Y. Suzuki, H. Tomioka, H. Nakagami and Y. Sato, Tohoku J. Exp. Med., 2023, 261, 239 CrossRef CAS PubMed.
- Y. Suzuki and Y. Sato, Tohoku J. Exp. Med., 2024, 262, 163 CrossRef CAS PubMed.
- X. Liu, Y. Zhuang, W. Huang, Z. Wu, Y. Chen, Q. Shan, Y. Zhang, Z. Wu, X. Ding, Z. Qiu, W. Cui and Z. Wang, Nat. Commun., 2023, 14, 4106 Search PubMed.
- E. Sah and H. Sah, J. Nanomater., 2015, 2015, 794601 Search PubMed.
- M. J. Shultz and T. H. Vu, J. Phys. Chem. B, 2015, 119, 9167 Search PubMed.
- J. Hao, H. Cheng, P. Butler, L. Zhang and C. C. Han, J. Chem. Phys., 2010, 132, 154902 Search PubMed.
- H. Tsuji, M. Nakahata, M. Hishida, H. Seto, R. Motokawa, T. Inoue and Y. Egawa, J. Phys. Chem. Lett., 2023, 14, 11235 CrossRef CAS PubMed.
- X. Hu, J. Yu and H. Liu, Water Sci. Technol., 2006, 53, 245 Search PubMed.
- F. Taemaitree, B. Fortuni, Y. Koseki, E. Fron, S. Rocha, J. Hofkens, H. Uji-i, T. Inose and H. Kasai, Nanoscale, 2020, 12, 16710 RSC.
- D. S. Talaga, W. L. Lau, H. Roder, J. Tang, Y. Jia, W. F. DeGrado and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13021 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |