
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2-Substituted oxazolyl and thiazolyl amino acids are suitable P1 surrogates for new SARS-CoV-2 main protease inhibitors
Zijie
Liu
,
Tayla J.
Van Oers
 ,
Conrad
Fischer
,
Anna
Nguyen
,
Johnson
Chew
and
John C.
Vederas
*
,
Conrad
Fischer
,
Anna
Nguyen
,
Johnson
Chew
and
John C.
Vederas
*
Department of Chemistry, University of Alberta, Edmonton, AB T6G 2G2, Canada. E-mail: john.vederas@ualberta.ca
First published on 2nd June 2026
Abstract
The SARS-CoV-2 main protease is a cysteine protease that can bind in a reversible covalent fashion to the warhead of peptidic inhibitors. The most commonly used P1 amino acid subunit in SARS-CoV-2 therapeutics is a cyclic glutamine derivative, and this moiety has been shown to be relatively quickly metabolized, but contains heteroatoms that interact with key residues in the active site. Here, a structure–activity study has been explored with the synthesis of oxazolyl and thiazolyl unnatural amino acids as P1 moieties. This investigation involved different substitution patterns of these heteroaromatic rings, in an effort to maintain a scaffold that can form the important interactions with residues in the active site. IC50 values with the SARS-CoV-2 main protease reveal the 2-oxazolyl and 2-thiazolyl derivatives are similarly effective (290 nM and 270 nM, respectively) to compounds with the cyclic glutamine derivative, demonstrating alternative P1 moieties that can be used in the efficient design of novel coronavirus main protease inhibitors.
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease (Mpro) is a cysteine protease that is key to the replication of the SARS-CoV-2 virus. This protease is one of two proteases that cleaves the two polyproteins (pp1a and pp1ab) to release several functional proteins.1 Due to the important role the Mpro plays in the replication cycle of the virus, this protease is a desirable therapeutic target. The main protease has a specific recognition sequence (Leu-Gln↓Ser-Ala-Gly), which allows it to be selective in the cleavage process.1 This recognition sequence is unique, and not found among human proteases, which makes this enzyme a selective target, with likely fewer off-target effects. The protease works using a catalytic dyad (Cys145 and His41), where the cysteine thiol/thiolate acts as a nucleophile attacking the substrate amide carbonyl, with the histidine acting as a general base.1,2Most of the current therapeutics that target the main protease are designed to mimic this recognition sequence, three literature examples are shown in Fig. 1. The protease can recognize the compound, and forms a reversible covalent bond with the warhead, which is an electrophilic motif that can react with a thiolate nucleophile. Three examples of commonly used warheads in this type of inhibitor are aldehydes, nitriles, and hydroxymethyl ketones.1,3–6 In this work, we have decided to focus primarily on compounds with bisulfites and aldehydes. The bisulfites are pro-drugs of aldehydes, as bisulfites convert to aldehydes when under physiological conditions (neutral aqueous conditions), but are more soluble, and often result in lower and better IC50 values.7 Aldehydes and bisulfites are easy to handle, and the synthetic route is shorter and yields two analogues in the same route (one step longer to the bisulfite).
 | ||
| Fig. 1 Chemical structure of literature SARS-CoV-2 main protease inhibitors and their respective IC50 values.1,3 | ||
Development of potent main protease inhibitors in the past has mostly revolved around improving P2 and P3 residues to fit tightly into the S2 and S3 subunits of the Mpro active site. It was a longstanding dogma that P1 has to be a glutamine analogue to warrant recognition of respective inhibitors by the protease.8 This P1 is present in the FDA approved therapeutic nirmatrelvir (Paxlovid™), as well as in Pfizer's second-generation inhibitor in clinical trials, ibuzatrelvir.5,9 However, this cyclic glutamine analogue has been shown to be rapidly metabolized by the P450 enzymes, as it is in nirmatrelvir (Fig. 2), which is the reason nirmatrelvir is provided in combination with ritonavir, an inhibitor of the P450 enzyme responsible for the metabolism.5,10 This poses a problem, as many individuals cannot take ritonavir, and therefore cannot take Paxlovid™. Heteroaromatic rings may be more stable to metabolic enzymes and we initially believed they may be suitable replacements for the cyclic glutamine analogue.3 In addition, mutants of the SARS-CoV-2 main protease have appeared in studies and patient isolates that have developed resistance to the commonly used drugs, such as Pfizer's Paxlovid™.11,12 A different P1 scaffold may provide an alternative option that may be effective against mutants.
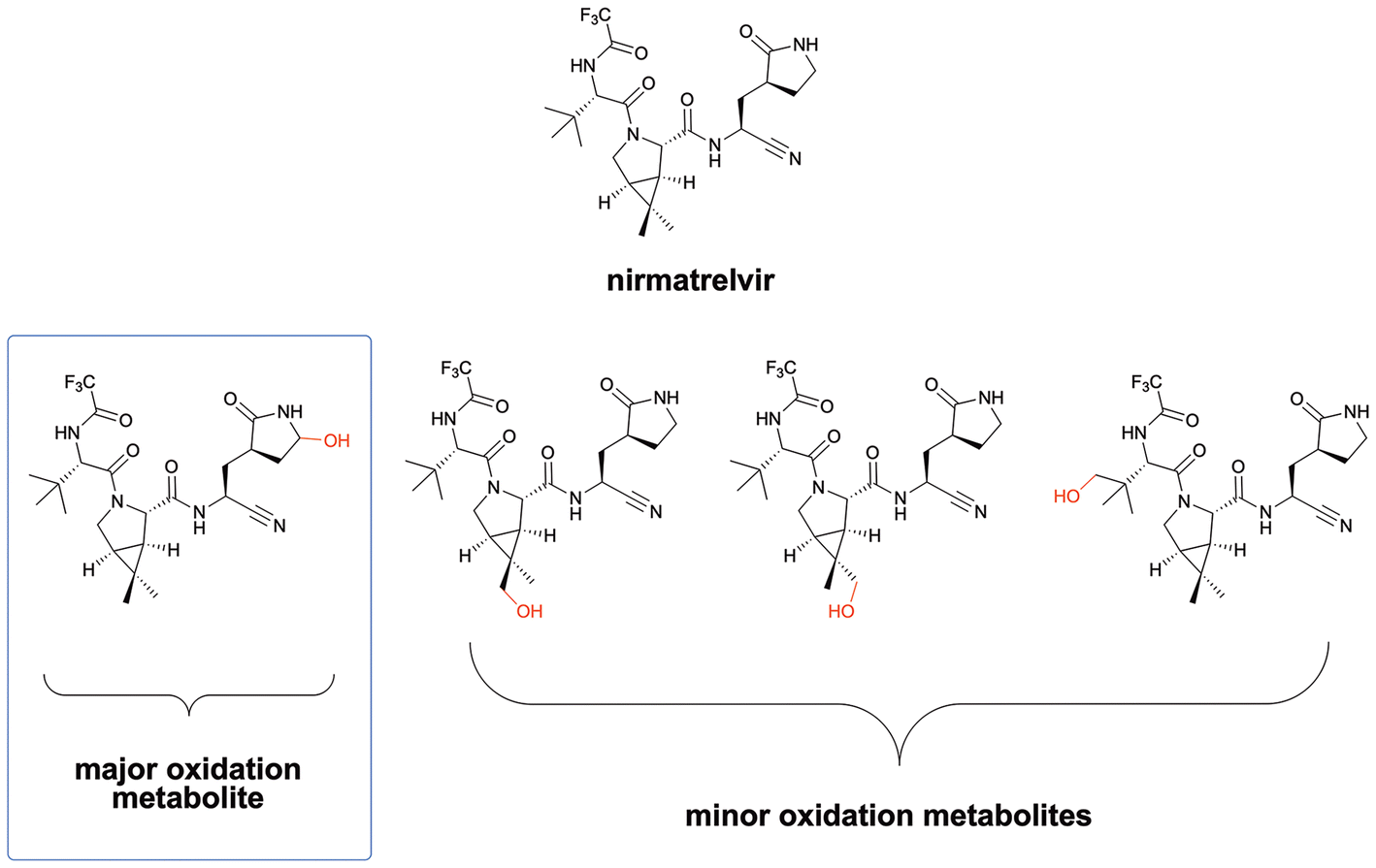 | ||
| Fig. 2 Pfizer reported the major oxidation metabolite to be the one resulting from oxidation on the lactam ring of the cyclic glutamine amino acid, with the minor metabolites resulting from oxidation on the methyl groups.5 | ||
Thanks to the abundance of previously acquired crystal structures of various inhibitors with the main protease, it was previously established that there are key interactions present between the P1 subunit and the residues in the S1 pocket that aid in the binding and effectiveness of the inhibitor.6,13–16 These key interactions are primarily hydrogen bonds to the carbonyl oxygen and the nitrogen of the side chain of the P1 amino acid. Only recently, alternative scaffolds including a 4-thiazole3 and a 4-oxazole moiety17 were challenged as P1 surrogates. Since both examples only included 4-substituted heteroazoles we were thus curious whether differently arranged heteroazoles (2- and 5-substituted rings) would potentially improve binding characteristics. We here report the synthesis and binding data of six novel aldehydes (4a–4f, Fig. 3) and their bisulfites (5a–5f, Fig. 3) that incorporate all three structural oxazole/thiazole isomers (permutations) in P1.
 | ||
| Fig. 3 Target compounds incorporating oxazolyl or thiazolyl amino acids with cyclopropyl alanine and 3F-Cbz. | ||
We wanted to keep the P2 and P3 residues the same throughout, and we selected cyclopropylalanine (P2) and 3-fluoro-benzyloxycarbonyl (3F-Cbz, P3) as those accommodate well the corresponding protease subunits and were generally found to give low IC50 values previously.14
Experimental
Materials and methods
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, v/v) with ninhydrin stain. The solvent was removed under reduced pressure after all the amino acid had been consumed, and this material was used directly in the next step.
:3). NaHSO3 (aq) solution (1 M, 1.0 equiv.) was added to the solution of the aldehyde, the reaction vessel was then sealed, and the reaction mixture was stirred at 50 °C for 3 h. Afterwards, the reaction mixture was filtered to remove any solids, and then the solids were washed with dry ethanol. The filtrate was concentrated under reduced pressure, followed by co-evaporation with diethyl ether (3×) to remove the remaining solvent, furnishing a solid. Diethyl ether was then added, the mixture was sonicated to afford a dispersion, after which, the solid was separated by centrifugation after removing the supernatant.
:1:1, v/v) with ninhydrin stain. The solvent was removed under reduced pressure after all the amino acid had been consumed, and this material was used directly in the next step.
:3). NaHSO3 (aq) solution (1 M, 1.0 equiv.) was added to the solution of the aldehyde, the reaction vessel was then sealed, and the reaction mixture was stirred at 50 °C for 3 h. Afterwards, the reaction mixture was filtered to remove any solids, and then the solids were washed with dry ethanol. The filtrate was concentrated under reduced pressure, followed by co-evaporation with diethyl ether (3×) to remove the remaining solvent, furnishing a solid. Diethyl ether was then added, the mixture was sonicated to afford a dispersion, after which, the solid was separated by centrifugation after removing the supernatant.
Results and discussion
The P1 surrogate unnatural amino acids were synthesized using robust methodology originally developed by Albertson and Archer in the 1940s, using diethyl acetamidomalonate (DEAM) as the glycine equivalent.22 This method begins with the alkylation of DEAM with an electrophilic building block to form the side-chain of the target amino acid. For this, we primarily used the mesylates but discovered that in some cases it was preferable to use the alkyl chloride to provide better yields. We were able to purchase the oxazole and thiazole substituted methanol building blocks to start our synthetic route (Scheme 1). From the alcohols, we could easily convert them to a reactive electrophile using methanesulfonyl chloride or thionyl chloride to yield the mesylate or alkyl chloride in one step each. Alkylation of DEAM was done using sodium ethoxide in freshly distilled ethanol and the electrophilic oxazole or thiazole building block. The crude material was then purified and used as soon as possible, to obtain the best yields. In addition, the 5-oxazole and 5-thiazole mesylate derivatives were found to be the least stable of all. An alternative method was used, which was to convert the alcohols to alkyl chlorides, and use the in situ Finkelstein method in the alkylation of these electrophiles. This modified route improved the yields significantly (about 2-fold).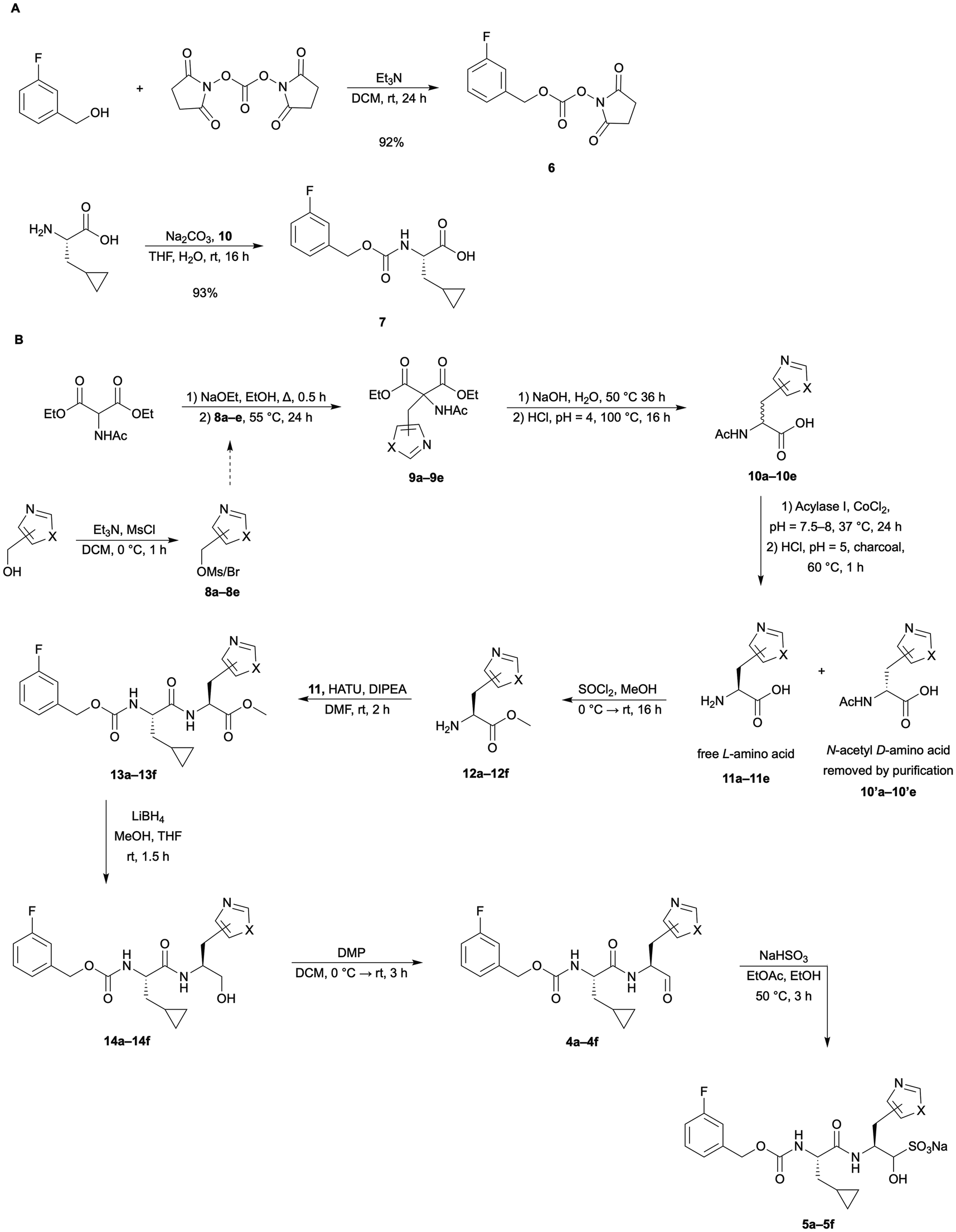 | ||
| Scheme 1 (A) Synthetic steps to intermediate 7, which is used to couple with the synthesized unnatural amino acid methyl esters. (B) Full general synthetic route from DEAM to the final inhibitor compound showing a general thiazole or oxazole side chain. While the aldehyde maintains the stereochemistry in organic solvents, it can epimerize in water at the stereocenter at the aldehyde. The bisulfite is synthesized as a pair of diastereomers, with the epimerization at the bisulfite carbon shown. Slight variations were made to this route for some analogues. See synthetic procedures and SI for specific procedures. | ||
The alkylated DEAM product then underwent hydrolysis and decarboxylation (Scheme 1) to yield the racemic N-acetyl amino acids, which could then be resolved by enzymatic enantioselective hydrolysis by Acylase I.23 Acylase I from Aspergillus melleus is a metalloenzyme that selectively removes the acetyl from the L-enantiomer only.23 After the reaction time is complete, the enzyme was adsorbed by activated charcoal addition, and then through a series of centrifugation, decantation, and extraction we were able to remove the enzyme and charcoal.
The resulting mixture of free L-amino acid and N-acetyl amino acid was then separated using an ion-exchange column loaded with Dowex 50WX8 (cation exchange resin). The resin was equilibrated with 0.01 M HCl, and the sample was acidified to pH = 2. The column was eluted first using MilliQ water to remove any uncharged species, which was followed by the elution of the N-acetyl amino acid and the inorganic salts with 0.5 M HCl. Then using MilliQ water only, the remaining HCl was washed from the column, and finally the free L-amino acid was eluted with an aqueous 2% ammonia solution. Solvent evaporation of the pure product fraction resulted in the pure L-amino acid as a solid. This purification procedure worked well for the oxazolyl derivatives but caused co-elution problems for the thiazolyl analogues. The free L-amino acids were subjected to Marfey's assay to determine if the presumed stereochemistry was correct, and we were pleased to see that the procedure worked as expected with only the pure L-amino acid present (Fig. S1–S5).
The N-acetyl thiazolyl amino acids co-eluted with the free L-thiazolyl amino acids. This is possibly because the nitrogen in the thiazole ring was basic enough to be protonated on the resin and caused it to be retained. It was likely protonated in both the N-acetyl and free forms, causing the separation to be unsuccessful. Reversed-phase high-performance liquid chromatography (HPLC) was then attempted, and while we were able to optimize the methods to obtain some separation, the scale was limited to very small amounts, and we determined that the separation at this step was not completely necessary. Because the N-acetyl amino acid amine was protected with the acetyl group (now an amide), it would not be nucleophilic enough to participate in a HATU-mediated amide bond formation, and we opted to proceed to the next steps with the mixture, with the understanding that only the free L-amino acid would react in the key coupling step.
The mixture of free L-thiazolyl amino acid and the N-acetyl thiazolyl amino acid or the pure free L-oxazolyl amino acid was subjected to thionyl chloride and methanol to convert the carboxylic acid to a methyl ester. The next step was the HATU-mediated coupling with 7, which proceeded as predicted, and at this point, the N-acetyl amino acid methyl ester impurity was able to be mostly extracted out in the work-up step for the thiazoles and fully separated out by flash column chromatography over silica gel. Following the coupling and purification, the remaining steps were reduction of the methyl ester with lithium borohydride and oxidation of the resulting alcohol to obtain the aldehyde. Column purification here yielded one of the target compounds as a pure peptide aldehyde. Upon addition of sodium bisulfite the adduct of each peptide aldehyde was obtained after lyophilisation. This route gave access to five target aldehydes, and their respective bisulfite adducts (4a–4e and 5a–5e, Fig. 2). Target candidate 4f and 5f were synthesised from a commercially available P1 building block following a slightly adapted procedure (see SI).
We used pure enzyme and a 10-amino acid Förster Resonance Energy Transfer (FRET) substrate to determine the IC50 values for each inhibitor. The FRET substrate is a linear sequence of amino acids that matches the natural recognition sequence of the main protease and contains a fluorophore (2-aminobenzoic acid) and a quencher (3-nitrotyrosine) moiety on either end of the peptide (Abz-SVTLQ↓SG-Y(NO2)-R).24 The main protease recognizes the peptide sequence and cleaves it, separating the fluorophore and quencher, which causes fluorescence at a certain wavelength that we can monitor. Concentration dependent monitoring of fluorescence for each inhibitor allows us to calculate the IC50 for each compound. The IC50 values we found (Table 1) reveal activity differences for the different substitution patterns of the side chain and the warhead of the inhibitor.
| Compound | IC50 (nM) | Compound | IC50 (nM) |
|---|---|---|---|
| 4a | 1220 ± 250 | 5a | 290 ± 70 |
| 4b | 20200 ± 2400 |
5b | 1300 ± 200 |
| 4c | 190 ± 45 | 5c | 420 ± 90 |
| 4d | 590 ± 120 | 5d | 270 ± 70 |
| 4e | 12200 ± 1800 |
5e | 11700 ± 2300 |
| 4f | 500 ± 100 | 5f | 380 ± 60 |
In nearly all cases, the bisulfite compound (5a–5f) has a lower IC50 value than its aldehyde analogue (4a–4f), indicating that the pro-drug versions of the compounds provide some benefit over the aldehydes. The substitution pattern also appears to play a large role in the efficiency of the inhibitor. Comparing compound sets 4a–4c and 5a–5c, all of these are oxazoles with the only difference being where the methylene is attached to the ring. Compounds 4b and 5b (with the substitution at the 5-position) appear to have a much higher IC50 value (about 3–4 fold higher) than the others. This same pattern appears with the thiazoles, though the 5-thiazole performs even worse than the 5-oxazole. The 2- and 4-substituted rings do not have as large of a difference, but the data does show that the 2-substituted rings may be the best performing examples of the bisulfite set, with compounds 5a and 5d exhibiting the lowest IC50 values of about 290 and 270 nM, respectively. This trend is supported by the Morrison Ki values determined for the bisulfite compounds (Table 2). The lowest Ki of the oxazoles is found for the 2-substituted heteroaromatic ring (5a), and the same is true of the thiazoles (5d). The lowest IC50 value overall was found for 4c with 190 nM, the 4-substituted oxazole aldehyde analogue. Because of the benefit of increased stability with the bisulfite, 5a and 5d would likely be preferable compounds, as the IC50 values are still very similar. In terms of oxazoles versus thiazoles, there does not seem to be a large difference, they exhibit similar values, although the oxazoles were more straightforward to synthesize, as they were easily purified with the ion-exchange column at the free amino acid stage.
| Compound | K i (nM) |
|---|---|
| 5a | 250 ± 45 |
| 5b | 970 ± 160 |
| 5c | 350 ± 75 |
| 5d | 185 ± 40 |
| 5e | 4530 ± 850 |
| 5f | 250 ± 70 |
We then used the best performing oxazole and thiazole bisulfite inhibitors, 5a and 5d, to test metabolic stability with cytochrome P450 3A4 (CYP3A4), with GC376 as a comparison. Each inhibitor was assayed in duplicate with controls to determine how much of the parent compound was remaining after 2 h of incubation with the oxidative enzyme (Table 3). GC376 was found to be the most stable with CYP3A4 of the three tested compounds. The oxazole, 5a appeared to be more stable than the thiazole 5d. Both 5a and 5d are similar to GC376, with more than 50% of the inhibitor still unoxidized after the 2 h incubation. In addition, we preformed liquid chromatography tandem mass spectrometry (LC-MS/MS) on the oxidized species detected by LC-MS (+16 m/z for the addition of an oxygen atom). Through analysis of the fragmentation patterns, we were able to determine that the oxidation was primarily taking place in the P1/warhead part of all three compounds. We have previously shown that oxidation of the aldehyde to the carboxylic acid occurs with the CYP3A4 enzyme, which may be occurring here as well.10
| Compound | % of compound remaining after 2 h |
|---|---|
| GC376 | 79 |
| 5a | 66 |
| 5d | 53 |
Conclusions
Based on our data, 2-substituted oxazole and thiazole moieties outperform 4- and 5-substituted heteroazoles as chemical P1 surrogates in SARS-CoV-2 Mpro inhibitor design, as illustrated by compounds 5a and 5d. These inhibitors are comparable to other SARS-CoV-2 inhibitors in literature in terms of IC50 data (270–290 nM), metabolic stability, and present alternative chemical motifs from the commonly used cyclic glutamine derivatives (as in 1 and 2). Because these new compounds stray from the commonly used scaffold of P1, to a novel P1 amino acid, these inhibitors may also have potential in the future, as an alternate therapeutic against mutants of the SARS-CoV-2 main protease that become immune to the current treatments. This approach adds to pandemic preparedness by expanding the chemical scope by usage of alternative scaffolds in the design of anti-coronaviral compounds.Author contributions
Zijie Liu: investigation, visualization, writing (review and editing). Tayla J. Van Oers: investigation, writing (original draft, review, and editing). Conrad Fischer: investigation, supervision, writing (review and editing). Anna Nguyen: investigation. Johnson Chew: investigation. John C. Vederas: supervision, funding acquisition, conceptualization.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: Fig. S1–S13, NMR spectra and further experimental details. See DOI: https://doi.org/10.1039/d6ob00697c.Ref. 25 is cited in the SI.
Acknowledgements
We would like to thank Nhat Pham for his assistance with IC50 testing of inhibitors. In addition, we thank Béla Reiz for his assistance with LC-MS/MS analysis. We gratefully acknowledge support by the Canadian Institutes of Health Research (CIHR) (J. C. V.: grant PS186126) and Alberta Ministry of Technology and Innovation through SPP-ARC (Striving for Pandemic Preparedness–The Alberta Research Consortium (J. C. V.)).References
- W. Vuong, M. B. Khan, C. Fischer, E. Arutyunova, T. Lamer, J. Shields, H. A. Saffran, R. T. McKay, M. J. van Belkum, M. A. Joyce, H. S. Young, D. L. Tyrrell, J. C. Vederas and M. J. Lemieux, Nat. Commun., 2020, 11, 4282 CrossRef CAS PubMed.
- Q. Hu, Y. Xiong, G. H. Zhu, Y. N. Zhang, Y. W. Zhang, P. Huang and G. B. Ge, Med. Commun., 2022, 3, 151 Search PubMed.
- J. R. Feys, K. Edwards, M. A. Joyce, H. A. Saffran, J. A. Shields, K. Garcia, D. L. Tyrrell and C. Fischer, ACS Med. Chem. Lett., 2024, 15, 2046–2052 CrossRef CAS PubMed.
- R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski, L. Y. Li, J. W. Lockner, J. Lou, M. T. Marra, L. J. Mitchell, B. W. Murray, J. A. Nieman, S. Noell, S. P. Planken, T. Rowe, K. Ryan, G. J. Smith, J. E. Solowiej, C. M. Steppan and B. Taggart, J. Med. Chem., 2020, 63, 12725–12747 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. A. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 Search PubMed.
- P. Chen, T. J. Van Oers, E. Arutyunova, C. Fischer, C. Wang, T. Lamer, M. J. van Belkum, H. S. Young, J. C. Vederas and M. J. Lemieux, JACS Au, 2024, 4, 3217–3227 CrossRef CAS PubMed.
- A. C. Galasiti Kankanamalage, Y. Kim, A. D. Rathnayake, K. R. Alliston, M. M. Butler, S. C. Cardinale, T. L. Bowlin, W. C. Groutas and K. O. Chang, J. Med. Chem., 2017, 60, 6239–6248 CrossRef CAS PubMed.
- Y. L. Janin, RSC Med. Chem., 2023, 15, 81–118 RSC.
- C. M. N. Allerton, J. T. Arcari, L. M. Aschenbrenner, M. Avery, B. M. Bechle, M. A. Behzadi, B. Boras, L. M. Buzon, R. D. Cardin, N. R. Catlin, A. A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, K. A. Farley, R. A. Ferre, S. S. Gernhardt, S. A. Gibson, S. E. Greasley, S. R. Greenfield, B. L. Hurst, A. S. Kalgutkar, E. Kimoto, L. F. Lanyon, G. H. Lovett, Y. Lian, W. Liu, L. A. Martínez Alsina, S. Noell, R. S. Obach, D. R. Owen, N. C. Patel, D. K. Rai, M. R. Reese, H. A. Rothan, S. Sakata, M. F. Sammons, J. G. Sathish, R. Sharma, C. M. Steppan, J. B. Tuttle, P. R. Verhoest, L. Wei, Q. Yang, I. Yurgelonis and Y. Zhu, J. Med. Chem., 2024, 67, 13550–13571 CrossRef CAS PubMed.
- T. J. Van Oers, A. Piercey, A. Belovodskiy, B. Reiz, B. L. Donnelly, W. Vuong, M. J. Lemieux, J. A. Nieman, K. Auclair and J. C. Vederas, Org. Lett., 2023, 25, 5885–5889 CrossRef CAS PubMed.
- D. Jochmans, C. Liu, K. Donckers, A. Stoycheva, S. Boland, S. K. Stevens, C. De Vita, B. Vanmechelen, P. Maes, B. Trüeb, N. Ebert, V. Thiel, S. De Jonghe, L. Vangeel, D. Bardiot, A. Jekle, L. M. Blatt, L. Beigelman, J. A. Symons, P. Raboisson, P. Chaltin, A. Marchand, J. Neyts, J. Deval and K. Vandyck, mBio, 2023, 14, e02815–e02822 Search PubMed.
- C. Fischer, J. Lu, M. J. Van Belkum, S. Demmon, P. Chen, C. Wang, T. J. Van Oers, T. Lamer, M. J. Lemieux and J. C. Vederas, RSC Med. Chem., 2025, 16, 5032–5040 RSC.
- E. Arutyunova, M. B. Khan, C. Fischer, J. Lu, T. Lamer, W. Vuong, M. J. van Belkum, R. T. McKay, D. L. Tyrrell, J. C. Vederas, H. S. Young and M. J. Lemieux, J. Mol. Biol., 2021, 433, 167003 CrossRef CAS PubMed.
- W. Vuong, C. Fischer, M. B. Khan, M. J. van Belkum, T. Lamer, K. D. Willoughby, J. Lu, E. Arutyunova, M. A. Joyce, H. A. Saffran, J. A. Shields, H. S. Young, J. A. Nieman, D. L. Tyrrell, M. J. Lemieux and J. C. Vederas, Eur. J. Med. Chem., 2021, 222, 113584 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- V. Kumar, J. Zhu, B. C. Chenna, Z. A. Hoffpauir, A. Rademacher, A. M. Rogers, C.-T. Tseng, A. Drelich, S. Farzandh, A. L. Lamb and T. D. Meek, J. Am. Chem. Soc., 2025, 147, 1631–1648 CrossRef CAS PubMed.
- Y. J. Lee, Y. Kim, H. Kim, J. Choi, G. H. Noh, K. S. Lee, J. Lee, C. H. Choi, S. H. Kim and J. Seo, Inorg. Chem., 2023, 62, 10279–10290 CrossRef CAS PubMed.
- D. Migulin, Y. Vysochinskaya, M. Buzin, A. Bakirov, G. Cherkaev and O. Shchegolikhina, J. Photochem. Photobiol., A, 2021, 407, 113003 Search PubMed.
- C. N. Hsiao, M. R. Leanna, L. Bhagavatula, E. De Lara, T. M. Zydowsky, B. W. Horrom and H. E. Morton, Synth. Commun., 1990, 20, 3507–3517 CrossRef CAS.
- H. K. Chenault, J. Dahmer and G. M. Whitesides, J. Am. Chem. Soc., 1989, 111, 6354–6364 CrossRef CAS.
- N. F. Albertson and S. Archer, J. Am. Chem. Soc., 1945, 67, 308 CrossRef CAS.
- S.-C. J. Fu and S. M. Birnbaum, J. Am. Chem. Soc., 1953, 75, 918–920 CrossRef CAS.
- J. E. Blanchard, N. H. Elowe, C. Huitema, P. D. Fortin, J. D. Cechetto, L. D. Eltis and E. D. Brown, Chem. Biol., 2004, 11, 1445–1453 CrossRef CAS PubMed.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |