
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guanidine-based azines from N-heterocyclic carbene (NHC)-derived selenoureas and diazo compounds: synthesis, structural diversification, and biological evaluation
Dimitrios K. Giannopoulosa,
Ilias Papantzimasb,
Spyridon A. Ferikogloua,
Efstathios Tonisa,
Nikolaos V. Tzouras ac,
Athanasios Pseftogasb,
Fady Nahracd,
Steven P. Nolanc,
Aristides G. Eliopoulos*b and
Georgios C. Vougioukalakis*a
ac,
Athanasios Pseftogasb,
Fady Nahracd,
Steven P. Nolanc,
Aristides G. Eliopoulos*b and
Georgios C. Vougioukalakis*a
aDepartment of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, 15771, Athens, Greece. E-mail: vougiouk@chem.uoa.gr
bDepartment of Biology, School of Medicine, National and Kapodistrian University of Athens, Mikras Asias 75, 11527, Athens, Greece
cDepartment of Chemistry and Centre of Sustainable Chemistry, Ghent University, Krijgslaan 289, S-3, 9000 Ghent, Belgium
dVITO (Flemish Institute for Technological Research), Boeretang 200, 2400 Mol, Belgium
First published on 26th May 2026
Abstract
Guanidine-based azines combine two functional groups in their structure, both of which are widely encountered in medicinal chemistry. Such molecules are accessible, among others, via the versatile reaction of N-heterocyclic carbene (NHC)-derived selenoureas with diazo compounds. Our present work introduces a highly efficient version of this transformation, simultaneously expanding its structural scope beyond the currently known ester-containing analogues. By utilizing a broad scope of diazo compounds, bearing amide, ketone, or heteroatom rich functionalities, we access various azine derivatives of biological significance. Key innovations of this new synthetic protocol include the use of polystyrene-supported triphenylphosphine as a recyclable reagent, enabling a streamlined purification process, eliminating the need for column chromatography, and allowing its repeated use under ambient conditions. Post-synthetic functionalization strategies further expand the structural diversity of the azine scaffold, and preliminary biological evaluation identifies several compounds with in vitro anti-cancer activity. The findings highlight guanidine-based azines as a promising scaffold for further chemical and biological exploration.
Introduction
Azines, formally known as N–N linked diimines (RR′C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NCRR′), represent a versatile and widely studied scaffold in modern synthetic chemistry. These compounds have found extensive applications in organic synthesis, materials science, and medicinal chemistry.1,2 More specifically, they exhibit a wide range of biological activities, including antibacterial,3 antiviral,4 and anticancer properties,5 making them valuable targets for pharmaceutical innovation.6 Azines are also commonly used as intermediates in the synthesis of more complex bioactive molecules, such as the generation of heterocycles.7 Moreover, they play a pivotal role in materials science, as chemical sensors,8 and as structural units of conducting polymers.9 As a result, the development of efficient synthetic methodologies towards azines remains an important goal in the field of organic chemistry.2,10,11
N–NCRR′), represent a versatile and widely studied scaffold in modern synthetic chemistry. These compounds have found extensive applications in organic synthesis, materials science, and medicinal chemistry.1,2 More specifically, they exhibit a wide range of biological activities, including antibacterial,3 antiviral,4 and anticancer properties,5 making them valuable targets for pharmaceutical innovation.6 Azines are also commonly used as intermediates in the synthesis of more complex bioactive molecules, such as the generation of heterocycles.7 Moreover, they play a pivotal role in materials science, as chemical sensors,8 and as structural units of conducting polymers.9 As a result, the development of efficient synthetic methodologies towards azines remains an important goal in the field of organic chemistry.2,10,11
In addition to the success of azines across diverse applications, their combination with a guanidine framework has increased potential in the context of biologically relevant molecules. Regarding the synthesis of such molecules, the Ma group has reported that 2,2,2-trifluorodiazoethane reacts with aldehydes or ketones under “aza-Wittig” type conditions.12,13 In a related study, they showed that the same diazo reagent also reacts with in situ generated NHCs (from azolium salts), providing a range of trifluoromethylated products.14 In order to target guanidine-based azine frameworks tailored towards biological applications, we have focused on developing a synthetic protocol with wide functional group tolerance, by utilizing the reactivity of selenoureas.15,16
Selenoureas exhibit unique reactivity, due to the presence of selenium,17 including enhanced nucleophilicity and strong coordination to metal centers.18–21 Given their useful chemical properties, selenoureas have attracted interest in various fields, including catalysis,22,23 medicinal chemistry,24 and the development of novel functional materials.25 N-Heterocyclic carbene (NHC)-derived selenoureas comprise a subclass of selenoureas, where a NHC moiety is directly bound to the selenium center.26 These compounds are typically synthesized via the reaction of NHCs with elemental selenium.27,28 The selenium center in these molecules serves as a spectroscopic probe for the electron-accepting properties of the parent NHCs, while also enabling synthetic and other applications.29–35
Among the various methodologies developed for the synthesis of azines,14,36–41 the use of N-heterocyclic carbene-derived selenoureas in combination with diazo reagents has recently emerged as a versatile strategy. Specifically, we introduced a methodology furnishing guanidine-based azines in air from diazo reagents and selenoureas, relying on a reaction reminiscent of the aza-Wittig reaction. This transformation is mediated by a phosphine, yet follows a mechanism that is distinct of the aza-Wittig reaction, while it is enabled by the presence of selenium (Scheme 1).15
 | ||
| Scheme 1 The known selenourea/diazo reagent-based protocol and the herein reported synthetic strategy. | ||
In our previous study (Scheme 1), we investigated the mechanistic details of this transformation using a combination of experimental and computational methods. Triphenylphosphine and the diazo compound were shown to reversibly generate an aza-ylide, while selenourea and phosphine react to produce a free N-heterocyclic carbene (NHC). The free carbene is then rapidly and irreversibly trapped by the diazo species to form the azine product. The transformation shown herein is expected to follow the same proposed mechanism.15
Our present work significantly broadens the structural diversity of azines, by introducing a wide range of functional groups, including amides, ketones and hetero-atom rich motifs, such as phosphorus-containing moieties and scaffolds bearing both an amide and an ester group. This demonstrates the remarkable functional group tolerance of the synthetic method, as the scope includes several base-sensitive functionalities. In addition, we explore the post functionalization of these scaffolds, introducing iodide or bromide substituents. Importantly, a polystyrene-supported triphenylphosphine (PS-PPh3) resin promotes the reaction, enabling recycling and reuse, in addition to product isolation without the requirement for column chromatography. Notably, the reaction works well under ambient conditions, without the need for dry solvents or an inert atmosphere, making it operationally simple. This synthetic approach advances the current state-of-the-art significantly, granting access to previously unexplored azine derivatives. The biological evaluation of the synthesized compounds shows very promising results, showcasing these molecules as potential pharmacologically relevant scaffolds and providing insight into the structure–activity relationship of functionalized azines.
Results and discussion
Our previous work,15 demonstrated that NHC-derived selenoureas couple efficiently with ester-functionalized diazo compounds leading to azine derivatives bearing a guanidine core. The optimal conditions of this protocol include the use of triphenylphosphine in toluene, at 110 °C for 16 h. These conditions comprise the starting point of our present approach that targets the construction of novel guanidine families.With the optimal conditions established, we turned to a systematic evaluation of the substrate scope of the reaction, by studying the influence of diazo compounds bearing electron-withdrawing and/or electron-donating groups. We initially targeted a diazo compound bearing a phosphorus (phosphonate ester group) functionality, which underwent the reaction smoothly to afford the desired product in high isolated yield (90%) (Scheme 2, 1). Introduction of a carbonyl group on the diazo substrate, was also well tolerated. Diazo compounds bearing a ketone or an ester group provided the desired azine products in very good isolated yields, as well (84–90%, Scheme 2, 2–5).
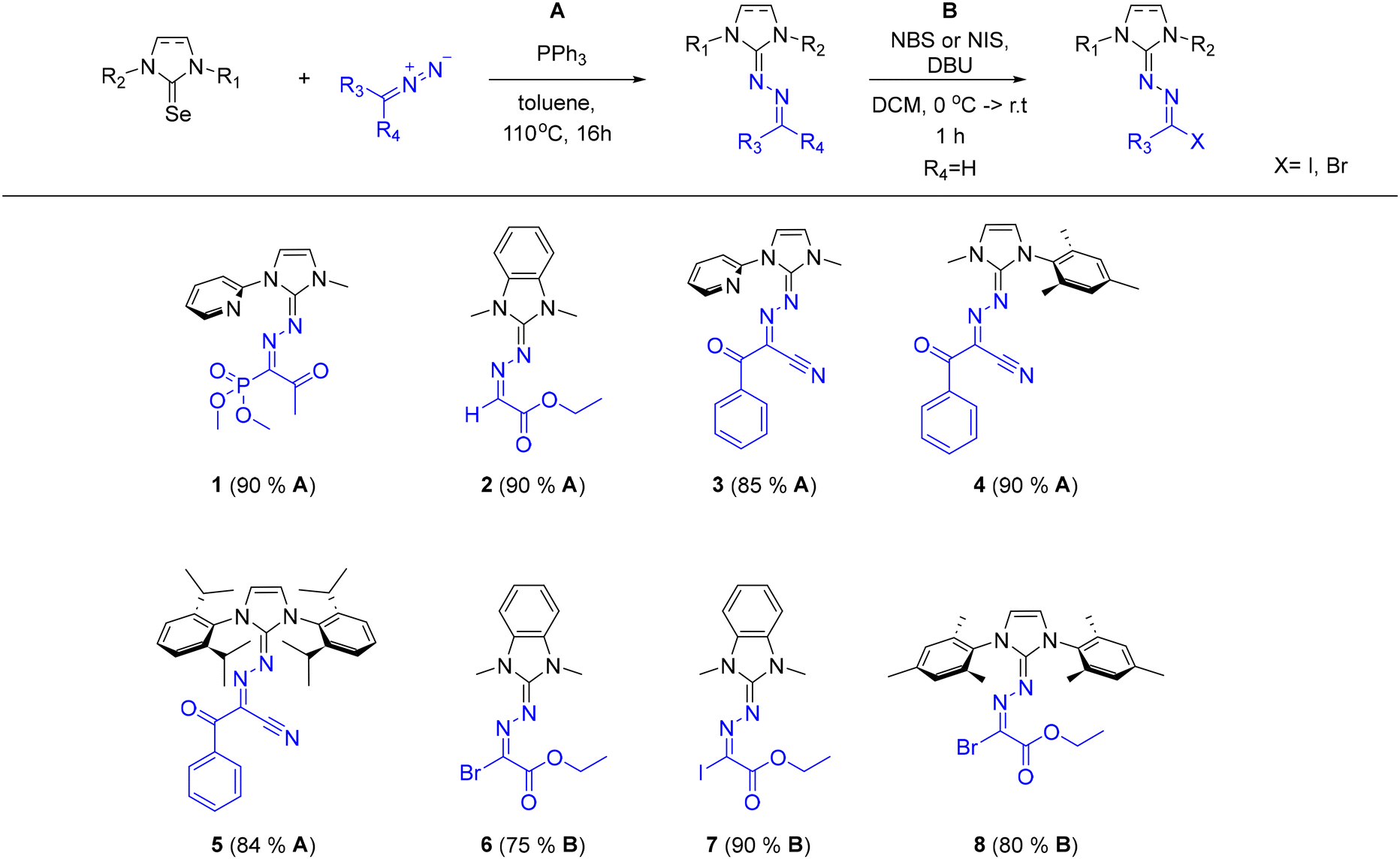 | ||
| Scheme 2 Final azine products bearing different functionalities. Conditions A: selenourea (0.125 mmol, 1 equiv.), diazo compound (1.2 equiv.), triphenylphosphine (3.0 equiv.), 0.5 mL of toluene, reflux, 16 h. Conditions B: azine product (0.046 mmol, 1 equiv.), DBU (3 equiv.), NIS or NBS (2 equiv.), 0.3 mL of DCM, 1 h. Isolated yields are shown in parentheses. | ||
In addition, following the synthesis of the latter products, post-functionalization of two azines derived from the same diazo precursor (2) was carried out using N-halosuccinimides (Scheme 2, 6–8). Specifically, halogenated azines bearing bromide or iodide substituents were efficiently obtained in very good to excellent isolated yields (75–90%), demonstrating the utility of the method in late-stage diversification. Encouraged by this reactivity, and the opportunity for further functionalization, we next sought to probe alternative diazo component, and define the structural limits of this transformation.
We continued our study by employing the in situ generated diazo compounds (1-(diazomethyl)-4-nitrobenzene, phenyldiazomethane and diazodiphenylmethane). However, these substrates, when reacted with the NHC-derived selenoureas to provide the corresponding azines, displayed low reactivity under the above reaction conditions. In only one case the product was identified in the reaction mixture by 1H NMR in a 20% conversion (Scheme S1 and Fig. S3, SI), but its isolation and purification via column chromatography was not possible. We attribute this low reactivity to rapid competitive decomposition pathways of these benzylic-diaryl diazo compounds under the conditions employed. This outcome highlighted the need for diazo partners bearing stabilizing electron-withdrawing substituents that could promote productive coupling.
Accordingly, a series of amide-containing diazo compounds bearing electron-withdrawing cyano and/or carbonyl groups were synthesized and investigated (Scheme 3). These compounds exhibit high reactivity, affording the desired azines in very good isolated yields (70–90%), demonstrating excellent tolerance towards amide-containing functionalities under the reaction conditions. Specifically, diazo compounds bearing a cyano-substituted amide motif perform particularly well, affording the corresponding products in high isolated yields (90%, Scheme 3, 9–12), while increasing heteroatom content. Changing the cyclic amide environment was also very well tolerated (Scheme 3, 13, 14).
 | ||
| Scheme 3 Substrate scope using amide-containing diazo compounds. Conditions: selenourea (0.125 mmol, 1 equiv.), diazo compound (1.2 equiv. or 2 equiv. for saturated Selenoureas), triphenylphosphine (3.0 equiv.), 0.5 mL of toluene, reflux, 16 h. | ||
In addition, a variety of saturated and unsaturated NHC-derived selenoureas were screened. Using unsaturated selenoureas, the azines were obtained in high yields (84–90%) by employing only 1.2 equivalents of diazo reagent (Scheme 3, 9–14, 18, 20–24). In contrast, saturated selenoureas required an increased diazo loading of 2.0 equivalents, to achieve good to very good yields (70–90%, Scheme 3, 15–17, 19). This comparison suggests that saturated NHC-derived selenoureas are less reactive under identical conditions, but the transformation remains broadly effective once diazo stoichiometry is adjusted.
Substrates incorporating an additional carbonyl functionality in the diazo counterpart (Scheme 3, 18–22), also reacted efficiently, affording azines in excellent yields (85–90%). Moreover, amide-containing diazo-compounds bearing an additional ester group remained fully compatible, affording the desired azines in very high isolated yields (85–90%, Scheme 4, 23, 24).
 | ||
| Scheme 4 Substrate scope using PS-PPh3. Conditions: selenourea (0.125 mmol, 1 equiv.), diazo compound (1.2 equiv.), polystyrene-supported triphenylphosphine (2.0 equiv.), 0.9 mL of toluene, reflux, 16 h. | ||
To streamline the purification process and render the azine synthesis more straightforward and user-friendly, we explored the use of a polystyrene-supported triphenylphosphine (PS-PPh3) resin, as a solid phase promoter for our system. This approach leads to the desired products in very good isolated yields (80–90%) under ambient atmosphere without the need for column-chromatography (Scheme 4, 25, 2, 6, 10, 24). Upon reaction completion, the resin was recovered by filtration and found to contain a mixture of phosphine oxide (PS-PPh3O) together with a selenium-bound species (PS-PPh3Se). A practical regeneration protocol was also established, by using TDMS (1,1,3,3-tetramethyldisiloxane) in the presence of Ti(OEt)4 as an additive, enabling efficient reduction of the oxidized/selenated resin. This treatment restored the active PS-PPh3 functionality and allowed the resin to be reintroduced into subsequent reactions (Scheme S3 in SI for recyclization process, Fig. 1 and Fig. S54–S56, SI). This recycling protocol was first validated using the synthesis of compound 25 (Fig. S52–S54, SI). The corresponding 1H and 31P-NMR spectra were recorded from the condensed filtrates obtained after removal of the solid-supported resin. These data show that the reaction proceeds successfully for at least four consecutive cycles. After the fourth cycle, a minor phosphorus-containing byproduct, assignable to oxidized phosphine-derived species, was detected in the filtrate (Fig. S55, S56, SI), likely due to the competitive oxidation of the phosphine under ambient atmosphere, which becomes increasingly difficult to reverse as the resin ages. By the fifth cycle, an increased proportion of phosphorus-containing byproducts was detected in the 1H-NMR and 31P-NMR spectra (Fig. S54–S56, SI), and further recycling was discontinued.
 | ||
| Fig. 1 Recycling performance of polystyrene-supported triphenylphosphine over five cycles for the synthesis of compound 25. | ||
In vitro anti-cancer activity of guanidine-based azines
To evaluate the biological activity of our new scaffolds, we performed an initial viability screen of the entire family of compounds at 10 and 30 μM in A2780 ovarian carcinoma cells, using an MTT conversion assay, with the chemotherapeutic agent cisplatin as a positive control. Compounds 5, 7, 14 and 16 each reduced the MTT signal by more than 50% at both concentrations (Fig. 2A and Fig. S57A–C, SI) and were selected for follow-up viability analysis in VM-CUB-1 bladder carcinoma cells. As shown in Fig. 2B and Fig. S57D in the SI, all four compounds also decreased MTT conversion in VM-CUB-1, albeit to a lesser extent than cisplatin. | ||
| Fig. 2 Effects of selected guanidine-based azines on cancer cell death. (A) & (B) Viability of A2780 ovarian carcinoma (A) and VM-CUB-1 bladder carcinoma (B) cell cultures following 48 hour exposure to 10 μM or 30 μM of compound 16 or, as control, 10 μM cisplatin. Viability was assessed by MTT conversion and is depicted as % reduction relevant to control untreated cultures which were given the arbitrary value of 100%. Data is the mean (±SD) of 4 independent assessments. (C) Compound 16 does not induce major changes in A2780 cell cycle distribution. Representative flow cytometry profiles of DNA content of A2780 cells treated for 24 h with compound 16 or cisplatin (CP) vs. untreated controls (CNT) (left panel) and collective data from 3 independent assessments (right panel) are shown. (D) Compound 16 induces apoptosis in A2780 cells. The left panel shows representative flow cytometry profiles of A2780 cells treated with compound 16 or cisplatin vs. untreated controls, following staining with FITC-labelled Annexin-V (x-axis) and PI (y-axis). Early apoptotic cells are Annexin-V+/PI− (bottom right quadrant), late apoptotic are Annexin-V+/PI+ (upper right), and necrotic are Annexin-V−/PI+ (upper left quadrant). Collective data from 3 independent experiments (mean values) is shown in the right panel. (E) Compound 16 induces p53 accumulation. A2780 cell cultures were exposed to 10 μM or 30 μM of 16 for 6 h or left untreated, followed by immunoblotting for p53, with β-actin as loading control. Cisplatin treatment (10 μM, 6 h) was used as positive control. Data shown is representative of 4 independent experiments. | ||
Because MTT cannot distinguish growth arrest from cell death, we assessed putative effects of compound 16 on A2780 cell cycle distribution using propidium iodide (PI) staining coupled with flow cytometry. Compound 16 did not produce a consistent accumulation of A2780 cells in any specific cell cycle phase (Fig. 2C). Similarly, compounds 5, 7 and 14 failed to elicit phase-specific accumulation despite lowering MTT readouts (Fig. S58, SI). As expected,42 cisplatin induced a robust G2-phase arrest (Fig. 2C).
To determine whether loss of viability was associated with apoptosis, we performed Annexin V/PI staining coupled with flow cytometric analysis. Compound 16 increased both early and late apoptotic fractions in A2780 cultures, with the overall extent of cell death at 10 μM remaining ∼50% lower than that observed with cisplatin (Fig. 2D).
Induction of p53 is a canonical response to DNA damage and ribosomal/nucleolar stress, capable of triggering cell-cycle arrest or apoptosis.43 We therefore examined p53 protein levels by immunoblotting in A2780 cells and found concentration-dependent p53 accumulation. At 30 μM, compound 16 increased p53 to levels comparable to those observed with 10 μM cisplatin (Fig. 2E).
Overall, compounds 5, 7, 14, and 16 reduced the viability of A2780 ovarian and VM-CUB1 bladder carcinoma cells, which harbor wild-type and mutated TP53, respectively. In A2780 cells treated with compound 16, p53 induction and cell death occurred in the absence of detectable accumulation in specific phases of the cell cycle (Fig. 2C & D), suggesting that compound 16 preferentially engages a pro-apoptotic program rather than a cytostatic checkpoint. Apoptosis induction in the absence of phase-specific arrest has been documented for other classes of anticancer agents, including TRAIL-receptor agonists,44 BCL-2 inhibitors,45,46 and proteasomal inhibitors such as epoxomicin.47 The structural modularity of the azine scaffold and the synthetic accessibility of diverse derivatives position this chemotype as a promising platform for systematic structure – activity exploration and mechanistic investigation.
Experimental information
Experimental procedure for the synthesis of diazo compounds
Diazo compounds were synthesized according to published procedures.48–50 An amide (1 mmol) was dissolved in 2 mL of dry CH3CN. Under an inert atmosphere, a solution of the appropriate azide (1 mmol) (triflic azide,48 4-acetamidobenzenesulfonyl azide,49 4-toluenesulfonyl azide50) was added dropwise at 0 °C (ice-water bath). Dry trimethylamine (2 mL) was then added, and the reaction mixture was stirred at room temperature for 18 h. The solvent was removed under reduced pressure, and the crude mixture was purified by silica gel column chromatography (petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylacetate 90:10) to afford the desired diazo compound.
:ethylacetate 90:10) to afford the desired diazo compound.
Experimental procedure for the synthesis of azine derivatives
To a 4 mL vial equipped with a magnetic stirrring bar and a septum sealed screwcap, were added the NHC-based selenourea (0.125 mmol, 1 equiv.), PPh3 (0.375 mmol, 3 equiv.), or PS-PPh3 (0.25 mmol, 2 equiv.), toluene (0.5 mL) and the diazo compound (0.15 mmol, 1.2 equiv.). The reaction mixture was stirred at 110 °C for 16 h in air. The solvent was then removed under reduced pressure, and the crude mixture was purified by flash column chromatography on silica gel (ethyl acetate:petroleum ether, 5:1 to 1:1) to afford the pure product.
Conclusions
We have developed a highly efficient synthetic methodology for the construction of guanidine-based azines via the reaction of diazo compounds with N-heterocyclic carbene (NHC)-derived selenoureas. We investigated the effects of different functionalities and broadened the substrate scope beyond ester-based diazo compounds, to construct a variety of new scaffolds. The ability to couple selenoureas with diazo compounds bearing enolizable ketones and other base-sensitive functional groups, is an important feature of this method. Additionally, attempts to introduce further complexity, through the functionalization of the azine core with halogens (Br, I), yielded the desired products in high yields, demonstrating the synthetic potential of the methodology. A significant finding is the use of recyclable polysterene-supported triphenylphosphine (PS-PPh3), which allow the synthesis of high purity products, without the need for column chromatography. Preliminary biological evaluation revealed that several azine derivatives reduce cancer cell viability in vitro. In particular, compound 16 induces p53 accumulation and apoptosis without detectable cell-cycle arrest, distinguishing its cellular response from that of canonical DNA-damaging agents such as cisplatin. These findings identify guanidine-based azines as promising scaffolds for further structure–activity studies and mechanistic investigation.Author contributions
D. K. G.: investigation, formal analysis, data curation, writing – original draft, I. P.: investigation, data curation, writing – original draft. S. A. F.: investigation, writing – original draft, E. T.: investigation, writing – review and editing, N. V. T.: conceptualization, writing – review and editing, A. P.: investigation, formal analysis, writing – review and editing, F. N.: conceptualization, writing – review and editing, S. P. N.: conceptualization, writing – review and editing A. G. E.: conceptualization, funding acquisition, resources, supervision, writing – review and editing, G. C. V.: conceptualization, funding acquisition, resources, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data underlying this study are available in the published article and its supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6ob00548a.Acknowledgements
The research project was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I) under the “1st Call for H.F.R.I. Research Projects to support Faculty Members & Researchers and the procurement of high-cost research equipment grant” (Project Number: 16). We also acknowledge Prof. Nikos Thomaides for the High Resolution Mass Spectra (HRMS) that were recorded at the core facility of the National and Kapodistrian University of Athens. We further thank Prof. David J. Nelson for valuable discussions regarding the use of PS-PPh3.References
- J. Safari and S. Gandomi-Ravandi, RSC Adv., 2014, 4, 46224–46249 RSC.
- S. S. Chourasiya, D. Kathuria, A. A. Wani and P. V. Bharatam, Org. Biomol. Chem., 2019, 17, 8486–8521 RSC.
- V. B. Kurteva, S. P. Simeonov and M. Stoilova-Disheva, Pharmacol. Pharm., 2011, 02, 1–9 CrossRef CAS.
- T. George, C. L. Kaul, R. S. Grewal and D. V. Mehta, J. Med. Chem., 1971, 14, 909–913 CrossRef CAS PubMed.
- C. Liang, J. Xia, D. Lei, X. Li, Q. Yao and J. Gao, Eur. J. Med. Chem., 2014, 74, 742–750 CrossRef CAS.
- S. M. N. Alasmari, A. Alam, F. Ur Rahman, A. A. Elhenawy, A. Ali, M. Ahmad and M. Khan, Mini-Rev. Med. Chem., 2025, 25, 425–439 CrossRef CAS PubMed.
- J. Boström, A. Hogner, A. Llinàs, E. Wellner and A. T. Plowright, J. Med. Chem., 2012, 55, 1817–1830 CrossRef.
- K. Tiwari, S. Kumar, V. Kumar, J. Kaur, S. Arora and R. K. Mahajan, Spectrochim. Acta, Part A, 2018, 191, 16–26 CrossRef CAS PubMed.
- B. Chaloner-Gill, W. B. Euler, P. D. Mumbauer and J. E. Roberts, J. Am. Chem. Soc., 1991, 113, 6831–6834 CrossRef CAS.
- A. Ramakrishnan, S. S. Chourasiya and P. V. Bharatam, RSC Adv., 2015, 5, 55938–55947 RSC.
- J. O. Bauer, G. Leitus, Y. Ben-David and D. Milstein, ACS Catal., 2016, 6, 8415–8419 CrossRef CAS PubMed.
- F. Zhang, N. Lv, Y. Zheng and J. Ma, Chin. J. Chem., 2018, 36, 723–730 CrossRef CAS.
- F. Zhang, J. Zeng, Y. Tian, Y. Zheng, D. Cahard and J. Ma, Chem. Eur. J., 2018, 24, 7749–7754 CrossRef CAS PubMed.
- R. Guo, Y. Zheng and J.-A. Ma, Org. Lett., 2016, 18, 4170–4173 CrossRef PubMed.
- E. Tonis, N. V. Tzouras, N. Bracho Pozsoni, M. Saab, S. Bhandary, K. Van Hecke, D. J. Nelson, F. Nahra, S. P. Nolan and G. C. Vougioukalakis, Chem. Eur. J., 2024, 30, e202401816 CrossRef CAS PubMed.
- R. Masuda, T. Yano and H. Kusama, Chem. Commun., 2025, 61, 4955–4958 RSC.
- R. A. Hussain, A. Badshah and A. Shah, Appl. Organomet. Chem., 2014, 28, 61–73 CrossRef CAS.
- L. Pruschinski, J. C. Namyslo and A. Schmidt, J. Org. Chem., 2024, 89, 15003–15019 CrossRef CAS PubMed.
- D. J. Nelson, F. Nahra, S. R. Patrick, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2014, 33, 3640–3645 CrossRef CAS.
- M. Saab, F. Nahra and K. Van Hecke, Molbank, 2022, 2022, M1344 CrossRef.
- M. Saab, D. J. Nelson, M. C. Leech, K. Lam, S. P. Nolan, F. Nahra and K. Van Hecke, Dalton Trans., 2022, 51, 3721–3733 Search PubMed.
- K. Srinivas, C. Naga Babu and G. Prabusankar, Dalton Trans., 2015, 44, 15636–15644 RSC.
- W.-G. Jia, Y.-B. Huang, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2008, 5612 RSC.
- M. De Franco, M. Saab, M. Porchia, C. Marzano, S. P. Nolan, F. Nahra, K. Van Hecke and V. Gandin, Chem. Eur. J., 2022, 28, e202201898 CrossRef CAS PubMed.
- D. J. Williams, K. M. White, D. VanDerveer and A. P. Wilkinson, Inorg. Chem. Commun., 2002, 5, 124–126 Search PubMed.
- M. S. S. Jamil and N. A. Endot, Molecules, 2020, 25, 5161 CrossRef CAS.
- M. Saab, D. J. Nelson, N. V. Tzouras, T. A. C. A. Bayrakdar, S. P. Nolan, F. Nahra and K. Van Hecke, Dalton Trans., 2020, 49, 12068–12081 RSC.
- T. Cauwenbergh, T. Scattolin, A. Simoens, N. V. Tzouras, C. V. Stevens and S. P. Nolan, Eur. J. Org. Chem., 2022, 2022, e202101296 Search PubMed.
- E. Tomás-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753–7756 Search PubMed.
- M. Yaqoob, M. Abbasi, H. Anwar, J. Iqbal, M. Asad, A. M. Asiri and M. A. Iqbal, Rev. Inorg. Chem., 2022, 42, 229–238 CrossRef CAS.
- A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS.
- T. Hölzel, M. Otto, H. Buhl and C. Ganter, Organometallics, 2017, 36, 4443–4450 CrossRef.
- G. P. Junor, J. Lorkowski, C. M. Weinstein, R. Jazzar, C. Pietraszuk and G. Bertrand, Angew. Chem., 2020, 132, 22212–22217 Search PubMed.
- M. Banerjee, R. Karri, K. S. Rawat, K. Muthuvel, B. Pathak and G. Roy, Angew. Chem., 2015, 127, 9455–9459 Search PubMed.
- J. Wang, Tetrahedron Lett., 2022, 108, 154135 Search PubMed.
- G. Maas, Angew. Chem., Int. Ed., 2009, 48, 8186–8195 Search PubMed.
- W. Kirmse and M. Kapps, Chem. Ber., 1968, 101, 994–1003 CrossRef CAS.
- Ł. W. Ciszewski, K. Rybicka-Jasińska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432–448 RSC.
- J. N. Gruhin, R. Kim, A. Vasilopoulos, E. A. Voight and E. J. Alexanian, J. Am. Chem. Soc., 2024, 146, 32919–32924 CrossRef CAS PubMed.
- M. P. Doyle, W. H. Tamblyn and V. Bagheri, J. Org. Chem., 1981, 46, 5094–5102 CrossRef CAS.
- T. Toma, J. Shimokawa and T. Fukuyama, Org. Lett., 2007, 9, 3195–3197 CrossRef CAS PubMed.
- A. G. Eliopoulos, D. J. Kerr, J. Herod, L. Hodgkins, S. Krajewski, J. C. Reed and L. S. Young, Oncogene, 1995, 11, 1217–1228 CAS.
- I. Orsolic, D. Jurada, N. Pullen, M. Oren, A. G. Eliopoulos and S. Volarevic, Semin. Cancer Biol., 2016, 37–38, 36–50 CrossRef CAS PubMed.
- H. Ehrhardt, F. Wachter, M. Grunert and I. Jeremias, Cell Death Dis., 2013, 4, e661 Search PubMed.
- R. Kawakatsu, K. Tadagaki, K. Yamasaki and T. Yoshida, Sci. Rep., 2024, 14, 4975 CrossRef CAS PubMed.
- M. Konopleva, R. Contractor, T. Tsao, I. Samudio, P. P. Ruvolo, S. Kitada, X. Deng, D. Zhai, Y.-X. Shi, T. Sneed, M. Verhaegen, M. Soengas, V. R. Ruvolo, T. McQueen, W. D. Schober, J. C. Watt, T. Jiffar, X. Ling, F. C. Marini, D. Harris, M. Dietrich, Z. Estrov, J. McCubrey, W. S. May, J. C. Reed and M. Andreeff, Cancer Cell, 2006, 10, 375–388 CrossRef CAS PubMed.
- J. Sidor-Kaczmarek, M. Cichorek, J. H. Spodnik, S. Wójcik and J. Moryś, Cell Biol. Toxicol., 2017, 33, 557–573 Search PubMed.
- S. Mo, C. Xu and J. Xu, Adv. Synth. Catal., 2016, 358, 1767–1777 CrossRef CAS.
- N. Döben, H. Yan, M. Kischkewitz, J. Mao and A. Studer, Org. Lett., 2018, 20, 7933–7936 CrossRef PubMed.
- W.-W. Chan, T.-L. Kwong and W.-Y. Yu, Org. Biomol. Chem., 2012, 10, 3749–3755 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |