
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRacemising 3-aryl-3-hydroxy-2-oxindoles in a Pickering emulsion under acid catalysis and its application to dynamic kinetic resolution
Jihoon Moon,
Shuji Akai†
 * and
Kyohei Kanomata*
* and
Kyohei Kanomata*
Graduate School of Pharmaceutical Sciences, The University of Osaka, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: kanomata@phs.osaka-u.ac.jp; akai@phs.osaka-u.ac.jp
First published on 30th April 2026
Abstract
3-Aryl-3-hydroxy-2-oxindoles were racemised under acid-catalysis in a water-in-oil Pickering emulsion comprising toluene and aqueous H2SO4. The developed biphasic methodology overcomes the decomposition issues often encountered during alcohol racemisation and affords high substrate recoveries. Successfully combining racemisation with enantioselective acylation led to the first dynamic kinetic resolution of an oxindole derivative.
Introduction
Tertiary alcohols are fundamental structural motifs in a number of bioactive molecules, natural products, and pharmaceuticals, as their stereochemically defined centres are often associated with distinctive biological properties.1 However, synthesising tertiary alcohols enantioselectively remains a major challenge in modern organic synthesis.2,3 One promising strategy involves the kinetic resolution (KR)4–12 or dynamic kinetic resolution (DKR)13–21 of a racemic tertiary alcohol through the acylation of its hydroxyl groups. In particular, DKR overcomes the inherent yield limitation of KR (50%) by facilitating the in situ racemisation of the starting material in a one-pot process.22–24Racemisation, together with KR, constitutes the fundamental basis of DKR. Although the KR of tertiary alcohols using enzymes and organocatalysts has been extensively studied,4–12 catalytic racemisation processes remain underdeveloped.25 Consequently, most reported DKR processes for tertiary alcohols rely on spontaneous racemisation via reversible hemiacetal formation.13–17 Transition-metal-catalysed redox racemisation, which is widely used for the DKR of secondary alcohols,22,24 is not feasible owing to the absence of a carbinol hydrogen. Alternatively, a tertiary alcohol can be racemised through acid catalysis via the formation of a carbocation intermediate. However, such a carbocation is highly susceptible to side reactions, including dehydration and rearrangement. As a result, few examples have been reported which successfully use catalytic racemisation in synthesising enantioenriched tertiary alcohol derivatives.18–21 Developing mild and general racemisation protocols that minimise substrate decomposition and are compatible with KR catalysts is crucial for achieving efficient tertiary-alcohol DKR. Bäckvall and co-workers used acid resins in aqueous media to racemise tertiary alcohols (Scheme 1a),26 while Boyce and co-workers demonstrated that combining arylboronic acids and oxalic acid facilitates the racemisation of tertiary oxindole alcohols under relatively mild conditions (Scheme 1b).27 However, to the best of our knowledge, DKR has not been achieved by combining these racemisation methods with KR because the corresponding reaction conditions are incompatible.
 | ||
| Scheme 1 Study background and synopsis. | ||
Herein, we report the Brønsted-acid-catalysed racemisation of 3-aryl-3-hydroxy-2-oxindole derivatives, an important class of bioactive compounds containing tertiary-alcohol motifs (Scheme 1c).28 The developed method relies on a biphasic oil/water reaction medium, wherein racemisation occurs in the aqueous phase, thereby suppressing substrate decomposition. Moreover, we report the first successful DKR of a 3-hydroxy-oxindole derivative by integrating the developed racemisation protocol with an isothiourea-based organocatalyst.
Results and discussion
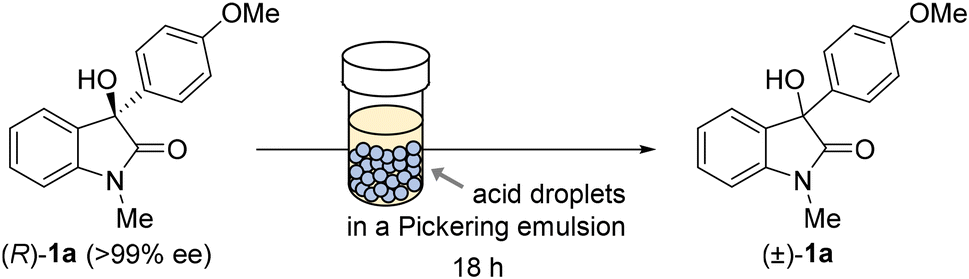
We selected a Pickering emulsion as a suitable reaction medium for our biphasic acid-catalysed racemisation protocol.29,30 A Pickering emulsion is an oil/water dispersion in which micrometre-sized droplets are stabilised by nanoparticles adsorbed at their interfaces.31–35 Racemisation proceeds in the aqueous phase of a Pickering emulsion prepared with aqueous acid and an organic solvent. Consequently, the carbocation intermediate is stabilised by hydration and rapidly trapped by a water molecule to regenerate the alcohol in racemic form, thereby suppressing the substrate decomposition typically associated with carbocations formed in organic media.26,29 Furthermore, the biphasic nature of a Pickering emulsion offers an additional advantage for DKR because the aqueous acid catalyst can be physically separated from the KR catalyst, thereby mitigating catalyst incompatibility.Our study commenced by investigating the racemisation of optically pure (R)-1a in various Pickering emulsions (Table 1; see SI for emulsion-preparation details). Racemisation proceeded smoothly when the reaction was carried out at 50 °C using HCl (5.0 M) as the aqueous phase to afford 1a with 3% ee (84% recovery) after 18 h (entry 1). While the use of HNO3 (5.0 M) under otherwise identical conditions led to comparable racemisation, less 1a (46%) was recovered (entry 2) and the reaction mixture became yellowish, indicative of the partial decomposition of 1a. In contrast, a significantly improved recovery and a high racemisation rate was obtained with H2SO4 (5.0 M), which resulted in complete racemisation and the quantitative recovery of 1a after 18 h (entry 3). The Pickering emulsions were stable under the aforementioned reaction conditions using H2SO4 (5.0 M), with no colour changes or degradation observed during the entire reactions. Lowering either the acid concentration or the reaction temperature led to considerably less racemisation (entries 4 and 5). Conversely, further increasing the acid concentration prevented the formation of a stable Pickering emulsion, which is probably ascribable to the high viscosity of the aqueous acid. For comparison, similar racemisation reactions were also performed using a conventional emulsion prepared with the Triton X-100 surfactant, as well as in a non-emulsified biphasic mixture of toluene and aqueous H2SO4. Both scenarios led to less racemisation than that observed for the corresponding Pickering emulsion (entries 6 and 7 vs. 3), which demonstrates that Pickering emulsions serve as highly efficient media for promoting biphasic reactions. This racemisation method was scalable without any significant loss in the reaction rate or recovery (entry 8).
|
||||
|---|---|---|---|---|
| Entry | Acid (conc.) | Temp. (°C) | % ee of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
% recovery of 1ac |
| a Unless otherwise stated, reactions were carried out with (R)-1a (0.10 mmol, >99% ee) for 18 h at the indicated temperature without stirring in a Pickering emulsion prepared using silica nanoparticles (60 mg), toluene (1.2 mL), and the indicated aqueous acid (0.6 mL).b Determined by HPLC using a chiral stationary phase.c Determined by 1H NMR spectroscopy of the crude reaction mixture using 1,1,2,2-tetrachloroethane as an internal standard.d The isolated yield given in parentheses.e Reaction was conducted in an emulsion prepared with Triton X-100 (12 mg), toluene (1.2 mL), and aqueous H2SO4 (0.6 mL).f A similar reaction was conducted in a vigorously stirred non-emulsified biphasic mixture of toluene (1.2 mL) and aqueous H2SO4 (0.6 mL).g A similar reaction was carried out with (R)-1a (3.0 mmol, >99% ee), silica nanoparticles (1.8 g), toluene (36 mL), and aqueous H2SO4 (5.0 M, 18 mL). | ||||
| 1 | HCl (5.0 M) | 50 | 3 | 84 |
| 2 | HNO3 (5.0 M) | 50 | <3 | 46 |
| 3 | H2SO4 (5.0 M) | 50 | <3 | >95 (97)d |
| 4 | H2SO4 (2.5 M) | 50 | 42 | >95 |
| 5 | H2SO4 (5.0 M) | 35 | 33 | >95 |
| 6e | H2SO4 (5.0 M) | 50 | 14 | >95 |
| 7f | H2SO4 (5.0 M) | 50 | 42 | >95 |
| 8g | H2SO4 (5.0 M) | 50 | <1 | (91)d |
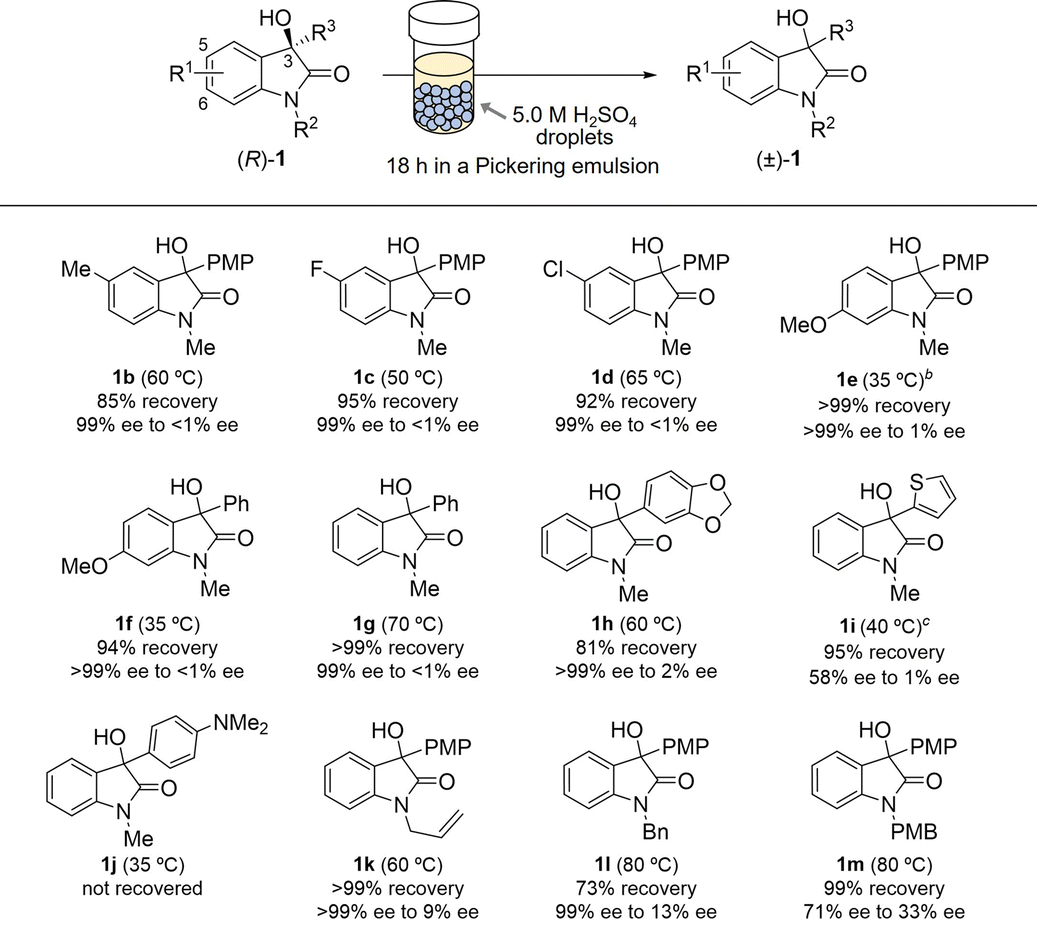
With the optimised racemisation conditions established for the Pickering emulsion, we next explored the scope and limitations of this protocol (Table 2). Racemisation proceeded smoothly when reactions were conducted for 18 h at an appropriate temperature for each substrate. A range of R1 oxindole benzene-ring substituents were well tolerated under the present conditions. 5-Me, 5-F, and 5-Cl-substituted oxindoles 1b–1d underwent racemisation with high recoveries (>85%) at 50–65 °C. Notably, substrate 1e bearing the electron-donating methoxy group [R1 = 6-MeO, R3 = p-methoxyphenyl (PMP)] underwent complete racemisation under somewhat milder conditions (35 °C, 1.0 M H2SO4). Racemisation of 1f (R1 = 6-MeO, R3 = Ph) also proceeded at 35 °C but required a higher acid concentration (5.0 M). These results reveal that the electron density at the C3 position of the oxindole markedly affects racemisation, which is ascribable to stabilisation of the carbocation intermediate generated under acid catalysis. We next examined the effect of the R3 substituent (at C3) by reacting substrates 1g–1j. A higher temperature (70 °C) was required to achieve complete racemisation when the PMP group in 1a was replaced with a phenyl group (as in 1g). The introduction of electron-rich aromatic groups, such as benzodioxol-5-yl (as in 1h) and 2-thiophenyl (as in 1i), resulted in racemisation rates and recoveries comparable to those observed for 1a. In contrast, 1j (R3 = C6H4-4-NMe2) was not recovered following the reaction, most likely because the substrate decomposed via protonation of its amino group. The substituent on the oxindole nitrogen (R2) was also found to influence the racemisation rate. Replacing the methyl group at the nitrogen atom with an allyl (as in 1k) or benzyl (as in 1l) group led to less racemisation while maintaining a high recovery ratio. Introduction of a p-methoxybenzyl (PMB) group at the N1 position (as in 1m) further decreased the racemisation rate, which is possibly attributable to the poor solubility of 1m in toluene.
a
| a Unless otherwise stated, reactions were carried out with (R)-1 (0.10 mmol) for 18 h at the indicated temperature without stirring in a Pickering emulsion prepared using silica nanoparticles (60 mg), toluene (1.2 mL), and aqueous H2SO4 (5.0 M, 0.6 mL). Recoveries were calculated based on the amount of isolated 1 obtained from the reaction. Enantiomeric excesses (% ees) were determined by HPLC using a chiral stationary phase.b Aqueous H2SO4 (1.0 M) was used.c Aqueous H2SO4 (2.0 M) was used. PMP = p-methoxyphenyl; PMB = p-methoxybenzyl. |
|---|
 |
The Pickering-emulsion-mediated racemisation method features an aqueous acid catalyst compartmentalised within nanoparticle shells, which enables it to cooperate with a Lewis base catalyst in a single reaction vessel. Accordingly, we finally explored the DKR of 3-hydroxy-2-oxindole by combining this racemisation method with acylative KR using the isothiourea based HyperBTM catalyst (Scheme 2).10–14,36 The DKR of (±)-1e with (2S,3R)-HyperBTM (10 mol%) was carried out in a Pickering emulsion comprising toluene and aqueous H2SO4 (5.0 M) in the presence of isobutyric anhydride (2) as the acyl donor, sodium isobutyrate as the base, and molecular sieves 4 Å (MS4A). Enantiomerically enriched (S)-3e (97% ee) was obtained in 58% yield, along with recovered (R)-1e (62% ee) in 28% yield after the emulsion was allowed to stand without stirring for 2 h at 25 °C. This outcome clearly demonstrates that DKR proceeds through the simultaneous racemisation and acylation of 1e.
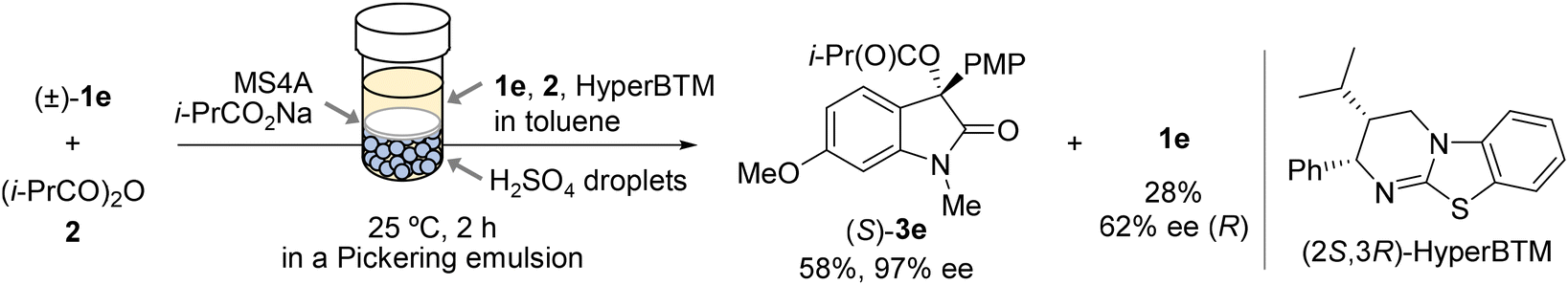 | ||
| Scheme 2 H2SO4/HyperBTM-co-catalysed DKR of (±)-1e in a Pickering emulsion. The reaction was carried out using (±)-1e (0.10 mmol), isobutyric anhydride (2, 3 equiv.), and (2S,3R)-HyperBTM (10 mol%) in the presence of i-PrCO2Na (4 equiv.) and MS4A (25 mg) in an unstirred Pickering emulsion comprising toluene (0.6 mL) and aqueous H2SO4 (5.0 M, 0.2 mL). | ||
Conclusions
Optically active 3-aryl-3-hydroxy-2-oxindoles were racemised in aqueous-H2SO4/toluene-based Pickering emulsions. The biphasic nature of the Pickering emulsion was found to be essential for efficient racemisation with minimal decomposition, which is often observed during the acid-catalysed racemisation of alcohols in organic solvents. The DKR of (±)-1e was achieved by combining the racemisation method with HyperBTM-catalysed enantioselective acylation in the oil phase of the Pickering emulsion. To the best of our knowledge, this represents the first reported DKR of this class of substrates. Ongoing efforts in our laboratory are focused on improving the Pickering-emulsion-mediated DKR method, with particular emphasis on expanding of the substrate scope and developing a reusable catalytic system using a solid-supported HyperBTM catalyst.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article are presented in the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6ob00544f.Acknowledgements
This work was supported by JST SPRING Grant Numbers JPMJSP2138 (J. M.), JSPS KAKENHI Grant Numbers 24K02148 (S. A.) and 24K08410 (K. K.), the Joint Usage/Research Center for Catalysis (23AY0215, 24AY0545) (K. K.), Amano Institute of Technology (K. K.), Tokuyama Science Foundation (K. K.), “Innovation inspired by Nature” Research Support Program, Sekisui Chemical Co., Ltd (K. K.), and Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP25ama121054.References
- D. Chiodi and Y. Ishihara, J. Med. Chem., 2025, 68, 7889–7913 CrossRef CAS PubMed.
- Y. L. Liu and X. T. Lin, Adv. Synth. Catal., 2019, 361, 876–918 CrossRef CAS.
- Y. P. He, D. Tian, X. Z. Li and H. Wu, Org. Chem. Front., 2023, 10, 3110–3129 RSC.
- S. H. Krishna, M. Persson and U. T. Bornscheuer, Tetrahedron: Asymmetry, 2002, 13, 2693–2696 CrossRef.
- S. Lu, S. B. Poh, W. Y. Siau and Y. Zhao, Angew. Chem., Int. Ed., 2013, 52, 1731–1734 CrossRef CAS PubMed.
- J. Löfgren, T. Görbe, M. Oschmann, M. S. Humble and J. E. Bäckvall, ChemBioChem, 2019, 20, 1438–1443 CrossRef PubMed.
- H. Mandai, R. Shiomoto, K. Fujii, K. Mitsudo and S. Suga, Org. Lett., 2020, 23, 1169–1174 CrossRef PubMed.
- S. Horino, K. Wagner, A. Hummel, K. Kanomata, H. Gröger and S. Akai, Eur. J. Org. Chem., 2024, e202400242 CrossRef CAS.
- K. Wagner, A. Hummel, J. Yang, S. Horino, K. Kanomata, S. Akai and H. Gröger, ChemBioChem, 2024, 25, e202400082 CrossRef CAS PubMed.
- M. D. Greenhalgh, S. M. Smith, D. M. Walden, J. E. Taylor, Z. Brice, E. R. T. Robinson, C. Fallan, D. B. Cordes, A. M. Z. Slawin, H. C. Richardson, M. A. Grove, P. H. Y. Cheong and A. D. Smith, Angew. Chem., Int. Ed., 2018, 57, 3200–3206 CrossRef CAS PubMed.
- S. Qu, S. M. Smith, V. Laina-Martín, R. M. Neyyappadath, M. D. Greenhalgh and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 16572–16578 CrossRef CAS PubMed.
- S. M. Smith, M. D. Greenhalgh, T. Feoktistova, D. M. Walden, J. E. Taylor, D. B. Cordes, A. M. Z. Slawin, P. H. Y. Cheong and A. D. Smith, Eur. J. Org. Chem., 2022, e202101111 CrossRef CAS.
- H. Zhu, A. Manchado, A. O. Farah, A. P. McKay, D. B. Cordes, P. H. Y. Cheong, K. Kasten and A. D. Smith, Angew. Chem., Int. Ed., 2024, 63, e202402908 CrossRef CAS PubMed.
- S. K. Agrawal, P. K. Majhi, A. S. Goodfellow, R. K. Tak, D. B. Cordes, A. P. McKay, K. Kasten, M. Bühl and A. D. Smith, Angew. Chem., Int. Ed., 2024, 63, e202402909 CrossRef CAS PubMed.
- Y. Liu, P. K. Majhi, R. Song, C. Mou, L. Hao, H. Chai, Z. Jin and Y. R. Chi, Angew. Chem., Int. Ed., 2020, 59, 3859–3863 CrossRef CAS PubMed.
- Y. Y. Gao, C. L. Zhang, L. Dai, Y. F. Han and S. Ye, Org. Lett., 2021, 23, 1361–1366 CrossRef CAS PubMed.
- M. Shan, Y. Yu, M. Sun, S. Yang, M. Wang, B. Zhu and J. Chang, Chin. Chem. Lett., 2025, 36, 109781 CrossRef CAS.
- F. Kühn, S. Katsuragi, Y. Oki, C. Scholz, S. Akai and H. Gröger, Chem. Commun., 2020, 56, 2885–2888 RSC.
- K. Wagner, S. Horino, A. Hummel, K. Kanomata, S. Akai and H. Gröger, Chem. Pharm. Bull., 2025, 73, 511–514 CrossRef CAS PubMed.
- T. Kin, S. Horino, J. Moon, M. Tsuda, H. Gröger, S. Akai and K. Kanomata, Green Chem., 2025, 27, 9672–9678 RSC.
- S. Horino, K. Wagner, A. Hummel, K. Kanomata, H. Gröger and S. Akai, Eur. J. Org. Chem., 2026, e202500980 CrossRef CAS.
- O. Verho and J. E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS PubMed.
- K. Kanomata and S. Akai, in Science of Synthesis: Dynamic Kinetic Resolution (DKR) and Dynamic Kinetic Asymmetric Transformations (DYKAT), ed. J. E. Bäckvall, Thieme, Stuttgart, 2023, pp. 181–217 Search PubMed.
- J. Zhang, Z. Zheng and C. Zhu, Chin. Chem. Lett., 2024, 35, 109160 CrossRef CAS.
- H. Gröger, S. Horino, K. Kanomata and S. Akai, Chem. – Eur. J., 2024, 30, e202304028 CrossRef PubMed.
- T. Görbe, R. Lihammar and J. E. Bäckvall, Chem. – Eur. J., 2018, 24, 77–80 CrossRef PubMed.
- G. R. Boyce, S. F. Musolino, J. Yang, A. D. Smith and J. E. Taylor, J. Org. Chem., 2022, 87, 13367–13374 CrossRef CAS PubMed.
- B. Yu, H. Xing, D. Q. Yu and H. M. Liu, Beilstein J. Org. Chem., 2016, 12, 1000–1039 CrossRef CAS PubMed.
- J. Moon, T. Kin, K. Mizuno, S. Akai and K. Kanomata, ChemCatChem, 2023, 15, e202300878 CrossRef CAS.
- T. Nishio, S. Akai and K. Kanomata, ACS Catal., 2025, 15, 6565–6571 CrossRef CAS.
- L. Ni, C. Yu, Q. Wei, D. Liu and J. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202115885 CrossRef CAS PubMed.
- Y. Li, J. Xu and H. Yang, Langmuir, 2023, 39, 5621–5630 CrossRef CAS PubMed.
- E. Guzmán, ChemCatChem, 2024, 16, e202400856 CrossRef.
- J. Tang, B. Zhang, M. Zhang and H. Yang, J. Phys. Chem. Lett., 2024, 15, 8973–8983 CrossRef CAS PubMed.
- Y. Wang, H. Shi, Y. Sun, X. Zhang, Y. Li and H. Yang, J. Am. Chem. Soc., 2025, 147, 31049–31059 CrossRef CAS PubMed.
- A. Kinens, S. Balkaitis, O. K. Ahmad, D. W. Piotrowski and E. Suna, J. Org. Chem., 2021, 86, 7189–7202 CrossRef CAS PubMed.
Footnote |
| † Current address: SANKEN, The University of Osaka, 8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan. |
| This journal is © The Royal Society of Chemistry 2026 |