
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong-range isomerisation/cyclopropane isomerisation/cycloisomerisation/aromatisation reactions using multifunctional rhodium catalysts
Momoko
Nagafuchi
a,
Takumi
Mizumachi
a,
Yuta
Sato
a,
Masaharu
Takatsuki
a,
Tsuyoshi
Matsuzaki
b,
Takeyuki
Suzuki
 b,
Atsushi
Nakayama
a,
Makoto
Sako
a and
Mitsuhiro
Arisawa
*a
b,
Atsushi
Nakayama
a,
Makoto
Sako
a and
Mitsuhiro
Arisawa
*a
aGraduate School of Pharmaceutical Sciences, The University of Osaka, Yamada-oka 1-6, Suita, Osaka 565-0871, Japan. E-mail: arisaw@phs.osaka-u.ac.jp
bComprehensive Analysis Center, SANKEN, The University of Osaka, 8-1, Mihogaoka, Ibaraki, Osaka 567-0047, Japan
First published on 29th April 2026
Abstract
Transition-metal-catalyzed chain-walking reactions have emerged as a powerful strategy for remote functionalization of alkenes; however, their integration with C–C bond activation remains largely unexplored. Herein, we report a rhodium-catalyzed reaction that combines long-range alkyl chain-walking with selective cyclopropyl C–C bond cleavage within a single catalytic system. In this transformation, migratory alkene isomerisation across extended carbon frameworks enables distal activation of a cyclopropylmethyl unit, leading to the efficient formation of substituted benzofurans under mild conditions. Unlike conventional chain-walking processes that terminate in remote functionalization, the present system exploits catalyst migration as a prerequisite for subsequent C–C bond activation at a spatially separated site. Mechanistic investigations, including temperature-controlled and time-resolved NMR studies, support a sequential pathway involving alkene chain-walking followed by cyclopropane activation. This work demonstrates a cooperative catalytic strategy that orchestrates spatially separated reaction events and expands the conceptual scope of chain-walking catalysis in organic synthesis.
Multitasking catalysts are catalysts capable of sequentially promoting multiple distinct chemical transformations within a single catalytic system, which reduces the number of extraction and purification steps required, thereby decreasing the consumption of reagents, solvents, and other materials needed for these processes. They are of significant synthetic chemical utility as they enable the one-pot synthesis of complex molecules, typically requiring multiple reactions, from simple starting materials (Scheme 1).1,2 Particularly, the ability to catalyse multiple chemical reactions involving rare elements using a single multi-task catalyst holds significant environmental conservation value.
 | ||
| Scheme 1 Multitasking catalytic reaction and conventional reaction. | ||
Among multitasking catalytic reactions, methodologies promoting three or more reactions to construct bicyclic heterocyclic structures, a key constituent structure of functional molecular compounds, via cycloisomerisation are currently limited to the rhodium-catalysed examples we have reported in recent years (Scheme 2A).3
 | ||
| Scheme 2 Multitasking catalytic system to give 2-alkyl-3-isopropylbenzofurans using multitasking single rhodium catalyst. | ||
Inert C(sp3)–H bond is functionalized after the alkene is isomerized and the double bond is moved along the carbon chain. Various reports have detailed the remote alkylations,4 arylations,5 aminations,6 borylations,7 carbonylations,8etc., using palladium, nickel, rhodium, iridium, cobalt, iron, etc., with alkene chain-walking.
In this context, we envisioned that substrates bearing a long-chain alkene tethered to an oxygen atom could enable a fundamentally different type of multitasking catalysis, in which long-range alkene chain-walking and cyclopropylmethyl C–C bond activation proceed concurrently under a single rhodium catalytic system.
Herein, we report a rhodium-catalysed transformation that orchestrates four chemically distinct reaction modes—long-range alkene isomerisation, cyclopropane isomerisation involving C–C bond cleavage, cycloisomerisation, and subsequent aromatisation—within a single catalytic manifold. Notably, the alkene migration occurs over extended carbon chains, significantly beyond the scope of previously reported multitasking cycloisomerisation reactions.
Furthermore, time-resolved NMR experiments reveal that these multiple isomerisation processes do not occur as an uncontrolled cascade but instead proceed in a defined temporal order, providing rare experimental insight into how a single rhodium catalyst mediates and differentiates multiple elementary steps. This study therefore expands the conceptual framework of multitasking catalysis by demonstrating the controlled integration of long-range chain-walking with C–C bond activation chemistry.
As an initial study, substrate 1a bearing cyclopropane and a long-chain alkene, Wilkinson's catalyst, and silver trifluoromethanesulfonate in a p-xylene solution were stirred at 120 °C for 24 hours. As a result, the desired benzofuran 2a was obtained in a 17% yield (Table 1, entry 1). Additionally, dihydrobenzofuran 3a was obtained as a by-product in a 13% yield. As the desired long-distance isomerisation reaction proceeded under the above conditions, further optimisation of the reaction conditions was carried out.
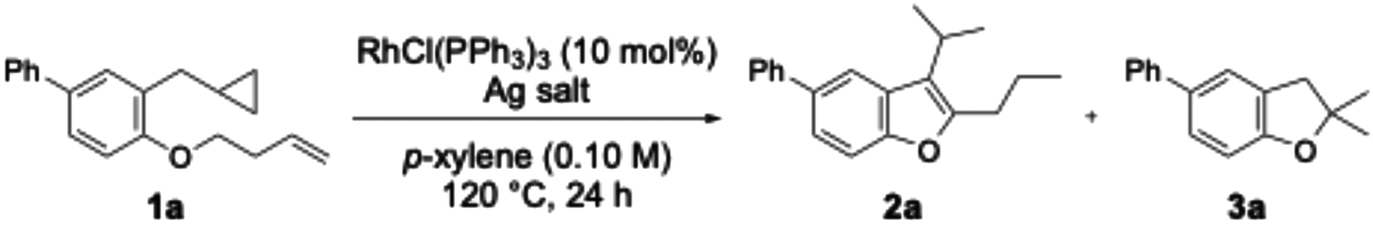
Next, the choice of silver salt and its equivalent amount were examined (Table 1, entries 2–6). When silver hexafluoroantimonate or silver tetrafluoroborate was used, compounds 2a and 3a were scarcely obtained (entries 2 and 3). Increasing the silver trifluoromethanesulfonate equivalent from 10 mol% to 15 mol% improved the yield of 2a to 44% (entry 4). Neither reducing the silver salt equivalent to 5 mol% (entry 5) nor increasing it to 30 mol% (entry 6) improved the yield of 2a. In the experiments where NaBArF was added, neither the isomerisation nor the cycloisomerisation of cyclopropane proceeded, and the desired benzofuran was not obtained.
Next, the rhodium catalyst was changed to [Rh(C2H4)2Cl]2, and various ligands were investigated (Table 2). Initially, using triphenylphosphine yielded benzofuran 2a in 75% yield (entry 1). Using tri(o-tolyl)phosphine, tri(m-tolyl)phosphine, and tri(p-tolyl)phosphine yielded 2a in 10%, 70%, and 64% yields, respectively; the yield of 2a decreased significantly when tri(o-tolyl)phosphine was employed (entries 2–4). These results suggested that the cone angle, arising from the steric bulkiness of the ligand, significantly influenced the reaction. Therefore, ligands bearing multiple substituents on the benzene ring were investigated. Using tri(2,6-xylyl)phosphine and tris(2,4,6-trimethylphenyl)phosphine, chain-walking proceeded, but cyclopropane did not open, and 2a was not obtained (entries 5 and 6). Next, ligands bearing two substituents in the meta position on the benzene ring were investigated. Using tri(3,5-xylyl)phosphine, the yield for 2a improved to 81% (entry 7). Replacing the methyl group with the more bulky ethyl group reduced the yield for 2a to 55% and yielded 3a in 22% (entry 8). Furthermore, replacing the methyl group with a methoxy group also reduced the yield of 2a (entry 9).
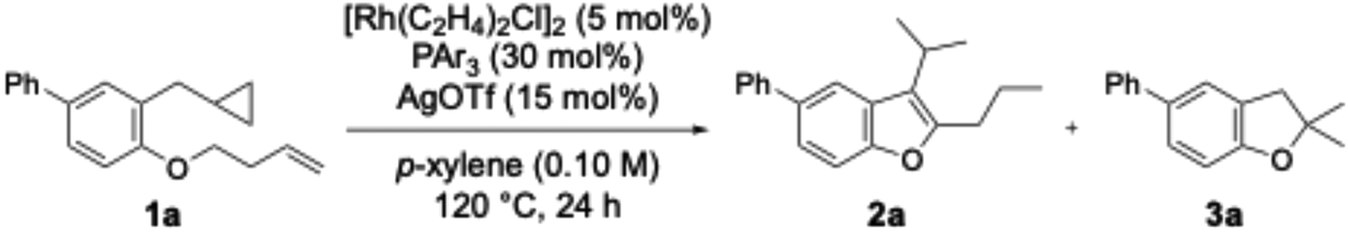
Furthermore, the reaction temperature and reaction time were investigated (Table 3, entries 1–5). When the reaction temperature was lowered to 80 °C, the yield of 2a decreased significantly, and dihydrobenzofuran 2a′ was obtained in a 28% yield (entry 2). At 100 °C, the yield of 2a improved slightly to 45%, but 2a′ was obtained similarly to entry 2 (entry 3). Furthermore, no yield improvement was observed even when the temperature was increased to 130 °C (entry 4), thus the optimum temperature was determined to be 120 °C. Moreover, shortening the reaction time to 12 hours reduced the yield of 2a to 68% (entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | AgOTf (mol%) | Temp. (°C) | Time (h) |
2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
2a′a (%) |
3aa (%) |
| a NMR yield (1,3,5-trimethoxybenzene was used as an internal standard). b cis/trans = 7/3. c cis/trans = 11/9. | ||||||
| 1 | 15 | 120 | 24 | 81 | 0 | 11 |
| 2 | 15 | 80 | 24 | 9 | 28b | 4 |
| 3 | 15 | 100 | 24 | 45 | 20c | 10 |
| 4 | 15 | 130 | 24 | 80 | 0 | 12 |
| 5 | 15 | 120 | 12 | 68 | 0 | 16 |
| 6 | 10 | 120 | 24 | 8 | 0 | 12 |
| 7 | 20 | 120 | 24 | 46 | 0 | 33 |

Finally, the equivalent amount of silver trifluoromethanesulfonate was re-examined (Table 3, entries 6–7). When 10 mol% of silver trifluoromethanesulfonate was added, the yield of the target benzofuran 2a was 8% (entry 6). At 15 mol%, 2a was obtained in 81% yield and 3a in 11% yield (entry 1). Further increasing the molar percentage to 20 mol% resulted in a decrease in the yield of 2a to 46%, while the yield of 3a increased to 33% (entry 7). Although it was considered that 10 mol% trifluoromethanesulfonate might be optimal as the counter anion for the rhodium catalyst, experimental results indicated that 15 mol% was optimal.9
Based on the above results, the optimal conditions were determined to be stirring at 120 °C with the addition of tri(3,5-xylyl)phosphine (30 mol%) and silver trifluoromethanesulfonate (15 mol%) using [Rh(C2H4)2Cl]2 (5 mol%) as the catalyst precursor (entry 1).
All reactions up to this stage were conducted on a 0.10 mmol scale. To investigate the feasibility of gram-scale reactions, 1 g (3.59 mmol) of 1a was used as the substrate, yielding benzofuran 2a in 83% yield and dihydrobenzofuran 3a in 17% yield. Given that the yields at the 0.10 mmol scale were 81% for 2a and 11% for 3a (Table 3, entry 1), this reaction was found to be scalable and not significantly affected by stirring efficiency, heating method, or heating efficiency.
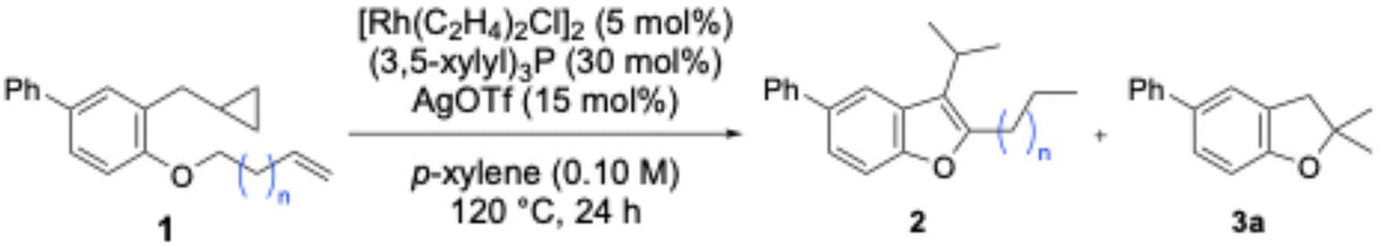
Under optimised reaction conditions, substrate generality was investigated. First, we investigated whether chain-walking and subsequent reactions proceeded when substrates with extended carbon chain lengths were used (Table 4). Although yields decreased with increasing chain length, the desired reaction proceeded for all substrates (1a–1h), yielding the corresponding benzofuran 2 in 39% to 81% yields (2a–2h). Furthermore, substrates bearing long-chain alkenes increased the yield of by-product 3a; using 1g and 1h yielded 3a in 32% and 39% yields, respectively. Additionally, when 1g and 1h were used as substrates, compounds were observed where chain-walking had halted prematurely. In long-chain substrates, chain-walking causes the double bond to migrate over a greater distance, increasing the number of stages involving the rhodium catalyst. This renders the catalytic cycle inefficient, resulting in a reduced yield of the desired benzofuran.
Subsequently, the effects of substituent groups on the benzene ring were investigated (Table 5). The results revealed that benzofuran derivatives bearing fluorine, chlorine, methyl, methoxy, or methoxycarbonyl groups at positions 4 to 7 were obtained in moderate to good yields.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 (R=) | 2/3 (%) | Entry | 1 (R=) | 2/3 (%) |
| 1 | 1a (5-Ph) | 81 (2a)/11 (3a) | 9 | 1p (5-CO2Me) | 64 (2p)/26 (3p) |
| 2 | 1i (4-F) | 50 (2i)/trace | 10 | 1q (5-CN) | 0/53 (3q) |
| 3 | 1j (4-OMe) | 57 (2j)/trace | 11 | 1r (6-F) | 70 (2r)/trace |
| 4 | 1k (5-F) | 75 (2k)/trace | 12 | 1s (6-Cl) | 51 (2s)/20 (3s) |
| 5 | 1l (5-Cl) | 55 (2l)/14 (3l) | 13 | 1t (6-OMe) | 70 (2t)/trace |
| 6 | 1m (5-Br) | 0/46 (3m) | 14 | 1u (7-CH3) | 80 (2u)/trace |
| 7 | 1n (5-CH3) | 85 (2n)/trace | 15 | 1v (7-OMe) | 54 (2v)/37 (3v) |
| 8 | 1o (5-OMe) | 70 (2o)/trace | |||
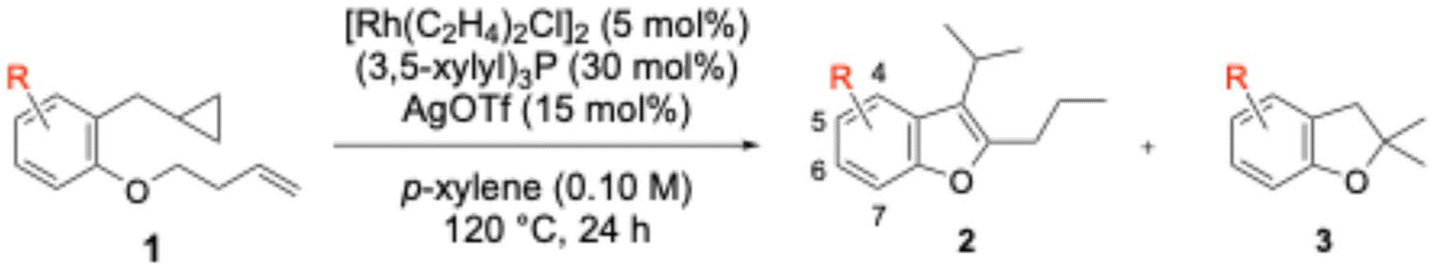
Although benzofuran derivatives bearing fluorine and methoxy groups at the 4-position were obtained, the yields remained moderate at 50% (2i) and 57% (2j). This is thought to arise from electronic effect and/or steric hindrance imposed by the 4-substituents.
The benzofuran bearing a fluorine group at the 5-position with electron-withdrawing properties, and electron-donating methyl, methoxy, or methoxycarbonyl groups, was obtained in good yields of 75% (2k), 85% (2n), 70% (2o), and 64% (2p). On the other hand, the desired benzofuran was not obtained from substrates bearing bromo or cyano groups (1m, 1q). Instead, dihydrobenzofuran derivatives 3m and 3q, bearing bromo and cyano groups at the 5-position respectively, were obtained in yields of 46% and 53%.
For benzofuran derivatives bearing substituents at the 7-position, introduction of a methyl group yielded 80%, while introduction of a methoxy group yielded 54%, revealing a significant yield difference.10
NMR experiments were conducted to analyse the reaction mechanism of this reaction. It is thought that this reaction proceeds through the stages of chain-walking of the terminal alkene, isomerisation involving cyclopropane cleavage, cycloisomerisation of the resulting diene, and aromatisation. To confirm that the reaction actually proceeds through these stages and to determine the order in which each reaction occurs, the reaction was monitored by 1H NMR. As the initial reaction stage proceeded rapidly, making detailed observation difficult, the temperature was lowered to 80 °C and subsequently raised to 120 °C. Specifically, 0.20 mmol of substrate 1a, 5 mol% [Rh(C2H4)2Cl]2, 30 mol% tri(3,5-xylyl)phosphine, and 15 mol% silver trifluoromethanesulfonate were added. The mixture was stirred at 80 °C for 6 hours, then heated to 120 °C and stirred for a further 6 hours. Reaction solutions were collected at each time point, diluted with dichloromethane, the solvent removed, and 1H NMR measurements performed in CDCl3. The 1H NMR spectra of the reaction solution at each time point are shown below (Fig. 1).
 | ||
| Fig. 1 Time-dependent changes in the reaction (1H NMR, 500 MHz, CDCl3, 80 °C to 120 °C). | ||
Throughout the conversion process from substrate 1a to the desired benzofuran 2a, several distinct peaks are observable. Here, we synthesised various anticipated intermediates and compared their characteristic peaks. First, we compared the spectra of the reaction mixture obtained from 1 minute to 1 hour after reaction initiation with those of the substrate and the various intermediates. The results are shown below (Fig. 2).
 | ||
| Fig. 2 Comparison of peaks in reaction solutions at each reaction time (1 min–1 h, 80 °C) concerning intermediates and products (1H NMR, 500 MHz, CDCl3). | ||
Focusing first on the cyclopropyl moiety, five minutes after reaction initiation, peaks ( ) originating from the terminal alkene of 8a appear around 4.7 and 4.8 ppm, while peaks corresponding to the terminal dimethyl groups of 9a and 9a′ appear around 1.9 and 2.0 ppm (
) originating from the terminal alkene of 8a appear around 4.7 and 4.8 ppm, while peaks corresponding to the terminal dimethyl groups of 9a and 9a′ appear around 1.9 and 2.0 ppm ( ). This confirms that isomerisation involving cyclopropane cleavage, followed by subsequent isomerisation to an internal alkene, has commenced. 9a′. After one hour, the cyclopropane-derived proton peaks (
). This confirms that isomerisation involving cyclopropane cleavage, followed by subsequent isomerisation to an internal alkene, has commenced. 9a′. After one hour, the cyclopropane-derived proton peaks ( ) near 0.2 and 0.5 ppm and the terminal alkene peaks (
) near 0.2 and 0.5 ppm and the terminal alkene peaks ( ) near 4.7 and 4.8 ppm had almost disappeared, confirming that isomerisation had progressed to the trisubstituted alkene. In our previous work, we confirmed that 1a was converted to 9a, when we choose an appropriate rhodium salt.3a
) near 4.7 and 4.8 ppm had almost disappeared, confirming that isomerisation had progressed to the trisubstituted alkene. In our previous work, we confirmed that 1a was converted to 9a, when we choose an appropriate rhodium salt.3a
Focusing next on the alkene side chain, observation of a double bond peak ( ) originating from the oxygen β-position of 9a around 5.4 ppm five minutes after reaction initiation indicated that chain-walking of the terminal alkene had commenced. Furthermore, one hour after reaction initiation, the proton peak (
) originating from the oxygen β-position of 9a around 5.4 ppm five minutes after reaction initiation indicated that chain-walking of the terminal alkene had commenced. Furthermore, one hour after reaction initiation, the proton peak ( ) originating from the oxygen α-position of substrate 1a, appearing around 4.1 ppm, had disappeared, confirming that the terminal double bond had isomerised internally.
) originating from the oxygen α-position of substrate 1a, appearing around 4.1 ppm, had disappeared, confirming that the terminal double bond had isomerised internally.
Thus, it became clear that isomerisation of both side chains proceeded almost simultaneously, with the majority converting to 9a under 80 °C conditions.
Subsequently, the spectra of the reaction mixtures obtained at 6 and 8 hours after reaction initiation were compared with those of the respective intermediates and products, as shown below (Fig. 3).
 | ||
| Fig. 3 Comparison of peaks in reaction solutions at each reaction time (6 h, 8 h, 80 °C to 120 °C) concerning intermediates and products (1H NMR, 500 MHz, CDCl3). | ||
Upon heating at 80 °C, a double bond peak ( ) originating from the oxygen β-position of 9a was observed around 5.4 ppm. However, proton peaks (
) originating from the oxygen β-position of 9a was observed around 5.4 ppm. However, proton peaks ( ,
,  ) originating from the benzyl position of 2a′ around 4.0 and 3.8 ppm, and the proton peak (
) originating from the benzyl position of 2a′ around 4.0 and 3.8 ppm, and the proton peak ( ) at the isopropyl group base around 3.1 ppm were scarcely detected. This indicated that cyclisation does not proceed at 80 °C.
) at the isopropyl group base around 3.1 ppm were scarcely detected. This indicated that cyclisation does not proceed at 80 °C.
Subsequently, upon raising the reaction temperature to 120 °C, the double bond peak ( ) near 5.4 ppm disappeared, and proton peaks (
) near 5.4 ppm disappeared, and proton peaks ( ,
,  ) originating from the benzyl position of 2a′ were observed near 3.8 and 4.0 ppm, confirming that cyclisation was proceeding. Furthermore, a proton peak (
) originating from the benzyl position of 2a′ were observed near 3.8 and 4.0 ppm, confirming that cyclisation was proceeding. Furthermore, a proton peak ( ) at approximately 3.1 ppm was observed at the isopropyl group site, indicating that aromaticisation proceeded rapidly, yielding benzofuran 2a.
) at approximately 3.1 ppm was observed at the isopropyl group site, indicating that aromaticisation proceeded rapidly, yielding benzofuran 2a.
We then proposed a reaction mechanism for this reaction (Scheme 3). NMR experiments indicate that chain-walking and cyclopropane isomerisation proceed almost simultaneously. For explanatory purposes, however, we will depict cyclopropane isomerisation on the inner side of the ring and chain-walking on the outer side, describing each sequentially. Furthermore, the tips of the wavy lines for A–C represent the structure where the butenyl group, the lower side chain of D–G, has isomerised, while the tips of the wavy lines for D–G represent the structure where the cyclopropyl group, the upper side chain of A–C, has isomerised.
 | ||
| Scheme 3 Plausible reaction mechanism to give compound 2a. | ||
First, focusing on the portion depicted inside the ring, the cyclopropane moiety undergoes oxidative addition to the rhodium catalyst (A), followed by β-hydrogen elimination and subsequent reductive elimination to form 8a. Subsequently, isomerisation of 8a converts it to H. Simultaneously, as shown on the outer side of the ring, after the terminal alkene coordinates to rhodium hydride,11 alkene insertion generates E, followed by β-hydrogen elimination to form F. Thus, through repeated cycles of alkene insertion and β-hydrogen elimination, when the double bond reaches the oxygen α-position, cyclisation is achieved via the alkyl rhodium intermediate I. Subsequent β-hydrogen elimination yields the dihydrobenzofuran 2a′. Finally, it is proposed that the terminal alkene of 2a′ coordinates to rhodium, followed by aromatisation, converting to 2a.
Finally, the formation of the by-product of this reaction, 2,2-dimethyl-2,3-dihydrobenzofuran 3, will be discussed. It is postulated that 3 is formed by cyclisation following the elimination of an alkyl chain from the substrate (Scheme 4).
 | ||
| Scheme 4 Plausible reaction mechanism to give compound 3. | ||
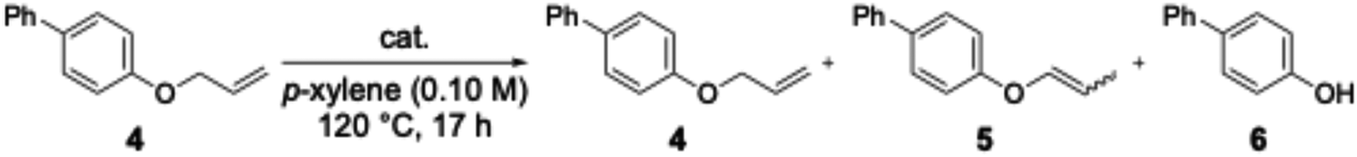
We therefore focused on the process by which the alkyl chain is eliminated. Corey et al. reported that, following the isomerisation of allyl ethers to enol ethers under rhodium catalysis, hydrolysis proceeds rapidly upon addition of acidic conditions, yielding alcohols (Scheme 5).12 Then, we hypothesised that in this reaction, silver trifluoromethanesulfonate acts as a Lewis acid, leading to the cleavage of the C–O bond in the enol ether; consequently, we conducted investigations into the conditions for the elimination of the alkyl chain using allyl ether 4 as the substrate (Table 6). The results of the experiments showed that the addition of a rhodium ethylene complex and a ligand resulted only in the isomerisation of allyl ether 4 to enol ether 5, with no cleavage of the C–O bond occurring (entry 1). When silver trifluoromethanesulfonate was added alone, allyl ether 4 was obtained in 12% yield and phenol 6 in 26% yield (entry 2). It was found that, in addition to the isomerisation of the allyl ether to an enol ether, cleavage of the C–O bond occurs under the action of silver trifluoromethanesulfonate. Furthermore, when the rhodium ethylene complex, the ligand and silver trifluoromethanesulfonate were all added, phenol 6 was obtained in 70% yield (entry 3). It is thought that phenol 6 is obtained in high yield because, after the isomerisation to enol ether proceeds efficiently under the action of the rhodium catalyst, silver trifluoromethanesulfonate acts as a Lewis acid, causing cleavage of the C–O bond in the enol ether.
 | ||
| Scheme 5 Reported cleavage of allyl ether using Rh catalyst. | ||
Next, we investigated the cyclisation process following the elimination of the alkyl chain. Specifically, using compound 7a—formed after the cyclopropane moiety had opened and the alkyl chain had been eliminated—we examined the effects of temperature and the presence or absence of silver trifluoromethanesulfonate (Table 7). When a solution of substrate 7a in paraxylene was stirred for 24 hours at 80 °C, 100 °C or 120 °C, the cyclised product 3a was not obtained at any of these temperatures (entries 1–3). It was therefore found that the cyclisation reaction of 7a does not proceed under heat alone.

Next, silver trifluoromethanesulfonate was added and the mixture was subjected to the same heating conditions (entries 4–6). As a result, whilst cyclised product 3a was not obtained under conditions of 80 °C or 100 °C, it was obtained in a yield of 68% under conditions of 120 °C. Consequently, it was found that silver trifluoromethanesulfonate and heat are required for the cyclisation process to yield by-product 3a.
These results indicate that by-product 3a is formed by cyclisation following the elimination of the alkyl chain from the substrate, and that this process requires silver trifluoromethanesulfonate and heat. This finding is consistent with the result shown in entry 7 of Table 3, where the yield of 3a increased to 33% when silver trifluoromethanesulfonate was used at 20 mol%.
Furthermore, Table 4 shows that the yield of by-product 3 increases when long-chain alkenes are used as substrates. This can be explained as follows. After the double bond moves to the α-position of the oxygen via chain-walking, the substrate proceeds along one of two pathways: (i) a pathway involving rhodium, via cyclo-isomerisation and aromatisation, yielding benzofuran 2; or (ii) a pathway without rhodium involvement, where the alkyl chain departs and cyclises to yield by-product 3. When long-chain alkenes 1g and 1h were used, the distance over which the double bond migrates increased, and the number of steps involving the rhodium catalyst rose; consequently, the efficiency of the catalytic cycle decreased, and pathway (ii), which does not require rhodium, was favoured over pathway (i), which does. It is thought that this resulted in an increase in the yield of by-product 3.
In conclusion, we conducted research into the development of long-range isomerisation/cyclopropane isomerisation/cycloisomerisation/aromatisation reactions using a multitasking rhodium catalyst, yielding the following results: [Rh(C2H4)2Cl]2, tri(3,5-xylyl)phosphine, and trifluoromethanesulfonate, four distinct reactions proceeded consecutively to yield benzofuran. The ability of a single rhodium catalyst to regulate long-range alkene migration and cyclopropyl C–C bond activation in a controlled manner highlights an underexplored dimension of chain-walking catalysis.
Through investigations into substrate generality, the application was successfully extended to eight substrates with elongated carbon chains and twelve substrates bearing substituents on the benzene ring.
NMR experiments were conducted to identify the intermediates in the sequential reaction and their order of formation, enabling the proposed reaction mechanism.
Author contributions
M. A. conceived the study, M. N., T. M., Y. S., M. T., T. M., T. S., A. N., M. S. and M. A. designed the experiments and analyzed the data. M. N., T. M. and T. M. performed the experiments. M. A. and M. N. prepared the draft of the manuscript. M. A. and M. N. performed the final editing. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, characterization data, NMR spectra of compounds. See DOI: https://doi.org/10.1039/d6ob00534a.Acknowledgements
This work was supported in parts by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Numbers JP23H02608 (M. A.)), JST CREST (No. JPMJCR20R1) (M. A.), The Iwatani Naoji Foundation (M. A.), Takahashi Industrial and Economic Research Foundation (M. A.), The Nippon Foundation – Osaka University Project for Infectious Disease Prevention (M. A.), and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP25ama121054 (M. A.).References
- A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- G. Szőllősi, Catal. Sci. Technol., 2018, 8, 389–422 RSC.
- (a) Y. Sato, T. Matsuzaki, T. Takehara, M. Sako, T. Suzuki and M. Arisawa, Chem. Commun., 2022, 58, 415–418 RSC; (b) M. Takatsuki, Y. Sato, T. Matsuzaki, M. Sako, A. Nakayama, T. Suzuki and M. Arisawa, Adv. Synth. Catal., 2026, 368, e70276 CrossRef CAS.
- (a) F. Zhou, J. Zhu, Y. Zhang and S. Zhu, Angew. Chem., Int. Ed., 2018, 57, 4058–4062 CrossRef CAS PubMed; (b) F. Zhou, Y. Zhang, X. Xu and S. Zhu, Angew. Chem., Int. Ed., 2019, 58, 1754–1758 CrossRef CAS PubMed.
- (a) Y. He, Y. Cai and S. Zhu, J. Am. Chem. Soc., 2017, 139, 1061–1064 CrossRef CAS PubMed; (b) Y. He, C. Liu, L. Yu and S. Zhu, Angew. Chem., Int. Ed., 2020, 59, 9186–9191 CrossRef CAS PubMed; (c) A. J. Borah and Z. Shi, J. Am. Chem. Soc., 2018, 140, 6062–6066 CrossRef CAS PubMed.
- (a) N. Wagner-Carlberg and T. Rovis, ACS Catal., 2023, 13, 16337–16343 CrossRef CAS PubMed; (b) S. Ding, J. Liu, C. Bu and L. Huang, Org. Lett., 2025, 27, 8504–8509 CrossRef CAS PubMed; (c) J. Xiao, Y. He, F. Ye and S. Zhu, Chem, 2018, 4, 1645–1657 CrossRef CAS; (d) Y. Zhang, J. He, P. Song, Y. Wang and S. Zhu, CCS Chem., 2021, 3, 2259 CrossRef CAS; (e) C. Han, Z. Fu, S. Guo, X. Fang, A. Lin and H. Yao, ACS Catal., 2019, 9, 4196–4202 CrossRef CAS.
- (a) N. G. Léonard, W. N. Palmer, M. R. Friedfeld, M. J. Bezdek and P. J. Chirik, ACS Catal., 2019, 9, 9034–9044 CrossRef; (b) M. Hu and S. Ge, Nat. Commun., 2020, 11, 1–10 Search PubMed; (c) J. Li, S. Qu and W. Zhao, Angew. Chem., Int. Ed., 2020, 59, 2360–2364 CrossRef CAS PubMed; (d) M. Zhang, Z. Liu and W. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202215455 CrossRef CAS PubMed.
- (a) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84–88 CrossRef PubMed; (b) J. He, P. Song, X. Xu, S. Zhu and Y. Wang, ACS Catal., 2019, 9, 3253–3259 CrossRef CAS.
- AgOTf might work to change the counter anion of the Rh catalyst system.
- Unfortunately, an NTs-substituted substrate was not succeeded to give the corresponding indole derivatives under the same reaction conditions.
- Although we do not have a witness that Rh-H is an actual active species in this reaction system, we can easily understand the reaction mechanism with Rh–H as a one candidate.
- E. J. Corey and J. W. Suggs, J. Org. Chem., 1973, 38, 3224 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |