DOI:
10.1039/D6OB00305B
(Paper)
Org. Biomol. Chem., 2026, Advance Article
Investigations toward a unified reaction pathway of thermal and TBSOTf-mediated oxidopyrylium-alkene (5 + 2) cycloadditions
Received
20th February 2026
, Accepted 16th April 2026
First published on 16th April 2026
Abstract
Oxidopyrylium-based (5 + 2) cycloadditions are crucial reactions to construct seven-membered carbocycles containing an ether bridge (i.e. oxabicyclo[3.2.1]octanes). Intramolecular silyloxypyrone-based (5 + 2) cycloadditions were investigated and revealed several features: (1) the TBDPS thermal process proceeds via a zwitterionic oxidopyrylium intermediate similar to previously reported TBS variants; (2) the TBSOTf-mediated reaction proceeds through a cationic oxidopyrylium intermediate; (3) quantum chemical calculations predict a stepwise process for an electron-rich dipolarophile for each set of conditions. The thermal silyloxypyrone-based (5 + 2) cycloadditions were extremely dependent on the nature of the dipolarophile and the silyl transfer group. The TBDPS enhances the rate compared to the TBS variant but only for less polarized alkenes. Relatively neutral alkenes were the least reactive for both, whereas electron-deficient and electron-rich dipolarophiles were more reactive providing evidence for ambident oxidopyrylium intermediates. TBSOTf-mediated cycloadditions, however, revealed evidence for a cationic intermediate that follows a more consistent mechanistic trend. Qualitative rate studies, Hammett linear free energy relationships, and theoretical calculations combine to provide evidence for both mechanistic scenarios.
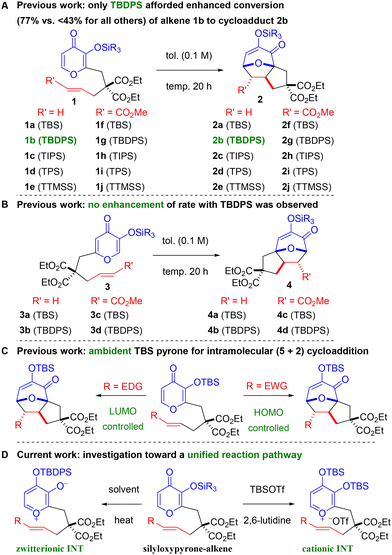
Cycloadditions can be initiated by the input of thermal energy or an activating agent to access key transition states en route to a desired product.1 Oxidopyrylium-based (5 + 2) variants2 can be mediated by different pathways, but two widely utilized strategies involve acetoxypyranones or silyloxypyrones (Scheme 1). Activation of acetoxypyranones, established by Hendrickson3 and Sammes,4 typically requires a base to initiate deprotonation followed by release of the acetoxy (or related leaving group) to deliver the oxidopyrylium. Activation of silyloxypyrones (or related starting material), pioneered by Wender and Mascareñas,5 takes advantage of either an internal group transfer process or an external Lewis acid to afford the oxidopyrylium moiety. Several variations of these methods have been reported6 and our group has contributed to both activation pathways,7 which are useful toward the total synthesis of natural products8 and other applications.9 The generally accepted mechanism7b,10 involves silyl transfer to the oxidopyrylium followed by concerted cycloaddition. We previously reported silyloxypyrone-based cycloadditions that revealed crucial data regarding three components: silyl group transfer,7g tether proximity to the transfer group,7g and dipolarophile electronics.7b Relative rate comparisons of a wide variety of silylated maltol-derived substrates 1a–j revealed an interesting result: only TBDPS-terminal olefin 1b exhibited enhanced reactivity and transfer group dependence (Scheme 2A).7g Furthermore, several silylated kojic acid-derived substrates 3a–d showed no dependence on the transfer group (Scheme 2B).7g Although enoates 1f–j and 3c–d were not affected by the silyl transfer group, enhanced conversion was observed compared to the terminal alkenes confirming the expected electronic effect of the ester that reduces the energy of the lowest unoccupied molecular orbital (LUMO). These combined results revealed a subtle interplay between transfer group, tether, and alkene. More recently, we undertook a detailed investigation of TBS-pyrones that revealed the substantial impact of dipolarophile electronics on the cycloaddition that illuminated a spectrum of reactivity7b passing through borderlands11 of concerted and stepwise (Scheme 2C). Herein, we disclose TBDPS-thermal and TBSOTf-mediated6o (5 + 2) cycloadditions that proceed via zwitterionic and cationic intermediates, respectively (Scheme 2D). Taken together with our previous work,7b,g these qualitative rate studies, Hammett linear free energy relationship (LFER) findings, and theoretical calculations provide a coherent framework for a unified mechanistic pathway of silyloxypyrone-based (5 + 2) cycloadditions (vide infra) and lay the foundation for continued exploration of this intriguing reaction.
 |
| | Scheme 1 Acetoxypyranone vs. silyloxypyrone activation. | |
 |
| | Scheme 2 Summary of transfer group, tether, and dipolarophile effects on silyloxypyrone-based (5 + 2) cycloaddition leading to further understanding toward a unified reaction pathway. | |
Results and discussion
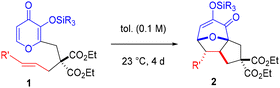
Building on the previous observation that the TBDPS transfer group showed no enhancement of enoate conversion (vide supra),7g we undertook a more thorough comparison of TBS- and TBDPS-pyrones7b with a range of substituents on the dipolarophile (Table 1 and Scheme 3). Ambient temperature qualitative rate studies afforded good mass recovery and, as expected, terminal olefins 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7b and 1b both afforded no reaction (Table 1, entries 1 and 2). As the electron-withdrawing ability of the substituent increased, enhanced conversion was observed. Enoates 1f–g7g gave minimal conversion (Table 1, entries 3 and 4),7b enones 1k7b and 1l12 provided more substantial quantities of adducts 2k–l (Table 1, entries 5 and 6), and nitro-enes 1m–n afforded complete conversion (Table 1, entries 7 and 8),12 thus confirming the effect of lowering the energy of the LUMO. These newly reported nitro-ene starting materials 1m–n and corresponding cycloadducts 2m–n were less stable than carbonyl substrates 1f–l. Therefore, we do not believe that conversions of nitro-enes 1m–n (entries 7 and 8) are reflective of rate enhancement by the TBDPS but rather reveal that the TBDPS is simply more stable and thus both are functioning similarly as transfer groups in the case of electron-poor dipolarophiles. Enamine 1o was previously reported7b to proceed at ambient temperature (in situ via aldehyde 5) and newly investigated variant 1p further demonstrates that enhancement with the TBDPS transfer group is limited to less polarized alkenes (vide infra). Unique parameters were necessary to probe this variant below ambient temperature (Scheme 3). When a cycloaddition proceeds at room temperature, the substrate is likely to continue reacting upon isolation and purification, potentially skewing the results. By simply passing the reaction mixture over silica gel, in situ generation of enamines 1o–p (not shown) from aldehydes 5/5′ is reversible, and the reaction is quenched. Therefore, enamine 1o was formed at 0 °C to give cycloadduct 2o (64% yield) and aldehyde 5 (28% yield) after column chromatography of the reaction mixture.7b A similar result in this study was obtained with the TBDPS variant 2p.12
7b and 1b both afforded no reaction (Table 1, entries 1 and 2). As the electron-withdrawing ability of the substituent increased, enhanced conversion was observed. Enoates 1f–g7g gave minimal conversion (Table 1, entries 3 and 4),7b enones 1k7b and 1l12 provided more substantial quantities of adducts 2k–l (Table 1, entries 5 and 6), and nitro-enes 1m–n afforded complete conversion (Table 1, entries 7 and 8),12 thus confirming the effect of lowering the energy of the LUMO. These newly reported nitro-ene starting materials 1m–n and corresponding cycloadducts 2m–n were less stable than carbonyl substrates 1f–l. Therefore, we do not believe that conversions of nitro-enes 1m–n (entries 7 and 8) are reflective of rate enhancement by the TBDPS but rather reveal that the TBDPS is simply more stable and thus both are functioning similarly as transfer groups in the case of electron-poor dipolarophiles. Enamine 1o was previously reported7b to proceed at ambient temperature (in situ via aldehyde 5) and newly investigated variant 1p further demonstrates that enhancement with the TBDPS transfer group is limited to less polarized alkenes (vide infra). Unique parameters were necessary to probe this variant below ambient temperature (Scheme 3). When a cycloaddition proceeds at room temperature, the substrate is likely to continue reacting upon isolation and purification, potentially skewing the results. By simply passing the reaction mixture over silica gel, in situ generation of enamines 1o–p (not shown) from aldehydes 5/5′ is reversible, and the reaction is quenched. Therefore, enamine 1o was formed at 0 °C to give cycloadduct 2o (64% yield) and aldehyde 5 (28% yield) after column chromatography of the reaction mixture.7b A similar result in this study was obtained with the TBDPS variant 2p.12
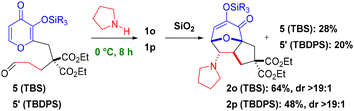 |
| | Scheme 3 Low temperature enamine (5 + 2) cycloaddition (reproduced in part with permission from ref. 7b). | |
Table 1 Qualitative rate comparison of various dipolarophiles in silyloxypyrone-based (5 + 2) cycloadditions
|
| Entry |
SiR3 |
R′ |
% rec.a (1) |
% yielda (2) |
| Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. Not detected. Previously reported in ref. 7b (J. Org. Chem. 2023, 88, 5972); included for comparison with new TBDPS data. Previously reported in ref. 7g (J. Org. Chem. 2019, 84, 10306); included for comparison with new TBDPS data. |
| 1 |
TBSc |
H |
>95 (1a) |
<5 (2a)b |
| 2 |
TBDPS |
H |
>95 (1b) |
<5 (2b)b |
| 3 |
TBSd |
CO2Me |
77 (1f) |
7 (2f) |
| 4 |
TBDPSd |
CO2Me |
78 (1g) |
8 (2g) |
| 5 |
TBSc |
C(O)Me |
54 (1k) |
36 (2k) |
| 6 |
TBDPS |
C(O)Me |
56 (1l) |
37 (2l) |
| 7 |
TBS |
NO2 |
<5 (1m)b |
73 (2m) |
| 8 |
TBDPS |
NO2 |
<5 (1n)b |
84 (2n) |
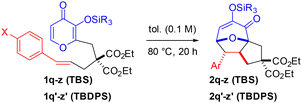
Linear free energy relationship (LFER) studies (i.e., Hammett plots) offer a powerful avenue to probe reaction pathways via substituent effects.13 Plotting the substituent constant (σ) vs. the corresponding log [k/k0] (or related rate parameter) typically reveals a linear trend that provides support for a consistent mechanism in operation, and non-linear Hammett plots afford compelling evidence for a change in mechanism across substituents.14 We previously synthesized various styrenes with TBS7b (1q–z) and herein with TBDPS12 (1q′–z′) transfer groups and subjected them to uniform conditions (Table 2). With both transfer groups, electron-donating and electron-deficient substrates were more reactive than less polarized variants. The TBDPS group slightly enhanced the reactivity as compared to the TBS further indicating that this phenomenon is limited to less polarized alkenes. For example, 4-dimethylaminostyrenes 1q/1q′ (Table 2, entry 1) gave similar conversion as the electron deficient ester-substituted 1x/1x′ (Table 2, entry 8) and 4-cyano-substituted 1y/1y′ (Table 2, entry 9), while the 4-nitrostyrenes 1z/1z′ afforded the highest conversion (Table 2, entry 10). Less-polarized derivatives such as phenyl 1u/1u′ and 4-fluoro 1v/1v′ were far less reactive by comparison (Table 2, entries 5 and 6). When log rate/rate0 was plotted against σp constants, non-linear Hammett plots were revealed (Fig. 1 and 2). These plots are consistent with a shift in reaction pathway with an inflection point near the phenyl 1u/1u′ (Table 2, entry 5) and 4-fluoro 1v/1v′ (Table 2, entry 6). In both cases, upon separation of the trendlines by Sigma (σ) value, two distinctly linear Hammett plots are revealed. The negative slope (ρ) correlates to electron-donating group acceleration, whereas electron-withdrawing group acceleration is indicated by the positive slope (ρ).
 |
| | Fig. 1 Hammett analysis: TBS thermal non-LFER (reproduced with permission from ref. 7b; J. Org. Chem. 2023, 88, 5972). | |
 |
| | Fig. 2 Hammett analysis: TBDPS Thermal non-LFER. | |
Table 2 Linear free-energy relationship comparison of TBS and TBDPS styrenes (reproduced in part with permission from ref. 7b)
|
| Entry |
X |
% yielda (2)b |
σp |
% yielda (2′) |
| Determined by the average of two trials as measured by 1H NMR analysis with 1,3,5-trimethoxybenzene as an internal standard. Previously reported in ref. 7b (J. Org. Chem. 2023, 88, 5972); included for comparison with new TBDPS data. |
| 1 |
NMe2 |
69 (2q) |
−0.83 |
87 (2q′) |
| 2 |
OiPr |
48 (2r) |
−0.45 |
65 (2r′) |
| 3 |
OMe |
45 (2s) |
−0.27 |
65 (2s′) |
| 4 |
Me |
44 (2t) |
0.17 |
58 (2t′) |
| 5 |
H |
44 (2u) |
0.00 |
47 (2u′) |
| 6 |
F |
37 (2v) |
0.06 |
46 (2v′) |
| 7 |
Cl |
54 (2w) |
0.23 |
68 (2w′) |
| 8 |
CO2Me |
67 (2x) |
0.39 |
77 (2x′) |
| 9 |
CN |
68 (2y) |
0.66 |
85 (2y′) |
| 10 |
NO2 |
87 (2z) |
0.78 |
94 (2z′) |
An alternative to thermal-mediated silyl transfer, TBSOTf-promoted (5 + 2) cycloadditions6o were explored with a subset of styrenes 2s,t,u,w,z (Table 3) and 4e–i (Table 4). Due to the proposed cationic intermediate (cf. Scheme 2D), these investigations afforded evidence of a more typical mechanistic pathway regardless of dipolarophile electronics. In each series, the reaction rate increased with enhanced electron density of the styrene substituent. Kojic acid substrates (Table 4) were more reactive than maltol variants (Table 3), with timeframes of 30 minutes vs. 20 hours, respectively. In both cases, Hammett plots revealed a straightforward linear trend (Fig. 3 and 4) providing evidence for a rate-determining transition state involving accumulation of positive charge on the nucleophilic styrene via the proposed cationic oxidopyrylium species (vide supra). It was a challenge to ascertain an appropriate time course for all substrates since methoxy-substituted styrenes 1s and 3e were completely consumed with only minimal conversion of the nitro-substituted styrenes 1z and 3i. Thus, the results were slightly skewed, and the R2-value improved upon calculation without the methoxy-substituted styrenes.
 |
| | Fig. 3 Hammett analysis: TBSOTf (maltol substrates). | |
 |
| | Fig. 4 Hammett analysis: TBSOTf (kojic acid substrates). | |
Table 3 Linear free-energy relationship of styrenes with TBSOTf

|
| Entry |
X |
% conv.a (2) |
σp+ |
Log (rate/rate0) |
| Determined by the average of two trials as measured by 1H NMR analysis with 1,3,5-trimethoxybenzene as an internal standard. |
| 1 |
OMe |
100 (2s) |
−0.78 |
0.569 |
| 2 |
Me |
98 (2t) |
−0.31 |
0.560 |
| 3 |
H |
27 (2u) |
0.00 |
0.000 |
| 4 |
Cl |
22 (2w) |
0.11 |
−0.089 |
| 5 |
NO2 |
4 (2z) |
0.79 |
−0.829 |
Table 4 Linear free-energy relationship of styrenes with TBSOTf
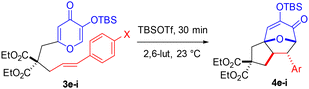
|
| Entry |
X |
% conv.a (4) |
σp+ |
Log (rate/rate0) |
| Determined by the average of two trials as measured by 1H NMR analysis with 1,3,5-trimethoxybenzene as an internal standard. |
| 1 |
OMe |
100 (4e) |
−0.78 |
0.467 |
| 2 |
Me |
89 (4f) |
−0.31 |
0.415 |
| 3 |
H |
33 (4g) |
0.00 |
0.000 |
| 4 |
Cl |
16 (4h) |
0.11 |
−0.301 |
| 5 |
NO2 |
1 (4i) |
0.79 |
−1.480 |
To confirm the stereospecific nature of the thermal and TBSOTf-mediated (5 + 2) cycloadditions, two cis alkenes 1s and 1g were synthesized.12 Cycloadditions utilizing these cis alkenes 1g and 1s (Scheme 4) complimented the stereospecific nature that was observed previously with various trans olefins.7b,g First, enoate cis-1g underwent smooth cycloaddition at 60 °C with complete stereospecificity (eqn (1)). Styrenes 1s were subjected to both sets of conditions (i.e. thermal and TBSOTf)12 to investigate relative reactivity and stereospecificity (eqn (2)–(3)). Styrene cis-1s was significantly less reactive than the corresponding styrene trans-1s and required increased heating (i.e. 110 °C) but afforded cis-2s, also with complete stereospecificity (eq (2)). To confirm this, a mixture of trans/cis (1:1) was utilized and, upon heating to only 80 °C, trans-1s underwent significant conversion, but minimal conversion to cis-2s was observed (not shown).12 Upon activation with TBSOTf a similar trend was observed after 2 hours with an initial trans/cis ratio of 1:1. Nearly complete conversion of trans-1s (<5% remaining) to trans-2s (41% yield; maximum 50%) was detected while only trace conversion to cis-2s (<5%) and nearly complete recovery of cis-1s (40% yield; maximum 50%) was observed (eqn (3)). Further conversion of cis-1s under these conditions, albeit slowly, was detected (not shown).12
 |
| | Scheme 4 Stereospecific cycloadditions with cis-alkenes 1 g per 1 s. | |
Quantum chemical calculations were undertaken to investigate the mechanism of these silyloxypyrone-based (5 + 2) cycloadditions. Initial conformational searches were carried out using XTB-CREST15 and subsequent density functional theory (DFT) calculations were carried out with Gaussian16.16 The M06-2X functional with the D3(0) dispersion correction17 was used to locate stationary points, since this functional is known to perform well for main group thermochemistry and kinetic studies.18 We employed the 6-31+G(d,p) basis set and the SMD continuum solvation model for geometry optimizations.19 The larger 6-311+G(2df,2p) basis set was used for computing single point energies. Reported Gibbs free energies include thermal corrections from frequency calculations at the M06-2X-D3(0)/6-31+G(d,p) level, which was benchmarked with other functional/basis set combinations to confirm that qualitative conclusions did not change.
For reactions in the absence of TBSOTf, silyl transfer via a pentacoordinate species was calculated to be fast and non-rate determining in each case.7b,12 The overall barrier via rate-determining concerted synchronous (5 + 2) cycloaddition for terminal alkene 1a was predicted to be 1.9 kcal mol−1 higher in energy than the corresponding synchronous concerted (5 + 2) cycloaddition for terminal alkene 1b (Fig. 5). These predictions correlate to experimental results in which the TBDPS variant reacts faster than the TBS (cf. Scheme 2A), although an even larger difference (2.4 kcal mol−1) is predicted for the kojic acid-derived analogs, which is not borne out in experiments (cf. Scheme 2B). These results with the TBDPS should be viewed with some caution (the issue is likely related to challenges in modeling the conformational properties of the TBDPS group). Nonetheless, barriers were predicted to be higher for kojic acid-derived substrates than for maltol-derived systems with both TBS (Fig. 5) and TBDPS12 transfer groups.
 |
| | Fig. 5 TSs of (a) maltol-derived terminal olefin (TBS);7b (b) maltol-derived terminal olefin (TBDPS); (c) kojic acid-derived terminal olefin (TBS). | |
Concerted but asynchronous (5 + 2) cycloaddition TSs were found for oxidopyrylium intermediates derived from 1q–z and the plot of computationally predicted free energy barriers vs. substituent constants (σp) was previously shown7b to be non-linear in qualitative agreement with experimental results (cf. Fig. 1). The same effect was found for TBDPS-containing systems 1q′, 1u′, 1z′ (Fig. 6). Again, lower barriers were predicted for the TBDPS variants by ∼2 kcal mol−1, reinforcing the influence of this bulkier group on less polarized dipolarophiles. DFT calculations for cationic intermediates derived from TBSOTf show that the methoxy substituent promotes a stepwise (5 + 2) cycloaddition (Fig. 7) while both H and NO2 substituents promote an asynchronous concerted process.12
 |
| | Fig. 6 TSs of (a) dimethylaminostyrene (TBDPS); (b) styrene (TBDPS); (c) nitrostyrene (TBDPS). | |
 |
| | Fig. 7 TSs of stepwise TBSOTf-mediated cationic (5 + 2) cycloaddition of methoxy styrene variant. | |
Comparison of previously calculated transition states7b for TBS variants with the newly calculated12 enoate 2f and nitro-ene 2m reveals an interesting trend (Fig. 8). The terminal olefin 1a is the closest to a synchronous concerted transition state 2a with an energy barrier of 25.9 kcal mol−1. As electron-deficient substituents are introduced, the energy decreases and the mechanism shifts to concerted, but asynchronous along the following trend: enoate 2f (23.4 kcal mol−1), enone 2k (23.1 kcal mol−1), and nitro-ene 2m (21.6 kcal mol−1). This trend correlates to experimental results in which more electrophilic dipolarophiles react faster than the terminal olefin 1a (cf. Table 1). However, enamine 1o, which was far more reactive experimentally (cf. Scheme 3), was predicted to be stepwise with an initial rate-determining transition state 2o energy of 19.7 kcal mol−1 followed by the second step which is only slightly lower in energy (18.8 kcal mol−1).7b Interestingly, nitro-ene 2m is not quite electrophilic enough to promote formation of an analogous stepwise process with an anionic intermediate, but decreased bond lengths suggest that the transition structure is progressively more asynchronous as the electron withdrawing ability of the substituent increases.
 |
| | Fig. 8 TSs of substitute olefins: (a) nitro; (b) ketone;7b (c) ester; (d) H;7b (e) amine7b (TBS only for simplicity). | |
Based on these experiments and calculations, a unified mechanistic pathway is envisioned for both modes of activation (Scheme 5). Silyloxypyrones can be activated by either thermal or Lewis acid-mediated (i.e. TBSOTf) conditions. Thermal activation with either TBS or TBDPS as transfer group promotes the formation of zwitterionic oxidopyrylium intermediates and can be achieved at temperatures as low as 0 °C in the case of electron-rich enamines (cf. Scheme 3). Ample evidence suggests that the TBDPS transfer group enhances the rate as compared to the TBS, but only for less polarized alkenes (i.e. terminal 1b and styrenes 1q-z). Likely this is due to an entropic effect that results from steric interactions of the TBDPS and the diester functionality leading to slightly lower energy barriers for cycloaddition. When the alkenes are more polarized, however, the dipolarophile substituent effect is more significant than this TBDPS acceleration and lowers the activation energy regardless. By contrast, TBSOTf promotes the formation of a cationic oxidopyrylium intermediate, which proceeds more rapidly in the case of electron-rich methoxy styrenes (cf. Tables 3 and 4). Both intermediates (i.e. zwitterionic or cationic) readily undergo concerted (5 + 2) cycloaddition (synchronous or asynchronous depending on the dipolarophile substituent) with neutral or electron-deficient dipolarophiles. However, quantum chemical calculations revealed evidence of LUMO-controlled, inverse electron demand stepwise pathways for electron-rich dipolarophiles for each set of activation parameters. Hammett plots also afforded crucial insight into both sets of conditions (cf. Fig. 1–4). In the case of the thermal-mediated zwitterionic-based cycloadditions, neutral alkenes were the least reactive, whereas both electron-deficient and electron-rich dipolarophiles were more reactive thus providing non-linear free energy relationships indicative of a change in mechanism. Hammett plots of the TBSOTf-mediated cycloadditions, however, revealed linear free energy relationships that are more in-line with a consistent mechanism throughout the reaction.
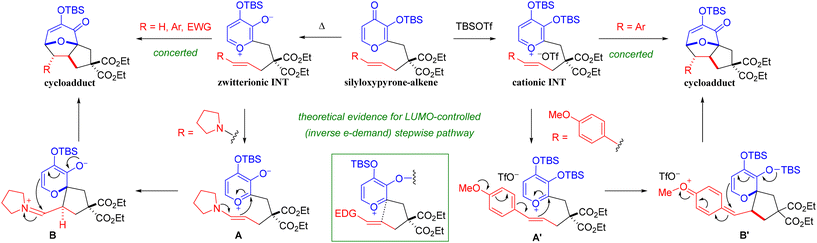 |
| | Scheme 5 Unified mechanism for thermal (TBS only for simplicity) and TBSOTf-mediated (5 + 2) cycloadditions. | |
Conclusions
Silyloxypyrone-based (5 + 2) cycloadditions were investigated using both thermal and TBSOTf-mediated processes that revealed similarities and differences between these modes of activation. Thermal-mediated (5 + 2) cycloaddition proceeds through a zwitterionic oxidopyrylium intermediate, whereas TBSOTf-mediated (5 + 2) cycloaddition proceeds via cationic, bis-silylated intermediates. Qualitative rate studies, Hammett plots, and quantum calculations combined to illustrate a dichotomy between these two pathways. In the case of thermally induced variants with both TBS and TBDPS, both electron-deficient and electron-rich dipolarophiles were more reactive than neutral alkenes thus providing evidence for ambident zwitterionic oxidopyrylium intermediates. Several examples reveal that the TBDPS lowers the activation barrier, but only for less polarized olefins. Although most thermal-initiated reactions were concerted, theoretical evidence for a stepwise reaction pathway of electron-rich enamines was predicted. In TBSOTf-mediated cycloadditions, by contrast, electron-rich dipolarophiles are the most reactive and electron-deficient dipolarophiles are the least reactive. This observation is suggestive of cationic intermediates that demonstrate a more linear free energy relationship. A stepwise reaction pathway of the electron-donating methoxy styrene was also predicted with DFT. Overall, several dipolarophiles were investigated utilizing both modes of activation to probe the range of reactivity, which revealed a variety of mechanistic nuances between concerted and stepwise borderlands. A deep appreciation of these important mechanistic details is enabling opportunities to discover novel transformations to be reported in due course.
Author contributions
The manuscript was written through contributions of all authors. A. J. Y., S. N. R., and W. G. contributed equally to this research. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). See DOI: https://doi.org/10.1039/d6ob00305b.
All structures are available in the ioChem-BD repository: https://doi.org/10.19061/iochem-bd-6-585.
Acknowledgements
Acknowledgment is made to the National Science Foundation: individual research awards (CHE-1954588 and CHE-2350125) and XSEDE program for computational resources.
References
-
(a) N. Nishiwaki, Methods and Applications of Cycloaddition Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2014 CrossRef;
(b) S. Kobayashi and K. A. Jørgensen, Cycloaddition Reactions in Organic Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed;
(c) W. Carruthers, Cycloaddition Reactions in Organic Chemistry, Tetrahedron Organic Chemistry, Pergamon, Oxford, 1990, vol. 8 Search PubMed.
-
(a) L. P. Bejcek and R. P. Murelli, Oxidopyrylium [5 + 2] cycloaddition chemistry: Historical Perspective and recent advances (2008–2018), Tetrahedron, 2018, 74, 2501–2521 CrossRef CAS PubMed;
(b) V. Singh, U. M. Krishna, Vikrant and G. K. Trivedi, Cycloaddition of oxidopyrylium species in organic synthesis, Tetrahedron, 2008, 64, 3405–3428 CrossRef CAS;
(c) J. L. Mascareñas, The [5 + 2] Cycloaddition Chemistry of β-Alkoxy-γ-pyrones, Adv. Cycloaddit., 1999, 6, 1–54 Search PubMed.
- J. B. Hendrickson and J. S. Farina, A New 7-Ring Cycloaddition Reaction, J. Org. Chem., 1980, 45, 3359–3361 CrossRef CAS.
- P. G. Sammes and L. J. Street, Intramolecular Cycloadditions with Oxidopyrylium Ylides, J. Chem. Soc., Chem. Commun., 1982, 1056–1057 RSC.
-
(a) A. Rumbo, A. Mouriño, L. Castedo and J. L. Mascareñas, 3-Hydroxy-4-Pyrones as Precursors of 4-Methoxy-3-Oxidopyridinium Ylides. An Expeditious Entry to Highly Substituted 8-Azabicyclo[3.2.1]octanes, J. Org. Chem., 1996, 61, 6114–6120 CrossRef CAS PubMed;
(b) A. Rumbo, L. Castedo, A. Mouriño and J. L. Mascareñas, Temporary Tethering Strategies for [5 + 2] Pyrone-Alkene Cycloadditions, J. Org. Chem., 1993, 58, 5585–5586 CrossRef CAS;
(c) P. A. Wender and J. L. Mascareñas, Preparation and Cycloadditions of a 4-Methoxy-3-Oxidopyrylium Ylid: A Reagent for the Synthesis of Highly Substituted Seven-Membered Rings and Tetrahydrofurans, Tetrahedron Lett., 1992, 33, 2115–2118 CrossRef CAS;
(d) P. A. Wender and F. E. McDonald, Studies on Tumor Promoters. 9. A Second-Generation Synthesis of Phorbol, J. Am. Chem. Soc., 1990, 112, 4956–4958 CrossRef CAS;
(e) P. A. Wender and J. L. Mascareñas, Studies on Tumor Promoters. 11. A New [5 + 2] Cycloaddition Method and its Application to the Synthesis of BC Ring Precursors of Phoboids, J. Org. Chem., 1991, 56, 6267–6269 CrossRef CAS.
-
(a) L. Yang, K. L. Chan, K. K. Cheung, P. Chiu, Z. Lin and J. Sun, Enantioselective type II intramolecular [5 + 2] cycloadditions of oxidopyrylium ylides using chiral-phosphoric-acid catalysis, Nat. Synth., 2025, 4, 1223–1231 CrossRef CAS;
(b) A. K. Ghosh and M. Yadav, Highly Diastereoselective Intramolecular Asymmetric Oxidopyrylium-olefin [5 + 2] Cycloaddition and Synthesis of 8-Oxabicyclo[3.2.1]oct-3-enone Containing Ring Systems, J. Org. Chem., 2021, 86, 8127–8142 CrossRef CAS PubMed;
(c) L. Zhang, Q. Shi, T. Cao and S. Zhu, Catalytic regio- and stereoselective intermolecular [5 + 2] cycloaddition via conjugative activation of oxidopyrylium, Chem. Commun., 2020, 56, 9533–9536 RSC;
(d) C. Zhao, D. A. Glazier, D. Yang, D. Yin, I. A. Guzei, M. M. Aristov, P. Liu and W. Tang, Intermolecular Regio- and Stereoselective Hetero-[5 + 2] Cycloaddition of Oxidopyrylium Ylides and Cyclic Imines, Angew. Chem., Int. Ed., 2019, 58, 887–891 CrossRef CAS;
(e) Y. Toda, M. Shimizu, T. Iwai and H. Suga, Triethylamine Enables Catalytic Generation of Oxidopyrylium Ylides for [5 + 2] Cycloadditions with Alkenes: An Efficient Entry to 8-Oxabicyclo[3.2.1]octane Frameworks, Adv. Synth. Catal., 2018, 360, 2377–2381 CrossRef CAS;
(f) H. Suga, T. Iwai, M. Shimizu, K. Takahashi and Y. Toda, Efficient generation of an oxidopyrylium ylide using a Pd catalyst and its [5 + 2] cycloadditions with several dipolarophiles, Chem. Commun., 2018, 54, 1109–1112 RSC;
(g) K. N. Fuhr, D. R. Hirsch, R. P. Murelli and S. E. Brenner-Moyer, Catalytic Enantioselective Intermolecular [5 + 2] Dipolar Cycloadditions of a 3-Hydroxy-4-pyrone-Derived Oxidopyrylium Ylide, Org. Lett., 2017, 19, 6356–6359 CrossRef CAS PubMed;
(h) S. M. Wilkerson-Hill, S. Sawano and R. Sarpong, Bis(1-cyanovinyl acetate) Is a Linear Precursor to 3-Oxidopyrylium Ions, J. Org. Chem., 2016, 81, 11132–11144 CrossRef CAS PubMed;
(i) M. P. D'Erasmo, C. Meck, C. A. Lewis and R. P. Murelli, Discovery and Development of a Three-Component Oxidopyrylium [5 + 2] Cycloaddition, J. Org. Chem., 2016, 81, 3744–3751 CrossRef PubMed;
(j) A. Orue, U. Uria, E. Reyes, L. Carrillo and J. L. Vicario, Catalytic Enantioselective [5 + 2] Cycloaddition between Oxidopyrylium Ylides and Enals under Dienamine Activation, Angew. Chem., Int. Ed., 2015, 54, 3043–3046 CrossRef CAS;
(k) G. Mei, X. Liu, C. Qiao, W. Chen and C.-C. Li, Type II Intramolecular [5 + 2] Cycloaddition: Facile Synthesis of Highly Functionalized Bridged Ring Systems, Angew. Chem., Int. Ed., 2015, 54, 1754–1758 CrossRef CAS PubMed;
(l) M. R. Witten and E. N. Jacobsen, Catalytic Asymmetric Synthesis of 8-Oxabicyclooctanes by Intermolecular [5 + 2] Pyrylium Cycloadditions, Angew. Chem., Int. Ed., 2014, 53, 5912–5916 CrossRef CAS PubMed;
(m) K. Tchabanenko, C. Sloan, Y.-M. Bunetel and P. Mullen, Diastereoselective 1,3-dipolar cycloaddition of Pyrylium ylides with chiral enamides, Org. Biomol. Chem., 2012, 10, 4215–4219 RSC;
(n) N. Z. Burns, M. R. Witten and E. N. Jacobsen, Dual Catalysis in Enantioselective Oxidopyrylium-Based [5 + 2] Cycloadditions, J. Am. Chem. Soc., 2011, 133, 14578–14581 CrossRef CAS;
(o) N. Ohmori, M. Yoshimura and K. Ohkata, Asymmetric Induction in Intramolecular [5 + 2] Cycloaddition of 2-(4-Alkenyl)-5-Benzoyloxy(or 5-Silyloxy)-4-Pyrones Involving Migration of the Pyrone O-5 Group to O-4, Heterocycles, 1997, 45, 2097–2100 CrossRef CAS.
-
(a) S. N. Angles, W. Guo, K. Darko, M. Erzuah, K. G. Pauley, I. E. Promise, J. R. Goodell, D. J. Tantillo and T. A. Mitchell, Net Intermolecular Silyloxypyrone-based (5 + 2) Cycloadditions Utilizing Amides as Enabling and Cleavable Tethers, Org. Lett., 2023, 25, 7137–7141 CrossRef CAS PubMed;
(b) A. J. Youman, S. N. Rokey, J. P. Grabowski, W. Guo, Q. Sun, S. N. Angles, J. R. Goodell, D. J. Tantillo and T. A. Mitchell, Experimental and Theoretical Investigation of the Synchronicity of Ambident Silyloxypyrone-Based (5 + 2) Cycloadditions, J. Org. Chem., 2023, 88, 5972–5981 CrossRef CAS PubMed;
(c) S. I. Corrie, A. C. Pearce, J. P. Grabowski, K. A. Randolph, W. Guo, D. J. Tantillo and T. A. Mitchell, Boron-tethered oxidopyrylium-based [5 + 2] cycloadditions, Tetrahedron Lett., 2022, 107, 154094 CrossRef CAS;
(d) Y. Lu and D. J. Tantillo, Comparison of (5 + 2) Cycloadditions Involving Oxidopyrylium and Oxidopyridinium Ions – Relative Reactivities, J. Org. Chem., 2021, 86, 8652–8659 CrossRef CAS PubMed;
(e) S. N. Rokey, J. A. Simanis, C. M. Law, S. Pohani, S. Willens Behrends, J. J. Bulandr, G. M. Ferrence, J. R. Goodell and T. A. Mitchell, Intramolecular asymmetric oxidopyrylium-based (5 + 2) cycloadditions, Tetrahedron Lett., 2020, 61, 152377 CrossRef CAS;
(f) J. P. Grabowski, G. M. Ferrence and T. A. Mitchell, Efforts toward the total synthesis of (±)-toxicodenane A utilizing an oxidopyrylium-based [5 + 2] cycloaddition of a silicon-tethered BOC-pyranone, Tetrahedron Lett., 2020, 61, 152324 CrossRef CAS;
(g) J. J. Bulandr, J. P. Grabowski, C. M. Law, J. L. Shaw, J. R. Goodell and T. A. Mitchell, Investigation of Transfer Group, Tether Proximity, and Alkene Substitution for Intramolecular Silyloxypyrone-Based (5 + 2) Cycloadditions, J. Org. Chem., 2019, 84, 10306–10320 CrossRef CAS PubMed;
(h) R. H. Kaufman, C. M. Law, J. A. Simanis, E. L. Woodall, C. R. Zwick III, H. B. Wedler, P. Wendelboe, C. G. Hamaker, J. R. Goodell, D. J. Tantillo and T. A. Mitchell, Oxidopyrylium-Alkene
(5 + 2) Cycloaddition Conjugate Addition Cascade (C3) Sequences: Scope, Limitation, and Computational Investigations, J. Org. Chem., 2018, 83, 9818–9838 CrossRef CAS PubMed;
(i) J. A. Simanis, C. M. Law, E. L. Woodall, C. G. Hamaker, J. R. Goodell and T. A. Mitchell, Investigation of Oxidopyrylium-Alkene [5 + 2] Cycloaddition Conjugate Addition Cascade (C3) Sequences, Chem. Commun., 2014, 50, 9130–9133 RSC;
(j) E. L. Woodall, J. A. Simanis, C. G. Hamaker, J. R. Goodell and T. A. Mitchell, Unique Reactivity of anti- and syn-Acetoxypyranones en route to Oxidopyrylium Intermediates Leading to a Cascade Process, Org. Lett., 2013, 15, 3270–3273 CrossRef CAS PubMed;
(k) S. C. Wang and D. J. Tantillo, Theoretical Studies on Synthetic and Biosynthetic Oxidopyrylium-Alkene Cycloadditions: Pericyclic Pathways to Intricarene, J. Org. Chem., 2008, 73, 1516–1523 CrossRef CAS PubMed.
-
(a) N. Wang, Y. Bai, Q. Zeng, T. Zhang and J. Deng, Recent progress of [5 + 2] cycloaddition reactions in natural product synthesis, Nat. Prod. Rep., 2025, 42, 1755–1785 RSC;
(b) K. Gao, Y.-G. Zhang, Z. Wang and H. Ding, Recent development on the (5 + 2) cycloadditions and their application in natural product synthesis, Chem. Commun., 2019, 55, 1859–1878 RSC;
(c) X. Liu, Y.-J. Hu, J.-H. Fan, J. Zhao, S. Li and C.-C. Li, Recent synthetic studies toward natural products via, (5 + 2) cycloaddition reactions, Org. Chem. Front., 2018, 5, 1217–1228 RSC;
(d) K. E. O. Ylijoki and J. M. Stryker, (5 + 2) Cycloaddition Reactions in Organic and Natural Product Synthesis, Chem. Rev., 2013, 113, 2244–2266 CrossRef CAS PubMed.
-
(a) A. Aimon, G. Karageorgis, J. Masters, M. Dow, P. G. E. Craven, M. Ohsten, A. Willaume, R. Morgentin, N. Ruiz-Llamas, H. Lemoine, T. Kalliokoski, A. J. Eatherton, D. J. Foley, S. P. Marsden and A. Nelson, Realisation of small molecule libraries based on frameworks distantly related to natural products, Org. Biomol. Chem., 2018, 16, 3160–3167 RSC;
(b) C. V. Credille, B. L. Dick, C. N. Morrison, R. W. Stokes, R. N. Adamek, N. C. Wu, I. A. Wilson and S. M. Cohen, Structure-Activity Relationships in Metal-Binding Pharmacophores for Influenza Endonuclease, J. Med. Chem., 2018, 61, 10206–10217 CrossRef CAS PubMed;
(c) D. R. Hirsch, D. V. Schiavone, A. J. Berkowitz, L. A. Morrison, T. Masaoka, J. A. Wilson, E. Lomonosova, H. Zhao, B. S. Patel, S. H. Datla, S. G. Hoft, S. J. Majidi, R. K. Pal, E. Gallicchio, L. Tang, J. E. Tavis, S. F. J. Le Grice, J. A. Beutler and R. P. Murelli, Synthesis and biological assessment of 3,7-dihydroxytropolones, Org. Biomol. Chem., 2018, 16, 62–69 RSC;
(d) D. J. Foley, P. G. E. Craven, P. M. Collins, R. G. Doveston, A. Aimon, R. Talon, I. Churcher, F. von Delft, S. P. Marsden and A. Nelson, Chem. – Eur. J., 2017, 23, 15227–15232 CrossRef CAS PubMed;
(e) T. Masaoka, H. Zhao, D. R. Hirsch, M. P. D'Erasmo, C. Meck, B. Varnado, A. Gupta, M. J. Meyers, J. Baines, J. A. Beutler, R. P. Murelli, L. Tang and S. F. J. Le Grice, Characterization of the C-Terminal Nuclease Domain of Herpes Simplex Virus pUL15 as a Target of Nucleotidyltransferase Inhibitors, Biochemistry, 2016, 55, 809–819 CrossRef CAS PubMed.
-
(a) R. J. Zaragoza, M. J. Aurell and L. R. Domingo, The role of the transfer group in the intramolecular (5 + 2) cycloadditions of substituted β-hydroxy-γ-pyrones: a DFT analysis, J. Phys. Org. Chem., 2005, 18, 610–615 CrossRef CAS;
(b) F. Lopez, L. Castedo and J. L. Mascarenas, A Theoretical Rationalization of the Asymmetric Induction in Sulfinyl-Directed [5C + 2C] Intramolecular Cycloadditions, J. Org. Chem., 2003, 68, 9780–9786 CrossRef CAS PubMed;
(c) L. R. Domingo and R. J. Zaragoza, Toward an Understanding of the Mechanisms of the Intramolecular (5 + 2) Cycloaddition Reaction of γ-Pyrones Bearing Tethered Alkenes. A Theoretical Study, J. Org. Chem., 2000, 65, 5480–5486 CrossRef CAS.
- D. J. Tantillo, Recent Excursions to the Borderlands between the Realms of Concerted and Stepwise: Carbocation Cascades in Natural Products Biosynthesis, J. Phys. Org. Chem., 2008, 21, 561–570 CrossRef CAS.
- See electronic supporting information.
- C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. M. Crawford, C. Kingston, F. D. Toste and M. S. Sigman, Data Science Meets Physical Organic Chemistry, Acc. Chem. Res., 2021, 54, 3136–3148 CrossRef CAS PubMed.
- S. Grimme, Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, Density functional theory with London dispersion corrections, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CAS.
-
(a) Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2006, 120, 215–241 Search PubMed;
(b) Y. Zhao and D. G. Truhlar, Construction of a generalized gradient approximation by restoring the density-gradient expansion and enforcing a tight Lieb–Oxford bound, J. Chem. Phys., 2008, 128, 184109–184118 CrossRef PubMed.
-
(a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS;
(b) A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † A. J. Y., S. N. R., and W. G. contributed equally to this research. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Samantha N. Rokey†
a,
Samantha N. Rokey†















