
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold-catalysed N-allenamide cyclisation as a platform for the construction of indole-fused quinoxaline and quinoline scaffolds
Silvia Meraviglia ,
Mehri Goudarzi,
Simone Borsi,
Giorgio Abbiati and
Valentina Pirovano*
,
Mehri Goudarzi,
Simone Borsi,
Giorgio Abbiati and
Valentina Pirovano*
Dipartimento di Scienze Farmaceutiche, Sezione di Chimica Generale e Organica “A. Marchesini”, Università degli Studi di Milano, Via Venezian, 21, 20133 Milano, Italy. E-mail: valentina.pirovano@unimi.it
First published on 13th January 2026
Abstract
We report a gold-catalysed cyclisation of N-allenamides derived from 1- and 2-(2-aminoaryl)indoles, providing easy access to 5,6-dihydroindolo[1,2-a]quinoxalines and 6,11-dihydro-5H-indolo[3,2-c]quinolines. The reaction proceeds under mild conditions, tolerates diverse functional groups, and enables the synthesis of previously unexplored indole-fused heterocycles, whose versatility was demonstrated through selected post-functionalisation reactions.
Introduction
Polycyclic indoles are ubiquitous in natural products and pharmaceuticals, where they play key roles in modulating biological activity.1 Among them, tetracyclic 5,6-dihydroindolo[1,2-a]quinoxalines and indolo[3,2-c]quinolines stand out as privileged scaffolds with diverse medicinal applications (Fig. 1).2,3 | ||
| Fig. 1 Biologically relevant indoloquinoxalines/quinolines. | ||
For example, indolo[1,2-a]quinoxaline A has shown promising antifungal activity against phytopathogenic fungi in vitro,2a whereas derivative B has been identified as an inhibitor of vascular endothelial growth factor receptor 3 (VEGFR-3), a target associated with cancer cell invasion and migration.2b In addition, Zheng and co-workers reported the anti-HIV properties of the 7-methyl-5,6-dihydroindolo[1,2-a]quinoxaline C, further underscoring the therapeutic relevance of this structural class.2c On the other hand, the 6,11-dihydro-indolo[3,2-c]quinoline derivative D has been investigated as a potential androgen receptor ligand,3a while the related indolo[3,2-c]quinoline E has displayed notable antimalarial properties.3b More recently, in vitro and in vivo studies revealed that indoloquindoline F exhibits a broad spectrum of antitumor activities.3c
Given their broad biological profiles, considerable effort has been devoted to developing efficient and versatile synthetic routes to these heterocycles. The most widely used approach to access 5,6-dihydroindolo[1,2-a]quinoxalines and 6,11-dihydro-5H-indolo[3,2-c]quinolines, relies on the Pictet–Spengler reaction between (2-aminoaryl)indoles and a carbonyl compound.2a,b,3c,4 Complementary methods based on transition metal-catalysis have been explored, including systems based on ruthenium,5 platinium,6 palladium,7 copper,8 molybdenum,9 scandium10 and rhodium.11 Gold catalysis has likewise emerged as a powerful tool for constructing these frameworks (Scheme 1). For example, Patil and co-workers reported the cyclization of 2-(1H-indol-1-yl)anilines and 2-(1H-indol-2-yl)anilines with phenylacetylene under cationic gold(I) catalysis (Scheme 1a),12 while Liu employed a gold(I)-catalysed domino processes with alkynoic acids to generate related polycyclic derivatives (Scheme 1b).13 A gold(III) catalyst has also been used to prepare 6,6-disubstituted 6,11-dihydro-5H-indolo[3,2-c]quinolones from 2-[(2-aminophenyl)ethynyl]phenylamines and ketones (Scheme 1c).14 Beyond these precedents, gold-catalysed additions of N-, O-, and C-based nucleophiles to allenes have proven particularly attractive, owing to their high atom economy, excellent regioselectivity, and mild reaction conditions.15 Despite the extensive study of these transformations, their application to the synthesis of indoloquinoxalines and indoloquinolines remains underexplored. Building on these premises and motivated by our group's long-standing interest in the assembly of complex indole architectures under gold catalysis,16 we report herein a mild and efficient gold(I)-catalysed cyclization of N-allenamides derived from 1- and 2-(2-aminoaryl)indoles. This unified strategy provides streamlined access to both indolo[1,2-a]quinoxaline and indolo[3,2-c]quinoline derivatives (Scheme 1d).
 | ||
| Scheme 1 Gold-catalysed syntheses of 5,6-dihydroindolo[1,2-a]quinoxalines and 6,11-dihydro-5H-indolo[3,2-c]quinolines and our work. | ||
Results and discussion
For the catalyst screening, a series of gold complexes bearing different ligands and counterions were evaluated (Table 1). In the initial experiments (entries 1–4), N-allenamide 1a was reacted in dichloromethane at room temperature with cationic gold(I) complexes. In every case, the reaction proceeded to full conversion, affording product 2a as the sole identifiable compound, although in variable yields. The phosphine complex [Au(PPh3)NTf2] gave the lowest yield (23%, entry 4), while [Au(JohnPhos)NTf2] and a gold(I) phosphite complex afforded 48% and 52% yield, respectively (entries 2 and 3). The highest efficiency was obtained with the cationic N-heterocyclic carbene complex [Au(IPr)NTf2], which delivered 2ain 74% yield (entry 1). Besides 2a, only unidentified degradation products were detected. To probe the influence of the counterion, [Au(IPr)(MeCN)SbF6] was employed (entry 5), resulting in a slightly improved yield of 75%. Lowering the reaction temperature to −20 °C (entry 6) did not provide any advantage, instead giving a diminished yield of 62%. Changing the solvent had a more pronounced effect: in toluene, the yield increased to 84% (entry 7). Based on these results, the optimal conditions were established as those in entry 7: [Au(IPr)(MeCN)SbF6], toluene, room temperature, 1 h. These parameters were then adopted for the subsequent scope studies.
|
|||
|---|---|---|---|
| Entry | [Au] (5 mol%) | Solvent (0.1 M) | 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
| a Reaction conditions: 1a (0.1 mmol), [Au] (5 mol%), in anhydrous solvent (1 ml, 0.1 M) at rt for 1 h.b Isolated yield.c Reaction performed at −20 °C. IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene; JohnPhos = (2-biphenyl)di-tert-butylphosphine; Ar = 2,4-di-tert-butylphenyl. | |||
| 1 | [Au(IPr)NTf2] | CH2Cl2 | 74 |
| 2 | [Au(JohnPhos)NTf2] | CH2Cl2 | 48 |
| 3 | [Au(ArO)3PNTf2] | CH2Cl2 | 52 |
| 4 | [Au(PPh3)NTf2] | CH2Cl2 | 23 |
| 5 | [Au(IPr)(MeCN)SbF6] | CH2Cl2 | 75 |
| 6c | [Au(IPr)(MeCN)SbF6] | CH2Cl2 | 62 |
| 7 | [Au(IPr)(MeCN)SbF6] | Toluene | 84 |

Having established the optimal conditions, we next explored the generality of the transformation with a series of differently substituted N-allenamides 1b–s (Scheme 2). We first examined modifications to the indole scaffold, focusing on substituents at the C3-position. The methyl group of 1a could be successfully replaced with an ethyl (1b), iso-propyl (1c), phenyl (1d), or naphthyl group (1e), and in all cases the corresponding products were obtained in good to excellent yields, reaching up to 99% for the ethyl derivative. Notably, substitution at C3 proved essential: the unsubstituted C3–H derivative decomposed completely under the optimised conditions, without affording any detectable cyclic product. We then investigated electronic effects at the C5- and C6-positions of the indole ring. Allenamides 1f–j, bearing either electron-donating or electron-withdrawing groups, were synthesised and tested. Methyl- and methoxy-substituted substrates (1f and 1g) delivered the corresponding indolo[1,2-a]quinoxalines 2f and 2g in comparable yields (78% and 79%, respectively). In contrast, the introduction of a fluorine atom at C5 resulted in a slightly diminished yield of 2h (70%). More pronounced effects were observed with C6-substitution: the 6-methoxy-substituted allenamide 1j reacted efficiently to furnish 2j in 93% yield, whereas the bromo derivative 1i gave a lower 72% yield. We further evaluated modifications on the aniline core. An ortho-substituent on the aniline ring negatively impacted reactivity, as observed for 1k, which furnished a reduced yield of the corresponding product 2k. In contrast, the tosyl group could be successfully replaced with alternative sulfonyl protecting groups, including mesitylenesulfonyl (2l), 2,4,6-triisopropylbenzenesulfonyl (2m), 3,5-difluorobenzenesulfonyl (2n), and 4-nitrobenzenesulfonyl (2o), all affording the corresponding indolo[1,2-a]quinoxalines in excellent yields. The methanesulfonyl derivative 2p was obtained in quantitative yield, further underscoring the tolerance of the transformation toward sulfonyl substituents. Finally, the aniline nitrogen could also be protected with a carbobenzyloxy (Cbz) group, with allenamide 1q undergoing smooth cyclisation to provide 2q in 82% yield. Extension to allenamides 1r and 1s, bearing non-terminal allenes, delivered the desired products 2r and 2s as single stereoisomers in 97% and 88% yield, respectively.
 | ||
| Scheme 2 Scope of the reaction. Reaction conditions: 1b–s (0.1 mmol), [Au(IPr)(MeCN)SbF6] (5 mol%), in anhydrous toluene (1 ml, 0.1 M) at rt for 1 h. Isolated yields are reported. | ||
To expand the scope of our methodology and to assess its potential for constructing other indole-fused heterocyclic scaffolds, we synthesised the isomeric allenamides 3 and 5, in which the aniline core was relocated from the indole nitrogen to the C2- and C3-positions, respectively. Both substrates were subjected to the optimised cyclisation conditions, and the results are summarised in Scheme 3. The C2-functionalised allenamide 3a (R1 = H, R2 = Me) underwent smooth cyclisation to afford the 6,11-dihydro-5H-indolo[3,2-c]quinoline derivative 4a in 82% yield. Encouraged by this result, we briefly explored the reaction scope further. Substitution of the N-methyl group with a benzyl group furnished the corresponding product 4b in 92% yield. Introduction of a methyl substituent at the C5-position afforded compound 4c in 91% yield. Modifications on the C2-aryl ring were also tolerated: allenamides 3d–f, bearing methyl, chloro, or naphthyl substituents, delivered the corresponding indolo[3,2-c]quinolines 4d–f in yields ranging from moderate (4d, 66%) to excellent (4e, 99%). In contrast, the use of the C3-functionalized allenamide 5 proved less successful. Under the optimised gold-catalysed conditions, this substrate underwent cyclisation to give a mixture of the seven-membered and six-membered derivatives 6 and 7, which were isolated in an overall 80% yield (6:7 ratio = 5.6:1). These results highlight the sensitivity of the cyclization outcome to the substitution pattern of the indole framework.
 | ||
| Scheme 3 Expansion of the reaction scope to C2- and C3-allenamides 3 and 5. Reaction conditions: 3a–f or 5 (0.1 mmol), [Au(IPr)(MeCN)SbF6)] (5 mol%), in anhydrous toluene (1 ml, 0.1 M) at rt for 1 h. Isolated yields are reported. | ||
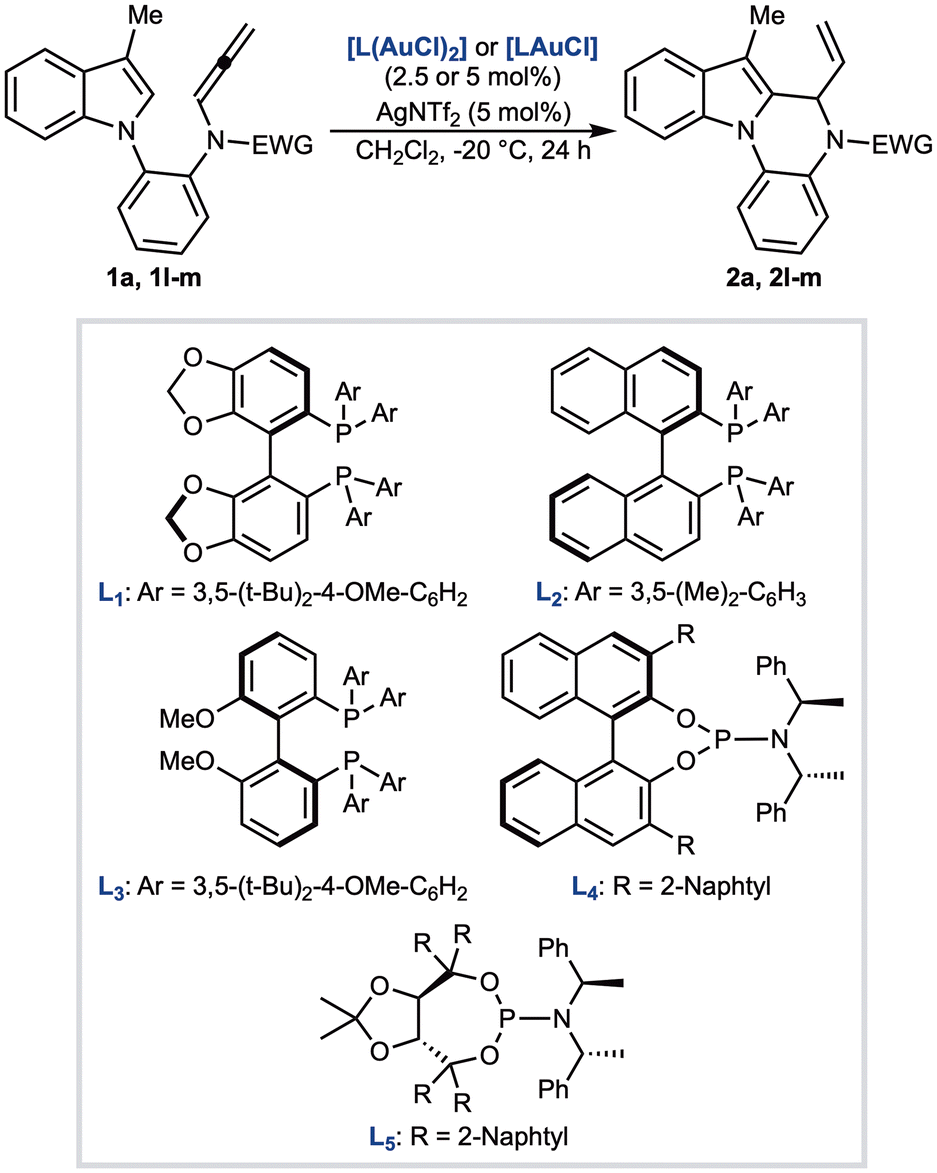
The presence of a chiral center in indolo[1,2-a]quinoxalines 2 prompted us to develop an enantioselective version of the reaction. To this end, the cyclization of 1a was examined in the presence of different chiral gold complexes (Table 2; see SI for the full screening). Bidentate complexes derived from (R)-DTBM-SEGPHOS (L1) or (R)-DM-BINAP (L2) generally afforded 2a in low to moderate yields, while the enantioinduction was limited, with the best results obtained using L1 (77:23 er, entries 1 and 2). Improved results were achieved with L3, a member of the BIPHEP family, which delivered 2a in 72% yield and 79:21 er (entry 3). Monodentate ligands such as the BINOL- and TADDOL-derived phosphoramidites L4 and L5 did not perform better: L4 gave racemic 2a in modest yield (entry 4), whereas L5 furnished 2a in 72% yield and 72:28 er (entry 5). Further variations of ligands, solvents, and counterions were explored, but none of them provided superior results compared to those obtained with L3 (entry 3). To address this limitation, we replaced the tosyl group of the allenamide with bulkier aryl sulfonyl rings. Accordingly, allenes 1l and 1m were reacted in the presence of L3(AuCl)2/AgNTf2 catalytic system at −20 °C for 24 hours. In both cases, we observed a decrease of the yield (31%), and only for 2l the er was improved up to 86:14 (entries 6 and 7).
|
||||
|---|---|---|---|---|
| Entry | 1 | [Au] | 2b (%) |
erc |
| a Reaction conditions: 1 (0.1 mmol), [Au] (2.5 or 5 mol%), AgNTf2 (5 mol%) in anhydrous CH2Cl2, (0.1 M) at −20 °C for 24 h.b Isolated yield.c Enantiomeric ratios (er) determined by chiral HPLC. See SI for full experimental details. | ||||
| 1 | 1a | [L1(AuCl)2] | 40 | 77:23 |
| 2 | 1a | [L2(AuCl)2] | 31 | 69:31 |
| 3 | 1a | [L3(AuCl)2] | 72 | 79:21 |
| 4 | 1a | [L4AuCl] | 39 | 53:47 |
| 5 | 1a | [L5AuCl] | 72 | 72:28 |
| 6 | 1l | [L3(AuCl)2] | 31 | 86:14 |
| 7 | 1m | [L3(AuCl)2] | 31 | 53:47 |
Finally, we explored the synthetic utility of the methodology by subjecting product 2a to a series of functional group transformations (Scheme 4). To this end, its synthesis was scaled up to 1.5 mmol, affording 2a in 82% yield. Removal of the tosyl group was achieved using magnesium in ethanol, and the corresponding NH-free derivative 8 was isolated in 70% yield. Subsequent treatment of 8 with 2-chloroethane-1-sulfonyl chloride under basic conditions provided 9 in 73% yield. An intramolecular olefin metathesis reaction, promoted by the Hoveyda–Grubbs II catalyst, furnished 10, characterized by the presence of a cyclic sulfone group. Then, the exocyclic double bond of 2a could be smoothly hydrogenated to give 11 quantitatively, while an intermolecular ruthenium-catalysed metathesis with styrene afforded 12 in 76% yield. Similarly, also tosyl group of 4a, could be easily removed to give NH-free derivative 13, while treatment with potassium t-butoxide led to indolo[3,2-c]quinoline 14 in 84% yield.
 | ||
| Scheme 4 Synthetic transformations of 2a and 4a. | ||
On the basis of established reactivity patterns of N-allenamides under gold catalysis,15b,d a plausible mechanistic pathway is proposed in Scheme 5 to rationalise the formation of indoloquinoxalines 2 and indoloquinolines 4. Coordination of the allene moiety in substrates 1 or 3 to the electrophilic gold species generates the corresponding aurated iminium intermediates I or III. Intramolecular nucleophilic attack of the indole core (at C2 for 1, and at C3 for 3) onto the α-carbon of the activated allene then furnishes intermediates II or IV, respectively. Subsequent aromatisation and protodeauration deliver the corresponding cyclised products 2 and 4
 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed a gold-catalysed cyclization of N-allenamides derived from 1- and 2-(2-aminoaryl)indoles that provides efficient access to 5,6-dihydroindolo[1,2-a]quinoxalines and 6,11-dihydro-5H-indolo[3,2-c]quinolines. The methodology features mild conditions, broad substrate scope, and high functional group tolerance, while also enabling the synthesis of previously unexplored indole-fused heterocyclic scaffolds. The synthetic utility of the products was further demonstrated through a variety of post-functionalisation reactions, highlighting their potential as versatile building blocks. Given the prevalence of indole-based polycyclic structures in bioactive molecules and functional materials, we anticipate that this methodology will find broad application in the development of new heteroaromatic architectures.Author contributions
S. M.: conceptualisation, investigation, validation, writing – original draft, data curation. M. G.: investigation, validation, writing – original draft, data curation. S. B.: investigation, data curation. G. A.: conceptualisation, writing – review & editing. V. P.: conceptualisation, funding acquisition, methodology, supervision, writing – original draft.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: synthesis and characterisation of products, NMR spectra and full screening of enantioselective reaction. See DOI: https://doi.org/10.1039/d5ob01867f.Acknowledgements
We would like to thank Mrs Donatella Nava for the NMR analyses, and Dr Stefano Fedeli for the MS analyses. This project has been founded by the Italian MIUR (PRIN 20227Z3BL8). This research was supported by Regione Lombardia – “Collabora&Innova” – Project 6154644 – GREEN-TECH – (“Applicazione di tecnologie innovative a basso impatto ambientale per lo sviluppo e l'ottimizzazione di processi chimici sostenibili”).References
- (a) N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159 Search PubMed; (b) S. M. Umer, M. Solangi, K. M. Khan and R. S. Z. Saleem, Molecules, 2022, 27, 7586 Search PubMed; (c) S.-F. Duan, L. Song, H.-Y. Guo, H. Deng, X. Huang, Q.-K. Shen, Z.-S. Quan and X.-M. Yin, RSC Med. Chem., 2023, 14, 2535 Search PubMed; (d) D. C. Holland and A. R. Carroll, Nat. Prod. Rep., 2023, 40, 1595 Search PubMed; (e) W. Zeng, C. Han, S. Mohammed, S. Li, Y. Song, F. Sun and Y. Du, RSC Med. Chem., 2024, 15, 788 Search PubMed.
- (a) H. Xu and L. Fan, Eur. J. Med. Chem., 2011, 46, 1919 Search PubMed; (b) P. T. Lin, D. B. Salunke, L. H. Chen and C. M. Sun, Org. Biomol. Chem., 2011, 9, 2925–2921 Search PubMed; (c) L.-L. Fan, N. Huang, R.-G. Yang, S.-Z. He, L.-M. Yang, H. Xu and Y.-T. Zheng, Lett. Drug Des. Discovery, 2012, 9, 44 Search PubMed.
- (a) X. Zhang, X. Li, G. F. Allan, A. Musto, S. G. Lundeen and Z. Sui, Bioorg. Med. Chem. Lett., 2006, 16, 3233 Search PubMed; (b) N. Wang, K. J. Wicht, K. Imai, M.-Q. Wang, T. A. Ngoc, R. Kiguchi, M. Kaiser, T. J. Egan and T. Inokuchi, Bioorg. Med. Chem., 2014, 22, 2629 Search PubMed; (c) S. Li, Z. Hu, W. Pan, H. Wu, W. Peng, Y. Wu, F. Jiang and X. Peng, J. Med. Chem., 2025, 68, 3212 Search PubMed.
- (a) P. K. Agarwal, D. Sawant, S. Sharma and B. Kundu, Eur. J. Org. Chem., 2009, 292 Search PubMed; (b) A. K. Verma, R. R. Jha, V. K. Sankar, T. Aggarwal, R. P. Singh and R. Chandra, Eur. J. Org. Chem., 2011, 6998 Search PubMed; (c) Y.-S. Fan, Y.-J. Jiang, D. An, D. Sha, J. C. Antilla and S. Zhang, Org. Lett., 2014, 16, 6112 Search PubMed; (d) H.-R. Huo, X.-Y. Tang and Y.-F. Gong, Synthesis, 2018, 2727 Search PubMed; (e) X.-W. Wang, X. Li, M.-W. Chen, B. Wu and Y.-G. Zhou, J. Org. Chem., 2021, 86, 6897 Search PubMed.
- C. S. Yi and S. Y. Yun, J. Am. Chem. Soc., 2005, 127, 17000 Search PubMed.
- N. T. Patil, R. D. Kavthe, V. S. Shinde and B. Sridhar, J. Org. Chem., 2010, 75, 3371 Search PubMed.
- L. Wang, W. Guo, X. X. Zhang, X. D. Xia and W. J. Xiao, Org. Lett., 2012, 14, 740 Search PubMed.
- (a) L. Zhang, F. Zhao, M. Zheng, Y. Zhai, J. Wang and H. Liu, Eur. J. Org. Chem., 2013, 5710 Search PubMed; (b) F. Zhao, L. Zhang, H. Liu, S. Zhou and H. Liu, Beilstein J. Org. Chem., 2013, 9, 2463 Search PubMed.
- R. Rubio-Presa, M. R. Pedrosa, M. A. Fernández-Rodríguez, F. J. Arnaíz and R. Sanz, Org. Lett., 2017, 19, 5470 Search PubMed.
- S. Yaragorla and A. Kumar, J. Org. Chem., 2025, 90, 4459 Search PubMed.
- P. J. Nagtilak, M. V. Mane, S. Prasad, L. Cavallo, D. J. Tantillo and M. Kapur, Chem. – Eur. J., 2023, 29, e202203055 Search PubMed.
- N. T. Patil, P. G. V. V. Lakshmi and V. Singh, Eur. J. Org. Chem., 2010, 4719 Search PubMed.
- Y. Zhou, X. Ji, G. Liu, D. Zhang, L. Zhao, H. Jiang and H. Liu, Adv. Synth. Catal., 2010, 352, 1711 Search PubMed.
- B. V. S. Reddy, M. Swain, S. M. Reddy, J. S. Yadav and B. Sridhar, Eur. J. Org. Chem., 2014, 3313 Search PubMed.
- (a) R. A. Widenhoefer, Chem. – Eur. J., 2008, 14, 5382 Search PubMed; (b) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 Search PubMed; (c) B. Alcaide and P. Almendros, Acc. Chem. Res., 2014, 47, 939 Search PubMed; (d) D. Campeau, D. F. L. Rayo, A. Mansour, K. Muratov and F. Gagosz, Chem. Rev., 2021, 121(14), 8756 Search PubMed; (e) T. Ghosh, J. Chatterjee and S. Bhakta, Org. Biomol. Chem., 2022, 20, 7151 Search PubMed.
- (a) V. Pirovano, L. Decataldo, E. Rossi and R. Vicente, Chem. Commun., 2013, 49, 3594 Search PubMed; (b) V. Pirovano, E. Arpini, M. Dell'Acqua, R. Vicente, G. Abbiati and E. Rossi, Adv. Synth. Catal., 2016, 358, 403 Search PubMed; (c) V. Pirovano, M. Borri, G. Abbiati, S. Rizzato and E. Rossi, Adv. Synth. Catal., 2017, 359, 1912 Search PubMed; (d) E. Brambilla, V. Pirovano, M. Giannangeli, G. Abbiati, A. Caselli and E. Rossi, Org. Chem. Front., 2019, 6, 3078 Search PubMed; (e) V. Pirovano, E. Brambilla, S. Rizzato, G. Abbiati, M. Bozzi and E. Rossi, J. Org. Chem., 2019, 84, 5150 Search PubMed; (f) E. Brambilla, M. Gugiatti, S. Rizzato, G. Abbiati and V. Pirovano, Eur. J. Org. Chem., 2024, e202400083 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |