
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable electrochemical synthesis of a new isoxazoline scaffold as turn inducer to build parallel β-hairpins
Zoe
Laface
a,
Gianluigi
Broggini
b,
Camilla
Loro
*b,
Davide
Di Lorenzo
a,
Giovanni
Macetti
 c,
Alessandro
Contini
a,
Maria Luisa
Gelmi
a,
Kaliroi
Peqini
a and
Raffaella
Bucci
*a
c,
Alessandro
Contini
a,
Maria Luisa
Gelmi
a,
Kaliroi
Peqini
a and
Raffaella
Bucci
*a
aDipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, via Venezian 21, 20133 Milan, Italy. E-mail: raffaella.bucci@unimi.it
bDipartimento di Scienza e Alta Tecnologia, Università degli Studi dell'Insubria, 22100 Como, Italy. E-mail: camilla.loro@uninsubria.it
cDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milan, Italy
First published on 22nd December 2025
Abstract
New diastereoisomeric isoxazoline scaffolds bearing two amino-alkyl chains were synthesized using a 1,3-dipolar cycloaddition reaction, with the aim to prepare N-to-N parallel β-hairpins. Two approaches were employed, both starting from methyl(azidomethyl)acrylate as dipolarophile, and an enantiopure chloroxime or oxime as dipoles derived from cheap L-phenylalanine. In the first method the chloroxime was treated with a base to in situ generate the corresponding nitrile oxide which then reacted with the dipolarophile. In the second approach, a more sustainable electrochemical cycloaddition was performed, enabling direct nitrile oxide generation from the oxime, avoiding one synthetic step. The regioselective cycloaddition allows the formation of two diastereoisomeric isoxazoline scaffolds, both used for the synthesis of model peptidomimetics. Comprehensive computational and NMR studies revealed that the R,S-isoxazoline more effectively stabilizes the desired parallel β-hairpin conformation.
Introduction
Nowadays, peptides are recognized as promising candidates for drug development, and an increasing number of peptide-based active pharmaceutical ingredients have been approved by the Food and Drug Administration.1 The increasing use of peptides as alternatives to small molecules is driven by their high selectivity for biological targets, efficacy, tolerability, predictable metabolism, as well as their short synthesis time and quick delivery to market.2 However, their therapeutic potential is limited by their proteolytic instability, poor absorption, and suboptimal transport properties.3Another limitation, particularly for short peptides, is their high flexibility, which lead them to adopt multiple conformations in solution. This conformational flexibility hinders the formation of stable secondary structures, which are crucial for their functional roles.4,5
To overcome these limitations, researchers focused on the design of peptidomimetics, molecules that mimic peptide chains, but with an unnatural backbone. Among the various synthetic strategies to prepare peptidomimetics, one approach involves incorporating non-natural amino acids (AAs) or non-aminoacidic scaffolds into the peptide chain. In general, these molecules exhibit greater metabolic stability and bioavailability compared to the parent peptides, while preserving the structural features, essential for their biological activity.6 The insertion of the non-natural portion could also improve their receptor affinity and selectivity.7
In general, designing a non-natural AA or scaffold to be incorporated into the peptide sequence poses significant synthetic challenges, i.e. the control of the secondary conformation of short peptides, which often depends on the absolute configuration of the non-natural portion.8–10
Between the secondary structures of biological relevance, β-hairpins are particularly important because they are widely utilized by various proteins in biomolecular recognition processes, as well as in stabilizing protein–protein interactions (PPIs) and contributing to enzymatic function.11 The design of new peptidomimetics with β-hairpin conformation formed by antiparallel or parallel β-strands is highly advantageous.12 The new motif often focuses on the turn region, as it enhances proteolytic resistance in this area and ensures the correct orientation of the two peptide arms, thereby stabilizing intra-strand hydrogen bonds.
Several synthetic turns have been reported in the literature, most of which can stabilize an antiparallel β-hairpin.13,14
Despite the abundance of scaffolds for antiparallel β-hairpins in literature, parallel β-hairpin mimics remain comparatively rare. In particular, scaffolds with a C-to-C connection of peptide strands are more common than those with N-to-N connections. For example, Nowick focused on creating artificial β-hairpins by utilizing an oligourea to organize multiple parallel peptide strands into hydrogen-bonded β-hairpins.15,16 The synthesis of aziridine-based parallel β-sheet mimetics was reported by Filigheddu et al.17 while Fisk et al. conducted a study on proline-containing linkers for both C-to-C and N-to-N connections of peptide strands.18 Few additional examples for N-to-N connections are reviewed by Ko et al.13
Recently, our group developed a bicyclic Δ2-isoxazoline scaffold fused with a pyrrolidine ring, able to stabilize parallel β-hairpin when used for the preparation of model peptidomimetics.8
Building on this knowledge and given the scarcity of scaffolds leading to an N-to-N connection of peptide strands, here we present the design and synthesis of novel diastereoisomeric isoxazoline compounds (S,S)-6/(R,S)-6′. This scaffold is functionalized with two amino-alkyl substituents and contains two stereocenters, which can influence the adoption of distinct conformations when integrated into peptide sequences (Scheme 1).
 | ||
| Scheme 1 Retrosynthetic scheme for the preparation of isoxazoline scaffolds and peptidomimetics. | ||
Typically, isoxazoline rings are synthesized via 1,3-dipolar cycloadditions, employing a nitrile oxide.19 The 1,3-dipole is usually generated in situ by the basic treatment of the corresponding halo-oxime, generally obtained by reaction between the oxime and a halo-succinimide.
Today, the development of new and sustainable procedures without the use of pre-activated substrates is particularly intriguing.20,21 In this context, electrochemical methods present an excellent alternative to traditional synthesis, enabling selective oxidations and reductions. The straightforward use of electricity allows the avoiding of expensive and hazardous reagents typically required in classical substrate conversions. A few electrochemical methods for the synthesis of isoxazolines from the corresponding oximes are reported in the literature.22–25 However, in these cases an excess of dipolarophile is often necessary, leading to unsatisfactory reaction yields.
In this work, the synthesis of isoxazoline scaffolds (S,S)-6/(R,S)-6′ was achieved using the (azidomethyl)acrylate 4 as the dienophile and the enantiopure oxime 1 or chloroxime 2, derived from the inexpensive L-phenylalanine (Scheme 1). The reaction conditions were optimized using both classical and electrochemical methods. Notably, the latter enabled more eco-friendly conditions by employing equimolar amounts of oxime 1, dipolarophile 4, and NaCl as a simple electrolyte. This approach avoided an additional synthetic step, achieved higher yields of the cycloadduct and eliminated the use of halogenating compounds, which pose significant safety and toxicity risks.
The two diastereoisomeric scaffolds, each in enantiopure form, served as key reagents for the preparation of peptide models (S,S)-11/(R,S)-11′ (Scheme 1) where the stereochemical descriptors are referred to the non-natural portion of the sequence. From this point on, they will be explicitly reported for all peptides containing the isoxazoline scaffold, for clarity. NMR experiments and computational studies confirmed that, depending on the S,S- or R,S-absolute configuration of the scaffold, the synthesized peptidomimetics adopt an extended or a parallel β-hairpin conformation, respectively.
Results
Synthesis of isoxazoline scaffold
The synthesis of the new isoxazoline scaffolds (S,S)-5/(S,R)-5′, bearing the azido and N-Boc moieties, was accomplished via a 1,3-dipolar cycloaddition reaction starting from chloroxime 2, the precursor of the nitrile oxide 3, and methyl 2-(azidomethyl)acrylate 4 (Method A, Scheme 2).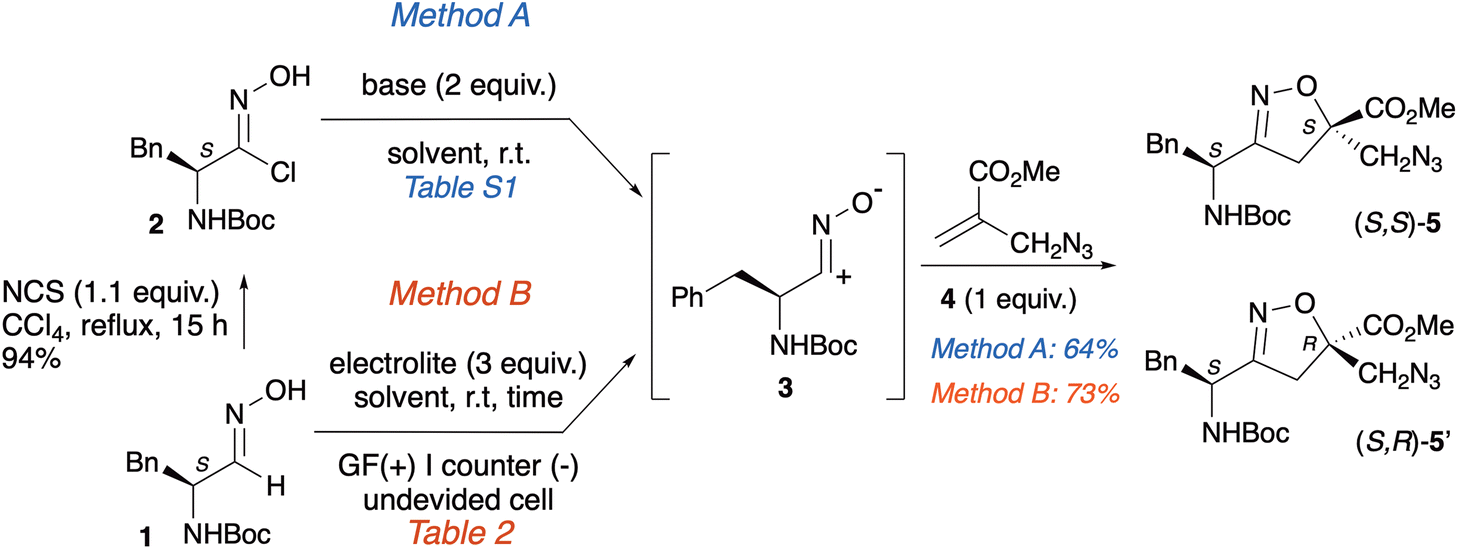 | ||
| Scheme 2 1,3-Dipolar cycloaddition reaction affording isoxazoline scaffolds (S,S)-5/(S,R)-5′. | ||
Chloroxime 2 (94%) was obtained according to a known procedure set-up by our group from the corresponding oxime 1 operating in the presence of N-chlorosuccinimide (NCS) in CCl4 at reflux overnight (Scheme 2).8 Azido methyl acrylate 4 was prepared following a known procedure from methyl 2-(bromomethyl)acrylate and NaN3 in a mixture of acetone/H2O (20 min, 25 °C, 90% yield).26
Using the conventional method, chloroxime 2 was in situ transformed into nitrile oxide 3 in the presence of a base. Since the enantiopure dipolarophile 2 was employed, the cycloaddition reaction with the dipolarophile 4 was expected to yield a pair of diastereoisomers.
Following Method A, the cycloaddition reaction was optimized by evaluating different solvents and bases to improve the yields and verify the potential influence of the base on the control of the diastereoselection. Details on the tested reaction conditions are reported in the SI (Table S1). Optimized conditions required the addition of a mixture of azido methacrylate 4 (1 equiv.; 0.5 M solution in dry THF) and TEA (2 equiv.) to a solution of the crude chloroxime 2 (1 equiv.; 0.1 M solution in dry THF) at 25 °C. After stirring overnight and flash chromatography, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers (S,S)-5/(S,R)-5′ was isolated in 64% yield as single regioisomers.
:1 mixture of diastereoisomers (S,S)-5/(S,R)-5′ was isolated in 64% yield as single regioisomers.
As previously highlighted, the use of NCS, a compound that presents significant safety risks, for the preparation of chloroxime is highly undesirable. For this reason, we investigate an ecofriendly approach, that also eliminates an additional synthetic step. Method B involves the electrochemical oxidation of oxime 1, enabling the direct generation of the corresponding 1,3-dipole 3, directly used in the cycloaddition reaction with azido-acrylate 4. To optimize the yields, several reaction conditions were tested (Table 1). The two substrates, utilized in equimolar amounts, underwent constant current electrolysis in the presence of NaCl as the electrolyte, using graphite as both the anode and cathode. The reaction was conducted in a THF/H2O solvent mixture (entry 1). The isoxazoline scaffold was obtained as a mixture of two diastereoisomers (S,S)-5/(S,R)-5′ in 26% yield. To improve the yield, some parameters were adjusted; however, neither the use of different cathodes (entries 2–5) nor the application of alternative solvents, such as tBuOMe, toluene, or EtOH (entries 6–8), influenced the reaction outcome. Conversely, the combination of NaCl in a CH2Cl2/H2O mixture as a solvent mixture, with graphite serving as both anode and cathode under a current of 14 mA, resulted in the formation of (S,S)-5 and (S,R)-5′with a yield of 73% (entry 9). This result is particularly interesting as the use of inexpensive electrodes (i.e. graphite)27 is combined with a biphasic solvent (i.e. CH2Cl2/H2O) which not only avoids undesired side reactions during the electrolysis process, but also allows a facile reaction workup.
| Entry | Cathode | Solvent | Time (h) | Electrolyte | Yields |
|---|---|---|---|---|---|
| a Reaction conditions: constant current, I = 15 mA, 1 (1 equiv.), 4 (1 equiv.), electrolyte (3 equiv.), solvent (4 mL), room temperature, undivided cell. b Reaction performed with I = 14 mA. c Reaction performed without applied current. | |||||
| 1 | GF | THF/H2O (1:1) |
2 | NaCl | 26 |
| 2 | Ni | THF/H2O (1:1) |
2 | NaCl | 9 |
| 3 | Ni foam | THF/H2O (1:1) |
2.30 | NaCl | — |
| 4 | Cu | THF/H2O (1:1) |
1 | NaCl | Trace |
| 5 | Pt | THF/H2O (1:1) |
1.30 | NaCl | 11 |
| 6 | GF |
t-BuOMe/H2O (1:1) |
3 | NaCl | 18 |
| 7 | GF | toluene/H2O (1:1) |
1.30 | NaCl | — |
| 8 | GF | EtOH/H2O (1:1) |
2 | NaCl | — |
| 9b | GF | CH2Cl2/H2O (1:1.5) |
1.30 | NaCl | 73 |
| 10 | GF | CH2Cl2/H2O (1:1.5) |
1.30 | LiCl | — |
| 11 | GF | CH2Cl2/H2O (1:1.5) |
2 | NaI | 7 |
| 12c | — | CH2Cl2/H2O (1:1.5) |
24 | NaCl | — |
Under the same conditions, replacing NaCl with LiCl or NaI as electrolytes (entries 10 and 11) led to inferior results. Lastly, a control experiment conducted without applying a current returned the unchanged substrates (entry 12), underscoring the crucial role of anodic oxidation.
The separation of (S,S)-5 and (S,R)-5′ proved to be quite challenging. Despite numerous attempts, column chromatography was found to be ineffective for this purpose. However, successful separation was achieved through crystallization in iPrOH at 80 °C (see HPLC chromatograms [Fig. S1] and NMR spectra in SI).
Unfortunately, even after several attempts, we were unable to grow a single crystal suitable for X-ray analysis, and therefore could not assign the absolute stereochemistry at this stage of the synthesis.
Fortunately, based on the computational data reported for peptidomimetics (S,S)-11/(S,R)-11′, which perfectly matched the NMR conformational study, compound 5 was found to possess the (S,S)- absolute configuration, whereas compound 5′ displayed the (S,R)-configuration, where R refers to the configuration of the isoxazoline stereocenter.
Peptidomimetics synthesis
To study the capability of our scaffold to stabilize a parallel turn conformation, two peptidomimetic models were synthesized, incorporating the N-Ac-Leu-Val and N-Ac-Met-Ile arms. The synthesis of the desired peptidomimetics was carried out in parallel, starting both from the pure diastereoisomer (S,S)-5, previously isolated via crystallization, and from the diastereoisomeric mixture of compounds (S,S)-5/(S,R)-5′.First, the azido group of (S,S)-5 was reduced to an amino group through a Staudinger reaction (Scheme 3). The enantiopure amino derivative (S,S)-6 was obtained in 91% yield by treating a 0.1 M solution of (S,S)-5 in THF with H2O (7 equiv.) and a 1 M solution of PMe3 (1.1 equiv.) in toluene for 16 h at 25 °C. The same procedure was applied to the diastereoisomeric mixture (S,S)-5/(S,R)-5′, resulting in a mixture of amines (S,S)-6/(S,R)-6′ in comparable yields.
 | ||
| Scheme 3 Staudinger azide reduction giving amines (S,S)-6/(S,R)-6′. | ||
In the first attempt the coupling of the diastereoisomeric mixture (S,S)-6/(S,R)-6′ with N-Ac-Leu-Val-H (7) was performed in CH2Cl2 using EDC (1.1 equiv.)/HOBT (1.1 equiv.) as coupling reagents and DIPEA (1.1 equiv.) as a base. Unfortunately, the use of a base caused epimerization of one stereocenter, resulting in the formation of four products.
To overcome this issue, the peptide coupling was performed in CH2Cl2 (0.1 M solution) using DIC (1.1 equiv.) and oxyma (1.1 equiv.) (Scheme 4). The resulting products were then purified by flash column chromatography, successfully separating diastereoisomers (S,S)-8 and (S,R)-8′, with an overall yield of 84%.
 | ||
| Scheme 4 Synthesis of peptidomimetics (S,S)-8, (S,S)-9, (S,S)-11 and (S,R)-8′, (S,R)-9', (S,R)-11′. | ||
The same protocol was applied to transform enantiopure (S,S)-6 into (S,S)-8, which was isolated with a 75% yield after purification. In this way, we had an enantiopure standard for HPLC (see Fig. S1 in SI).
The two diastereoisomers (S,S)-8 and (S,R)-8′ underwent Boc deprotection using a 1:1 mixture of TFA and CH2Cl2 (25 °C, 30 min), yielding compounds (S,S)-9 and (S,R)-9′ in quantitative yields after quenching with aqueous NaHCO3.
The coupling with the second peptide arm, N-Ac-Met-Ile (10), was carried out following the protocol used for the synthesis of (S,S)-8/(S,R)-8′. The reaction times varied depending on the starting material: (S,S)-11 (20% yield) required overnight reaction time, whereas (S,R)-11′ (40% yield) completed within 3 h. The difference in yields among these peptidomimetics is due to the varying solubility of the starting materials, and the poor solubility of compound (S,S)-11 significantly hampered its isolation, contributing to its lower yield.
NMR spectroscopy conformational studies for synthesized peptidomimetics
The conformational behavior of pure compounds (S,S)-8/(S,R)-8′ and (S,S)-11/(S,R)-11′ was explored through 1D and 2D homo- and heteronuclear NMR analyses (1H, 13C, TOCSY, NOESY, COSY, HSQC, HMBC, 400 MHz for (S,S)-8/(S,R)-8′ and 600 MHz for (S,S)-11/(S,R)-11′).NMR experiments for compounds (S,S)-8 and (S,R)-8′ were performed in CD3CN and CDCl3, respectively. The assigned chemical shift and NOEs are detailed in Tables S2 and S3a, b respectively (see SI). To confirm the formation of H-bonds, variable-temperature experiments (0–55 °C) were conducted for both compounds (Fig. 1C). Focusing on compound (S,S)-8, the NHs exhibited Δδ/ΔT values of 5 ppb or lower. This information suggests the transient formation of hydrogen bonds, but not a stable secondary structure, given the short peptide length, which precludes the involvement of all NH groups in stable hydrogen bonding. In contrast, compound (S,R)-8′ displayed only one NH (CH2NHIsox; Δδ/ΔT = −2.2 ppb) capable of forming a hydrogen bond, likely with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OBoc. The Δδ/ΔT values for the other NH groups exceeded 8 ppb (Fig. 1C).
OBoc. The Δδ/ΔT values for the other NH groups exceeded 8 ppb (Fig. 1C).
 | ||
| Fig. 1 NMR data for (S,R)-8′ (CD3Cl, 0.018 M, 400 MHz) and (S,R)-11′ (TFE-d2, 12 mM, 600 MHz): (A) and (B) NOEs and H-bonds for (S,R)-8′ and (S,R)-11′ (intrastrand NOEs, blue arrows; interstrand NOEs, red arrow; H-bonds: dotted line). Δδ/ΔT NH values for diastereoisomers (C) (S,S)-8/(S,R)-8′ (273–328 K) and (D) (S,S)-11/(S,R)-11′ (273–323 K). | ||
NMR analysis for tripeptide (S,S)-8 revealed that the NHs resonate within a similar range (δ 6.90–5.79 ppm) and the potential diastereotopic CH2NH and PhCH2CH are characterized by a small Δδ (0.16 ppm) chemical shift. Furthermore, Noesy experiments (Table S2 in SI) indicated no intrastrand spatial proximities, showing only CHα/NH (i, i + 1) NOEs in the longest arm. This information suggests that the isoxazoline scaffold present in peptide (S,S)-8 could induce an extended conformation.
On the other hand, diastereisomer (S,R)-8′ exhibits the presence of two conformers in a 2:1 ratio, which equilibrate, as clearly shown in the Noesy experiment (for details see SI). The full set of resonances is provided in Table S3a for the main isomer, while only selected resonances are reported for the minor isomer (Table S3b). The main isomer exhibits well-dispersed NH signals across a wide range (δ 8.56–6.05 ppm). Unlike compound (S,S)-8, the diastereotopic CH2NH groups of the isoxazoline substituent show a Δδ greater than 1 ppm, confirming their distinct orientation within a turn. Additionally, NHBoc resonates at a very low field (δ 7.50 ppm), likely due to its orientation, suggesting that it is well-aligned within the rigid turn and de-shielded by the Ph-ring.
The second conformer displays three overlapping NH signals (δ 6.25–6.22 ppm), with NHVal resonating in a nearby region (δ 6.78 ppm).
Based on this observation, we hypothesize that the first isomer adopts a more ordered conformation. This hypothesis is further supported by the Noesy experiment of the main conformer (Fig. 1A). The NH group linked to COVal along with PhCH2CH exhibited spatial proximity to only one of the two protons of CH2N (δ 3.36 ppm) and CH2-4Isox (δ 3.27 ppm) groups, respectively, further confirming their rigid orientation. The longest arm is characterized by the presence of all possible CHα/NH (i, i + 1) NOEs. The flexibility of the tBuO moiety, along with the dipeptide chain at the N-terminus, explain the intrastrand NOE observed between NHLeu and Boc.
For the longer peptides (S,S)-11 and (S,R)-11′, due to their low solubility, especially in the case of peptide (S,R)-11′, NMR experiments were conducted in TFE-d2. The assigned chemical shifts and ROEs are detailed in Tables S4 and S5, respectively (see SI).
NHs show similar resonances in both isomers, except for NHLeu, NHIle and most notably for PhCH2CHNH, which resonates at lower field in (S,R)-11′. This suggests that the last NH is well-oriented and de-shielded by the phenyl group, as observed for NHBoc in (S,R)-8′.
Although not as pronounced as in (S,R)-8′, the CH2NH diastereotopic protons display a greater Δδ value in (S,R)-11′ (0.25 ppm) compared to (S,S)-11 (0.19 ppm), supporting their defined orientation within a turn.
The formation of a turn in (S,R)-11′ is also confirmed by the Roesy experiment (Table S4 and Fig. 1B): spatial proximities between CH2-4Isox with both PhCH2CH and CH2NH characterize (S,R)-11′, as well as between NHs of the isoxazoline substituents with the side chain protons of the corresponding AA at position i + 1, indicating consistent orientation. Additionally, beyond the CHα/NH (i, i + 1) ROEs within each arm, an inter-strand ROE was observed between NHIle and HαVal.
These data support the formation of a parallel β-hairpin for compound (S,R)-11′ and a more extended conformation for (S,S)-11, which is characterized exclusively by CHα/NH (i, i + 1) ROEs (see Table S5 in SI).
The experiment at variable temperature (273–323 K; Fig. 1D) yielded similar results for both diastereoisomers (S,S)-11 and (S,R)-11′: only NHIle has a Δδ/ΔT lower then −4 ppb, indicating its potential to form a H-bonds. This conclusion is further corroborated by computational studies in case of (S,R)-11′ (see below).
It is worth noting that these NMR analyses were valuable not only for investigating the conformations of the synthesized peptides, but also for determining the absolute configuration of the unnatural segments within the sequences. In fact, the assignment of the absolute configuration of diastereoisomers (S,S)-5 and (S,R)-5′ was achieved indirectly by comparing the computational and NMR data obtained for peptides (S,S)-11/(S,R)-11′.
Molecular modelling
To gain insight into the conformational preferences of peptidomimetics (S,S)-11 and (S,R)-11′ connected with the different stereochemistry of the isoxazoline ring, replica exchange molecular dynamics (REMD) simulations were performed using the Amber24 software suite.28 After parameterization of the S,S- and R,S-isoxazoline scaffolds (details are given in the SI), models peptidomimetics M11 and M11′ containing respectively the above scaffolds, were built using an extended conformation and subjected to REMD simulations. These followed a protocol previously found effective in describing short peptides favouring β-hairpin conformations,29–32 which combines the ff96 Amber force field and the OBC(II) implicit solvent model.33,34 Twelve replicas of 800 ns each were performed, with temperatures spanning from 300.0 to 860.9 K. The 300 K trajectory was then extracted and analysed. The results from the H-bond and clustering analyses are discussed below, with key data summarized in Table 2 and Fig. 2. Complete methodological details and full clustering data (Tables S5 and S6) are available in the SI. | ||
| Fig. 2 Representative conformation of first and second highest populated clusters for M11 (A and B; pop. = 40.9% and 31.3%, respectively) and M11′ (C and D; pop. = 60.3% and 14.8%, respectively). Cluster analyses were performed on the 600–800 ns portion of the 800 ns REMD trajectories. | ||
| Donor | Acceptor | Occ.% | |
|---|---|---|---|
| a An H-bond was defined by a donor–acceptor distance ≤4.0 Å and a donor-H-acceptor angle ≥120. H-bonds with an occupancy equal or higher than 5%, and not involving the acetyl or NHMe caps, are reported. | |||
| M11 | IleNH | LeuCO |
5.0 |
| M11′ | IleNH | LeuCO |
37.1 |
The most representative conformations of the first and second most populated clusters (Fig. 2) suggest that both M11 and M11′ experience rotation of the bond between C3 of the isoxazoline ring and the C1 of the 1-ammino-2-phenylethyl moiety, leading to an equilibrium between an extended-like and a β-like conformation. However, both cluster and H-bond analyses show that the R,S-isoxazoline scaffold is more effective in stabilizing a β-hairpin conformation compared to S,S-scaffold. The most representative structure of the main cluster for M11 (Fig. 2A), though turn-like, does not form a well-structured β-hairpin, in contrast to the structure observed for M11′ (Fig. 2C). Additionally, H-bond analysis (Table 2) reveals a stable H-bond between NHIle and COLeu in M11′, with an occupancy of 37.1%. This finding is in excellent agreement with the NMR data, which identified an inter-strand ROE between NHIle and HαVal and a low temperature coefficient for the NHIle. Conversely, a very low occupancy (5.0%) is found for the same H-bond in M11, suggesting that a folded β-hairpin, although transiently sampled, is not stably maintained by the S,S-isoxazoline scaffold.
Based on the NMR and computational results obtained for (S,S)-11 and (S,R)-11′, we can conclude that the R,S-isoxazoline scaffold demonstrates significant potential for inducing a parallel turn, compared to the S,S-diastereoisomer. This characteristic enables the formation of parallel β-hairpins, highlighting its valuable role in structural design.
Conclusion
The synthesis of a new isoxazoline scaffold substituted with two amino-alkyl chains was achieved via a 1,3-dipolar cycloaddition reaction. This involved the nitrile oxide derived from the inexpensive, enantiopure L-phenylalanine and azido methyl acrylate. The reaction conditions were optimized to maximize yield. Additionally, we successfully employed an eco-friendly electrochemical cycloaddition reaction, starting directly from the oxime and avoiding the need for the corresponding chloroxime.In this way, we obtained a diastereoisomeric mixture of cycloadducts (S,S)-5/(S,R)-5′, which served as the starting materials for producing the orthogonally protected di-amino compounds (S,S)-6/(S,R)-6′. This enabled the preparation of peptidomimetics containing two distinct arms. The availability of both diastereoisomers, yielding the corresponding peptidomimetics (S,S)-11 and (S,R)-11′, provided the opportunity to define the correct stereochemistry required for the turn induction. Based on NMR and computational studies, the R,S-isoxazoline scaffold showed superior performance in forming a parallel β-hairpin, as observed with (S,R)-11′. Consequently, this scaffold holds significant potential for designing peptidomimetics with extended arms that stabilize the parallel structure.
Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ob01798j.Acknowledgements
All the authors thank the Mass Spectrometry facility of the Unitech COSPECT at the University of Milan for MS analyses. The authors thank the GREEN-TECH project, funded by Regione Lombardia, FESR 2021-2027, co-funded by the European Union (Grant Agreement ID: 6154644 - CUP E19I25001040007).References
- K. Sharma, K. K. Sharma, A. Sharma and R. Jain, Drug Discovery Today, 2023, 28, 103464 CrossRef CAS.
- J. H. Hamman, G. M. Enslin and A. F. Kotzé, BioDrugs, 2005, 19, 165–177 CrossRef CAS PubMed.
- G. Rossino, E. Marchese, G. Galli, F. Verde, M. Finizio, M. Serra, P. Linciano and S. Collina, Molecules, 2023, 28, 7165 CrossRef CAS PubMed.
- S. Ohtake, Y. Kita, R. Payne, M. Manning and T. Arakawa, Protein Pept. Lett., 2013, 20, 1308–1323 Search PubMed.
- V. Apostolopoulos, J. Bojarska, T. T. Chai, S. Elnagdy, K. Kaczmarek, J. Matsoukas, R. New, K. Parang, O. P. Lopez, H. Parhiz, C. O. Perera, M. Pickholz, M. Remko, M. Saviano, M. Skwarczynski, Y. Tang, W. M. Wolf, T. Yoshiya, J. Zabrocki, P. Zielenkiewicz, M. Alkhazindar, V. Barriga, K. Kelaidonis, E. M. Sarasia and I. Toth, Molecules, 2021, 26, 430 CrossRef CAS.
- E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262–3277 RSC.
- R. Bucci, L. De Rosa, G. Bertoni, R. Di Stasi, M. della Valle, D. Diana, S. Peppicelli, K. Peqini, M. L. Gelmi, F. Bianchini and L. D. D'Andrea, Bioorg. Chem., 2025, 165, 109039 CrossRef CAS.
- R. Bucci, S. Giofré, F. Clerici, A. Contini, A. Pinto, E. Erba, R. Soave, S. Pellegrino and M. L. Gelmi, J. Org. Chem., 2018, 83, 11493–11501 CrossRef CAS.
- R. Bucci, A. Contini, F. Clerici, S. Pellegrino and M. L. Gelmi, Org. Chem. Front., 2019, 6, 972 RSC.
- F. Vaghi, R. Bucci, F. Clerici, A. Contini and M. L. Gelmi, Org. Lett., 2020, 2020, 14 Search PubMed.
- R. B. Dyer, S. J. Maness, E. S. Peterson, S. Franzen, R. M. Fesinmeyer and N. H. Andersen, Biochemistry, 2004, 43, 11560–11566 CrossRef CAS PubMed.
- J. A. Robinson, Acc. Chem. Res., 2008, 41, 1278–1288 CrossRef CAS PubMed.
- E. Ko, J. Liu, L. M. Perez, G. Lu, A. Schaefer and K. Burgess, J. Am. Chem. Soc., 2011, 133, 462–477 CrossRef CAS.
- R. Bucci, F. Foschi, C. Loro, E. Erba, M. L. Gelmi and S. Pellegrino, Eur. J. Org. Chem., 2021, 2887–2900 CrossRef CAS.
- J. S. Nowick, Acc. Chem. Res., 2008, 41, 1319–1330 CrossRef CAS.
- J. S. Nowick, Acc. Chem. Res., 1999, 32, 287–296 CrossRef CAS.
- S. N. Filigheddu and M. Taddei, Tetrahedron Lett., 1998, 39, 3857–3860 CrossRef CAS.
- J. D. Fisk, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 2000, 122, 5443–5447 CrossRef CAS.
- M. Breugst and H. U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293–12307 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC.
- A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed.
- A. A. Al-Romema, H. Xia, K. J. J. Mayrhofer, S. B. Tsogoeva and P. Nikolaienko, Chem. – Eur. J., 2024, 30, e202402696 CrossRef CAS.
- T. Shono, Y. Matsumura, K. Tsubata, T. Kamada and K. Kishi, J. Org. Chem., 1989, 54, 2249–2251 CrossRef CAS.
- S. D. L. Holman, A. G. Wills, N. J. Fazakerley, D. L. Poole, D. M. Coe, L. A. Berlouis and M. Reid, Chem. – Eur. J., 2022, 28, e202103728 CrossRef CAS.
- S. Hofmann, J. Winter, T. Prenzel, M. d. J. Gálvez-Vázquez and S. R. Waldvogel, ChemElectroChem, 2023, 10, e202300434 Search PubMed.
- R. Bucci, F. Vaghi, D. Di Lorenzo, F. Anastasi, G. Broggini, L. Lo Presti, A. Contini and M. L. Gelmi, Eur. J. Org. Chem., 2022, e202200601 Search PubMed.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, D. S. Cerutti, G. A. Cisneros, V. W. D. Cruzeiro, N. Forouzesh, T. J. Giese, A. W. Götz, H. Gohlke, S. Izadi, K. Kasavajhala, M. C. Kaymak, E. King, T. Kurtzman, T.-S. Lee, P. Li, J. Liu, T. Luchko, R. Luo, M. Manathunga, M. R. Machado, H. M. Nguyen, K. A. O'Hearn, A. V. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, A. Risheh, S. Schott-Verdugo, A. Shajan, J. Swails, J. Wang, H. Wei, X. Wu, Y. Wu, S. Zhang, S. Zhao, Q. Zhu, T. E. Cheatham, D. R. Roe, A. Roitberg, C. Simmerling, D. M. York, M. C. Nagan and K. M. Merz, J. Chem. Inf. Model., 2023, 63, 6183–6191 CrossRef CAS.
- F. Dordoni, D. Scarpi, F. Bianchini, A. Contini and E. G. Occhiato, Eur. J. Org. Chem., 2020, 4371–4383 Search PubMed.
- L. Sernissi, L. Ricci, D. Scarpi, F. Bianchini, D. Arosio, A. Contini and E. G. Occhiato, Org. Biomol. Chem., 2018, 16, 3402–3414 RSC.
- L. Ricci, L. Sernissi, D. Scarpi, F. Bianchini, A. Contini and E. G. Occhiato, Org. Biomol. Chem., 2017, 15, 6826–6836 RSC.
- S. Pellegrino, N. Tonali, E. Erba, J. Kaffy, M. Taverna, A. Contini, M. Taylor, D. Allsop, M. L. Gelmi and S. Ongeri, Chem. Sci., 2017, 8, 1295–1302 RSC.
- A. Onufriev, D. Bashford and D. A. Case, Proteins:Struct., Funct., Bioinf., 2004, 55, 383–394 CrossRef CAS.
- P. A. Kollman, Acc. Chem. Res., 1996, 29, 461–469 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |