DOI:
10.1039/D5OB01668A
(Review Article)
Org. Biomol. Chem., 2026,
24, 1734-1757
Advances of well-defined alkali metal complexes in organic synthesis
Received
23rd October 2025
, Accepted 30th January 2026
First published on 2nd February 2026
Abstract
Alkali metal reagents in organic synthesis are ubiquitous within academia and industry due to the wide array of synthetic utility as bases, nucleophiles, and Lewis acids. While organolithium chemistry has been the most prominent, the development of analogous technologies with the later alkali metals has garnered recent interest. This review aims to develop an introductory primer on well-defined heavier alkali metal complexes and the corresponding contemporary utility in organic synthesis. Key developments will be highlighted from the past decade to provide direct comparison to early fundamental studies, aspiring to provide a template for future advances in the field of alkali metal-mediated methods and catalysis.
1. Introduction
Organic transformations mediated by alkali metals (AM) represent some of the foundational concepts taught in every introductory organic textbook as characteristic bases and nucleophiles. Although alkali metal-based intermediates have been studied since the early 1900s, the relationship between structure and activity has remained nebulous due to ionicity, aggregation, and instability.1–7 Of the alkali metals, organolithium compounds are at the forefront of organic chemistry as potent nucleophiles, Brønsted bases, and productive transmetalation and ligand-exchange agents. The wealth of available organolithium precursors has facilitated detailed mechanistic study of elusive intermediates, granting key insight into its metal-based reactivity.8–13 As a result, the usage of organolithiums as organometallic reagents has become commonplace in synthesis. Moving forward, growing environmental and sustainability concerns warrant investigation into alternative avenues.14
In this vein, the heavier alkali metals (AM = Na–Cs) represent an attractive substitute to lithium, maintaining analogous high nucleophilicity and Brønsted basicity coupled with increased earth abundance and biocompatibility. Contrastingly, the heavier alkali metals have larger ionic radii, enabling expanded coordination spheres (CN = 6–12) resulting in weaker and reactive bonds (Fig. 1).15–18 The use of heavier alkali metal salts and alkoxides has been prominent in organic chemistry as the increased basicity and proton affinity engenders enhanced rate and selectivity. Simultaneously, these attributes make the structural and mechanistic analysis of well-defined organometallic complexes of the alkali metals challenging to study. Considering their versatility, this review seeks to highlight reported systems that provide thorough structural and mechanistic elucidation, extending to well-defined complexes and intermediates whose identity is supported by spectroscopic data, X-ray diffraction, or reactivity studies.
 |
| | Fig. 1 General trends of the alkali metals. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Shannon radii for most common coordination number, bmost common coordination numbers, cexperimental data, dcomputational data calculated using PBE0/CRENBL level of theory with ECP basis set.15,16,19 Shannon radii for most common coordination number, bmost common coordination numbers, cexperimental data, dcomputational data calculated using PBE0/CRENBL level of theory with ECP basis set.15,16,19 | |
Given the effectiveness of the heavier alkali metal reagents in organic synthesis, initial work was focused on isolation and speciation of reactive intermediates of organosodium analogues, building upon existing studies with organolithium complexes. Aggregation and solvent effects in lithium systems have been well studied, but initial investigations of organosodium complexes were not as straightforward considering the changes in radii and coordination sphere. In this regard, the Collum group has extensively studied both lithium and sodium-based systems, specifically targeting alkali metal enolates to understand aggregation processes and enable entry to study these reactive intermediates.20 By utilizing the method of continuous variation (MCV) and NMR spectroscopy, the structure of metal-based aggregates was determined and demonstrated that solvent plays a key role in aggregate formation. It should be mentioned that although 6/7Li and 23Na NMR spectroscopy can be used to characterize the formation of in situ metal complexes, the 23Na NMR nuclei is quadrupolar and exhibits broad resonances that make it difficult to extract useful structural and mechanistic information.
Despite these challenges, Collum and coworkers utilized these spectroscopic methods to determine the structures of sodium amides, with NaN(SiMe3)2 represented as a titular example below (Fig. 2A). Solvation effects on sodium complexes result in a wider array of different solvent-based aggregates and ion pair complexes (1–5) than are normally observed with lithium counterparts, ultimately emphasizing the stark contrast of solution- and solid-state reactivity.21,22 Further studies continued to expand on the structure of alkali metal amides and reported the solution-state structures of KN(SiMe3)2, which exists predominantly as a dimer (6–8) in non-coordinating solvents, but transitions to a monomer (9) with strongly coordinating solvents and ligands (Fig. 2B).23 Subsequent titration studies, isotopic labelling experiments, and thorough 2D and heteronuclear NMR spectroscopic analysis enabled rigorous solution-state investigation, providing a precise basis for the heavier alkali metal amide chemistry. As a result, NaN(SiMe3)2 and KN(SiMe3)2 based reactivity are the most studied systems and represent the framework for comparison amongst many alkali metal complexes.24
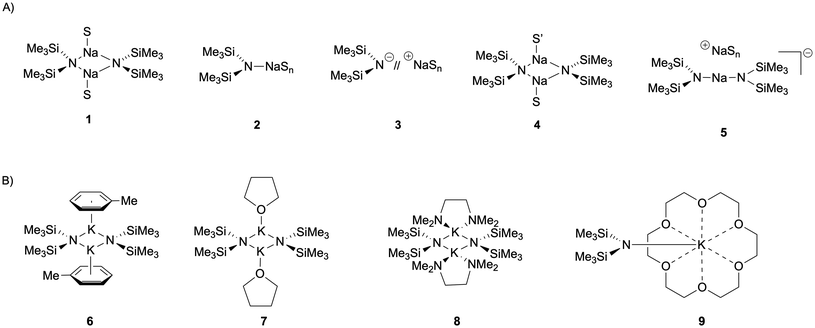 |
| | Fig. 2 (A) Common solvated structures of NaN(SiMe3)2, (B) various solution state structures of KN(SiMe3)2. | |
In contrast, rubidium and cesium complexes have been much less explored, though literature reports indicate enhanced reactivity in line with general periodic trends.25 Interestingly, organocesium reagents show increased solubility and decreased aggregation, in comparison to sodium and potassium, resulting in faster reaction rates and chemodivergent activity, coined as the ‘Cesium effect’ which has been reviewed previously.26,27 As such, recent studies demonstrate new developments for the heavier organoalkali compounds in rising areas of interest in organic chemistry such as C–C bond formation, C–H activation, and hydrofunctionalization. The main challenges for advancement of the later alkali metals are staunchly entranced in the decreased solubility of these reagents in hydrocarbon and ethereal solvents, the increased thermodynamic preference for forming aggregates, and the incompatibility or deleterious reactivity with certain functionalities or substrates.
With this heightened reactivity, methods to tame these metals have included the use of Lewis donor-based solvents or additives as exogeneous supports in solution, and the incorporation of N- or O-based ligand scaffolds to minimize aggregation and maximize stability at the metal center. In addition to enhanced stability, the incorporation of ligands provides spectroscopic handles by which solution-state reactivity can be deciphered and compared to isolated solid-state complexes. Progress towards the isolation of well-defined AM complexes sets the stage for systematic mechanistic study and demonstrates significant differences in reactivity that can be exploited for new organic transformations.
This review focuses on recent advances of the heavier alkali metals over the last ten years, and the resultant applications of these reagents within organic synthesis, methods, and catalysis. Distinct emphasis on synthetic availability and mechanistic investigations of key transformations is highlighted and contextualized. Within this scope, reports on the use of alkali metal dispersions, hydrides, salts, alkoxides, and carbonates have been omitted, as well as discussion of francium complexes due to their limited syntheses.28,29
2. Donor assisted deprotonation
2.1. Deprotonation
The deprotonation chemistry of AM reagents is expansive and commonly mediated using prototypical AM hydroxides, with other examples including alkoxides, aryloxides and carbonates. Selectivity in deprotonation for these systems is seldom controlled due to a lack of synthetic tunability at the alkali metal center. The addition of exogeneous donors, in the form of mono- or multidentate ligands, represents a new avenue to promote site-selective deprotonation. Few well-defined analogues that enable selectivity exist, and current studies have focused on developing structural elucidation of metalated intermediates to understand how to modulate basicity and selectivity in deprotonation. Solvent choice plays a key role in deprotonation reactivity as Lewis basic donor solvents readily engage AM complexes in metalation and aggregation, inducing changes in the Brønsted basicity of the metal center.
2.1.1. Sodium.
In alkali metal chemistry, solvent incompatibility results in diminished reactivity, wherein the basicity of the AM center has also been shown to induce decomposition of the solvent or reagent. Within this context, Collum and coworkers studied the metalation/deprotonation of THF with sodium amides, comparing the reactivity to that of established lithium systems.30 Particularly, the comparison of sodium diisopropylamide (NaDA) in solvents such as THF and N,N-dimethylethylamine (DMEA) to that of lithium diisopropylamide (LDA) in THF was explored by comparing the relative rates of metalation reactions between NaDA and LDA (krel). The use of NaDA has been limited due to the scarcity of solvents that allow for high solubility; however, NaDA–DMEA demonstrates higher metalation rates than those of LDA–THF with comparable yields and selectivity.1
Further investigation into the sodium mediated decomposition of THF showed that NaDA forms a tetra-solvated monomer in solution, which is then coordinatively primed to perform the α-deprotonation of THF (Scheme 1). The proposed mechanism for the decomposition of THF is shown below where intermediate 10 deprotonates at the α-position, followed by the loss of the diisopropylamide group and formation of the oxacarbenoid-precursor to the carbene-Na structure. Alternatively, an E2-like β-metalation pathway may occur; however, isotopic studies showed that scrambling prevents clear differentiation between the two mechanisms. Comparisons with THF and THF-d4 results in a kH/kD ∼ 6, suggesting that the C–H cleavage is the rate limiting step. The decomposition of DME by NaDA affords sodium methoxide and methyl vinyl ether proceeding through a di-solvated-monomer-based transition state.
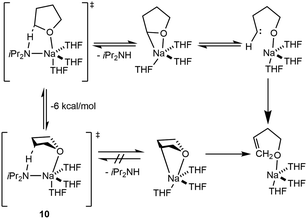 |
| | Scheme 1 Mechanism of THF decomposition by NaDA. | |
The sodiation of a series of unactivated arenes was achieved through the application of highly basic 2,2,6,6-tetramethylpiperidide (NaTMP) and a polyamine donor, either N,N,N′,N′-tetramethylethylenediamine (TMEDA) or N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA), by the Hevia group.31,32 Although the arylation step resulted in low isolated yields, subsequent arene borylations done in a one-pot procedure resulted in 80–83% yields (Fig. 3). Mechanistic studies with anisole as a representative substrate for ortho-metalation were explored. In the presence of either NaTMP/PMDETA (36% yield) or NaTMP/TMEDA (35% yield), a bimetallic intermediate forms with two sodium centers bridged by one TMP and one anisole, flanked by one donor molecule each. This represents a rare example of a well-defined mixed sodium-aryl/sodium-amide complex, elucidated by NMR spectroscopy and X-ray crystallographic studies (Fig. 3c).
 |
| | Fig. 3 Proposed pathway for the borylation of anisole by NaTMP. | |
2.2. Metal-directed deprotonation
Falling under the umbrella of general deprotonation, examples of metal-directed deprotonation involve the formation of discrete metal–substrate bonds which are integral for subsequent bond formation or rearrangement.
2.2.1. Sodium.
The utility of NaDA extends to the conversion of aryl carbamates (11) to ortho-acylated phenols in THF.33 The ortho-metalation of the phenols is proposed to form via an arylsodium intermediate (12a–g) which undergoes a Snieckus-Fries rearrangement (13a–g) to form the ortho-acylated product (14, Scheme 2). DFT calculations suggest that the metalation and rearrangement steps both proceed through THF-solvated, monomeric NaDA pathways. The rearrangement reactions occur in good yields; however, aryl carbamates with halogen substituents may undergo either rearrangement or competitive halide eliminations, depending on the substituents, forming benzyne preferentially.
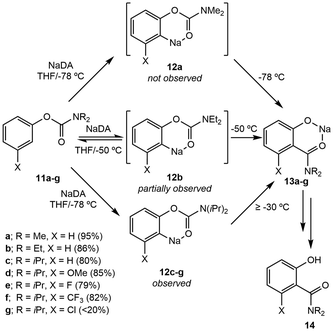 |
| | Scheme 2 The synthesis of ortho-acylated phenols (14a–g) from aryl carbamates (11a–g) using NaDA in THF through arylsodium intermediates (12a–g). | |
Based on prior work with lithium Evans enolates, the Collum group generated a series of enolates using NaDA or NaN(SiMe3)2 in the presence of TMEDA, trans-N,N,N′,N′-tetramethylcyclohexanediamine (R,R)-TMCDA, or (S,S)-TMCDA (Scheme 3).34 The sodiated Evans enolates formed mixed dimers, each of which was examined for aldol additions, stereoselective quaternizations, and azaaldol additions. Although similar results were produced using either NaDA or NaN(SiMe3)2, it was found that NaN(SiMe3)2 was the preferred reagent due to its commercial availability and versatility. The TMEDA-solvated sodiated Evans enolates (16) demonstrate improved structural control compared to THF-solvated counterparts and maintain comparable selectivity (Scheme 3). This diverges from lithium enolates, with which THF has been the solvent of choice, and the potential utility of sodium enolates for transformations with imines and aldehydes further widens this divide.
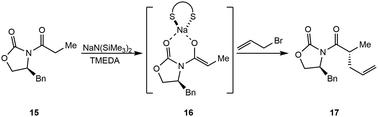 |
| | Scheme 3 Synthesis of sodiated Evans enolates using NaN(SiMe3)2. | |
Application of NaTMP to halothiophenes (18, X = Cl or Br) by Mori and coworkers results in metalation, forming sodium thiophenes (19) as precursors for polymerization.35 Quenching the metalated thiophene product with iodine afforded 2-chloro-3-hexyl-5-iodothiophene (85%) and 2-bromo-3-hexyl-5-iodothiophene (17%, 20, Scheme 4), with the latter resulting in lower yields due to deleterious reactivity by NaTMP at the C–Br bond.
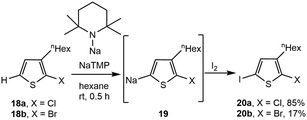 |
| | Scheme 4 Deprotonative metalation of halothiophenes (18) with NaTMP to yield sodiated halothiophenes (19). | |
2.2.2. Potassium, rubidium, cesium.
Examples of deprotonation with potassium, rubidium, and cesium alkoxides, aryloxides, and carbonates are known, similar to the lithium and sodium analogues. While there are well-defined examples where deprotonation is utilized with these metals, the transformations have been classified under an alternative reaction type (see 3.3.1, 6.1.2 and 6.2.2) within this review.
3. Amination, enolization, and epoxide ring-opening
3.1. Amination and Mannich reactions
The formation of new C–N bonds enables incorporation of nitrogen into organic molecules, which is indispensable in pharmaceutical chemistry and total synthesis of natural products. The improved biocompatibility of alkali metals compared to transition metals increases the appeal of pursuing AM-mediated routes for installing nitrogen centers in organic moieties.
3.1.1. Sodium.
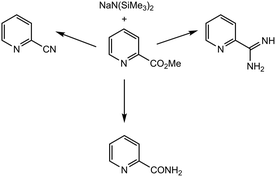
Solvent-dependent control over reactivity, commercial availability, and heightened tolerance to a variety of conditions render NaN(SiMe3)2 the preferred sodium amide for amination reactions. The Collum group explored NaN(SiMe3)2 as a strong base toward carbon-centered electrophiles for a series of C–N bond-forming reactions.36 The reaction of NaN(SiMe3)2 with electrophiles (ester-substituted arenes, pyridines, and epoxides) in various solvents resulted in pristine products of generally high yields (55%–96%). Discrepancies in reactivity and yield were explored with mechanistic studies in which it was found that the solvent choice or reaction conditions heavily influenced the reaction outcomes, delineated in Table 1. Although THF and DMEA preferentially form monomeric NaN(SiMe3)2 (1), reactivity was dominated by dimeric NaN(SiMe3)2 (2) in the presence of substrates. Solvent choice dictated chemoselectivity, whereby in THF the release of NaOSiMe3 results in an imino ether intermediate, affording entry 5 or entry 6. However, in DMEA, MeOSiMe3 is released instead, forming the carboxamide product preferentially (entry 2), demonstrating the importance of solvent selection for generating the necessary aggregate intermediate.
Table 1 Reaction of NaN(SiMe3)2 with electrophiles in various solvents
|
|
| Reaction of NaN(SiMe3)2 with electrophiles in various solvents |
| Entry |
Substrate |
Conditions |
Product |
Yield (%) |
Entry |
Substrate |
Conditions |
Product |
Yield (%) |
| 1 |

|
2.0 equiv. |
|
72 |
10 |
|
3.0 equiv. |
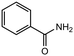
|
78 |
| NaN(SiMe3)2 |
NaN(SiMe3)2 |
| 25 °C, 5 h |
50 °C, 1 h |
| Toluene |
DMEA |
| 2 |
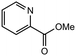
|
3.0 equiv. |

|
95 |
11 |
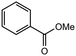
|
3.0 equiv. |
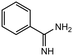
|
92 |
| NaN(SiMe3)2 |
NaN(SiMe3)2 |
| 25 °C, 0.3 h |
50 °C, 2 h |
| DMEA |
25 °C, 24 h |
| THF |
| 3 |
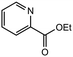
|
3.0 equiv. |
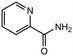
|
90 |
12 |
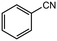
|
2.0 equiv. |
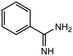
|
95 |
| NaN(SiMe3)2 |
NaN(SiMe3)2 |
| 25 °C, 0.3 h |
25 °C, 0.05 h |
| Toluene |
DMEA |
| 4 |
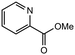
|
1.0 equiv. |
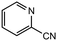
|
86 |
13 |
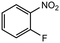
|
2.0 equiv. |
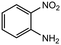
|
85 |
| NaN(SiMe3)2 |
NaN(SiMe3)2 |
| 50 °C, 0.1 h |
25 °C, 2 h |
| THF |
Toluene |
| 5 |
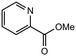
|
3.0 equiv. |
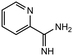
|
92 |
14 |
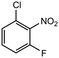
|
2.0 equiv. |
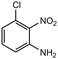
|
77 |
| NaN(SiMe3)2 |
NaN(SiMe3)2 |
| 50 °C, 0.3 h |
25 °C, 1 h |
| THF |
Toluene |
| 6 |
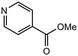
|
3.0 equiv. |
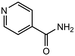
|
85 |
15 |
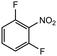
|
2.0 equiv. |
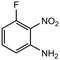
|
83 |
| NaN(SiMe3)2 |
NaN(SiMe3)2 |
| 70 °C, 1 h |
25 °C, 1 h |
| Toluene |
Toluene |
| 7 |
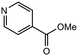
|
3.0 equiv. |
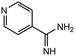
|
96 |
16 |
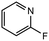
|
2.0 equiv. |
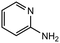
|
55 |
| NaN(SiMe3)2 |
NaN(SiMe3)2 |
| 70 °C, 1 h |
110 °C, 2 h |
| THF |
Toluene |
| 8 |
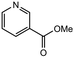
|
2.0 equiv. |
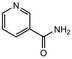
|
76 |
17 |
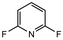
|
2.0 equiv. |
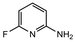
|
76 |
| NaN(SiMe3)2 |
NaN(SiMe3)2 |
| 70 °C, 1 h |
110 °C, 3 h |
| Toluene |
Toluene |
| 9 |
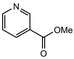
|
3.0 equiv. |
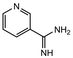
|
95 |
18 |
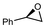
|
2.0 equiv. |
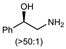
|
86 |
| NaN(SiMe3)2 |
NaN(SiMe3)2 |
| 70 °C, 1 h |
60 °C, 24 h |
| THF |
Toluene |
| 19 |
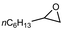
|
2.0 equiv. |
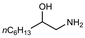
|
76 |
| NaN(SiMe3)2 |
| 25 °C, 24 h |
| THF |
Diversifying the landscape of sodium reagents through rigorous mechanistic and coordination studies, the Collum group extended similar investigations to include sodium alkylsilazides such as sodium isopropyl(trimethylsilyl)amide (NaPTA) and sodium tert-butyl(trimethylsilyl)amide (NaBTA). Reactivity such as N-alkylations, epoxide openings, and aminations were achieved with similar efficacy to NaN(SiMe3)2 while maintaining the lability of ethereal solvents demonstrated by NaDA. Despite these enhanced modifications, sodium alkylsilazides are less synthetically accessible than the parent NaN(SiMe3)2.37
3.1.2. Potassium.
Proceeding down the group, potassium exhibits a larger ionic radius and increased reactivity compared to sodium, making it suitable for more challenging transformations. Kobayashi and colleagues present a chiral potassium salt catalyst system (K-box) for catalytic, asymmetric Mannich reactions.38 K-box catalysts (21) are prepared from KN(SiMe3)2 and chiral bis(oxazoline) ligands, and have demonstrated efficiency in targeting imines (22) and weakly acidic amides (23) to produce β-amino acid derivatives (24) with high diastereo- and enantioselectivity (Scheme 5A). KN(SiMe3)2 interacts with the chiral K-box ligand to form a potassium enolate-K-box complex (25), which facilitates imine addition (Scheme 5B). The complex stabilizes the potassium enolate and promotes highly efficient deprotonation of the amide, enabling precise asymmetric control during imine addition. High yields and enantioselectivities (up to 94% ee) were observed, with substrate-specific modifications further enhancing selectivity. Applications included gram-scale reactions and β-lactam synthesis, demonstrating the system's scalability and practical utility with a heavier alkali metal.
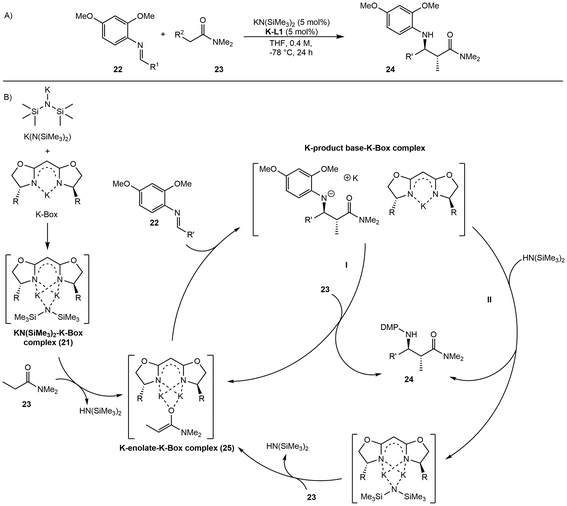 |
| | Scheme 5 A) Optimized reaction conditions for Mannich reaction using K-box catalyst system (21), (B) proposed catalytic cycle. | |
3.2. Enolizations
Alkali metal bases have been routinely applied towards enolizations under basic conditions, but the structural conformation of the alkali metal intermediates has been minimally investigated. Comparatively, organic systems using lithium amides (LDA, LiN(SiMe3)2) have demonstrated reactive competency with limited efficiency and selectivity.39–42
3.2.1. Sodium.
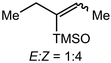
In comparison, the shortcomings of lithium amides are addressed through the use of NaN(SiMe3)2 for ketone enolization, whereby aggregation and conformation are directly influenced by solvent and substrate, with E:Z selectivity and kinetic rates varying heavily based on the system. The Collum group has identified several plausible mechanistic pathways, wherein NaN(SiMe3)2 can exist as a monomer (26), dimer (27), ion pair (28), or free ion (29) depending on the solvent, which then reacts with the ketone substrate in any variation of these forms to generate the enolate products 30-E and 30-Z (Scheme 6).43 The effect on E:Z selectivity was shown to range from 20:1 (Et3N/toluene) to 1:90 (THF). Et3N and methyl-t-butyl ether (MTBE) show the highest selectivity towards the E-product, with an E:Z ratio greater than 10:1. Toluene, TMEDA, and PMDETA have an E:Z ratio less than 10:1 but still exhibit preference for the E-enolate. Diglyme is the only example studied with a 1:1 ratio. Both THF and DME (1:20) selectively form the Z-product. Small changes in substrate were shown to impact the mechanism drastically, as changing the substrate from 2-methyl-3-pentanone to 2-methylcyclohexanone causes the mechanism to change from a monomer-promoted pathway to one involving a triple-ion pair system. In comparison to lithium reagents, superior selectivity and faster reactivity were reported, emphasizing NaN(SiMe3)2 as a competent, accessible reagent choice.
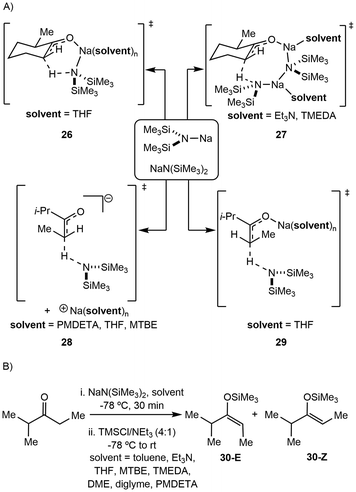 |
| | Scheme 6 A) Plausible mechanistic pathways for the enolization of ketones with NaN(SiMe3)2 (B) E:Z selectivity by solvent system. | |
3.3. Epoxide ring-opening
Epoxides represent useful chemical feedstocks for polymerizations, alcohol formation, and building larger organic molecules. Epoxide ring-opening can be performed under both basic and acidic conditions, commonly employing alkali salts and alkoxides for base-promoted openings.44 In addition, there are few routes utilizing well-defined AM precursors for epoxide ring-opening reactions, though limited examples with sodium complexes have been previously reported (see section 3.1.1).36,44,45
3.3.1. Sodium and potassium.
The Shirakawa group employed well-defined glycol-stabilized sodium and potassium salts for the catalytic conversion of epoxides and atmospheric CO2 into cyclic carbonates (31) under mild conditions (Scheme 7A).46 The highest performing complexes all consisted of iodide salts and glycols with either three (33, 67%) or four (32, 68%; 34, 70%) ethylene linkers (Scheme 7B). Mechanistic investigation using 34 suggests that tetraethylene glycol (tEG) activates epoxides via hydrogen bonding, while the iodide anion from KI facilitates nucleophilic attack. This process leads to the formation of cyclic carbonates through a three-step catalytic cycle: epoxide fixation (35) and activation (36), CO2 fixation (37), and intramolecular ring closure (31, Scheme 7C). The reaction tolerates a broad substrate scope, including simple and functionalized epoxides, in addition to enantiopure substrates without loss of stereochemical integrity. Large-scale syntheses and catalyst recycling experiments achieved over 10 catalytic cycles with no loss of efficiency of the glycol-potassium salt pair. Subsequent computational analysis (DFT) from Butera and Detz exploring the mechanism reveals that the rate-determining step involves the nucleophilic attack of iodide on the epoxide, with a calculated energy barrier significantly reduced compared to the uncatalyzed process.47 Natural bond order (NBO) and Hirshfeld charge analyses confirm that tEG's coordination with potassium weakens the K–I bond, freeing iodide ions for nucleophilic activity.
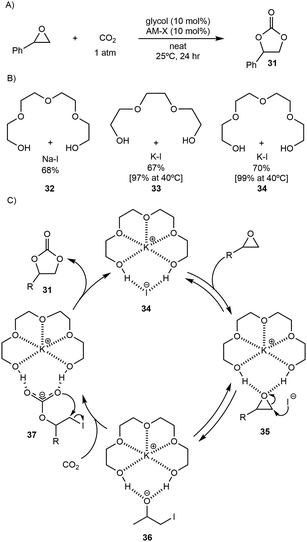 |
| | Scheme 7 A) Conversion of epoxides into cyclic carbonates using atmospheric CO2 and a catalytic potassium iodide-tetraethylene glycol complex, (B) highest performing glycol and alkali salt combinations, (C) proposed mechanism of cyclic carbonate formation. | |
4. Isomerization and olefination
4.1. Isomerization
Isomerization is primarily mediated by transition metals that are able to access metal hydride or allyl intermediates.48 However, there have been reports of stoichiometric alkali metal bases, such as hydrides, alkoxides, and amides, performing isomerization reactions with both ill- and well-defined systems.49–51 Chemical tunability with well-defined systems is part of ongoing efforts to improve AM-isomerization methods, taking advantage of their basicity and reactivity in stoichiometric and catalytic avenues.
4.1.1. Sodium.
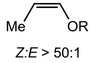
Sodium diisopropylamide (NaDA) serves as an effective Brønsted base for the isomerization of C(sp2)–C(sp2) bonds, aided by cation–π interactions. The Collum group has explored the isomerization of alkenes and the metalation of dienes using NaDA in THF (Scheme 8).52 The reaction of NaDA with 1,4-dienes afforded the dienyl sodium product via metalation (38) while isomerization with alkene substrates demonstrated high selectivity for the Z-isomer, with the exception of 1-pentene (39). Treatment of allyloxy ethers with NaDA afforded enol ethers with good selectivity (>50:1 Z:E, 40a–d), while substituted allyl ethers underwent 1,4-elimination rather than isomerization (41a–b). In the case of allyloxytrimethylsilane, the addition of catalytic (6.5 mol%) NaDA resulted in fast conversion to the isomerized product 40b with 80% isolated yield. In most cases, isomerization utilizing dimeric [NaDA(THF)2]2 requires lower temperatures.
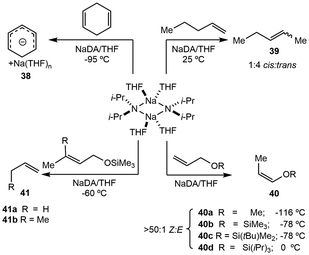 |
| | Scheme 8 Summary of NaDA reactivity with alkenes. | |
Super-basic NaTMP has been implemented for alkene isomerization, with the Hevia group exploring the use of catalytic NaTMP in conjunction with the tridentate PMDETA ligand to increase basicity.53,54 At 10 mol% loading for both NaTMP and PMDETA, the isomerization of a larger scope of alkenes was observed at room temperature (Fig. 4). The isomerization of allylsilanes yielded predominately E-isomer products in good yields at much lower temperatures and durations than comparable reactions using B(C6F5)3. Activated allylbenzenes and internal silyl-substitued olefins showed efficient isomerization to the internal alkene product (43a–43h). Longer and more substituted alkyl chains resulted in slightly decreased yields (43g–h), inclusion of both N- and O- in the allyl unit resulted in good conversion to the target enamines and vinyl ethers (43j–43n). The isomerization of halide-, carbonyl-, and nitrile-containing compounds was unsuccessful, though isomerization of some substituted alkynes and dienes were productive (43o–p). Longer chain α-olefins such as 1-octene lacked selectivity and provided a mixture of isomers (43q). When THF was used instead of hexane, rapid decomposition of the base prevented the isomerization reaction from occurring, in line with similar reports by Collum (see section 2.2.1). In C6D6, the incorporation of deuterium into cycloalkenes was observed in moderate to excellent yields rather than the isomerization exhibited in C6D12, demonstrating the high basicity of the mixed NaTMP/PMDETA system.
 |
| | Fig. 4 Substrate scope for the isomerization of alkenes using the catalytic NaTMP/PMDETA system. Grey circles indicate the initial position of the double bond. aIsolated yields, b50 °C, 4 h, cTMEDA (0.1 mL), 120 h, dTMEDA (10 mol%), 0 °C, 15 min, eTMEDA (0.1 mL), f80 °C, g96 h. | |
4.2. Olefination
Various types of olefinations are mediated by specialized reagents (e.g. Wittig reagent), often in conjunction with an alkali metal base, such as organolithium reagents for Julia olefinations and Shapiro reactions, or alkali metal hydrides and alkoxides for Horner–Wadsworth–Emmons olefinations and Peterson olefinations.55–57 In these types of olefinations, the identity of the alkali metal plays a role in the stability of the intermediate and the stereochemistry of the product, ultimately making it challenging to develop a universal system with common bases.58 Despite these challenges, there are contemporary reports of alkali metals complexes used for olefinations.59,60 Improved understanding of alkali metal coordination chemistry has led to purposeful design and synthetic control of well-defined systems for general olefinations, broadening the scope of reactivity with common substrates.
4.2.1. Sodium.
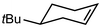
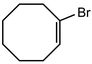
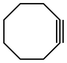
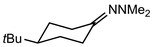
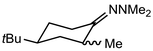
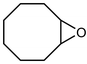
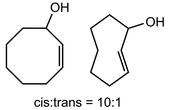
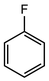
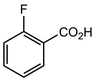
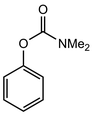
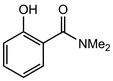
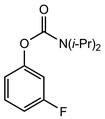
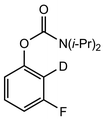
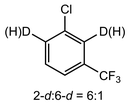
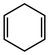
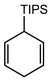
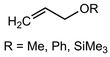
In their rigorous study of NaDA, Collum and coworkers studied the effect of solvent-ligands on sodium amides and competency for olefination.30 NaDA–DMEA reactions exhibited substantially faster krel values and an increased variety of transformations compared to that of LDA–THF. Dehalogenations (Table 2, entries 1–3) occur at a rate most similar to LDA–THF, with the exception of the equatorial conformation of entry 2, which is highly effective compared to the absence of equatorial elimination with LDA–THF. Entry 6 demonstrates a remarkable deviation from LDA-based reactivity, forming cyclooctenols with a 10:1 cis:trans selectivity without requiring additional heating or formation of side products. Facile ortho-metalations (entries 7–10) were observed without benzyne formation at low (less than −30 °C) temperatures. Metalation with subsequent silylation (entry 11) and ether rearrangement (entry 12) both proceed as expected and significantly faster than the analogous LDA reactions.
Table 2 Reaction of substrates using NaDA
| Entry |
Substrate |
Conditions |
E+ |
Product |
k
rel
|
Yield (%) |
| 1 |
nC8H17Br |
1.2 equiv. NaDA |
— |
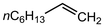
|
5 |
87 |
| 0 °C |
| 2 |
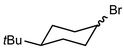
|
1.2 equiv. NaDA |
— |
|
>500 |
80 |
| 0 °C |
| 3 |
|
1.2 equiv. NaDA |
— |
|
5 |
87 |
| 0 °C |
| 4 |
|
1.1 equiv. NaDA |
TMSCl |
|
N/A |
61 |
| −78 °C |
| 5 |
|
1.2 equiv. NaDA |
CH3I |
|
>300 |
78 |
| −78 °C |
| 6 |
|
1.3 equiv. NaDA |
H2O |
|
>500 |
80 |
| rt |
| 7 |
|
1.1 equiv. NaDA |
CO2 |
|
N/A |
60 |
| −78 °C |
| 8 |
|
1.2 equiv. NaDA |
H2O |
|
>200 |
92 |
| −78 °C |
| 9 |
|
1.2 equiv. NaDA |
CH3OD |
|
>500 |
94 |
| −78 °C |
| 10 |
|
1.2 equiv. NaDA |
CH3OD |
|
1000 |
92 |
| −78 °C |
| 11 |
|
1.1 equiv. NaDA |
TIPSCl |
|
>100 |
82 |
| 0 °C |
| 12 |
|
1.1 equiv. NaDA |
— |
|
>1000 |
83 |
| 0 °C |
R = Ph |
More recently, the utility of NaDA–DMEA for olefinations was demonstrated by the Collum group through the addition of catalytic PMDETA (10 mol%) to NaDA (Table 3).61 PMDETA was compared to the bidentate TMEDA using both kinetic and computational studies which showed the inherent hemilability of the different ligand scaffolds was crucial for enabling reactivity. Olefin products in most cases were achieved in high yields (82–95%) and experienced rate acceleration upon the addition of PMDETA; however, for entries 16–18, PMDETA did not result in acceleration, with the authors predicting that the tridentate coordination prevents substrate addition to the sodium center.
Table 3 Reactivity of NaDA in DMEA and PMDETA using different substrates
Metalated benzylsilanes have demonstrated the effective in situ olefination of ketones, aldehydes and amides by well-defined lithium (44) and sodium (45) silylbenzyl complexes, affording trisubstituted alkenes in excellent yields.62 High conversion (>95%) of both bulky and simple carbonyls under mild conditions in C6D6 presents these alkali metal complexes as competitive alternatives in comparison to traditional olefination methods. Moderate E:Z selectivity was observed for entries 2, 7, 8, and 9 and suggests the potential use of chiral ligands for manipulating selectivity (Table 4).
Table 4 Olefination of carbonyls by alkali–metal silylbenzyl complexes
Despite the similarity between 44 and 45 for olefination, (Me6Tren)Li(CH2SiMe3) and (Me6Tren)Na(CH2SiMe3) (compounds 46 and 47, respectively) exhibit divergent reactivity for the olefination of ketones as highlighted by the Lu group.63 Treatment of ketones with 46 results in nucleophilic addition at the O-atom upon the addition of the ketone substrate; however, reaction with 47 proceeds through three possible pathways (Scheme 9). Methylenation of the carbonyl was the predominant transformation across 10 substrates, including benzophenone (>95%), electron deficient fluorinated ketones (>90%), and ketones that do not readily undergo enolization: dicyclohexyl ketone (∼70%), phenyl cyclohexyl ketone (>95%), phenyl tert-butyl ketone (>95%). Nucleophilic addition was only the major reaction path (>95%) when benzaldehyde was the substrate, but after three days, full conversion to the methylenated product was observed. Deprotonation to form the enolate was only the primary path when acetophenone was used (>95%). It was determined that catalytic amounts of the Me6Tren ligand (5 mol%) could be added in situ to stoichiometric equivalents of [Na(CH2SiMe3)]∞ and substrate, resulting in >95% conversion to the methylenated product.
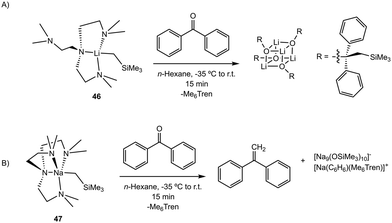 |
| | Scheme 9 A) Divergent reactivity of (Me6Tren)Li(CH2SiMe3) (46) with benzophenone, and (B) divergent reactivity of (Me6Tren)Na(CH2SiMe3) (47) with benzophenone. | |
4.2.2. Potassium.
Although potassium is commonly used as a base additive in olefinations, specifically as an alkoxide, Essman and Jacobsen report a potassium–isothiourea-boronate ion pair complex as an efficient chiral Lewis acid catalyst for enantioselective Wittig olefinations (Scheme 10).64 The system enables the asymmetric synthesis of axially chiral alkenes via olefination of 4-substituted cyclohexanones with non-stabilized phosphorus ylides, achieving high enantioselectivity (up to 92% ee). The reaction proceeds through a Lewis acidic mechanism involving a stepwise cycloaddition, where the potassium center coordinates to the macrocyclic amide-boronate framework (48), stabilizing the intermediate oxaphosphetane in turn facilitating enantioselective bond formation. Steric and electronic tuning of the arylpyrrolidine moiety on the potassium complex significantly impacts selectivity, with the 3-phenanthryl derivative affording the highest enantioselectivity. Upon comparison of the analogous Li, Na, and K complexes, the authors proposed that potassium's unique coordination to the framework, emulating an amide metal boronate instead of an amide metal isothiurea, makes 48 much more effective than its lighter congeners. Electron-deficient aromatic substrates enhanced reaction rates and selectivity, while sterically hindered ylides reduced enantioselectivity.
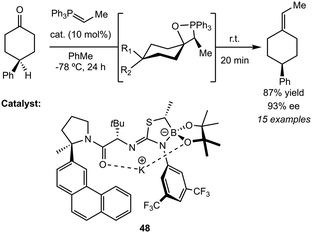 |
| | Scheme 10 Enantioselective Wittig olefinations by chiral potassium–isothiourea-boronate catalyst (48). | |
5. Alkylation, benzylic functionalization, and allylic activation
5.1. Alkylation
Alkali metal reagents, specifically organolithium reagents, have been widely utilized as alkylating reagents since their discovery due to their nucleophilic nature.2 However, the availability and commonplace usage of organolithium reagents have overshadowed the study of its heavier congeners (see section 3.2.1 for further discussion).
5.1.1. Sodium.
Sodium enolates, also known as Oppolzer enolates, are commonly used in organic syntheses as potent nucleophiles, which exhibit solvent-dependent structural conformation and reactivity. To generate the sodium enolate, the Collum group used solutions of either sodium isopropyl(trimethylsilyl)amide (NaPTA) or NaN(SiMe3)2 in a variety of solvents (Scheme 11A).65 The conformation of the sodium amide was explored through in situ kinetic studies. It was found that enolate and allyl bromide were first-order suggesting a monomer-based alkylation (50), with second-order dependence on hexamethylphosphoramide (HMPA) further inferring the formation of a complex solvated ion pair (49-HMPA, Scheme 11B). In the absence of HMPA, alkylations were performed in PMDETA and TMEDA without success, while THF resulted in a 100-fold decrease in rate. In the case of MeI, alkylation (51) occurred sufficiently fast to allow for the omission of HMPA (49-THF, Scheme 11C). Further alkylation rate studies suggest the reaction involves [Na(THF)6]+ as the cationic half of a hexa-solvated ion pair intermediate en route towards alkylation.
 |
| | Scheme 11 A) Comparison of NaDA and NaN(SiMe3)2 (B) alkylation in the presence of HMPA (C) alkylation using MeI in the absence of HMPA. | |
The investigation of NaDA continued with the synthesis of pseudoephedrine-derived (52) Myers enolates (54) by forming the disodium salt (53, Scheme 12).66 Poor yield and incomplete reactions were found to be associated with deleterious aggregation of NaDA and O-alkylations in the second step.
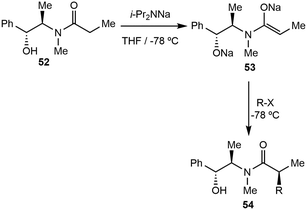 |
| | Scheme 12 Synthesis of Myers enolate from acylated pseudoephedrine (52) with NaDA. | |
5.2. Benzylic functionalization
5.2.1. Sodium.
The influence of solvent-ligands additionally dictates the aggregation of alkali metal alkyl complexes. The benzylic activation and aroylation of toluenes with Weinreb amides by Na(CH2SiMe3)∞ has been demonstrated by the Hevia group using PMDETA to deaggregate polymeric Na(CH2SiMe3)∞ for C(sp3)–H activation.67,68 The addition of stoichiometric toluene, Na(CH2SiMe3)∞, and PMDETA results in the formation of discrete isolated complex (PMDETA)Na(CH2Ph) (55). Subsequent addition of a Weinreb amide to 55 affords the aroylated toluene product 56a–y (Fig. 5). This approach gave reasonable yields for simpler alkyl and aryl substituted toluenes (75–95% yield), and methyl-substituted pyridines were similarly compatible (59–93%). Tolerance towards other functional groups varied widely (28–78%), for some bisaryl substrates, metalated intermediates were not basic enough to react with the Weinreb amides. The efficacy of (PMDETA)Na(CH2Ph) insertions into C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN, and CC bonds were additionally investigated and explored. Under a CO2 atmosphere, phenylacetic acid (91%) was obtained after aqueous workup. Addition to N-benzylideneaniline affords amine N-(1,2-diphenylethyl)aniline (88%).
O, CN, and CC bonds were additionally investigated and explored. Under a CO2 atmosphere, phenylacetic acid (91%) was obtained after aqueous workup. Addition to N-benzylideneaniline affords amine N-(1,2-diphenylethyl)aniline (88%).
 |
| | Fig. 5 Substrate scope for the benzylic aroylation of toluenes by NaCH2SiMe3. aSolvent system: benzene/hexane, bmixture of tautomers, cT = −78 °C, dmixture of products (single:double addition, 57%:9%), eT = 0 °C, finseparable mixture of isomers (2:1). | |
Invoking complementary nucleophilicity and basicity, the Hevia group employs benzylic metal intermediate (57) with stoichiometric Na(CH2SiMe3)∞ and PMDETA with 1.5 equivalents of toluene, inducing subsequent C–H addition to diarylethenes and ketones to form the desired coupled product (Fig. 6).69 Initial studies showed that addition of one equivalent of 1,1-diphenylethylene to 57, followed by hydrolysis, afforded the coupled toluene-ethylene product 58. The crystallographic data shows products from both the sodiation, PMDETA[Na(CH(NMe)2Ph)], and CC insertion steps prior to hydrolysis [{ArCH2CH2C(Ph)2}Na(PMDETA)]. The same approach was additionally applied for the deprotonative coupling of toluenes and aromatic ketones.
 |
| | Fig. 6 Substrate scope for the coupling of substituted toluenes and olefins by NaCH2SiMe3. aHexane/benzene 1:1 solvent system. bA 2:1 ratio of two diastereomers observed. | |
5.3. Allylic activation
Allylic activation and functionalization are typically only seen with transition metals, with palladium complexes being the state-of-the-art, due to the ability to access the necessary π-allyl complexes needed for activation.70,71
5.3.1. Sodium.
The Schneider group explored the C(sp3)–H activation of alkenes with NaN(SiMe3)2 for C–C bond formation.72 The addition of catalytic NaN(SiMe3)2 (10 mol%) to a terminal alkene and protected imine lead to the desired amine with good yields, selectivity, and tolerance to a variety of functionalities on the imine (Fig. 7). Mechanistic studies indicate that under the reaction conditions, the sodium amide forms a η3 Na-allyl intermediate in situ, akin to that observed with Pd π-allyl complexes, readily observed by 23Na NMR spectroscopy (−5.3 ppm). The observed η3 Na-allyl intermediate then forms the sodiated product (4.9 ppm) upon the addition of imine, which is considered catalytically active. It was found that both intermediates are essential for the catalysis. Both LiN(SiMe3)2 and KN(SiMe3)2 were also explored, resulting in lower conversions alongside isomerized starting material, indicating that the stability of the AM-allyl intermediate is necessary for the transformation.
 |
| | Fig. 7 Substrate scope for the coupling of terminal alkenes and imines by catalytic NaN(SiMe3)2.109 All yields are isolated yields after preparative thin layer chromatography. aUse of 3 equiv. of alkene. bThe reaction was conducted at 40 °C. cUse of 1.8 equiv. of alkene (successive addition). dUse of 2.5 equiv. of alkene (successive addition). eUse of 2 equiv. of alkene (successive addition). fThe reaction was conducted with 1.5 equiv. of alkene at 60 °C for 72 h. | |
6. Hydrogen isotope exchange (HIE) and transfer hydrogenation
6.1. HIE
Hydrogen Isotope Exchange (HIE) is the process of exchanging protons for a heavier isotope of hydrogen (2H/D or 3H/T), which is important in the pharmaceutical industry for drug design and development.73,74 Transition metal complexes have prevailed as typical HIE catalysts, although acid/base catalytic systems have also been explored.75 The cost effectiveness of the latter makes acid/base mediated HIE and the development of well-defined alkali metal catalysts an engaging and exciting area of research.
6.1.1. Sodium.
Taking advantage of the basicity of NaTMP, Tortajada and Hevia illustrate HIE via the perdeuteration of arenes. NaTMP, in combination with polyamine PMDETA and C6D6 as the deuterium source, showed catalytic deuteration of non-activated arenes (60) in C(sp2)–H and C(sp3)–H positions (Fig. 8).76 Under these conditions, simple arenes (60a–d) showed excellent deuterium incorporation, while alkylbenzenes showed a pronounced decrease in deuteration ortho- to the alkyl. Symmetric substrates like hexamethylbenzene (95%), ferrocene (94%), and diphenylacetylene (97%) showed equal deuteration at all positions. Substrates that were incompatible with this deuteration system fell into two classes: those with functional groups prone to decomposition from organosodium reagents, and those with more acidic C–H bonds (diphenylmethane and benzofuran) that rapidly form the metalated product. Further utility for HIE was demonstrated through the addition of ferrocene-d10 to C6H6, which afforded the 94% conversion to proteo-ferrocene with <5% deuteration.
 |
| | Fig. 8 Substrate scope for the perdeuteration of arenes by NaTMP. a48 h, b0.1 mmol scale, caverage deuteration reported due to overlapping signals in 1H NMR spectrum, d0.05 mmol scale. | |
The investigation into the structure–reactivity relationship for sodium amide reagents has led to progress in well-defined HIE reactions using sodium. This area of interest has encouraged the use of more sterically confined, super-basic, sodium amides such as NaTMP, NaNCy2, NaNAd2, and NaNAdTMS in tandem with TMEDA and PMDETA (Scheme 13).77 The synthesis of the sodium amides was performed via the addition of nBuNa to the appropriate amine in hexanes. Deuteration studies were conducted using anisole as a model compound, with 10 mol% sodium amide and 10 mol% of an amine donor in C6D6 over 16 hours. The degree of deuteration by NaNAdTMS was very low, with less than 5% of deuterium incorporation across all positions in anisole using both PMDETA and TMEDA. Both NaTMP and NaNCy2, using PMDETA, saw deuterium incorporation of greater than 95% at all positions. NaTMP and TMEDA showed slight preference for the meta-position (>95%) compared to the ortho- or para-positions (82%) alongside selective deuteration at the methoxy group (85%). NaNCy2 was significantly less effective at all three positions (meta-: <5%, ortho- or para-: 60%, methoxy: 24%).
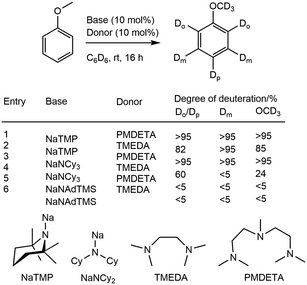 |
| | Scheme 13 Summary of deuterium incorporation with different base/donor systems. | |
Deuteration is not limited to extremely bulky amides, as the Hevia group demonstrated the potential for NaN(SiMe3)2 and NaCH2SiMe3 to selectively deuterate heterocycles, N-heterocyclic carbenes (NHCs), fluoroarenes, and other substrates using DMSO-d6 as the deuterium source (Scheme 14A).78,79 When using NaN(SiMe3)2, nitrogen-based heterocycles showed selective deuteration at the more acidic protons (up to 95% exchange in many substrates) and other heterocycles (furan, thiophene) showed selectivity for the α-proton. Characterization of catalytic intermediates utilizing 2-methylpyridine revealed formation of the C(sp3)–H activated product as a sodiated dimer (61), which was predisposed to HIE. Additional crystallographic studies reveal a separated ion pair 62 that showed increased deuteration than both the in situ conditions and exclusive NaN(SiMe3)2 (Scheme 14B). Methyl-substituted pyridines preferentially deuterated the C(sp3)–H bonds of the methyl with more than >95% exchange (Scheme 14C). Faster exchange activity was observed using NaN(SiMe3)2 with toluene (37% compared to 68% after 16 hours), which was then applied to several less-activated substrates, yielding deuteration up to 95% in some cases.
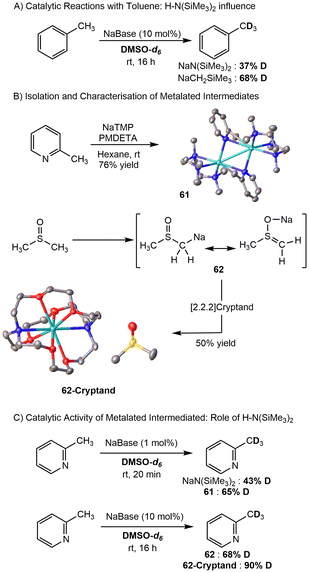 |
| | Scheme 14 Summary of approaches for HIE using various sodium bases in DMSO-d6. | |
6.1.2. Potassium, rubidium and cesium.
Potassium bases (–OR, –OH, –CO3) have been commonly used for HIE with D2O and DMSO-d6 as the deuterium source.80 However, any potassium base in DMSO forms what is reported as “superbase media” with no well-defined intermediate, but still demonstrates HIE capabilities.81
The Guan group showed that both RbN(SiMe3)2 (91% D enrichment, 98% yield) and CsN(SiMe3)2 (97% D enrichment, 98% yield) complexes can selectively deuterate benzylic C–H bonds using D2 gas at 10 mol% catalyst loading.82 The same was observed for several drug molecules, with tritiation using T2 gas performed on Tesmilifene, Imipramine, and Vortioxetine. The authors propose that AMN(SiMe3)2 (AM = Rb, Cs) reacts with benzylic substrates through σ-bond metathesis to form the benzylic metal intermediate, which is then reacted with D2 to afford the deuterated product. Subsequently, a metal hydride intermediate is formed that is protonated again to continue the cycle, although the authors state that the metal hydride intermediate can be accessed first to afford the same deuterated product.
Further extension towards the ortho-directed HIE of various aromatic ethers and fluorides was also explored.83 Comparatively, LiN(SiMe3)2 and NaN(SiMe3)2, along with other potassium bases exhibited no exchange, while KN(SiMe3)2 (60 mol%) showed 95% deuterium incorporation to 4-phenylanisole. However, the heavy alkali metals showed enhanced incorporation with CsN(SiMe3)2 (30 mol%) affording up to 97% deuterium incorporation (Scheme 15). Mechanistic experiments and DFT calculations reveal AMN(SiMe3)2 (AM = Na, K, Cs), in the dimeric form, coordinate three anisole molecules, in which one of the three is deprotonated by the metal, leaving an unstable carbanion intermediate that reacts with D2. The previously released HN(SiMe3)2 reacts with the dimer, allowing for regeneration of the catalyst.
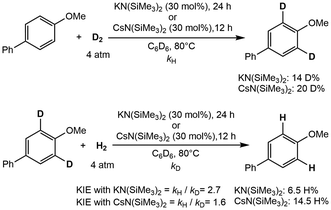 |
| | Scheme 15 KIE studies of KN(SiMe3)2 and CsN(SiMe3)2. | |
6.2. Transfer hydrogenation
Transfer hydrogenation is an alternative to direct hydrogenation, which removes the hazards of using hydrogen gas. There have been few reports of alkali metal bases (–OR, –OH) that have been shown to do transfer hydrogenation on carbonyls.84–87
6.2.1. Sodium.
Mulvey and coworkers focused on generating a “masked” sodium-hydride analogue via the dihydropyridylsodium compound (Na-1,2-tBu-DH(DMAP)) (64), and its monomeric variant, [Na-1,2-tBu-DH(DMAP)]Me6TREN for catalytic transfer hydrogenation (Scheme 16).88 Mechanistically, reduction of DPE was found to be facile from Na-1,2-tBu-DH(DMAP) to generate the sodiated diphenylmethane en route to the Meisenheimer intermediate. The reactivity of these sodium complexes proved useful in the reduction of alkenes, outperforming those of NaN(SiMe3)2, NaTMP, and NaH with similar mechanistic profiles.
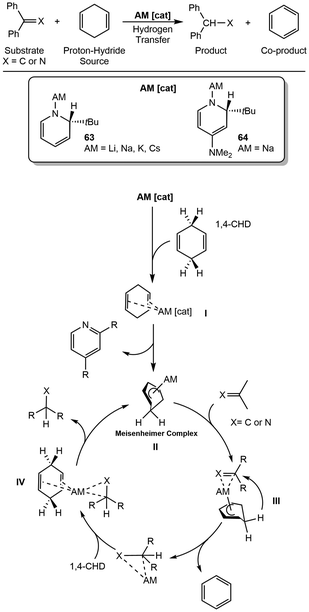 |
| | Scheme 16 Transfer hydrogenation of imines and alkenes using 1,4-cyclohexadiene (CHD) and the general mechanism undergoing a Meisenheimer-based intermediate. | |
6.2.2. Potassium, rubidium and cesium.
In follow-up studies, Mulvey and coworkers synthesized a series of monometallic alkali metal (AM = Li–Cs) dihydropyridines [AM(tBuDHP)] complexes (63) and explored their utility for transfer hydrogenation (Scheme 16).89,90 Catalytic competency for each metal complex was tested using 1,4-cyclohexadiene (1,4-CHD) as the hydride source and N-benzylideneaniline as an initial substrate. The heavier congeners (Rb and Cs) were found to be higher yielding (92 and 97%) with fast reaction times. Similarly, full conversion and high yields (91%) with Cs(tBuDHP) was reported upon changing the substrate to the more polar N-benzylidene-tert-butylamine, in stark contrast to Li(tBuDHP), which showed minimal conversion (15%). Mechanistic studies and DFT allude to the mechanism of transfer hydrogenation for Cs(tBuDHP) following a base mediated initiated pathway by generation of a π-complex I, followed by deprotonation to form the key Meisenheimer intermediate II. This intermediate then coordinates with the substrate for hydride transfer III, releasing benzene as a byproduct in the process. Upon coordination of an additional equivalent of the 1,4-CHD to the metal center IV, the hydrogenated product is then released, regenerating the active catalyst.
Mulvey and coworkers synthesized a series of bimetallic magnesium and alkali metal complexes for exploring the catalytic transfer hydrogenation of alkenes, utilizing 1,4-CHD as the hydrogen source.91 A series of alkali metal magnesiates are synthesized by reacting Mg(N(SiMe3)2)2 with AM(N(SiMe3)2) (AM = Li–Cs) to form AMMg(N(SiMe3)2)3 (65, Scheme 17). CsMg(N(SiMe3)2)3 shows the best conversion for the hydrogenation of styrene to ethylbenzene with >98% conversion with 5 or 10 mol% at 75 °C within 30 minutes. The proposed mechanism for transfer hydrogenation utilizing these bimetallic manifolds (65) begins by hydride formation from the sacrificial 1,4-CHD to form the AM–Mg bridging hydride intermediate 66. Intermediate 66 can then undergo insertion of styrene into the AM-stabilized Mg–H bond, forming 67, followed by protonation by free HN(SiMe3)2, product release, and regeneration of the active catalyst. Oligomerization was observed as a major side reaction that the authors further investigated by utilizing 1,1-diphenylethylene (DPE) as a substrate. The studies with DPE demonstrated that the potassium analogue was selective for the oligomerization of DPE, with full conversion in a shorter period than its larger congeners, Rb and Cs.
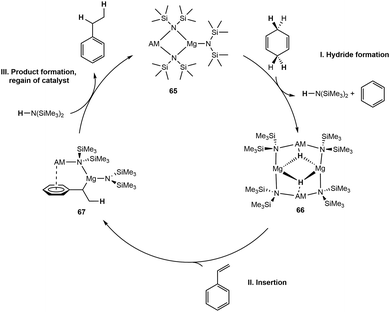 |
| | Scheme 17 Proposed catalytic cycle for the transfer hydrogenation of styrene. | |
7. Ring opening polymerization
Ring opening polymerization (ROP) is the main method in which to produce industrially relevant polymers from cyclic monomers, such as lactones, lactams, and epoxides, and is mediated by a wide array of organo- and organometallic catalysts. Alkali metal reagents have been incorporated in base-catalyzed ROP, but the use of well-defined alkali systems is relatively new.92 The ring opening polymerization of cyclic rac-lactide (rac-LA, 68) to form polylactic acid (PLA) is one of the most application-based polymerizations available to alkali metal systems. Extenstion to other cyclic lactones (69, 70) and strainless macrolactones (71, 72) has also been explored (Scheme 18). The usage of well-defined sodium and potassium metal precatalysts in this area over the last decade is summarized by ligand-supported classes.
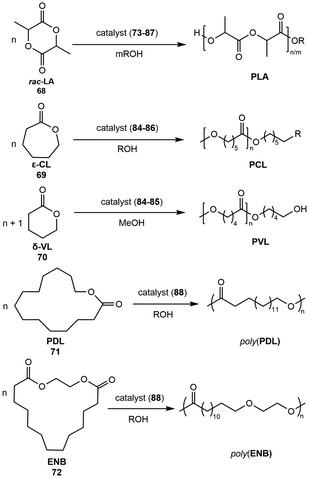 |
| | Scheme 18 General summary of ring-opening polymerizations discussed within. | |
7.1. Naphthalenolate catalysts
Crown ether-stabilized sodium and potassium catalysts were synthesized by the Wu group for the ROP of rac-lactide. In the case of the naphthalenolate complexes (73), the potassium complex showed higher activity than the sodium counterpart, achieving high isoselectivity (Pm = 0.73) and controlled molecular weights at room temperature, with rapid monomer conversion under optimized conditions (Fig. 9A).93
 |
| | Fig. 9 (A) Naphthalenolate-supported catalysts, (B) phenoxide-supported catalysts, (C) iminophenoxide-supported catalysts, (D) quinoline-8-olate-supported catalysts, and (E) binolate-supported catalysts, (F) potassium phenolate catalysts, (G) aminobisphenolate ion-pair catalysts, (H) TrenSal-supported catalysts, (I) phosphoselenoic amide-supported catalysts, (J) potassium N-arylbenzimidate catalysts, (K) amidinate–potassium separated ion-pair catalysts, (L) 15-C-5 supported sodium phenolate catalysts. | |
7.2. Phenoxide catalysts
Using phenol ligands treated with different crown ethers and AMN(SiMe3)2 (AM = Na and K, 74) allowed access to a series of precatalysts.94 The polymerization of rac-lactide using these catalysts proceeds rapidly at room temperature, achieving high conversion rates (90–97%) within one minute. Potassium-based systems demonstrated superior performance, with high isoselectivity (Pm = 0.89, −60 °C) indicating the efficacy of these alkali–metal catalysts, achieving the formation of multiblock isotactic copolymers with an average isotactic length of nine lactic acid units (Fig. 9B). Single crystal X-ray diffraction studies and experimental results demonstrate the impact steric hindrance and electronic effects have on this transformation, wherein it was determined that larger crown ethers and increased steric hindrance near the active center enhanced isoselectivity, while electron-donating groups diminished it.
Similarly, sodium and potassium iminophenoxide complexes supported by crown ethers have demonstrated successful ROP of rac-lactide (Fig. 9C). For both metals, Wang and coworkers observed that the presence of ortho-tert-butyl groups on the phenoxy-moiety of the ligand hindered the rate of ROP.95 The potassium complex 75d produced polylactides with the highest isoselectivity (Pm = 0.75); however, the sodium complex 75a proved to be the most active catalyst. Similar potassium quinolin-8-olate complexes (76) were investigated by the same group (Fig. 9D).96 Unlike 75d, the 76a–e exhibit O,N-chelation, which restricts rotation of the O–K bond, thereby influencing selectivity.
7.3. Binolate catalysts
Moving away from O,N-chelating scaffolds, Pan and coworkers demonstrated the activity of two enantiopure potassium binolate complexes (77) for the isoselective ROP of rac-lactide using (R)-2,2′-dihydroxy-1,1′-dinaphthyl (R-BINOL) ligands (Fig. 9E).97 The presence of intramolecular hydrogen bonding in 77a reduces catalytic activity but enhances stereoselectivity (Pm = 0.80 at room temperature). Although sodium complexes often show high activity for ROP, transesterification can occur as a side reaction; providing additional stabilization at the metal center reduces this deleterious reactivity. Lin and coworkers utilized NNO-tridentate Schiff base ligands with a pendant amino arm to minimize transesterification while also retaining a high catalytic activity at the sodium center for the ROP of L-lactide.98
7.4. Aminobisphenolate catalysts
Wu and coworkers have also investigated the application and synthesis of bench-stable potassium complexes with multidentate phenolate ligands for the ROP of rac-lactide (78a–c, Fig. 9F).99 Isoselectivity improved significantly at low temperatures due to the suppression of epimerization side reactions, reaching a maximum value of Pm = 0.83 at −70 °C. To increase ligand denticity, the tetradentate aminobisphenolate ligand was employed by Wu and coworkers to stabilize sodium and potassium metal centers.100 Benzyl alcohol coordinates to 79 as both a Lewis donor at the sodium and through hydrogen bonding with the ligand O-atoms (Fig. 9G). This coordination does not occur in the case of potassium, which instead forms dimeric 80. The best molecular weight control and stereoselectivity was observed after 6 hours at −70 °C using sodium catalyst 79 (Pm = 0.82, 87% conversion, 0.05 mol% catalyst).
7.5. Salen-based catalysts
In an attempt to balance activity with polymerization control, the Garden group has implemented the salen-derived TrenSal ligand to access three sodium-based catalysts for the ROP of rac-lactide (Fig. 9H).101 Of the three catalysts, 81 was the least active but demonstrated the best control (77% conversion in 40 min, Đ = 1.25–1.49). Slightly more active (76% conversion in 10 min), 82 provided improved control (Đ = 1.75); however, late-stage transesterification led to dispersity broadening. The improved activity of precatalyst 83 (83% conversion in 4 min) is attributed to the increased number of metal centers, but resulted in poor control over polymerization (Đ = 2.00). In addition to the activated monomer pathway demonstrated by 81 and 82, MALDI-ToF end group analysis of PLA generated by 83 is consistent with that produced by a coordination–insertion mechanism, suggesting that 83 proceeds via two separate mechanisms, resulting in poor control. Tetradentate amino-phenolate potassium complexes were applied to the ROP of rac-lactide by Ma and coworkers, resulting in high catalytic activity but very little stereoselectivity. Solid state structures were obtained by SC-XRD as dimeric potassium complexes, while DOSY and variable temperature NMR were used to determine monomeric complexes existed in solution.102
7.6. Phosphoselenoic amide catalysts
Anionic ROP provides the benefit of improved control. Panda and coworkers applied phosphinoselenoic amide alkali metal complexes (84, 85) toward the ROP of rac-lactide, ε-caprolactone, and δ-valerolactone (Fig. 9I).103 The potassium catalysts displayed the highest activity with 85c, featuring a nitro-functionalized ligand, achieving the highest isoselectivity (Pm = 0.78) and converting 1000 equivalents of rac-LA to PLA within 30 minutes at room temperature. The steric and electronic properties of the ligands significantly influence the reaction, with nitro-substituted ligands enhancing catalytic activity and stereocontrol.
7.7. Arylbenzimidate and amidinate catalysts
Potassium N-arylbenzimidates demonstrate efficient anionic ROP in the presence of benzyl alcohol for ε-caprolactone and L-lactide.104 Structural characterization revealed diverse coordination geometries across the eight complexes; some uniquely formed two-dimensional polymeric assemblies (86a–h, Fig. 9J), stabilized by π-aryl and σ-donor interactions. For ε-caprolactone, conversions exceeded 95% under optimized conditions (0.4 mol% catalyst, 50 °C, 30 minutes), affording polycaprolactones with controlled molecular weights and narrow dispersity. Electron-donating groups improved catalysis, while electron-withdrawing groups hindered it. The polymerization of L-lactide showed even greater efficiency, achieving nearly 100% conversion at optimized conditions (0.2 mol% catalyst loading, 60 °C, 30 minutes).
Wu and colleagues report the isoselective anionic ROP of rac-lactide using ion-paired potassium amidinate complexes (Fig. 9K).105 Three potassium amidinate pairs (87a–c) were accessed, with differing degrees of steric hindrance present at the amidinate side arm. Single crystal X-ray diffraction studies were utilized to confirm the formation of separated ion-pair complexes, wherein the potassium is not bound to the amidinate. Reduced temperature (−30 °C or −70 °C) was found to improve polymer conversion for all three catalysts, which in addition to the presence of BnOH as an initiator, minimized unfavorable side reactions. Under optimized conditions, 87b showed 92% conversion and low dispersity (Đ = 1.05).
7.8. Phenolate catalysts
Compared to lactide monomers, which can easily undergo ROP, macrolactones are generally harder to open due to reduced ring strain. During the ROP process, transesterification rates are similar to those of chain propagation rates. Liu and coworkers report the ROP of macrolactones (71, 72) using sterically hindered phenoxide sodium complexes with 15-crown-5 (88a–d) wherein steric repulsion between the ligand and polymer chain inhibits transesterification, allowing for polymerization to proceed in a controlled manner (Fig. 9L).106
8. Conclusions and perspectives
From the initial study of organolithiums in the 1930′s, organoalkali chemistry has evolved significantly in the context of organic transformations. Simple organoalkali reagents have been extensively studied using modern and updated techniques, such as multinuclear NMR spectroscopy and single crystal X-ray diffraction, allowing these reagents to be used more effectively in a variety of applications. Taken together, these recent advances illustrate the dynamic role of AMs in modern organic chemistry, which are now central to strategies spanning ligand design, small-molecule activation, asymmetric catalysis, and flow chemistry. A recent example is the design of a chiral phenanthroline-potassium catalyst that enables the enantioselective ring opening alcoholysis of biaryl lactams to produce axially chiral δ-amino acid derivatives.107,108 Additionally, Hilt and coworkers have demonstrated that organosodium intermediates can also be generated and used under continuous-flow conditions for the synthesis of alkyl aryl ketones from Weinreb amides.109 As these studies demonstrate, the reactivity of alkali metals is set to inspire even broader applications in organic chemistry.
Conflicts of interest
There are no conflicts to declare.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Acknowledgements
This work was supported by the Cancer Prevention and Research Institute of Texas (Grant No. RR220055).
References
-
J. Clayden, Organolithiums: selectivity for synthesis, Elsevier, 2002, vol. 23 Search PubMed.
-
Z. Rappoport and I. Marek, The chemistry of organolithium compounds, John Wiley & Sons, 2004 Search PubMed.
-
B. J. Wakefield, The Chemistry of Organolithium Compounds, Pergamon, 2013 Search PubMed.
- V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204–14210 Search PubMed.
- T. L. Rathman and J. A. Schwindeman, Org. Process Res. Dev., 2014, 18, 1192–1210 CrossRef CAS.
- J. García-Álvarez, E. Hevia and V. Capriati, Eur. J. Org. Chem., 2015, 6779–6799 Search PubMed.
- U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194–204 Search PubMed.
- K. Inoue and K. Okano, Asian J. Org. Chem., 2020, 9, 1548–1561 Search PubMed.
- M. Power, E. Alcock and G. P. McGlacken, Org. Process Res. Dev., 2020, 24, 1814–1838 Search PubMed.
- A. Nagaki, Y. Ashikari, M. Takumi and T. Tamaki, Chem. Lett., 2021, 50, 485–492 Search PubMed.
- Z. Ye, J.-B. Liao and L. Gong, Chem. Lett., 2024, 53, upae103 Search PubMed.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247–9262 Search PubMed.
-
P. Christmann, E. Gloaguen, J.-F. Labbé, J. Melleton and P. Piantone, in Lithium Process Chemistry, ed. A. Chagnes and J. Światowska, Elsevier, Amsterdam, 2015, pp. 1–40 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 Search PubMed.
- O. C. Gagné and F. C. Hawthorne, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 602–625 Search PubMed.
- A. Stokłosa, J. Zajęcki and S. S. Kurek, Mater. Sci.-Pol., 2004, 22, 35–45 Search PubMed.
-
R. F. Koby and T. P. Hanusa, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr., Elsevier, Oxford, 2021, pp. 2–48 Search PubMed.
- P. Burk and S. Tamp, J. Mol. Struct.: THEOCHEM, 2003, 638, 119–128 CrossRef CAS.
- L. L. Tomasevich and D. B. Collum, J. Am. Chem. Soc., 2014, 136, 9710–9718 CrossRef CAS PubMed.
- R. F. Algera, Y. Ma and D. B. Collum, J. Am. Chem. Soc., 2017, 139, 7921–7930 Search PubMed.
- R. A. Woltornist and D. B. Collum, J. Org. Chem., 2021, 86, 2406–2422 Search PubMed.
- J. A. Spivey and D. B. Collum, J. Am. Chem. Soc., 2024, 146, 17827–17837 Search PubMed.
- R. Sreedharan and T. Gandhi, Chem. – Eur. J., 2024, 30, e202400435 Search PubMed.
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2024, 63, e202313556 Search PubMed.
- R. Rabie, M. M. Hammouda and K. M. Elattar, Res. Chem. Intermed., 2017, 43, 1979–2015 Search PubMed.
- S. Biswas, W. B. Hughes, L. D. Angelis, G. C. Haug, R. Trevino, S. O. Fremin, H. D. Arman, O. V. Larionov and M. P. Doyle, Chem. Sci., 2024, 15, 5277–5283 Search PubMed.
- A. Sudalai, A. Khenkin and R. Neumann, Org. Biomol. Chem., 2015, 13, 4374–4394 RSC.
- S. Asako, H. Nakajima and K. Takai, Nat. Catal., 2019, 2, 297–303 Search PubMed.
- Y. Ma, R. F. Algera and D. B. Collum, J. Org. Chem., 2016, 81, 11312–11315 Search PubMed.
- L. J. Bole, A. Tortajada and E. Hevia, Angew. Chem., 2022, 134, e202204262 Search PubMed.
- L. J. Bole, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2022, 61, e202204262 CrossRef CAS PubMed.
- Y. Ma, R. F. Algera, R. A. Woltornist and D. B. Collum, J. Org. Chem., 2019, 84, 10860–10869 CrossRef CAS PubMed.
- Z. Zhang and D. B. Collum, J. Am. Chem. Soc., 2019, 141, 388–401 CrossRef CAS PubMed.
- T. Inoue, S. Yamamoto, Y. Sakagami, M. Horie, K. Okano and A. Mori, Organometallics, 2021, 40, 3506–3510 CrossRef CAS.
- Q. You and D. B. Collum, J. Am. Chem. Soc., 2023, 145, 23568–23584 Search PubMed.
- Q. You, Y. Ma, R. A. Woltornist, N. M. Lui, J. A. Spivey, I. Keresztes and D. B. Collum, J. Am. Chem. Soc., 2024, 146, 30397–30421 Search PubMed.
- Y. Yamashita, A. Noguchi, S. Fushimi, M. Hatanaka and S. Kobayashi, J. Am. Chem. Soc., 2021, 143, 5598–5604 Search PubMed.
- X. Sun and D. B. Collum, J. Am. Chem. Soc., 2000, 122, 2459–2463 CrossRef CAS.
- K. A. Mack, A. McClory, H. Zhang, F. Gosselin and D. B. Collum, J. Am. Chem. Soc., 2017, 139, 12182–12189 Search PubMed.
- G. J. Reyes-Rodríguez, R. F. Algera and D. B. Collum, J. Am. Chem. Soc., 2017, 139, 1233–1244 Search PubMed.
- D. B. Collum, Synthesis, 2025, 3158–3178 Search PubMed.
- R. A. Woltornist and D. B. Collum, J. Am. Chem. Soc., 2021, 143, 17452–17464 CrossRef CAS PubMed.
- A. Kummari, S. Pappuru, S. S. Roy and D. Chakraborty, Polym. Chem., 2022, 13, 4684–4691 RSC.
- S. Bonollo, D. Lanari and L. Vaccaro, Eur. J. Org. Chem., 2011, 2587–2598 Search PubMed.
- S. Kaneko and S. Shirakawa, ACS Sustainable Chem. Eng., 2017, 5, 2836–2840 Search PubMed.
- V. Butera and H. Detz, ACS Omega, 2020, 5, 18064–18072 Search PubMed.
- C. K. Patel, S. Banerjee, K. Kant, R. Sengupta, N. Aljaar and C. C. Malakar, Asian J. Org. Chem., 2023, 12, e202300311 CrossRef CAS.
- M. Hassam, A. Taher, G. E. Arnott, I. R. Green and W. A. L. Van Otterlo, Chem. Rev., 2015, 115, 5462–5569 Search PubMed.
- S. R. Macaulay, J. Org. Chem., 1980, 45, 734–735 Search PubMed.
- S. R. Abrams, D. D. Nucciarone and W. F. Steck, Can. J. Chem., 1983, 61, 1073–1076 CrossRef CAS.
- R. F. Algera, Y. Ma and D. B. Collum, J. Am. Chem. Soc., 2017, 139, 11544–11549 Search PubMed.
- A. Tortajada, G. L. Righetti, A. McGinley, M. Mu, M. García-Melchor and E. Hevia, Angew. Chem., Int. Ed., 2024, 63, e202407262 Search PubMed.
- A. Tortajada, G. L. Righetti, A. McGinley, M. Mu, M. García-Melchor and E. Hevia, Angew. Chem., 2024, 136, e202407262 CrossRef.
- V. N. Korotchenko, V. G. Nenajdenko, E. S. Balenkova and A. V. Shastin, Russ. Chem. Rev., 2004, 73, 957–989 CrossRef CAS.
-
Y. Gu and S.-K. Tian, in Stereoselective Alkene Synthesis, ed. J. Wang, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, vol. 327, pp. 197–238 Search PubMed.
- D. J. Peterson, J. Org. Chem., 1968, 33, 780–784 Search PubMed.
- D. L. Hooper, S. Garagan and M. M. Kayser, J. Org. Chem., 1994, 59, 1126–1128 Search PubMed.
- K. Izod, L. J. Bowman, C. Wills, W. Clegg and R. W. Harrington, Dalton Trans., 2009, 3340–3347 Search PubMed.
- K. A. Ouzounthanasis, S. R. Rizos and A. E. Koumbis, Eur. J. Org. Chem., 2023, e202300626 Search PubMed.
- Y. Ma, R. A. Woltornist, R. F. Algera and D. B. Collum, J. Am. Chem. Soc., 2021, 143, 13370–13381 CrossRef CAS PubMed.
- J. Barker, N. Davison, P. G. Waddell and E. Lu, Chem. Commun., 2023, 59, 8083–8086 RSC.
- N. Davison, C. L. McMullin, L. Zhang, S.-X. Hu, P. G. Waddell, C. Wills, C. Dixon and E. Lu, J. Am. Chem. Soc., 2023, 145, 6562–6576 Search PubMed.
- J. Z. Essman and E. N. Jacobsen, J. Am. Chem. Soc., 2024, 146, 7165–7172 CrossRef CAS PubMed.
- N. M. Lui and D. B. Collum, Org. Chem. Front., 2023, 10, 4750–4757 Search PubMed.
- Y. Zhou, I. Keresztes, S. N. MacMillan and D. B. Collum, J. Am. Chem. Soc., 2019, 141, 16865–16876 CrossRef CAS PubMed.
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202218498 CrossRef CAS PubMed.
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., 2023, 135, e202218498 CrossRef.
- D. E. Anderson, A. H. N. Truong and E. Hevia, Chem. – Eur. J., 2024, 30, e202400492 Search PubMed.
- F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 5863–5883 CrossRef CAS.
- R. Wang, Y. Luan and M. Ye, Chin. J. Chem., 2019, 37, 720–743 CrossRef CAS.
- W. Bao, H. Kossen and U. Schneider, J. Am. Chem. Soc., 2017, 139, 4362–4365 CrossRef CAS PubMed.
- Q.-K. Kang and H. Shi, Synlett, 2022, 329–338 Search PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- A. Tortajada and E. Hevia, J. Am. Chem. Soc., 2022, 144, 20237–20242 CrossRef CAS PubMed.
- M. S. Tschopp, A. W. J. Platten, E. Hevia and A. Tortajada, Eur. J. Inorg. Chem., 2024, 27, e202400200 CrossRef CAS.
- M. S. Tschopp, A. Tortajada and E. Hevia, Angew. Chem., 2025, 137, e202421736 Search PubMed.
- M. S. Tschopp, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2025, 64, e202421736 CrossRef CAS PubMed.
- A. Tortajada and E. Hevia, Catal. Sci. Technol., 2023, 13, 4919–4925 Search PubMed.
- Y. Yuan, I. Thomé, S. H. Kim, D. Chen, A. Beyer, J. Bonnamour, E. Zuidema, S. Chang and C. Bolm, Adv. Synth. Catal., 2010, 352, 2892–2898 CrossRef CAS.
- H.-Z. Du, J.-Z. Fan, Z.-Z. Wang, N. A. Strotman, H. Yang and B.-T. Guan, Angew. Chem., Int. Ed., 2023, 62, e202214461 Search PubMed.
- H.-Z. Du, J. Li, S. Christodoulou, S.-Y. Li, Y.-S. Cui, J. Wu, S. Ren, L. Maron, Z.-J. Shi and B.-T. Guan, ACS Catal., 2024, 14, 9640–9647 Search PubMed.
- R. Radhakrishan, D. M. Do, S. Jaenicke, Y. Sasson and G.-K. Chuah, ACS Catal., 2011, 1, 1631–1636 CrossRef CAS.
- A. Ouali, J.-P. Majoral, A.-M. Caminade and M. Taillefer, ChemCatChem, 2009, 1, 504–509 CrossRef CAS.
- I. D. Alshakova, H. C. Foy, T. Dudding and G. I. Nikonov, Chem. – Eur. J., 2019, 25, 11734–11744 CrossRef CAS PubMed.
- V. Polshettiwar and R. S. Varma, Green Chem., 2009, 11, 1313–1316 RSC.
- P. A. Macdonald, A. R. Kennedy, C. E. Weetman, S. D. Robertson and R. E. Mulvey, Commun. Chem., 2024, 7, 1–7 Search PubMed.
- P. A. Macdonald, S. Banerjee, A. R. Kennedy, A. van Teijlingen, S. D. Robertson, T. Tuttle and R. E. Mulvey, Angew. Chem., 2023, 135, e202304966 CrossRef.
- P. A. Macdonald, S. Banerjee, A. R. Kennedy, A. van Teijlingen, S. D. Robertson, T. Tuttle and R. E. Mulvey, Angew. Chem., Int. Ed., 2023, 62, e202304966 Search PubMed.
- T. X. Gentner, A. R. Kennedy, E. Hevia and R. E. Mulvey, ChemCatChem, 2021, 13, 2371–2378 Search PubMed.
- E. Fazekas, P. A. Lowy, M. A. Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2022, 51, 8793–8814 RSC.
- J. Xiong, J. Zhang, Y. Sun, Z. Dai, X. Pan and J. Wu, Inorg. Chem., 2015, 54, 1737–1743 CrossRef CAS PubMed.
- Z. Dai, Y. Sun, J. Xiong, X. Pan, N. Tang and J. Wu, Catal. Sci. Technol., 2016, 6, 515–520 RSC.
- B.-B. Wu, L.-L. Tian and Z.-X. Wang, RSC Adv., 2017, 7, 24055–24063 Search PubMed.
- B.-B. Wu and Z.-X. Wang, RSC Adv., 2017, 7, 11657–11664 RSC.
- Y. Cui, C. Chen, Y. Sun, J. Wu and X. Pan, Inorg. Chem. Front., 2017, 4, 261–269 Search PubMed.
- H.-W. Ou, K.-H. Lo, W.-T. Du, W.-Y. Lu, W.-J. Chuang, B.-H. Huang, H.-Y. Chen and C.-C. Lin, Inorg. Chem., 2016, 55, 1423–1432 CrossRef CAS PubMed.
- F. Ren, X. Li, J. Xian, X. Han, L. Cao, X. Pan and J. Wu, J. Polym. Sci., 2022, 60, 2847–2854 CrossRef CAS.
- X. Li, Z. Jia, X. Pan and J. Wu, Chem. – Asian J., 2019, 14, 662–669 CrossRef CAS PubMed.
- Y. Zhou, G. S. Nichol and J. A. Garden, Eur. J. Inorg. Chem., 2022, e202200134 CrossRef CAS.
- C. Yao, Y. Yang, S. Xu and H. Ma, Dalton Trans., 2017, 46, 6087–6097 RSC.
- A. Harinath, J. Bhattacharjee, A. Sarkar and T. K. Panda, New J. Chem., 2019, 43, 8882–8891 RSC.
- J. Gao, W. Zhang, F. Cao, G. A. Solan, X. Zhang, Y. Jiang, X. Hao and W.-H. Sun, Mol. Catal., 2020, 498, 111280 Search PubMed.
- C. Chen, J. Jiang, X. Mao, Y. Cong, Y. Cui, X. Pan and J. Wu, Inorg. Chem., 2018, 57, 3158–3168 CrossRef CAS PubMed.
- X. Wang, X. Wang, N. Zhen, J. Gu, H. Zhang, B. Dong, F. Wang and H. Liu, Polym. Chem., 2021, 12, 1957–1966 RSC.
- S. Cen, S. Li, Y. Zhao, M. Zhao and Z. Zhang, Angew. Chem., 2024, 136, e202407920 Search PubMed.
- S. Cen, S.-S. Li, Y. Zhao, M.-X. Zhao and Z. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202407920 Search PubMed.
- P. Knupe-Wolfgang, B. Mahn and G. Hilt, Org. Lett., 2024, 26, 6972–6976 CrossRef CAS PubMed.
Footnote |
| † Joint authorship. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Dinora N.
Rodriguez†
,
Dinora N.
Rodriguez†





















































































































































