
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quinic acid as a chiron in total synthesis†
Manuel J.
Verganista
a,
Iago C.
Vogel
a and
Nuno R.
Candeias
 *ab
*ab
aLAQV-REQUIMTE, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: ncandeias@ua.pt
bFaculty of Engineering and Natural Sciences, Tampere University, Korkeakoulunkatu 8, 33101 Tampere, Finland
First published on 3rd March 2026
Abstract
Covering: 1980 up to 2025.
Quinic acid became a versatile, accessible chiral building block for asymmetric synthesis, offering a functionalized, stereochemically rich scaffold that enables the creation of many natural products and bioactive compounds. This review covers synthetic strategies from the 1980s to 2025, highlighting quinic acid's ongoing importance in organic synthesis. It discusses its role in synthesizing carbocyclic frameworks, vitamin D analogues, carbasugars, cyclitols, aminocyclitols, lactones, alkaloids, and macrocyclic fragments. The review summarizes four decades of progress and emerging trends that reinforce quinic acid's status as a key chiral building block for complex molecule synthesis.
1. Introduction
(−)-Quinic acid is the most naturally occurring stereoisomer of quinic acid and is systematically named as (1R,3R,4S,5R)-1,3,4,5-tetrahydroxycyclohexane-1-carboxylic acid. The D/L symbology developed by Fischer, in which the descriptor is based on its relation to a reference carbon with one of the D-(+)-glyceraldehyde, has resulted in the widespread use of “D-quinic acid” to describe such a levorotatory substance. This nomenclature issue was covered in multiple articles,1–3 and the natural levoratory isomer (−)-quinic acid was recently identified as L.2 Nevertheless, the natural isomer is frequently described in the recent literature and by chemical suppliers as D-quinic acid. Fig. 1 shows the IUPAC numbering system for cyclitols,4 where the lowest numbered carbon is assigned to the substituent other than an unmodified hydroxyl group. Additionally, numbers 3 and 5 follow Maquenne's fractional system.5 | ||
| Fig. 1 Structure and numbering of (–)-quinic acid. | ||
The molecular structure of quinic acid features four stereocenters, three of them being contiguous, and with four hydroxyl groups arranged on a cyclohexane ring. This dense stereochemistry provided synthetic chemists with a ready-made, enantiopure platform for constructing complex molecules, particularly those requiring strict stereochemical control, allowing avoidance of challenging asymmetric synthesis steps. Its fixed configuration allows for regio- and stereoselective modifications using established protection, oxidation, reduction, elimination, and substitution protocols. Its value as a chiral starting material was soon recognized by the synthetic community, as demonstrated by several reviews on the use of quinic acid as a starting material for the synthesis of natural products.6–9
Despite the above-mentioned existence of different literature reviews demonstrating the synthetic value of quinic acid, namely the 1998 report by Barco and co-workers6 and Enev's reviews,8,9 there has not been a compilation on the topic since 2012. The present literature review aims to comprehensively cover all the uses of quinic acid as a starting point for total or formal synthesis of natural products and some selected related compounds, namely vitamin D derivatives. To facilitate readability, and in recognition of the impact that introducing acyl or other substituents to quinic acid's hydroxyl groups may have on the IUPAC systematic nomenclature—particularly the carbon skeleton numbering—the numbering system of unsubstituted quinic acid will be adopted throughout this manuscript. The manuscript is organized according to the type of natural product synthesised.
Quinic acid (1) is a secondary metabolite of the shikimate pathway (Scheme 1).10,11 This metabolic pathway is responsible for the biosynthesis of aromatic amino acids in plants and microorganisms. Although not a direct precursor in the main shikimate pathway, quinic acid can be reconverted into 3-dehydroquinate in microorganisms,12 serving as an alternative carbon source and establishing regulatory links with the biosynthesis of aromatic compounds. The shikimic acid pathway initiates with the condensation of phosphoenolpyruvate (3, PEP) and erythrose-4-phosphate (4, E4P), both derived from the metabolism of glucose (2) or other carbohydrates. This aldol-type reaction is catalyzed by DAHP synthase, an enzyme subject to feedback inhibition by the three aromatic amino acids ultimately produced through this pathway.
 | ||
| Scheme 1 Biosynthetic pathway for the production of quinic acid (shikimate pathway). Abbreviations: PEP, phosphoenolpyruvate; E4P, erythrose-4-phosphate; DAHP, 3-deoxy-D-arabino-heptulosomate-7-phosphate; DHQ, 3-dehydroquinic acid. | ||
The product, 3-deoxy-D-arabino-heptulosonate 7-phosphate (5, DAHP), undergoes a complex transformation into 3-dehydroquinic acid (6). This conversion involves a sequence of oxidation, β-elimination of phosphate, reduction, ring opening, and intramolecular aldol condensation—all orchestrated by a single enzyme, 3-dehydroquinase synthase. 3-Dehydroquinic acid (6, DHQ) can either reversibly convert into quinic acid (the biosynthetic quinate pathway) or proceed towards shikimic acid (8) formation (shikimate pathway) via dehydration and reduction, catalyzed by DHQ dehydratase and shikimate dehydrogenase, respectively. Shikimic acid then undergoes a series of transformations into chorismic acid, ultimately taken into the synthesis of the aromatic amino acids.
Quinic acid was first isolated as an impurity in crude quinine in 1790,13 but its structure and stereochemistry were fully assigned only in 1932, by Fischer and Dangschat14 when isolating 3-O-caffeoylquinic acid from green coffee beans. Structural elucidation was further complemented by Haslam and Turner in 1971, with NMR studies15 and by Allen and co-workers in 1988 with a crystal structure.3
2. Synthesis of carbacyles
Carbacycles, which are ring systems made exclusively of carbon atoms, are fundamental in organic chemistry and natural product synthesis. Quinic acid, featuring a cyclohexane ring heavily substituted with hydroxyl groups and a carboxyl group, offers a valuable starting point for such frameworks. This chapter discusses approaches that utilize quinic acid as a chiral template for carbacycle synthesis, highlighting the synthesis of carbacycles with biological relevance.Usami and coworkers conducted extensive research on the total synthesis of pericosines (A to E) from quinic acid. Pericosines (Scheme 2) are natural products isolated from fungus Periconia byssoides, found in the gastrointestinal tract of Aplysia kurodai sea hare, and display in vitro antitumor activity.16,17
 | ||
| Scheme 2 Structures of some naturally occurring pericosines. | ||
Usami's research on the total synthesis of (+)-pericosine A resulted in a revision of its original stereochemical assignment, as well as clarifying the structures of other pericosines due to spectral discrepancies between the synthetic sample and the naturally isolated molecule.16,18 The group designed another synthetic strategy to obtain the de facto (+)-pericosine A from quinic acid (Scheme 3a), as well as two other diastereomers.19,20 The synthetic strategy relied first on the oxidation followed by reduction, aiming at the inversion of the configuration of C5 and regioselective TFAA-mediated dehydration; however, the large number of protection steps highly impacted the overall yield. Later, Usami and his team devised a more efficient route with fewer protection steps, thus significantly increasing the overall yield of the natural product from quinic acid from 0.57% to 11.7% (Scheme 3b).21
 | ||
| Scheme 3 (a) Total synthesis of (+)-pericosine from quinic acid; (b) improved total synthesis of (+)-pericosine A. | ||
While initially thought that only (+)-pericosine B existed in nature, it was later discovered that both pericosines B and C exist as enantiomeric mixtures when isolated from Periconia byssoides.22 The synthesis of (−)-pericosine B (Scheme 4) was accomplished within seven steps from cyclohexylidene-protected methyl quinate 21, where mCPBA demonstrated low conversion and regioselective epoxidation of 22. The acid-catalysed opening of the epoxide 23 was accomplished with moderate yield. As the final significant step, the stereocenter at C5 was inverted with oxidation of 24 with DMP, followed by reduction with NaBH4.23,24
 | ||
| Scheme 4 Total synthesis of (−)-pericosine B. | ||
Both enantiomers of pericosine C were synthesized from quinic acid-derived epoxide 26, where, after cyclohexylidene ketal deprotection, the acid- and base-catalysed epoxide opening yielded (+) and (−)-pericosine C, respectively (Scheme 5).21
 | ||
| Scheme 5 Total synthesis of (+) and (−)-pericosine C. | ||
The synthesis of the diastereomers of pericosine A mentioned above proved useful in the confirmation of the relative configuration of pericosine D.25 Although the synthetic strategy was not optimized, it provided enough material to compare to the spectroscopical data (aside from specific rotation) of (−)-pericosine D (Scheme 6).26
 | ||
| Scheme 6 Total synthesis of pericosine D. | ||
Chlorohydrin 29 was prepared from QA, which, following its reaction with an epoxide derived from shikimic acid, permitted the synthesis of (+)-pericosine E – the only O-linked carbohydrate identified in pericosines (Scheme 7).27 The formation of the ether bond was achieved in 52% by using BF3·Et2O as a catalyst (10 mol%), whilst the oxidation-reduction sequence resulted in a more modest yield of 34%.
 | ||
| Scheme 7 Synthesis of (+)-pericosine E. | ||
Shing and colleagues have reported the total synthesis of cyclophellitol, initially isolated from the culture filtrates of the mushroom Phenellinus sp. and known as a potent inhibitor of β-D-glucosidase.28,29 The main steps for the synthesis of cyclophellitol involved regiospecific sulphate opening, regiospecific oxidative elimination, and epoxidation in 15 steps from quinic acid (Scheme 8). In the same work, the synthesis of the (1R,6S) and (1R,2S,6S) diastereomers was also reported, similarly achieved in 15 steps from quinic acid. Later, Shing and coworkers prepared 6-deoxy-1,2-anhydro analogs of cyclophetilol and evaluated their anti-glycosidase activity.30
 | ||
| Scheme 8 Total synthesis of cyclophellitol. | ||
(+)-Crotepoxide and (+)-boesenoxide are naturally occurring cyclohexene oxides found in the fruits of Croton macrostachys and Piper futokadzura.31,32 (+)-β-Senepoxide and (+)-pipoxide, have been isolated from Piper hookeri and Uvaria catocarpa, respectively.33 These cyclohexene oxides and related analogues display significant biological properties, such as tumour inhibition, anti-leukemic effects, and antibiotic activity. They were synthesized from quinic acid, primarily through a strategy where the main steps of the synthetic strategy involved Corey–Winter olefination with thiocarbonyldiimizadole (TCDI) and trimethyl phosphite, singlet photooxygenation mediated by tetraphenylporphyrin (TPP) and endoperoxide rearrangement facilitated by cobalt-TPP (Scheme 9).33,34
 | ||
| Scheme 9 (a) Synthesis of (+)-crotepoxide, (−)-iso-crotepoxide and (+)-boesenoxide; (b) synthesis of (+)-β-senepoxide, (+)-pipoxide acetate, senepoxide and (−)-tingtanoxide. | ||
The mycosporin-like amino acids represent a structurally unique class of metabolites produced by various aquatic organisms, making them very attractive to the cosmetic industry, particularly as UV blockers.35 Quinic acid was used as a chiral scaffold for the synthesis of Mycosporin-Gly and Mycosporin-l, where the key steps were the Staudinger reaction followed by Aza–Wittig reaction (Scheme 10).36,37
 | ||
| Scheme 10 Synthesis of Mycosporin-Gly and Mycosporin-l. | ||
Barros and coworkers presented the synthesis of a metabolite isolated from the culture medium of the fungus Eutypa lata, (+)-eutypoxide B. The key step was the epoxidation of TBDMS-enone (55), where the bulkiness of the silyl-protecting group and the oxidizing agent were crucial for achieving good diastereoselectivity at the epoxide intermediate (Scheme 11a).38 Additionally, reports from the same group described the total synthesis of (+)-epoformin (Scheme 11b), an antibiotic derived from the culture broth of Penicillium claviforme. The key step, again, was the epoxidation, and in this case, using a less bulky oxidizer (H2O2) has been shown to yield better diasteroselectivity.39
 | ||
| Scheme 11 Natural products synthesized by Barros et al. (a) total synthesis of (+)-eutypoxide B; (b) total synthesis of epoformin. | ||
The gabosines are secondary metabolites isolated from various strains of Streptomyces bacteria. Quinic acid was used in the enantioselective synthesis of gabosines A and B (Scheme 12) and the synthesis of enantiomers of gabosines D and E. The key steps involved a Mislow–Evans rearrangement to reach the exo-methylene alcohol 65 and a DBU-assisted epimerization in the synthesis of gabosine B.40
 | ||
| Scheme 12 Total synthesis of gabosines A and B. | ||
3-Deoxy-D-glycero-D-galacto-2-nonulosonic acid (Kdn) is a member of the sialic acid family, which plays a crucial role in mediating and regulating a wide range of physiological and pathological processes.41 Banwell and co-workers synthesized (−)-Kdn and two of its epimers from quinic acid, where the key step was the photoisomerization of the unsaturated aldehyde 71 to its Z-isomer. The enal 72 was submitted to Luche reduction, OsO4-catalysed dihydroxylation and, after removal of the protecting group, (−)-Kdn was obtained as a single diastereomer (Scheme 13).42
 | ||
| Scheme 13 Total synthesis of (−)-Kdn. | ||
Holmstedt et al. reported the use of quinic acid for the total synthesis of a metabolite43 isolated from the African ant Crematogaster nigriceps.44 The main step in this process was the base-promoted silyl migration, where three types of migration could be obtained and optimized by varying the silyl group, base, and solvent. Two other key steps were the Barton–McCombie deoxygenation and the epoxide opening with oleyl lithium (Scheme 14).
 | ||
| Scheme 14 Total synthesis of a metabolite isolated from the African ant Crematogaster nigriceps. | ||
(−)-Zeylanone, a natural product isolated from Uvaria grandiflora,45 demonstrated to be a potent antitumor agent, suppressing myelogenous leukemia, cervical carcinoma, prostate and gastric cancer.46,47 Motivated by the need to clarify the structure-activity relationship of zeylanones, Sun and co-workers developed a synthesis of the (+)-enantiomer in 13 steps from quinic acid with a 10% overall yield. The main step was the Sharpless asymmetric dihydroxylation (Scheme 15).48
 | ||
| Scheme 15 Total synthesis of (+)-zeylenone. | ||
Pseudohygrophorones A12 and B12 were derived from the fungus Hygrophorus abieticola, and showed notable activity against the plant pathogenic fungi Septoria tritici and Botrytis cinerea, as well as the oomycete Phytophthora infestans.49 A total synthesis of those compounds, starting from quinic acid, had as key steps the diastereoselective 1,2-addition of dodecylmagnesium bromide to a quinic acid-derived cyclohexanone 85, producing a single diastereomer of the tertiary alcohol. The use of mCPBA afforded good chemoselectivity toward Rubottom oxidation over epoxidation of the less activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (Scheme 16). The removal of the acetonide with FeCl3 resulted in the formation of the epimeric forms of pseudohygrophorones A12 and B12.50
C bond (Scheme 16). The removal of the acetonide with FeCl3 resulted in the formation of the epimeric forms of pseudohygrophorones A12 and B12.50
 | ||
| Scheme 16 Total synthesis of (−)-pseudohygrophorones A12 and B12. | ||
Barco et al. reported the formal synthesis of (−)-ovalicin,51 a sesquiterpene isolated from the culture of the fungus Pseudorotium ovalis Stolk.52 The main steps in this synthetic sequence for the formation of synthetic intermediate 95 were the debromination of 90 with Bu3SnH and the formation of the epoxide by intramolecular Williamson ether synthesis (Scheme 17).51 The epoxide 95 had been previously converted into (−)-ovalicin by Barton and co-workers in three additional steps (oxidation, Shapiro reaction with the corresponding vinyl lithium, and Sharpless epoxidation).53
 | ||
| Scheme 17 Formal synthesis of (−)-ovallicin. | ||
Guignardones A, B, and C were first isolated by Tan et al. from Guignardia mangiferae cultures, and they possess a characteristic 6-oxabicyclo[3.2.1]octane framework.54 There are at least 12 types of guignardones with a wide range of bioactivities, but guignardone B stood out among the others by having the most potent inhibition of Candida albicans, with an MIC of 15.5 µg mL−1.55 QA was used as a chiral scaffold for the synthesis of guignardones A and B, in 19 and 20 steps, respectively. The main stages of this synthetic route involved constructing the 6-oxabicyclo[3.2.1] octane core and performing the Knoevenagel condensation-6π-electrocyclization with the unsaturated aldehyde. Dehydration of guignardone A with Burgess reagent yielded guignardone B in 73% yield (Scheme 18).56
 | ||
| Scheme 18 Total syntheses of guignardones A and B. | ||
Stagonosporynes can be isolated from Parastagonospora nodorum fungus, being the major pathogen of wheat. The synthesis of stagonosporyne G was recently achieved from quinic acid-derived enone 54 in 8 steps, with the key steps being carefully studied, namely the conjugate addition of lithium TMS-acetylene to the enone and the Rubottom oxidation with TMSOTf and mCPBA (Scheme 19).57
 | ||
| Scheme 19 Total synthesis of stagonosporyne G. | ||
3. Synthesis of vitamin D and derivatives
Vitamin D, in its active form, calcitriol, broadly affects calcium metabolism and immune regulation. Novel analogs can be engineered to selectively target specific pathways, to improve certain biological functions while reducing side effects. This is especially vital for diseases like osteoporosis, rickets, and chronic kidney disease, where vitamin D is essential for maintaining balance in calcium and phosphate levels.58 The reader is directed to other reviews on the synthesis of 19-nor type vitamin D derivatives, in which QA takes a role other than the one covered in the present article.59,60DeLuca and his team have significantly contributed to the synthesis of new vitamin D analogs since the early 1990s. Quinic acid has been extensively utilized as a chiral scaffold for constructing the ring A of vitamin D. In their initial research, quinic acid was transformed into di-TBS cyclohexanone 112 by deoxygenating the hydroxyl groups on C1 and C4 through oxidative cleavage using NaIO4 and Barton–McCombie conditions, respectively (Scheme 20). Cyclohexene phosphine oxide 113 was synthesized through a laborious yet effective method, which, under Wittig–Horner conditions, followed by TBDMS deprotection, yielded 1α,25-dihydroxy-19-nor-vitamin D3 114.61 The same coupling was used for the synthesis of both pure diastereomers of triol 1α,2,25-trihydroxy-19-norvitamin D3. The diastereomers were subjected to the same transformations after separation by HPLC, which was followed by the Peterson reaction with methyl (trimethylsilyl)acetate (Scheme 21).62
 | ||
| Scheme 20 Synthesis of 1α,25-dihydroxy-19-nor-vitamin D3. | ||
 | ||
| Scheme 21 Synthesis of 1α,2β,25-trihydroxy-19-norvitamin D3. | ||
An alternative method for modifying QA during the preparation of 1α,25-dihydroxy-19-nor-vitamin D3 was reported by Vandewalle and co-workers (Scheme 22).63 The silyl-protected lactone 120 was submitted to Barton–McCombie deoxygenation of both exposed hydroxyl groups to provide 121. The lactone ring was opened by methanolysis, and the hydroxy group was transformed into brosylate, followed by an intramolecular alkylation of the ester-enolate formed by treatment with tBuOK. The carboxylic ester 122 was converted into the corresponding aldehyde, to follow a Seyferth–Gilbert homologation to the terminal alkyne 123. Coupling of the two fragments, followed by reduction and solvolysis of the obtained allylic alcohol under acidic conditions, afforded the 1α,25-dihydroxy-19-nor-vitamin D3 124. The bulkiness from the ring derived from quinic acid was found to be crucial to obtain the desired trans-configuration.
 | ||
| Scheme 22 Vandewalle synthesis of 19-nor-1α,25-dihydroxy-vitamin D3. | ||
The preparation of norvitamin D3 derivatives containing 2-hydroxymethyl, 2-methyl, and 2-methylene substituents was achieved starting with the synthesis of the QA-derived ketone 125, and its conversion into 2-methylene unit 127 (Scheme 23). The coupling of the QA and the 9,10-seco steroid skeletons was achieved through the same Peterson olefination of the cyclohexanone derivative, followed by installation of the phosphine oxide moiety and Wittig–Horner reaction. The 2-methylene-substituted analogue 129 was hydrogenated or hydroborated with 9-borabicyclo[3.3.1]nonane (BBN) to afford the 2-methyl or 2-hydroxymethyl moieties, respectively.64 The same synthetic pathway was used for the introduction of other substituents at position 2, such as 3′-alkoxypropylidene,65 3′-alkoxypropylidene,66 pegylated alkylidene chains67 and the formation of an additional dihydropyran68,69 and dihydrofuran70 rings. The 9,10-seco steroid moiety has also been modified in its alkyl side chain, to have the hydroxyl group removed or replaced by a methyl substituent,71 to include an alkyne72 or ether73 moieties, and in the synthesis of the (22Z)-isomer of paricalcitol.74
 | ||
| Scheme 23 Synthesis of 2-methylene-19-norvitamin D3. | ||
A series of 2-methylene-substituted vitamin D derivatives has been produced by the same research group (Scheme 24). The introduction of the additional exomethylene group was achieved by dehydration of the tertiary hydroxyl in 131 using Martin's sulfurane, followed by 1,3-dipolar cycloaddition of diazomethane to yield a single diastereomer of the diazole 132, which was submitted to thermolytic nitrogen extrusion, forming the tetrasubstituted cyclic olefin derivative 133 with 34% of the cyclopropyl isomer. The carboxylic ester was converted to the aldehyde functionality by a full reduction-oxidation sequence, and then the carbonyl functionality reacted with the anion of trimethylsilyldiazomethane to provide the dienyne 134. Coupling of the two carbon skeletons was achieved by Sonogashira reaction with the corresponding vinyl triflates.75–77
 | ||
| Scheme 24 Synthesis of 2-methylene analogues of calcitriol. | ||
In their quest for the preparation of locked analogues of 6-s-cis conformer of 1α, 25-dihydroxyprevitamin D3, Gotor and co-workers78–80 have prepared the four stereoisomers of the QA-derived enyne, namely 139, 141, 143, 145 (Scheme 25). Those were coupled with vinyl triflates in the same way as later explored by DeLuca.75–77 The preparation of such isomers was based on the inversion of the hydroxyl stereocenters through Mitsunobu reactions, whilst the installation of the alkyne moiety followed the abovementioned reaction of the aldehyde functionality with the lithiated anion of trimethylsilyldiazomethane.
 | ||
| Scheme 25 Preparation of QA-derived enyne stereoisomers. | ||
The synthesis of a similar type of 2-modified 1α, 25-dihydroxy-19-norvitamin D3 derivatives has been explored by Kittaka and co-workers. Faced with failures in using the Wittig–Horner reaction for the coupling of the QA-derived cyclohexane moiety and the 9,10-seco steroid skeletons, the authors have developed an alternative route using the Julia olefination reaction (Scheme 26). The introduction of the alkyl moiety in the C4 position of QA was achieved through allylation of thioimidazolide 146. The QA derivative was then reduced to the diol to follow C–C oxidative cleavage and reduction of the olefin to yield 148a, or the olefin was first hydroborated, then reduced and cleaved with periodate. Different cyclohexanones 148 were then reacted with sulfone 149 using LiHMDS to provide a mixture of 19-norvitamin D3 epimers at C2.81–85 The same olefination procedure was extended to the preparation of 25-hydroxy-19-norvitamin D3 (Scheme 27),86 in which cyclic cis-carbonothioate was heated in P(OMe)3 to provide the cyclohexene derivative 152. The synthesis of 1,3-cis-25-dihydroxy-19-norvitamin D3, having a similar Julia olefination step, was also reported.87
 | ||
| Scheme 26 Synthesis of 2-substituted-19-norvitamin D3 through Julia olefination. | ||
 | ||
| Scheme 27 Synthesis of 25-hydroxy-19-norvitamin D3. | ||
Another approach for the installation of the methylene substituent was the one reported by Reddy and co-workers (Scheme 28). Ketone 112 was alkylated in the α-position through an aldol condensation with formaldehyde, followed by elimination of the primary mesylate. The enone was converted into its enol triflate and coupled with a stannylacetylene through Stille coupling. The enyne 156 was then coupled with the triflate in similar conditions to the above-mentioned Sonogashira coupling.88
 | ||
| Scheme 28 α-Methylenation of QA-derived cyclohexanone. | ||
4. Synthesis of cyclitols, aminocyclitols and carbasugars
Cyclitols, a class of cyclic polyols, are widely distributed in nature and play crucial roles in biological and chemical systems. Aminocyclitols are a subclass of cyclitols, where the cyclohexane ring includes at least one amine moiety.89,90 Considering the high number of hydroxyls and the cyclic nature of quinic acid, it is not surprising that it has been widely explored as a starting material for the synthesis of cyclitols and aminocyclitols.A practical synthesis of positional stereoisomers of valiolamine 161 and 166 and their corresponding polyhydroxy γ-amino acids 163 and 167 has been obtained from quinic acid. The main steps include the stereocontrolled synthesis of the epoxides 159 and 164 and respective azidolysis, which afforded the azido-alcohols 160 and 165 (Scheme 29).91 The opening of the lactone 164 was tested using methanol or methoxide as nucleophiles, resulting in the formation of the methyl ester in low yields, likely due to relactonization.
 | ||
| Scheme 29 Synthesis of aminocyclitols 161 and 166 and their corresponding γ-amino acids 163 and 167. | ||
Two glycosidase inhibitors, 1,4,5-trideoxy-1,5-imino-D-ribo-hexitol 170 and (+)-proto-quercitol 172, were synthesized from enones 168. Luche reduction of the enone provided a mixture of the corresponding allylic alcohols 168a and 168b in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.4 ratio (Scheme 30). The piperidine ring was formed via the dihydroxylation of cyclohexene, followed by oxidative cleavage with NaIO4, ending with reduction steps. The primary step in constructing the quercitol ring was the dihydroxylation by KMnO4, with some complications arising in the previous steps due to aromatization of the synthetic intermediates at high temperatures.92
:1.4 ratio (Scheme 30). The piperidine ring was formed via the dihydroxylation of cyclohexene, followed by oxidative cleavage with NaIO4, ending with reduction steps. The primary step in constructing the quercitol ring was the dihydroxylation by KMnO4, with some complications arising in the previous steps due to aromatization of the synthetic intermediates at high temperatures.92
 | ||
| Scheme 30 Synthesis of trihydroxypiperidine 170 and (+)-proto-quercitol 172. | ||
Murugan et al. reported efficient syntheses of carba-α-L-rhamnose 176, (−)-gala177, and (+)-proto-quercitols 172 from shikimic acid derivative 173 (Scheme 31). The unsaturated methyl ester was converted to the MOM-protected cyclitol 174 and, through a Corey–Winter protocol for installation of exocyclic olefin followed by hydrogenation and deprotection, yielded 176. The ketone 175 was obtained via NaIO4-oxidative cleavage of 174. Cyclitols 177 and 172 were produced through the diastereoselective reduction of the ketone with K-Selectride and NaBH4, respectively.93
 | ||
| Scheme 31 Preparation of carba-α-L-rhamnose 176, (−)-gala177, (+)-proto-quercitols 172. | ||
Before the synthesis of carba-α-L-rhamnose, some of its derivatives have been prepared from the deacetylated analogue 178 (Scheme 32). The key steps for synthesizing 181 and 182 involved the highly stereoselective hydroboration of 179 (c.a. 97%), prepared upon reduction of the alkyl halide formed by the Appel reaction, and the regioselective benzylation facilitated by Bu2SnO in the last step.94
 | ||
| Scheme 32 Preparation of regioisomers of protected carba-α-L-rhamnose. | ||
Following a similar approach for the synthesis of allyl alcohols 183, Shih and co-workers reported the preparation of (+)-vibo, and (−)-talo, muco- and (+)-epi-quercitols, through epoxidation, followed by acetolysis and deprotection (Scheme 33). Starting from allyl alcohol 188, in a similar approach, allowed the synthesis of (−)-gala-quercitol.95 (+)-Gala-quercitol 189 was later prepared by the same authors through syn-dihydroxylation of allylic alcohol 183b.96
 | ||
| Scheme 33 Synthesis of various quercitols from quinic acid-derived allylic alcohols. | ||
A stereospecific synthesis of neo and (−)-epi-quercitols was also reported by Shih et al. (Scheme 34), upon deacetylation of 196 and 197, respectively. When reacted with KMnO4, allylic alcohol 190 and allyl acetate 191 gave significantly different diastereomeric ratios, which were attributed to the influence of the hydroxyl group in directing the dihydroxylation through intermolecular hydrogen bonding in the case of 190.96 Non-natural quercitol derivatives, namely deoxyfluoro quercitols, have been prepared from quinic acid using DAST-mediated fluorination of allylic alcohols.97
 | ||
| Scheme 34 Synthesis of neo and (−)-epi-quercitols. | ||
Gero and co-workers have reported the synthesis of dideoxy fortamine analogue 200 (Scheme 35).98 The ditosylation analogue of diol 199 was reported to undergo selective replacement of the tosylate at C5 under carefully controlled conditions with sodium azide in DMF. Treatment with sodium methoxide resulted in the formation of epoxide 137, upon benzoylation of the free hydroxy group, which underwent opening by a second azide moiety to provide the diamine dihydrochloride 200 after hydrogenation.
 | ||
| Scheme 35 Synthesis of a fortamine analogue 200. | ||
D-myo-Inositol-1,4,5-trisphosphate 206 was synthesized straightforwardly from quinic acid in 12 steps, through intermediate allylic alcohol 201 (Scheme 36). The additional hydroxyls were introduced through the stereoselective [2,3]-sigmatropic rearrangement of the allylic selenoxide 202 and hydroboration of the silyl-ether 204.99,100
 | ||
| Scheme 36 Synthesis of D-myo-inositol-1,4,5-trisphosphate 206. | ||
Under the same work, other inositol phosphates were prepared (Scheme 37).100 Starting with shikimic acid derivative 207, the additional oxygens necessary for the inositol core were introduced by Mislow–Evans rearrangement of 210 and hydroboration of the enol-silyl ether of 212. The protected triphosphate 214 was the key intermediate to obtain the inositol phosphates 215–217 with different substitution patterns and degrees of phosphorylation. Of note, the introduction of a 1,2-di-O-stearoyl-glycerophosphate moiety in 214, followed by hydrogenolysis, allowed the formation of 217. A distinct protection strategy to selectively modify the inositol core, allowing for regioselective phosphorylation, was further explored.101,102
 | ||
| Scheme 37 Synthesis of inositol phosphates with different substitution patterns and degrees of phosphorylation. | ||
The cyclitol 6-O-(2-amino-2-deoxy-α-D-glucopyranosyl)-D-chiro-inositol-1-phosphate 224, an insulin mimetic, was prepared from enone 218 in 15 steps (Scheme 38).103 The additional hydroxyl groups were added through stereoselective silylation of 218 with TBDMSOTf and OsO4-mediated dihydroxylation of 220. The disaccharide core was assembled by glycosylation with a sugar imidate, achieving good yield. A lengthy but effective deprotection route, along with phosphorylation between the sequences, led to 224.
 | ||
| Scheme 38 Synthesis of D-chiro-inositol. | ||
Penta-N,O,O,O,O-acetyl-(+)-validamine 228 and penta-N,O,O,O,O-acetyl-(+)-2-epi-validamine 229 were prepared from the sulfonate 225, prepared from quinic acid, in 9 steps (Scheme 39). The azido alcohol was submitted to a two-step sequence of activation of the hydroxy group, followed by SN2-type displacement of the triflate with tetrabutylammonium acetate in THF. Deacetylation by methanolysis to provide 227, followed by azide reduction, debenzylation, and acetylation, provided the fully acetylated validamine 228. As for the synthesis of the epi-analogue, the same azide reduction, debenzylation, and acetylation sequence was applied to azido alcohol 226.104
 | ||
| Scheme 39 Synthesis of pentaacetyl (+)-validamine 228 and pentaacetyl-(+)-2-epi-validamine 229. | ||
Valiolamine, along with three of its diastereomers, was synthesized by Shing and Wan (Scheme 40). Key steps in this process included azidolysis and OsO4-catalyzed oxidations, whilst inversion of hydroxy-containing stereocenters was achieved through a sequence of activation with triflic anhydride followed by reaction with tetrabutylammonium acetate and methanolysis.105,106 The same authors later reported the enantiospecific synthesis of valienamine using two different approaches. The first approach explored the regio and stereospecific ring opening of cyclic sulfite 237 with azide anion and also allowed the synthesis of 2-epi-valienamine (Scheme 41).107 The second approach used a regio- and stereospecific palladium-catalysed allylic amination (Scheme 42), after the regioselective dehydration of 241.108 Specifically, Martin's sulfurane in refluxing benzene provided the allyl acetate through elimination of the more acidic ring methylene proton over the exocyclic methylene proton. The allyl acetate underwent amination with benzylamine in acetonitrile to afford 242 upon methanolysis, whilst the use of THF hindered the desired palladium-catalyzed amination of the allyl acetate. Full deprotection of the allyl amine provided the cyclitol in 11% overall yield and 20 steps from quinic acid.
 | ||
| Scheme 40 Synthesis of valiolamine 161 and three of its diastereomers. | ||
 | ||
| Scheme 41 Synthesis of pentaacetylvalienamine 241 and its 2-epimer 239. | ||
 | ||
| Scheme 42 Synthesis of valienamine. | ||
An enantiospecific synthesis of 1,1′-bis-valienamine 250 and a first synthesis of 1,1′-bis-2-epi-valienamine 249 were achieved in 14 and 15 steps from QA with overall yields of 12% and 24%, respectively (Scheme 43). Both syntheses involved the stereospecific Pd-catalysed coupling reaction between an allylamine and the corresponding allyl chloride as the key steps, using tris(2,4,6-trimethoxyphenyl)phosphine (TMPP) as the ligand.109
 | ||
| Scheme 43 Synthesis of 1,1′-bis-valienamine 250. | ||
Shih et al. detail the efficient synthesis of four aminocyclitols from quinic acid (Scheme 44).110 A significant aspect of their method is the highly regioselective ring opening of epoxides with NaN3, which allows for precise functionalization of the cyclitol framework. Interestingly, the opening of epoxides 251 and 252 by azide was regioselective, whilst the opening of epoxide 255 provided epimeric tetraols 256 and 257 upon deprotection. The latter was rationalized to be formed upon nucleophilic attack by the hydroxyl at C5 to form a new epoxide, opened by the azide nucleophilic attack at C4. This approach offers a streamlined pathway to producing structurally diverse aminocyclitols.
 | ||
| Scheme 44 Synthesis of aminocyclitols 253, 254, 256, and 257. | ||
4.1 Synthesis of carbasugars
Pseudosugars, also known as carbasugars, are carbohydrate analogs that have garnered significant interest from medicinal chemists due to their potential as active pharmaceutical ingredients. These compounds exhibit structural resemblance to carbohydrates but contain a methylene group rather than a ring oxygen. Their structural similarity to conventional monosaccharides suggests they can bind to and inhibit the same protein targets.111 A key advantage of carbasugar-containing compounds is their resistance to enzymatic degradation, as they lack a glycosidic linkage, which results in enhanced stability. Quinic acid provides a privileged skeleton and richness in stereocenters for synthesizing these compounds.112 The first example of the synthesis of carbasugars from (−)-quinic acid was reported by Shin et al., on its conversion into pseudo-β-D-mannopyranose 259 and pseudo-β-D-fructopyranose 262 (Scheme 45). The 4-epi-3-dehydro-shikimate 207 served as a precursor for the preparation of both polyols.111,113 | ||
| Scheme 45 Synthesis of pseudo-β-D-mannopyranose and pseudo-β-D-fructopyranose. | ||
Later,114,115 the same authors adopted a similar strategy for the synthesis of pseudo-α-D-glucopyranose 265 and pseudo-α-D-mannopyranose 266 (Scheme 46), through dibenzylated intermediate 263. Removal of the acetal followed by Corey–Winter deoxygenation resulted in the formation of the cyclohexene derivative 264 that could then be transformed into both pyranoses through cis or trans dihydroxylations.
 | ||
| Scheme 46 Synthesis of pseudo-α-D-glucopyranose and pseudo-α-D-mannopyranose. | ||
Carballido et al. presented the synthesis of carbasugars, outlining a strategy that incorporates selective functionalization and ring modifications to convert quinic acid into carbocyclic sugar analogs. The primary step involved OsO4-catalyzed dihydroxylation of the unsaturated quinic acid derivative 269, and in this work, 15 different carbasugars were prepared. The face selectivity for the dihydroxylation of 269 and analogs was controlled by the use of different co-oxidants, with NaIO4 and NMO favoring the formation of Si-hydroxylation products. In the absence of the lactone ring, the sole diastereomer 274 was obtained independently of the reaction conditions (Scheme 47).116
 | ||
| Scheme 47 Synthesis of (1S, 2R, 3R, 4S, 5R)-(270) and (1S, 2S, 3S, 4S, 5R)-1,2,3,4,5-pentahydroxy-1-hydroxymethylcyclohexane (272) and their corresponding carboxylic acids (271 and 273). | ||
Shan et al. reported the synthesis of various cyclitol sugar phosphates (Scheme 48).117 Hydroxyenone 277 was prepared from enone 54, where the critical steps included the Simmons–Smith cyclopropanation and diastereospecific ring opening of cyclopropanol under Pd/C hydrogenation conditions to obtain the corresponding α-methyl ketone 276. The one-pot hydrosilylation of 54, followed by Simmons–Smith cyclopropanation, resulted in the formation of a separable mixture of cyclopropanes 275, in a ratio that varied from 1:1 (at 0 °C) to 2:1 (at −20 °C). Deprotection of cyclopropanol 275a, followed by hydrogenolysis, resulted in opening of the cyclopropane ring to provide ketone 276 that was dehydrated to the enone 277, serving as a key intermediate to prepare 2,3-dideoxy-4-oxo-5a-carba-α-D-rhamnopyranose phosphate 280, 5a-carba-α-D-rhamnopyranose phosphate 281, and 2,3-dideoxy-5a-carba-α-D-rhamnopyranose phosphate 282.
 | ||
| Scheme 48 Synthesis of carbasugar phosphates 280, 281 and 282. | ||
Cyclopropane derivative 275b served as a synthetic intermediate for the synthesis of two other sugar phosphates (Scheme 49).117 The synthesis of 285 started with the deprotection of the isopropylidene group of 275b, followed by diastereoselective cyclopropane hydrogenolysis and deoxygenation at C3, which was achieved via carbonate formation and subsequent elimination. The stereocenter at C4 was inverted using the Mitsunobu reaction with p-nitrobenzoic acid, followed by reduction of the ketonic carbonyl with lithium aluminium hydride, with high diastereoselectivity (11:1). Compound 284 was obtained by TBS protection and hydrolysis of the nitrobenzoyl ester. In the final step, the free alcohol was phosphorylated with dibenzyl phosphate. The endocyclic double bond was dehydroxylated under Upjohn's oxidation conditions, and after deprotection of TBS and Bn groups, 5a-carba-α-L-rhamnopyranose phosphate 285 was obtained.
 | ||
| Scheme 49 Synthesis of 5a-carba-α-L-rhamnopyranose phosphate 285 and 5a-carba-β-D-digitoxopyranose phosphate 291. | ||
For 291, its synthetic route started with an enone with butane bisacetal protecting group on the trans-vicinal diol 79. This was converted to cyclopropanol 287via a one-pot process comprising hydrosilylation, cyclopropanation, and desilylation, yielding a 1:1 diastereomeric mixture. Pd/C-mediated isomerization did not achieve diastereoselectivity, but epimerization with concentrated HCl led to the (R)-methyl ketone 288 in 57% yield. The allylic alcohol 289 was produced by reduction with LiAlH4 and deoxygenation of C5, which involved removing the bisacetal protecting group, addition of Boc group, and then elimination under basic conditions. After two additional steps—similar to the last two steps used to prepare 285—5a-carba-β-D-digitoxopyranose phosphate 291 was obtained.
Quinic acid has been explored in the synthesis of other non-natural carbasugar derivatives. Karukurichi et al. reported the synthesis of D-carbofructopyranosyl-1,2-diamines, for the preparation of chiral Co(III)-salen catalysts,118 whilst Li et al. presented a synthesis of two analogues of the flavonoid glycoside natural product SL0101, with the carbasugar constructed using QA as a chiral scaffold.119
In a related note, quinic acid can also be converted to de facto sugar derivatives (Scheme 50). A careful stereoselective synthesis of a β-D-ribo-hept-6-ulopyranosuronamide derivative 296, is a valuable intermediate for the synthesis of other higher sugars, as has been reported by Baptistella and Cerchiaro. The transformation of carbacycle to carbohydrate, an essential phase in this process, occurred in a one-pot reaction involving ozonolysis of the double bond of the α,β-unsaturated amide 294, carried out under mild conditions.120
 | ||
| Scheme 50 Synthesis of a β-D-ribo-hept-6-ulopyranosuronamide derivative. | ||
5. Synthesis of other carbacycles
Two carbasugars isolated from Streptomyces lincolnensis have been synthesised from quinic acid, upon reductive opening of a quinide derivative. The acetonide removal readily provided one of the natural carbasugars, whilst its epimer 299 (Scheme 51) could be obtained by oxidation of the primary hydroxyl, epimerization upon treatment with base, followed by reduction and acetonide removal.121 | ||
| Scheme 51 Synthesis of Streptomyces lincolnensis carbasugar. | ||
Maycock and co-workers have developed the first synthesis of (−)-asperpentyn, based on the conversion of quinic acid into epoxide 304, followed by its conversion into the corresponding α-iodoenone, further submitted to Stille coupling with tetramethylstannane (Scheme 52). The acetonide elimination from 300 resulted in the formation of interconvertible isomers 301 and 302, due to O,O-silyl migration. The enone isomers were epoxidized and, after separation, 304 was iodinated to be further submitted to Stille coupling conditions. Luche reduction of the enone resulted in the formation of both epimers of the allyl alcohol with some preference for the desired compound, which, upon deprotection and separation, allowed the isolation of enantiomerically pure (−)-asperpentyn. Some of the synthetic intermediates were taken in the synthesis of other cyclitols, namely (+)-harveynone, (+)-epieporformin, and (−)-theobroxide.122
 | ||
| Scheme 52 Synthesis of (−)-asperpentyn 307. | ||
In an attempt to develop the first total synthesis of ribisin F, previously characterized upon isolation from the fruiting bodies of Phellinus ribis,123 Badwell and co-workers took the acetonide-protected quinic acid as a starting material (Scheme 53). A mismatch in the spectral characterization of the synthesized compound (assisted by single crystal X-ray analysis) with the one from the natural isolate pointed to a wrongly assigned structure. The synthesis of the enantiomer of the envisioned natural product consisted on the formation of allyl alcohol 308, followed by Suzuki coupling, oxidation to the corresponding enone, phenol deprotection, palladium-catalyzed oxidative cyclization, and acetonide removal.124
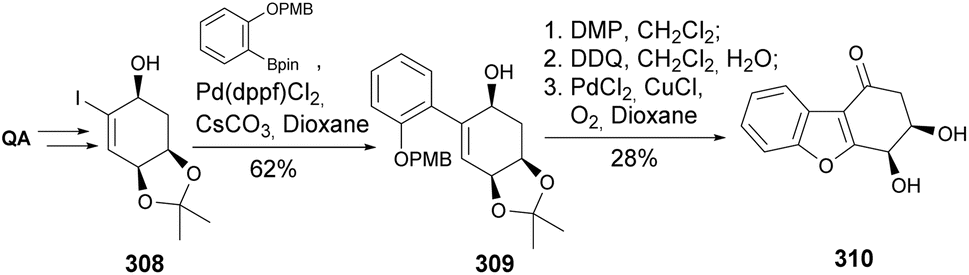 | ||
| Scheme 53 Synthesis of previously assigned ent-ribisin F. | ||
6. Lactones
Altenuene 313 and isoaltenuene 314 are minor toxins produced by various Alternaria fungi, and Altemöller reported their total synthesis from quinic acid (Scheme 54a). The iodocyclohexenone 311 was treated with MeMgBr at low temperatures to obtain a diastereomeric mixture of the alcohols 312, which were separated by chromatographic methods.125 In a different work,126 the enone 316 was α-iodinated and then reduced stereospecifically to provide epimers 318a and 318b depending on the reducing agent. The four isomers of the vinyl iodide were submitted to Suzuki coupling with the same boronic ester, to provide toxins altenuene 313, isoaltenuene 314, neoaltenuene 319 and epi-neoaltenuene 320, and (Scheme 54b). | ||
| Scheme 54 Total synthesis of Alternaria fungi toxins: (a) altenuene and isoaltenuene; (b) neoaltenuene and epi-neoaltenuene. | ||
Trichodermamides A 333 and B 334 are modified heterocyclic dipeptides isolated from cultures of the marine-derived fungus Trichoderma virens. Wan and coworkers prepared both natural products from 321 (Scheme 55).127,128 Spiro-lactone 323 was obtained by the addition of MeCN conjugate base to epoxide 322, followed by treatment with NaOMe. A modified Rubottom oxidation, followed by protection, afforded the α-silyloxylactone 325. The oxazine ring was formed by cyclization of the corresponding oxime of 328 in a basic medium. Allyl oxidation of 329 proved challenging, eventually resulting in the formation of the enone in ≈30% yield using CrO3/3,5-dimethoxypyrazole. Luche reduction provided both diastereomers of allyl alcohol 330 that could be separated by chromatography. Protection of the allylic alcohol and installation of the carboxylic acid group provided 331, which was coupled with aminocoumarin mediated by EDCI. Compounds 332 were subjected to deprotection conditions, resulting in 333 and 334, with the latter requiring an additional step to convert the secondary alcohol to the corresponding chloride.
 | ||
| Scheme 55 Total synthesis of trichodermamides A 333 and B 334. | ||
(−)-Malyngolide 341 and (R)-(+)-tanikolide 345 are two metabolites isolated from the lipid extract of a blue-green algae cyanobacterium, Lyngbia mujuscula.129 (−)-Malyngolide 341 is an antibiotic, whilst (R)-(+)-tanikolide is an antifungal that can be prepared from the same synthetic intermediate. Matsuo and co-workers developed a synthetic route for the preparation of lactol 338 based on NaIO4-mediated oxidative cleavages of the quinic acid triol moiety, followed by Wittig reactions and hydrogenation of the prepared olefins. The use of different Wittig ylides and adjustments on the synthetic sequence allowed obtaining both metabolites130,131 from the same lactol 338 (Scheme 56).
 | ||
| Scheme 56 Total synthesis of (−)-malyngolide and (R)-(+)-tanikolide. | ||
(−)-Bactobolin A 351 is a polyketide-peptide natural product first isolated in 1979 as a secondary metabolite of Pseudomonas sp. Vojáčková et al. reported a highly stereoselective synthesis of 351 from quinic acid (Scheme 57). The key transformations in this approach include a vinylogous aldol reaction of 346 to introduce the dichloromethyl group, a rhodium(II)-catalyzed C–H amination of 348, and an intramolecular alkoxycarbonylation to obtain the bicyclic lactone 350.132 The final amide coupling step required screening different coupling agents due to the occupation of the amino group at the axial position. COMU ((1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate)) delivered the amide in good 73% yield that could be deprotected with TFA to yield target 351.
 | ||
| Scheme 57 Total synthesis of (−)-bactobolin A. | ||
Švenda and co-workers have reported an improved synthesis path for the preparation of (+)-actinobolin (Scheme 58).133 Starting from the diol-protected QA-derived enone as radical acceptors, and exploring oxazolidinone derivatives as a source of carbon-centered radicals, the photocatalyzed Giese addition was used for the preparation of epimeric ketones 352 and 353. Changing the diol protecting groups to a silyl-protected enone had some effect on the diastereoselectivity towards the desired epimer, although at the expense of the reaction yield. The N-sulfonylation of 352 triggered an intramolecular alkoxycarbonylation and, after nosyl removal, the amine group was coupled with Boc-protected L-alanine. The removal of the diol group with TFA resulted in the preparation of the trifluoroacetate salt of (+)-actinobolin in 9 steps and 18% overall yield from quinic acid.
 | ||
| Scheme 58 Total synthesis of (+)-actinobolin. | ||
(+)-Rubellin C 361 is an anthraquinoid with a distinct scaffold, characterized by a 6-5-6 ring system. Rubellins can inhibit and reverse tau protein aggregation, a therapeutically relevant target for Alzheimer's disease. The high synthetic complexity of this molecule, with five contiguous stereocenters, was addressed by Gartman et al.,134,135 who converted the quinic acid-derived enone 54 in 8 steps into the stereochemically rich allyl alcohol 356 by Saegusa–Ito oxidation with addition of organomagnesium reagent, followed by oxidation of the ketone to the corresponding enone with Pd(OAc)2. An additional ring was introduced through the borylation of the alcohol, followed by the addition of methyl 2-formyl-6-methoxy-4-methylbenzoate, resulting in a single diastereomer of the lactone 357. Another crucial step involved forming the anthraquinone ring, where TBS-deprotection and oxidation of the phenolic moiety with phenyliodine(III) diacetate (PIDA) yielded the p-quinone monoketal 358. Under Hauser–Kraus annulation conditions, anthraquinoid 359 was obtained. The final key step entailed an intramolecular Heck reaction of the corresponding triflate of 359 with the endocyclic double bond, followed by total removal of the oxygen protecting groups (Scheme 59).
 | ||
| Scheme 59 Total synthesis of (+)-rubellin C. | ||
The 10-membered macrolide (+)-Sch 642305 366, isolated from Penicillium verrucosum and later found to inhibit bacterial DNA primase, was synthesized from quinic acid (Scheme 60).136 The synthesis relies on a Mukaiyama–Michael addition between enone 362 and a silyl ketene acetal in the presence of TBSOTf as catalyst. The mixture of the two diastereomers was submitted to allylation with tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) to provide the diallyl compound that was readily submitted to ring-closing metathesis reaction, resulting in the formation of 10-membered ring lactone 364. Hydrogenation followed by dehydrogenation and deprotection with buffered TBAF finalized the synthesis of the antibiotic.
 | ||
| Scheme 60 Synthesis of (+)-Sch 642305. | ||
Gao and co-workers have recently reported the total synthesis of cephanolides A (370) and B (374), exploiting quinic acid as the starting material for the construction of ring C of the natural products (Scheme 61).137,138 The cyclohexene derivatives were obtained upon arylation of the vinyl triflates with the corresponding boronic acid. Two different methods were developed for the installation of the quaternary stereocenter in C-5. A Sc(OTf)3-mediated hydroxymethylation was successful in forming the quaternary stereocenter in decent diastereoselectivity of 7:1 for the formation of 368, taken for the total synthesis of chephanolide A. The other route consisted in C-5 carboxylation, C-4 methylation, and C-5 allylation to provide 372 as an equimolar stoichiometric mixture. After arylation, epimers of 373 were separated, and epimer 373a was used in the total synthesis of cephanolide B 374.
 | ||
| Scheme 61 Synthesis of cephanolides A and B. | ||
The vinyl triflate derived from 368 (Scheme 61) was later used in the design of a synthetic scheme that could provide the preparation of several Cephalotaxus diterpenoids (Scheme 62). The synthetic scheme encompassed a Nicholas/Hosomi–Sakurai cascade reaction and an intramolecular Pauson–Khand reaction to form the 7-5-6 tricyclic system. The vinyl triflate 375 was used both in the preparation of aldehyde 378 and in the synthesis of linear fragment 379. Lactone 380 was further reacted with Co2(CO)8 to afford an organometallic cluster that was used in the synthesis of diterpenoids 381–386.139
 | ||
| Scheme 62 Synthesis of Cephalotaxus diterpenoids. | ||
7. Alkaloids
Alkaloids represent a significant class of molecules due to their structural complexity and potent biological activities. Trost et al. reported the synthesis of an optically active isoquinuclidine, that could allow further cyclization to either enantiomeric series of the iboga alkaloids (Scheme 63). The crucial steps for controlling stereochemistry included the hydroboration of the exocyclic double bond with AlMe3, which was essential for diastereoselectivity. The synthesis route involved tosylation of the primary hydroxy group, followed by installation of an azide at the primary carbon, and reduction to provide an amino group prone to condensation with a carboxylic acid. The amide 390 was reduced, and the resulting amine was protected with 2-trimethylsilyl chloroformate. Deprotection of the secondary alcohol, followed by mesylation, acetonide hydrolysis, and epoxide formation, followed by Collins oxidation and Wittig methylenation, afforded the vinyl epoxide 392. Its cyclization using palladium catalysis allowed the synthesis of the isoquinuclidine fragment 394.140 | ||
| Scheme 63 Synthesis of optically active isoquinuclidine. | ||
Manzamines are alkaloids that consist of a β-carboline heterocycle paired with polycyclic diamine moieties containing an isoquinoline ring.141 Kamenecka et al.142 reported the synthesis of the cis-decahydroisoquinoline substructure from quinic acid (Scheme 64). Key steps include the stereoselective addition of allyltributyltin to enone 395, an acid-catalyzed Mannich cyclization with formaldehyde in formic acid, and lastly, the formation of the tricyclic system through debenzylation with methyl chloroformate, followed by cleavage of the p-methoxybenzyl group and an acid-catalyzed dehydration that afforded the enamide 398. The oxidation of the enamide by magnesium monoperoxyperphthalic acid, followed by epoxide rearrangement and elimination, resulted in the formation of the tetracyclic core of (+)-manzamine A, although its final synthesis has not been reported through this route.
 | ||
| Scheme 64 Synthesis of the cis-decahydroisoquinoline core Manzanine 399. | ||
Albertini et al. developed a synthetic strategy to obtain a precursor of epibatidine,143,144 an alkaloid with a 7-azabicyclo[2.2.1]heptane ring, isolated from the skin of the Ecuadorian poison frog Epipedobates tricolor.145 The uncommon 7-azabicyclo[2.2.1]heptane ring was built by an intramolecular transannular cyclization of a 1,4-trans disubstituted cyclohexane azido sulfate 403 (Scheme 65). The sulfate derivative was obtained upon diastereoselective reduction of 400 in a 1:11 ratio, mesylation, azide introduction by nucleophilic substitution, followed by reaction with thionyl chloride to afford a diastereomeric mixture of sulfites, readily oxidized by NaIO4 using RuCl3 as catalyst. Reduction of the 403's azido group was followed by the intended cyclization, N-protection and Swern oxidation to provide 405, a synthetic intermediate previously reported for the synthesis of (−)-epibatidine 406.146
 | ||
| Scheme 65 Synthesis of a precursor of (−)-epibatidine 406. | ||
Maycock and co-workers have later expanded the utility of quinic acid on the synthesis of both enantiomers of epibatidine (Scheme 66). The key step in this route was the diastereoselective reduction of the 4-chloropyridyl cyclohexanone 408 with NaBH4, for which the most selective diastereoselective conditions were searched for.147,148 Interestingly, reduction with NaBH4 in the absence or presence of CeCl3 did not affect the diastereoselectivity, whilst the presence of DMSO was determined to be more relevant for the desired selectivity. Mesylation of the hydroxy group and installation of the amino group through the Mitsunobu reaction provided intermediate 410 that could be taken up into the synthesis of (+)-epibatidine, similarly to the previously reported racemic routes.149,150 For the synthesis of the other enantiomer, the authors have started from the trans-diol protected derivative of quinic acid 313 through a sequence of dehydration, hydrogenation, another dehydration, followed by silylation to provide 413. Analogous iodination of 413 followed by a similar synthetic sequence as described for 407 would provide (−)-epibatidine.
 | ||
| Scheme 66 Synthetic approaches to (+)-epibatidine, extendable to (−)-epibatidine. | ||
White and co-workers151,152 have reported a novel synthesis of (−)-huperzine starting from enone 362 (Scheme 67), prepared following Danishefsky's protocol.153 The presented synthesis started with the installation of a cyclobutane unit by an intramolecular [2 + 2] cycloaddition, followed by the construction of tetracyclic compound 416, oxidation, and methylation. Opening of the cyclic ether ring, oxidation, and carbonyl methylation provided secondary alcohol 418 that was further treated with Comins reagent, and the obtained enol triflate was submitted to Stille reaction. Treatment with anhydrous PTSA in hot benzene provided rearranged product 420, to be taken up in the previously established synthetic route of (−)-huperzine.154
 | ||
| Scheme 67 Synthetic approaches to (−)-huperzine A. | ||
(−)-Balanol is a fungal metabolite derived from the fungus Verticillium balanoides155 and was identified amid the search for novel PKC inhibitors. Barco and co-workers used the same quinic acid-derived precursor 400 used in the synthesis of epibatidine for the synthesis of (−)-balanol, relying on the Beckmann rearrangement of oxime sulfonate 422 (Scheme 68).144 Despite the lack of regioselectivity of the rearrangement, both lactams could be separated by crystallization and the reduction of the isomeric mixture resulted in the preferred formation of hexahydroazepine 424a over its meso isomer 424b. After acetonide deprotection, the sulfate moiety was installed in the same manner as for the previous synthesis, followed by regioselective nucleophilic substitution with ammonium benzoate acid treatment for removal of the sulfate group. Epoxide installation followed by regioselective opening with sodium azide provided the cyclic skeleton in the form of synthetic intermediate 427, prone to being included in the previously described synthesis of (−)-balanol upon stereoinversion.156
 | ||
| Scheme 68 Synthesis of a precursor to (−)-balanol. | ||
Following the chiron approach, Hanessian and co-workers have explored quinic acid as the chiral template for the construction of ring E in the enantioselective synthesis of (−)-reserpine 437 (Scheme 69).157 Benzylated lactone 429 was transformed into its dimethoxy derivative, followed by debenzylation and oxidation to provide ketone 430, prone to diastereoselective installation of a vinyl moiety in the conjugated ester to yield tertiary alcohol 431. The alcohol was submitted to an intramolecular free-radical cyclization of the α-iodoacetate ester, resulting in the formation of both epimers of bicyclic lactone 432, which could all be transformed into the β-epimer by treatment with DBU. Ozonolysis followed by oxidation and esterification resulted in the formation of lactone 433, containing the carbon skeleton of rings D and E. The assembly of the pentacyclic ring skeleton was accomplished upon differentiation of the three carbonyl groups by conversion of 433 into its hemiacetal with disiamylborane, followed by reaction with the tryptamine derivative. The hexacyclic epimers of 434 were separated, and the desired one was submitted to lactam carbonyl reduction, upon the needed protection of the tertiary hydroxyl. Removal of the silyl ether was required for effective deoxygenation of 435 with SmI2, and the natural product 437 was obtained upon silyl protecting group removal and installation of the trimethoxybenzoyl unit.
 | ||
| Scheme 69 Hanessian's total synthesis of (−)-reserpine. | ||
(+)-Pancratistatin occurs naturally in the Hawaiian spider lily with antineoplastic activity.158 Due to its low natural abundance, efforts have been made toward its total synthesis and the synthesis of congeners. Pandey and coworkers developed a synthetic strategy for (+)-2,7-dideoxypancratistatin 442, with a key step being the control of a trans cyclization via photo-induced electron transfer cyclization of silylenol 439 to an electron-rich aromatic ring 440 (Scheme 70).159 Quinic acid was first converted to silyl ether-protected enone 438, which undergoes conjugate addition of N-lithiated piperonylamine carbonate, followed by trapping of the enolate as TBS ether. The photoinduced cyclization of silylenol was achieved by using a >280 nm medium-pressure lamp in the presence of 2 mol% of 1,4-dicyanonaphthalene (DCN). The tetracyclic compound was reduced and protected as TBS ether, followed by benzylic oxidation and complete deprotection to provide the natural product.
 | ||
| Scheme 70 Synthesis of (+)-2,7-dideoxypancratistatin. | ||
The abovementioned strategy was in part adapted for the total synthesis of 1,10b-epi-7-deoxypancratistatin 449 (Scheme 71).160 The key step was the Suzuki-cross coupling for the formation of the aza–Michael precursor that was readily cyclized upon base treatment, to provide the tetracyclic 447 as a single diastereomer. Attempts to epimerize the α-carbonyl position to reach the trans-fused ring junction resulted in retro-Michael reaction and ring aromatization.
 | ||
| Scheme 71 Synthesis of 1,10b-epi-7-deoxypancratistatin. | ||
Dragmacidins are a family of alkaloids that can be obtained from marine sponges, and they possess a wide range of biological activities.161 The total syntheses of both enantiomers of dragmacidin F starting from quinic acid were developed by Stoltz and co-workers (Scheme 72).162,163 The divergent approach started with the preparation of carboxylic acid 451 through oxidation of quinide derivative 450, installation of an exocyclic olefin, followed by diastereoselective hydrogenation and base-promoted elimination of the carboxylate functionality. Upon conversion of the carboxylic moiety to the corresponding Weinreb amide, a pyrrole moiety was installed to provide 453, prone to palladium-catalyzed intramolecular carbocyclization when employing DMSO as a palladium ligand. The pyrrole-fused bicycle was hydrogenated and O-methylated to provide 454, to which the pinacol boronate was installed to be used in the Suzuki coupling with a brominated indole derivative. The final steps of the synthesis, namely the removal of all protecting groups and the installation of the aminoimidazole moiety, followed the lessons previously learned from the total synthesis of dragmacidin D,164 resulting in the use of a late-stage Neber rearrangement to provide (+)-dragmacidin F 457. The conversion of quinic acid to the enantiomer of 453, through desymmetrization of a pseudo-C2-symmetric compound, followed by formation of a bicyclic carbonate and hydrogenation, provided a new route for the synthesis of the antipode molecule.
 | ||
| Scheme 72 Total synthesis of (+)-dragmacidin F 457.163 | ||
Montanine-type alkaloids found in the Amaryllidaceae family exhibit a broad range of biological activities, including antiviral, anxiolytic, antidepressant, and anticonvulsant effects. (−)-Brunsvigine 463 and (−)-manthine 464 were synthesized from quinic acid (Scheme 73), after its conversion into common enone 54, iodination, and diastereoselective reduction of the iodoenone resulted in the formation of epimers 458 that were separated, and the undesired isomer 458a was easily transformed to its epimer 458bvia a Mitsunobu reaction. The bicyclic structure 460 was obtained upon introduction of the Weinreb amide side chain, followed by anionic cyclization by treatment with nBuLi. The reduction of amine 460 with NaBH4 in the presence of hydrated cerium chloride was shown to be diastereoselective, thus allowing the formation of pivaloate 461, readily converted to synthetic intermediate 462 through a copper-mediated SN2 reaction with an arylmagnesium bromide.165
 | ||
| Scheme 73 Total synthesis of (−)-brunsvigine 463 and (−)-manthine 464. | ||
8. Macrocyle fragments
Hanessian et al. disclosed a new synthetic method for the hexahydrobenzofuran core found in avermectins and milbemycins (Scheme 74).166 Such an approach utilized a radical-induced intramolecular Michael cyclization of a bromovinyl ether 467, for which the installation of the vinyl bromide moiety was accomplished using thallium ethoxide in DMF, after failure with more common alkylation procedures. | ||
| Scheme 74 Synthetic route to tetrahydrobenzofurans. | ||
The synthesis of the macrocyclic motif of espearmicin A1, an antitumor antibiotic, can be done by having quinic acid as the starting material (Scheme 75). The formation of the enediyne macrocycle occurred with the addition of an organocerium reagent to 472 as the first step, with the cyclohexylidene ketal being essential to promote anti-addition. Subsequently, the addition of vinylenthynyl chloride was aided by Pd-catalysis, while the intramolecular cyclization of 472a was achieved by introducing the deprotected terminal alkyne to the aldehyde.167
 | ||
| Scheme 75 Synthesis of the macrocyclic motif of espearmicin A1. | ||
Mulzer and coworkers have conducted extensive research on the synthetic pathways to the antibiotic branimycin 476 (Fig. 2), using quinic acid as a chiral scaffold for the synthesis of the cis-decalin system.168–171
 | ||
| Fig. 2 Structure of branimycin. | ||
In their studies, the cis-isoxazoline subunit can be obtained by an intramolecular nitrile oxide olefin cycloaddition (INOC) (mediated by either tBuOCl168,169 or N-chlorosuccinimide169,170) or by ring-closing metathesis.169,170 The former approach using tBuOCl (Scheme 76) was successfully applied to the preparation of model isoxazoline 483 from quinic acid, which included the diastereoselective hydroboration of 478 and the formation of allylic alcohol 480 using a Corey–Hopkins protocol that included the use of 1,3-dimethyl-2-phenyl-1,3,2-diazaphospholidene (DPD) for the elimination of a thionocarbonate. Upon successful formation of the desired isoxazoline, a similar approach was applied to the preparation of a more substituted derivative 489 (Scheme 77). The cis-isoxazoline 488 had its N–O bond cleaved to proceed with an Eschenmoser–Claisen rearrangement for the installation of the amide moiety.170
 | ||
| Scheme 76 Synthesis of highly substituted cis-decalin by INOC reaction mediated by tBuOCl. | ||
 | ||
| Scheme 77 Cis-decalin by INOC reaction mediated by NCS. | ||
The ring closing metathesis strategy encompassed the Mukaiyama-type condensation with dimethoxymethane, a diastereoselective process due to the conformational rigidity imposed by the trans-diequatorial-protected 1,2-diol (Scheme 78).171 Ester 491 undergoes an Ireland–Claisen rearrangement, through a silylketene acetal, to provide 492 as a β:α = 3:1 epimeric mixture. Installation of the alkene moiety was achieved via three steps, including a Wittig olefination, to provide 493. A second Claisen–Ireland rearrangement to provide 495 was achieved upon desilylation of 493, followed by ester installation through Mitsunobu inversion. The installation of the second cyclohexene moiety was achieved by RCM with Hoveyda–Grubbs catalyst, after the 2nd generation Grubbs failed to withstand 75 °C. PMB group was removed and the allyl alcohol oxidized to provide 498, which was stereo- and regioselectively epoxidized to yield 499, whose structure was confirmed by X-ray. Eventually, the first synthesis of branimycin was achieved by the same authors, starting from diepoxynaphthalene.172
 | ||
| Scheme 78 Preparation of cis-decalin by ring-closing metathesis. | ||
Quinic acid has been converted into a dienyne and used in the preparation of taxadiene analogues by ring-closing metathesis (Scheme 79).173 Enone 500 was converted into the α-iodoketone 501 and then treated with ethylmagnesium bromide and the aldehyde containing the required unsaturations. The ring closing metathesis was achieved with second-generation Grubbs catalyst in refluxing dichloromethane to provide the desired tricyclic structure 503 together with the bicyclic derivative 504 (in 40%). The latter could be easily converted into the desired taxadiene analogue 503 by further treatment with the same catalyst at higher temperatures.
 | ||
| Scheme 79 Preparation of taxadiene analog. | ||
9. Others
Although not so explored, QA has also been used in the total synthesis of linear natural products. One example is the synthesis of antibiotic (+)-negamycin 510 (Scheme 80). The oxidative ring cleavage of the cis diol was achieved using lead tetraacetate, followed by dialdehyde reduction. A series of protections, deprotections, nucleophilic substitution with azide, and reduction led to the synthesis of the linear natural product.174 | ||
| Scheme 80 Synthesis of (−)-negamycin 510. | ||
A total synthesis of diterpene (+)-aphidicolin was reported by Fukumoto and co-workers,175 by converting quinic acid into a synthetic intermediate used in the synthetic route developed by Smith.176 A rather long synthetic sequence, starting from enone 511, leads to the formation of enyne 512, which undergoes a palladium-catalyzed cycloisomerization to provide bicyclic fragment 515. This transformation enables the preparation of intermediate 516, which can then be advanced via the reported Smith route (Scheme 81).
 | ||
| Scheme 81 Preparation of synthetic fragments for (+)-aphidicolin 517. | ||
Notwithstanding the multiple previous syntheses of aquayamycin, Toshima and co-workers explored a new synthetic route in which the A ring fragment could be obtained from quinic acid (Scheme 82). This allowed shortening the previous routes in 16 synthetic steps, having as a key step the diastereoselective 1,2-addition of a C-glycosyl naphthyllithium to the QA-derived ketone 520. The construction of the B ring was achieved by an intramolecular pinacol coupling of a ketoaldehyde.177
 | ||
| Scheme 82 Synthesis of aquayamycin 522. | ||
The first total asymmetric synthesis of (+)-nodulisporiviridin E, reported by Gao and co-workers (Scheme 83),178 started from the QA-derived enone 523, obtained as previously reported by Maycock and co-workers.147 The synthesis of this furanosteroid from the enone was based on an intramolecular Heck reaction for the construction of the tetracyclic core and the furan E ring built upon an intramolecular oxonium trapping reaction.
 | ||
| Scheme 83 Synthesis of (+)-nodulisporividin E. | ||
Quinic acid was used in the synthesis of the protected 4-epimer of shikimic acid, which, together with (−)-shikimic acid itself, was engaged in the synthesis of scytolide and multiple isomers (Scheme 84).179 The scytolide epimer 532, obtained from quinic acid, was submitted to Diels–Alder cycloaddition in the presence of sorbiclinol as the enophile to yield compound 533 in 25%, the endo compound, although with different spectral characterization than the previously reported natural product.
 | ||
| Scheme 84 Synthesis of spirosorbiclinol C. | ||
10. Conclusions and future perspectives
Quinic acid has established itself as a privileged chiral building block in total synthesis, enabling the efficient construction of structurally diverse and biologically relevant molecules. Its densely functionalized cyclohexane core, bearing four stereocenters, offers an inherent stereochemical advantage that reduces the need for complex asymmetric induction steps. Over the past decades, synthetic strategies have relied on key intermediates such as quinide lactones, decarboxylation-derived enones, and shikimic acid analogues, combined with transformations including oxidation-reduction epimerization, deoxygenation, and selective installation of exocyclic functionalities. These approaches have facilitated the synthesis of natural products ranging from carbacycles and alkaloids to vitamin D analogues, carbasugars, and cyclitols.Despite these achievements, the chemoselective differentiation of hydroxyl groups remains the most significant challenge, often requiring extensive protection–deprotection sequences. To date, the most common methods rely on the selective protection of the trans diol moiety with butanedione180,181 or the cis moiety with acetonide.127,182
While intramolecular protection and selective diol masking have improved efficiency, future progress will depend on the development of protecting-group-free methodologies and site-selective catalytic transformations. Such advances would dramatically reduce step count, improve atom economy, and align with the principles of sustainable synthesis.
Looking ahead, the adoption of greener processes, including catalytic and solvent-minimized reactions, will be essential to reduce environmental impact and improve scalability. Chemoenzymatic strategies hold promise for achieving unparalleled selectivity in hydroxyl functionalization and epimerization, complementing traditional chemical methods. Furthermore, the exploration of quinic acid derivatives in medicinal chemistry—especially for glycosidase inhibitors, vitamin D analogues, and alkaloid frameworks—will continue to drive innovation, as these scaffolds provide unique opportunities for tuning biological activity and pharmacokinetic profiles.
By combining these emerging approaches with the intrinsic stereochemical advantages of quinic acid, future synthetic strategies will not only enhance efficiency and sustainability but also expand the chemical space accessible from this versatile chiral building block, reinforcing its role as a key enabler in modern organic synthesis.
11. Author contributions
NRC performed the bibliographic search, curated the articles, organized the contents and revised the manuscript. MJV curated the articles, wrote the text and drew the schemes. ICV participated in the bibliographic search and revised the text.12. Conflicts of interest
There are no conflicts to declare.13. Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.14. Acknowledgements
Financial support from PT national funds (FCT/MCTES, Fundação para a Ciência e Tecnologia, and Ministério da Ciência, Tecnologia e Ensino Superior) through the projects PTDC/QUI-QOR/1131/2020, and UID/50006/2025 – Laboratório Associado para a Química Verde – Tecnologias e Processos Limpos) is acknowledged. MJV acknowledges FCT/MCTES for the PhD Fellowship (2024.04089.BDANA).15. Notes and references
- D. Kremr, T. Bajer, P. Bajerová, S. Surmová and K. Ventura, Quim. Nova, 2016, 39, 530–533 CAS.
- L. Abranko and M. N. Clifford, J. Agric. Food Chem., 2017, 65, 3602–3608 CrossRef CAS PubMed.
- C. Abell, F. H. Allen, T. D. H. Bugg, M. J. Doyle and P. R. Raithby, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 1988, 44, 1287–1290 CrossRef.
- IUPAC Commission on the Nomenclature of Organic chemistry (CNOC) and IUPAC-IUB Commission on Biochemiocal Nomenclature (CBN), Biochem. J., 1976, 153, 23–31 Search PubMed.
- R. Meldola, Nature, 1900, 61, 461–462 CrossRef.
- A. Barco, S. Benetti, C. D. Risi, P. Marchetti, G. P. Pollini and V. Zanirato, Tetrahedron: Asymmetry, 1997, 8, 3515–3545 CrossRef CAS.
- P. Q. Huang, Chin. J. Org. Chem., 1999, 19, 371–373 Search PubMed.
- V. Enev, Compr. Chirality, 2012, 325–345, DOI:10.1016/b978-0-08-095167-6.00221-4.
- J. Mulzer, M. Drescher and V. Enev, Curr. Org. Chem., 2008, 12, 1613–1630 CrossRef CAS.
- V. V. Shende, K. D. Bauman and B. S. Moore, Nat. Prod. Rep., 2024, 41, 604–648 RSC.
- N. R. Candeias, B. Assoah and S. P. Simeonov, Chem. Rev., 2018, 118, 10458–10550 CrossRef CAS PubMed.
- N. H. Giles, M. E. Case, C. W. Partridge and S. I. Ahmed, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 1453–1460 CrossRef CAS PubMed.
- F. C. Hofmann, Crell's Ann., 1790, 2, 314 Search PubMed.
- H. O. L. Fischer and G. Dangschat, Ber. Dtsch. Chem. Ges., 1932, 65, 1037–1040 CrossRef.
- E. Haslam and M. J. Turner, J. Chem. Soc. C, 1971, 1496–1500 RSC.
- T. Yamada, M. Iritani, H. Ohishi, K. Tanaka, K. Minoura, M. Doi and A. Numata, Org. Biomol. Chem., 2007, 5, 3979 RSC.
- Y. Usami, H. Ichikawa and M. Arimoto, Int. J. Mol. Sci., 2008, 9, 401–421 CrossRef CAS PubMed.
- Y. Usami and Y. Ueda, Chem. Lett., 2005, 34, 1062–1063 CrossRef CAS.
- Y. Usami, I. Takaoka, H. Ichikawa, Y. Horibe, S. Tomiyama, M. Ohtsuka, Y. Imanishi and M. Arimoto, J. Org. Chem., 2007, 72, 6127–6134 CrossRef CAS PubMed.
- Y. Usami and Y. Ueda, Synthesis, 2007, 2007, 3219–3225 CrossRef.
- Y. Usami, M. Ohsugi, K. Mizuki, H. Ichikawa and M. Arimoto, Org. Lett., 2009, 11, 2699–2701 CrossRef CAS PubMed.
- Y. Usami, Y. Okada and T. Yamada, Chirality, 2011, 23, E7–E11 CrossRef CAS PubMed.
- Y. Usami, C. Hatsuno, H. Yamamoto, M. Tanabe and A. Numata, Chem. Pharm. Bull., 2004, 52, 1130–1133 CrossRef CAS PubMed.
- Y. Usami, K. Suzuki, K. Mizuki, H. Ichikawa and M. Arimoto, Org. Biomol. Chem., 2009, 7, 315–318 RSC.
- Y. Usami, K. Mizuki, H. Ichikawa and M. Arimoto, Tetrahedron: Asymmetry, 2008, 19, 1461–1464 CrossRef CAS.
- Y. Usami and K. Mizuki, J. Nat. Prod., 2011, 74, 877–881 CrossRef CAS PubMed.
- Y. Usami, K. Mizuki, R. Kawahata, M. Shibano, A. Sekine, H. Yoneyama and S. Harusawa, Mar. Drugs, 2017, 15, 22 CrossRef PubMed.
- T. K. M. Shing and V. W. F. Tai, J. Chem. Soc., Chem. Commun., 1993, 995–997 RSC.
- T. K. M. Shing and V. W. F. Tai, J. Chem. Soc., Perkin Trans. 1 1, 1994, 2017–2025 RSC.
- V. W.-F. Tai, P. H. Fung, Y. S. Wong and T. K. M. Shing, Tetrahedron: Asymmetry, 1994, 5, 1353–1362 CrossRef CAS.
- S. M. Kupchan, R. J. Hemingway and R. M. Smith, J. Org. Chem., 1969, 34, 3898–3902 CrossRef CAS PubMed.
- S. Takahashi, Phytochemistry, 1969, 8, 321–322 CrossRef CAS.
- T. K. M. Shing and E. K. W. Tam, J. Org. Chem., 1998, 63, 1547–1554 CrossRef CAS.
- T. K. M. Shing and E. K. W. Tam, Tetrahedron: Asymmetry, 1996, 7, 353–356 CrossRef CAS.
- E. Chrapusta, A. Kaminski, K. Duchnik, B. Bober, M. Adamski and J. Bialczyk, Mar. Drugs, 2017, 15, 326 CrossRef PubMed.
- J. D. White, J. H. Cammack, K. Sakuma, G. W. Rewcastle and R. K. Widener, J. Org. Chem., 1995, 60, 3600–3611 CrossRef CAS.
- J. D. White, J. H. Cammack and K. Sakuma, J. Am. Chem. Soc., 2002, 111, 8970–8972 CrossRef.
- M. T. Barros, C. D. Maycock and M. R. Ventura, J. Org. Chem., 1997, 62, 3984–3988 CrossRef CAS.
- M. Teresa Barros, C. D. Maycock and M. R. Ventura, Tetrahedron, 1999, 55, 3233–3244 CrossRef.
- Y. Ohfune, T. Shinada, T. Fuji, Y. Ohtani and Y. Yoshida, Synlett, 2002, 14, 1341–1343 Search PubMed.
- G. N. Guerrero-Flores, F. M. Butler, V. L. Martinez Marignac, G. Zhang, F. J. Pacheco and D. S. Boskovic, Biologics, 2025, 5, 10 CrossRef.
- M. G. Banwell, N. L. Hungerford and K. A. Jolliffe, Org. Lett., 2004, 6, 2737–2740 CrossRef CAS PubMed.
- S. Holmstedt, A. Efimov and N. R. Candeias, Org. Lett., 2021, 23, 3083–3087 CrossRef CAS PubMed.
- P. Laurent, A. Hamdani, J.-C. Braekman, D. Daloze, L. A. Isbell, J.-C. De Biseau and J. M. Pasteels, Tetrahedron Lett., 2003, 44, 1383–1386 CrossRef CAS.
- L. Yong-Hong, X. Li-Zhen, Y. Shi-Lin, D. Jie, Z. Yong-Su, Z. Min and S. Nan-Jun, Phytochemistry, 1997, 45, 729–732 CrossRef.
- X. Huo, Y. Liao, Y. Tian, L. Gao and L. Cao, RSC Adv., 2016, 6, 114096–114108 RSC.
- S. Yang, Y. Liao, L. Li, X. Xu and L. Cao, Molecules, 2018, 23, 2149 CrossRef PubMed.
- Z. Sun, S. Yang, C. Xu, F. Yi, L. Cao, Y. Tian, J. Lin and X. Xu, Bioorg. Chem., 2021, 116, 105333 CrossRef CAS PubMed.
- A. Otto, A. Porzel, J. Schmidt, W. Brandt, L. Wessjohann and N. Arnold, J. Nat. Prod., 2016, 79, 74–80 CrossRef CAS PubMed.
- S. Das, A. Dalal and S. L. Gholap, Synth. Commun., 2020, 50, 580–586 CrossRef CAS.
- A. Barco, S. Benetti, C. De Risi, P. Marchetti, G. P. Pollini and V. Zanirato, Tetrahedron: Asymmetry, 1998, 9, 2857–2864 CrossRef CAS.
- H. P. Sigg and H. P. Weber, Helv. Chim. Acta, 1968, 51, 1395–1408 CrossRef CAS PubMed.
- D. H. R. Barton, S. Bath, D. C. Billington, S. D. Gero, B. Quiclet-Sire and M. Samadi, J. Chem. Soc., Perkin Trans. 1 1, 1995, 1551–1558 RSC.
- W. H. Yuan, M. Liu, N. Jiang, Z. K. Guo, J. Ma, J. Zhang, Y. C. Song and R. X. Tan, Eur. J. Org Chem., 2010, 2010, 6348–6353 CrossRef.
- T.-X. Li, M.-H. Yang, X.-B. Wang, Y. Wang and L.-Y. Kong, J. Nat. Prod., 2015, 78, 2511–2520 CrossRef CAS PubMed.
- Z. Yan, C. Zhao, J. Gong and Z. Yang, Org. Lett., 2020, 22, 1644–1647 CrossRef CAS PubMed.
- K. Koyama, H. Okamura and H. Takikawa, Tetrahedron, 2025, 173, 134488 CrossRef CAS.
- M. A. Maestro, F. Molnar and C. Carlberg, J. Med. Chem., 2019, 62, 6854–6875 CrossRef CAS PubMed.
- Y. Akagi, K. Yasui and K. Nagasawa, J. Steroid Biochem. Mol. Biol., 2017, 173, 64–68 CrossRef CAS PubMed.
- S. Fernandez and M. Ferrero, Pharmaceuticals, 2020, 13, 159 CrossRef CAS PubMed.
- K. L. Perlman, R. E. Swenson, H. E. Paaren, H. K. Schnoes and H. F. DeLuca, Tetrahedron Lett., 1991, 32, 7663–7666 CrossRef CAS.
- R. R. Sicinski, K. L. Perlman and H. F. DeLuca, J. Med. Chem., 1994, 37, 3730–3738 CrossRef CAS PubMed.
- P.-q. Huang, K. Sabbe, M. Pottie and M. Vandewalle, Tetrahedron Lett., 1995, 36, 8299–8302 CrossRef CAS.
- R. R. Sicinski, J. M. Prahl, C. M. Smith and H. F. DeLuca, J. Med. Chem., 1998, 41, 4662–4674 CrossRef CAS PubMed.
- A. Glebocka, R. R. Sicinski and H. F. DeLuca, J. Steroid Biochem. Mol. Biol., 2004, 89–90, 25–30 CrossRef CAS PubMed.
- A. Glebocka, R. R. Sicinski, L. A. Plum, M. Clagett-Dame and H. F. DeLuca, J. Med. Chem., 2006, 49, 2909–2920 CrossRef CAS PubMed.
- A. Fabisiak, P. Brzeminski, K. Berkowska, E. Marcinkowska and R. R. Sicinski, J. Steroid Biochem. Mol. Biol., 2019, 185, 251–255 CrossRef CAS PubMed.
- R. R. Sicinski, A. Glebocka, L. A. Plum and H. F. DeLuca, J. Steroid Biochem. Mol. Biol., 2007, 103, 293–297 CrossRef CAS PubMed.
- R. R. Sicinski, A. Glebocka, L. A. Plum and H. F. DeLuca, J. Med. Chem., 2007, 50, 6154–6164 CrossRef CAS PubMed.
- A. Glebocka, R. R. Sicinski, L. A. Plum and H. F. DeLuca, J. Steroid Biochem. Mol. Biol., 2010, 121, 46–50 CrossRef CAS PubMed.
- P. Grzywacz, L. A. Plum, W. Sicinska, R. R. Sicinski, J. M. Prahl and H. F. DeLuca, J. Steroid Biochem. Mol. Biol., 2004, 89–90, 13–17 CrossRef CAS PubMed.
- D. R. Laplace, M. Van Overschelde, P. J. De Clercq, A. Verstuyf and J. M. Winne, Eur. J. Org Chem., 2012, 2013, 728–735 CrossRef.
- B. Chen, M. Kawai and J. R. Wu-Wong, Bioorg. Med. Chem. Lett., 2013, 23, 5949–5952 CrossRef CAS PubMed.
- R. Samala, S. Sharma, M. K. Basu, K. Mukkanti and F. Porstmann, Tetrahedron Lett., 2016, 57, 1309–1312 CrossRef CAS.
- I. K. Sibilska, M. Szybinski, R. R. Sicinski, L. A. Plum and H. F. Deluca, J. Steroid Biochem. Mol. Biol., 2013, 136, 9–13 CrossRef CAS PubMed.
- I. K. Sibilska, R. R. Sicinski, J. T. Ochalek, L. A. Plum and H. F. DeLuca, J. Med. Chem., 2014, 57, 8319–8331 CrossRef CAS PubMed.
- I. K. Sibilska, M. Szybinski, R. R. Sicinski, L. A. Plum and H. F. DeLuca, J. Med. Chem., 2015, 58, 9653–9662 CrossRef CAS PubMed.
- M. Diaz, M. Ferrero, S. Fernandez and V. Gotor, J. Org. Chem., 2000, 65, 5647–5652 CrossRef CAS PubMed.
- M. Dìaz, M. Ferrero, S. Fernández and V. Gotor, Tetrahedron Lett., 2000, 41, 775–779 CrossRef.
- L. Sanchez-Abella, S. Fernandez, A. Verstuyf, L. Verlinden, V. Gotor and M. Ferrero, J. Med. Chem., 2009, 52, 6158–6162 CrossRef CAS PubMed.
- K. Ono, A. Yoshida, N. Saito, T. Fujishima, S. Honzawa, Y. Suhara, S. Kishimoto, T. Sugiura, K. Waku, H. Takayama and A. Kittaka, J. Org. Chem., 2003, 68, 7407–7415 CrossRef CAS PubMed.
- A. Kittaka, A. Yoshida, K. Ono, Y. Suhara, N. Saito and H. Takayama, Synlett, 2003, 15, 1175–1179 CrossRef.
- M. A. Arai and A. Kittaka, Anticancer Res., 2006, 26, 2621–2631 CAS.
- T. Sakaki, A. Kittaka, H. Hara, K. Yasuda, M. Takano, M. A. Arai, D. Sawada, H. Saito, K. Takenouchi and T. C. Chen, Heterocycles, 2010, 82, 649 CrossRef PubMed.
- A. Kittaka, H. Hara, M. Takano, D. Sawada, M. A. Arai, K. Takagi, T. Chida, Y. Harada, H. Saito, K. Takenouchi, S. Ishizuka, K. Hayashi, S. Ikushiro, T. Sakaki, T. Sugiura and T. C. Chen, Anticancer Res., 2009, 29, 3563–3569 CAS.
- A. Kittaka, M. A. Arai, R. Tsutsumi, H. Hara, T. C. Chen, T. Sakaki, N. Urushino and K. Inouye, Heterocycles, 2005, 66, 469–479 CrossRef PubMed.
- K. Usuda, T. Biswas, T. Yamaguchi, Y. Akagi, K. Yasui, M. Uesugi, I. Shimizu, S. Hosokawa and K. Nagasawa, Chem. Pharm. Bull., 2016, 64, 1190–1195 CrossRef CAS PubMed.
- N. E. Lee, G. S. Reddy, A. J. Brown and P. G. Williard, Biochemistry, 1997, 36, 9429–9437 CrossRef CAS PubMed.
- N. Simsek Kus, Chem. Biodivers., 2024, 21, e202301064 CrossRef CAS PubMed.
- L. Diaz and A. Delgado, Curr. Med. Chem., 2010, 17, 2393–2418 CrossRef CAS PubMed.
- M. Carballido, L. Castedo and C. González-Bello, Eur. J. Org Chem., 2004, 2004, 3663–3668 CrossRef.
- T.-L. Shih, W.-S. Kuo and Y.-L. Lin, Tetrahedron Lett., 2004, 45, 5751–5754 CrossRef CAS.
- A. Murugan, A. K. Yadav and M. K. Gurjar, Tetrahedron Lett., 2005, 46, 6235–6238 CrossRef CAS.
- L. Panza, E. Valsecchi, A. Tacchi, D. Prosperi and F. Compostella, Synlett, 2004, 16, 2529–2532 CrossRef.
- T. L. Shih and Y. L. Lin, Synth. Commun., 2005, 35, 1809–1817 CrossRef CAS.
- T.-L. Shih, Y.-L. Lin and W.-S. Kuo, Tetrahedron, 2005, 61, 1919–1924 CrossRef CAS.
- T.-L. Shih, W.-Y. Liao and W.-C. Yen, Tetrahedron, 2014, 70, 9621–9627 CrossRef CAS.
- L. Castellanos, J. Cléophax, C. Colas, S. D. Gero, J. Leboul, D. Mercier, A. Olesker, A. Rolland, B. Quiclet-Sire and A.-M. Sepulchre, Carbohydr. Res., 1980, 82, 283–301 CrossRef CAS.
- A. Abdali, S. J. Wittenberger, P. Yadagiri, J. R. Falck and A. Pap, Am. Chem. Soc., 1990, 200, 145–154 Search PubMed.
- J. R. Falck and P. Yadagiri, J. Org. Chem., 1989, 54, 5851–5852 CrossRef CAS.
- J. R. Falck and A. Abdali, Bioorg. Med. Chem. Lett., 1993, 3, 717–720 CrossRef CAS.
- J. R. Falck and A. Abdali, ACS Symp. Ser., 1991, 463, 145–154 CrossRef CAS.
- K. K. Reddy, J. R. Falck and J. Capdevila, Tetrahedron Lett., 1993, 34, 7869–7872 CrossRef CAS.
- T. K. M. Shing and V. W. F. Tai, J. Org. Chem., 1995, 60, 5332–5334 CrossRef CAS.
- T. K. M. Shing and L. H. Wan, Angew Chem. Int. Ed. Engl., 1995, 34, 1643–1645 CrossRef CAS.
- T. K. M. Shing and L. H. Wan, J. Org. Chem., 1996, 61, 8468–8479 CrossRef CAS.
- T. K. Shing, T. Y. Li and S. H. Kok, J. Org. Chem., 1999, 64, 1941–1946 CrossRef CAS PubMed.
- S. H. Kok, C. C. Lee and T. K. Shing, J. Org. Chem., 2001, 66, 7184–7190 CrossRef CAS PubMed.
- T. K. Shing, C. S. Kwong, A. W. Cheung, S. H. Kok, Z. Yu, J. Li and C. H. Cheng, J. Am. Chem. Soc., 2004, 126, 15990–15992 CrossRef CAS PubMed.
- T. L. Shih and S. Y. Yang, Molecules, 2012, 17, 4498–4507 CrossRef CAS PubMed.
- T. K. M. Shing and Y. Tang, J. Chem. Soc., Chem. Commun., 1990, 748–749 RSC.
- A. Zorin, L. Klenk, T. Mack, H. P. Deigner and M. S. Schmidt, Top. Curr. Chem., 2022, 380, 12 CrossRef CAS PubMed.
- T. K. M. Shing and Y. Tang, Tetrahedron, 1991, 47, 4571–4578 CrossRef CAS.
- T. K. M. Shing, Y.-X. Cui and Y. Tang, J. Chem. Soc., Chem. Commun., 1991, 756–757 RSC.
- T. K. M. Shing, Y.-X. Cui and Y. Tang, Tetrahedron, 1992, 48, 2349–2358 CrossRef CAS.
- M. Carballido, L. Castedo and C. González, Tetrahedron Lett., 2001, 42, 3973–3976 CrossRef CAS.
- M. Shan and G. A. O'Doherty, Org. Lett., 2008, 10, 3381–3384 CrossRef CAS PubMed.
- K. R. Karukurichi, X. Fei, R. A. Swyka, S. Broussy, W. Shen, S. Dey, S. K. Roy and D. B. Berkowitz, Sci. Adv., 2015, 1, e1500066 CrossRef PubMed.
- M. Li, Y. Li, K. A. Ludwik, Z. M. Sandusky, D. A. Lannigan and G. A. O'Doherty, Org. Lett., 2017, 19, 2410–2413 CrossRef CAS PubMed.
- L. H. Baptistella and G. Cerchiaro, Carbohydr. Res., 2004, 339, 665–671 CrossRef CAS PubMed.
- S. Holmstedt and N. R. Candeias, Tetrahedron, 2020, 76, 131346 CrossRef CAS.
- M. T. Barros, C. D. Maycock and M. R. Ventura, Chem. – Eur. J., 2000, 6, 3991–3996 CrossRef CAS PubMed.
- M. Y. Dong, Y. Zhang, H. Q. Jiang, W. J. Ren, L. C. Xu, Y. Q. Zhang and Y. H. Liu, Nat. Prod. Res., 2021, 35, 5145–5152 CrossRef CAS PubMed.
- Y. Zhang, P. Lan and M. G. Banwell, Eur. J. Org Chem., 2023, 26, e202300010 CrossRef CAS.
- M. Altemöller, J. Podlech and D. Fenske, Eur. J. Org Chem., 2006, 2006, 1678–1684 CrossRef.
- M. Altemöller and J. Podlech, Eur. J. Org Chem., 2009, 2009, 2275–2282 CrossRef.
- X. Wan, G. Doridot and M. M. Joullie, Org. Lett., 2007, 9, 977–980 CrossRef CAS PubMed.
- X. Wan and M. M. Joullie, J. Am. Chem. Soc., 2008, 130, 17236–17237 CrossRef CAS PubMed.
- I. P. Singh, K. E. Milligan and W. H. Gerwick, J. Nat. Prod., 1999, 62, 1333–1335 CrossRef CAS PubMed.
- K. Matsuo, J. Hikita and K. Nishiwaki, Heterocycles, 2011, 83, 2601–2605 CrossRef CAS.
- K. Matsuo, T. Matsumoto and K. Nishiwaki, Heterocycles, 1998, 48, 1213–1220 CrossRef CAS.
- P. Vojackova, L. Michalska, M. Necas, D. Shcherbakov, E. C. Bottger, J. Sponer, J. E. Sponer and J. Svenda, J. Am. Chem. Soc., 2020, 142, 7306–7311 CrossRef CAS PubMed.
- P. R. Tharra, A. A. Mikhaylov, J. vejkar, M. Gysin, S. N. Hobbie and J. venda, Angew Chem. Int. Ed. Engl., 2022, 61, e202116520 CrossRef CAS PubMed.
- J. A. Gartman and U. K. Tambar, Org. Lett., 2020, 22, 9145–9150 CrossRef CAS PubMed.
- J. A. Gartman and U. K. Tambar, J. Org. Chem., 2021, 86, 11237–11262 CrossRef CAS PubMed.
- E. M. Wilson and D. Trauner, Org. Lett., 2007, 9, 1327–1329 CrossRef CAS PubMed.
- H. Zhang, H. He and S. Gao, Angew Chem. Int. Ed. Engl., 2020, 59, 20417–20422 CrossRef CAS PubMed.
- H. Y. Zhang, H. B. He and S. H. Gao, Org. Chem. Front., 2021, 8, 555–559 RSC.
- H. Wang, Y. Liu, H. Zhang, B. Yang, H. He and S. Gao, J. Am. Chem. Soc., 2023, 145, 16988–16994 CrossRef CAS PubMed.
- B. M. Trost and A. G. Romero, J. Org. Chem., 1986, 51, 2332–2342 CrossRef CAS.
- C. K. Kim, R. Riswanto, T. H. Won, H. Kim, B. Elya, C. J. Sim, D. C. Oh, K. B. Oh and J. Shin, J. Nat. Prod., 2017, 80, 1575–1583 CrossRef CAS PubMed.
- T. M. Kamenecka and L. E. Overman, Tetrahedron Lett., 1994, 35, 4279–4282 CrossRef CAS.
- E. Albertini, A. Barco, S. Benetti, C. De Risi, G. P. Pollini and V. Zanirato, Tetrahedron Lett., 1997, 38, 681–684 CrossRef CAS.
- E. Albertini, A. Barco, S. Benetti, C. De Risi, G. P. Pollini and V. Zanirato, Tetrahedron, 1997, 53, 17177–17194 CrossRef CAS.
- T. F. Spande, H. M. Garraffo, M. W. Edwards, H. J. C. Yeh, L. Pannell and J. W. Daly, J. Am. Chem. Soc., 1992, 114, 3475–3478 CrossRef CAS.
- S. R. Fletcher, R. Baker, M. S. Chambers, S. C. Hobbs and P. J. Mitchell, J. Chem. Soc., Chem. Commun., 1993, 1216–1218 RSC.
- M. T. Barros, C. D. Maycock and M. R. Ventura, J. Chem. Soc., Perkin Trans. 1 1, 2001, 166–173 RSC.
- M. T. Barros, C. D. Maycock and M. R. Ventura, Tetrahedron Lett., 1999, 40, 557–560 CrossRef CAS.
- E. Albertini, A. Barco, S. Benetti, C. De Risi, G. P. Pollini, R. Romagnoli and V. Zanirato, Tetrahedron Lett., 1994, 35, 9297–9300 CrossRef CAS.
- C. A. Broka, Tetrahedron Lett., 1993, 34, 3251–3254 CrossRef CAS.
- J. D. White, Y. Li, J. Kim and M. Terinek, Org. Lett., 2013, 15, 882–885 CrossRef CAS PubMed.
- J. D. White, Y. Li, J. Kim and M. Terinek, J. Org. Chem., 2015, 80, 11806–11817 CrossRef CAS PubMed.
- J. E. Audia, L. Boisvert, A. D. Patten, A. Villalobos and S. J. Danishefsky, J. Org. Chem., 1989, 54, 3738–3740 CrossRef CAS.
- Y. Xia and A. P. Kozikowski, J. Am. Chem. Soc., 1989, 111, 4116–4117 CrossRef CAS.
- P. Kulanthaivel, Y. F. Hallock, C. Boros, S. M. Hamilton, W. P. Janzen, L. M. Ballas, C. R. Loomis, J. B. Jiang and B. Katz, J. Am. Chem. Soc., 1993, 115, 6452–6453 CrossRef CAS.
- D. Tanner, L. Tedenborg, A. Almario, I. Pettersson, I. Csöregh, N. M. Kelly, P. G. Andersson and T. Högberg, Tetrahedron, 1997, 53, 4857–4868 CrossRef CAS.
- S. Hanessian, J. Pan, A. Carnell, H. Bouchard and L. Lesage, J. Org. Chem., 1997, 62, 465–473 CrossRef CAS PubMed.
- A. McLachlan, N. Kekre, J. McNulty and S. Pandey, Apoptosis, 2005, 10, 619–630 CrossRef CAS PubMed.
- G. Pandey, A. Murugan and M. Balakrishnan, Chem. Commun., 2002, 2, 624–625 RSC.
- G. Pandey, M. Balakrishnan and P. S. Swaroop, Eur. J. Org Chem., 2008, 2008, 5839–5847 CrossRef.
- R. J. Capon, F. Rooney, L. M. Murray, E. Collins, A. T. R. Sim, J. A. P. Rostas, M. S. Butler and A. R. Carroll, J. Nat. Prod., 1998, 61, 660–662 CrossRef CAS PubMed.
- N. K. Garg, D. D. Caspi and B. M. Stoltz, J. Am. Chem. Soc., 2005, 127, 5970–5978 CrossRef CAS PubMed.
- N. K. Garg, D. D. Caspi and B. M. Stoltz, J. Am. Chem. Soc., 2004, 126, 9552–9553 CrossRef CAS PubMed.
- N. K. Garg, R. Sarpong and B. M. Stoltz, J. Am. Chem. Soc., 2002, 124, 13179–13184 CrossRef CAS PubMed.
- A. W. Hong, T. H. Cheng, V. Raghukumar and C. K. Sha, J. Org. Chem., 2008, 73, 7580–7585 CrossRef CAS PubMed.
- S. Hanessian, P. Beaulieu and D. Dubé, Tetrahedron Lett., 1986, 27, 5071–5074 CrossRef CAS.
- G. Ulibarri, H. Audrain, W. Nadler, H. Lhermitte and D. S. Grierson, Pure Appl. Chem., 1996, 68, 601–604 CrossRef CAS.
- J. Mulzer, V. S. Enev, M. Drescher and H. Kählig, Synlett, 2005, 17, 2227–2229 Search PubMed.
- J. Mulzer, D. Castagnolo, W. Felzmann, S. Marchart, C. Pilger and V. S. Enev, Chem. – Eur. J., 2006, 12, 5992–6001 CrossRef CAS PubMed.
- V. S. Enev, M. Drescher and J. Mulzer, Tetrahedron, 2007, 63, 5930–5939 CrossRef CAS.
- S. Marchart, J. Mulzer and V. S. Enev, Org. Lett., 2007, 9, 813–816 CrossRef CAS PubMed.
- V. S. Enev, W. Felzmann, A. Gromov, S. Marchart and J. Mulzer, Chem. – Eur. J., 2012, 18, 9651–9668 CrossRef CAS PubMed.
- S. Pérez-Estrada, N. Sayar and J. R. Granja, Org. Chem. Front., 2016, 3, 1331–1336 RSC.
- C. D. Maycock, M. T. Barros, A. G. Santos and L. S. Godinho, Tetrahedron Lett., 1992, 33, 4633–4636 CrossRef CAS.
- M. Toyota, Y. Nishikawa and K. Fukumoto, Tetrahedron, 1996, 52, 10347–10362 CrossRef CAS.
- C. J. Rizzo and A. B. Smith, J. Chem. Soc., Perkin Trans. 1 1, 1991, 969–979 RSC.
- S. Kusumi, H. Nakayama, T. Kobayashi, H. Kuriki, Y. Matsumoto, D. Takahashi and K. Toshima, Chem. – Eur. J., 2016, 22, 18733–18736 CrossRef CAS PubMed.
- Y. Ji, Z. Xin, Y. Shi, H. He and S. Gao, Org. Chem. Front., 2020, 7, 109–112 RSC.
- T. M. Milzarek and T. A. M. Gulder, Commun. Chem., 2023, 6, 187 CrossRef CAS PubMed.
- J. L. Montchamp, F. Tian, M. E. Hart and J. W. Frost, J. Org. Chem., 1996, 61, 3897–3899 CrossRef CAS PubMed.
- C. Le Sann, C. Abell and A. D. Abell, Synth. Commun., 2003, 33, 527–533 CrossRef CAS.
- S. Jiang, G. Singh, D. J. Boam and J. R. Coggins, Tetrahedron: Asymmetry, 1999, 10, 4087–4090 CrossRef CAS.
Footnote |
| † In memory of Professor Ana M. Lobo (1945–2024). |
| This journal is © The Royal Society of Chemistry 2026 |