
Hybrid azine–oxamic acid molecular design: synthesis, structure, and supramolecular implications
Ketlyn Wolfart
Borth
 a,
Letícia dos Santos
Leal
a,
Verônica de Carvalho
Teixeira
b,
Grazielli
da Rocha
a and
Tatiana Renata Gomes
Simões
*a
a,
Letícia dos Santos
Leal
a,
Verônica de Carvalho
Teixeira
b,
Grazielli
da Rocha
a and
Tatiana Renata Gomes
Simões
*a
aDepartment of Chemistry, Federal University of Paraná, Curitiba, 81531-980, Brazil. E-mail: tatiana.renata@ufpr.br
bBrazilian Synchrotron Light Laboratory (LNLS), Brazilian Center for Research in Energy and Materials (CNPEM), Campinas, 13083-100, Brazil
First published on 18th March 2026
Abstract
Since the 1960s, oxamic acid derivatives and their coordination compounds have been extensively studied, with applications ranging from protective agents for marble to biologically relevant systems. However, no reports exist on hybrid molecules combining an azine group with an oxamic acid-derived group. The azine group is notable for its photochromic properties and π-conjugation, which make it valuable in fields such as biomedicine, optoelectronics, and chemical sensing. This work reports the synthesis and structural characterization of two novel molecules incorporating both azine and oxamic acid-derived groups: the ester form, named NN-Et2H2oba, and the acid form, NN-H4oba. The synthesis was initiated by forming the azine group from 2-nitrobenzaldehyde and hydrazine dichlorohydrate, followed by a reduction of the nitro groups and subsequent addition of the oxamic acid-derived group. Based on the structures obtained by single-crystal X-ray diffraction (SXRD), correlations were established with data from FTIR, 1H NMR, and elemental analysis. Additionally, given the critical role of intermolecular interactions in determining material properties, a comprehensive study of nature and energies of the non-covalent interactions was conducted using computational methods, including Hirshfeld surface analysis, fingerprint plots, energy framework diagrams, and crystal lattice energy calculations. Intramolecular N–H⋯N hydrogen bonds, present in both molecules, as well as intermolecular C–H⋯O interactions observed in NN-Et2H2oba, were found to significantly contribute to the stabilization of molecular planarity. This structural conformation endows the molecules with potential for photophysical applications. Moreover, the supramolecular organization of NN-Et2H2oba is primarily stabilized by London dispersive forces, while that of NN-H4oba is stabilized by both dispersive forces and hydrogen bonds.
1 Introduction
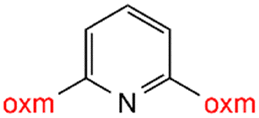
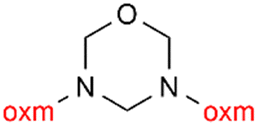
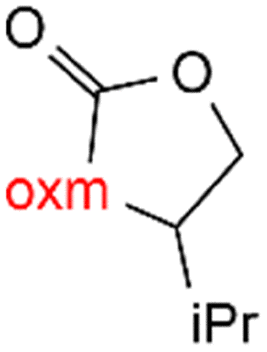
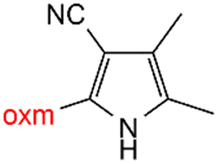
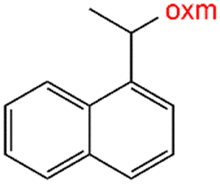
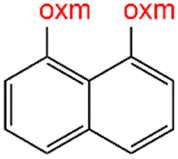
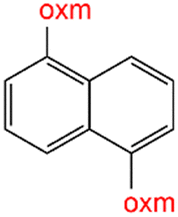
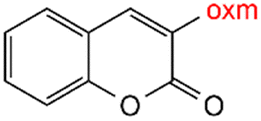
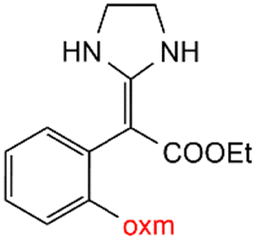
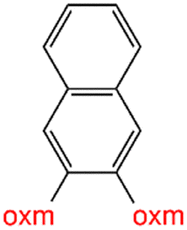
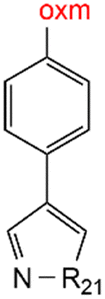
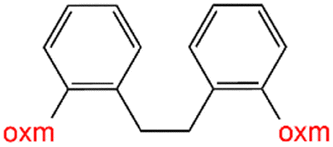
Oxamic acid derivatives and coordination compounds containing such molecules have been reported in the Cambridge Crystallographic Data Center (CCDC) database since the 1960s.1 Oxamic acid consists of both an amide and a carboxylic acid functional group, and its derivatives (oxm) are formed by the substitution of hydrogen atoms with organic moieties (Fig. 1a).2 A detailed database search revealed a wide variety of structural motifs, particularly when substitutions at the amide nitrogen were considered (Table 1). These compounds have attracted interest due to their structural versatility and their ability to participate in multiple intermolecular interactions. | ||
| Fig. 1 Structural drawings of the ester NN-Et2H2oba and acid form NN-H4oba. | ||
|
|
|||||
|---|---|---|---|---|---|
| Structure | Ref. | Structure | Ref. | Structure | Ref. |
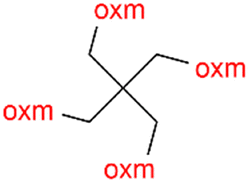
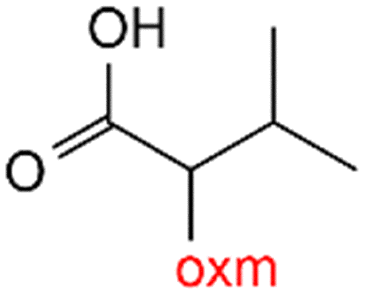
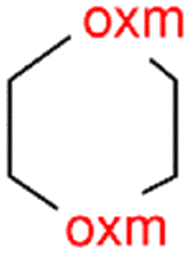
R1 = H, Me, Et, Ph, NH2, CH2Ph, COOtBu, (CH2)2NH2, (CH2)2oxm, (CH2)3oxm, CH2CH(Ph)2, CH2COOH, CONH2, COOCH2Ph, CH2COHCH2oxm, pTol, CH2NH3, CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, CH2C(CH3)2CH2oxm, CH(CH3)COOH, CH2CF3, CH2C(F2CH3), NHC(NH2)2, (CH2)2NHCH3, cy-hexyloxm, CH2CH(NH3)COOH, CH(PhCH3); R2 = OH, COOH, oxm, Cl, Br, F, CONH(CH2)3COOMe, SO2Phoxm, oxmPhoxm, C C, CH2C(CH3)2CH2oxm, CH(CH3)COOH, CH2CF3, CH2C(F2CH3), NHC(NH2)2, (CH2)2NHCH3, cy-hexyloxm, CH2CH(NH3)COOH, CH(PhCH3); R2 = OH, COOH, oxm, Cl, Br, F, CONH(CH2)3COOMe, SO2Phoxm, oxmPhoxm, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPhoxm, CCPhoxm, NNPhoxm, SCH3, OCMe, OMe, NH(CO)2NHCH3, N(Phoxm)2, CH2R1, SO2NH3, COOEt, CHNNC(SNH2); R3 = H, Br, OMe, Me, NO2, CCPhF; R4 = Cl, Me, NO2, CH(Ph)2, iPr; R5 = H, Me, CH(Ph)2, iPr; R6 = NH; R7 = Br; R8 = R9 = R10 = Me; R11 = H, Me, Cl, NO2, N(CH3)2, NCHPhOH; R12 = Me, Cl, NO2, N(CH3)2, COOH, NCHPhOH, CF3, COOH; R13 = Cl, COOH; R14 = OH, CF3; R15 = R16 = R17 = R18 = Me; R19 = R20 = COOH; R21 = NH, O; R22 = H, tBu; R23 = Me, OMe; R24 = C, N; R25 = S; R26 = Me; R27 = oxmPh(CH3)oxm; oxmPh(CH3)oxmPh(CH3)oxm. CPhoxm, CCPhoxm, NNPhoxm, SCH3, OCMe, OMe, NH(CO)2NHCH3, N(Phoxm)2, CH2R1, SO2NH3, COOEt, CHNNC(SNH2); R3 = H, Br, OMe, Me, NO2, CCPhF; R4 = Cl, Me, NO2, CH(Ph)2, iPr; R5 = H, Me, CH(Ph)2, iPr; R6 = NH; R7 = Br; R8 = R9 = R10 = Me; R11 = H, Me, Cl, NO2, N(CH3)2, NCHPhOH; R12 = Me, Cl, NO2, N(CH3)2, COOH, NCHPhOH, CF3, COOH; R13 = Cl, COOH; R14 = OH, CF3; R15 = R16 = R17 = R18 = Me; R19 = R20 = COOH; R21 = NH, O; R22 = H, tBu; R23 = Me, OMe; R24 = C, N; R25 = S; R26 = Me; R27 = oxmPh(CH3)oxm; oxmPh(CH3)oxmPh(CH3)oxm. |
|||||
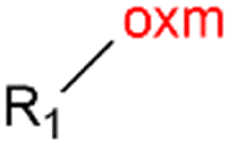
|
15–38 |
|
39 |
|
40 |
|
41 |
|
42–63 |
|
64–70 |
|
71 |
|
12 |
|
72,73 |
|
74 |
|
75 |
|
76 |
|
77 |
|
78 |
|
79 |
|
80 |
|
81 |
|
82 |
|
83 |
|
84 |
|
85 |
|
86 |
|
87 |

|
88 |
|
89 |
|
90 |
|
91 |
|
92,93 |
|
94 |
|
95 |
|
96 |
|
97 |
|
98 |
|
99,100 |
|
101 |
|
102 |
|
103 |
|
104 |
|
105 |
|
106 |
|
107 |
|
108 |

|
109,110 |

|
111 |
|
112 |

|
113 |

|
114 |
|
115 |
|
116 |
|
117 |

|
110 |
|
118 |
|
119 |

|
120 |
|
121 |
|
122 |
|
123 |

As a direct consequence of this structural versatility, these compounds have been widely explored in different contexts. Maiore et al.3 demonstrated that molecules such as ammonium N-phenyloxamate, belonging to this class, act as effective protective agents for Carrara marble and biomicritic limestone against acidic agents by reducing the water solubility of calcium salts deposited on the surface. In addition, molecular docking studies revealed strong interactions between the oxamic acid derivative, 2-guanylhydrazido-oxomethanesulfonic acid, and the human butyrylcholinesterase (BChE) receptor – a critical step towards understanding the biological relevance of aminoguanidine derivatives.4 Chloroxaloterpin B, another oxamic acid derivative, exhibited potent inhibitory activity against Botrytis cinerea spore germination.5 Furthermore, investigations into the linear and nonlinear optical properties of oxamic acid derivatives – such as sulfanilamide [(4-sulfamoylphenyl)carbamoyl]formic acid salt – have demonstrated significant potential for use in advanced optical devices.6
Despite the wide variety of crystal structures reported for oxamic acid derivatives, no studies to date have described the presence of azine groups in combination with oxamic acid moieties. The azine group has a general structure represented by R1R2CN–NCR3R4 and is particularly notable for its photochromic properties, undergoing thermal and photochemical isomerization at the CN bond.7 Azines – especially those with nitrogen-containing aromatic systems – constitute a class of compounds that promote π-conjugation, enabling efficient light absorption and energy transfer. Additionally, they participate in π–π stacking interactions, which have given rise to a significant degree of research interest in azine-containing molecules as building blocks in supramolecular chemistry and functional molecular architectures.8 Azine derivatives have also been employed in the design of multifunctional aggregation-induced emission luminogens (AIEgens), owing to their high selectivity and sensitivity for picric acid detection via hydrogen bonding interactions – highlighting their potential in biomedicine, optoelectronics, and chemical sensing.9 Furthermore, a study by Diyali et al. reported antidiabetic properties in azine derivatives.10
Given the growing interest in multifunctional molecules, the integration of azine groups with oxamic acid-derived compounds represents a promising strategy for the development of hybrid compounds with enhanced structural and functional versatility.11–14 In this context, the present work reports the synthesis and characterization of two novel molecules incorporating both functional groups: the ester NN-Et2H2oba (Fig. 1b) and the acid form NN-H4oba (Fig. 1c). These species exhibit conformational versatility and can adopt antiperiplanar (NN-EtxH4−xobaanti) or gauche (NN-EtxH4−xobgauche) arrangements due to free rotation around the central single nitrogen–nitrogen bond of the azine group (Fig. 1d and e). In addition, this study investigates the nature and energies of intermolecular interactions within their crystal structure – an aspect of relevance, as numerous studies have demonstrated the critical role such interactions play in determining material properties and potential applications. These interactions were analyzed using Hirshfeld surface mapping, fingerprint plots, and lattice energy–structure diagrams. As these are newly synthesized compounds, the study also includes characterization of their thermal properties (melting points) and spectroscopic features using 1H NMR (nuclear magnetic resonance) and IR (infrared) spectroscopy.
2 Experimental section
2.1 Materials
Hydrazine dihydrochloride (≥98%, CAS: 5341-61-7), sodium hydroxide (97%, CAS: 1310-73-2), 2-nitrobenzaldehyde (98%, CAS: 552-89-6), ethanol (99.5%, CAS: 64-17-5), metallic zinc (98%, CAS: 7440-66-6), ammonium chloride (99%, CAS: 12125-02-9), methanol (99.8%, CAS: 67-56-1), dichloromethane (99.5%, CAS: 75-09-2), ethyl chlorooxoacetate (98%, CAS: 4755-77-5), tetrahydrofuran (99%, CAS: 109-99-9), potassium hydroxide (>90%, CAS: 1310-58-3), hydrochloric acid (37%, CAS: 7647-01-0), and dimethyl sulfoxide (99.9%, CAS: 67-68-5) were used. All chemicals and solvents used were of analytical grade and used without further purification in the syntheses.2.2 Synthesis
Oester)], 1710 [ν(COamide)], 1602, 1583 and 1535 [ν(C–Carom)], 1283 [ν(C–N)], 1175 and 1110 [ν(C–O)], 757 [δ(C–Hortho)]. 1H NMR (200 MHz, CDCl3): 13.49 (s, 2H), 9.04 (s, 2H), 8.82 (d, J = 8.3 Hz, 2H), 7.57 (m, 4H), 7.28 (ddd, J = 0.9, 7.3 Hz, 2H). 13C NMR (400 MHz, dmso-d6): 164.2, 160.0, 154.9, 137.4, 133.8, 132.7, 120.7, 120.2, 62.8, 13.8 ppm. Anal. calcd for C22H22N4O6 (NN-Et2H2oba) (%): C, 60.3; H, 5.1; N, 12.8. Found (%): C, 59.2; H, 5.0; N, 12.7.
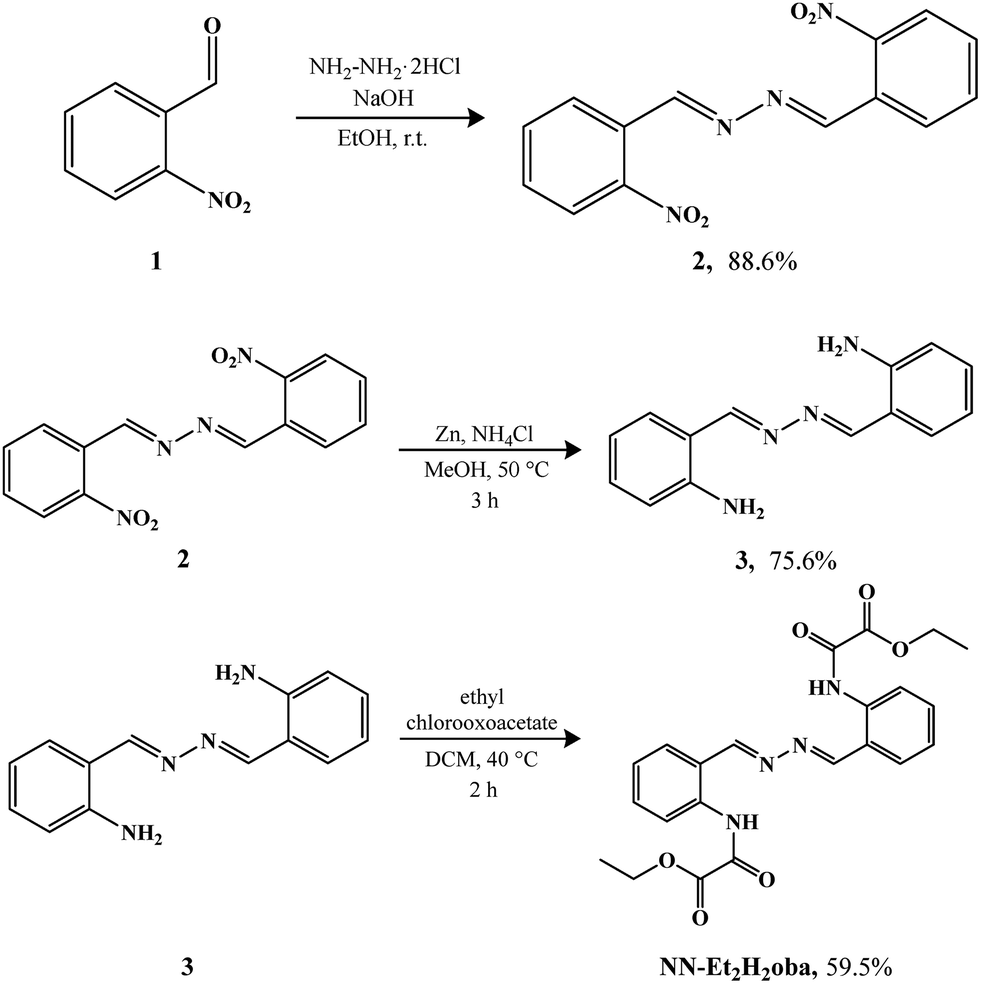 | ||
| Scheme 1 Synthesis steps of NN-Et2H2oba. | ||
Oacid)], 1679 [ν(COamide)], 1605, 1579 and 1543 [ν(C–C)arom], 1354 [ν(C–N)], 1310 [ν(C–O)], 767 [δ(C–Hortho)]. 1H NMR (200 MHz, dmso-d6, TMS), δ (ppm): 13.11 (s, 2H), 8.96 (s, 2H), 8.60 (d, J = 8.4 Hz, 2H), 7.67 (m, 4H) 7.36 (ddd, J = 1.0, 7.5 Hz, 2H). Anal. calcd for C18H14N4O6·dmso (NN-H4oba) (%): C, 52,2; H, 4,4; N, 12.2. Found (%): C, 51.8; H, 4.1; N, 12.9.
 | ||
| Scheme 2 Hydrolysis of the NN-Et2H2oba to obtain compound NN-H4oba. | ||
2.3 Instrumentation
Melting points (uncorrected) were determined using an MP90 Melting Point System (Mettler Toledo) in open capillary tubes. The IR spectrum of NN-Et2H2oba was recorded on a Bomen Michelson MB100 spectrometer using KBr pellets, with a resolution of 4 cm−1 and 16 scans. For NN-H4oba, the IR spectrum was acquired in attenuated total reflectance (ATR) mode at the IMBUIA beamline of the Brazilian Synchrotron Light Laboratory (LNLS) that is part of the Brazilian Center for Research in Energy and Materials (CNPEM), with a resolution of 2 cm−1 and 256 scans. 1H NMR spectra were recorded at 200 MHz on a Bruker DPX-200 spectrometer at ambient temperature using CDCl3 for NN-Et2H2oba and dmso-d6 for NN-H4oba. The 13C NMR spectrum of the NN-Et2H2oba was recorded at 400 MHz on a Bruker Avance III spectrometer at ambient temperature using dmso-d6. Chemical shifts were expressed in ppm and referenced to tetramethylsilane (δTMS = 0.00). Elemental analyses (C, H, and N) were performed using a Thermo Fisher Scientific Flash EA 1112 Series elemental analyzer.2.4 X-ray data collection and refinements
Single crystals of NN-Et2H2oba and NN-H4oba were mounted on Micro-mesh supports and cooled under a nitrogen stream on a Bruker D8 Venture diffractometer equipped with a Photon II C7 detector, a Cu-Kα µS-microsource radiation (λ = 1.54178 Å), and a Göbbel mirror monochromator. Intensity data were measured by thin-slice ω- and ϕ-scans and processed with the APEX4 software package.124 The structure was refined by full-matrix least-squares methods on F2's using SHELXL125 and solved by the intrinsic phasing methods implemented in SHELXT.126 Absorption corrections were applied using a multi-scan method.127 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were located in different Fourier maps and refined isotropically without constraints. Neutral atom scattering factors were taken from standard literature sources.128 All crystallographic calculations were carried out using the WinGX129 suite, and structural figures were generated using Mercury.130 Crystallographic data and structure refinement parameters for NN-Et2H2oba and NN-H4oba are summarized in Table 2. The crystallographic data have been deposited in the Cambridge Structural Database under deposition numbers CCDC 2478688 and 2478689.| Compound | NN-Et2H2oba | NN-H4oba |
|---|---|---|
| Formula | C22H22N4O6 | C18H14N4O6, 4(C2H6OS) |
| Formula weight | 438.43 | 694.86 |
| Crystal system | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2(1)/c |
| a (Å) | 7.4167(2) | 13.269(4) |
| b (Å) | 11.8025(3) | 5.3039(6) |
| c (Å) | 13.2319(4) | 24.374(4) |
| α (°) | 114.5540(10) | 90.00 |
| β (°) | 92.4900(10) | 105.583(15) |
| γ (°) | 96.2670(10) | 90.00 |
| Z | 2 | 2 |
| Wavelength (Å) | 1.54178 | 1.54178 |
| Volume (Å3) | 1042.16(5) | 1652.3(6) |
| T (K) | 100(2) | 100(2) |
| D calc (g cm−3) | 1.397 | 1.397 |
| Crystal size (mm3) | 0.336 × 0.111 × 0.061 | 0.798 × 0.163 × 0.152 |
| F (000) | 460 | 732 |
| µ (mm−1) | 0.865 | 3.144 |
| θ range (°) | 4.2–74.0 | 3.5–70.9 |
| Refl. collected | 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 270 270 |
73279 |
| Refl. indep. (Rint) | 4192 (0.038) | 3183 (0.044) |
| Refl. obs. [I > 2σ(I)] | 3631 | 3106 |
| Restraints/parameters | 0/377 | 0/251 |
| Goodness-of-fit | 1.121 | 1.061 |
| R, wR [I > 2σ(I)] | 0.038, 0.086 | 0.033, 0.088 |
| R, wR (all data) | 0.047, 0.091 | 0.034, 0.089 |
| Largest diff. peak and hole (e Å−3) | 0.24, −0.20 | 0.43, −0.38 |
| Reflections weighted | w = [s2(Fo2) + (0.0211P)2 + 0.6738P]−1 | w = [σ2(Fo2) + (0.0516P)2 + 0.9103P]−1 |
| where P = (Fo2 + 2Fc2)/3 | where P = (Fo2 + 2Fc2)/3 |
2.5 Hirshfeld surface (HS) analysis, two-dimensional fingerprint plots, and energy-framework diagrams
The Hirshfeld surface analyses131,132 and the non-covalent interaction energy133 calculations for NN-H4oba and NN-Et2H2oba were performed using the crystallographic information files (CIF) obtained from single-crystal X-ray diffraction. All computations were carried out with CrystalExplorer 21.5.134 The energy framework analyses were based on density functional theory (DFT) calculations using the B3LYP/6-31G(d,p) level of theory.3 Results and discussion
3.1 General characterization
The 1H NMR and 13C NMR spectra of NN-Et2H2oba are shown in Fig. S1, while the 1H NMR spectrum of NN-H4oba is presented in Fig. S2. For NN-Et2H2oba, the 1H NMR spectrum displays eight distinct signals, consistent with the proposed molecular structure. The singlets at 9.04 and 13.49 ppm are attributed to hydrogens 1 and 6, respectively. Hydrogen 2 appears as a doublet centered at 8.82 ppm (J = 8.2 Hz, 2H). The aromatic region (hydrogens 3–5) shows hydrogen 3 as a triplet of doublet (J = 0.9, 7.4 Hz, 2H), while hydrogens 4 and 5 appear as a triplet and a doublet, respectively, overlapping into a single broadened signal. The quartet at 4.48 ppm and the triplet at 1.48 ppm correspond to the ethyl group, further supporting ester formation. In the 13C NMR spectrum, eleven signals corresponding to the characteristic carbon atoms of the NN-Et2H2oba ester are observed. Resonances in the 150–165 ppm region are attributed to the carbon atoms of the oxamic acid-derived group and to the CN carbon of the azine unit. The signals associated with the aromatic ring carbons appear in the 120–140 ppm range. The formation of the ester is confirmed by the presence of characteristic CH3 and CH2 carbon signals of the ethyl group at 13.8 and 62.8 ppm, respectively. In the spectrum of the acid NN-H4oba, hydrogens 3 and 5 appear as a triplet of doublets at 7.36 ppm (J = 1.0, 7.5 Hz, 2H) and a doublet at 8.60 ppm (J = 8.43 Hz, 2H), respectively. The multiplet centered at 7.67 ppm corresponds to hydrogens 2 and 4. The azine –CN– hydrogen appears at 8.96 ppm and the amide N–H at 13.11 ppm.135
The IR spectra of NN-Et2H2oba and NN-H4oba are presented in Fig. S3. In both spectra, the band corresponding to the out-of-plane angular deformation of the ortho-substituted aromatic ring hydrogen is observed at 757 cm−1 for NN-Et2H2oba and at 767 cm−1 for NN-H4oba. It is also possible to identify the bands that indicate the presence of the oxamate group corresponding to the C–O and C–N stretching vibrations (1110 cm−1, 1175 and 1283 cm−1 for NN-Et2H2oba and 1310 cm−1 and 1354 cm−1 for NN-H4oba). Furthermore, the most intense bands in the spectrum are associated with the CO stretching of the oxamate groups (1710 and 1735 cm−1 for NN-Et2H2oba; 1676 and 1766 cm−1 for NN-H4oba). The bands corresponding to C–Carom stretching vibrations were identified at 1535, 1583, and 1602 cm−1 for the ester, and at 1543, 1579, and 1605 for the acid. In both spectra, the presence of bands associated with C–Harom stretching vibrations was observed (3073 and 3114 cm−1 for NN-Et2H2oba and 3108 cm−1 for NN-H4oba) while the bands originating from C–Haliph stretching vibrations were observed only in the NN-Et2H2oba spectrum (2976 and 2935 cm−1), confirming the presence of the ethyl ester group. Spectra exhibit N–H stretching vibrations at 3235 cm−1 for the ester and at 3255 cm−1 for the acid.136 The observed bands are consistent with the structures determined by SXRD.
3.2 The molecular and supramolecular structures of NN-Et2H2oba and NN-H4oba
The molecular structures of NN-Et2H2oba and NN-H4oba determined from SXRD are shown in Fig. 2. A summary of the crystallographic data and refinement parameters is provided in Table 2. Both molecules adopt an antiperiplanar conformation (NN-EtxH4−xobaanti), probably due to the steric hindrance imposed by the oxamic acid-derived groups. Furthermore, a center of symmetry is located at the midpoint of each molecule, between the N–N bonds of the azine group.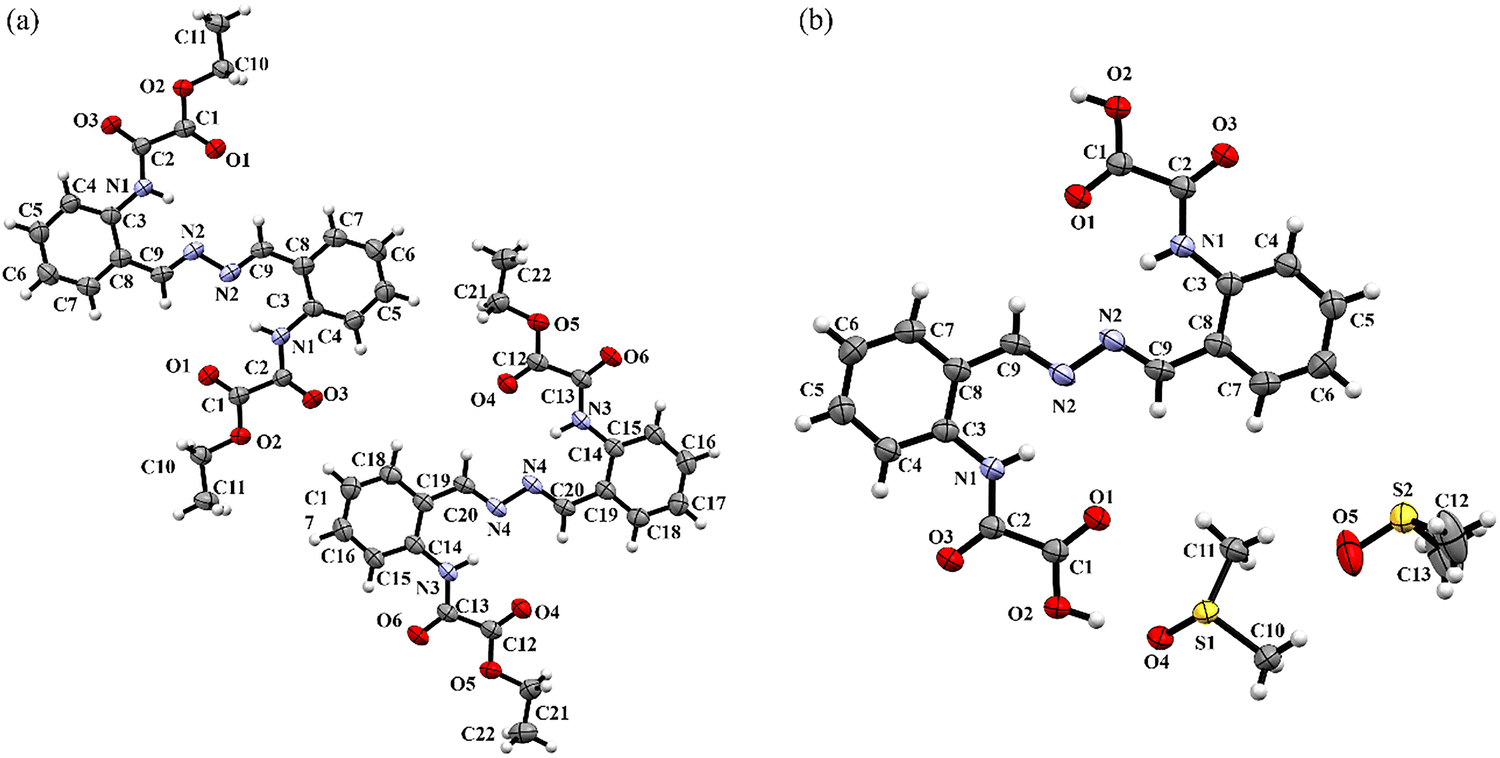 | ||
| Fig. 2 (a) NN-Et2H2oba; (b) NN-H4oba. Thermal ellipsoids are drawn at the 80% probability level. | ||
NN-Et2H2oba crystallizes in the triclinic space group P. The crystal structure reveals two crystallographically independent molecules in the asymmetric unit, each represented by half a molecule, with no solvent molecules present. Bond lengths within the azine group agree with the values reported for similar molecules (see Table S1).137 Similarly, the bond lengths in the oxamic acid-derived group are consistent with literature data, particularly with the values reported by Pim et al.138 for aromatic oxamic acid derived in ester form (H2Et2L) (see Table S1). Among the two crystallographically distinct units, one is essentially planar (lower unit – Fig. 3a and b), whereas the other exhibits a slight deviation from planarity, with an interplanar angle of α = 8.38° between the plane of the azine group (C20–N4–N4–C20) and that of the oxamic acid-derived group (N3–C13–C12–O4) (upper unit – Fig. 3a and b).
 | ||
| Fig. 3 Representation of the plane formed by the azine group (gray) in (a) and (c), and the angle formed between this plane and the plane defined by the oxamic acid-derived group (red) in (b) and (d), for NN-Et2H2oba and NN-H4oba, respectively. | ||
The acid form, NN-H4oba, crystallizes in the monoclinic space group P2(1)/c, with an inversion center imposed by symmetry within the azine group. As a result, the asymmetric unit comprises half of the NN-H4oba molecule together with two dmso molecules. The C9–N2 and N2–N2 bond lengths are very similar to those in NN-Et2H2oba (see Table S1), resulting in similar structural features. The same trend is observed for the oxamic acid-derived groups, whose bond lengths show minimal deviations from those in the ester and from the literature data for H3opba139 (Table S1). The molecule adopts a quasiplanar conformation (Fig. 3c), with a dihedral angle of α = 3.22° (Fig. 3d).
The three-dimensional packing of NN-Et2H2oba (Fig. 4 and Fig. S4) is governed by intra- and intermolecular hydrogen bonds together with dispersive interactions. Intramolecular N–H⋯N interactions involving amide and azine groups are observed in both crystallographically independent molecules in the bc plane (N1–H⋯N2 and N3–H⋯N4; Fig. 4a), with distances of 1.88(2) and 1.931(19) Å and angles of 140.0(2)° and 136.9(2)° (Table 3), consistent with moderate interactions.140 This interaction restricts intramolecular rotation in azine bonds, which may be useful in applications such as aggregation-induced emission.9 Peng et al.141 demonstrated the application of a salicylaldehyde azine as a fluorophore, in which these properties originated in part from hydrogen bonding between the hydroxyls of the aromatic ring and the nitrogen of the azine, forming interactions similar to N–H⋯N. Intermolecular C–H⋯O hydrogen bonds (C4–H⋯O3 and C15–H⋯O6; Fig. 4a) connect adjacent molecules between the oxygen from oxamic acid-derived groups and the aromatic rings, forming a supramolecular chain. This results in a hetero-R22(12) ring motif, with donor–acceptor distances of 2.413(18) Å and 2.390(18) Å, and angles of 133.9(13)° and 136.4(14)°, respectively. Together with the intramolecular N–H⋯N contacts, these interactions stabilize the molecular planarity. The supramolecular chains pack along the a-axis through London dispersion forces (Fig. 4b), involving hydrogen, carbon, and oxygen contacts. Among these, a weak carbonyl–carbonyl n → π* interaction142,143 is observed, which is attributed to the partial donation of electron density from the non-bonding electron pair (n) located on the oxygen atom of the carbonyl group to the antibonding π* orbital of the neighboring carbon atom from carbonyl. These contacts adopt the geometry of a sheared-parallel motif.142 Specifically, interactions between C12–C13⋯O3 and C12ii–C13ii⋯O3i (Fig. 4b) are identified, with contact distances of 3.157(2) and 3.653(2) Å [symmetry code: (i) 1 − x, 1 − y, 1 − z; (ii) −x, 1 − y, 1 − z]. In a previous work by Morales–Santana et al.,144 a molecule containing the oxamate group showed a similar interaction between carbon and sulfur, classified as a π–hole interaction.
 | ||
| Fig. 4 (a) Hydrogen bonds and (b) dispersion interactions stabilizing the supramolecular structure in NN-Et2H2oba. | ||
| Interactions | Distance H⋯A (Å) | Angle (°) |
|---|---|---|
| Symmetry code: (i) −1 + x, −1 + y, z; (ii) x, y, 1 + z; (iii) x, 1 + y, z; (iv) x, 1.5 − y, 0.5 + z; (v) 2 − x, 0.5 + y, 1.5 − z. | ||
| NN-Et2H2oba | ||
| C4–H5⋯O3i | 2.390(18) | 136.4(14) |
| C15–H16⋯O6ii | 2.413(18) | 140.0(17) |
| N1–H6⋯N2 | 1.88(2) | 133.9(13) |
| N3–H17⋯N4 | 1.931(19) | 136.9(17) |
| NN-H4oba | ||
| C11–H⋯O1 | 2.52(2) | 136.4(15) |
| C11–H⋯O1iii | 2.44(2) | 154.7(17) |
| C12–H⋯O2iv | 2.39 | 155.5 |
| C13–H⋯O3iv | 2.48 | 149.5 |
| C10v–H⋯O3iv | 2.59(2) | 152.2(17) |
| O2–H⋯O4 | 1.54(3) | 179(2) |
| C10–H⋯O5 | 2.43(2) | 155.0(16) |
| C11v–H⋯O5iii | 2.44(2) | 148.8(16) |
| C11–H⋯O5 | 2.44(2) | 148.8(16) |
| C11v–H⋯O5v | 2.44(2) | 148.8(16) |
| C12–H⋯O5v | 2.63 | 178.5 |
| N1–H2⋯N2 | 1.97(2) | 139.7(17) |
The supramolecular structure of NN-H4oba (Fig. 5a) exhibits the same intramolecular N1–H⋯N2 hydrogen bond between amide and azine groups, as in NN-Et2H2oba, with a bond length of 1.97(2) Å and an angle of 139.7(17)°, which contributes to the molecular planarity and similar functional implications. The packing in the bc plane is further stabilized by an extended hydrogen-bond network involving oxamic acid oxygen atoms and crystallization dmso molecules (Fig. 5a and Fig. S5a), comprising both strong and moderate interactions (Table 3). In the b direction (Fig. S5a), these hydrogen bonds are also involved; however, the molecular units within the layers are connected through interactions similar to those observed for the ester form [C2–C1⋯O4iv 3.006(2) and C1–C2⋯O4iv 3.104(3) Å; symmetry code: (iv) x, −1 + y, z], forming a hetero-R21(3) ring.145 In this arrangement, the carbonyl group interacts with the sulfoxide group, with the oxygen atom from the sulfoxide group acting as an electron-density donor toward the π* orbital carbon from the carbonyl group. This interaction gives rise to a zigzag-type supramolecular arrangement (Fig. 5b). In this case, the n → π* interaction occurs between the NN-H4oba unit and the dmso molecule, in contrast to NN-Et2H2oba, where the interaction takes place between the units themselves through carbonyl groups. Furthermore, the adopted geometry of the interaction is perpendicular.142 Consequently, the interaction in the acid form is stronger than that in the ester form, due to reduced steric hindrance.
 | ||
| Fig. 5 (a) Hydrogen bonds that form the bc plane. (b) n → π* interactions stabilizing the supramolecular structure in the b direction in NN-H4oba. | ||
The Hirshfeld surfaces (HSs) and fingerprint plots of NN-Et2H2oba and NN-H4oba were mapped using the normalized contact distance (dnorm), defined from de (external distance) and di (internal distance). The color coding on the HSs indicates the proximity of interatomic contacts relative to the sum of the van der Waals radii: red = smaller, blue = larger, and white = similar distance. The 2D fingerprint plots and percentage contributions are shown in Fig. S6 and S7. In NN-Et2H2oba, the absence of crystallization solvents results in interactions exclusively between the NN-Et2H2oba units. HSs (Fig. 6a) display bright red spots associated with medium-strength hydrogen bonds between amide oxygen and the aromatic hydrogen – previously identified in Fig. 4. These O⋯H/O⋯H interactions contribute 19.5% to the overall supramolecular characteristics (Fig. S6). The remaining interactions are mainly dispersive (Fig. 6b and c), involving H⋯H, C⋯C, H⋯C, and C⋯O atoms, represented by small light-red spots. Among these, an n → π* interaction (Fig. 6c) – previously described in Fig. 4b – is also identified. Ethyl groups enable additional hydrogen-based interactions. Major contributions are H⋯H/H⋯H (43.2%) and C⋯C/C⋯C (19.7%), while C⋯O/C⋯O (5.3%) and C⋯C/C⋯C (3.0%) are less significant. Similar results have been reported in the literature for molecules containing the azine moiety, in which H⋯H/H⋯H contacts are likewise identified as the dominant contributors to the crystal packing, accounting for 44.8%146 and 48.0%147 of the total intermolecular interactions.
 | ||
| Fig. 6 Views of the Hirshfeld surfaces mapped over dnorm for NN-Et2H2oba. Emphasis on (a) hydrogen-bonding interactions and (b) and (c) dispersive-type interactions. | ||
In the NN-H4oba (Fig. 7), bright red spots indicate strong hydrogen bonds between the NN-H4oba and dmso1a. The dmso molecules are identified according to the sulfur symmetry code (1 refers to Si and 2 to Sii): 1a (1 − x, 1 − y, 1 − z), 1b (1 − x, 2 − y, 1 − z), 1c (−1 + x, y, z) and 2a (1 − x, −1/2 + y, 1.5 − z). Additional interactions with the dmso1b, dmso1c, and dmso2a molecules appear as light red spots (Fig. 7). These visible O⋯H/O⋯H and C⋯O/C⋯O interactions contribute 25.7% and 5.0%, respectively, to the supramolecular features (Fig. S7). Other major contributions are H⋯H/H⋯H (33.4%) and C⋯H/C⋯H (20.2%), followed by N⋯H (5.6%) and C⋯C (4.5%), which, although smaller, still influence the supramolecular organization.
 | ||
| Fig. 7 Views of the Hirshfeld surfaces mapped over dnorm for NN-H4oba. The dmso molecules are identified according to the symmetry code of the S atom (1 corresponds to Si and 2 to Sii): 1a (1 − x, 1 − y, 1 − z), 1b (1 − x, 2 − y, 1 − z), 1c (−1 + x,y,z) and 2a (1 − x, −1/2 + y, 1.5 − z). | ||
The interaction energy results for NN-Et2H2oba and NN-H4oba are summarized in Table 4 (lines 1–10). Only contacts with total interaction energy (Etot) values greater than −15.0 kJ mol−1 are discussed. The molecular environments are shown in Fig. S8 and S9. For NN-Et2H2oba, the highest stabilization energy (Etot = 104.6 kJ mol−1 – line 5) is mainly due to London dispersion involving hydrogen and carbon atoms, as well as n → π* interactions between the carbonyl oxygen of the oxamic acid-derived moiety and the carbon atoms of the same group. The second most stabilizing interaction is the R22(12) heterocyclic ring formed by hydrogen bonds between aromatic hydrogen atoms and the carbonyl oxygen of the amide, with Etot = 37.4 kJ mol−1 (line 3). In all these interactions, as well as in the remaining three interactions, dispersion energy (Edis) is greater than electrostatic energy (Eele). Dispersion contributions (%Edis) range from 50.1% to 85.6%, confirming that London dispersion dominates the stabilization of the NN-Et2H2oba structure.
| Line | Units | Interaction | −Eele | −Epol | −Edis | E rep | −Etot | %Eelec | %Edisc | R |
|---|---|---|---|---|---|---|---|---|---|---|
| a Asymmetric unit 1. b Asymmetric unit 2. c The % contribution of the component (electrostatic or dispersion) (%Ecomp) is calculated through (Ecomp/Estab) × 100, in which Estab = Eele + Epol + Edis. d R = distance between centroids in Å; Etot = keleEele + kpolEpol + kdisEdis + krepErep (scale factors: kele: 1.057; kpol: 0.740; kdis: 0.871; krep: 0.618). | ||||||||||
| NN-Et2H2oba | ||||||||||
| 1 | NN-Et2H4obaa⋯NN-Et2H4obab |
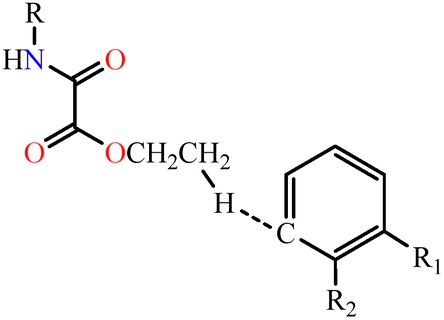
|
5.4 | 1.1 | 25.2 | 15.2 | 19.1 | 17.0 | 79.5 | 11.98 |
| 2 | NN-Et2H4obab⋯NN-Et2H4obaa |
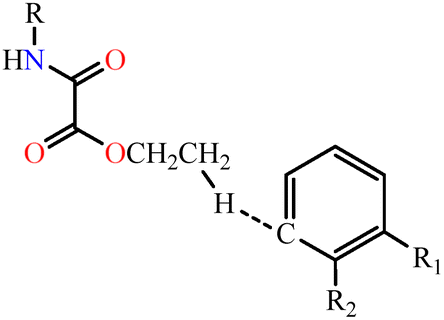
|
5.3 | 1.8 | 15.9 | 0.0 | 20.8 | 23.0 | 69.1 | 13.90 |
| 3 | NN-Et2H4obaa⋯NN-Et2H4obaa |
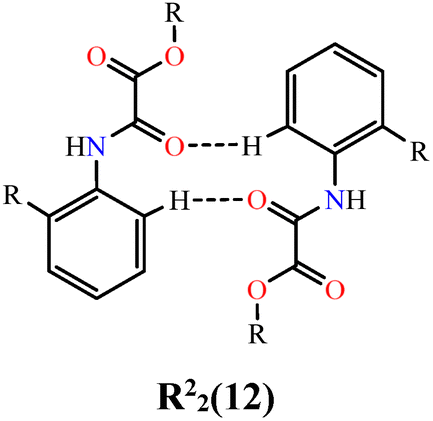
|
13.5 | 7.0 | 20.6 | 0.0 | 37.4 | 32.8 | 50.1 | 13.23 |
| 4 | NN-Et2H4obaa⋯NN-Et2H4obaa |
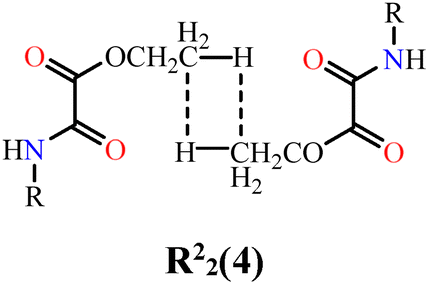
|
3.4 | 1.4 | 28.6 | 0.0 | 29.6 | 10.2 | 85.6 | 13.24 |
| 5 | NN-Et2H4obaa⋯NN-Et2H4obab |
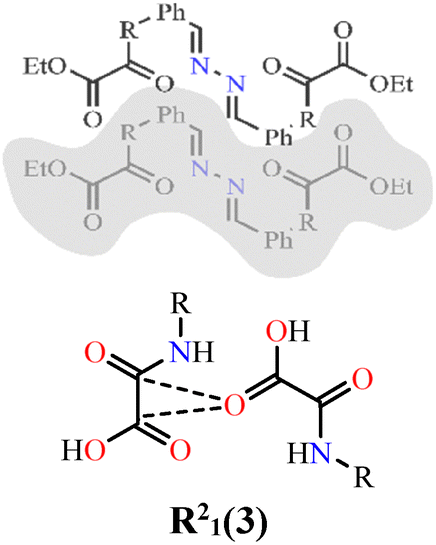
|
27.4 | 6.4 | 134.9 | 75.5 | 104.6 | 16.2 | 80.0 | 3.71 |
| NN-H4oba | ||||||||||
| 6 | NN-H4oba⋯dmso1a |
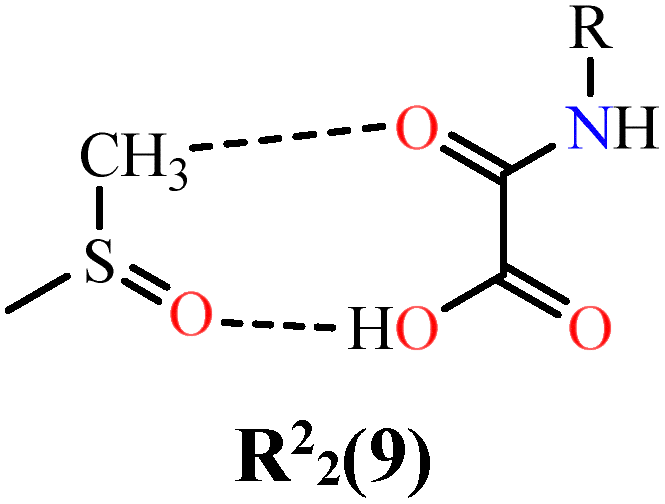
|
110.2 | 24.8 | 19.1 | 143.3 | 62.8 | 71.5 | 12.4 | 6.57 |
| 7 | NN-H4oba⋯dmso1b |
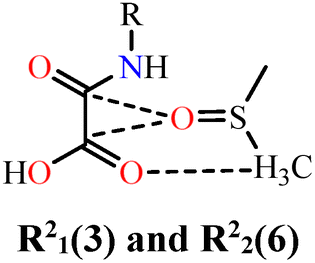
|
12.0 | 5.7 | 27.7 | 26.2 | 24.8 | 26.4 | 61.0 | 4.96 |
| 8 | NN-H4oba⋯dmso2a |
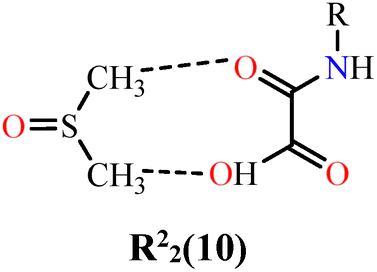
|
7.6 | 2.9 | 79.1 | 34.2 | 58.0 | 8.5 | 88.3 | 9.02 |
| 9 | NN-H4oba⋯dmso1c |
|
13.5 | 2.4 | 10.4 | 11.0 | 18.3 | 51.3 | 39.5 | 8.66 |
| 10 | NN-H4oba⋯NN-H4oba |
|
7.6 | 2.9 | 79.1 | 34.2 | 57.9 | 8.5 | 88.3 | 5.30 |
In the case of NN-H4oba, the shortest hydrogen bond corresponds to the most stabilizing interaction, the heterocyclic ring R22(9) formed between NN-H4oba and dmso1a (Etot = 62.8 kJ mol−1; line 6), consistent with literature on similar oxamic acid-derived groups and dmso molecules rings.144 Two further interactions contribute significantly to supramolecular stabilization: (i) hydrogen bonding between the oxygen atoms of the oxamic acid-derived and the methyl hydrogen atoms of dmso2a, forming R22(10) (Etot = 58.0 kJ mol−1; line 8), with Edis = 88.3% and e Eele = 8.5%; and (ii) a purely dispersive contact between NN-H4oba units themselves (Etot = 57.9 kJ mol−1; line 10), also dominated by 88.3% dispersion forces. Additional contributions arise from the R21(3) and R22(6), and R21(6) motifs, with Etot values of 24.8 kJ mol−1 (line 7) and 18.3 kJ mol−1 (line 9). Therefore, both systems benefit from dispersive contributions. NN-Et2H2oba is mainly stabilized by dispersive interactions and steric effects from ethyl groups. In contrast, NN-H4oba presents the largest contribution from directional hydrogen bonds with active participation of the crystalline solvent, complemented by London dispersion energies comparable to hydrogen bonds. Small changes in the crystalline environment can have a substantial impact on the supramolecular energy balance.
The data in Table 3 are represented in the energy framework diagrams in Fig. 8, constructed for clusters of nearest-neighbor molecules within 3.8 Å of a central NN-Et2H2oba or NN-H4oba fragment. In both crystals, dispersion energy (green) predominates over electrostatic energy (red), as also reflected in the total energy frameworks (blue). This dominance agrees with the fingerprint plots, which show high frequency of H⋯H and C⋯H contacts. Moreover, most hydrogen bonds in both structures involve carbon atoms as donors, highlighting the key role of weak, cumulative noncovalent interactions in stabilizing the supramolecular structures. These results contrast with the findings reported by Morales et al.,144 in which systems containing only the oxamate group and solvent molecules were predominantly electrostatic interactions. One hypothesis to explain our results is that the planarity of the molecules—favored by the intramolecular hydrogen bonds between the azine nitrogen and the amide hydrogen—also contributes to spatially enhancing dispersive interactions.
 | ||
| Fig. 8 Energy-framework diagrams for Edis, Eele, and Etot for a cluster of nearest-neighbor molecules in (a) NN-Et2H2oba and (b) NN-H4oba. All diagrams use the same cylinder scale of 50 for energies and no interaction was cut off. | ||
4 Conclusion
The synthetic routes for hybrid compounds containing azine and oxamic acid derivatives, NN-Et2H2oba and NN-H4oba, have been successfully established. Their crystal structures, elucidated by single-crystal X-ray diffraction, reveal that both molecules adopt an antiperiplanar conformation. Notably, the acidic form of the molecule incorporates solvent molecules within the crystal lattice, while the ester form crystallizes as solvent-free molecular units, which significantly altered the forces involved in stabilizing the supramolecular structure. Hirshfeld surface analysis, two-dimensional fingerprint plots, and energy framework diagrams collectively indicate that, for NN-Et2H2oba, the structure stability is partly governed by hydrogen bonding but is predominantly dictated by dispersive interactions. In contrast, the supramolecular stabilization of NN-H4oba is mainly attributed to hydrogen bonding with the crystallization dmso molecules. Despite these differences, energy framework analyses confirm that dispersion forces are the dominant contributors to the overall stabilization in both structures, significantly favored by high molecular planarity and intramolecular N-H⋯N interactions, which restrict rotational freedom in the azine portion. This conformational restriction, previously associated with aggregation-induced emission in azine-based systems, underscores the relevance of these structural characteristics for potential photophysical applications. A detailed understanding of these intermolecular interactions is crucial for the rational design of new molecular architectures with tailored physicochemical properties, particularly in fields such as biomedicine, sensing technologies, and optoelectronic materials.Author contributions
Ketlyn W. Borth: methodology, formal analysis, data curation, investigation, writing – original draft, and software. Letícia dos S. Leal: methodology. Grazielli da Rocha: formal analysis and data curation. Verônica de C. Teixeira: writing – review & editing, supervision, project administration, funding acquisition, and conceptualization. Tatiana R.G. Simões: writing – review & editing, supervision, project administration, funding acquisition, and conceptualization.Conflicts of interest
No conflicts of interest exist.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary information (SI). Supplementary information: 1H NMR spectra, IR spectra, SXRD structural data, fingerprints, and data from calculations of intermolecular interactions. Crystallographic data for compounds NN-Et2H2oba and NN-H4oba were deposited at the Cambridge Crystallographic Data Center (CCDC), with numbers CCDC 2478688 (NN-Et2H2oba) and 2478689 (NN-H4oba). See DOI: https://doi.org/10.1039/d5nj04656d.CCDC 2478688 and 2478689 contain the supplementary crystallographic data for this paper.148a,b
Acknowledgements
The authors acknowledge the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Araucária, Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, #2021/08111-2), and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) Projects 1 – Finance Code 001, for financial support. They also thank the IMBUIA beamline from the LNLS/CNPEM for the support during the IR measurements under proposal #20232001.References
- B. Beagley and R. W. H. Small, Proc. R. Soc. London, Ser. A, 1963, 275, 469–491 CAS.
- H. O. Stumpf, Y. Pei, O. Kahn, J. Sletten and J. P. Renard, J. Am. Chem. Soc., 1993, 115, 6738–6745 CrossRef CAS.
- L. Maiore, M. C. Aragoni, G. Carcangiu, O. Cocco, F. Isaia, V. Lippolis, P. Meloni, A. Murru, A. M. Z. Slawin, E. Tuveri, J. D. Woollins and M. Arca, New J. Chem., 2016, 40, 2768–2774 RSC.
- R. Selvakumar, S. J. Geib, A. Muthu Sankar, T. Premkumar and S. Govindarajan, J. Phys. Chem. Solids, 2015, 86, 49–56 CrossRef CAS.
- Y. Bi and Z. Yu, J. Agric. Food Chem., 2016, 64, 8525–8529 CrossRef CAS PubMed.
- J. Wojnarska, M. Gryl, T. Seidler and K. M. Stadnicka, CrystEngComm, 2018, 20, 3638–3646 RSC.
- J. Safari and S. Gandomi-Ravandi, RSC Adv., 2014, 4, 46224–46249 RSC.
- S. Kotowicz, M. Siwy, S. Golba, J. G. Malecki, H. Janeczek, K. Smolarek, M. Szalkowski, D. Sek, M. Libera, S. Mackowski and E. Schab-Balcerzak, J. Lumin., 2017, 192, 452–462 CrossRef CAS.
- M. Sathiyaraj, K. Pavithra and V. Thiagarajan, New J. Chem., 2020, 44, 8402–8411 RSC.
- N. Diyali, M. Chettri, A. De and B. Biswas, Eur. J. Chem., 2022, 13, 234–240 CrossRef CAS.
- C. O. C. da Silveira, S. H. M. Santos, T. T. da Cunha, W. X. C. Oliveira, K. C. V. Nolasco, W. D. do Pim, A. A. de Almeida, M. Knobel, C. B. Pinheiro, J. C. D. Figueiredo-Júnior, L. C. A. Oliveira, E. F. Pedroso and C. L. M. Pereira, J. Mol. Struct., 2023, 1294, 136472 CrossRef CAS.
- L.-N. Ma, Y.-K. Lu, W.-J. Shi, L. Hou and Y.-Y. Wang, Inorg. Chem. Commun., 2019, 107, 107490 CrossRef CAS.
- I. Kuźniarska-Biernacka, M. M. M. Raposo, R. M. F. Batista, O. S. G. P. Soares, M. F. R. Pereira, P. Parpot, C. Oliveira, E. Skiba, E. Jartych, A. M. Fonseca and I. C. Neves, J. Mol. Struct., 2020, 1206, 127687 CrossRef.
- F. Mircheraghali, A. Mesbah, S. Sharifi, E. Iravani, B. Neumüller and S. M. Bafrouei, J. Mol. Struct., 2025, 1321, 140092 CrossRef CAS.
- A. Hammershøi and M. Magnussen, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 2009, 65, m201–m204 CrossRef PubMed.
- J. Tercero, C. Diaz, J. Ribas, J. Mahía and M. A. Maestro, Inorg. Chem., 2002, 41, 5373–5381 CrossRef CAS PubMed.
- Y. Maruno, K. Yabe, H. Hagiwara, T. Fujinami, N. Matsumoto, N. Re and J. Mrozinski, Bull. Chem. Soc. Jpn., 2010, 83, 1511–1517 CrossRef CAS.
- D. J. Price and M. Martinez-Belmonte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m3158–m3158 CrossRef CAS.
- H. Wyss, F. Brisse and S. Hanessian, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 1984, 40, 1894–1897 CrossRef.
- J. F. do Prado, A. K. Valdo, F. T. Martins, R. C. de Santana and D. Cangussu, J. Mol. Struct., 2020, 1203, 127468 CrossRef CAS.
- G. Türkoglu, C. Pubill Ulldemolins, R. Müller, E. Hübner, F. W. Heinemann, M. Wolf and N. Burzlaff, Eur. J. Inorg. Chem., 2010, 2962–2974 CrossRef.
- H. S. Shieh and D. Voet, Acta Crystallogr., Sect. B, 1975, 31, 2192–2201 CrossRef.
- R. Marx, S. J. Glaser and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o1425–o1426 CrossRef CAS.
- O. Kahn, Y. Pei, M. Verdaguer, J. P. Renard and J. Sletten, J. Am. Chem. Soc., 1988, 110, 782–789 CrossRef CAS.
- L. Maiore, M. C. Aragoni, G. Carcangiu, O. Cocco, F. Isaia, V. Lippolis, P. Meloni, A. Murru, A. M. Z. Slawin, E. Tuveri, J. D. Woollins and M. Arca, New J. Chem., 2016, 40, 2768–2774 RSC.
- F. R. Fortea-Pérez, I. Schlegel, M. Julve, D. Armentano, G. De Munno and S.-E. Stiriba, J. Organomet. Chem., 2013, 743, 102–108 CrossRef.
- X. Liu, W. Liu, Z. Ju, J. Jiang and W. Liu, Inorg. Chem., 2022, 61, 19658–19662 CrossRef CAS PubMed.
- A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391–4394 CrossRef CAS PubMed.
- T. Grancha, J. Ferrando-Soria, D. Armentano and E. Pardo, J. Coord. Chem., 2019, 72, 1204–1221 CrossRef CAS.
- R. Selvakumar, S. J. Geib, T. Premkumar and S. Govindarajan, Polyhedron, 2015, 87, 321–328 CrossRef CAS.
- X. Yu, H. Zhang, Y. Cao, Y. Chen and Z. Wang, J. Solid State Chem., 2006, 179, 247–252 CrossRef CAS.
- J. Köhling, N. Kalinovich, R. Pajkert, S. N. Tverdomed, E. Lork, V. Wagner and G. Röschenthaler, ChemistrySelect, 2021, 6, 1882–1886 CrossRef.
- R. Selvakumar, S. J. Geib, A. Muthu Sankar, T. Premkumar and S. Govindarajan, J. Phys. Chem. Solids, 2015, 86, 49–56 CrossRef CAS.
- C. Duan, D. Luo, H. Zeng, M. Kang and Z. Lin, CrystEngComm, 2012, 14, 5734 RSC.
- A. J. Davis, M. B. Hursthouse, M. Motevalli, P. O’Brien and P. B. Nunn, Phytochemistry, 1991, 30, 3635–3638 CrossRef CAS.
- J. Bálint, G. Egri, M. Czugler, J. Schindler, V. Kiss, Z. Juvancz and E. Fogassy, Tetrahedron: Asymmetry, 2001, 12, 1511–1518 CrossRef.
- G. S. Papaefstathiou, A. Peeters, A. T. H. Lenstra, H. O. Desseyn and S. P. Perlepes, J. Mol. Struct., 2001, 559, 167–177 CrossRef CAS.
- X.-W. Li, X.-H. Zhao, Y.-T. Li and Z.-Y. Wu, Inorg. Chim. Acta, 2019, 488, 219–228 CrossRef CAS.
- M. Liang, L. Zhu, S. Liu, D. Liao, Z. Jiang, S. Yan and P. Cheng, Z. Anorg. Allg. Chem., 2004, 630, 1655–1658 CrossRef CAS.
- T. Grancha, J. Ferrando-Soria, D. M. Proserpio, D. Armentano and E. Pardo, Inorg. Chem., 2018, 57, 12869–12875 CrossRef CAS PubMed.
- F. J. Martínez-Martínez, R. E. Rojas-Pérez, E. V. García-Báez, H. Höpfl and I. I. Padilla-Martínez, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, o699–o701 CrossRef PubMed.
- M. Yu, L. Wang, L. Wang and Z. Wu, IUCrdata, 2020, 5, x200603 CrossRef CAS PubMed.
- Y. Bi and Z. Yu, J. Agric. Food Chem., 2016, 64, 8525–8529 CrossRef CAS PubMed.
- S. Rasapalli, Y. Huang, V. R. Sammeta, R. Alshehry, F. Anver, J. A. Golen, S. Krishnamoorthy and S. P. Chavan, Tetrahedron Chem, 2024, 9, 100062 CrossRef CAS PubMed.
- T. Kundu, K. Manna, A. K. Jana and S. Natarajan, New J. Chem., 2019, 43, 13263–13270 RSC.
- M.-C. Dul, E. Pardo, R. Lescouëzec, L.-M. Chamoreau, F. Villain, Y. Journaux, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, J. Pasán and C. Ruiz-Pérez, J. Am. Chem. Soc., 2009, 131, 14614–14615 CrossRef CAS PubMed.
- T. T. da Cunha, V. M. M. Barbosa, W. X. C. Oliveira, E. F. Pedroso, D. M. A. García, W. C. Nunes and C. L. M. Pereira, Inorg. Chem., 2020, 59, 12983–12987 CrossRef CAS PubMed.
- M. A. Abdulmalic, A. Aliabadi, A. Petr, Y. Krupskaya, V. Kataev, B. Büchner, T. Hahn, J. Kortus and T. Rüffer, Dalton Trans., 2012, 41, 14657 RSC.
- A. Li, L.-M. Chamoreau, B. Baptiste, Y. Li, Y. Journaux and L. Lisnard, Dalton Trans., 2021, 50, 681–688 RSC.
- F. R. Fortea-Pérez, B. L. Rothenpieler, N. Marino, D. Armentano, G. De Munno, M. Julve and S.-E. Stiriba, Inorg. Chem. Front., 2015, 2, 1029–1039 RSC.
- F. R. Fortea-Pérez, M. L. El Idrissi Moubtassim, D. Armentano, G. De Munno, M. Julve and S.-E. Stiriba, Inorg. Chem. Front., 2018, 5, 2148–2156 RSC.
- X. Ma, S. Deng, J. Liang, J. Chen, J. Su, H. Huang and Q. Song, Tetrahedron Chem, 2022, 3, 100026 CrossRef CAS.
- L. C. Cabrera-Pérez, E. V. García-Báez, M. O. Franco-Hernández, F. J. Martínez-Martínez and I. I. Padilla-Martínez, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 381–385 CrossRef PubMed.
- F. R. Fortea-Pérez, F. Neve, D. Armentano, G. De Munno, S.-E. Stiriba and M. Julve, Inorg. Chim. Acta, 2016, 443, 267–273 CrossRef.
- L. H. G. Kalinke, D. Cangussu, M. Mon, R. Bruno, E. Tiburcio, F. Lloret, D. Armentano, E. Pardo and J. Ferrando-Soria, Inorg. Chem., 2019, 58, 14498–14506 CrossRef CAS PubMed.
- T. Rüffer, B. Bräuer, F. Eya’ane Meva, B. Walfort, G. Salvan, A. K. Powell, I. J. Hewitt, L. Sorace and A. Caneschi, Inorg. Chim. Acta, 2007, 360, 3777–3784 CrossRef.
- R. C. A. Vaz, I. O. Esteves, W. X. C. Oliveira, J. Honorato, F. T. Martins, L. F. Marques, G. L. dos Santos, R. O. Freire, L. T. Jesus, E. F. Pedroso, W. C. Nunes, M. Julve and C. L. M. Pereira, Dalton Trans., 2020, 49, 16106–16124 RSC.
- J. Wojnarska, M. Gryl, T. Seidler and K. M. Stadnicka, CrystEngComm, 2018, 20, 3638–3646 RSC.
- J. He, X. Song, H. Yan and R. Zhong, J. Heterocycl. Chem., 2012, 49, 1357–1361 CrossRef CAS.
- J. Zhang, Q.-X. Liu, A.-H. Li, B.-L. Liu and R.-J. Tao, Z. Naturforsch. B, 2014, 69, 49–54 CrossRef CAS.
- H. O. Stumpf, Y. Pei, O. Kahn, J. Sletten and J. P. Renard, J. Am. Chem. Soc., 1993, 115, 6738–6745 CrossRef CAS.
- I. Fernández, R. Ruiz, J. Faus, M. Julve, F. Lloret, J. Cano, X. Ottenwaelder, Y. Journaux and M. C. Muñoz, Angew. Chem., Int. Ed., 2001, 40, 3039–3042 CrossRef.
- E. Pardo, J. Faus, M. Julve, F. Lloret, M. C. Muñoz, J. Cano, X. Ottenwaelder, Y. Journaux, R. Carrasco, G. Blay, I. Fernández and R. Ruiz-García, J. Am. Chem. Soc., 2003, 125, 10770–10771 CrossRef CAS PubMed.
- F. R. Fortea-Pérez, M. Julve, E. V. Dikarev, A. S. Filatov and S.-E. Stiriba, Inorg. Chim. Acta, 2018, 471, 788–796 CrossRef.
- F. R. Fortea-Pérez, N. Marino, D. Armentano, G. De Munno, M. Julve and S.-E. Stiriba, CrystEngComm, 2014, 16, 6971 RSC.
- Z. Yin, L. Jiang, J. He and J.-P. Cheng, Chem. Commun., 2003, 2326–2327 RSC.
- M. Bołt, A. Mermela and P. Żak, Eur. J. Inorg. Chem., 2023, e202200622 CrossRef.
- E. H. Wi, J. Y. Ryu, S. G. Lee, U. Farwa, M. Pait, S. Lee, S. Cho and J. Lee, Inorg. Chem., 2019, 58, 11493–11499 CrossRef CAS PubMed.
- J. Ferrando-Soria, E. Pardo, R. Ruiz-García, J. Cano, F. Lloret, M. Julve, Y. Journaux, J. Pasán and C. Ruiz-Pérez, Chem. – Eur. J., 2011, 17, 2176–2188 CrossRef CAS PubMed.
- W. Liu, S. Liu, H. Xie, Z. Qing, J. Zeng and P. Cheng, RSC Adv., 2015, 5, 17383–17388 RSC.
- B. Lassalle-Kaiser, R. Guillot and A. Aukauloo, Tetrahedron Lett., 2007, 48, 7004–7006 CrossRef CAS.
- J. S. González-González, O. Zúñiga-Lemus, F. J. Martínez-Martínez, J. Gonzalez, E. V. García-Báez and I. I. Padilla-Martínez, J. Chem. Crystallogr., 2015, 45, 244–250 CrossRef.
- J. S. González-González, F. J. Martínez-Martínez, A. L. Peraza Campos, M. de Jesus Rosales-Hoz, E. V. García-Báez and I. I. Padilla-Martínez, CrystEngComm, 2011, 13, 4748 RSC.
- J. Ferrando-Soria, J. Pasán, C. Ruiz-Pérez, Y. Journaux, M. Julve, F. Lloret, J. Cano and E. Pardo, Inorg. Chem., 2011, 50, 8694–8696 CrossRef CAS PubMed.
- G. D. Tarlas, A. D. Katsenis and G. S. Papaefstathiou, Eur. J. Inorg. Chem., 2018, 4458–4464 CrossRef CAS.
- X. Ottenwaelder, J. Cano, Y. Journaux, E. Rivière, C. Brennan, M. Nierlich and R. Ruiz-García, Angew. Chem., Int. Ed., 2004, 43, 850–852 CrossRef CAS PubMed.
- D. L. Browne, I. R. Baxendale and S. V. Ley, Tetrahedron, 2011, 67, 10296–10303 CrossRef CAS.
- M. Tejeda-Serrano, M. Mon, B. Ross, F. Gonell, J. Ferrando-Soria, A. Corma, A. Leyva-Pérez, D. Armentano and E. Pardo, J. Am. Chem. Soc., 2018, 140, 8827–8832 CrossRef CAS PubMed.
- E. Pardo, J. Ferrando-Soria, M. Dul, R. Lescouëzec, Y. Journaux, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, L. Cañadillas-Delgado, J. Pasán and C. Ruiz-Pérez, Chem. – Eur. J., 2010, 16, 12838–12851 CrossRef CAS PubMed.
- H. Hu, C. Yeh and J. Chen, Eur. J. Inorg. Chem., 2004, 4696–4701 CrossRef CAS.
- T. S. Fernandes, W. D. C. Melo, L. H. G. Kalinke, R. Rabelo, A. K. Valdo, C. C. da Silva, F. T. Martins, P. Amorós, F. Lloret, M. Julve and D. Cangussu, Dalton Trans., 2018, 47, 11539–11553 RSC.
- M. O. Plutenko, M. Haukka, A. O. Husak, T. S. Iskenderov and N. U. Mulloev, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2021, 77, 298–304 CrossRef CAS PubMed.
- G. Tan, M. Das, H. Keum, P. Bellotti, C. Daniliuc and F. Glorius, Nat. Chem., 2022, 14, 1174–1184 CrossRef CAS PubMed.
- D. J. Duchamp, E. C. Olson, B. V. Cheney, J. A. Ryan and R. E. Christoffersen, J. Mol. Struct. THEOCHEM, 1983, 104, 179–189 CrossRef.
- L. Bereczki, P. Bombicz, J. Bálint, G. Egri, J. Schindler, G. Pokol, E. Fogassy and K. Marthi, Chirality, 2009, 21, 331–338 CrossRef CAS PubMed.
- B. Cervera, J. L. Sanz, M. J. Ibáñez, G. Vila, F. LLoret, M. Julve, R. Ruiz, X. Ottenwaelder, A. Aukauloo, S. Poussereau, Y. Journaux and M. C. Muñoz, J. Chem. Soc., Dalton Trans., 1998, 781–790 RSC.
- M. Castellano, W. P. Barros, J. Ferrando-Soria, M. Julve, F. Lloret, J. Pasán, C. Ruiz-Pérez, L. Cañadillas-Delgado, R. Ruiz-García and J. Cano, J. Coord. Chem., 2018, 71, 675–692 CrossRef CAS.
- M. F. Iskander, T. E. Khalil and S. Foro, Inorg. Chim. Acta, 2016, 453, 633–646 CrossRef CAS.
- E. V. García-Báez, F. J. Martínez-Martínez, H. Höpfl and I. I. Padilla-Martínez, Cryst. Growth Des., 2003, 3, 35–45 CrossRef.
- F. Saczewski, T. Debowski, M. Gdaniec and Z. Gdaniec, J. Org. Chem., 1996, 61, 5425–5429 CrossRef CAS.
- T. Rüffer, B. Bräuer, F. Eya’ane Meva and L. Sorace, Inorg. Chim. Acta, 2009, 362, 563–569 CrossRef.
- W. X. C. Oliveira, C. B. Pinheiro, Y. Journaux, M. Julve and C. L. M. Pereira, CrystEngComm, 2024, 26, 647–665 RSC.
- W. X. C. Oliveira, W. D. do Pim, C. B. Pinheiro, Y. Journaux, M. Julve and C. L. M. Pereira, CrystEngComm, 2019, 21, 2818–2833 RSC.
- W. D. do Pim, W. X. C. Oliveira, M. A. Ribeiro, É. N. de Faria, I. F. Teixeira, H. O. Stumpf, R. M. Lago, C. L. M. Pereira, C. B. Pinheiro, J. C. D. Figueiredo-Júnior, W. C. Nunes, P. P. de Souza, E. F. Pedroso, M. Castellano, J. Cano and M. Julve, Chem. Commun., 2013, 49, 10778 RSC.
- E. Pardo, J. Faus, M. Julve, F. Lloret, M. C. Muñoz, J. Cano, X. Ottenwaelder, Y. Journaux, R. Carrasco, G. Blay, I. Fernández and R. Ruiz-García, J. Am. Chem. Soc., 2003, 125, 10770–10771 CrossRef CAS PubMed.
- M. Castellano, J. Ferrando-Soria, E. Pardo, M. Julve, F. Lloret, C. Mathonière, J. Pasán, C. Ruiz-Pérez, L. Cañadillas-Delgado, R. Ruiz-García and J. Cano, Chem. Commun., 2011, 47, 11035 RSC.
- L. H. G. Kalinke, M. S. Silva, R. Rabelo, A. K. Valdo, F. T. Martins, N. Moliner, M. Julve, F. Lloret, J. Cano and D. Cangussu, New J. Chem., 2024, 48, 1971–1982 RSC.
- K. V. Tan, Z. Li, R. E. Karmis and P. J. Barnard, ChemistrySelect, 2018, 3, 2830–2836 CrossRef CAS.
- M. Castellano, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, Y. Journaux, G. De Munno and D. Armentano, Chem. Commun., 2013, 49, 3534 RSC.
- T. Rüffer, B. Bräuer, F. E. Meva and B. Walfort, Dalton Trans., 2008, 5089 RSC.
- J. Ferrando-Soria, D. Cangussu, M. Eslava, Y. Journaux, R. Lescouëzec, M. Julve, F. Lloret, J. Pasán, C. Ruiz-Pérez, E. Lhotel, C. Paulsen and E. Pardo, Chem. – Eur. J., 2011, 17, 12482–12494 CrossRef CAS PubMed.
- M. Milek, A. Witt, C. Streb, F. W. Heinemann and M. M. Khusniyarov, Dalton Trans., 2013, 42, 5237 RSC.
- G. Pelizzi, A. Bacchi, I. Ivanovic-Burmazovic, M. Gruden and K. Andjelkovic, Inorg. Chem. Commun., 2001, 4, 311–314 CrossRef CAS.
- M. Adib, M. Hosein Sayahi, H. Ziyadi, H. R. Bijanzadeh and L.-G. Zhu, Tetrahedron, 2007, 63, 11135–11140 CrossRef CAS.
- J. Navarro-Alapont, D. Armentano, E. Pardo and J. Ferrando-Soria, J. Coord. Chem., 2022, 75, 2413–2423 CrossRef CAS.
- E. M. Scanlan, A. M. Z. Slawin and J. C. Walton, Org. Biomol. Chem., 2004, 2, 716–724 RSC.
- J. Elguero, M. Garcia-Rodriguez, E. Gutierrez-Puebla, A. De la Hoz, M. A. Monge, C. Pardo and M. del M. Ramos, J. Org. Chem., 1992, 57, 4151–4155 CrossRef CAS.
- Y. Zhao, G. Wang, Y. Li, S. Wang and Z. Li, Chin. J. Chem., 2010, 28, 475–479 CrossRef CAS.
- M. Adib, M. Nosrati, M. Mahdavi, L.-G. Zhu and P. Mirzaei, Synlett, 2007, 2703–2706 CrossRef CAS.
- M. Bao, B. Jiang, H. Wang and L. Li, Tetrahedron, 2016, 72, 1011–1017 CrossRef CAS.
- J. F. Zhang, L. E. Guo, T. N. Zang, Y. L. Duan, X. Y. Liu, Z. Yang, P. Verwilst, K. Luo, G. K. Wang, J. F. Kou, Y. Zhou and J. S. Kim, Chem. – Asian J., 2015, 10, 1165–1169 CrossRef CAS PubMed.
- V. Kettmann, J. Světlík and L. Veizerová, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o2607–o2608 CrossRef CAS.
- G. A. El-Hiti, K. Smith, M. Alamri, C. A. Morris, B. M. Kariuki and P. Kille, Z. Kristallogr. - New Cryst. Struct., 2017, 232, 333–335 CrossRef CAS.
- K. Yabe, Y. Maruno, M. Towatari, T. Hamamatsu, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, Chem. Lett., 2007, 36, 1452–1453 CrossRef CAS.
- I. A. Smetanin, A. V. Agafonova, N. V. Rostovskii, A. F. Khlebnikov, D. S. Yufit and M. S. Novikov, Org. Chem. Front., 2020, 7, 525–530 RSC.
- C. Peifer, D. Ott, D. Schollmeyer and S. Laufer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o1415–o1417 CrossRef CAS.
- M. A. Abdulmalic and T. Rüffer, Bull. Chem. Soc. Jpn., 2013, 86, 724–728 CrossRef CAS.
- M. Luisa Gelmi, F. Clerici, R. Destro, E. Erba and D. Pocar, Heterocycles, 1988, 27, 1411 CrossRef.
- C.-H. Mao, H.-B. Song, Q.-M. Wang, R.-Q. Huang, L. Chen and J. Shang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o2522–o2524 CrossRef CAS.
- J. W. Yoon, M. J. Chang, S. Hong and M. H. Lee, Tetrahedron Lett., 2017, 58, 3887–3893 CrossRef CAS.
- T. N. Zang, R. R. Zhao, X. Z. Yang, Y. Gao, G. K. Wang, Y. Zhou, B. Liu and J. F. Zhang, RSC Adv., 2015, 5, 71756–71759 RSC.
- L. Cui, J. Ge, C. F. Leong, D. M. D’Alessandro and J.-L. Zuo, Dalton Trans., 2017, 46, 3980–3988 RSC.
- K. E. Berg, Y. Pellegrin, G. Blondin, X. Ottenwaelder, Y. Journaux, M. M. Canovas, T. Mallah, S. Parsons and A. Aukauloo, Eur. J. Inorg. Chem., 2002, 323–325 CrossRef CAS.
- Bruker AXS Inc., Suite of Crystallographic Software, 2021, preprint, Suite of Crystallographic Software: Version 2021–10.0.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- Bruker AXS, 2015, preprint.
- A. J. C. Wilson, International Tables for X-ray Crystallography, Kluwer Academic Publishers, Dordrecht, Netherlands, C., 1992 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267, 215–220 CrossRef CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- W. Tang, Y. Xiang and A. Tong, J. Org. Chem., 2009, 74, 2163–2166 CrossRef CAS PubMed.
- C. A. Simosono, R. M. R. da Silva, N. R. De Campos, M. A. R. Silva, A. C. Doriguetto, L. S. Flores, C. C. Correa, T. R. G. Simões, A. K. S. M. Valdo, F. T. Martins, F. Garcia, G. P. Guedes, B. R. L. Galvão, J. Cancino-Bernardi, R. D. dos Reis, H. O. Stumpf, D. D. Justino, P. F. R. Ortega, W. D. do Pim, M. Julve and M. V. Marinho, Molecules, 2023, 28, 2086 CrossRef CAS PubMed.
- P. Megha, V. Kumar, P. Kaur and K. Singh, Spectrochim. Acta, Part A, 2023, 290, 122239 CrossRef PubMed.
- W. D. do Pim, W. X. C. Oliveira, M. A. Ribeiro, É. N. de Faria, I. F. Teixeira, H. O. Stumpf, R. M. Lago, C. L. M. Pereira, C. B. Pinheiro, J. C. D. Figueiredo-Júnior, W. C. Nunes, P. P. de Souza, E. F. Pedroso, M. Castellano, J. Cano and M. Julve, Chem. Commun., 2013, 49, 10778 RSC.
- G. P. Souza, C. Konzen, T. R. G. Simões, B. L. Rodrigues, A. F. C. Alcântara and H. O. Stumpf, J. Mol. Struct., 2012, 1016, 13–21 CrossRef CAS.
- N. Nazwir, A. Yanuar and R. R. Syahdi, Int. J. Appl. Pharm., 2020, 12, 51–55 CrossRef CAS.
- L. Peng, L. Xiao, Y. Ding, Y. Xiang and A. Tong, J. Mater. Chem. B, 2018, 6, 3922–3926 RSC.
- F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320–329 CrossRef.
- A. Rahim, P. Saha, K. K. Jha, N. Sukumar and B. K. Sarma, Nat. Commun., 2017, 8, 78 CrossRef PubMed.
- M. Morales-Santana, S. Chong-Canto, J. M. Santiago-Quintana, F. J. Martínez-Martínez, E. V. García-Báez, A. Cruz, S. Rojas-Lima and I. I. Padilla-Martínez, CrystEngComm, 2022, 24, 1017–1034 RSC.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Phys. Chem., 2015, 16, 2496–2517 CrossRef PubMed.
- S. Ramkumar and R. Rajalakshmi, Mater. Today Proc., 2023 DOI:10.1016/j.matpr.2023.05.638.
- C. Chiter, A. Bouchama, T. N. Mouas, H. Allal, M. Yahiaoui, I. Warad, A. Zarrouk and A. Djedouani, J. Mol. Struct., 2020, 1217, 128376 CrossRef CAS.
- (a) CCDC 2478688: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2p68pw; (b) CCDC 2478689: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2p68qx.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2026 |