
Introducing CLipPA lipid chemodiversity to enzymatically truncated polymyxin B: a soft drug strategy to combat Gram-negative pathogens
Tae-Ung
Na
 abc,
Yann O.
Hermant
abc,
Andrew
Siow
abc,
Jeremy G.
Owen
f,
Susanna T. S.
Chan
g,
Gavin F.
Painter
g,
Cameron C.
Hanna
abc,
Zillah
Daysh
bc,
Beatrix L.
Goggin
bc,
Jane R.
Allison
bc,
Veronika
Sander
cd,
Alan J.
Davidson
cd,
Georgia
Campbell
ce,
Scott A.
Ferguson
ce,
Gregory M.
Cook
ce,
Paul W. R.
Harris
*abc,
Margaret A.
Brimble
*abc and
Alan J.
Cameron
*abc
abc,
Yann O.
Hermant
abc,
Andrew
Siow
abc,
Jeremy G.
Owen
f,
Susanna T. S.
Chan
g,
Gavin F.
Painter
g,
Cameron C.
Hanna
abc,
Zillah
Daysh
bc,
Beatrix L.
Goggin
bc,
Jane R.
Allison
bc,
Veronika
Sander
cd,
Alan J.
Davidson
cd,
Georgia
Campbell
ce,
Scott A.
Ferguson
ce,
Gregory M.
Cook
ce,
Paul W. R.
Harris
*abc,
Margaret A.
Brimble
*abc and
Alan J.
Cameron
*abc
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds Street, Auckland 1010, New Zealand. E-mail: alan.cameron@auckland.ac.nz
bSchool of Biological Sciences, The University of Auckland, 3A Symonds Street, Auckland 1010, New Zealand
cMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, 3 Symonds Street, Auckland 1010, New Zealand
dDepartment of Molecular Medicine and Pathology, The University of Auckland, 85 Park Road, Auckland 1023, New Zealand
eDepartment of Microbiology and Immunology, Faculty of Biomedical Sciences, The University of Otago, 720 Cumberland Street, Dunedin 9054, New Zealand
fSchool of Biological Sciences, Victoria University of Wellington, TTR 410, Te Toki A Rata, Gate 7, Kelburn Parade, Wellington 6012, New Zealand
gFerrier Research Institute, Victoria University of Wellington, GIQ, Gracefield Innovation Quarter, Lower Hutt, Wellington 5010, New Zealand
First published on 6th February 2026
Abstract
A chemodiverse library of new semi-synthetic antimicrobial peptides is reported herein. The polymyxin B nonapeptide (PMBN) holds promise as a developmental platform to access potent antimicrobial lipopeptide derivatives that circumvent the serious side effects of natural polymyxins used in clinical settings. By combining our unique radical thiol–ene lipidation strategy (CLipPA) with a chemoenzymatic synthesis, we achieved the chemoselective re-functionalisation of semi-synthetic PMBN, affording a family of potent “soft drugs” decorated with linear, cyclo- and branched alkyl and aromatic esters of varying steric properties. The high efficiency of this late-stage diversification strategy is demonstrated for lipopeptide semi-synthesis. In this process, the significance of the lipid orientation on the observed bioactivity of the molecule was determined and rationalised using molecular dynamics (MD) simulations, wherein, unlike on the parent PMB scaffold, D-Cys lipid handle was found to be superior to L-Cys in its ability to confer antimicrobial potency. The stability of linear chain and hindered ester derivatives was also evaluated in human serum, demonstrating the appropriate hydrolytic stability of a highly branched pivalate ester derivative.
Introduction
The early promise of conquering bacterial infections with so-called “magic bullets” has thus far remained unfulfilled, primarily due to the rapid development of antibiotic resistance in these pathogens. Moreover, the emergence of infectious diseases caused by Gram-negative bacteria that are largely insusceptible to hydrophobic small molecules further warrants the need for a new antimicrobial armoury.1Due to their unique structural motifs, antimicrobial peptides (AMPs) have been proposed as a promising approach to tackle the current antimicrobial resistance (AMR) crisis.1 One representative example is the polymyxin family of AMPs, amongst which two members, polymyxins B and E (colistin), have been approved for clinical use.2,3 In light of their superior antimicrobial activities to combat multi-drug resistant (MDR) Gram-negative pathogens, these lipopeptides are the current last-line therapies for infections unresponsive to all other antibiotics.4,5
Strict regulatory control of polymyxin use is needed due to nephrotoxicity occurring in up to half of patients, resulting from their toxic accumulation in the renal proximal tubule epithelial cells.6,7 One strategy to alleviate this toxicity by reducing the drug accumulation in vivo, is to use “soft drugs”, which have emerged as a favourable strategy for structural redesign of the polymyxins.8 The principle of “soft drugs” is to exploit the metabolic/hydrolytic susceptibility of newly introduced, more labile chemical entities.8 Relevant examples include MicuRx analogues with ester-stitched lipids,9,10 a benzyl ester linked analogue we recently reported,11 ‘Barcelona analogues’ that contain a disulfide-linked macrocycle,12–15 mixed-disulfide stitched lipo-derivatives,16 and ‘thiol–ene’-derived lactone analogues17 (Fig. 1). Our group18 reported a focused library of chemically synthesised polymyxin B (PMB, 1) analogues bearing ester-linked lipids, installed using a photochemically driven thiol–ene reaction between vinyl esters and peptide-incorporated Cys residues, termed “CLipPA” (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ysteine
ysteine ![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif)
![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif)
![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) idation on
idation on ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) eptide or
eptide or ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) mino acid). Importantly, these S-lipopeptide analogues were potent towards Gram-negative species from the WHO priority 1 (Critical) list, with greatly reduced nephrotoxicity. Polymyxin B nonapeptide (PMBN, 2) is a PMB-derived cyclic peptide which lacks the N-terminal lipid unit and Dab1 residue present in native PMB. Although the nonapeptide derivative retains most of the structural features of the parent compounds, it lacks antimicrobial activity and exhibits significantly reduced cytotoxicity towards kidney cells.19,20 The introduction of new lipid motifs onto the PMBN scaffold offers a potential strategy to reinstate the desired antimicrobial activity while uncoupling this beneficial effect from toxicity. We recently demonstrated a late-stage chemoselective peptide lipidation strategy for lipid re-functionalisation of PMBN, deployed in partnership with a facile and low-cost chemoenzymatic preparation of the PMBN framework, affording highly potent antimicrobial candidates in respectable overall yields.11 Based on our prior success with CLipPA-mediated lipidation of synthetic PMB derivatives, we also decided to apply this chemoselective lipidation to re-lipidate PMBN, which was, in turn, prepared chemoenzymatically thereby furnishing a small library of “soft drug” truncated PMBN analogues. Given that the toxicity of the polymyxins is believed to be associated with both the N-terminal acyl group as well as the cationic nature of the peptide,21–23 we postulated that utilising the combination of a soft drug approach powered by CLipPA and the reduced charge of the PMBN scaffold, might yield S-lipopeptide analogues with an improved therapeutic window.
mino acid). Importantly, these S-lipopeptide analogues were potent towards Gram-negative species from the WHO priority 1 (Critical) list, with greatly reduced nephrotoxicity. Polymyxin B nonapeptide (PMBN, 2) is a PMB-derived cyclic peptide which lacks the N-terminal lipid unit and Dab1 residue present in native PMB. Although the nonapeptide derivative retains most of the structural features of the parent compounds, it lacks antimicrobial activity and exhibits significantly reduced cytotoxicity towards kidney cells.19,20 The introduction of new lipid motifs onto the PMBN scaffold offers a potential strategy to reinstate the desired antimicrobial activity while uncoupling this beneficial effect from toxicity. We recently demonstrated a late-stage chemoselective peptide lipidation strategy for lipid re-functionalisation of PMBN, deployed in partnership with a facile and low-cost chemoenzymatic preparation of the PMBN framework, affording highly potent antimicrobial candidates in respectable overall yields.11 Based on our prior success with CLipPA-mediated lipidation of synthetic PMB derivatives, we also decided to apply this chemoselective lipidation to re-lipidate PMBN, which was, in turn, prepared chemoenzymatically thereby furnishing a small library of “soft drug” truncated PMBN analogues. Given that the toxicity of the polymyxins is believed to be associated with both the N-terminal acyl group as well as the cationic nature of the peptide,21–23 we postulated that utilising the combination of a soft drug approach powered by CLipPA and the reduced charge of the PMBN scaffold, might yield S-lipopeptide analogues with an improved therapeutic window.
 | ||
| Fig. 1 Chemical structures of native polymyxin B and selected analogues containing semi-stable “soft drug” moieties (highlighted with a light blue circle).10,11,14,16–18 | ||
Herein, we report semi-synthetic PMBN soft drug antibiotics that effectively target Gram-negative bacteria. Combining enzymatic peptidolysis of commercial PMB with late-stage chemoselective CLipPA “click” lipidation, we achieved the facile preparation of 27 PMBN analogues by effecting S-lipidation with structurally diverse vinyl esters. Antimicrobial screening revealed important stereochemical effects of the lipid-anchoring Cys and excellent potency of several of these semi-synthetic S-lipidated analogues against a panel of Gram-negative pathogens. The stereochemical effects of the Cys handle were further validated by molecular dynamics simulations using model membrane structures, which revealed key design insights for the PMBN scaffold. Pleasingly, nephrotoxicity towards a human kidney organoid model11,18,24 was markedly attenuated and human serum stability assays revealed that the degree of lipid chain branching provided a powerful strategy to equip these less significantly accumulating soft drugs with the optimal hydrolytic stability.
Results and discussion
Our initial attention focused on the preparation of tetra-Boc-PMBN (3), which is easily accessed from a commercially available mixture of PMBs (Scheme 1). Enzymatic cleavage of PMB using papain and selective Nγ-Boc-protection of PMB (1) were undertaken according to previous reports,11,16,19,25–27 yielding tetra-Boc-PMBN (3). Subsequent lipidation of the PMBN scaffold using CLipPA required the initial solution-phase introduction of an N-terminal Cys handle. Two low racemisation procedures were used to effect this transformation: procedure A. utilised Boc-L-thiazolidine-OH (Boc-L-Thz-OH), which avoids the inherent propensity of Cys to racemise, HATU and DIPEA in CH2Cl2 or procedure B. utilised Boc-L-Cys(Trt)-OH, in a base-free DIC/HOAt-mediated coupling in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN/CH2Cl2 (v/v). Gratifyingly, the reaction proceeded without notable epimerisation in either case (see SI Fig. S29–S32). Use of Boc-L-Cys(Trt)-OH was the preferred method to provide key L-Cys-coupled PMBN intermediate 4, due to simultaneous deprotection of the Cys(Trt) group taking place during TFA-catalysed Boc removal from the Dab side chains, thereby avoiding the need for a separate step to effect ring opening of the thiazolidine. We also decided to prepare D-Cys intermediate 5 for our SAR studies. Given that our previous study on PMB decapeptides yielded the most promising results with the L-Cys analogues18 and the exocyclic linear chain of PMBN is shorter than that of PMB, we hypothesised that stereoinversion of the lipid handle from the L-configuration to the D congener may have a potentially beneficial effect on antimicrobial activity. This hypothesis is based on the work from Slingerland et al.,16 who reported superior antimicrobial activity in general of PMBN analogues that incorporated a D-Cys thiol as the lipid attachment site. The intermediate 5 was prepared by analogous coupling of Boc-D-Cys(Trt)-OH to 3, with a difference in the column retention time between diastereomers 5 and 4 being observed (see SI Fig. S32).
:1 MeCN/CH2Cl2 (v/v). Gratifyingly, the reaction proceeded without notable epimerisation in either case (see SI Fig. S29–S32). Use of Boc-L-Cys(Trt)-OH was the preferred method to provide key L-Cys-coupled PMBN intermediate 4, due to simultaneous deprotection of the Cys(Trt) group taking place during TFA-catalysed Boc removal from the Dab side chains, thereby avoiding the need for a separate step to effect ring opening of the thiazolidine. We also decided to prepare D-Cys intermediate 5 for our SAR studies. Given that our previous study on PMB decapeptides yielded the most promising results with the L-Cys analogues18 and the exocyclic linear chain of PMBN is shorter than that of PMB, we hypothesised that stereoinversion of the lipid handle from the L-configuration to the D congener may have a potentially beneficial effect on antimicrobial activity. This hypothesis is based on the work from Slingerland et al.,16 who reported superior antimicrobial activity in general of PMBN analogues that incorporated a D-Cys thiol as the lipid attachment site. The intermediate 5 was prepared by analogous coupling of Boc-D-Cys(Trt)-OH to 3, with a difference in the column retention time between diastereomers 5 and 4 being observed (see SI Fig. S32).
 | ||
| Scheme 1 Retrosynthetic analysis of CLipPA PMBN analogues 6 and 7 from a commercial PMB mixture 1 using the actual chemical structure of polymyxin B1 as an example. | ||
CLipPA analogues prepared from commercially available vinyl esters
With the thiol handle successfully installed onto the PMBN scaffold, CLipPA S-lipidation of intermediates 4 and 5 was effected using excess vinyl ester in the presence of 2,2-dimethoxy-2-phenylacetophenone (DMPA) and radical scavengers in a similar manner to our previously reported procedure28 (Scheme 2, Table 1). The L- and D-Cys intermediates 4 and 5 were either used as crude material or after a simple enrichment step using a semi-preparative HPLC column. The focused library of S-lipopeptides was prepared using a structural variety of commercially available vinyl esters as lipid surrogates, spanning linear alkyl chains of various lengths, branched alkyl groups and benzoyl groups. Pleasingly, the S-lipopeptides 6a–i and 7a–h were afforded in up to 94% conversion over three steps, with final isolated yields of up to 23.4% over five or six steps (Table 1). All analogues were obtained in ≥95% purity, after purification by semi-preparative reversed phase HPLC. | ||
| Scheme 2 Preparation of L- or D-Cys coupled intermediates 4 and 5. | ||
| Entry | Product | Handle orientation | R | Reaction time | % conv.a | Yieldb [%] |
|---|---|---|---|---|---|---|
| a Percentage conversion estimated (over three steps, due to overlapping intermediate compound and impurity signals) from crude RP-HPLC chromatogram (210 nm or 214 nm); entries 1–4 and 7–9 over three steps from Thz-coupled Boc4-PMBN (8), considering the ratio of Thz-coupled Boc4-PMBN (8) to Boc5-PMBN (S2), as estimated from the HPLC trace following Thz coupling; entries 5, 6 and 10–17 over three steps from Boc4-PMBN (3), considering the ratio of Boc4-PMBN (3) to Boc5-PMBN (S2), as estimated from the HPLC trace following Boc-protection. b Overall percentage yield over five or six steps from PMB (1). | ||||||
| 1 | 6a | l |

|
60 min | 89 | 21.5 |
| 2 | 6b | l |

|
90 min | 75 | 9.1 |
| 3 | 6c | l |

|
40 min | 70 | 15.1 |
| 4 | 6d | l |

|
60 min | 65 | 13.1 |
| 5 | 6e | l |

|
60 min | 75 | 8.5 |
| 6 | 6f | l |

|
60 min | 38 | 7.1 |
| 7 | 6g | l |

|
60 min | 83 | 5.5 |
| 8 | 6h | l |

|
80 min | 92 | 6.2 |
| 9 | 6i | l |

|
80 min | 94 | 5.6 |
| 10 | 7a | d |

|
80 min | 84 | 23.4 |
| 11 | 7b | d |

|
80 min | 74 | 12.2 |
| 12 | 7c | d |

|
60 min | 72 | 12.3 |
| 13 | 7d | d |

|
60 min | 82 | 16.0 |
| 14 | 7e | d |

|
80 min | 81 | 17.5 |
| 15 | 7f | d |

|
80 min | 87 | 16.9 |
| 16 | 7g | d |

|
60 min | 87 | 9.6 |
| 17 | 7h | d |

|
80 min | 90 | 14.2 |
Antimicrobial activity
Minimum inhibitory concentration (MIC) assays of the resultant S-lipopeptides (6a–i, 7a–h) against a reference strain of E. coli (ATCC 25922), were undertaken to evaluate their antimicrobial activity (Table 2). PMBN analogue 6a bearing an ester-linked propionyl lipid tail was inactive (MIC = 64 μM) towards E. coli. Interestingly, this represented a significant change from our previously reported PMB analogue,18 which bore an equivalent acyl moiety (MIC ≈ 2 μM). We postulated that this is a result of the decrease in the total penetration of the lipid tail in the bacterial membrane or a change in its spatial orientation due to the removal of the N-terminal amino acid in the truncated PMBN scaffold. This hypothesis was supported by the observed recovery of activity upon increasing the length of the lipid chain. However, a parabolic relationship (Fig. 2) was observed with respect to alkyl chain length with activity then decreasing after reaching optimal activity (MIC = 1 μM) for the octanoic analogue 6d. Pleasingly, stereochemical inversion for the linear chain analogues of the D-Cys series resulted in substantially greater potency compared to those of the L-Cys series. Similar to the L-Cys analogues 6a–f, linear alkyl chain D-Cys derivatives 7a–f also produced a characteristic parabolic trend (Fig. 2) in potency towards E. coli. However, optimal activity was shifted towards shorter chain lengths, C6-chain (7d, MIC = 0.0625 μM), and substantial potency was retained even with the short propionyl lipid (7a, MIC = 2 μM), which was essentially inactive for the equivalent L-Cys analogue 6a. Importantly, the most active compound 7d was 16-fold more potent than the most active L-Cys analogue 6d and contained a shorter C6 lipid, which would likely render it less nephrotoxic than the C8-containing L-Cys analogues 6d, based on our prior observations with the PMB decapeptide scaffold.18 These results support the hypothesis that the angular orientation of lipid relative to the core structure of polymyxin influences the antimicrobial activity, in addition to the absolute length of the lipid. Of particular interest was the observation that even the least active compounds 7a, 7b and 7f from the D-Cys series retained activity towards E. coli (MIC = 1–2 μM), similar to the most active L-Cys derivative, 6d (MIC = 1 μM). Gratifyingly, the pivaloyl D-Cys PMBN analogue (7g, MIC = 0.25 μM) demonstrated near equipotency to our previously reported pivaloyl PMB analogue (MIC ≈ 0.25 μM),18 despite the reduction in overall net cationic charge. Interestingly, the aromatic L-Cys derivatives 6h and 6i appeared to be less impacted by the stereochemistry of the Cys residue, in that they exhibited notable activities towards E. coli (8 μM and 1 μM respectively), particularly the 4-tBu-benzoate 6i, which was found to be near equipotent to an equivalently lipidated PMB decapeptide analogue18 (1 μM vs. 1 μg mL−1 [ca. 1 μM]). The 4-tBu-benzoyl group incorporated into the D-Cys scaffold afforded a slight improvement in activity (7h, 0.5 μM) compared to those on L-Cys PMBN and PMB decapeptide.18|
|
|||||
|---|---|---|---|---|---|
| Compound | MIC (μM) | ||||
| Polymyxin B (1) | 0.125 | ||||

| L-Cys CLipPA library | D-Cys CLipPA library | ||||
|---|---|---|---|---|---|
| Compound | MIC (μM) | Compound | MIC (μM) | ||
| a Indicates the equivalently lipidated L-Cys PMB decapeptide analogue. | |||||
| 6a |

|
64 | 7a |

|
2 |
| Decapeptidea ≈ 2 | |||||
| 6b |

|
16 | 7b |

|
1 |
| Decapeptidea ≈ 0.5 | |||||
| 7c |

|
0.25 | |||
| 6c |

|
4 | 7d |

|
0.0625 |
| 6d |

|
1 | 7e |

|
0.25 |
| 6e |
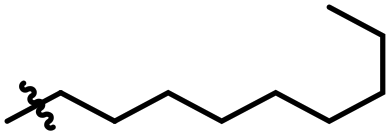
|
2 | 7f |

|
1 |
| Decapeptidea ≈ 2 | |||||
| 6f |

|
8 | |||
| 6g |

|
32 | 7g |

|
0.25 |
| Decapeptidea ≈ 0.25 | |||||
| 6h |

|
8 | |||
| Decapeptidea ≈ 0.25 | |||||
| 6i |

|
1 | 7h |

|
0.5 |
| Decapeptidea ≈ 1 | |||||
 | ||
| Fig. 2 The relationship between the chain length of linear alkyl lipids and MIC towards E. coli ATCC 25922 (μM). Left) D-Cys-PMBN linear chain analogues 7a–f and right) L-Cys analogues 6a–f and D-Cys analogues 7a–f overlaid. Blue and red plots depict L-Cys analogues 6a–f and D-Cys analogues 7a–f, respectively. | ||
Molecular dynamics simulations
To further test the hypothesis that the orientation of the lipid tail drives the significantly different activity profiles of the L-Cys and D-Cys analogues, we used molecular dynamics (MD) simulations to characterise the conformational preferences in solution of one equivalently lipidated peptide from each series, namely 6b (L-Cys) and 7b (D-Cys), carrying an ester-linked butyl chain.Clustering of the conformations sampled during a 500 ns MD simulation of each analogue showed the L-Cys analogue 6b to be less conformationally dynamic (26 vs. 39 clusters, 60% vs. 26% in cluster 1, see SI Table S6) and tends to sample more compact conformations compared to the D-Cys analogue 7b (Fig. 3A). The most common L-Cys conformation (cluster 1, occupied for 60% of the simulation, respectively) has the lipid tail and residues L-Cys1 and Thr2 of the exocyclic chain bent back towards the cycle formed by residues 4–10 (Fig. 3B), a conformation which is stabilised by formation of multiple hydrogen bonds (Fig. 3B, SI Table S7, Fig. S112). In contrast, the most common D-Cys conformation (cluster 1, occupied for 26% of the simulation) has the exocyclic chain and lipid tail extended away from the cycle. Although the second most common D-Cys conformation (cluster 2, 24%) has the lipid tail and residues D-Cys1, Thr2, and Dab3 bent back towards the cycle, no stable hydrogen bonds are formed between the exocyclic chain and the cycle, making it far more mobile.
 | ||
| Fig. 3 Conformational differences between 6b (L-Cys) and 7b (D-Cys). A. Radius of gyration (Rg) measures the size of the smallest sphere that contains each conformation sampled during the simulation. The Rg distribution for 6b (L-Cys) is biased towards more compact conformations compared to 7b (D-Cys). B. The most common conformation of 6b (L-Cys), occupied for 60% of the simulation, has the lipid folded back against the cycle, held in place by hydrogen bonds occupied for 0.01–11.69% of the simulation (the most occupied representative is indicated). The lipid points in the same direction as D-Phe6 and Leu7. The most common conformation of 7b (D-Cys), occupied for 26% of the simulation, has the lipid extended away from the cycle pointing in the opposite direction as D-Phe6 and Leu7. The second most common conformation of 7b (D-Cys), occupied for 24% of the simulation, has the lipid folded back against the cycle, held in place by a variety of unstable hydrogen bonds occupied for 0.01–4.63% of the simulation (one representative shown as grey dashed line). The L-Cys1-lipid moiety, D-Phe6 and Leu7 are rendered opaquely, and the remainder of the structure is transparent, with hydrogen bonds shown as black or grey dashed lines. C. 2D probability densities of the angle between the lipid (red dashed arrow) and either the normal to the plane of the cycle (solid arrow emanating from shaded triangle) or D-Phe (red dashed arrow). For 6b (L-Cys), the angle values are concentrated in a region corresponding to lipid-cycle normal angles around 120°, corresponding to the lipid being almost parallel to the plane of the cycle, and lipid-D-Phe angles around 30°. In contrast, the lipid of 7b (D-Cys) samples a wide range of both angles, indicating less strong conformational preferences. These angles include smaller lipid-cycle normal angles that correspond to the lipid tail lying perpendicular to the plane of the cycle, which are seldom sampled by 6b (L-Cys). | ||
Polymyxin activity is believed to be correlated with the conformations in which the lipid tail and, potentially, also the D-Phe or Leu side chains are oriented perpendicular to the cycle.29–31 Such conformations allow the cycle to lie flat on the membrane surface, where the positively charged Dab side chain amines can form electrostatic interactions with the negatively charged lipid head groups, while the hydrophobic lipid tail and amino acid side chains penetrate into the membrane. To determine whether 6b (L-Cys) and 7b (D-Cys) exhibit conformational differences that might explain their respective activities, we calculated the angles between the normal of a plane representing the cycle and the lipid tail, D-Phe6 or Leu7 side chains (see SI Fig. S113). For both 6b (L-Cys) and 7b (D-Cys), the D-Phe6 and Leu7 side chains tend to be perpendicular to the cycle normal, indicating that they lie in the same plane as the cycle itself. However, for both variants, the lipid tail samples a wider range of angles with respect to the cycle normal, and the distribution of angles is more uniform for 7b (D-Cys), indicating that its lipid tail is less often in the plane of the cycle.
As this analysis does not distinguish between conformations in which the lipid tail is folded against the cycle or extended away from the cycle, we also calculated the angles between vectors representing the lipid tail and the D-Phe or Leu side chains; the latter tend to lie in the plane of the cycle and point away from the cycle interior (see SI Fig. S114). For 7b (D-Cys), the lipid tail uniformly samples angles relative to the D-Phe or Leu side chains from 45–135°, which corresponds to the lipid tail being somewhat perpendicular to the cycle. In contrast, the distribution of angles for 6b (L-Cys) shows that the lipid tail prefers angles between 30–60°, which corresponds to it lying flat against the ring, pointing in a similar direction to the D-Phe and Leu side chains. The difference in conformational preferences between the 6b (L-Cys) and 7b (D-Cys) analogues is particularly obvious when the lipid-cycle normal and lipid-D-Phe angle distributions are combined (Fig. 3C).
Together, our simulation results suggest that 7b (D-Cys) is more likely to sample conformations in which the lipid tail is somewhat perpendicular to the cycle, which relate to favourable membrane interactions and bioactivity,29–31 whereas 6b (L-Cys) is more likely to sample conformations in which the lipid tail is folded against the cycle, which would inhibit both the formation of hydrogen bonds with the membrane and membrane penetration. These conformational preferences may therefore explain the observed differences in activity between the L-Cys and D-Cys analogue series.
Lipidation with cyclo- and branched alkyl vinyl esters
Encouraged by the potency of the D-Cys PMBN alkyl ester-linked analogues 7a–f towards E. coli, further derivatisation was investigated. It has been demonstrated that the lipid portion comprising branched esters, provides superior stability in human serum compared to ester-linked linear chains.9,10 Using this knowledge, we therefore envisioned preparing a series of branched/cyclo-alkyl chain-containing vinyl esters for introduction by CLipPA. Given the inherent safety concerns using a mercuric salt-catalysed trans-vinylation reaction32 to prepare the desired vinyl esters, we were motivated to find a safer alternative for vinyl ester synthesis. After considering several alternatives,33–36 a Pd(OAc)2-catalysed protocol was chosen to prepare ten vinyl esters 9a–j that were not readily commercially available (Scheme 3). | ||
| Scheme 3 Preparation of cyclo- and branched alkyl vinyl esters via a transvinylation reaction using [Pd(OAc)2]3.35 The percentage isolated yields are shown in brackets. | ||
The carboxylic acids of interest (as lipid source) were left to stir overnight in excess vinyl acetate, which was employed as the reaction solvent. The reaction was catalysed by [Pd(OAc)2]3 (0.156 equiv.) with KOH (0.1 equiv.) to afford vinyl esters 9a–j in yields ranging ca. 11–76%. The isolated yield varied largely due to challenges associated with the high volatility of the vinyl esters 9a–j.
With the desired cyclo- and branched alkyl esters 9a–j in hand, CLipPA was then effected to afford D-Cys analogues 7i–r (Table 3). Pleasingly, all of the building blocks 9a–j proved amenable to the CLipPA lipidation reaction, affording S-lipidated peptides 7i–7r. The reaction conversion and overall yield largely depended on the quality of crude D-Cys precursor 5, where the final products 7j, 7k and 7m–r prepared using slightly impure starting material resulted in lower conversion (25–53%) and final yield (2.4–8.4%, over five steps) compared to cyclopropyl and cyclohexyl derivatives 7i and 7l, which gave ca. 75% conversion and 11.6–13.6% yield over five steps.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Product | Reaction time | % conv.a | Yieldb [%] |
| a Percentage conversion estimated from crude RP-HPLC chromatogram (214 nm) over the three steps from Boc4-PMBN (3), considering the ratio of Boc4-PMBN (3) to Boc5-PMBN (S2), as estimated from the HPLC trace following Boc-protection. b Overall percentage yield over five steps from PMB (1). | |||||
| 1 | 7i |

|
1 h | 75 | 11.6 |
| 2 | 7j |

|
1 h | 25 | 4.0 |
| 3 | 7k |

|
1 h | 42 | 4.4 |
| 4 | 7l |

|
1 h | 76 | 13.6 |
| 5 | 7m |

|
1 h | 45 | 4.5 |
| 6 | 7n |

|
1 h | 34 | 4.1 |
| 7 | 7o |

|
1 h | 31 | 3.3 |
| 8 | 7p |

|
1 h | 38 | 8.4 |
| 9 | 7q |

|
1 h | 28 | 2.4 |
| 10 | 7r |

|
1 h | 53 | 6.3 |

Antimicrobial activity of D-Cys CLipPA analogues towards a broader spectrum of pathogens
Curious to investigate the activity spectrum of the cyclo- and branched alkyl D-Cys analogues 7g, and 7i–r, we decided to screen these against a broader panel of Gram-negative pathogens, which included two polymyxin-resistant strains (Table 4). In doing so, linear alkyl and aromatic analogues, which were found to be promising against E. coli, were also subjected to a broader spectrum MIC screening.| Compound | MIC (μM) | MIC (μg mL−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| E. coli ATCC 25922 | A. baumannii ATCC 19606 | P. aeruginosa ATCC 27853 | K. pneumoniae ATCC 33495 | K. pneumoniae ATCC 700603 | E. coli MS8345 | K. pneumoniae MS6671 | ||
| Polymyxin B (1) | 0.125 | 0.5 | 0.25 | 0.5 | 0.25 | 4 | >32 | |
| 3 carbon lipid tail | ||||||||
| 7a |

|
2 | — | — | — | — | — | — |
| 4 carbon lipid tail | ||||||||
| 7b |

|
1 | 16 | 4 | 2 | 4 | >32 | >32 |
| 7i |

|
0.5 | 16 | 4 | 2 | 4 | >32 | >32 |
| 5 carbon lipid tail | ||||||||
| 7c |

|
0.25 | 4 | 2 | 1 | 1 | 32 | >32 |
| 7j |

|
0.125 | 4 | 2 | 1 | 2 | >32 | >32 |
| 7g |

|
0.25 | 4 | 2 | 1 | 2 | 32 | >32 |
| 7m |
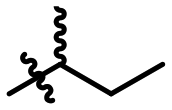
|
0.5 | 4 | 1 | 1 | 2 | 16 | >32 |
| 7n |

|
0.25 | 4 | 1 | 1 | 1 | 16 | >32 |
| 6 carbon lipid tail | ||||||||
| 7d |

|
0.0625 | 4 | 2 | 1 | 2 | 16 | >32 |
| 7k |

|
0.125 | 16 | 2 | 1 | 1 | 16 | >32 |
| 7o |

|
0.25 | 4 | 4 | 2 | 2 | 32 | >32 |
| 7p |

|
0.5 | 4 | 2 | 1 | 2 | 16 | >32 |
| 7q |

|
0.125 | 4 | 2 | 1 | 2 | 16 | >32 |
| 7r |
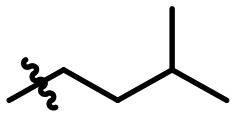
|
0.125 | 2 | 1 | 1 | 1 | 4 | >32 |
| 7 carbon lipid tail | ||||||||
| 7l |

|
0.125 | 4 | 2 | 2 | 2 | 16 | >32 |
| 8 carbon lipid tail | ||||||||
| 7e |

|
0.25 | 2 | 2 | 2 | 2 | 16 | >32 |
| 10 carbon lipid tail | ||||||||
| 7f |

|
1 | — | — | — | — | — | — |
| Aromatic lipid tail | ||||||||
| 7h |

|
0.5 | 4 | 2 | 2 | 4 | 2 | >32 |
In comparison to the near equipotency to PMB (1) observed for many analogues towards E. coli, alkyl chain analogues of C5 to C87c–e were for the most part less active against other bacterial species including A. baumannii (2–16 μg mL−1vs. 0.5 μg mL−1 by PMB), P. aeruginosa (2–4 μg mL−1vs. 0.25 μg mL−1 by PMB) and K. pneumoniae (1–4 μg mL−1vs. 0.25–0.5 μg mL−1 by PMB). They were also almost inactive against polymyxin-resistant strains (MIC = 16 to >32 μg mL−1), including against the mcr-1 gene-acquired strain of E. coli towards which native PMB (1) displayed moderate activity (MIC = 4 μg mL−1). Compared to our previously reported PMB decapeptide analogues18 that contained the additional Dab1 residue, equivalently lipidated compounds were generally less potent (ca. 2–16-fold). While this was not the case for E. coli, this suggests that the overall net charge conferred by the Dab1 charged position is quite species-specific. The 4-tBu-benzoate analogue 7h, which is the only aromatic candidate in the D-Cys series, was found to be potent towards non-resistant species and showed a moderate MIC value (MIC = 4 μg mL−1) towards A. baumannii. Pleasingly, this analogue demonstrated a 2-fold increase in activity towards the mcr-1 strain of E. coli (MIC = 2 μg mL−1), relative to PMB (1) (MIC = 4 μg mL−1). However, this represents a 2-fold decrease compared to our equivalently lipidated decapeptide analogue (MIC = 1 μg mL−1).18
While the cyclo-alkyl derivatives 7i–l exhibited significant activity profiles towards E. coli (MIC = 0.125–0.5 μM) and moderate activity towards other non-resistant isolates (MIC = 1–4 μg mL−1), relatively insignificant activities were shown towards A. baumannii (MIC = 4–16 μg mL−1). The cyclic lipids (7i–l) also failed to afford improvement towards polymyxin-resistant strains (MIC = 16 to >32 μg mL−1), compared to the linear chain analogues with the same or similar number of carbons (7b–e). Interestingly, cyclo-alkyl chains and linear alkyl chains with the equivalent number of carbons (e.g., 7bvs.7i; 7cvs.7j, etc.), conferred surprisingly similar activities across the panel of bacteria, suggesting the significance of the overall hydrophobic moment in determining the overall potency. Among the seven branched alkyl chain analogues screened (7g and 7m–r), 4-Me-valerate 7r exhibited the most robust activity across the board, including against A. baumannii (MIC = 2 μg mL−1), which was found to be the most challenging target amongst polymyxin-susceptible isolates. Branched alkyl chain derivative 7r was also the only non-aromatic compound that possessed activity equivalent to PMB (1) towards the mcr-1 harbouring strain of E. coli (MS8345, MIC = 4 μg mL−1). Of the branched alkyl derivatives with the same number of carbons in the lipid unit (e.g., 7mvs.7netc.), the position of branching relative to the carbonyl group had minimal influence on activity, although derivatives branched furthest from the carbonyl group (i.e., 7n [5 carbons] and 7r [6 carbons]) were generally the most active compounds. Again, this finding aligns with the notion that overall hydrophobicity is the primary contributor, rather than the precise arrangement of lipid structure. The branched alkyl analogues typically resulted in near equipotency to PMB towards E. coli ATCC 25922, but exhibited up to 8-fold reduced activity towards other polymyxin-susceptible bacterial isolates, except for 7o against P. aeruginosa ATCC 27853 (a 16-fold reduction). Despite some general reduction in potency towards pathogens other than E. coli, the low micromolar MIC values and “soft drug” nature of these PMBN analogues, combined with their reduced net cationic charge, still hold promise for an overall improved therapeutic window.
Nephrotoxicity and serum stability
The most promising candidates, 7b–e, 7g, 7h, 7l and 7p were subjected to nephrotoxicity assays in human kidney organoids derived from induced pluripotent stem cells, which retain the physiological and morphological properties of the live organ (Fig. 4).18,39,40 An increasing trend of nephrotoxicity was observed for the linear chain compounds as the total carbon count increased, where the analogues 7b–d pleasingly displayed approximately 3-fold reduced nephrotoxicity, with the C6-analogue 7d causing mild deterioration at 300 μM, which was not observed for 7b and 7c. However, analogue 7e with a longer lipid (C8), showed a somewhat surprisingly equivalent nephrotoxic effect to that of native PMB (1) at 300 μM and 1 mM, despite its soft-drug ester linkage. Interestingly, cyclohexanoate derivative 7l, with a cyclic acyl group comprising a similar number of carbons (C7) to hexanoate, not only resulted in a similar antimicrobial profile, but also similar toxicity towards human kidney organoids (ca. 3-fold reduced compared to PMB, 1). This further suggests that the overall hydrophobicity, rather than the arrangement of carbon atoms is the key predictor of both desirable and toxic bioactivities. The branched analogues 7g and 7p, which are also relatively short, gave rise to roughly 3-fold reduced toxicity, while the 4-tBu-benzoate derivative 7h was found to be equally nephrotoxic to PMB (1) at all three concentrations screened. Overall, alkyl lipids with lengths of C5–6 in any configuration appear to provide the most optimal balance between activity and toxicity, offering improved therapeutic indices towards a polymyxin-susceptible reference strain of E. coli. | ||
| Fig. 4 Bright field images of A control kidney organoids and B organoids treated with native PMB (1) and lead analogues 7b–e, 7g, 7h, 7l and 7p, with the table underneath demonstrating the antimicrobial activity against the panel of Gram-negative bacteria and the magnitude of tissue deterioration associated with each compound at different concentrations. Representative images of two independent assays are shown. Scale bars, 400 μm. | ||
The stability of D-Cys analogues bearing linear, branched and cyclo-alkyl lipid chains (Fig. 5) was also assessed in human serum and Dulbecco's phosphate-buffered saline (DPBS). The stability of test compounds remained similar in DPBS, whereby they were relatively stable (63–89%) over 24 h. However, in human serum, quite dramatic differences in stability were evident. While near complete cleavage of the ester soft drug linkage was observed over the initial 4 h for linear, cyclic and branched analogues bearing a secondary or tertiary carbon centre adjacent to the ester carbonyl group (7c, 7d, 7j, 7l and 7p), the pivalate analogue 7g bearing a quaternary carbon centre in this position demonstrated a much more favourable stability profile. Pleasingly, PMBN analogue 7g remained largely intact after 4 h (ca. 70%), gradually degrading over 24 h to reach 22% remaining. We hypothesised this may account for its ca. 3-fold reduced toxicity towards kidney organoids by preventing toxic accumulation in proximal tubule cells. Native polymyxin B (1), on the other hand, proved entirely insusceptible to degradation in human serum in our prior studies and is highly toxic.18 The antimicrobial activity profile of pivalate analogue 7g alongside its moderate stability as a soft drug may provide the optimum balance of properties amongst the series of analogues investigated in this study.
 | ||
| Fig. 5 Stability assessments of D-Cys analogues 7c, 7d, 7g, 7j, 7l and 7p in above) human serum (from human male AB plasma) and below) DPBS, over 24 h. | ||
Conclusions
CLipPA was successfully implemented to effect the re-functionalisation of semi-synthetic PMBN, utilising both commercially available and synthetically prepared vinyl esters decorated with linear, cyclo-, and branched alkyl, as well as aromatic chemical entities. This semi-synthetic route to access S-lipidated PMBN analogues utilising a combination of enzymatic truncation of native PMB with solution-phase radical thiol–ene-mediated lipidation (CLipPA) was demonstrated to be robust and facile, affording final compounds in overall yields as high as 23.4% over five or six steps. A focused library of 27 S-lipidated analogues was prepared using two stereochemically distinct L-Cys and D-Cys handles. This S-lipopeptide family comprises chemodiverse novel macrocyclic lipo-AMPs with potential to provide significant therapeutic benefits. In screening assays for biological profiles, a vast majority of compounds from the D-Cys series were found to exhibit near equipotency to the natural polymyxin (1) particularly towards E. coli ATCC 25922, and MD simulations demonstrated that incorporation of D-Cys as a handle likely leads to a more desirable lipid orientation and molecular conformation for activity. Many of the potent analogues also showed reduced nephrotoxicity in human kidney organoids. Furthermore, the pivaloyl analogue 7g shows potential as a “soft drug” by exhibiting optimal in vitro half-life in human serum and reduced nephrotoxicity. The PMBN analogue 7g, equipped with a highly branched pivalate lipid was potent towards E. coli, and its favourable stability profile offers opportunities for further fine-tuning. Our previously reported pivalate-equipped PMB analogue,18 bearing the additional charged Dab1 residue, exhibited potency near equivalent to that of native PMB towards a wider array of pathogens and may also offer a similarly desirable stability profile.Author contributions
Conceptualisation: TN, AC, JO, MB, PH; funding acquisition: AC, PH, MB, SF, VS, AD, GrCo; investigation: all authors; project administration: AC, MB, PH; resources: AC, PH, MB, GrCo, JO, GP, JA, AD; writing: TN, AC, JA, MB.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 16th February, which contained errors in the notation used to report the MIC ranges.Data availability
The data supporting this article have been included as part of the supplementary information (SI).Additional ref. 41–54 cited in the SI have been listed in the article's reference list.
Supplementary information: Table S1–S7, Fig. S1–S127 and further experimental details. See DOI: https://doi.org/10.1039/d5md01141h.
Acknowledgements
The authors acknowledge the following organisations for generous financial support: the Ministry of Business, Innovation and Employment (contract UOAX2010), and the Maurice Wilkins Centre for Molecular Biodiscovery (Project Number 3726524).Notes and references
- S. M. Hickson, E. L. Ledger and T. J. Wells, npj Antimicrob. Resist., 2025, 3, 16 CrossRef PubMed.
- A. P. Zavascki, L. Z. Goldani, J. Li and R. L. Nation, J. Antimicrob. Chemother., 2007, 60, 1206–1215 CrossRef CAS PubMed.
- M. Vaara, Front. Microbiol., 2019, 10, 1689 CrossRef PubMed.
- J. Yin, G. Wang, D. Cheng, J. Fu, J. Qiu and Z. Yu, Antimicrob. Agents Chemother., 2019, 63 DOI:10.1128/aac.02378-18.
- S. Yang, H. Wang, D. Zhao, S. Zhang and C. Hu, Front. Pharmacol., 2024, 15, 1424765 CrossRef CAS PubMed.
- R. L. Nation, M. H. P. Rigatto, D. R. Falci and A. P. Zavascki, Antibiotics, 2019, 8, 24 Search PubMed.
- X.-L. Wu, W.-M. Long, Q. Lu, X.-Q. Teng, T.-T. Qi, Q. Qu, G.-F. He and J. Qu, Front. Pharmacol., 2022, 13, 672543 CrossRef CAS PubMed.
- N. Bodor and P. Buchwald, Med. Res. Rev., 2000, 20, 58–101 Search PubMed.
- M. F. Gordeev, J. Liu, X. Wang and Z. Yuan, WO2016/100578A3, 2016.
- M. F. Gordeev, J. Liu, X. Wang and Z. Yuan, US9771394B2, 2017.
- T. Na, V. Sander, A. J. Davidson, R. Lin, Y. O. Hermant, M. T. Hardie Boys, D. Pletzer, G. Campbell, S. A. Ferguson, G. M. Cook, J. R. Allison, M. A. Brimble, B. H. Northrop and A. J. Cameron, Angew. Chem., 2024, 63, e202407764 CrossRef CAS PubMed.
- A. Clausell, F. Rabanal, M. Garcia-Subirats, M. Asunción Alsina and Y. Cajal, J. Phys. Chem. B, 2006, 110, 4465–4471 CrossRef CAS PubMed.
- F. Rabanal, A. Grau-Campistany, X. Vila-Farrés, J. Gonzalez-Linares, M. Borràs, J. Vila, A. Manresa and Y. Cajal, Sci. Rep., 2015, 5, 10558 CrossRef PubMed.
- F. Rabanal Anglada, WO2017/093210A1, 2017.
- F. Rabanal Anglada, EP3173421A1, 2017.
- C. J. Slingerland, C. M. J. Wesseling, P. Innocenti, K. G. C. Westphal, R. Masereeuw and N. I. Martin, J. Med. Chem., 2022, 65, 15878–15892 CrossRef CAS PubMed.
- C. Smith, A. Siow, R. Kowalczyk, S. A. Ferguson, M. J. B. Smith, G. M. Cook, V. Sander, A. J. Davidson, M. A. Brimble and P. W. R. Harris, Chem. Commun., 2026 10.1039/D5CC03871E , Advance Article.
- P. W. R. Harris, A. Siow, S.-H. Yang, A. D. Wadsworth, L. Tan, Y. Hermant, Y. Mao, C. An, C. C. Hanna, A. J. Cameron, J. R. Allison, A. Chakraborty, S. A. Ferguson, S. Mros, K. Hards, G. M. Cook, D. A. Williamson, G. P. Carter, S. T. S. Chan, G. A. Painter, V. Sander, A. J. Davidson and M. A. Brimble, ACS Infect. Dis., 2022, 8, 2413–2429 CrossRef CAS PubMed.
- R. L. Danner, K. A. Joiner, M. Rubin, W. H. Patterson, N. Johnson, K. M. Ayers and J. E. Parrillo, Antimicrob. Agents Chemother., 1989, 33, 1428–1434 CrossRef CAS PubMed.
- N. D. Keirstead, M. P. Wagoner, P. Bentley, M. Blais, C. Brown, L. Cheatham, P. Ciaccio, Y. Dragan, D. Ferguson, J. Fikes, M. Galvin, A. Gupta, M. Hale, N. Johnson, W. Luo, F. McGrath, M. Pietras, S. Price, A. G. Sathe, J. C. Sasaki, D. Snow, R. L. Walsky and G. Kern, Toxicol. Sci., 2014, 137, 278–291 CrossRef CAS PubMed.
- K. Abdelraouf, K.-T. Chang, T. Yin, M. Hu and V. H. Tam, Antimicrob. Agents Chemother., 2014, 58, 4200–4202 CrossRef PubMed.
- F. Rabanal and Y. Cajal, Nat. Prod. Rep., 2017, 34, 886–908 RSC.
- S. N. Avedissian, J. Liu, N. J. Rhodes, A. Lee, G. M. Pais, A. R. Hauser and M. H. Scheetz, Antibiotics, 2019, 8, 31 CrossRef CAS PubMed.
- V. Sander, A. Przepiorski, A. E. Crunk, N. A. Hukriede, T. M. Holm and A. J. Davidson, STAR Protoc., 2020, 1, 100150 CrossRef PubMed.
- H. Paulus and E. Gray, J. Biol. Chem., 1964, 239, 865–871 CrossRef CAS PubMed.
- H. O'Dowd, B. Kim, P. Margolis, W. Wang, C. Wu, S. L. Lopez and J. Blais, Tetrahedron Lett., 2007, 48, 2003–2005 CrossRef.
- K. M. Hamill, L. S. McCoy, E. Wexselblatt, J. D. Esko and Y. Tor, Chem. Sci., 2016, 7, 5059–5068 RSC.
- S.-H. Yang, Y. O. J. Hermant, P. W. R. Harris and M. A. Brimble, Eur. J. Org. Chem., 2020, 2020, 944–947 CrossRef CAS.
- D. Weerakoon, J. K. Marzinek, C. Pedebos, P. J. Bond and S. Khalid, J. Biol. Chem., 2024, 300, 107754 CrossRef CAS PubMed.
- J. Mares, S. Kumaran, M. Gobbo and O. Zerbe, J. Biol. Chem., 2009, 284, 11498–11506 CrossRef CAS PubMed.
- T. Velkov, P. E. Thompson, R. L. Nation and J. Li, J. Med. Chem., 2010, 53, 1898–1916 CrossRef CAS PubMed.
- P. Magrone, F. Cavallo, W. Panzeri, D. Passarella and S. Riva, Org. Biomol. Chem., 2010, 8, 5583–5590 RSC.
- A. Nakamura and M. Tokunaga, Tetrahedron Lett., 2008, 49, 3729–3732 CrossRef CAS.
- H. Nakagawa, Y. Okimoto, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2003, 44, 103–106 CrossRef CAS.
- M. Mastihubová and V. Mastihuba, Bioorg. Med. Chem. Lett., 2013, 23, 5389–5392 CrossRef PubMed.
- S. Kaliyaperumal Appaye, S. Pandurang Nikumbh, R. Reddy Govindapur, S. Banerjee, D. S. Bhalerao and U. K. Syam Kumar, Helv. Chim. Acta, 2014, 97, 1115–1122 CrossRef CAS.
- B. M. Forde, H. M. Zowawi, P. N. A. Harris, L. Roberts, E. Ibrahim, N. Shaikh, A. Deshmukh, M. A. Sid Ahmed, M. Al Maslamani, K. Cottrell, E. Trembizki, L. Sundac, H. H. Yu, J. Li, M. A. Schembri, D. M. Whiley, D. L. Paterson and S. A. Beatson, mSphere, 2018, 3, e00486-18 CrossRef PubMed.
- H. M. Zowawi, B. M. Forde, M. Alfaresi, A. Alzarouni, Y. Farahat, T.-M. Chong, W.-F. Yin, K.-G. Chan, J. Li, M. A. Schembri, S. A. Beatson and D. L. Paterson, Sci. Rep., 2015, 5, 15082 CrossRef CAS PubMed.
- A. Przepiorski, V. Sander, T. Tran, J. A. Hollywood, B. Sorrenson, J.-H. Shih, E. J. Wolvetang, A. P. McMahon, T. M. Holm and A. J. Davidson, Stem Cell Rep., 2018, 11, 470–484 CrossRef CAS PubMed.
- J. L. M. Digby, T. Vanichapol, A. Przepiorski, A. J. Davidson and V. Sander, Am. J. Physiol. Renal Physiol., 2020, 318, F971–F978 CrossRef CAS PubMed.
- V. Yim, I. Kavianinia, M. K. Knottenbelt, S. A. Ferguson, G. M. Cook, S. Swift, A. Chakraborty, J. R. Allison, A. J. Cameron, P. W. R. Harris and M. A. Brimble, Chem. Sci., 2020, 11, 5759–5765 RSC.
- Performance Standards for Antimicrobial Susceptibility Testing, CLSI Document M100S; Clinical and Laboratory Standards Institute, Wayne, PA, 26th edn, 2016 Search PubMed.
- J. K. Oh, A. Przepiorski, H.-H. Chang, R. C. Dodd, V. Sander, B. Sorrenson, J.-H. Shih, J. A. Hollywood, J. R. de Zoysa, P. R. Shepherd, A. J. Davidson and T. M. Holm, J. R. Soc. N. Z., 2022, 52, 54–67 CrossRef PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 Search PubMed.
- N. Schmid, A. P. Eichenberger, A. Choutko, S. Riniker, M. Winger, A. E. Mark and W. F. van Gunsteren, Eur. Biophys. J., 2011, 40, 843–856 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Aust. J. Chem., 2012, 4, 17 CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, Interaction Models for Water in Relation to Protein Hydration, in Intermolecular Forces: Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry Held in Jerusalem, ed. B. Pullman, Springer Netherlands, Dordrecht, 1981 Search PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |