
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and optimization of simplified inhibitors targeting Escherichia coli and Klebsiella pneumoniae IspE
Danica J.
Walsh
 ab,
Rawia
Hamid
abc,
Tim
Giele
ab,
Norbert
Reiling
de,
Matthias
Rottmann
fg,
Mostafa M.
Hamed
ab and
Anna K. H.
Hirsch
*abc
ab,
Rawia
Hamid
abc,
Tim
Giele
ab,
Norbert
Reiling
de,
Matthias
Rottmann
fg,
Mostafa M.
Hamed
ab and
Anna K. H.
Hirsch
*abc
aHelmholtz Institute for Pharmaceutical Research Saarland (HIPS) – Helmholtz Centre for Infection Research (HZI), Campus Building E8.1, 66123 Saarbrücken, Germany. E-mail: anna.hirsch@helmholtz-hips.de
bPharmaScienceHub, Campus Building E 2.1, 66123 Saarbrücken, Germany
cDepartment of Pharmacy, Saarland University, 66123 Saarbrücken, Germany
dMicrobial Interface Biology, Research Center Borstel, Leibniz Lung Center, Parkallee 1, Borstel 23845, Germany
eGerman Center for Infection Research, DZIF, Partner Site Hamburg-Lübeck-Borstel-Riems, Borstel 23845, Germany
fSwiss Tropical and Public Health Institute, Medical Parasitology and Infection Biology, Kreuzstrasse 2, CH-4123 Allschwil, Switzerland
gUniversity of Basel, 4001 Basel, Switzerland
First published on 8th January 2026
Abstract
The methylerythritol phosphate (MEP) pathway is essential for isoprenoid biosynthesis in many pathogenic bacteria but is absent in humans, making its enzymes attractive antibacterial targets. IspE catalyzes the ATP-dependent phosphorylation of 4-diphosphocytidyl-2-C-methylerythritol, a key step in this pathway. Using a previously identified optimized hit as a starting point, we designed and synthesized a focused library of twelve simplified analogues that retained essential pharmacophoric features while improving synthetic accessibility. Docking studies with Escherichia coli IspE guided the design and predicted binding orientations consistent with known ligand interactions. Biochemical evaluation of the library against E. coli and Klebsiella pneumoniae IspE revealed several low-micromolar inhibitors, confirming the predicted binding interactions. Structure–activity relationships indicated that the hydrophobic pocket adjacent to the cytidine-binding region is a key determinant of potency. Although the compounds showed limited whole-cell activity, these results demonstrate that simplified amide analogues can effectively engage the IspE active site and highlight the importance of the hydrophobic pocket in ligand binding. Overall, this work combines structure-based design, synthesis, and biochemical validation to provide a foundation for further optimization of simplified IspE inhibitors as potential antibacterial leads.
Introduction
The rapid rise of antimicrobial resistance has resulted in a substantial decrease in the ability to treat infections and illnesses.1 This presents significant challenges in healthcare, food security, and global development.2 Established resistance is difficult to displace, while new resistance continues to emerge among diverse bacteria across the globe. There has been a sharp decline in new antibiotic development since the 1960s and an increase in the spread of resistance.3 Consequently, over a million lives are lost each year due to infections that can no longer be treated with existing antibiotics. As a result, there is an urgent need for the identification of new drug targets and inhibitors with unprecedented modes of action.The 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway is a metabolic pathway responsible for the biosynthesis of the universal isoprenoid precursors isopentenyl diphosphate (IDP) and dimethylallyl diphosphate (DMADP). Isoprenoids are vital to cell life as they are used in diverse processes such as hormone synthesis, cell–membrane maintenance, protein anchoring and N-glycosylation.4 The MEP pathway is essential for a wide range of clinically relevant pathogenic bacteria, such as Klebsiella pneumoniae, Escherichia coli, but is absent in humans.5 Although the MEP pathway features several viable drug targets, few inhibitors have been reported so far.6
Although IspE is highly conserved among Gram-negative pathogens, subtle amino acid differences exist between orthologs that can influence ligand binding and inhibitory activity. For instance, K. pneumoniae and E. coli IspE contain Pro182 and Cys211, whereas in Acinetobacter baumannii IspE these residues are replaced by Gln174 and Phe205, respectively. This substitution alters the local binding environment, meaning that different types of molecular interactions may occur at this site (Fig. 1).
Previously, we identified and developed inhibitor 1, which demonstrated activity towards K. pneumoniae IspE (KpIspE), E. coli IspE (EcIspE) and Acinetobacter baumannii IspE (AbIspE).7–9 In our previous reports, it was shown that this inhibitor occupies the substrate binding site and a small, hydrophobic sub-pocket at the active site of IspE where CDP–ME binds, bypassing the ATP binding site (Fig. 2A). In the most recent study, we attempted to simplify inhibitor 1, by removal of the tetrahydrothiophene ring (2) (Fig. 1). This was done in an effort to simplify the synthesis of this class of inhibitors by eliminating a low-yielding and time-consuming step. While this ultimately decreased potency, compound 2 maintained significant activity towards both KpIspE and EcIspE.
 | ||
| Fig. 1 IC50 values for compounds 1 and 2 obtained from the biochemical assay with various IspE homologues. | ||
In this study, we aimed to optimize activity, while maintaining the simplified structure. This was done through a docking study that led to the synthesis of a library of amide inhibitors (Table 1). Based on a docking study using EcIspE (PDB ID: 1OJ4), we predicted that the simplified amide derivatives would be accommodated within the substrate pocket in a manner similar to compounds 1 and 2 (Fig. 2). After our initial modeling was completed, we deposited high-resolution crystal structures of E. coli IspE in complex with ligands (PDB IDs 8QCC, 8QCN).9 We therefore compared our docking poses to these X-ray crystal structures and found the key binding features to be conserved: the cytidine/cytosine group occupies the same general region and engages the same nearby residues (including His26 and interactions consistent with Asn12/Asp141), and the hydrophobic sub-pocket that accommodates the cyclopentyl/cyclopropyl moieties in our models is present in the crystal structures. These comparisons validate that the docking models used to design the amide library captured the principal features of ligand binding observed in the experimental structures. In Fig. 2, it can be observed that the cytosine group of compounds 1 (B), 2 (C) and 3 (D) interact with His26, while the cyclopropyl moiety is in a slightly different position in 2 and 3. The sulfonamide group of compound 1 hydrogen bonds with Asn12 and Asp141, while 2 interacts with Val156 and the carbonyl group of 3 with Asp141 and Ser108. These predicted contacts correspond well to those observed in the deposited E. coli IspE crystal structures (PDB IDs 8QCC and 8QCN) where the cytidine/cytosine fragment occupies the same pocket and establishes comparable polar contacts.
|
|
|||
|---|---|---|---|
| Inhibitor | R group | IC50KpIspE (μM) | IC50EcIspE (μM) |
| 3 | Cyclopropyl | 1.9 ± 0.1 | 1.3 ± 0.1 |
| 4 | Methyl | 18 ± 4.4 | 8.5 ± 4.3 |
| 5 | Ethyl | 1.3 ± 0.1 | 1.3 ± 0.1 |
| 6 | n-Bu | 11.6 ± 1.8 | 5.0 ± 1.5 |
| 7 | t-Bu | 7.5 ± 1.5 | 1.3 ± 0.1 |
| 8 | Cyclohexyl | 19.4 ± 1.5 | 14.8 ± 1.2 |
| 9 | –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
23.3 ± 6.9 | 5.9 ± 1.1 |
| 10 | –CH2CF3 | 4.6 ± 0.5 | 4.5 ± 1.8 |

 | ||
| Fig. 2 Overlay of docked pose of compound 1 and CDP–ME (A), compound 1 (B), compound 2 (C), and compound 3 (D) in Escherichia coli IspE (PDB ID: 1OJ4). Green spheres indicate favorable contributions to the binding affinity, while red spheres indicate unfavorable contributions as calculated by the HYDE scoring function in SeeSAR. The size of each sphere reflects the magnitude of the interaction. | ||
The synthesis was carried out in three steps (Scheme 1). First, propargylamine was reacted with the appropriate acid chloride. Iodo-cytosine was synthesized by reacting iodic acid with iodine in acetic acid at room temperature overnight. Final products were obtained via a Sonogashira cross-coupling reaction, involving a terminal acetylene and aryl or alkenyl halides in the presence of Pd(0) and copper(I) catalysts with base.
 | ||
| Scheme 1 General synthesis for amide derivatives. a) TEA, DCM, 0 °C – R.T., 30 min b) I2, HIO3, AcOH, 40 °C, 12 c) [PdCl2(PPh3)2], CuI, TEA, DMF, r.t., 12 h | ||
Results
First, we synthesized compound 3, which retains the cyclopropyl group present in compounds 1 and 2 on the amide linker. All compounds were evaluated for inhibitory activity (S1.1). This compound showed a significant boost in activity compared to compound 2 towards both KpIspE and EcIspE with an IC50 of 1.9 ± 0.1 μM and 4.7 ± 1.2 μM, respectively. Although all activity towards AbIspE was lost, we focused on Kp and EcIspE here due to the similarities in the substrate binding pocket.10 It has been shown that EcIspE and KpIspE possess an 82% sequence identity, with conserved residues, which are involved in binding to ATP and CDP–ME.9 By contrast, AbIspE shares only 39% sequence identity with EcIspE and 42% with KpIspE. Subsequently, we synthesized a library of nine amide derivatives to target Ec and KpIspE in order to establish a structure–activity relationship (SAR) that would help to optimize this novel library of inhibitors.Several other aliphatic chains or rings, including methyl (4), ethyl (5), n-butyl (6), t-butyl (7), vinyl (9) and cyclohexyl (8) were evaluated. The ethyl group had comparable activity as the cyclopropyl against KpIspE and improved activity against EcIspE. Based on this observation, we synthesized the methyl (4) and n-butyl (6) derivatives but, neither compound was as active as the ethyl derivative (5). The larger cyclohexyl group (8) also showed a decrease in activity compared to 3. Although these compounds showed activity towards KpIspE and EcIpsE, they lost activity towards AbIspE at concentrations up to 500 μM. Compounds 3 and 5 emerged as the most active against KpIspE and 7 and 5 against EcIspE. To further evaluate this simplified class of derivatives, we synthesized compounds 11–15 (Table 2). In derivatives 11 and 12, we omitted the amide group and a tertiary amine was employed instead. For compound 13 we maintained the amide and removing the carbonyl of the cytosine ring, while in 14 and 15 the linker placement was modified. In compound 15, the linker was also modified to make it more flexible by eliminating the triple bond. In compound 16, the alkyne was also reduced, but with the linker in the original position.
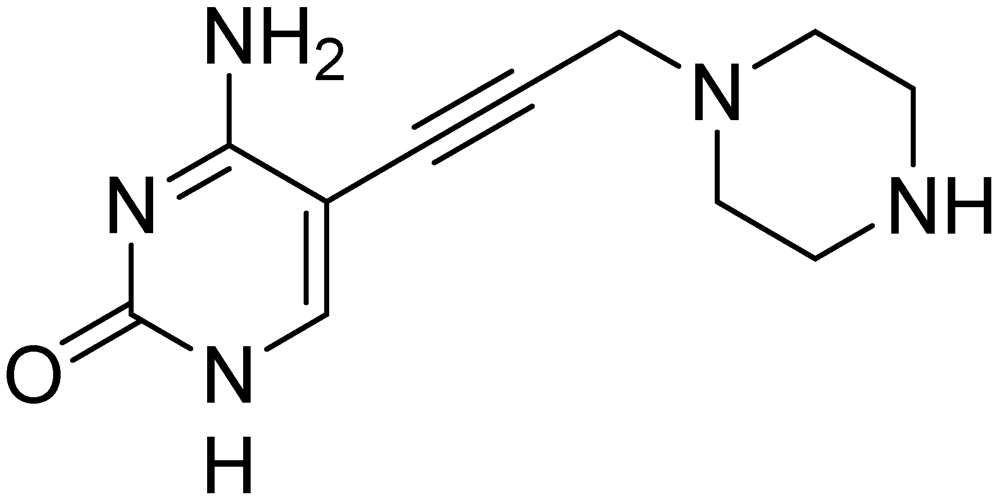




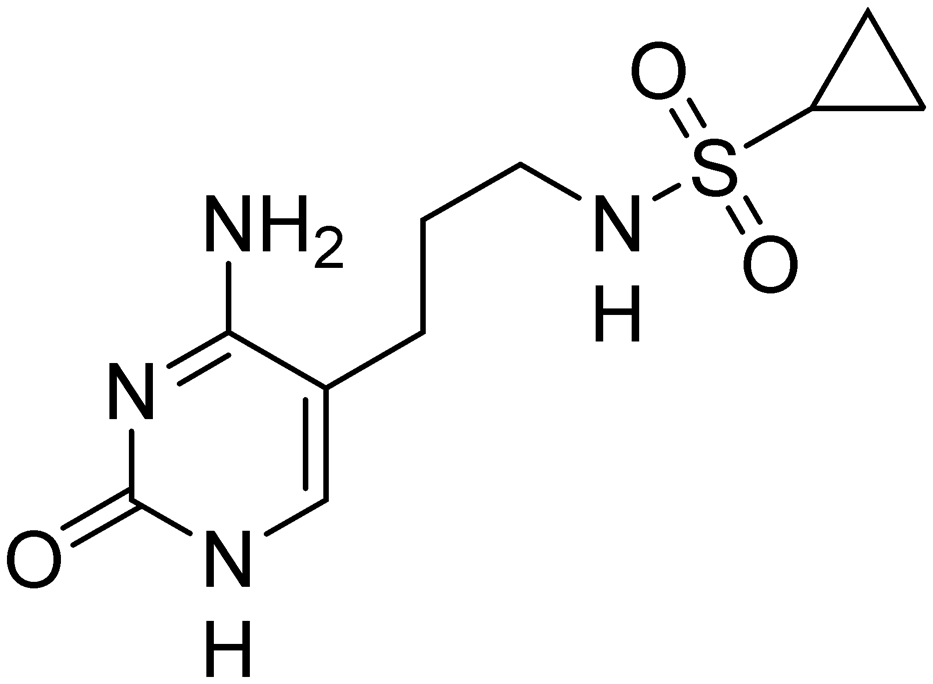
The tertiary amine 11 showed a significant decrease in activity towards EcIspE and a complete loss of activity towards KpIspE at 500 μM. Next, we synthesized compound 13 to assess the importance of the carbonyl group on the aryl ring, while maintaining aromaticity leading to a complete loss of activity. Additionally, we synthesized compounds 14 and 15 to determine how the placement of the alkyne moiety would affect activity. Unfortunately, 14 was only slightly soluble in DMSO and insoluble in the 5% DMSO buffer solution used for IC50 assays, hindering its biological evaluation. Compound 15, which lacks the alkyne and has an alkane linker, also showed a complete loss of activity. Lastly, compound 16 was synthesized to determine if the placement of the saturated linker was the major reason for loss of activity. No activity was observed in compound 16. In conclusion, we have assessed that the placement and the rigidity of the linker as well as the presence of the amide, are important for the activity.
As a next step, the tetrahydrothiophenyl moiety was reinstated to give compound 17 (Fig. 3), aiming to restore the potency lost upon its removal in compound 2. Since removal of this group from 1 to 2 resulted in decreased activity across all orthologs, we anticipated that reinstating it would enhance potency for EcIspE and KpIspE as well. However, compound 17 displayed activity comparable to 3 against EcIspE and a slight decrease against KpIspE, while showing improved inhibition of AbIspE. This unexpected trend suggests that the tetrahydrothiophenyl group contributes differently depending on the overall binding orientation of the scaffold. The results may indicate that compound 3, or its derivatives, adopt a slightly altered binding mode compared to the parent sulfonamide 1, which could explain why reinstating the tetrahydrothiophenyl ring did not yield the anticipated potency increase. Therefore, while the data confirm that this group can modulate activity, they do not conclusively demonstrate that it is more critical for AbIspE than for EcIspE or KpIspE. Further structural or co-crystallization studies will be required to clarify these binding-mode differences.
 | ||
| Fig. 3 IC50 values for compounds 1, 2, 3 and 17 obtained from the biochemical assay with various IspE homologues. | ||
We determined the dissociation constant (Kd) for six selected compounds for the target protein KpIspE (Table 3). Kd values were assessed using microscale thermophoresis (MST) (S1.2) and IC50 values were determined using the enzymatic assay as described (S1.1). As expected, the Kd values found were in line with their IC50 values. Compound 8 was chosen because of its relatively lower activity to serve as a comparison.
| Compound | K d KpIspE | IC50KpIspE |
|---|---|---|
| 1 | 7.7 ± 1.5 μM | 3.5 ± 0.2 μM |
| 3 | 2.0 ± 0.2 μM | 1.9 ± 0.1 μM |
| 5 | 1.1 ± 0.1 μM | 1.3 ± 0.1 μM |
| 8 | 30.0 ± 7.2 μM | 19.4 ± 1.5 μM |
| 17 | 8.4 ± 0.5 μM | 5.5 ± 3.1 μM |
To further evaluate these compounds as early drug candidates, StarDrop (version: 7.0.1.29911) was used to predict parameters in Lipinski's rule of 5 such as number of rotatable bonds, number of hydrogen bond donors (HBD), number of hydrogen bond acceptors, clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, and polar surface area (PSA, Table 4).
P, and polar surface area (PSA, Table 4).
P, PSA and most basic pKa as well as the number of rotatable bonds, hydrogen bond donating and accepting groups and IC50 values from the biochemical assay
| Compound | IC50KpIspE (μM) | logD |
logP |
Rotatable bonds | HBD | HBA | PSA | Most basic pKa |
|---|---|---|---|---|---|---|---|---|
| 1 | 7.7 ± 1.5 | 0.09669 | 1.004 | 6 | 2 | 7 | 107.1 | 6.5 |
| 2 | 36.5 ± 1.7 | −1.026 | −0.3395 | 5 | 3 | 7 | 117.9 | 6.6 |
| 3 | 1.9 ± 0.1 | −0.3387 | −0.2886 | 5 | 3 | 6 | 100.9 | 6.2 |
| 4 | 18 ± 4.4 | −0.9024 | −0.6713 | 4 | 3 | 6 | 100.9 | 6.4 |
| 5 | 1.3 ± 0.1 | −0.5209 | −0.2316 | 5 | 3 | 6 | 100.9 | 6.4 |
| 6 | 11.6 ± 1.8 | 0.02716 | 0.3068 | 7 | 3 | 6 | 100.9 | 6.5 |
| 7 | 7.5 ± 1.5 | 0.3219 | 0.2242 | 5 | 3 | 6 | 100.9 | 6.1 |
| 8 | 19.4 ± 1.5 | 0.3153 | 0.5164 | 5 | 3 | 6 | 100.9 | 6.5 |
| 9 | 23.3 ± 6.9 | −0.8195 | −0.5079 | 5 | 3 | 6 | 100.9 | 6.3 |
| 10 | 4.6 ± 0.5 | 0.427 | 0.8066 | 6 | 3 | 6 | 100.9 | 5.9 |
| 11 | >500 | −1.079 | −0.7783 | 3 | 3 | 6 | 87.04 | 9.6 |
| 12 | >500 | −0.6512 | −0.4294 | 3 | 2 | 6 | 78.25 | 9.0 |
| 13 | >500 | −0.6978 | 1.275 | 5 | 2 | 5 | 80.9 | 4.6 |
| 14 | N/A | −0.3724 | −0.1803 | 5 | 2 | 6 | 90.01 | 5.2 |
| 15 | >500 | −0.2503 | −0.4501 | 6 | 2 | 6 | 90.01 | 6.1 |
| 16 | >500 | −0.832 | −0.4789 | 6 | 3 | 7 | 117.9 | 7.3 |
| 17 | 5.5 ± 3.1 | 0.6873 | 0.9077 | 6 | 2 | 6 | 90.01 | 6.3 |
All active compounds conformed to the rule of 5, having fewer than 5 HBD, fewer than 10 HBA, a molecular weight under 500 Dalton, and a clogP of under 5.
We also evaluated the in vitro ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties (Table 5). Optimizing ADMET properties is important in the drug-design and development process to create safer and more efficacious drug candidates and enable a true multiparameter optimization. This evaluation also helps to explain how pharmacokinetic processes happen. Compounds 3, 17, and 2 were chosen. Compound 3 was selected because it was among the most active and has a close structural resemblance to the simplified sulfonate 2. Compound 17 was selected to directly compare the sulfonamide 1 to the amide derivative.
| Kinetic solubility | LogD7.4 |
Toxicity | Mouse plasma | Mouse liver S9 | Mouse liver microsomes | ||
|---|---|---|---|---|---|---|---|
| Compound | Mean (μM) | SD | HepG2 | t 1/2 [min] | t 1/2 [min]/Clint [μL mg−1 min−1] | t 1/2 [min]/Clint [μL mg−1 min−1] | |
| 1 | >1000 | 0 | 0.34 | <10% at 100 μM | >240 | >120/<5.8 | >120/<11.6 |
| 2 | 520.1 | 13.57 | −1.41 | <10% at 100 μM | >240 | >120/<5.8 | >120/<11.6 |
| 3 | 115.1 | 55.29 | −1.62 | <10% at 100 μM | >240 | >120/<5.8 | >120/<11.6 |
| 17 | 206.3 | 140.3 | 0.14 | <10% at 100 μM | >240 | >120/<5.8 | >120/<11.6 |
We observed that 1 has a higher solubility compared to its simplified structure 2. When the sulfonamide is replaced with an amide, we observed a decrease in solubility. Like the sulfonamides 1 and 2, the evaluated amides showed no cytotoxicity towards HepG2 cells and were stable in mouse liver S9 fraction, liver microsomes and plasma (Table 5).
Next, we evaluated the same four compounds towards the wild type E. coli strain K12 (S1). Since these compounds showed no activity, they were then evaluated for inhibitory activity towards E. coli delta-acrB. An important factor in drug resistance are mechanisms that actively remove antibiotics from the inside of the cell, such as efflux pumps. The acrB strain was selected to assess, if this compound class lacks antibacterial activity due to efflux in E. coli. None of the five selected compounds demonstrated inhibitory activity towards E. coli delta-acrB (Table S1), suggesting that permeability is the main bottleneck.
Next, several compounds were selected for evaluation towards Mycobacterium tuberculosis and Plasmodium falciparum because IspE inhibitors have been previously studied on these organisms as well (Fig. S1).11 Unfortunately, the compounds showed no antimicrobial activity towards either pathogen.
Conclusion
Our study aimed to restore potency towards Ec and KpIspE of the previously simplified derivatives of IspE inhibitors. To achieve this, we kept the simplified scaffold and through a docking–guided study developed an amide derivative library. We were able to restore potency towards Ec and KpIspE for multiple compounds and towards AbIspE for compound 17. Our findings provide a synthetically simple but potent IspE inhibitors. Although it was found that this class of compounds lacks inhibitory activity in whole-cells assays, there is still potential for augmentation in order to gain activity. We note that co-crystal structures of E. coli IspE with ligands have now been deposited (PDB IDs 8QCC, 8QCN), which already provide high-resolution structural context for ligand binding and confirm the presence of the hydrophobic subpocket exploited by our designs.Thus, the next step in the project is the elucidation of co-crystal structures with 2 and 3 in order to gain a better understanding of their binding mode, and to conduct further experiments to confirm the reason for the lack of antibacterial activity.
Author contributions
Synthesis was performed by D. J. W. and T. G. Biological evaluation of compounds was performed by R. H., M. R., N. R. and D. J. W. D. J. W. performed the MST assays for the determination of Kd values. D. J. W. performed the docking studies. M. M. H. and A. K. H. H. designed and coordinated the study, which was supervised by A. K. H. H. D. J. W. wrote the manuscript and M. M. H. and A. K. H. H. proofread it, with the contribution of all authors. All authors have given approval of the final version.Conflicts of interest
There are no conflicts to declare.Data availability
All data supporting the findings of this study are available within the article and its supplementary information (SI) files. Supplementary information is available. See DOI: https://doi.org/10.1039/d5md00874c.Acknowledgements
The authors would like to thank the technicians at HIPS for the great work, specifically Dr. Andreas Kany, Dr. Jörg Haupenthal, Simone Amann, Jeannine Jung and Jannine Seelbach. The authors would also like to recognize Dr Boris Illarionov and Prof. Markus Fischer at Hamburg School of Food Science, Institute of Food Chemistry for their contribution of reagents necessary for biological assays. D. J. W. received funding from the Marie Skłodowska-Curie Action Postdoctoral Fellowship (Grant agreement number: 101103471) and R. H. from the Schlumberger Foundation Faculty for the Future programme. A. K. H. H. and N. R. from the MepAnti: MSCA Innovative Training network (Grant agreement number: 860816).References
- C. S. Ho, C. T. H. Wong, T. T. Aung, R. Lakshminarayanan, J. S. Mehta, S. Rauz, A. McNally, B. Kintses, S. J. Peacock, C. de la Fuente-Nunez, R. E. W. Hancock and D. S. J. Ting, Antimicrobial resistance: a concise update, Lancet Microbe, 2025, 6(1), 100947 CrossRef PubMed.
- A. George, Antimicrobial resistance, trade, food safety and security, One Health, 2018, 5, 6–8 CrossRef PubMed.
- L. Bo, H. Sun, Y.-D. Li, J. Zhu, J. N. D. Wurpel, H. Lin and Z.-S. Chen, Combating antimicrobial resistance: the silent war, Front. Pharmacol., 2024, 15, 1347750 CrossRef CAS PubMed.
- L. Zhao, W. C. Chang, Y. Xiao, H. W. Liu and P. Liu, Methylerythritol phosphate pathway of isoprenoid biosynthesis, Annu. Rev. Biochem., 2013, 82, 497–530 CrossRef CAS PubMed.
- A. Frank and M. Groll, The Methylerythritol Phosphate Pathway to Isoprenoids, Chem. Rev., 2017, 117(8), 5675–5703 CrossRef CAS PubMed.
- T. Masini and A. K. Hirsch, Development of inhibitors of the 2C-methyl-D-erythritol 4-phosphate (MEP) pathway enzymes as potential anti-infective agents, J. Med. Chem., 2014, 57(23), 9740–9763 CrossRef CAS PubMed.
- A. K. H. Hirsch, S. Lauw, P. Gersbach, W. B. Schweizer, F. Rohdich, W. Eisenreich, A. Bacher and F. Diederich, Nonphosphate Inhibitors of IspE Protein, a Kinase in the Non-Mevalonate Pathway for Isoprenoid Biosynthesis and a Potential Target for Antimalarial Therapy, ChemMedChem, 2007, 2(6), 806–810 CrossRef CAS PubMed.
- A. P. Schütz, S. Osawa, J. Mathis, A. K. H. Hirsch, B. Bernet, B. Illarionov, M. Fischer, A. Bacher and F. Diederich, Exploring the Ribose Sub-Pocket of the Substrate-Binding Site in Escherichia coli IspE: Structure-Based Design, Synthesis, and Biological Evaluation of Cytosines and Cytosine Analogues, Eur. J. Org. Chem., 2012, 2012(17), 3278–3287 CrossRef.
- R. Hamid, D. J. Walsh, E. Diamanti, D. Aguilar, A. Lacour, M. M. Hamed and A. K. H. Hirsch, IspE kinase as an anti-infective target: Role of a hydrophobic pocket in inhibitor binding, Structure, 2024, 32(12), 2390–2398.e2 Search PubMed.
- S. Walesch, J. Birkelbach, G. Jézéquel, F. P. J. Haeckl, J. D. Hegemann, T. Hesterkamp, A. K. H. Hirsch, P. Hammann and R. Müller, Fighting antibiotic resistance—strategies and (pre)clinical developments to find new antibacterials, EMBO Rep., 2023, 24(1), e56033 Search PubMed.
- S.-R. Choi and P. Narayanasamy, Investigating Novel IspE Inhibitors of the MEP Pathway in Mycobacterium, Microorganisms, 2024, 12(1), 18 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |