
Unlocking the therapeutic potential of HDAC8-degrading PROTACs: progress, challenges, and future directions
Suvankar
Banerjee
ab,
Nilanjan
Adhikari
 *a and
Balaram
Ghosh
*c
*a and
Balaram
Ghosh
*c
aNatural Science Laboratory, Division of Medicinal and Pharmaceutical Chemistry, Department of Pharmaceutical Technology, Jadavpur University, Kolkata, India. E-mail: nilanjan_juphar@rediffmail.com
bSchool of Pharmacy, The Neotia University, Sarisa, Diamond Harbour Road, 24 Parganas (South), West Bengal 743368, India
cEpigenetic Research Laboratory, Department of Pharmacy, Birla Institute of Technology and Science-Pilani Hyderabad Campus, Shamirpet, Hyderabad, 500078, India. E-mail: balaram@hyderabad.bits-pilani.ac.in
First published on 5th January 2026
Abstract
Histone deacetylase 8 (HDAC8) is a class I enzyme associated with various diseases, including cancer and neurological disorders. Although small-molecule HDAC inhibitors have been developed, their lack of selectivity often leads to off-target effects and toxicities. Alternatively, targeting specific HDAC isoforms for their degradation represents a more precise therapeutic strategy. This review focuses on the design and development of proteolysis-targeting chimeras (PROTACs) that selectively degrade HDAC8. We explore how existing selective HDAC8 inhibitors can be leveraged as warheads in PROTACs to effectively eliminate the enzyme. Recent studies have successfully designed HDAC8-selective PROTACs by linking HDAC8 inhibitors to E3 ubiquitin ligase recruiters such as VHL and CRBN. These PROTACs have demonstrated high potency in degrading HDAC8 in various cancer cell lines with single-digit nanomolar DC50 values, showing superior anti-proliferative effects compared to their parent inhibitors. Therefore, apart from these handful of reports, more research related to HDAC8-PROTAC should provide a better therapeutic development technology for HDAC8-associated disorders while avoiding any therapy-related adversities and complications.
Suvankar Banerjee is a Research Scholar at Jadavpur University under the guidance of Dr. Nilanjan Adhikari and Professor Tarun Jha (Retd.). He is also an Assistant Professor at the School of Pharmacy, The Neotia University. His research focuses on computational drug discovery, molecular modeling, and AI-driven drug design, with expertise in fragment-based drug design, molecular docking, MD simulations, and ML applications for drug-protein interactions. With 52 research articles published in international peer-reviewed journals, his work extensively covers metalloenzyme inhibitors, epigenetic modulators, and advanced chemoinformatic strategies. |
Dr. Nilanjan Adhikari is an Assistant Professor in the Department of Pharmaceutical Technology, Jadavpur University, Kolkata, India. He completed his B.Pharm. (2007), M.Pharm. (2009), and Ph.D. (2018) at Jadavpur University, Kolkata. His research interests include the development of anticancer small molecules, especially inhibitors of metalloenzymes. He also works on molecular modeling of different bioactive compounds using rigorous computational techniques including QSAR and machine learning approaches. Dr. Adhikari has been recognized among the world's top 2% scientists for the past four years. He has published more than 150 articles in different international peer-reviewed journals. |
Dr. Balaram Ghosh has been actively involved in epigenetic drug discovery for the past 18 years. After obtaining his Ph.D. from Wayne State University, USA, he joined Harvard University, USA (2009–2013) and developed a couple of isoform-selective HDAC inhibitors as drug candidates. His expertise lies in the design and synthesis of selective small molecules, target validation, and druggable new chemical entities. Dr. Ghosh has been recognized among the world's top 2% scientists (2021–2023). He has published 180 papers in peer-reviewed journals. |
1. Introduction
A change in normal epigenetic mechanisms can lead to abnormal biological conditions that can allow the manifestation of pathophysiological conditions and deadly diseases, including neurological disorders, cancer, and genetic disorders.1,2 Additionally, the post-translational modification of histone proteins can induce the functional alteration of gene expression, which in turn can lead to epigenetic disorders.3 Histone deacetylases (HDACs) are a group of enzymes that eliminate the N-terminal ε-lysine-bound acetyl moiety of histone proteins. In tandem with histone acetyl transferases (HATs), HDACs govern chromatin condensation, thus affecting the epigenetic machinery.4,5 Depending on their functional variability and sequential homology to yeast Rpd3, Hda1, and Sir2, the 18 discovered HDAC isoforms are classified into four classes, i.e., class I (HDAC1–3 and HDAC8), class II (HDAC4–7 and HDAC9–10), class III (SIRT1–7), and class IV (HDAC11). Based on their structural, functional, and locational variability and similarities, class II HDAC isoforms are also categorized into several sub-classes, which include class Ia (HDAC1 and HDAC2), class Ib (HDAC3), class Ic (HDAC8), class IIa (HDAC4, HDAC5, HDAC7, and HDAC9), and class IIb (HDAC6 and HDAC10).4,5 Another fascinating fact regarding HDAC isoforms is their structural dependency on catalytic activity. Based on their catalytic reliability, these HDACs can be grouped into two major classes, i.e., Zn2+-dependent HDACs and nicotinamide adenine dinucleotide (NAD+)-dependent HDACs. Given that all the isoforms of class I, II, and IV require a catalytic Zn2+ ion for their deacetylation activity, these isoforms are aggregated as Zn2+-dependent metalloenzymes, whilst class III HDACs, which are NAD+-dependent isoforms popularly known as sirtuins (SIRT1–7).5,6 Given that they are associated with epigenetic mechanisms and post-translational modifications of the histone schemes, the Zn2+-dependent HDACs are associated with a wide spectrum of diseases and pathophysiological conditions, including neuronal disorders, cancer conditions, parasitic and viral infections, cardiac diseases, and epigenetic disorders, including cancer.7Due to this involvement and contribution of HDAC metalloenzymes, the development of small-molecule drug candidates has become a key medium for the treatment of HDAC-associated diseases. Although a number of studies have been performed to develop effective drug candidates against HDAC activity, only a handful of HDAC inhibitors have been approved by the US-FDA and CFDA for the development and treatment of diseases related to HDAC activity. Similar to the other HDAC metalloenzymes, the class-I HDAC isoform, also known as histone deacetylase 8 (HDAC8), is associated with several disease conditions.4,6 Despite their approval, these HDAC inhibitors, due to their pan-HDAC inhibitory capability, are associated with off-target binding and adverse toxicities, thus restricting their use to a few specific HDAC-related diseases.4,6
A handful of HDAC inhibitors have already been established and approved by the FDA. These HDAC inhibitors can weaken and/or inhibit the HDAC activity and allow the development of effective drug candidates against several diseases, including neurological, inflammatory and autoimmune diseases, metabolic and cardiovascular diseases, rheumatoid arthritis, asthma, diabetes, HIV infection, Alzheimer's disease, Parkinson's disease, Huntington's disease, Schizophrenia, and several cancers.4–6Vorinostat was the first of the HDAC inhibitors approved by the US-FDA for the treatment of relapse/refractory T-cell lymphoma, followed by romidepsin and belinostat, to treat patients with T-cell lymphoma. In recent advancements, the HDAC inhibitor givinostat has been approved by the US-FDA to treat Duchenne muscular dystrophy (DMD).1 Also, the HDAC inhibitor entinostat has been approved by the National Medical Products Association (NMPA) to treat breast cancer.1 Therefore, chidamide/tucidinostat and entinostat are the only two HDAC inhibitors approved by CFDA (in 2014) and Chinese National Medical Products for the treatment of cutaneous T-cell lymphoma and cancer, respectively.1 Additionally, the HDAC inhibitors approved by the US-FDA (vorinostat, romidepsin, and belinostat, Fig. 1) are already available in the market for the treatment of refractory T-cell lymphoma and multiple myeloma. In recent advancements, HDAC inhibitors have been approved by the US-FDA to treat breast tumors (vorinostat, romidepsin, and belinostat, Fig. 1), followed by the Chinese National Medical Products Administration (NMPA, 2021) for the treatment of hormone receptor-positive (HR+), negative breast cancer, hepatocellular carcinoma, and endometrial carcinoma.1 Similar to other HDAC isoforms, the class Ic HDAC8 is also known as an oncogenic isoenzyme, as it is associated with a wide range of abnormal diseases.4–6
 | ||
| Fig. 1 Structure of the FDA-approved HDAC inhibitors and the most selective HDAC8 inhibitors OJI-1 and PCI-34051. | ||
In contrast, there are a few potential inhibitors available, such as NBM-BMX (Fig. 2), which was identified as a novel and selective HDAC8 inhibitor and is a first-in-class small molecule inhibitor of HDAC8 activity and has been tested against glioblastoma.8 Besides, PCI-34051 and OJI-1 (Fig. 1) are two hydroxamate derivatives that have highly potent HDAC8 inhibitory activity with isoform selectivity for HDAC8.4,9,10 In contrast, NBM-BMX (Fig. 2) is a semisynthetic derivative of osthole (Fig. 2). It is the only HDAC8-selective inhibitor to complete a phase I clinical trial and can be tested in phase II.8 Therefore, the development of selective, isoform-specific inhibition/distribution of HDAC8 activity can provide a breakthrough for the treatment of diseases associated with HDAC8. Also, OJI-1 and PCI-34051 (Fig. 1) are two hydroxamate derivatives that are not only highly potent but also have remarkable selectivity for HDAC8.4
 | ||
| Fig. 2 Structure of the potent HDAC8 inhibitory small molecules. | ||
Besides these inhibitors, to maximize the success rate in the development of HDAC8-specific therapeutic interventions, other methods, such as monoclonal antibodies (mAbs), have shown promising outcomes when used for the treatment of cancer.11 However, in contradiction to these approaches, the targeted degradation of the HDAC8 enzyme via proteolysis can be a viable yet familiar approach for the treatment of HDAC8-related diseases. In this scenario, Proteolysis Targeting Chimera (PROTAC) technology can be a quite useful tool that can be used in conjunction with selective HDAC8 inhibitors to not only avoid the mechanism of action (MOA)-based temporary inhibition of HDAC8 but to degrade the enzyme actively to disrupt its pathophysiological contribution in patients. Therefore, this study encompasses reported HDAC8-PROTACs and their outcomes, along with the analysis of the possibility of novel HDAC8-selective PROTAC development using existing selective HDAC8 inhibitors to aid the development of HDAC8-related therapeutics for disease and pathophysiological conditions in the future.
2. Proteolysis activating chimeras (PROTACs)
The proteolysis activating chimeras (PROTACs), also known as degraders, protein degradation brake (PDB), proteolysis targeting peptides, specific and non-genetic IAP-dependent protein erasers (SNIPER), as well as degradomers.12The PROTACs are small hetero-bifunctional molecules/compounds capable of degrading a specific protein via simultaneous binding to the protein and ubiquitin, subsequently leading to the degradation of the targeted protein molecule.13,14
PROTACs contain three major pharmacophoric features in their structure. The two main components are two ligand groups responsible for binding with the targeted protein of interest (POI) and another ligand group to recruit E3 ubiquitin ligase (E3) connected through a linker motif.12
PROTACs mediate protein degradation by forming a ternary complex with the POI and the E3 ubiquitin ligase, thereby allowing ubiquitination, and leading to increased promiscuity to the POI, and the 26S proteasome, as a part of the ubiquitin-proteasome system (UPS), identifies the polyubiquitinated POI and degrades it.13,14 The timeline of the development of PROTACs is shown in Fig. 3.
 | ||
| Fig. 3 Graphical representation of the timeline of PROTAC development, with the initiation of HDAC-related PROTAC development. | ||
Although the development of different PROTACs is quite recent, the historical development of PROTACs dates back to 2001, where the first reported PROTACs were able to induce proteolysis of methionine aminopeptidase-2 (met-AP-2) by recruiting SCFB-TRCP E3.15 The subsequent reports of PROTAC development targeted the degradation of nuclear receptors (NR) such as androgen receptor (AR) and estrogen receptor (ER),16 which were more capable of functioning inside intact cells through micro injection. The FKBP-targeting PROTACs were designed to target and degrade the von Hippel–Lindau tumor suppressor protein (VHL) E3, containing a cell-penetrating peptide with an HIF-1 peptide fragment, which showed efficacy in intact cells without microinjection.17
Interestingly, the initially designed PROTACs suffered drawbacks due to their low permeability and lower micromolar range activity, where the identification of E3 recruiting small-molecule ligands paved the way for the development of small-molecule PROTACs.18 The first reported small-molecule-based PROTACs targeted the mouse double minute 2 homologue (MDM2) along with E3 ubiquitin ligase and demonstrated the viability of cell-permeant ability of PROTAC and the significance of micromolar concentration, while inducing AR degradation.19 Simultaneously, the PROTAC targeting the cellular inhibitor of apoptosis protein-1 (cIAP1) showed cIAP1 auto-ubiquitination and degradation of cIAP1.20 The development of cIAP1-recruiting PROTAC targeting the cellular retinoic acid-binding protein (CRABP-1 and II) via reshuffling was reported. Later, in 2012, the development of peptide-mimetic VHL–E3 targeting PROTACs with high activity was reported by the Department of Pharmacology, Yale University, USA.21–23
Further significant advancement in PROTAC technology was noticed with the report of phospho-PROTACs demonstrating PROTAC efficacy in in vivo mouse models.24 The isoform-specificity of a receptor tyrosine kinase (RTK) further enabled the selectivity of phospho-PROTACs as the POI-targeting moiety was designed to incorporate peptide sequences similar to RTKs for phosphorylation.24 Then, Halo-PROTAC was reported, which is a novel class of PROTACs consisting of small-molecule VHL ligands targeted for Halo-Tag 7 fusion protein degradation.25
Interestingly, to host potent Halo-PROTAC, targeting GFP-HT7 demonstrated the degradation of 90% (Dmax) with a low DC50 of 19 nM.25 The CLIPTAGs (in-cell link-based proteolysis-activating chimeras) were reported in 2016 as tetrazine-tagged chlorotrion haloalkane derivatives that crosslink covalently with the protein in cells to form PROTACs to recruit Carbon E3 ligase. These CLIPTAGs were able to degrade various cancer targets such as BRD4 and ER-1/2.25 The first application of a developed HDAC-targeting small-molecule PROTAC was reported by Young and co-workers, which is the first small molecule of HDAC6 E3 PROTAC, selectively targeting the zinc-dependent metalloenzyme HDAC6.26
Apart from this development, over the last 2 years, more than half a dozen PROTACs have been tested in different phases of clinical trials for the therapy of breast cancer, associated disorders-mediated insomnia, sensory processing disorder, childhood fibrolamellar carcinoma, head and neck cancer, and solid malignant neoplasm/recurrent fibrolamellar carcinoma.8
3. HDACs in diseases: a summary
As discussed earlier, HDAC8 is the only class I, Zn2+-dependent metalloenzyme that participates in chromatin condensation via histone deacetylation.4–6 Moreover, the HDAC8 gene is located on an X-linked region, and thus the gene of HDAC8 is located in the q13 region, precisely from the X-inactivation centre and pre-leukemic condition-related chromosomal breakpoints on the X chromosome.26 It is observed that HDAC8 is capable of deacetylating the histone proteins. Apart from the histone proteins, p53, ERK, α-tubulin, SMC3, and inv(16) are also substrates of HDAC8.1 Due to the functionality of HDAC8 on histone and non-histone proteins, it is associated with both epigenetic modification and disease conditions, including cancer and neuronal disorders.4–6The previously unknown extra-proteasomal actions of the protein hRpn13 are described in another study that aimed to modify the transcriptome and proteome via the epigenetic factors HDAC8 and PADI4, and transcription factor NF-kB-p50.27 It was discovered that a PROTAC that targets hRpn13 also co-depletes other crucial components, such as PADI4 and HDAC8, proving that the impact of hRpn13 goes beyond its conventional function in protein degradation.
Apart from transcriptional modifications, HDAC8 is associated with several diseases and pathophysiological disorders,4,6,28,29 including breast cancer,4 cervical cancer,30 non-small cell lung cancer (NSCLC),31 colorectal cancer,32 gastric adenocarcinoma,33,34 hepatocellular carcinoma,35 acute myeloid leukemia (AML),36 neuroblastoma,37 child hood acute lymphoblastic leukemia (ALL),38 other squamous cell carcinomas,39 lymphoma,40 melanoma,41 glioblastoma,42 and pancreatic cancer.43 Apart from different cancer conditions, HDAC8 is also associated with multiple diseases and epigenetic disorders, including Cornelia de Lange Syndrome,6 as well as viral and parasitic disorders such as schistosomiasis,44 foot-and-mouth disease,45Influenza A virus,46 and Uukuniemi virus.6
4. Development of HDAC8 PROTACs
Despite the reports of different HDAC-targeting PROTACs, the targeting of selective HDAC8 degraders was first reported by Chotitumnavee and coworkers in 2022, two decades after the development of the first PROTAC.47 Based on the molecular docking study of their previously reported selective HDAC8 inhibitors NCC-149 (Fig. 2), the researchers deduced that the substitution at the meta-position of NCC-149 (Fig. 2) is more preferable for selective HDAC8 inhibition compared to para/ortho-group substitutions due to steric clashes of the linker with the active site. Additionally, as the linker groups are an essential part for PROTAC development, considering the observations of NCC-149, the researchers developed a series of NCC-149-based PROTACs with variable linker lengths, such as C5, C8, and C11, while using pomalidomide as a ligand for E3 ubiquitination.To evaluate the effects of the newly designed selective HDAC8 degraders, the researchers tested these molecules (PROTAC 1–4, Fig. 2 and Table 1) against the Jurkat T-cell leukemia cell line. Among these compounds, PROTAC 4 (Fig. 2 and Table 1) with a C11 linker chain attached at the para-position of the phenyl hydroxamate moiety was found inactive, where substitution at the meta-position with a C8 and C11 chain in its linker, which is attached at the meta-position of the phenyl ring (PROTAC 2 and 3, Fig. 2 and Table 1), showed a dose-dependent reduction of the HDAC8 levels, suggesting the beneficial effect of meta-phenyl substitution for selective HDAC8 degradation. Also, between the molecules having the linker motif substituted at the meta-position of the phenyl hydroxamate moiety, the compound PROTAC 3 (Fig. 2 and Table 1) with a C11 carbon chain in its linker moiety showed the most potent HDAC8 degradation (HDAC8 DC50 = 0.702 μM). Also, the in vitro enzyme inhibition assay of compound PROTAC 3 (Fig. 2) showed its strong inhibition against HDAC8 (IC50 = 0.372 μM), while demonstrating no inhibition against HDAC1 and 2, as well as weak inhibition against HDAC6 (IC50 >30 μM).
|
|
|||||
|---|---|---|---|---|---|
| PROTAC ID | n | * (position) | HDAC8 IC50 (μM) | HDAC8 DC50 (μM) | Jurkat GI50 (μM) |
| PROTAC 1 | 5 | meta | — | — | — |
| PROTAC 2 | 8 | meta | — | — | — |
| PROTAC 3 | 11 | meta | 0.372 | 0.702 | 0.780 |
| PROTAC 4 | 11 | para | — | — | — |

In another study on the design and development of selective HDAC8 degraders, Darwish et al.48 utilized the earlier reported hydroxamate-based inhibitors in combination with different E3 ligands such as CRBN, VHL, and MDM2 to design a novel series of PROTACs. The structure–activity relationship (SAR) study of these hydroxamates has shown that their SAR involves small hydrophobic substitution at the para-position of the phenyl hydroxamate and hydrophobic capping (HyT) to design a novel series of PROTACs. It is interesting to note that the presence of this moiety showed potent HDAC8 inhibition (Fig. 4 and 5).
 | ||
| Fig. 4 SAR of the NCC-149 and NCC-172 derivatives. | ||
 | ||
| Fig. 5 SAR of the PROTACs reported by Darwish and co-workers. | ||
It is interesting to note that the presence of electronegative groups such as chlorine at the para-position of the phenyl ring increased the selectivity toward HDAC8 over HDAC1 but diminished the HDAC8/6 selectivity. Also, replacement of the electronegative atom at R with hydrogen has been shown to improve the HDAC8 selectivity over HDAC1 and HDAC6. Again, regarding the substitution at the meta-position of the phenyl ring, it is clearly observed that 4-methoxy benzyl amino group substitution also improves the HDAC8 selectivity over HDAC1 but suppresses the selectivity over HDAC6. Further, substitution with a p-methoxy benzyl amino group at the meta-position of the phenyl ring destroys the HDAC1 inhibitory potency (Fig. 5). Interestingly, the benzoyloxycarbonylmethyl group substitution at the meta-position of the phenyl ring was found to be more valuable for HDAC8 selectivity by drastically decreasing the HDAC inhibitory potency, while diminishing the HDAC1 inhibitory activity if any electronegative element is present at R (i.e., chlorine), HDAC8 IC50 = 0.41 μM, HDAC1 IC50 > 20 μM, and HDAC6 IC50 = 7.4 μM.
A similar observation was also noticed for the molecule with methoxy and 4-methoxy benzyl groups at R and the meta-position of the phenyl ring, respectively, showing inactivity against HDAC1 (IC50 > 20 μM) but was able to diminish the HDAC8 selectivity over HDAC6 (HDAC8 IC50 = 0.01 μM, HDAC6 IC50 = 0.15 μM) (Fig. 6). It is interesting to acknowledge that the compound with a benzyl amino group at the meta-position of the phenyl ring and a methoxy function at R showed quite an improvement in the HDAC8 selectivity over both HDAC6 and HDAC1, despite being unable to show its inactivity against HDAC1 (HDAC8 IC50 = 0.07 μM, HDAC1 IC50 = 14.5 μM, and HDAC6 IC50 = 51 μM) (Fig. 6). This clearly suggests the affirmative contribution of the benzyl amino substitution for HDAC8 selectivity over HDAC1 and HDAC6.
 | ||
| Fig. 6 SAR of the PROTACs comprising methoxy and 4-methoxy benzyl groups. | ||
Based on the enzyme inhibitory assay in vitro against HDAC1, HDAC6, and HDAC8, it is observed that the VHL E3-ligand function containing an ester moiety with a C-aryloxy moiety, despite its weak activity against HDAC1, showed a highly reduced HDAC6/8 dual active PROTACs (Fig. 7). Interestingly, only the VHL E3-ligand-containing prodrug with a 4-(n-butyl) amino carbonyl methoxy benzyl amino linker and chloro substitution at the para-position of the phenyl ring showed quite improved HDAC8 selectivity over HDAC1 and HDAC6, with potent HDAC8 inhibition. Additionally, the VHL-based E3-ligand showed considerably better HDAC8 selectivity over HDAC1 and HDAC6 (PROTAC 5, Fig. 7).
 | ||
| Fig. 7 Structure of PROTAC 5–10. | ||
In the case of the CRBN-based pomalidomide–E3 ligand-containing PROTACs, it is observed that both a change in the linker and linker length, and the R-substitution present in the para position of the phenyl ring of the HDAC8 inhibitor part (NCC-149, Fig. 2) had a significant effect on both the HDAC8 inhibitory potency as well as selectivity over HDAC1 and HDAC6. It was noticed that for CRBN-based pomalidomide-containing PROTACs, the linker length of these PROTAC degraders was reported to have a correlation between their linker length and HDAC8 selectivity over HDACs in the in vitro enzyme inhibition studies (PROTAC 7–9, Fig. 7). It is observed that although both the PROTACs containing a 5-carbon and 7-carbon chain linker (PROTAC 7 and PROTAC 8, respectively, Fig. 7) showed similar selectivity (2-fold) for HDAC8 over HDAC6, an increase in carbon chain length (n = 9) (PROTAC 9, Fig. 7) showed a drastic decrease in HDAC8 selectivity (HDAC6/8 = 0.7). Additionally, these CRBN-based degraders, with a pomalidomide cap and variable substitutions such as methyl, chloro, and methoxy groups at the R position showed quite effective and selective HDAC8 inhibition over HDAC1 and 6, except the chloro-substituted degrader, which showed a detrimental HDAC8 selectivity due to its potent HDAC6 inhibition. Furthermore, it is also observed that replacement of the n-allyl/n-alkyl triazolyl methoxy benzyl amino linker moiety with allyl polyglycol–ethylene glycol-based linker moiety showed a detrimental effect on the HDAC8 inhibition, where chloro substitution at the R-position, though showing potent HDAC8 inhibitory activity, unlike its hydrogen atom containing counterpart (Fig. 8).
 | ||
| Fig. 8 SAR of pomalidomide-based HDAC8-targeting PROTACs. | ||
In another pioneer effort to design selective HDAC8-degraders, Huang and co-workers49 reported a series of HDAC8-selective PROTACs with HDAC8 as the HDAC8 warhead, in combination with allyl and polyethylene glycol (PEG) linkers, as well as CRBN–pomalidomide/CRBN and VHL(von Hippel–Lindau)-based E3 ligands (PROTAC 11–13, Fig. 8 and Table 2). Regarding their HDAC8 degradation ability, the VHL–E3 ligand-containing compounds have shown a comparatively greater extent of degradation compared to their CRBN–pomalidomide-based analogs. Interestingly, regarding the changes in the linker motif of these degraders, the n-pentyl linker-containing degrader (PROTAC 12, Fig. 9) showed almost similar HDAC8 degradation (39.5% at 200 μM and 85.3% at 2 μM) compared to its diethylene glycol (DEG)-containing analog having the same pomalidomide-based CRBN–E3 warhead (PROTAC 13, 39.7% at 200 μM, 85.4% at 2 μM). However, for these CRBN-based molecules, both an increase (n = 6) and decrease (n = 4) in the alkyl linker length were found to reduce their HDAC8 degradation capability. However, although the n-butyl linker-containing PROTAC 11 (Fig. 10) was unable to show similar degradation ability at 200 μM, it was capable of showing an almost similar degradation of HDAC8 at 2 μM conc (30.6% at 200 μM, 83.6% at 2 μM). Interestingly, although for the allyl linker-containing degraders, it was noticed that the CRBN-based degraders showed marginally better degradation of HDAC8, for the VHL-based E3 ligand-containing degraders, the n-butyl linker as well as diethylene glycol linker motifs showed effective HDAC8 degradation for both CRBN and VHL–E3 ligand substitutions (PROTAC 11: 34.6% at 200 μM and 75.7% at 2 μM, PROTAC 15: 45.7% at 200 μM and 91.3% at 2 μM, Table 2). Besides the primary determination, the researchers also tested the degradation capability of these PROTACs to induce degradation in different cancer cells, including A549, HeLa, and HCT-116, where the PROTAC with CRBN–E3 ligand and n-hexyl linker was found to cause an effective dose-dependent reduction in the HDAC8 protein levels with >90% degradation at 10 μM, while almost sparing other class I (HDAC1, HDAC2, HDAC3), class II (HDAC6, HDAC9), and class III SIRT 7.
|
|
||||||
|---|---|---|---|---|---|---|
| PROTAC ID | L | R | HDAC8 DC50 (μM) | Degradation of HDAC8 | HDAC8 IC50 (μM) | |
| 0.2 μM | 2 μM | |||||
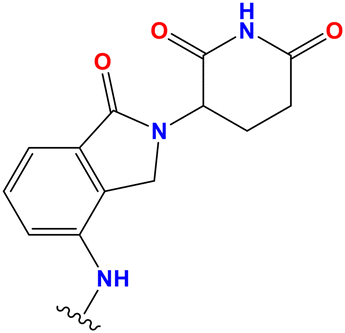
| PROTAC 11 | (CH2)4 |

|
0.47 | 30.6% | 83.6% | 7.11 |
| PROTAC 12 | (CH2)5 |
|
0.62 | 39.5% | 85.9% | 12.52 |
| PROTAC 13 | CH2CH2OCH2CH2 |

|
0.58 | 39.7% | 85.4% | 9.55 |
| PROTAC 14 | (CH2)4 |

|
— | 34.6% | 75.7% | — |
| PROTAC 15 | CH2CH2OCH2CH2 |
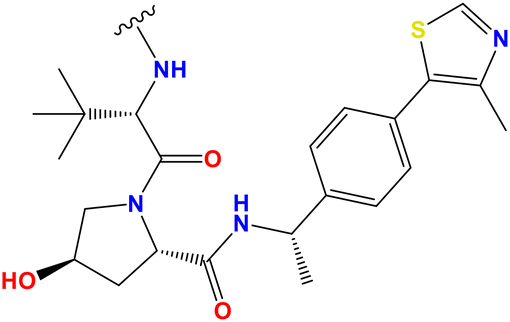
|
— | 45.7% | 91.3% | — |
 | ||
| Fig. 9 Structure of the HDAC8-targeting PROTACs. | ||
 | ||
| Fig. 10 Structure of PROTAC 19–24. | ||
Also, an added degradation of IREF protein was noticed for compounds PROTAC 11, 12, and 13 (Fig. 8) to a lower extent at 10 μM conc in the A549 cell line. Upon testing the HCT-116 and HeLa cell lines, all the compounds (PROTAC 11–13, Fig. 8) showed a near-complete degradation of HDAC8. Furthermore, the most potent VHL-based PROTAC 11 also showed similar results when evaluated against the HCT-116, HeLa, and A549 cell lines. Through further validation of the observations, the post-immunofluorescence assay revealed the dose-dependent inhibition of HDAC8 foci formation by compound PROTAC 15 (Fig. 9) for around 60 h in both the A549 and HCT-116 cell lines. The ELISA-based determination of the DC50 of these compounds showed selective/preferential HDAC8 degradation over HDACs 1–3, while displaying a DC50 of 0.58 μM for HDAC8 with 95% HDAC8 degradation at 10 μM conc. The other CRBN–alkyl linker-based compounds, PROTAC 11 and PROTAC 12, also showed a similar degradation (>90%) at 10 μM with a DC50 value of 0.47 μM and 0.62 μM, respectively. The cancer cell growth and anti-proliferative activity studies also revealed the concentration-dependent inhibition of cancer cell proliferation for compounds PROTAC 11–13 with IC50 values of 7.11 μM, 12.52 μM, and 9.55 μM, respectively. Additionally, the combined treatment of PROTAC 12 and irradiation showed an even stronger anti-proliferative effect with an IC50 of 6.04 μM, which is higher in comparison to the combined treatment of PCI-34051 (Fig. 1) and irradiation therapy.
In the journey to develop selective HDAC8 PROTACs, Sun et al.50 developed a series of pomalidomide-based degraders with a warhead containing BZD-7354, an HDAC 6/8 dual inhibitor that showed decreased activity in the HCT-116 cell line. Interestingly, though the warhead and the E3 ligand remained unchanged, a change in the linker length of these compounds showed a dramatic change in their HDAC8 degradation capability. The SAR study of these compounds showed that the n-alkyl moiety present in the linker function (Fig. 9), a 5C containing n-alkyl chain (n-pentyl) between the two amino groups (PROTAC 16, % HDAC8 degradation in HCT116 at 5 μM = 85%), and n-butyl group between the phenoxy and carbonyl groups provided potent HDAC8 degradation. Also, the presence of a pentanoyl–piperazine group in place of the PROTAC 16 linker motif drastically improved the HDAC8 degradation (PROTAC 17, HDAC8 degradation in HCT116 at 5 μM = 74.4%, PROTAC 16) but decreased the HDAC8 DC50, where the amino n-pentyl amino carbonyl–n-butyl linker (PROTAC 18, % HDAC8 degradation in HCT116 at 5 μM = 68.9%) showed moderate HDAC8 degradation and HDAC8 DC50.
A crucial investigation was outlined in the reports of Xiao and colleagues,51 introducing YX968 (PROTAC 16, Fig. 9), a dual PROTAC degrader of HDAC3 and HDAC8, which marked a substantial development in this sector. PROTAC 19 was identified as the most effective HDAC8 degrader containing a CRBN-based E3 recruiter with a hydrazide-ZBG-based HDAC-targeting warhead, demonstrating robust activity that is superior to previously reported single-target PROTACs and the ability to induce rapid and comprehensive degradation of both HDAC3 and HDAC8, as well as effective growth suppression of aggressive breast tumor cell lines, including MDA-MB-231. Biologically, the selective and powerful dual-degradation of HDAC3/8 by YX968 (PROTAC 19) is particularly significant. Unlike pan-HDAC inhibitors, which cause widespread histone hyperacetylation and associated side effects, the tailored degradation by YX968 (PROTAC 19, Fig. 10) produces anti-cancer benefits without this global alteration. The findings of this study revealed that YX968 (PROTAC 19, Fig. 10) caused HDAC8 inhibition with IC50 values of 0.591 μM, 0.283 μM, and 0.740 μM for HDAC1, HDAC3, and HDAC8, respectively, whilst providing HDAC8 degradation with a DC50 value of approximately 0.0061 μM in MDA-MB-231 cells.
In another study, researchers used click chemistry to generate and screen vast libraries of chemicals.52 This strategy enabled them to quickly identify and optimize lead compounds. Their initial screening revealed NCC-149 to be a strong and specific HDAC8 inhibitor. Then, they ran a structural optimization campaign, changing the triazole linker in NCC-149 (Fig. 2). This resulted in the discovery of NCC-172 (Fig. 2), a molecule with high potency and selectivity. The NCC-172-based PROTAC 20 (Fig. 10) incorporates an E3 ligase recruiter pomalidomide via a linker. The creation of this PROTAC was a significant step given that it resulted in a molecule capable of eliciting HDAC8 degradation, thus blocking both its catalytic and scaffolding roles. The SAR research gives key insights, such as how replacing the triazole ring in NCC-149 with other aromatic rings drastically affected both potency and selectivity due to slight geometric variations between the rings. It was discovered that the meta-position on the phenyl ring of NCC-172 was the best site for linker attachment, whereas analogs with the linker at the para-position (PROTAC 21, Fig. 10) showed no degrading activity, supporting the importance of exact spatial orientation for successful ternary complex formation.
In the in vitro enzyme inhibition study (PROTAC 21, Fig. 10), it selectively destroyed HDAC8, while leaving other HDACs (HDAC1, HDAC2, and HDAC6) alone in the cell. PROTAC 21 (Fig. 10) was much more effective in reducing cell growth. Despite having lower in vitro inhibitory activity than its parent inhibitor (IC50 of 0.372 μM vs. 0.053 μM for NCC-172), the PROTAC-induced degradation pathway led to an approximately 9-fold increase in anti-cancer activity, with a GI50 of 0.78 μM. This significant difference suggests that targeting both the catalytic and scaffolding activities of HDAC8 is a more effective cancer treatment strategy than simply decreasing its enzymatic activity.
In another study, the researchers created a series of PROTACs that are intended to specifically break down HDAC8.53 This work investigated a more promising strategy than simple inhibition by causing protein degradation, given that abnormalities in HDAC8 functions are connected to a number of disorders. This research effectively created (PROTAC 22, Fig. 10) a strong and specific HDAC8 degrader, which was demonstrated in both Jurkat T-cell leukemia cells and MDA-MB-231 triple-negative breast cancer cells, as well as single-digit nanomolar DC50 values (MDA-MB-231 HDAC8 DC50 = 0.0047 μM, MDA-MB-231 HDAC8 Dmax = 95% μM, Jurkat HDAC8 DC50 = 0.0018 μM, Jurkat HDAC8 Dmax = 97%). Strong anti-migration and apoptotic cell death were seen in PROTAC 22, indicating the therapeutic potential of HDAC8 degradation for associated disorders.
The discovery of unidentified binding sites appropriate for the synthesis of allosteric covalent inhibitors of HDAC8 influenced the develop potent HDAC8 degrader.54 Using this knowledge, the researchers created a novel, lead-like covalent inhibitor (PROTAC 23, Fig. 10) by combining an electrophilic hit fragment with a recognized HDAC8 inhibitor. The linker length, which has been determined to be crucial for optimizing the binding pose, and consequent inhibitory activity, is the primary driver of the SAR. Long linkers caused a change in the binding mode and activity loss, whereas the compounds with medium to short linkers had noticeably greater binding and inhibitory effects.
During the creation of selective hydrazide-based HDAC8 degraders, a number of novel HDAC8-selective PROTACs were created using an HDAC8 warhead. The researchers rationally constructed a new chemical, YX862 (PROTAC 24, Fig. 10), by tweaking the YX968 (PROTAC 19, Fig. 10) warhead. This redesign successfully eliminated HDAC3 degradation activity, yielding a highly potent and selective HDAC8-PROTAC degrader capable of inducing apoptosis and killing cancer cells; however, its dual-targeting nature limited its ability to serve as a selective probe for HDAC8 functions alone. To solve the selectivity issue, the researchers created YX862 (PROTAC 24, Fig. 10), and its selectivity for HDAC8 was achieved by altering the warhead to give HDAC3-sparing activity.
This modification allowed the creation of a tool that could selectively degrade HDAC8 without the confounding effects of also degrading HDAC3, thereby enabling a more precise study of the unique biological functions of HDAC8. Regarding the SAR of these hydrazide-based PROTACs, key observation implicates that the use of a hydrazide group as a zinc-binding group (ZBG) to address the issues of instability, toxicity, and low bioavailability associated with more common hydroxamic acid-based ZBGs. The hydrazide-based design is central to the potency and selectivity of the HDAC8 degraders.
The SAR investigations discovered that a hexyl-substituted hydrazide group was especially successful because its unusual structure caused it to protrude from the active sites of off-target HDACs such as HDAC2 and HDAC6, making it extremely selective for binding to HDAC8. This emphasizes the value of accurate linker and warhead design in obtaining target specificity. YX862 (PROTAC 24, Fig. 10) exhibited highly effective and selective HDAC8 degradation. It was demonstrated to be a powerful degrader with a DC50 value in the single-digit nanomolar range and a 400-fold selectivity for HDAC3 degradation. In terms of biological activity, YX862 degradation of HDAC8 resulted in a considerable increase in the acetylation of nonhistone substrates, especially SMC3, while not significantly causing global histone hyperacetylation. This finding lends credence to the concept that the primary in vivo role of HDAC8 is in nonhistone protein modification, rather than a broad effect on gene expression via histones. Furthermore, YX862 showed promising antiproliferative efficacy. In DLBCL cell lines, YX862 (PROTAC 24, Fig. 10) outperformed the typical HDAC8 inhibitor, PCI-34051, in terms of cell growth suppression. This implies that PROTAC-mediated degradation, which removes both the enzymatic and scaffolding activities of HDAC8, is a more effective treatment strategy than simple inhibition.55
In another study, four pomalidomide E3 ligand-containing HDAC8 degraders were introduced by Zhao and co-workers and their HDAC8 degrading capability evaluated, along with their apoptotic and anti-migration properties (PROTAC 25–27, Table 3).56 Among these degraders, PROTAC 27 with a carboxamido n-octyl linker showed the most effective HDAC8 degradation, besides its potent apoptosis-inducing and anti-migration efficacy.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| PROTAC | LINKER | Cell line | HDAC8 DC50 (nM)/degradation | D max (%) | % HDAC8 degradation | Key notes | |
| At 1 μM | At 10 μM | ||||||
| PROTAC 25 | –CO–NH–(CH2)2– | MDA-MB-231 (TNBC) | 53 | 95 | 90 | 93 | Reduced potency compared to CT-4 due to shorter linker length |
| PROTAC 26 | –CO–NH–(CH2)6– | MDA-MB-231 (TNBC) | 14 | 91 | 94 | 44 | Reduced potency compared to CT-4 due to shorter linker length |
| PROTAC 27 | –CO–NH–(CH2)8– | MDA-MB-231 (TNBC) | 1.8 | 97 | 94 | 62 | Most potent in series. Exhibited potent anti-migration activity but limited anti-proliferative activity |
| Jurkat (T-cell leukemia) | 4.7 | 95 | Potent anti-proliferative activity (GI50 = 2.4 μM) with effective apoptosis induction | ||||

Among them, the novel PROTAC 27 emerged as a very potent and efficient HDAC8 degrader, outperforming earlier analogs (PROTAC 25–26, Table 3) and previously described HDAC8-targeting PROTACs. It had single-digit nanomolar degradation potency, with DC50 values of 1.8 nM in MDA-MB-231 cells and 4.7 nM in Jurkat cells, and it increased the maximum degradation Dmax by more than 95% across both cell lines. PROTAC 27 demonstrated considerable selectivity for HDAC8 over HDAC6 (with a ∼20-fold DC50 difference in MDA-MB-231 cells) and good selectivity over HDAC1 and HDAC3.
The SAR of these novel degraders demonstrates that their degradation potency is predominantly determined by their linker length and successful CRBN interaction. The PROTACs were created by attaching the HDAC8 inhibitor to the CRBN recruiter pomalidomide through an aliphatic linker. Systematic variation found that the longest linker (PROTAC 27, with C8 carbon chain, n = 8, Table 3) yielded the most powerful HDAC8 degradation (DC50 = 1.8 nM). PROTAC 26 (n = 6), and PROTAC 27 (n = 2) had progressively shorter linkers, which resulted in ≥10-fold decrease in potency, suggesting that the C8 linker is best for effective ternary complex production.
5. Conclusion
Regarding the development of PROTACs, the findings reveal a significant shift in therapeutic strategy from blocking to degrading HDAC8. Traditional small-molecule inhibitors have exhibited some effectiveness, but their lack of selectivity frequently results in off-target effects and systemic damage. This review argues that HDAC8 PROTACs are a superior choice. By connecting a powerful HDAC8 warhead (such as NCC-149 or hydroxamate derivatives) to an E3 ubiquitin ligase recruiter (such as VHL or CRBN), these bifunctional compounds can produce a ternary complex that causes polyubiquitination and subsequent proteasomal destruction of HDAC8. This approach is especially helpful given that it not only removes the catalytic function of the enzyme but also destroys its scaffolding and non-enzymatic activities, resulting in a more profound and long-lasting therapeutic effect. Studies have revealed that these PROTACs degrade HDAC8 nearly completely in numerous cancer cell lines at single-digit nanomolar concentrations and outperform their parent inhibitors in terms of anti-proliferative efficacy.Moreover, future work should focus on gaining a better understanding of the SAR for both the linker and the HDAC8-targeting warhead. This includes looking into new linker chemistries to improve the length, stiffness, and hydrophilicity, all of which are important for increasing the cell permeability and ternary complex formation. Furthermore, discovering new, highly selective HDAC8 warheads other than the present hydroxamate-based chemicals, such as OJI-1 and PCI-34051 (Fig. 1),4,9,10 could result in more effective and selective degraders.
Furthermore, all these HDAC8-targeting degraders contained a POI-targeting warhead containing a zinc binding group (ZBG). Due to their reliability and potency, ZBG-containing warheads (e.g., hydrazide and hydroxamate) have been extensively used. However, a series of weak ZBGs are available as HDAC8 inhibitors (e.g., thiol, thiophene, oxazole, and triazole),57 while selective HDAC8 inhibitors without any ZBG can be tested for the development of HDAC8-targeting degraders. Besides, Zhao and co-workers have shown that despite targeting HDAC8, PROTAC27 was able to not only exhibit apoptotic activity in the Jurkat cell line, but also demonstrated anti-migratory activity in the MDA-MB-231 cell line.56 Therefore, targeting non-catalytic functions can be exploited for the treatment of other cancer conditions to provide effective therapeutic efficacy.
Furthermore, the existing PROTACs rely heavily on VHL and CRBN. Future studies should look into different E3 ligases, as this could provide alternative degradation pathways, lower the chance of resistance, and improve therapeutic effects. For example, enlisting E3 ligases that are abundantly expressed in specific tumor types may improve targeted degradation, while minimizing off-target effects. Also, the strong anti-proliferative action of HDAC8 PROTACs highlights their potential in combination therapy.
It is also quite interesting to note that almost all the HDAC8-targeting degraders with effective HDAC8 degrading effects have shown an optimal linker length between C5–C11 carbon chain, especially the C8 and C11 linker motifs in combination with CRBN and VHL-based E3 ligands.
Future research should investigate the synergistic effects when the successful preclinical development of these degraders, as well as continuing clinical studies on other PROTACs, demonstrate the enormous potential of this technology for treating HDAC8-related disorders including the development of PROTAC-based nano formulations and PROTAC-antibody conjugates to minimize therapeutic adverse effects, while providing efficacy.58 The field of HDAC8 PROTACs is still in its early stages, and several critical areas require additional research to fully realize their therapeutic potential, which can point toward a bright future for effective therapy development.
Conflicts of interest
There are no conflicts to declare.Data availability
No new data were generated or analyzed in this review article. All data necessary to support this review are available within the published literature cited herein.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5md00871a.
Acknowledgements
The authors are sincerely thankful to the Department of Pharmaceutical Technology, Jadavpur University, Kolkata, India, and the Department of Pharmacy, Birla Institute of Technology and Science-Pilani, Hyderabad Campus, for providing the necessary research facilities.References
- Q. Zhao, H. Liu, J. Peng, H. Niu, J. Liu, H. Xue, W. Liu, X. Liu, H. Hao, X. Zhang and J. Wu, HDAC8 as a target in drug discovery: function, structure and design, Eur. J. Med. Chem., 2024, 280, 116972 CrossRef PubMed.
- A. Chakrabarti, J. Melesina, F. R. Kolbinger, I. Oehme, J. Senger, O. Witt and W. Sippl, Jung M. Targeting histone deacetylase 8 as a therapeutic approach to cancer and neurodegenerative diseases, Future Med. Chem., 2016, 8, 1609–1634 Search PubMed.
- L. Verdone, M. Caserta and E. Di Mauro, Role of histone acetylation in the control of gene expression, Biochem. Cell Biol., 2005, 83, 344–353 Search PubMed.
- S. Banerjee, B. Ghosh, T. Jha and N. Adhikari, A patent review of histone deacetylase 8 (HDAC8) inhibitors (2013-present), Expert Opin. Ther. Pat., 2024, 34, 1019–1045 CrossRef.
- S. Banerjee, S. K. Baidya, T. Jha, B. Ghosh and N. Adhikari, Arylcarboxamide derivatives as promising hdac8 inhibitors: an overview in light of structure-activity relationship and binding mode of interaction analysis, Med. Chem., 2025, 21, 471–500 CrossRef.
- S. Banerjee, N. Adhikari, S. A. Amin and T. Jha, Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: An overview, Eur. J. Med. Chem., 2019, 164, 214–240 CrossRef.
- M. Minisini, M. Mascaro and C. Brancolini, HDAC-driven mechanisms in anticancer resistance: epigenetics and beyond, Cancer Drug Resist., 2024, 7, 46 Search PubMed.
- https://clinicaltrials.gov/ .
- S. Balasubramanian, J. Ramos, W. Luo, M. Sirisawad, E. Verner and J. J. Buggy, A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas, Leukemia, 2008, 22, 1026–1034 CrossRef PubMed.
- O. J. Ingham, R. M. Paranal, W. B. Smith, R. A. Escobar, H. Yueh, T. Snyder, J. A. Porco Jr, J. E. Bradner and A. B. Beeler, Development of a potent and selective HDAC8 Inhibitor, ACS Med. Chem. Lett., 2016, 7, 929–932 CrossRef PubMed.
- S. Banerjee, S. A. Amin and T. Jha, Therapies of hematological malignancies: An overview of the potential targets and their inhibitors, Curr. Chem. Biol., 2021, 15, 19–49 CrossRef.
- M. Pettersson and C. M. Crews, PROteolysis TArgeting Chimeras (PROTACs) — past, present and future, Drug Discovery Today:Technol., 2019, 31, 15–27 CrossRef.
- M. Hochstrasser, Ubiquitin, proteasomes, and the regulation of intracellular protein degradation, Curr. Opin. Cell Biol., 1995, 7, 215–223 CrossRef.
- A. Navon and A. Ciechanover, The 26 S proteasome: from basic mechanisms to drug targeting, J. Biol. Chem., 2009, 284, 33713–33718 CrossRef CAS.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, PROTACs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS.
- K. M. Sakamoto, K. B. Kim, R. Verma, A. Ransick, B. Stein, C. M. Crews and R. J. Deshaies, Development of PROTACs to target cancer-promoting proteins for ubiquitination and degradation, Mol. Cell. Proteomics, 2003, 2, 1350–1358 CrossRef CAS.
- J. S. Schneekloth, F. N. Fonseca, M. Koldobskiy, A. Mandal, R. Deshaies, K. Sakamoto and C. M. Crews, Chemical genetic control of protein levels: selective in vivo targeted degradation, J. Am. Chem. Soc., 2004, 126, 3748–3754 CrossRef CAS.
- A. R. Schneekloth, M. Pucheault, H. S. Tae and C. M. Crews, Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics, Bioorg. Med. Chem. Lett., 2008, 18, 5904–5908 CrossRef CAS.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2, Science, 2004, 303, 844–848 CrossRef CAS PubMed.
- K. Sekine, K. Takubo, R. Kikuchi, M. Nishimoto, M. Kitagawa, F. Abe, K. Nishikawa, T. Tsuruo and M. Naito, Small molecules destabilize cIAP1 by activating auto-ubiquitylation, J. Biol. Chem., 2008, 283, 8961–8968 CrossRef CAS.
- D. L. Buckley, J. L. Gustafson, I. Van Molle, A. G. Roth, H. S. Tae, P. C. Gareiss, W. L. Jorgensen, A. Ciulli and C. M. Crews, Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1α, Angew. Chem., Int. Ed., 2012, 51, 11463–11467 CrossRef CAS.
- D. L. Buckley, I. Van Molle, P. C. Gareiss, H. S. Tae, J. Michel, D. J. Noblin, W. L. Jorgensen, A. Ciulli and C. M. Crews, Targeting the von Hippel–Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction, J. Am. Chem. Soc., 2012, 134, 4465–4468 CrossRef CAS PubMed.
- I. Van Molle, A. Thomann, D. L. Buckley, E. C. So, S. Lang, C. M. Crews and A. Ciulli, Dissecting fragment-based lead discovery at the von Hippel–Lindau protein: hypoxia inducible factor 1α protein–protein interface, Chem. Biol., 2012, 19, 1300–1312 CrossRef CAS.
- J. Hines, J. D. Gough, T. W. Corson and C. M. Crews, Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8942–8947 CrossRef CAS.
- D. L. Buckley, K. Raina, N. Darricarrere, J. Hines, J. L. Gustafson, I. E. Smith, A. H. Miah, J. D. Harling and C. M. Crews, HaloPROTACs: use of small molecule PROTACs to induce degradation of HaloTag fusion proteins, ACS Chem. Biol., 2015, 10, 1831–1837 CrossRef CAS.
- I. Van den Wyngaert, W. de Vries, A. Kremer, J. Neefs, P. Verhasselt, W. H. Luyten and S. U. Kass, Cloning and characterization of human histone deacetylase 8, FEBS Lett., 2000, 478, 77–83 CrossRef CAS.
- V. Osei-Amponsa, M. Chandravanshi, X. Lu, V. Magidson, S. Das, T. Andresson, M. Dyba, V. R. Sabbasani, R. E. Swenson, C. Fromont, B. Shrestha, Y. Zhao, M. E. Clapp, R. Chari and K. J. Walters, hRpn13 shapes the proteome and transcriptome through epigenetic factors HDAC8, PADI4 and transcription factor NF-κB p50, Mol. Cell, 2024, 84, 522–537.e8 CrossRef CAS.
- M. M. Rahman and T. O. Tollefsbol, Combinatorial phenethyl isothiocyanate and withaferin A targets multiple epigenetics pathways to inhibit MCF-7 and MDA-MB-231 human breast cancer cells, Cancer Cell Int., 2024, 24, 422 CrossRef CAS PubMed.
- P. An, F. Chen, Z. Li, Y. Ling, Y. Peng, H. Zhang, J. Li, Z. Chen and H. Wang, HDAC8 promotes the dissemination of breast cancer cells via AKT/GSK-3β/Snail signals, Oncogene, 2020, 39, 4956–4969 CrossRef CAS PubMed.
- G. R. Vanaja, H. G. Ramulu and A. M. Kalle, Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6, Cell Commun. Signaling, 2018, 16, 20 CrossRef CAS.
- S. Liu, S. Ma, G. Liu, L. Hou, Y. Guan, L. Liu, Y. Meng, W. Yu, T. Liu, L. Zhou, Z. Yuan, S. Pang, S. Zhang, J. Li, X. Ren and Q. Sun, CK2B induces CD8+ T-cell exhaustion through HDAC8-mediated epigenetic reprogramming to limit the efficacy of anti-PD-1 therapy in non-small-cell lung cancer, Adv. Sci., 2025, 12, e2411053 CrossRef PubMed.
- J. Chen, L. Cao, J. Ma, C. Yue, D. Zhu, R. An, X. Wang, Y. Guo and B. Gu, HDAC8 promotes liver metastasis of colorectal cancer via inhibition of IRF1 and upregulation of SUCNR1, Oxid. Med. Cell. Longevity, 2022, 2815187 CrossRef CAS.
- Y. Wang, P. Xu, J. Yao, R. Yang, Z. Shi, X. Zhu, X. Feng and S. Gao, MicroRNA 216b is down regulated in human gastric adenocarcinoma and inhibits proliferation and cell cycle progression by targeting oncogene HDAC8, Target. Oncol., 2016, 11, 197–207 CrossRef.
- S. Song, Y. Wang, P. Xu, R. Yang, Z. Ma, S. Liang and G. Zhang, The inhibition of histone deacetylase 8 suppresses proliferation and inhibits apoptosis in gastric adenocarcinoma, Int. J. Oncol., 2015, 47, 1819–1828 CrossRef CAS.
- W. Yang, Y. Feng, J. Zhou, O. K. W. Cheung, J. Cao, J. Wang, W. Tang, Y. Tu, L. Xu, F. Wu, Z. Tan, H. Sun, Y. Tian, J. Wong, P. B. S. Lai, S. L. Chan, A. W. H. Chan, P. B. O. Tan, Z. Chen, J. J. Y. Sung, K. Y. L. Yip, K. F. To and A. S. L. Cheng, A selective HDAC8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma, Sci. Transl. Med., 2021, 13, eaaz6804 CrossRef CAS.
- J. Qi, S. Singh, W. K. Hua, Q. Cai, S. W. Chao, L. Li, H. Liu, Y. Ho, T. McDonald, A. Lin, G. Marcucci, R. Bhatia, W. J. Huang, C. Chang and Y. H. Kuo, HDAC8 inhibition specifically targets inv(16) acute myeloid leukemic stem cells by restoring p53 acetylation, Cell Stem Cell, 2015, 17, 597–610 CrossRef CAS PubMed.
- I. Oehme, H. E. Deubzer, D. Wegener, D. Pickert, J. P. Linke, B. Hero, A. Kopp-Schneider, F. Westermann, S. M. Ulrich, A. von Deimling, M. Fischer and O. Witt, Histone deacetylase 8 in neuroblastoma tumorigenesis, Clin. Cancer Res., 2009, 15, 91–99 CrossRef CAS PubMed.
- D. A. Moreno, C. A. Scrideli, M. A. A. Cortez, R. de Paula Queiroz, E. T. Valera, V. da Silva Silveira, J. A. Yunes, S. R. Brandalise and L. G. Tone, Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia, Br. J. Haematol., 2010, 150, 665–673 CrossRef CAS PubMed.
- M. Y. Ahn and J. H. Yoon, Histone deacetylase 8 as a novel therapeutic target in oral squamous cell carcinoma, Oncol. Rep., 2017, 37, 540–546 Search PubMed.
- T. Higuchi, T. Nakayama, T. Arao, K. Nishio and O. Yoshie, SOX4 is a direct target gene of FRA 2 and induces expression of HDAC8 in adult T cell leukemia/lymphoma, Blood, 2013, 121, 3640–3649 CrossRef CAS.
- M. F. Emmons, R. L. Bennett, A. Riva, K. Gupta, L. A. Da Costa Carvalho, C. Zhang, R. Macaulay, D. Dupéré-Richér, B. Fang, E. Seto, J. M. Koomen, J. Li, Y. A. Chen, P. A. Forsyth, J. D. Licht and K. S. M. Smalley, HDAC8 mediated inhibition of EP300 drives a transcriptional state that increases melanoma brain metastasis, Nat. Commun., 2023, 14, 7759 CrossRef CAS.
- A. Farid, N. Beri, D. Torres Barba and C. Whitcomb, A young man with acute chest pain, Cleve. Clin. J. Med., 2019, 86, 586–594 CrossRef.
- Y. Sixto-López, J. A. Gómez-Vidal, N. de Pedro, M. Bello, M. C. Rosales-Hernández and J. Correa-Basurto, Hydroxamic acid derivatives as HDAC1, HDAC6 and HDAC8 inhibitors with antiproliferative activity in cancer cell lines, Sci. Rep., 2020, 10, 10462 CrossRef.
- A. Chakrabarti, I. Oehme, O. Witt, G. Oliveira, W. Sippl, C. Romier, R. J. Pierce and M. Jung, HDAC8: a multifaceted target for therapeutic interventions, Trends Pharmacol. Sci., 2015, 36, 481–492 CrossRef CAS PubMed.
- H. Zhang, X. Wang, M. Qu, Z. Li, X. Yin, L. Tang, X. Liu and Y. Sun, Foot and mouth disease virus structural protein VP3 interacts with HDAC8 and promotes its autophagic degradation to facilitate viral replication, Autophagy, 2023, 19, 2869–2883 CrossRef.
- B. Xia, J. Lu, R. Wang, Z. Yang, X. Zhou and P. Huang, miR 21 3p regulates influenza A virus replication by targeting histone deacetylase 8, Front. Cell. Infect. Microbiol., 2018, 8, 175 CrossRef.
- J. Chotitumnavee, Y. Yamashita, Y. Takahashi, Y. Takada, T. Iida, M. Oba, Y. Itoh and T. Suzuki, Selective degradation of histone deacetylase 8 mediated by a proteolysis targeting chimera (PROTAC), Chem. Commun., 2022, 58, 4635–4638 RSC.
- S. Darwish, E. Ghazy, T. Heimburg, D. Herp, P. Zeyen, R. Salem-Altintas, J. Ridinger, D. Robaa, K. Schmidtkunz, F. Erdmann, M. Schmidt, C. Romier, M. Jung, I. Oehme and W. Sippl, Design, Synthesis and Biological Characterization of Histone Deacetylase 8 (HDAC8) Proteolysis Targeting Chimeras (PROTACs) with Anti-Neuroblastoma Activity, Int. J. Mol. Sci., 2022, 23, 7535 CrossRef.
- J. Huang, J. Zhang, W. Xu, Q. Wu, R. Zeng, Z. Liu, W. Tao, Q. Chen, Y. Wang and W. G. Zhu, Structure based discovery of selective histone deacetylase 8 degraders with potent anticancer activity, J. Med. Chem., 2023, 66, 1186–1209 CrossRef PubMed.
- Z. Sun, B. Deng, Z. Yang, R. Mai, J. Huang, Z. Ma, T. Chen and J. Chen, Discovery of pomalidomide based PROTACs for selective degradation of histone deacetylase 8, Eur. J. Med. Chem., 2022, 239, 114544 CrossRef.
- Y. Xiao, S. Hale, N. Awasthee, C. Meng, X. Zhang, Y. Liu, H. Ding, Z. Huo, D. Lv, W. Zhang, M. He, G. Zheng and D. Liao, HDAC3 and HDAC8 PROTAC dual degrader reveals roles of histone acetylation in gene regulation, Cell Chem. Biol., 2023, 30, 1421–1435.e12 CrossRef.
- Y. Yamashita, Y. Takada, Y. Itoh and T. Suzuki, Discovery of HDAC8-selective inhibitors and their application to PROTACs, MEDCHEM NEWS, 2023, 33, 181–186 Search PubMed.
- C. Zhao, D. Chen, F. Suo, R. Setroikromo, W. J. Quax and F. J. Dekker, Discovery of highly potent HDAC8 PROTACs with anti tumor activity, Bioorg. Chem., 2023, 136, 106546 CrossRef PubMed.
- C. Zhao, J. Zhang, H. Zhou, R. Setroikromo, G. J. Poelarends and F. J. Dekker, Exploration of hydrazide based HDAC8 PROTACs for the treatment of hematological malignancies and solid tumors, J. Med. Chem., 2024, 67, 14016–14039 CrossRef.
- C. Zhao, J. Zhang, H. Zhou, R. Setroikromo, G. J. Poelarends and F. J. Dekker, Hydrazide-based HDAC8 PROTACs: optimization and evaluation, J. Med. Chem., 2024, 67, 12784–12806 Search PubMed.
- C. Zhao, D. Chen, F. Suo, R. Setroikromo, W. J. Quax and F. J. Dekker, Discovery of highly potent HDAC8 PROTACs with anti-tumor activity, Bioorg. Chem., 2023, 136, 106546 Search PubMed.
- S. A. Amin, N. Adhikari and T. Jha, Structure–activity relationships of HDAC8 inhibitors: non-hydroxamates as anticancer agents, Pharmacol. Res., 2018, 131, 128–142 CrossRef PubMed.
- M. S. Hussain, L. Zhang, A. J. Rana, M. Maqbool, S. Ashique, Y. Khan, V. Jakhmola, A. Hanbashi, W. Mawkili and G. Khan, Epigenetic therapy meets targeted protein degradation: HDAC-PROTACs in cancer treatment, Future Med. Chem., 2025, 17, 1725–1737 CrossRef.
| This journal is © The Royal Society of Chemistry 2026 |