DOI:
10.1039/D5MD00810G
(Review Article)
RSC Med. Chem., 2026,
17, 105-131
Prodrug strategies in developing antiviral nucleoside analogs
Received
11th September 2025
, Accepted 6th December 2025
First published on 2nd January 2026
Abstract
Prodrug strategies are used to enhance the physicochemical and pharmaceutical properties of drug candidates that may not be suitable for specific delivery or are limited by formulation options. A prodrug derivative is converted into its active pharmaceutical ingredient (drug) through enzymatic or chemical reactions within the body. Antiviral nucleoside prodrugs have garnered considerable interest in drug discovery, leading to the approval of key drugs such as remdesivir (SARS-CoV-2), Sovaldi (hepatitis C virus, HCV), and tenofovir disoproxil fumarate [hepatitis B virus (HBV) and human immunodeficiency viruses (HIV)]. Their success lies in improving the oral bioavailability and delivering the parent drug to the targeted tissues. This review focuses on the prodrugs of antiviral nucleosides evaluated in humans (approved, in development or terminated), providing an overview of the different approaches utilized and discussing their in vitro and in vivo benefits.
 Group from left to right: Sijia Tao, Shaoman Zhou, Longhu Zhou, HongWang Zhang, Mahesh Kasthuri, Rama Suresh, Raymond F. Schinazi, Tuniyazi Abuduani, Zhe Chen, Mohammed Loubidi, Zahira Tber, Chengwei Li, and Franck Amblard | Dr. Raymond F. Schinazi is the Frances Winship Walters Professor of Pediatrics at Emory University. He is the founder and director of the Laboratory of Biochemical Pharmacology (LOBP) and serves as a Senior Advisor at the Center for ViroScience and Cure (CVC). He has published over 600 peer-reviewed papers, hold over 100 issued US patents and received five honorary degrees from institutions around the globe. Dr. Schinazi's group focuses on designing and developing new antiviral agents targeting human viral infections, including HIV, herpesviruses, hepatitis B virus (HBV), norovirus, enteroviruses and emerging viruses such as dengue, West Nile, yellow fever or chikungunya viruses. He is best known for his pioneering work on HIV and hepatitis B & C drugs, including d4T (stavudine), 3TC (lamivudine), FTC (emtricitabine/Emtriva), LdT (telbivudine), sofosbuvir (Sovaldi), which are now approved by the FDA, as well as for his work on inflammation leading to the approval of baricitinib for COVID-19. |
1. Introduction
Medical professionals commonly prescribe antiviral nucleoside analogs (NA) to treat herpes simplex virus (HSV), human immunodeficiency virus type 1 (HIV-1), hepatitis B and C (HBV, HCV), cytomegalovirus (CMV), and SARS-CoV-2 infections.1–4 For these NAs to become active, they must be phosphorylated within the cell by various cellular kinases, converting them into their respective NA-triphosphate forms (NTP). The NTPs then compete with the natural nucleotide triphosphates for incorporation by the viral polymerases, ultimately inhibiting the virus's genetic replication.
However, some nucleoside analogs can exhibit poor oral absorption, and low bioavailability. Therefore, prodrugs have been developed to enhance oral drug absorption, improve cellular uptake, and ensure the selective phosphorylation of nucleoside analogs in tissues vulnerable to viral infections. The term prodrug was coined in 1958 by Adrien Albert; however, the concept has historical roots from earlier medical applications, such as methenamine (prodrug of formaldehyde, discovered in 1859), phenacetin (prodrug of acetaminophen, discovered in 1887), and prontosil (prodrug of sulfanilamide, discovered in 1932). A prodrug is a biologically inert or weakly active molecule that, upon biotransformation in vivo, releases the pharmacologically active parent compound.5
When considering the development of nucleoside prodrugs, several approaches have been successfully used over the years including; 1) derivatization of the free hydroxy groups at the nucleoside 2′, 3′ and 5′ positions of the sugar moiety, with simple aliphatic acetates, amino acid esters; In general, simple ester prodrugs (e.g., isobutarate) increase passive permeation or active transport (e.g., amino acid prodrugs via PEPT-1) to alter pharmacokinetic parameters. Amino acid esters increase bioavailability and solubility in addition to modular properties such as quick release (e.g., glycine) or extended release patterns (e.g., isoleucine)6,7 2) functionalization of the nucleoside heterobase using enzymatically labile groups (Fig. 1). These lipophilic prodrugs intend to reduce pre-systemic metabolism and enhance tissue- and cell-selective delivery of the NA. This selective delivery enables subsequent phosphorylation by intracellular kinases, generating the active NTP form.
 |
| | Fig. 1 Generic structures of representative prodrug types discussed in this review. | |
Since the initial monophosphorylating step of a nucleoside analogs can be rate-limiting, nucleoside monophosphate (MP) prodrugs, such as phosphoramidates (ProTides) or phosphonates [(Bis (POM), and Bis (POC) derivatives] containing “masked” and intracellularly cleavable monophosphate moieties, have also been developed2 (Fig. 1).
This review focuses on these different strategies, discussing the benefits and their success in improving pharmacokinetics, targeting properties, and antiviral in vivo efficacy of NA, emphasizing the most recent preclinical or clinical developments (Table 1).
Table 1 Advanced and approved antiviral nucleoside prodrugs (1980–2025)
| Drug name, and structure |
Pharmacokinetics (PK) properties |
Mechanism of action |
Approved date and medical uses |
Clinical status |
Ref. |
 Valacyclovir (Valtrex)
Valacyclovir (Valtrex) |
Improved aqueous solubility, absorption, and oral bioavailability |
HSV DNA polymerase inhibitor |
Approved in June 1995 for herpes simplex virus (HSV) infections |
Zoster eye disease study (phase 4) |
121 page 3 |
 Valganciclovir
Valganciclovir
|
Improved aqueous solubility, absorption, and oral bioavailability |
Viral DNA polymerase inhibitor |
Approved in March 2001 for the treatment of CMV retinitis in patients with acquired immunodeficiency syndrome (AIDS) |
Glioblastoma (phase 2) |
122, 123 page 4 |
| Kidney transplant complication, and cytomegalovirus (CMV) (phase 4) |
| Hepatocellular carcinoma (HCC) (phase 2) |
| Major depressive disorder (phase 2) |
 Famciclovir (Famvir)
Famciclovir (Famvir)
|
Improved oral bioavailability |
HSV DNA polymerase inhibitor |
Approved in June 1994 for herpes simplex virus (HSV) and varicella zoster virus (VZV) infections |
Multiple sclerosis (phase 1) |
124 page 4 |
 Valopicitabine (NM-283)
Valopicitabine (NM-283)
|
Improved aqueous solubility, absorption, and oral bioavailability |
HCV RNA-dependent RNA polymerase (NS5B) inhibitor |
Hepatitis C virus (HCV) |
No longer in active development |
125 page 5 |
| Discontinued due to the safety concerns |
| Hepatitis C virus (HCV) (phase 2) |
 Valnivudine hydrochloride (FV-100)
Valnivudine hydrochloride (FV-100) |
Improvedaqueous solubility, absorption, and oral bioavailability |
Viral DNA polymerase inhibitor |
Herpes zoster |
Shingles |
17 page 6 |
| Herpes zoster |
| Postherpetic neuralgia (phase 3) |
| Discontinued because of availability of other effective therapies and market dynamics |
 Molnupiravir (Lagevrio)
Molnupiravir (Lagevrio)
|
Improved oral bioavailability |
Viral RNA-dependent RNA polymerase (RdRp) inhibitor |
FDA granted an emergency use authorization (EUA) in 2021 for the treatment of mild-to-moderate COVID-19 |
Coronavirus disease (COVID-19) (phase 3), influenza, and acute respiratory infections (ARIs) |
23, 25 page 6 |
 Lumicitabine (ALS-8176)
Lumicitabine (ALS-8176)
|
Improved oral bioavailability |
Viral RNA-dependent RNA polymerase (RdRp) inhibitor |
Respiratory syncytial virus (RSV) and human metapneumovirus (hMPV) infections |
Metapneumovirus (phase 2); discontinued due to lack of sufficient clinical efficacy and safety concerns associated with neutropenia |
29 page 7 |
 Mericitabine
Mericitabine
|
Improved oral bioavailability |
HCV RNA-dependent RNA polymerase (NS5B) inhibitor |
Hepatitis C virus (HCV) infection |
Chronic hepatitis C (phase 2) |
126, 127 page 8 |
 Balapiravir (R1626)
Balapiravir (R1626)
|
Improved oral bioavailability |
NS5B RNA polymerase inhibitor |
Hepatitis C virus (HCV); discontinued due to the safety concerns (mitochondrial toxicity) |
Dengue (phase 1) |
39 page 9 |
 Obeldesivir (GS-5245, ATV006)
Obeldesivir (GS-5245, ATV006)
|
Improved permeability, and oral bioavailability |
Viral RNA-dependent RNA polymerase (RdRp) inhibitor |
Hepatitis C virus (HCV) and COVID-19 |
COVID-19 (phase 3) (terminated due to low rate of exposure) |
128 page 10 |
 Deuremidevir (VV116)
Deuremidevir (VV116)
|
Improvedpermeability, and oral bioavailability |
Viral RNA-dependent RNA polymerase (RdRp) inhibitor |
Approved for COVID-19 treatment in China and Uzbekistan |
Chinese infants hospitalized with RSV (phase 2) |
129 page 11 |
 Brincidofovir (CMX001, Tembexa)
Brincidofovir (CMX001, Tembexa)
|
Improved cellular uptake and lower plasma concentrations of cidofovir |
Viral DNA polymerase inhibitor |
Approved in June 2021 for the treatment of smallpox |
Adenoviruses (AdV) and cytomegalovirus (CMV) (phase 2) |
130 page 12 |
 Adefovir dipivoxil (Hepsera)
Adefovir dipivoxil (Hepsera)
|
Improved permeability (absorption) and cellular uptake |
Viral reverse transcriptase (HIV) or viral DNA polymerase (HBV) inhibitor |
Approved in September 2002 for the treatment of chronic hepatitis B |
Liver cirrhosis, and portal hypertension (unknown status) |
131 page 13 |
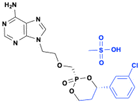 Pradefovir mesylate (MB-06886)
Pradefovir mesylate (MB-06886)
|
Improvedoral bioavailability and target selectivity (hepatocytes) |
HBV reverse transcriptase inhibitor |
Chronic hepatitis B virus (HBV) infection |
Chronic hepatitis B (phase 2 and 3) (terminated due to nonclinical carcinogenicity) |
132, 133 page 13 |
 Tenofovir disoproxil fumarate (Viread, TDF)
Tenofovir disoproxil fumarate (Viread, TDF)
|
Improved intestinal absorption and oral bioavailability |
Viral reverse transcriptase (HIV) or viral DNA polymerase (HBV) inhibitor |
Approved in June 2002 for the treatment of HIV-1 infection |
Observational study: impact of fatty liver on hepatitis B therapy |
134 page 14 |
| Approved in August 2008 for the treatment of chronic hepatitis B |
Parkinson's disease (phase I) |
 Besifovir dipivoxil maleate (LB80380)
Besifovir dipivoxil maleate (LB80380)
|
Improved intestinal absorption and oral bioavailability |
Inhibitor of the HBV DNA polymerase |
Approved in September 2017 in South Korea for the treatment of HBV, and in China (January 2019) |
Renal impairment (phase 1) |
135 page 15 |
| Hepatitis B (phase 4) |
 Tenofovir alafenamide (Vemlidy, TAF)
Tenofovir alafenamide (Vemlidy, TAF)
|
Improved potency and target selectivity (lymphoid tissues and hepatocytes/liver cells) |
Viral reverse transcriptase in HIV and the HBV polymerase in hepatitis B inhibitor |
Approved in November 2015 for the treatment of HIV-1 and for chronic hepatitis B virus infection in April 2016 |
Observational study: impact of fatty liver on hepatitis B therapy, NCT06535048 |
136–138 page 18 |
| Accelerated ART initiation for protocol for people with HIV who are out of care (phase 4) |
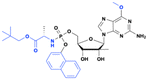 BMS-986094 (INX-08189)
BMS-986094 (INX-08189)
|
Improved cellular uptake and 500 times more potent than the parent nucleoside |
NS5B RNA-dependent RNA polymerase inhibitor |
HCV infection |
No longer in active development |
76 page 19 |
| Discontinued due to the safety concerns associated with heart and kidney toxicity |
| Phase 2 (terminated) |
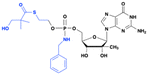 IDX-184
IDX-184
|
Improved potency and target selectivity (liver cells) |
HCV NS5B RNA- polymerase inhibitor |
Discontinued due to the safety concerns (serious cardiac adverse events) |
HCV (phase 2)No longer in active development |
79 page 19 |
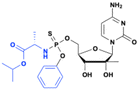 ALS-2200 (epimeric mixture)
ALS-2200 (epimeric mixture) |
Improved potency and target selectivity (liver cells) |
NS5B RNA-dependent RNA polymerase inhibitor |
Discontinued because of availability of other effective therapies for HCV |
HCV (phase 2) |
83 page 20 |
| No longer in active development |
|
VX-135 (single diastereomer of ALS-2200) |
Improved potency and target selectivity (liver cells) |
NS5B RNA-dependent RNA polymerase inhibitor |
Discontinued because of availability of other effective therapies for HCV and market dynamics |
HCV (phase 2) |
127 page 21 |
| No longer in active development |
 Sofosbuvir (Sovaldi)
Sofosbuvir (Sovaldi)
|
Improved potency and target selectivity (liver cells) |
NS5B RNA-dependent RNA polymerase inhibitor |
First approved in September 2013 in South Korea for the treatment of HCV, HCV, and cirrhosis |
443 clinical trials for HCV, liver cancer, Covid-19, and male infertility |
87, 88 page 21 |
| Binds to two Mg2+ ions present in HCV NS5B polymerase's GDD active site motif and preventing further replication of HCV |
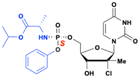 Uprifosbuvir (MK-3682)
Uprifosbuvir (MK-3682)
|
Improved potency and target selectivity (hepatocytes) |
HCV NS5B inhibitor |
Hepatitis C virus (HCV) infjection |
Phase 2 (terminated due to lack of clinical efficacy as a standalone drug) |
139 page 22 |
 ATI-2173
ATI-2173
|
Improved oral bioavailability with better safety |
HBV polymerase inhibitor |
HBV infection |
HBV, phase 2 (terminated due to lack of clear regulatory pathway) |
140 page 23 |
 AL-335 (JNJ-4178, and adafosbuvir)
AL-335 (JNJ-4178, and adafosbuvir)
|
Improved oral bioavailability, and higher levels of TP compared to the parent nucleoside |
NS5B RNA polymerase inhibitor |
HCV infection |
HCV (phase 1) (J&J withdrawn because of availability of other effective therapies for HCV) |
93 page 23 |
| Phase 2, NCT02993250 (completed) |
| Renal impairment (phase 1) |
 Remdesivir (Veklury)
Remdesivir (Veklury)
|
Improved potency and target selectivity (Calu-3 cells) |
Viral RNA-dependent RNA polymerase (RdRp) inhibitor |
Approved in October 2020 for hospitalized COVID-19 patients |
Respiratory syncytial virus (RSV) (phase 2) |
141 page 24 |
| COVID-19, SARS-CoV-2 infection, and influenza (observational study) 167 clinical trials |
 Bemnifosbuvir (BEM; AT-527)
Bemnifosbuvir (BEM; AT-527)
|
Improved solubility, and plasma concentration of the active guanosine TP metabolite (AT-9010) |
Viral RNA-dependent RNA polymerase (RdRp) inhibitor |
HCV infection and COVID-19 |
HCV (phase 2) |
105, 142 page 25 |
| COVID-19, and SARS-CoV-2 (phase 3) |
 AT-752 (Rp-diastereomer)
AT-752 (Rp-diastereomer) |
Improved solubility, and plasma concentration of the active guanosine TP metabolite (AT-9010) |
Viral RNA-dependent RNA polymerase (RdRp) inhibitor |
A range of RNA viruses, including COVID-19 and dengue |
Dengue (phase 2; terminated) |
104 page 25 |
| Development of AT-752 for COVID-19 has been discontinued due to business strategic considerations |
 2′-α-Fluoro-2′-β-C-(fluoromethyl)guanosine
2′-α-Fluoro-2′-β-C-(fluoromethyl)guanosine
|
Improved potency and target selectivity (lung epithelial cells) |
RNA-dependent viral RNA polymerase (RdRp) |
Preclinical drug for RSV, human metapneumovirus (hMPV), and human rhinovirus (HRV) |
Preclinical candidate for RSV African green monkey (AGM) challenge model |
97 page 26 |
 PSI-352938 (PSI-938, GS-0938)
PSI-352938 (PSI-938, GS-0938)
|
Improved potency and target selectivity (hepatocytes) |
HCV NS5B RNA-polymerase inhibitor |
Discontinued due to the safety concerns (elevations in liver enzymes (ALT/AST) causing to hepatotoxicity) |
HCV (phase 2) |
143, 144 page 16 |
| No longer in active development |
 Taribavirin/hydrochloride (Viramidine)
Taribavirin/hydrochloride (Viramidine)
|
Improved solubility and target selectivity (liver tissues) – less exposure to red blood cells |
DNA and RNA viral polymerases inhibitor |
HCV |
Phase 2, NCT00446134 (completed) for HCV |
113, 145 page 29 |
| HCV (phase 3) |
| RSV (phase 2) |
| Crimean-Congo hemorrhagic fever (phase 2), and Lassa fever (phase 2) |
 Abacavir
Abacavir
|
Improved oral bioavailability, and CNS permeability |
HIV-1 reverse transcriptase inhibitor |
Approved in December 1998 for the treatment of HIV-1 |
Amyotrophic lateral sclerosis (phase 3, terminated not due to the safety issues) |
58 page 30 |
| High grade glioma (phase 1) |
 Amdoxovir/DAPD
Amdoxovir/DAPD
|
Improve solubility, selectivity (liver and intestine) and oral bioavailability |
Inhibits viral reverse transcriptase (for HIV) or DNA polymerase (for HBV) |
HIV-1/HBV |
HIV-1 (phase 2; terminated due to poor recruitment) |
146 page 28 |
2. Ester prodrugs
Valacyclovir
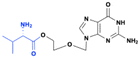
Acyclovir, a purine acyclic nucleoside, is used to treat herpes simplex, chickenpox, and shingles virus infections.8 It is activated by the viral thymidine kinase, which converts it to acyclovir monophosphate. This monophosphate is further converted to active acyclovir triphosphate, which inhibits viral DNA synthesis by competitive inhibition of the viral DNA polymerase and acts as a viral DNA chain terminator. However, the drug has poor oral bioavailability (10–20%) and a short half-life of approximately 2.5–3.0 h.8 Therefore, a prodrug approach was adopted for improving the PK parameters of acyclovir. Valacyclovir is an L-valyl ester prodrug that is converted to acyclovir during first-pass metabolism, thus enhancing the bioavailability of the active drug by three to four times, while retaining its safety profile9 (Fig. 2).
 |
| | Fig. 2 Structures of acyclovir and valaciclovir. | |
Valganciclovir
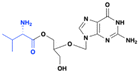
Valganciclovir is the L-valine amino acid prodrug of ganciclovir. Valganciclovir was FDA-approved in 1988 for the treatment of cytomegalovirus in HIV infected persons as well as persons having undergone organ transplants. Valganciclovir is not a curative treatment. The 5′-amino acid adduct of ganciclovir enhances oral bioavailability by improving absorption in the intestines and liver, where it is subsequently cleaved by esterases to release the active drug (Fig. 3). While ganciclovir has an oral bioavailability of only 8.5%, valganciclovir significantly increases this to 82.5%.10,11
 |
| | Fig. 3 Structures of valganciclovir and ganciclovir. | |
Famciclovir
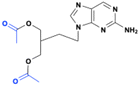
Famciclovir is a diacetyl ester prodrug of penciclovir, used to treat herpes zoster (shingles) infections. Esterases in the intestine quickly metabolize the ester moieties, and the purine base is subsequently oxidized in the liver by an aldehyde oxidase to deliver the active drug, penciclovir which gets phosphorylated by the viral thymidine kinase (TK). Phosphorylation of one of the hydroxyl groups of penciclovir generates a chiral center, leading to either R- or S-enantiomer. It was found that the S-enantiomer appeared to be amplified for the active triphosphate form, which acts as a DNA polymerase inhibitor in the infected herpes simplex virus cells12 (Fig. 4). Clinical studies demonstrated that the prodrug significantly improves the absorption and the oral bioavailability of penciclovir, achieving up to 77% following a single dose of famciclovir.13
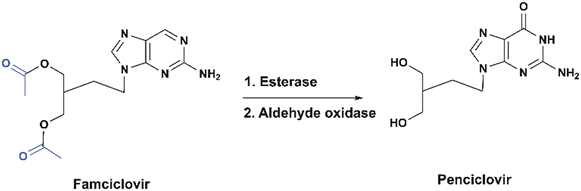 |
| | Fig. 4 Structures of famciclovir and penciclovir. | |
Valopicitabine (NM283)
Although 2′-C-methylcytidine exhibited potent in vitro activity, its clinical development for the treatment of chronic hepatitis C virus (HCV) infection was impeded by poor oral bioavailability and formulation challenges.14 To address these limitations, valopicitabine, a 3′-O-L-valinyl ester prodrug of 2′-C-methylcytidine was designed, resulting in improved oral bioavailability by 34% in rats (Fig. 5).13 Valopicitabine is cleaved by esterases in the gut and/or liver to deliver 2′-C-methylcytidine (Fig. 5). A phase 2 study showed that valopicitabine in conjunction with pegylated interferon-alpha (PEG-IFN-α) led to significant reductions in HCV RNA levels compared to PEG-IFN-α alone, with viral load decreases surpassing −3 log10 IU per mL in some cases by week 12.15 However, valopicitabine's development faced setbacks due to dose-dependent gastrointestinal side effects and hematological toxicity, such as neutropenia and anemia, leading to the discontinuation of its clinical development.
 |
| | Fig. 5 Structures of valopicitabine (NM-283) and 2′-C-methylcytidine (NM-107). | |
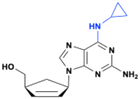
FV-100
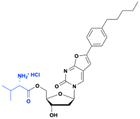
Bicyclic nucleoside analog (BCNA), CF-1743, is a highly specific inhibitor of varicella-zoster virus (VZV) replication (EC50 ∼ 440 pM).16 Despite its potency, CF-1743 has poor oral bioavailability (<14%) due to its high lipophilicity and very low water solubility (0.9 μg mL−1). To overcome these limitations, a 5′-valyl ester prodrug (FV-100) was developed as a hydrochloride salt (Fig. 6). This modification significantly increases water solubility, bioavailability, and systemic absorption, enabling effective delivery of CF-1743 to the infected tissues. After oral administration, FV-100 is rapidly hydrolyzed into CF-1743, which is then selectively phosphorylated in infected cells by VZV thymidine kinase (TK) to generate its active triphosphate metabolite (CF-1743-TP).17 The prodrug exhibits 500 times greater solubility and a 10-fold increase in bioavailability compared to its parent drug.18 Additionally, FV-100 shows approximately a 10-fold improvement in both AUC and Cmax compared to CF-1743.19 FV-100 has successfully completed phase I and II clinical trials for the treatment of herpes zoster (shingles).
 |
| | Fig. 6 Structures of FV-100 and CF1743. | |
Molnupiravir
Nucleoside derivative β-D-N4-hydroxycytidine (NHC, PSI-0194, EIDD-1931) shows antiviral activity against several viruses including HCV, zika virus, chikungunya virus, yellow fever virus, influenza, Jamestown Canyon virus, Cache Valley virus and SARS-CoV-2.20–22
Because of its ability to tautomerism, NHC-TP can substitute for either cytidine-TP or uridine-TP in the elongating RNA strand and subsequently pairs with either guanosine or adenosine in the RNA template.23 This unique property results in an accumulation of nucleotide errors in viral RNA/genome and probably explain the broad-spectrum antiviral activity of NHC/molnupiravir.24 Although NHC exhibited good oral availability in mice (56%),25 its plasma concentration was low in cynomolgus monkeys, causing poor bioavailability (8.4%). To improve the oral bioavailability of NHC, a 5′-isobutyl ester prodrug was developed (molnupiravir – EIDD-2801), which is quickly absorbed and hydrolyzed by esterases to NHC during absorption and first-pass metabolism, resulting in an increased bioavailability of NHC (45% in cynomolgus monkeys)26 (Fig. 7).
 |
| | Fig. 7 Structures of molnupiravir and EIDD-1931. | |
Molnupiravir showed excellent pharmacokinetic properties in cynomolgus monkeys and ferrets, and the single ascending dose oral pharmacokinetic (PK) profile in ferrets provided information on dose selection for clinical trials.27 Molnupiravir received authorization from regulators in the United States, the United Kingdom, and Australia in late 2021 and early 2022. The US Food and Drug Administration (FDA) granted emergency use authorization on December 23, 2021, for the treatment of mild-to-moderate COVID-19 in adults with positive SARS-CoV-2 viral testing who are at high risk for progression to severe COVID-19. Although the drug exhibits activity against pathogenic RNA viruses, NHC was shown to display host mutational activity in an animal cell culture assay indicating that NHC/molnupiravir could hold long terms risks for the patients.22,28
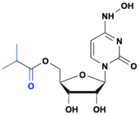
Lumicitabine
4-Chloromethyl-2-deoxy-2-fluorocytidine (ALS-8112) is a potent inhibitor of RSV replication in vitro (EC50 = 0.15 μM., IC50 = 0.02 μM).29 However, the cytidine analog ALS-8112 does not achieve good oral bioavailability due to its high polarity. Therefore, lumicitabine (ALS-8176), a 3′,5′-O-diester prodrug approach was developed,29 and eventually considered as a drug candidate to treat respiratory syncytial virus (RSV) and human metapneumovirus (hMPV) (Fig. 8).30,31 The diester prodrug increased the oral bioavailability of ALS-8112 from 3% to 44% in monkeys, allowing for the delivery of a high level of ALS-8112-TP in vivo.29 ALS-8176 demonstrated excellent anti-RSV efficacy and safety in a phase 2 clinical RSV challenge study,32 but was discontinued due to safety concerns associated with neutropenia.33
 |
| | Fig. 8 Structures of lumicitabine and ALS-8112. | |
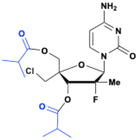
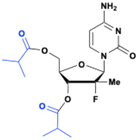
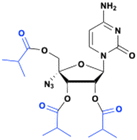
Mericitabine (RG7128)
Mericitabine (RG7128) is a 3′,5′-diisobutyrate ester prodrug of the 2′-F-2′-C-methyl cytidine nucleoside analog PSI-6130, specifically designed to combat HCV infections. PSI-6130 demonstrated antiviral activity by selectively inhibiting HCV replication in sub genomic replicon assays with an EC90 of 4.6 μM.34 Preclinical studies highlighted its strong safety profile, showing no cytotoxicity at the highest concentrations tested and a no-effect dose of 100 mg kg−1 in a 6 day mouse toxicity study.35 Despite having a satisfactory pharmacological profile, PSI-6130 faced significant challenges related to its metabolism and low bioavailability (25% in rhesus monkeys). In vitro studies revealed that PSI-6130 is a substrate for cytidine deaminase (CDA), which deaminates it into the inactive uridine analog PSI-6206 (ref. 36) (Fig. 9).
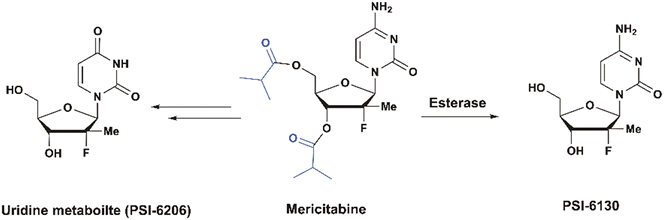 |
| | Fig. 9 Structures of PSI-6130, PSI-6206, and mericitabine. | |
To overcome these limitations, mericitabine (RG7128) was developed, which improved its pharmacokinetic properties and enhanced bioavailability. In clinical trials, mericitabine demonstrated remarkable efficacy. During a 14 day monotherapy study in patients with HCV genotype 1, a dosage of 1500 mg twice daily resulted in an average reduction of 2.7 log10 IU per mL in HCV RNA levels.37 Importantly, no adverse effects or viral rebound were observed, underscoring the safety and effectiveness of this approach. However, mericitabine (RG7128) was discontinued in 2013 during phase II clinical trials. Although it demonstrated promising antiviral activity and safety in early studies, its development was halted due to its limited efficacy compared to other direct-acting antivirals such as sofosbuvir.
Balapiravir (R-1626)
Balapiravir (R-1626) is the tri-isobutyrate prodrug of nucleoside R-1479, a highly potent and selective inhibitor of NS5B HCV RNA polymerase.38 This lipophilic prodrug (R-1626) was developed to increase permeability/bioavailability and improve antiviral activity of R-1479 (Fig. 10).39 R-1626 is efficiently converted to R-1479 after oral administration by esterases in the cellular membranes before the first-pass metabolism.40 The mean elimination half-life ranged from 20 to 25 h, while the effective half-life of R-1479 ranged from 4.62 to 5.87 h. Phase 1b study showed that doses up to 3000 mg twice a day were well tolerated after 14 days of treatment. The clinical development of balapiravir for HCV infection was halted due to safety concerns identified in patients receiving extended courses (2–3 months). It is worth noting that balapiravir was also evaluated for the treatment of dengue virus but did not produce significant changes in viral load, viremia kinetics, cytokine expression levels, or hematological and biochemical parameters in dengue-infected patients.40–42
 |
| | Fig. 10 Structures of balapiravir and R-1479. | |
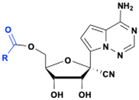
Obeldesivir (GS-5245, ATV006)
The 1′-CN adenosine analog GS-441512 was designed to avoid adenine deamination by adenosine deaminase (ADA) and improve nucleoside stability by circumventing the breakdown of the C–N bond by purine nucleoside phosphorylase (PNP) enzymes. Although remdesivir, a phosphoramidate prodrug of GS-441524 (see below), has been approved for the treatment of COVID-19 patients, its partial efficacy and intravenous (IV) administration restricts its use in clinical settings. In addition, a meta-analysis of nonhuman primates model study, showed that remdesivir is quickly metabolized and that the parent compound GS-441524 may contribute to the major clinical antiviral efficacy. Because the GS-441524 bioavailability is about 5% in Sprague–Dawley (SD) rats when administrated intravenous or intragastric (IG), the development of GS-441524 as an oral drug was hampered.43 Subsequently, the monoester prodrug of GS-441524 Obeldesivir (GS-5245) was evaluated (Fig. 11). When administered orally at a dose of 25 mg kg−1 to Sprague Dawley rats, the bioavailability of obeldesivir (ATV006) was 81.5%, an improvement over GS-441524 at 21.7%. Furthermore, the oral bioavailability of GS-441524 after oral administration of obeldesivir in mice, rats, ferrets, dogs, and cynomolgus monkeys was also found to be 41%, 63.9%, 154%, 94%, and 38%, respectively.43
 |
| | Fig. 11 Structures of obeldesivir and Shen26. | |
The ester prodrug demonstrates promising PK values that balance solubility, stability, and permeability, thus enhancing the plasma concentration and bioavailability of GS-441524. It also reduced viral loads and alleviated lung damage when administered to K18-hACE2 mice challenged with the Delta variant of SARS-CoV-2.44 In a phase 3 clinical trial, obeldesivir reduced SARS-CoV-2 infectious titers. Viral load levels were reduced by 30% and 24% at days 3 and 5, respectively, compared to placebo.45 Very recently, clinical studies were terminated for both RSV (NCT06784973) and COVID-19 (NCT05603143) due to low infection rates and challenging patient recruitment.
Shen26 (ATV014)
Shen26 (ATV014) is another 5′-ester prodrug of GS-441524 developed by the Southern University of Science and Technology (Shenzhen, China) demonstrating anti-SARS-CoV-2 activity. After a single oral administration of Shen26, 25 mg kg−1 to SD rats, the oral bioavailability of GS-441524 was found to be 53.4% whereas GS-441524 had 21.7%.46 In comparison, the oral bioavailability of GS-441524 after administration of remdesivir (RDV) was only 14% in rats.
GS-621763
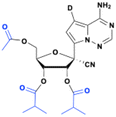
GS-621763, the triisobutyl ester prodrug of GS-441524, was also considered as an alternative to obeldesivir, based on its high oral exposure (57% and 28% oral bioavailability in rats and non-human primates, respectively (Fig. 12).47 However, its poor aqueous solubility and issues to obtain crystalized forms effectively complicated its formulation for drug development.
 |
| | Fig. 12 Structures of GS-621763 and VV116. | |
Mindeudesivir (VV116)
Junshi Biosciences developed Mindeudesivir (VV116), a deuterated version of GS-621763. As expected, the deuterated parent nucleoside performed the same as its non-deuterated version GS-441524, showing in vitro anti-SARS-CoV-2 activity but low oral bioavailability (F = 21.7% in rats) due to its poor aqueous solubility and permeability. However, the corresponding tri-isobutyrate ester VV116 shows improved oral bioavailability (F = ∼50%) in rats, with a half-life ranging from 4.80 h to 6.95 h.48,49 After significant effort to find the suitable crystallization salt, the hydrobromide salt (VV116) was selected due to its good oral bioavailability (F = ∼80% in rats).48 Oral administration of VV116 demonstrated dose-dependent anti-SARS-CoV-2 efficacy in mice.48 Mindeudesivir received conditional approval in January 2023 for the treatment of adult patients with mild to moderate COVID-19.50
3. Phosphonate prodrugs
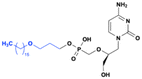
Brincidofovir (CMX001)
Cidofovir is an acyclophosphonate nucleoside analog that was approved by the FDA in 1996 for the treatment of cytomegalovirus (CMV) retinitis in HIV/AIDS patients.51,52 Brincidofovir (CMX001) is a lipid conjugate prodrug of cidofovir, developed by Chimerix to address the limitations of cidofovir, particularly its nephrotoxicity and poor oral bioavailability.51 Brincidofovir contains a hexadecyloxypropyl moiety that masks the phosphate group of cidofovir (Fig. 13).53 This 3-hexadecyloxy-1-propanol (HDP) lipid modification is adopted from the endogenous lysophosphatidylcholine, but with an ether linkage at the sn-1 position, instead of an acyl group. This modification prevents the hydrolysis of acyl group by lysophospholipase during absorption. The (HDP) lipid moiety significantly enhances lipophilicity, plasma stability, and allowing the prodrug to cross cellular membranes more efficiently, thereby improving oral bioavailability (approximately 25–35%).54 Once inside the cells, phospholipase C cleaves the lipid moiety, releasing cidofovir, which is then phosphorylated into its active form, cidofovir diphosphate.55 Brincidofvir was approved by FDA for the treatment of smallpox in 2021, and it is also being investigated for the prevention and treatment of cytomegalovirus (CMV), BK Virus (BKV), adenoviruses (AdV) (NCT04706923), monkeypox, and Epstein–Barr virus (EBV).56
 |
| | Fig. 13 Structures of brincidofovir (CMX001) and cidofovir. | |
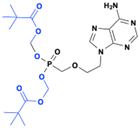
Adefovir dipivoxil
Adefovir dipivoxil is a prodrug of adefovir and was approved by the FDA in 2002 for the treatment of HBV (Fig. 14). The di-tert-butyl ester prodrug exhibits better lipophilicity and cellular permeability compared to adefovir. It is rapidly absorbed through the intestine and hydrolyzed by esterases to adefovir, which is then metabolized by the cellular adenylate kinase to the active adefovir triphosphate. The triphosphate competes with 2′-deoxyadenosine triphosphate for incorporation into the growing viral DNA strand, thereby blocking the HBV DNA polymerase (reverse transcriptase). Notably, the prodrug is effective against lamivudine-resistant HBV and increases the bioavailability of adefovir by 3 to 4-fold.57,58
 |
| | Fig. 14 Structures of adefovir dipivoxil and adefovir. | |
Pradefovir mesylate (MB-06886)
Pradefovir mesylate is an orally administered liver-targeted prodrug of adefovir used to treat chronic hepatitis B virus (HBV) infections. It was specifically designed to improve the pharmacokinetic and safety profile of its parent compound, adefovir (Fig. 15). Adefovir is associated with significant systemic toxicity, particularly nephrotoxicity, due to its non-selective distribution. The kidney-to-liver PMEA ratio of adefovir dipivoxil is approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while pradefovir achieves a substantially improved kidney-to-liver ratio of 1:20, which reduces the risk of kidney damage while maintaining therapeutic efficacy. Pradefovir is mainly metabolized to the active antiviral agent adefovir diphosphate by the CYP3A4 enzyme in the liver,59,60 where HBV replication primarily occurs.
:1, while pradefovir achieves a substantially improved kidney-to-liver ratio of 1:20, which reduces the risk of kidney damage while maintaining therapeutic efficacy. Pradefovir is mainly metabolized to the active antiviral agent adefovir diphosphate by the CYP3A4 enzyme in the liver,59,60 where HBV replication primarily occurs.
 |
| | Fig. 15 Structures of pradefovir mesylate and adefovir. | |
Pradefovir improves oral bioavailability compared to adefovir dipivoxil, allowing for effective viral suppression at lower doses. Clinical studies have demonstrated that patients treated with pradefovir exhibit greater reductions in viral load and improved safety outcomes, making the drug a promising option for the long-term treatment of chronic HBV infections.59,60 Pradefovir is currently under investigation in phase 3 trials (NCT04543565) for treating chronic HBV infections.
Tenofovir-disoproxil fumarate (TDF)
Tenofovir (TFV) is an acyclic nucleoside that is poorly absorbed orally [mice (<2%), and monkeys (5.3%)] due to the hydrophilic nature of the negatively charged phosphonate group. Therefore, tenofovir disoproxil fumarate (TDF) was developed (Fig. 16) and is widely used for treating HIV and HBV. TDF has two alkyl methyl carbonate ester moieties that can be cleaved by esterases to release the TFV in vivo. These carbonate ester groups mask the charged phosphonate moiety in TFV, significantly improving intestinal absorption and oral bioavailability (25% in fasting patients, and 39% after a high-fat meal).61,62
 |
| | Fig. 16 Structures of tenofovir-disoproxil fumarate (TDF) and tenofovir (TFV). | |
Besifovir dipivoxil maleate
Besifovir dipivoxil maleate (BSV, LB80380 maleate) is a prodrug designed to increase the oral absorption and bioavailability of besifovir (LB80331) (Fig. 17). BSV displays strong antiviral activity against HBV with a better safety profile (low renal and bone toxicities) compared to TDF, and a high barrier to resistance during long-term treatment.63 BSV is actually a double prodrug which is activated through esterase cleavage in the intestine and liver, followed by oxidation, to yield the corresponding guanosine intermediate LB-80317, which is further phosphorylated to the active diphosphate form.64 It was approved for the treatment of CHB patients in Korea in May 2017 after a successful phase 3 trial (NCT01937806).65
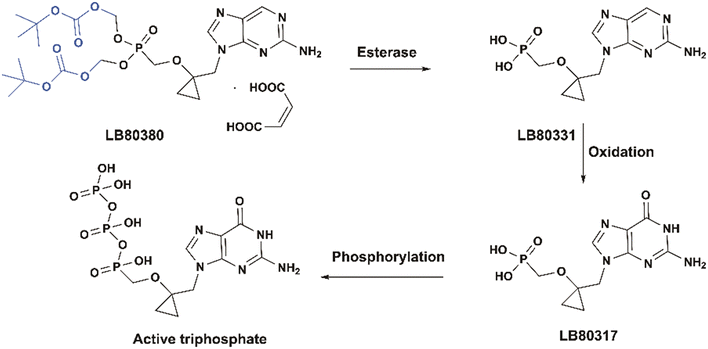 |
| | Fig. 17 Structures of LB80380 and LB80317. | |
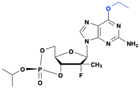
PSI-352938
Based on the structure of PSI-6130, Pharmasset developed several 2′-Me, 2′-F purine nucleoside derivatives as a second-generation anti-HCV agent. Among these, the guanosine derivative PSI-352938 demonstrated broad genotype coverage and was 60 times more active than the parent nucleoside. The prodrug generated high levels of active triphosphate in both primary human hepatocytes and rat liver tissues, and demonstrated prolonged stability in gastrointestinal, intestinal fluids, human plasma, and human liver S9 (an in vitro model for liver stability). Further cytotoxicity testing revealed no cytotoxicity across a range of cell lines and no mitochondrial toxicity. The prodrug is activated in three steps (Fig. 18). First, P450 3A4 (CYP3A4) removes the isopropyl group from the 3′,5′-cyclic phosphate. Then, liver phosphodiesterases (PDEs) open the cyclic phosphate ring, followed by adenosine deaminase-like protein 1 cleaving the O6-ethyl group on the nucleobase. This CYP3A4-dependent metabolism highlights the prodrug's effectiveness in targeting the liver.66
 |
| | Fig. 18 Structures of PSI-352938 and 2′-methylguanosine-5′-monophosphate. | |
Ultimately, PSI-352938 was chosen as the clinical candidate for further investigation due to its favorable PK profile.67 In different clinical studies, PSI-352938 exhibited excellent antiviral activity, showing an average reduction of 4 log10 IU per mL in HCV RNA from baseline and consistent decline in HCV RNA levels without any instances of viral breakthrough.68 A phase I trial confirmed its safety, with no reported adverse events.69 However, routine safety monitoring revealed laboratory abnormalities associated with liver function in 235 participants receiving PSI-352938, either as monotherapy or in combination with sofosbuvir or sofosbuvir and ribavirin (RBV)70 leading to the end of the trial.
4. ProTides/phosphoramidate prodrugs
To exhibit antiviral activity, a nucleoside analog must be metabolized to its active 5′-TP form. This mechanism involves a series of enzymatical phosphorylation steps, with the first phosphorylation often being rate-limiting step. Due to their negative charges and relatively low stability, nucleosides monophosphate cannot be used as a drug, as they cannot efficiently cross the cellular membranes. Therefore, several monophosphate approaches have been developed over the years to mask the monophosphate charges using intracellularly cleavable groups.71 Among these methods, phosphoramidate prodrug (ProTide), originally developed by Christopher McGuigan quickly gained popularity and led to the approval of several key antivirals such as sofosbuvir (HCV), TAF (HIV), remdesivir (Cov2). Once inside the cell, the carboxyl ester bond of the ProTide is cleaved by two liver enzymes, cathepsin A (CatA), and carboxylesterase 1 (CES1), initiating a cascade of chemical and enzymatic reactions. This sequence leads to the release of an alaninyl phosphate intermediate, which is subsequently converted into the active 5′-monophosphate form by human histidine triad nucleotide-binding protein 1 (Hint-1), followed by further phosphorylation by cellular kinases to generate the active nucleoside triphosphate (Fig. 19).
 |
| | Fig. 19 General mechanism for the metabolic activation of a nucleoside ProTide. | |
Tenofovir alafenamide (TAF)
Tenofovir disoproxil fumarate (TDF) and tenofovir alafenamide (TAF) are both used for the treatment of HIV and hepatitis B. While both are prodrugs designed to enhance the oral bioavailability of the parent drug tenofovir (TFV), their pharmacokinetic profiles differ significantly. The oral bioavailability of TFV is about 25–39% from TDF, resulting in higher levels of tenofovir in the circulation.61 In contrast, the aryloxy phosphonamidate moiety in tenofovir alafenamide (TAF, GS-7340) stabilizes the molecule against esterases in the intestine, resulting in delivery of intact prodrug into the hepatic circulation (Fig. 20).72,73 Consequently, plasma levels of TFV with TAF decreased by 90% while still reaching effective concentrations in target cells. This difference is key to TAF's improved renal and bone safety profile, as less tenofovir reaches the kidneys and bone when using TAF.74
 |
| | Fig. 20 Structures of tenofovir alafenamide (TAV) and tenofovir (TFV). | |
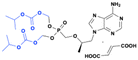
Prodrugs of 2′-C-methylguanosine
2′-C-Methylguanosine-5′-triphosphate (2′-C-MeG-TP) is a potent active inhibitor of HCV NS5B-mediated RNA synthesis (IC50 = 0.3 μM). However, the parent nucleoside (2′-C-methylguanosine) shows weak activity in a cell-based assay (EC50 = 1.9 μM) due to its poor uptake and metabolism to the active triphosphate in hepatoma cells.75 Therefore, several prodrugs have been developed to improve 2′-C-methylguanosine's pharmacokinetic and pharmacodynamic limitations and overall druglike properties.
BMS-986094 is a double prodrug of 2′-C-methylguanosine, showed high passive permeability and generates high intracellular concentrations of the active triphosphate metabolite within hepatocytes. This process is facilitated by hepatic uptake transporters OATP1B1 and OATP1B3. Furthermore, phosphoramidate derivates of 6-O-methyl-2′-C-methyl guanosine demonstrate improved potency, lipophilicity, cell permeability, and rapid intracellular conversion to the active triphosphate in human hepatocyte cultures.76 Preclinical data showed that BMS-986094 exhibits nanomolar EC50 values (0.9–12 nM), demonstrating superior potency compared to the parent nucleoside.76 During phase II trials, a dose-dependent reduction in HCV RNA levels was observed, with a median decline of 2.53 log10 IU per mL at a daily dose of 100 mg after seven days of treatment.77 However, despite its efficacy, the clinical development of BMS-986094 was discontinued due to significant safety concerns such as cardiovascular complications, including heart failure and myocardial injury, as well as renal toxicity, were reported.
IDX184 is an S-acyl-2-thioethyl (SATE) phosphoramidate prodrug developed by Idenix Pharmaceuticals78 that was shown to improve the cellular uptake, permeability, and selective delivery of the 5′-monophosphate (5′-MP) of 2′-C-methylguanosine to human liver cells. Although the metabolism of IDX-184 to 2′-C-methyl guanosine monophosphate is not fully understood, it primarily occurs in liver cells and involves both esterases and the intracellular phosphoramidase enzyme (Hint-1). In an HCV replicon assay, IDX184 demonstrated tenfold greater potency (EC50 = 0.4 μM) compared to the parent compound and exhibited promising antiviral activity and toxicity profiles in infected chimpanzees and monkeys.79 IDX184 was in several clinical trials for HCV, but the FDA imposed a clinical hold in 2010 for the phase 1 due to three serious adverse events related to elevated liver function tests. Nevertheless, the FDA later placed IDX184 under the partial clinical hold to both IDX184 and IDX19368 – another HCV nucleotide polymerase inhibitor for which Idenix had filed an IND but not initiated clinical trials (Fig. 21).
 |
| | Fig. 21 Structures of 2′-C-methylguanosine, BMS-986094, IDX-184, and IDX-19638. | |
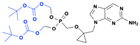
ALS-2200 and VX-135
ALS-2200, a monophosphate prodrug of 2′-C-methylcytidine licensed in June 2011 by Vertex, has emerged as a promising candidate for the treatment of hepatitis C virus (HCV) infection due to its potent antiviral activity across multiple genotypes (Fig. 22).80In vitro assays demonstrated an EC50 value of 150 nM in genotype 1b replicons and a broader activity range of 12–390 nM across other genotypes.81 Notably, ALS-2200 exhibits a high specificity for HCV and does not interfere with mitochondrial protein synthesis or human nucleic acid polymerases. Resistance studies identified the S282T mutation as the primary resistance-associated variant, conferring a >38-fold reduction in susceptibility.82 When evaluated in combination with other antivirals, ALS-2200 exhibited synergy with the nucleotide polymerase inhibitor ALS-2158 and the protease inhibitor telaprevir, while an additive effect was observed with ribavirin.81
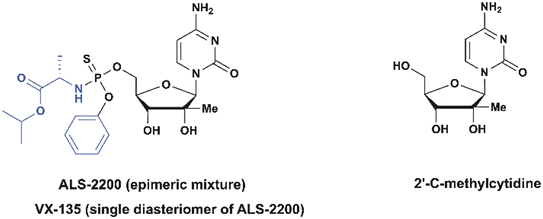 |
| | Fig. 22 Structures of ALS-2200, and VX-135 (a single phosphorus diastereomer). | |
A phase I study with healthy volunteers and HCV-infected individuals assessed its pharmacokinetics, revealing mean HCV RNA reductions of 0.97, 3.02, and 4.03 log10 IU per mL for doses of 15, 50, or 100 mg, respectively.83 No virological rebound was detected, and the compound was well tolerated with no severe adverse effects reported, even in patients with cirrhosis. A 200 mg dose resulted in median viral load reductions of approximately 4.2 log10 IU per mL.84
VX-135, a single phosphorus diastereomer of ALS-2200,85 exhibited comparable antiviral efficacy during HCV clinical trials (Fig. 23). Preclinical studies demonstrated similar hepatic drug accumulation between VX-135 and ALS-2200, supporting its potential as a liver-targeted antiviral agent. However, concerns regarding hepatotoxicity emerged during clinical development. In a study evaluating VX-135 in combination with ribavirin for 12 weeks in genotype 1b-infected patients, patients receiving the 200 mg dose had significant reductions in HCV RNA levels, with 80% achieving undetectable viral loads.86 Unfortunately, elevated liver enzyme levels were noted in patients on higher doses, leading to the discontinuation of the 400 mg dose arm. Lower doses were generally well tolerated without severe adverse events.
 |
| | Fig. 23 Structures of PSI-6206, and sofosbuvir (Sovaldi). | |
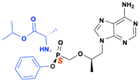
Sofosbuvir (Sovaldi)
Sofosbuvir (Sovaldi) is a phosphoramidate prodrug of 2′-α-F-2′-β-C-methyluridine nucleosides (PSI-6206) initially developed by Pharmasset (Fig. 23). It revolutionized the treatment of chronic HCV infections and was approved by the FDA in 2013 for use against HCV genotypes 1, 2, 3, and 4. This marked the introduction of the first interferon-free therapy for HCV. The parent compound PSI-6206 is inherently an inactive nucleoside analog, but its triphosphate form was shown to be a potent inhibitor of the HCV NS5B polymerase in biochemical assays. The inactivity of the parent uridine nucleoside is due to its inability to undergo the critical initial phosphorylation step necessary for activation. The ProTide moiety enhances cellular uptake and generates the parent nucleoside PSI-6206 monophosphate within the target cells by bypassing the first phosphorylation step required for activation. Sofosbuvir exhibited pan-genotypic efficacy against HCV genotypes 1 through 6 and is notable for its immaculate safety profile among nucleotide analogs.87 Even at high concentrations, it demonstrated minimal off-target effects, with no sign of cytotoxicity, mitochondrial toxicity or bone marrow toxicity.88 Today, sofosbuvir is usually used clinically in combination with an NS5A inhibitor such as velpatasvir, ledipasvir, or daclatasvir.
Uprifosbuvir (MK-3682)
Uprifosbuvir (MK-3682) is a 2′-Me, 2′-Cl uracil prodrug developed by Merck & Co. for the treatment of chronic hepatitis C virus (HCV) infection (Fig. 24). The parent nucleoside drug, 2′-α-chloro-2′-β-C-methyluridine exhibited only modest activity in the RPL cell-based assay, with an EC50 of 57 μM while its corresponding triphosphate form has shown excellent potency (IC50 = 0.154 μM).89 On the other hand, its monophospahte prodrug uprifosbuvir exhibits potent antiviral effect across various HCV genotypes.89
 |
| | Fig. 24 Structures of 2′-α-chloro-2′-β-C-methyluridine, and uprifosbuvir (MK-3682). | |
Like Sovaldi, the prodrug is intracellularly metabolized extensively in hepatocytes to release the active triphosphate. Preclinical studies demonstrated that MK-3682 generated high triphosphate levels in the liver with no signs of genotoxicity, mitochondrial toxicity or cardiotoxicity. A dosage of up to 300 mg QD × 7 days was safe and well tolerated in healthy subjects.90 Despite its promising safety profile, the development of uprifosbuvir was eventually discontinued in 2017 during phase II clinical trials. The termination of these trials was likely due to the emergence of more effective and simplified HCV therapies that set higher efficacy benchmarks that uprifosbuvir could not meet.91
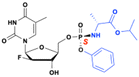
ATI-2173
ATI-2173 is an orally administered liver-targeted monophosphate prodrug of clevudine used to treat chronic HBV infections. It bypasses the initial phosphorylation step required by clevudine (L-FMAU), thereby reducing systemic exposure and minimizing the risk of extrahepatic toxicities, such as skeletal myopathy associated with prolonged clevudine use (Fig. 25). Once administered, ATI-2173 is converted to its active triphosphate within the liver cells. The triphosphate inhibits HBV DNA polymerase and reduces viral replication through a noncompetitive, non-chain-terminating mechanism, distinguishing it from traditional nucleos(t)ide analogs.
 |
| | Fig. 25 Structures of clevudine (L-FMAU), and ATI-2173. | |
Phase Ib clinical trial found that ATI-2173 was well-tolerated, with headache being the most reported adverse event. Patients treated with ATI-2173 experienced mean reductions in HBV DNA levels ranging from −2.72 to −2.78 log10 IU per mL by day 28, compared to a +0.17 log10 IU per mL increase in the placebo group. Many patients exhibited sustained off-treatment viral suppression and reductions in covalently closed circular DNA (cccDNA) biomarkers, with one patient maintaining undetectable HBV DNA levels 24 weeks after treatment cessation.92 However, despite these promising results from early-phase clinical trials, the clinical development of ATI-2173 was halted due to numerous regulatory hurdles.
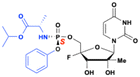
Adafosbuvir
Adafosbuvir (AL-335) is a ProTide prodrug of 2′-C-methyl-4′-F-uridine, developed by Alios BioPharma to treat HCV infections (Fig. 26). Adafosbuvir generates significantly higher levels of the active nucleoside triphosphate (TP) compared to the parent nucleoside, both in vitro in primary human hepatocytes and Huh-7 cells, and in dog liver tissues after a single oral dose. The triphosphates of the uridine analog demonstrated potent inhibition of HCV NS5B polymerase, with IC50 of about 140 nM. In an HCV subgenomic replicon assay, AL-335 showed strong activity, with an EC50 of 0.07 μM and no cytotoxicity (CC50 > 99 μM).93
 |
| | Fig. 26 Structures of adafosbuvir (AL-335), and 2′-C-methyl 4′-F-uridine. | |
AL-335 demonstrated promising results in phase 1 and phase 2 trials. A prospective 3-year follow-up study (NCT03099135, phase 3) was initiated in May 2017, involving HCV-infected subjects who had been treated in earlier phase 2 or phase 3 studies with a regimen containing NS5A inhibitor odalasvir and AL-335, with or without simeprevir.94 However, the efficacy of AL-335 was less compelling compared to other direct-acting antivirals as and it did not demonstrate the same level of effectiveness or rapid viral clearance as other treatments like sofosbuvir, when used in combination with other agents. Consequently, the phase 3 study (NCT03099135) was terminated.
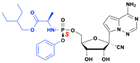
Remdesivir
GS-441524 has been shown to be active in vitro against many RNA viruses, including HCV, Ebola, RSV and coronaviruses. However, its physicochemical properties, such as polarity and solubility, impede oral bioavailability and permeability, limiting its clinical benefit as an oral drug for SARS-CoV-2.95 To improve the delivery, cellular uptake, and efficacy of GS-441512, a McGuigan ProTide was developed, Remdesivir. Remdesivir exhibits favorable cell permeability and was initially used to treat patients infected with HCV, Ebola and RSV viruses. Remdesivir was later approved for emergency treatment of COVID-19 infected persons96 (Fig. 27).
 |
| | Fig. 27 Structures of GS-441524, and remdesivir. | |
The bioactivation of this type of prodrug involves multiple enzymes, such as carboxylesterase (CES1), cathepsin A (CTSA), and phosphatase (histidine trinucleotide binding protein; Hint-1), which are highly expressed in the liver cells and type II alveolar cells in the lung tissues. The active metabolite, remdesivir triphosphate, could trigger mitochondrial toxicity, which can lead to elevated aminotransferase levels in the liver, causing liver injury.97 In addition, remdesivir is a substrate for the P-glycoprotein (P-gp) transporter, which may decrease the efflux of remdesivir out of the hepatocyte, resulting in a hepatocellular concentration above the toxic threshold.98 It is postulated that the kidney toxicity may occur due to the long-term treatment, although the exact injury mechanisms are unknown.97
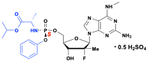
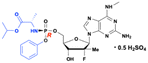
Bemnifosbuvir (AT-527) and AT-752
Bemnifosbuvir (AT-527) and AT-752, developed by Atea Pharmaceuticals, Inc., are orally available double prodrugs of 2′-C-Me, 2′-F-guanosine is 2′-C-Me, 2′-F-guanosine nucleoside analog (AT-273), with broad-spectrum antiviral activity against several RNA viruses, including HCV and COVID-19. Both compounds have a methyl group at the N6 position on the purine ring, which stabilizes the glycosidic bond and prevents the formation of toxic metabolites, such as mutagenic O6-alkylated nucleobases associated with earlier guanosine analogues (BMS-986094 and PSI-938). This structural feature enhances target cell uptake and promotes efficient formation of the active triphosphate metabolite (AT-9010).99
These two compounds are diastereomers, with the only structural difference is the chirality at the phosphorous atom – AT-752 is the Rp-diastereomer, while AT-527 is the Sp-diastereomer (Fig. 28). The hemisulfate salt forms influence the physiochemical properties such as solubility, and stability, allowing the prodrugs to be highly soluble and maintain a solid form suitable for oral formulation. Due to their structural similarity, both isomers produce the same active triphosphate (AT-9010) and its plasma surrogate nucleoside marker AT-273.
 |
| | Fig. 28 Structures of AT-273, bemnifosbuvir (AT-527), and AT-752. | |
Both AT-752 and AT-527, like other ProTide, are converted into the same 5′-monophosphate metabolite (AT-8001) in infected cells. This metabolite is then subsequently phosphorylated into the active 5′-triphosphate metabolite (AT-9010). This active triphosphate metabolite selectively inhibits the nonstructural DENV2 NS5 protein, and viral RdRp of HCV.100,101
Bemnifosbuvir (AT-527) is prominent for its potential activity against a range of RNA viruses, including HCV, and SARS-CoV-2 (Fig. 28). It advanced to phase 3 clinical trial for the treatment of SARS-CoV-2 infections (NCT04889040). While early clinical trials showed promising results with a unique mechanism targeting the SARS-CoV-2 RNA polymerase, the later stages yielded inconsistent results. The phase 3 study was terminated early as it failed to meet its primary endpoint of symptom alleviation and did not significantly reduce viral load. However, the drug was well-tolerated and reduced relative hospitalization risk by 71%.102
The AT-752 (Rp-diastereomer) demonstrates strong antiviral activity against all four serotypes of the dengue virus, with an EC50 of about 0.50 μM and low cytotoxicity.103 A completed phase 1 clinical trial confirmed its good tolerability, with no serious adverse events reported.104 The metabolite AT-273 (2′-fluoro-2′-C-methylguanosine) has a long half-life of approximately 15–25 h, with plasma levels increasing at higher doses, indicative of the long half-life of the active triphosphate (AT-9010) formed intracellularly.104,105 AT-752's safety and pharmacokinetic profiles make it a promising option for dengue treatment. Additionally, it showed efficacy against the yellow fever virus (YFV), with its free base, AT-281, exhibiting an EC50 of 0.31 μM.106
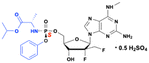
2′-α-Fluoro-2′-β-C-(fluoromethyl) purine nucleoside phosphoramidate prodrug
The phosphoramidate prodrug of 2′-α-fluoro-2′-β-C-(fluoromethyl)guanosine nucleoside demonstrated in vitro antiviral activity against the SARS-CoV-2 20SF107 strain (EC50 = 0.56 ± 0.06 μM) and the Omicron BA.5 variant (EC50 = 0.96 ± 0.23 μM) with minimal cytotoxicity (Fig. 29).107 This is a hemisulfate double prodrug similar to AT-527 and AT-752 with good solubility and oral bioavailability compared to its parent compound. Upon biotransformation, the active nucleoside triphosphate effectively blocked RNA synthesis by inhibiting SARS-CoV-2 RdRp. In vivo studies demonstrated that it was well-tolerated in rats at doses up to 2000 mg kg−1, and a single oral dose of 40 mg kg−1 resulted in high concentrations of the bioactive nucleoside triphosphate in the target organ, the lungs, with a prolonged half-life.107 These results highlight the potential of a prodrug as an orally available antiviral agent for the treatment of SARS-CoV-2 infections.
 |
| | Fig. 29 Structures of 2′-α-fluoro-2′-β-C-(fluoromethyl)guanosine, and its phosphoramidate prodrug. | |
GS-7682
GS-646089 is a 4′-CN-C-ribonucleoside that demonstrates activity against respiratory syncytial virus (RSV) in a cell-free RSV-A2 ribonucleoprotein (RNP) complex assay (IC50 = 46 nM) and in the RdRp enzymatic assay (IC50 = 43 nM). Enzyme kinetics show that GS-646939-triphosphate is more efficiently incorporated into the RdRp complex of several viruses, including human metapneumovirus (HMPV), human rhinovirus type 16 (HRV-16), and enterovirus 71 (EV-71) than its native ATP counterpart. Once incorporated into the nascent RNA chain, GS-646939 acts as an immediate chain terminator.108
To efficiently deliver the GS-646089-TP to the lung epithelial cells, the double prodrug GS-7682 was developed for a nebulized inhaled formulation (Fig. 30). Prodrug optimization identified 5-methyl [(S)-hydroxy(phenoxy)phosphoryl]-L-alaninate, in combination with 2,3-diisobutyrate as achieving high levels of intracellular triphosphate formation in lung cells both in vitro and in vivo.32 This phosphoramide prodrug exhibits high potency against RSV (EC50 < 7 nM) in human bronchial epithelial (NHBE) cells, demonstrating a 400-fold improvement compared to the corresponding parent nucleoside (GS-646089, ∼2.8 μM). However, oral administration of GS-7682 is subject to high first-pass hepatic extraction, leading to limited lung exposure with bioavailability of <1% in cynomolgus monkeys. In addition to inconvenient intravenous (IV) dosing for the treatment in the outpatient setting, GS-7682 also has a short half-life (t1/2 = 0.34 ± 0.17 h) from a 30 min IV infusion at 5 mg kg−1 in cynomolgus monkeys. This short half-life is due to the rapidly degradation in tissue and plasma, resulting in persistent plasma exposures of parent nucleoside GS-646089 (t1/2 = 8.71 ± 3.01 h).
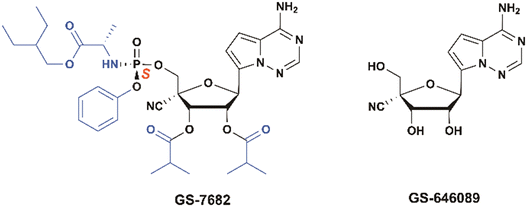 |
| | Fig. 30 Structures of GS-646089, and GS-7682. | |
When administered via inhalation, the prodrug GS-7682 showed high exposure and potent antiviral efficacy in the African green monkey (AGM) RSV model. Following intratracheal (IT) administration in cynomolgus monkeys (lung deposited dose of 4.0 mg kg−1), plasma concentrations of GS-7682 were maintained with a prolonged half-life (t1/2 = 13.0 ± 0.6 h). Moreover, the concentration levels of GS-646089 -TP in lung tissue were approximately 6-fold higher at 24 hours post-administration compared to intravenous infusion in cynomolgus monkeys. Thus, inhaled GS-7682 could become an effective treatment for RSV and potentially for pneumonia and picornaviruses.32
5. Miscellaneous: nucleoside analogs activated by purine degradation/metabolic enzymes
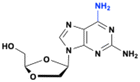
APD and DAPD
9-(β-D-1,3-Dioxolan-4-yl)guanine (DXG) is a nucleoside analog that exhibits potent activity against HIV and HBV in vitro.109,110 The limited aqueous solubility and low bioavailability of DXG renders the compound difficult to administer in vivo, resulting in the synthesis of several prodrugs of DXG such as (−)-β-D-2-aminopurine dioxolane (APD) and (−)-β-D-2,6-diaminopurine dioxolane (amdoxovir, DAPD) (Fig. 31).
 |
| | Fig. 31 Structures of nucleoside analogs activated by purine metabolic enzymes. | |
APD is more water soluble than DXG and is metabolized into DXG by xanthine oxidase, which is highly expressed in the liver and the intestine.111 The serum concentration of DXG is higher after the oral administration of APD in the woodchuck model of chronic HBV infection, indicating significant first-pass intestinal and/or hepatic metabolism. The reduction of woodchuck hepatitis virus (WHV) viremia is dose dependent.109
DAPD is converted to DXG through the hydrolytic action of adenosine deaminase at the 6-amino position. Although the Km value measured for DAPD is comparable to that of the natural substrate adenosine, the overall substrate efficiency of DAPD is substantially less due to a significantly slower kcat value. Nonetheless, DAPD is efficiently converted to DXG in whole human blood (half-life of ∼1 h). DAPD exhibits all the desirable preclinical attributes that a nucleoside analog must possess to be considered a viable clinical candidate.109 Although clinical trials with DAPD were scheduled in Argentina, the studies were stopped due to insufficient patient recruitment and competition from integrase inhibitors at that time.
Taribavirin and viramidine
Taribavirin and viramidine (taribavirin hydrochloride) are prodrugs of ribavirin. Although ribavirin exhibits broad antiviral properties both in vitro and in vivo, it is approved only for a limited range of conditions, such as HCV and severe RSV infections. The prodrug viramidine has reduced exposure to red blood cells due to the partial protonation of the 3-carboximidine group at the physiological pH,112 which decreases hematological toxicity. It is metabolized during the first-pass metabolism by liver cells and converted intracellularly by adenosine kinase113,114 to the active form ribavirin triphosphate. Thus, the active metabolite ribavirin is limited to the liver tissues, which is the site of HCV replication. Viramidine acts through a dual-action mechanism, serving as a prodrug of ribavirin and concurrently as an inhibitor (Ki = 2.5 μM) of nucleoside phosphorylase, which catalyzes the catabolism of ribavirin.115 Ribavirin has side effects, particularly hemolytic anemia, which often limits its dosage. In the phase III clinical trial, viramidine was orally absorbed and rapidly converted to ribavirin with a Tmax of 1.5 to 3.0 h for both viramidine and ribavirin in plasma.116
Abacavir
Abacavir, commonly known as Ziagen, was approved for use against HIV in 1998. It is a nucleoside analog reverse-transcriptase inhibitor typically used in combination with other HIV treatments. Abacavir is a prodrug of carbocyclic nucleoside carbovir, which is active/effective against HIV-1 and HIV-2. It is a lipophilic prodrug that has excellent central nervous system (CNS) penetration and has a great absolute bioavailability (105% in rat, and 83% in humans) compared to carbovir (26% and 23% in the rat and monkey, respectively).58 Unlike other nucleoside analogous, abacavir is metabolized to its active metabolite, carbovir triphosphate, through a unique phosphorylation pathway. After oral administration, abacavir is initially phosphorylated to abacavir monophosphate by adenosine phosphotransferase within the cell. It is then converted to carbovir monophosphate by a cytosolic deaminase and subsequently transformed to carbovir triphosphate by cellular kinases.117 This unique activation mechanism overcomes the pharmacokinetic and toxicological deficiencies of carbovir while preserving its potent and selective anti-HIV activity (Fig. 32).117
 |
| | Fig. 32 Pathway for the anabolism of abacavir to carbovir triphosphate in human cells. | |
6. Conclusions
Nucleoside prodrugs have significantly impacted the development of antiviral drugs. In fact, most current antiviral treatments include at least one nucleoside prodrug. (e.g., HIV: TDF; HBV: TDF, HSV: valacyclovir; HCV: SOF; SARS-CoV-2: remdesivir). Compared to their parent compounds, these prodrugs can lead to enhanced physiochemical, pharmacokinetics properties and improved intracellular phosphorylation. Furthermore, these prodrugs are targeted to specific cellular accumulation and tissues susceptible to the virus (or cancer), while also improving the oral bioavailability of the active nucleoside with minimal off-target effects, thereby enhancing the therapeutic index. Despite all these successes, the race toward novel prodrug approaches is still ongoing, including the development of nucleoside triphosphate prodrugs that can deliver the active triphosphate nucleoside directly into the cell, bypassing complete kinase activation.118,119 Alternatively, novel controlled release/long-acting technologies like implants are being developed for nucleoside analogs (i.e., islatravir). These analogs are designed to provide sustained drug release and prolonged therapeutic activity, with the goal of improving adherence and overall treatment outcomes in individuals living with HIV. By leveraging optimized pharmacokinetic profiles, these formulations aim to maintain effective drug concentrations in the body over extended periods. This has opened the door to dosing regimens that were previously unattainable with traditional therapies, including once-monthly intramuscular injections and even quarterly subcutaneous implants. This approach represents a significant step forward in simplifying HIV treatment and prevention strategies, potentially reducing the burden of daily medication and enhancing patient quality of life.120 Although significant progress has been made, the development of any nucleoside prodrugs remains a challenge as activation of these compounds can be limited and vary across different cell types, tissues, or patients, leading to suboptimal therapeutic outcomes.
Author contributions
Conceptualization, RRS, RFS, SJH, and FA; writing – original draft preparation, RRS, writing – review and editing, all authors.
Conflicts of interest
All authors have read and agreed to the published version of the manuscript. The authors declare no conflict of interest.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Acknowledgements
This work was supported in part by NIH support to the Emory CFAR (P30-AI-050409). This article is dedicated to the memory of Dr. Selwyn J. Hurwitz, who initiated this review and sadly passed away on November 23, 2024.
References
- S. J. Hurwitz and R. F. Schinazi, Drug Discovery Today:Technol., 2012, 9, e183–e193 CrossRef CAS.
- S. J. Hurwitz and R. F. Schinazi, Curr Opin HIV AIDS, 2013, 8, 556–564 CrossRef CAS.
- N. G. Rouphael, S. J. Hurwitz, M. Hart, A. Beck, E. J. Anderson, G. Deye, B. Osborn, S. Y. Cai, C. Focht, C. Amegashie, T. L. Bowlin, J. Brooks and M. J. Mulligan, Antimicrob. Agents Chemother., 2019, 63 DOI:10.1128/aac.00717-19.
- S. J. Hurwitz, R. De, J. C. LeCher, J. A. Downs-Bowen, S. L. Goh, K. Zandi, T. McBrayer, F. Amblard, D. Patel, J. J. Kohler, M. Bhasin, B. S. Dobosh, V. Sukhatme, R. M. Tirouvanziam and R. F. Schinazi, Viruses, 2024, 16, 651 CrossRef CAS.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS.
- H. Han, R. L. de Vrueh, J. K. Rhie, K. M. Covitz, P. L. Smith, C. P. Lee, D. M. Oh, W. Sadee and G. L. Amidon, Pharm. Res., 1998, 15, 1154–1159 CrossRef CAS.
- C. P. Landowski, P. L. Lorenzi, X. Song and G. L. Amidon, J. Pharmacol. Exp. Ther., 2006, 316, 572–580 CrossRef CAS.
-
(a)
M. G. Taylor and V. Gerriets, Acyclovir, StatPearls Publishing, 2025 Search PubMed;
(b) National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/books/NBK542180/, 2025.
- W. R. Lin, H. H. Lin, S. S. Lee, H. C. Tsai, C. K. Huang, S. R. Wann, Y. S. Chen, S. C. Chiang, M. Y. Yen and Y. C. Liu, J. Microbiol., Immunol. Infect., 2001, 34, 138–142 Search PubMed.
- R. D. Anderson, K. G. Griffy, D. Jung, A. Dorr, J. D. Hulse and R. B. Smith, Clin. Ther., 1995, 17, 425–432 CrossRef PubMed.
- A. Caldés, H. Colom, Y. Armendariz, M. J. Garrido, I. F. Troconiz, S. Gil-Vernet, N. Lloberas, L. Pou, C. Peraire and J. M. Grinyó, Antimicrob. Agents Chemother., 2009, 53, 4816–4824 CrossRef.
- M. R. Rashidi, J. A. Smith, S. E. Clarke and C. Beedham, Drug Metab. Dispos., 1997, 25, 805–813 Search PubMed.
- C. M. Perry and A. J. Wagstaff, Drugs, 1995, 50, 396–415 CrossRef.
- X.-L. Qiu, X.-H. Xu and F.-L. Qing, Tetrahedron, 2010, 66, 789–843 CrossRef.
- G. Liu-Young and M. J. Kozal, AIDS Patient Care STDS, 2008, 22, 449–457 CrossRef PubMed.
- S. K. Tyring, P. Lee, G. T. Hill, Jr., J. C. Silverfield, A. Y. Moore, T. Matkovits and J. Sullivan-Bolyai, J. Med. Virol., 2017, 89, 1255–1264 CrossRef PubMed.
- C. McGuigan and J. Balzarini, J. Antimicrob. Chemother., 2009, 64, 671–673 CrossRef PubMed.
- M. Migliore, Antiviral Chem. Chemother., 2010, 20, 107–115 CrossRef PubMed.
- E. De Clercq, Viruses, 2022, 14, 770 CrossRef PubMed.
- T. N. D. Do, K. Donckers, L. Vangeel, A. K. Chatterjee, P. A. Gallay, M. D. Bobardt, J. P. Bilello, T. Cihlar, S. De Jonghe, J. Neyts and D. Jochmans, Antiviral Res., 2021, 192, 105122 Search PubMed.
- Y. Li, M. Liu, Y. Yan, Z. Wang, Q. Dai, X. Yang, X. Guo, W. Li, X. Chen, R. Cao and W. Zhong, Viruses, 2022, 14, 1142 CrossRef PubMed.
- S. Zhou, C. S. Hill, S. Sarkar, L. V. Tse, B. M. D. Woodburn, R. F. Schinazi, T. P. Sheahan, R. S. Baric, M. T. Heise and R. Swanstrom, J. Infect. Dis., 2021, 224, 415–419 CrossRef PubMed.
- B. M. Maas, J. Strizki, R. R. Miller, S. Kumar, M. Brown, M. G. Johnson, M. Cheng, C. De Anda, M. L. Rizk and J. A. Stone, Clin. Transl. Sci., 2024, 17, e13732 CrossRef PubMed.
- F. Kabinger, C. Stiller, J. Schmitzová, C. Dienemann, G. Kokic, H. S. Hillen, C. Höbartner and P. Cramer, Nat. Struct. Mol. Biol., 2021, 28, 740–746 CrossRef.
- G. R. Painter, R. A. Bowen, G. R. Bluemling, J. DeBergh, V. Edpuganti, P. R. Gruddanti, D. B. Guthrie, M. Hager, D. L. Kuiper, M. A. Lockwood, D. G. Mitchell, M. G. Natchus, Z. M. Sticher and A. A. Kolykhalov, Antiviral Res., 2019, 171, 104597 CrossRef PubMed.
- M. Toots, J.-J. Yoon, R. M. Cox, M. Hart, Z. M. Sticher, N. Makhsous, R. Plesker, A. H. Barrena, P. G. Reddy, D. G. Mitchell, R. C. Shean, G. R. Bluemling, A. A. Kolykhalov, A. L. Greninger, M. G. Natchus, G. R. Painter and R. K. Plemper, Sci. Transl. Med., 2019, 11, eaax5866 CrossRef PubMed.
- L. Tian, Z. Pang, M. Li, F. Lou, X. An, S. Zhu, L. Song, Y. Tong, H. Fan and J. Fan, Front. Immunol., 2022, 13, 855496 CrossRef.
- R. Swanstrom and R. F. Schinazi, Science, 2022, 375, 497–498 CrossRef PubMed.
- G. Wang, J. Deval, J. Hong, N. Dyatkina, M. Prhavc, J. Taylor, A. Fung, Z. Jin, S. K. Stevens, V. Serebryany, J. Liu, Q. Zhang, Y. Tam, S. M. Chanda, D. B. Smith, J. A. Symons, L. M. Blatt and L. Beigelman, J. Med. Chem., 2015, 58, 1862–1878 CrossRef.
- J. P. DeVincenzo, M. W. McClure, J. A. Symons, H. Fathi, C. Westland, S. Chanda, R. Lambkin-Williams, P. Smith, Q. Zhang, L. Beigelman, L. M. Blatt and J. Fry, N. Engl. J. Med., 2015, 373, 2048–2058 CrossRef PubMed.
- A. Oey, M. McClure, J. A. Symons, S. Chanda, J. Fry, P. F. Smith, K. Luciani, M. Fayon, K. Chokephaibulkit, R. Uppala, J. Bernatoniene, K. Furuno, T. Stanley, D. Huntjens, J. Witek and R. S. V. S. Groups, PLoS One, 2023, 18, e0288271 CrossRef.
- D. S. Siegel, H. C. Hui, J. Pitts, M. S. Vermillion, K. Ishida, D. Rautiola, M. Keeney, H. Irshad, L. Zhang, K. Chun, G. Chin, B. Goyal, E. Doerffler, H. Yang, M. O. Clarke, C. Palmiotti, A. Vijjapurapu, N. C. Riola, K. Stray, E. Murakami, B. Ma, T. Wang, X. Zhao, Y. Xu, G. Lee, B. Marchand, M. Seung, A. Nayak, A. Tomkinson, N. Kadrichu, S. Ellis, O. Barauskas, J. Y. Feng, J. K. Perry, M. Perron, J. P. Bilello, P. J. Kuehl, R. Subramanian, T. Cihlar and R. L. Mackman, J. Med. Chem., 2024, 67, 12945–12968 CrossRef.
- M. Kasthuri, C. Li, K. Verma, O. O. Russell, L. Dickson, L. McCormick, L. Bassit, F. Amblard and R. F. Schinazi, Molecules, 2020, 25, 1258 CrossRef PubMed.
- J. L. Clark, L. Hollecker, J. C. Mason, L. J. Stuyver, P. M. Tharnish, S. Lostia, T. R. McBrayer, R. F. Schinazi, K. A. Watanabe, M. J. Otto, P. A. Furman, W. J. Stec, S. E. Patterson and K. W. Pankiewicz, J. Med. Chem., 2005, 48, 5504–5508 CrossRef.
- L. J. Stuyver, T. R. McBrayer, P. M. Tharnish, J. Clark, L. Hollecker, S. Lostia, T. Nachman, J. Grier, M. A. Bennett, M. Y. Xie, R. F. Schinazi, J. D. Morrey, J. L. Julander, P. A. Furman and M. J. Otto, Antiviral Chem. Chemother., 2006, 17, 79–87 CrossRef PubMed.
- G. Asif, S. J. Hurwitz, J. Shi, B. I. Hernandez-Santiago and R. F. Schinazi, Antimicrob. Agents Chemother., 2007, 51, 2877–2882 CrossRef CAS.
-
M. J. Sofia, P. A. Furman and W. T. Symonds, in Accounts in Drug Discovery, ed. J. Barrish, P. Carter, P. Cheng and R. Zahler, The Royal Society of Chemistry, 2010, 10.1039/9781849731980-00238.
- D. B. Smith, J. A. Martin, K. Klumpp, S. J. Baker, P. A. Blomgren, R. Devos, C. Granycome, J. Hang, C. J. Hobbs, W. R. Jiang, C. Laxton, S. Le Pogam, V. Leveque, H. Ma, G. Maile, J. H. Merrett, A. Pichota, K. Sarma, M. Smith, S. Swallow, J. Symons, D. Vesey, I. Najera and N. Cammack, Bioorg. Med. Chem. Lett., 2007, 17, 2570–2576 CrossRef CAS PubMed.
- S. K. Roberts, G. Cooksley, G. J. Dore, R. Robson, D. Shaw, H. Berns, G. Hill, K. Klumpp, I. Najera and C. Washington, Hepatology, 2008, 48, 398–406 CrossRef CAS.
- M. Brandl, X. Wu, M. Holper, L. Hong, Z. Jia, R. Birudaraj, M. Reddy, T. Alfredson, T. Tran, S. Larrabee, X. Hadig, K. Sarma, C. Washington, G. Hill and D. B. Smith, Drug Dev. Ind. Pharm., 2008, 34, 683–691 CrossRef CAS.
- Y. L. Chen, N. Abdul Ghafar, R. Karuna, Y. Fu, S. P. Lim, W. Schul, F. Gu, M. Herve, F. Yokohama, G. Wang, D. Cerny, K. Fink, F. Blasco and P. Y. Shi, J. Virol., 2014, 88, 1740–1747 CrossRef.
- N. M. Nguyen, C. N. Tran, L. K. Phung, K. T. Duong, A. Huynh Hle, J. Farrar, Q. T. Nguyen, H. T. Tran, C. V. Nguyen, L. Merson, L. T. Hoang, M. L. Hibberd, P. P. Aw, A. Wilm, N. Nagarajan, D. T. Nguyen, M. P. Pham, T. T. Nguyen, H. Javanbakht, K. Klumpp, J. Hammond, R. Petric, M. Wolbers, C. T. Nguyen and C. P. Simmons, J. Infect. Dis., 2013, 207, 1442–1450 CrossRef CAS.
- Y. Li, L. Cao, G. Li, F. Cong, Y. Li, J. Sun, Y. Luo, G. Chen, G. Li, P. Wang, F. Xing, Y. Ji, J. Zhao, Y. Zhang, D. Guo and X. Zhang, J. Med. Chem., 2022, 65, 2785–2793 CrossRef CAS PubMed.
- L. Cao, Y. Li, S. Yang, G. Li, Q. Zhou, J. Sun, T. Xu, Y. Yang, R. Liao, Y. Shi, Y. Yang, T. Zhu, S. Huang, Y. Ji, F. Cong, Y. Luo, Y. Zhu, H. Luan, H. Zhang, J. Chen, X. Liu, R. Luo, L. Liu, P. Wang, Y. Yu, F. Xing, B. Ke, H. Zheng, X. Deng, W. Zhang, C. Lin, M. Shi, C. M. Li, Y. Zhang, L. Zhang, J. Dai, H. Lu, J. Zhao, X. Zhang and D. Guo, Sci. Transl. Med., 2022, 14, eabm7621 CrossRef CAS.
- L. Rodriguez, A. Streinu-Cercel, J. M. Gonzalez Del Castillo, Y. Koullias, A. Mozaffarian, K. Etchevers, S. Chen, R. H. Hyland, J. Llewellyn, R. Geleziunas, L. Fouche, S. C. Chang, A. Castagna and C. Hedskog, Open Forum Infect. Dis., 2025, 12(1), ofae631.029 CrossRef.
- Q. Zhou, S. Yang, L. Cao, Y. Yang, T. Xu, Q. Chen, H. Lu, Y. Li, D. Guo and X. Zhang, Signal Transduction Targeted Ther., 2023, 8, 27 CrossRef CAS PubMed.
- R. L. Mackman, H. C. Hui, M. Perron, E. Murakami, C. Palmiotti, G. Lee, K. Stray, L. Zhang, B. Goyal, K. Chun, D. Byun, D. Siegel, S. Simonovich, V. Du Pont, J. Pitts, D. Babusis, A. Vijjapurapu, X. Lu, C. Kim, X. Zhao, J. Chan, B. Ma, D. Lye, A. Vandersteen, S. Wortman, K. T. Barrett, M. Toteva, R. Jordan, R. Subramanian, J. P. Bilello and T. Cihlar, J. Med. Chem., 2021, 64, 5001–5017 CrossRef CAS.
- Y. Xie, W. Yin, Y. Zhang, W. Shang, Z. Wang, X. Luan, G. Tian, H. A. Aisa, Y. Xu, G. Xiao, J. Li, H. Jiang, S. Zhang, L. Zhang, H. E. Xu and J. Shen, Cell Res., 2021, 31, 1212–1214 CrossRef CAS PubMed.
- H. J. Qian, Y. Wang, M. Q. Zhang, Y. C. Xie, Q. Q. Wu, L. Y. Liang, Y. Cao, H. Q. Duan, G. H. Tian, J. Ma, Z. B. Zhang, N. Li, J. Y. Jia, J. Zhang, H. A. Aisa, J. S. Shen, C. Yu, H. L. Jiang, W. H. Zhang, Z. Wang and G. Y. Liu, Acta Pharmacol. Sin., 2022, 43, 3130–3138 CrossRef CAS PubMed.
- China's NMPA conditionally approves two oral drugs for Covid-19.
- D. El-Haddad, F. El Chaer, J. Vanichanan, D. P. Shah, E. J. Ariza-Heredia, V. E. Mulanovich, A. M. Gulbis, E. J. Shpall and R. F. Chemaly, Antiviral Res., 2016, 134, 58–62 CrossRef CAS.
- M. Imran, M. K. Alshammari, M. K. Arora, A. K. Dubey, S. S. Das, M. Kamal, A. S. A. Alqahtani, M. A. Y. Sahloly, A. H. Alshammari, H. M. Alhomam, A. M. Mahzari, Abida, A. A. Rabaan and T. Dzinamarira, Biomedicines, 2023, 11, 278 CrossRef CAS.
- B. G. Wang, B. W. Cao, Z. C. Bei, L. K. Xu, D. N. Zhang, L. L. Zhao, Y. B. Song and H. Q. Wang, Eur. J. Med. Chem., 2023, 258, 115601 CrossRef CAS PubMed.
- Y. Zhang, C. Fan, J. Zhang, X. Tian, W. Zuo and K. He, Eur. J. Med. Chem., 2024, 275, 116614 CrossRef CAS PubMed.
- A. J. Wiemer and D. F. Wiemer, Top. Curr. Chem., 2015, 360, 115–160 CrossRef CAS.
- National Library of Medicine (US), National Center for Biotechnology Information, PubChem Compound Summary for CID 483477, Brincidofovir, 2004, https://pubchem.ncbi.nlm.nih.gov/compound/Brincidofovir.
- P. Barditch-Crovo, J. Toole, C. W. Hendrix, K. C. Cundy, D. Ebeling, H. S. Jaffe and P. S. Lietman, J. Infect. Dis., 1997, 176, 406–413 CAS.
- S. M. Daluge, S. S. Good, M. B. Faletto, W. H. Miller, M. H. St Clair, L. R. Boone, M. Tisdale, N. R. Parry, J. E. Reardon, R. E. Dornsife, D. R. Averett and T. A. Krenitsky, Antimicrob. Agents Chemother., 1997, 41, 1082–1093 CrossRef CAS PubMed.
- C. C. Lin, C. Fang, S. Benetton, G. F. Xu and L. T. Yeh, Antimicrob. Agents Chemother., 2006, 50, 2926–2931 CrossRef CAS.
- H. L. Tillmann, Curr. Opin. Invest. Drugs, 2007, 8, 682–690 CAS.
- B. P. Kearney, J. F. Flaherty and J. Shah, Clin. Pharmacokinet., 2004, 43, 595–612 CrossRef CAS PubMed.
- B. L. Robbins, R. V. Srinivas, C. Kim, N. Bischofberger and A. Fridland, Antimicrob. Agents Chemother., 1998, 42, 612–617 CrossRef CAS.
- H. J. Yim, W. Kim, S. H. Ahn, J. M. Yang, J. Y. Jang, Y. O. Kweon, Y. K. Cho, Y. J. Kim, G. Y. Hong, D. J. Kim, Y. K. Jung, S. H. Um, J. H. Sohn, J. W. Lee, S. J. Park, B. S. Lee, J. H. Kim, H. S. Kim, S. K. Yoon, M. Y. Kim, K. S. Lee, Y. S. Lim, W. S. Lee and K. H. Han, Am. J. Gastroenterol., 2020, 115, 1217–1225 CrossRef PubMed.
- M.-F. Yuen, S.-H. Lee, H.-M. Kang, C. R. Kim, J. Kim, V. Ngai and C.-L. Lai, Antimicrob. Agents Chemother., 2009, 53, 1779–1785 CrossRef CAS PubMed.
- D. S. Song, W. Kim, S. H. Ahn, H. J. Yim, J. Y. Jang, Y. O. Kweon, Y. K. Cho, Y. J. Kim, G. Y. Hong, D. J. Kim, Y. K. Jung, J. H. Sohn, J. W. Lee, S. J. Park, B. S. Lee, J. H. Kim, H. S. Kim, S. K. Yoon, M. Y. Kim, K. S. Lee, Y. S. Lim, W. S. Lee, J. M. Yang, K. H. Kim, K. H. Han and S. H. Um, Clin. Mol. Hepatol., 2021, 27, 346–359 CrossRef.
- C. Niu, T. Tolstykh, H. Bao, Y. Park, D. Babusis, A. M. Lam, S. Bansal, J. Du, W. Chang, P. G. Reddy, H.-R. Zhang, J. Woolley, L.-Q. Wang, P. B. Chao, A. S. Ray, M. J. Otto, M. J. Sofia, P. A. Furman and E. Murakami, Antimicrob. Agents Chemother., 2012, 56, 3767–3775 CrossRef CAS.
- P. G. Reddy, D. Bao, W. Chang, B. K. Chun, J. Du, D. Nagarathnam, S. Rachakonda, B. S. Ross, H. R. Zhang, S. Bansal, C. L. Espiritu, M. Keilman, A. M. Lam, C. Niu, H. M. Steuer, P. A. Furman, M. J. Otto and M. J. Sofia, Bioorg. Med. Chem. Lett., 2010, 20, 7376–7380 CrossRef CAS.
-
R. E. T. Smith, Pharmasset Reports Positive Preliminary Antiviral Data with PSI-938 for the Treatment of Hepatitis C, https://www.natap.org/2010/HCV/102810_03.htm.
-
R. E. T. Smith, Pharmasset Initiates QUANTUM, a Phase 2b Interferon-Free Trial of PSI-7977 and PSI-938 for All HCV Genotypes, https://www.natap.org/2011/HCV/091611_02.htm.
-
R. E. T. Smith, Pharmasset Announces Intent to Amend QUANTUM Trial - PSI938 liver function abnormality detected, https://www.natap.org/2011/HCV/121611_02.htm.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed.
- G. Birkus, R. Wang, X. H. Liu, N. Kutty, H. MacArthur, T. Cihlar, C. Gibbs, S. Swaminathan, W. Lee and M. McDermott, Antimicrob. Agents Chemother., 2007, 51, 543–550 CrossRef CAS.
- E. Murakami, T. Wang, Y. Park, J. Hao, E. I. Lepist, D. Babusis and A. S. Ray, Antimicrob. Agents Chemother., 2015, 59, 3563–3569 CrossRef CAS PubMed.
- G. Di Perri, Infez. Med., 2021, 29, 526–529 Search PubMed.
- G. Migliaccio, J. E. Tomassini, S. S. Carroll, L. Tomei, S. Altamura, B. Bhat, L. Bartholomew, M. R. Bosserman, A. Ceccacci, L. F. Colwell, R. Cortese, R. De Francesco, A. B. Eldrup, K. L. Getty, X. S. Hou, R. L. LaFemina, S. W. Ludmerer, M. MacCoss, D. R. McMasters, M. W. Stahlhut, D. B. Olsen, D. J. Hazuda and O. A. Flores, J. Biol. Chem., 2003, 278, 49164–49170 CrossRef CAS.
- J. H. Vernachio, B. Bleiman, K. D. Bryant, S. Chamberlain, D. Hunley, J. Hutchins, B. Ames, E. Gorovits, B. Ganguly, A. Hall, A. Kolykhalov, Y. Liu, J. Muhammad, N. Raja, C. R. Walters, J. Wang, K. Williams, J. M. Patti, G. Henson, K. Madela, M. Aljarah, A. Gilles and C. McGuigan, Antimicrob. Agents Chemother., 2011, 55, 1843–1851 CrossRef CAS PubMed.
- M. Rodriguez-Torres, E. Lawitz, L. Hazan, A. N. Barry, E. D. Wenzel, J. Alam, G. W. Henson and J. Patti, Hepatology, 2011, 54, 535A Search PubMed.
- E. Cretton, C. Perigaud, S. Peyrottes, L. Licklider, M. Camire, M. La Colla, E. Hildebrand, L. Lallos, J. Bilello, J. McCarville and M. Seifer, J. Hepatol., 2008, 48, 588 CrossRef.
- X. J. Zhou, K. Pietropaolo, J. Chen, S. Khan, J. Sullivan-Bólyai and D. Mayers, Antimicrob. Agents Chemother., 2011, 55, 76–81 CrossRef CAS PubMed.
- R. A. Vere Hodge, Antiviral Res., 2013, 100, 276–285 CrossRef CAS.
- H. Tan, H. Kang, C. Moy, M. Jiang, D. Reagan, J. Deval, N. Dyatkina, G. Wang, T. L. Kieffer, R. Rijnbrand, L. Beigelman, D. B. Smith, L. M. Blatt and J. A. Symons, Hepatology, 2012, 56, 1072A Search PubMed.
- H. Tan, A. Jekle, H. Kang, C. Moy, J. Deval, A. Tigges, E. Zhang, M. Jiang, A. Andzinski, N. Dyatkina, G. Wang, R. Rijnbrand, T. L. Kieffer, L. M. Blatt, L. Beigelman, D. B. Smith and J. A. Symons, J. Hepatol., 2013, 58, S496 CrossRef.
- P. Marcellin, S. Popa, A. Berliba, I. Moscalu, N. Boyer, R. S. Kauffman, M. J. Koziel, V. Garg, M. Tong, S. M. Chanda, Q. Zhang, C. Westland, L. Beigelman, L. M. Blatt and J. Fry, Hepatology, 2012, 56, 235A CrossRef PubMed.
- P. Marcellin, S. Popa, A. Streinu-Cercel, N. Boyer, D. Dospinoiu, A. Patat, N. Ghicavii, V. Garg, R. S. Kauffman, M. Koziel, M. J. Tong, S. Chanda, Q. Zhang, C. Westland, L. Beigelman, L. M. Blatt and J. Fry, J. Hepatol., 2013, 58, S355 Search PubMed.
- H. Tan, J. Deval, H. Kang, C. Moy, Z. Jin, M. Jiang, A. Ardzinski, R. Rijnbrand, T. Kieffer, L. M. Blatt, S. M. Chanda, N. Dyatkina, G. Wang, Q. Zhang, J. A. Symons, L. Beigelman and D. B. Smith, J. Hepatol., 2013, 58, S495 CrossRef.
- S. Popa, A. Berliba, N. Ghicavii, A. A. Patat, R. S. Kauffman, J. Krop, V. Garg, M. Tong, S. M. Chanda, Q. Zhang, C. Westland, M. W. McClure, L. Beigelman, L. M. Blatt and J. Fry, Hepatology, 2013, 58, 745A Search PubMed.
- A. M. Lam, C. Espiritu, S. Bansal, H. M. Micolochick Steuer, C. Niu, V. Zennou, M. Keilman, Y. Zhu, S. Lan, M. J. Otto and P. A. Furman, Antimicrob. Agents Chemother., 2012, 56, 3359–3368 CrossRef PubMed.
- J. Y. Feng, Y. Xu, O. Barauskas, J. K. Perry, S. Ahmadyar, G. Stepan, H. Yu, D. Babusis, Y. Park, K. McCutcheon, M. Perron, B. E. Schultz, R. Sakowicz and A. S. Ray, Antimicrob. Agents Chemother., 2016, 60, 806–817 CrossRef PubMed.
- F. R. Alexandre, E. Badaroux, J. P. Bilello, S. Bot, T. Bouisset, G. Brandt, S. Cappelle, C. Chapron, D. Chaves, T. Convard, C. Counor, D. Da Costa, D. Dukhan, M. Gay, G. Gosselin, J. F. Griffon, K. Gupta, B. Hernandez-Santiago, M. La Colla, M. P. Lioure, J. Milhau, J. L. Paparin, J. Peyronnet, C. Parsy, C. Pierra Rouviere, H. Rahali, R. Rahali, A. Salanson, M. Seifer, I. Serra, D. Standring, D. Surleraux and C. B. Dousson, Bioorg. Med. Chem. Lett., 2017, 27, 4323–4330 CrossRef PubMed.
-
E. J. Gane, E. Sicard, S. Popa, X.-J. Zhou, M.-F. Temam, J. Chen, D. Frank, E. F. Donovan, K. Pietropaolo and D. Mayers, 65th Annual Meeting of the American Association for the Study of Liver Diseases, Boston, MA, https://www.natap.org/2014/AASLD/AASLD_34.htm Search PubMed.
- Merck Press Release, https://www.merck.com/news/merck-discontinues-mk-3682b-and-mk-3682c-development-programs/.
- K. E. Squires, L. Ogilvie, A. Jucov, I. Anastasiy, N. Ghicavii, J. Huguet, R. Melara, M. Constantineau, A. De La Rosa and D. L. Mayers, J. Viral Hepatitis, 2023, 30, 19–28 CrossRef.
- G. Wang, N. Dyatkina, M. Prhavc, C. Williams, V. Serebryany, Y. Hu, Y. Huang, J. Wan, X. Wu, J. Deval, A. Fung, Z. Jin, H. Tan, K. Shaw, H. Kang, Q. Zhang, Y. Tam, A. Stoycheva, A. Jekle, D. B. Smith and L. Beigelman, J. Med. Chem., 2019, 62, 4555–4570 CrossRef PubMed.
- E. J. Gane, C. A. Stedman, C. Schwabe, L. Vijgen, S. Chanda, T. N. Kakuda, J. Fry, L. M. Blatt and M. W. McClure, Hepatology, 2018, 68, 2145–2157 CrossRef PubMed.
- Y. J. Li, L. Cao, G. Li, F. Cong, Y. F. Li, J. Sun, Y. Z. Luo, G. J. Chen, G. G. Li, P. Wang, F. Xing, Y. X. Ji, J. C. Zhao, Y. Zhang, D. Y. Guo and X. M. Zhang, J. Med. Chem., 2022, 65, 2785–2793 CrossRef PubMed.
-
https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19
.
- C. K. H. Wong, I. C. H. Au, W. Y. Cheng, K. K. C. Man, K. T. K. Lau, L. Y. Mak, S. L. Lui, M. S. H. Chung, X. Xiong, E. H. Y. Lau and B. J. Cowling, Aliment. Pharmacol. Ther., 2022, 56, 121–130 CrossRef PubMed.
- E. Leegwater, A. Strik, E. B. Wilms, L. B. E. Bosma, D. M. Burger, T. H. Ottens and C. van Nieuwkoop, Clin. Infect. Dis., 2020, 72, 1256–1258 CrossRef.
- X. J. Zhou, S. S. Good, K. Pietropaolo, Q. Huang, A. Moussa, J. M. Hammond and J. P. Sommadossi, Expert Opin. Invest. Drugs, 2024, 33, 9–17 CrossRef.
- A. Chazot, C. Zimberger, M. Feracci, A. Moussa, S. Good, J. P. Sommadossi, K. Alvarez, F. Ferron and B. Canard, PLoS Biol., 2024, 22, e3002743 Search PubMed.
- S. S. Good, A. Shannon, K. Lin, A. Moussa, J. G. Julander, P. La Colla, G. Collu, B. Canard and J. P. Sommadossi, Antimicrob. Agents Chemother., 2021, 65, e0098821 Search PubMed.
- A. Horga, R. Saenz, G. Yilmaz, A. Simon-Campos, K. Pietropaolo, W. J. Stubbings, N. Collinson, L. Ishak, B. Zrinscak, B. Belanger, C. Granier, K. Lin, X. J. Zhou, S. Wildum and J. Hammond, Future Virol., 2023, 18(13), 839–853 Search PubMed.
- E. Berliba, M. Bogus, F. Vanhoutte, P. J. Berghmans, S. S. Good, A. Moussa, K. Pietropaolo, R. L. Murphy, X. J. Zhou and J. P. Sommadossi, Antimicrob. Agents Chemother., 2019, 63(12), e01201-19 CrossRef PubMed.
- X. J. Zhou, J. Lickliter, M. Montrond, L. Ishak, K. Pietropaolo, D. James, B. Belanger, A. Horga and J. Hammond, Antimicrob. Agents Chemother., 2024, 68, e0161523 CrossRef.
- S. S. Good, A. Moussa, X. J. Zhou, K. Pietropaolo and J. P. Sommadossi, PLoS One, 2020, 15, e0227104 CrossRef PubMed.
- K. Lin, S. S. Good, J. G. Julander, A. E. Weight, A. Moussa and J. P. Sommadossi, PLoS Neglected Trop. Dis., 2022, 16, e0009937 Search PubMed.
- L. Liang, Y. Meng, X. Chang, E. Li, Y. Huang, L. Yan, Z. Lou, Y. Peng, B. Zhu, W. Yu and J. Chang, J. Med. Chem., 2025, 68, 1994–2007 CrossRef PubMed.
- C. J. Gordon, S. M. Walker, E. P. Tchesnokov, D. Kocincova, J. Pitts, D. S. Siegel, J. K. Perry, J. Y. Feng, J. P. Bilello and M. Gotte, J. Biol. Chem., 2024, 300, 107514 Search PubMed.
- S. Menne, G. Asif, J. Narayanasamy, S. D. Butler, A. L. George, S. J. Hurwitz, R. F. Schinazi, C. K. Chu, P. J. Cote, J. L. Gerin and B. C. Tennant, Antimicrob. Agents Chemother., 2007, 51, 3177–3184 CrossRef PubMed.
- L. Zhou, S. J. Coats, H. Zhang, S. Junxing, D. R. Bobeck and R. F. Schinazi, Tetrahedron, 2012, 68, 5738–5743 CrossRef PubMed.
- P. A. Furman, J. Jeffrey, L. L. Kiefer, J. Y. Feng, K. S. Anderson, K. Borroto-Esoda, E. Hill, W. C. Copeland, C. K. Chu, J. P. Sommadossi, I. Liberman, R. F. Schinazi and G. R. Painter, Antimicrob. Agents Chemother., 2001, 45, 158–165 CrossRef PubMed.
- J. Z. Wu, C.-c. Lin and Z. Hong, J. Antimicrob. Chemother., 2003, 52, 543–546 CrossRef.
- P. Deming and S. Arora, Expert Opin. Invest. Drugs, 2011, 20, 1435–1443 CrossRef PubMed.
- J. Z. Wu, G. Larson, H. Walker, J. H. Shim and Z. Hong, Antimicrob. Agents Chemother., 2005, 49, 2164–2171 CrossRef PubMed.
- J. Z. Wu, G. Larson and Z. Hong, Antimicrob. Agents Chemother., 2004, 48, 4006–4008 CrossRef.
- C. C. Lin, L. T. Yeh, D. Vitarella and Z. Hong, Antiviral Chem. Chemother., 2003, 14, 145–152 CrossRef PubMed.
- M. B. Faletto, W. H. Miller, E. P. Garvey, M. H. St Clair, S. M. Daluge and S. S. Good, Antimicrob. Agents Chemother., 1997, 41, 1099–1107 CrossRef CAS.
- C. Meier, Antiviral Chem. Chemother., 2017, 25, 69–82 CrossRef.
- X. Jia, D. Schols and C. Meier, J. Med. Chem., 2023, 66, 12163–12184 CrossRef CAS PubMed.
- R. P. Matthews, W. Ankrom, E. Friedman, D. Jackson Rudd, Y. Liu, R. Mogg, D. Panebianco, I. De Lepeleire, M. Petkova, J. A. Grobler, S. A. Stoch and M. Iwamoto, Clin. Transl. Sci., 2021, 14, 1935–1944 Search PubMed.
- D. B. Warner, B. H. Jeng, J. Kim, M. Liu, A. B. Troxel, J. S. Hochman, K. H. Baratz, S. I. Mian, M. Y. Choulakian, J. J. Meyer, Y. Lu, A. Twi-Yeboah, T. F. Lee, C. Lopez-Jimenez, S. C. Laury and E. J. Cohen, JAMA Ophthalmol., 2025, 143, 277–285 CrossRef PubMed.
- V. Lukacova, P. Goelzer, M. Reddy, G. Greig, B. Reigner and N. Parrott, AAPS J., 2016, 18, 1453–1463 CrossRef CAS.
-
R. Vardanyan and V. Hruby, in Synthesis of Best-Seller Drugs, ed. R. Vardanyan and V. Hruby, Academic Press, Boston, 2016, pp. 687–736, DOI:10.1016/B978-0-12-411492-0.00034-1.
- R. Dobson, D. Holden, N. Vickaryous, J. Bestwick, K. George, T. Sayali, L. Bianchi, M. Wafa, J. Gold and G. Giovannoni, Mult. Sclerosis, 2024, 30, 63–70 CrossRef CAS.
- C. Pierra, A. Amador, S. Benzaria, E. Cretton-Scott, M. D'Amours, J. Mao, S. Mathieu, A. Moussa, E. G. Bridges, D. N. Standring, J. P. Sommadossi, R. Storer and G. Gosselin, J. Med. Chem., 2006, 49, 6614–6620 CrossRef CAS.
- H. Ma, S. L. Pogam, S. Fletcher, F. Hinojosa-Kirschenbaum, H. Javanbakht, J.-M. Yan, W.-R. Jiang, N. Inocencio, K. Klumpp and I. Nájera, Antimicrob. Agents Chemother., 2014, 58, 2614–2625 CrossRef CAS.
- I. Gentile, N. Coppola, A. R. Buonomo, E. Zappulo and G. Borgia, Expert Opin. Invest. Drugs, 2014, 23, 1211–1223 CrossRef CAS.
- D. R. Martinez, F. R. Moreira, N. J. Catanzaro, M. V. Diefenbacher, M. R. Zweigart, K. L. Gully, G. De la Cruz, A. J. Brown, L. E. Adams, B. Yount, T. J. Baric, M. L. Mallory, H. Conrad, S. R. May, S. Dong, D. T. Scobey, C. Nguyen, S. A. Montgomery, J. K. Perry, D. Babusis, K. T. Barrett, A. H. Nguyen, A. Q. Nguyen, R. Kalla, R. Bannister, J. Y. Feng, T. Cihlar, R. S. Baric, R. L. Mackman, J. P. Bilello, A. Schäfer and T. P. Sheahan, Sci. Transl. Med., 2024, 16, eadj4504 CrossRef CAS PubMed.
- K. W. Zhu, ACS Pharmacol. Transl. Sci., 2023, 6, 1306–1309 CrossRef CAS PubMed.
- National Center for Biotechnology Information, PubChem Compound Summary for CID 483477, Brincidofovir, 2025, Retrieved from https://pubchem.ncbi.nlm.nih.gov/compound/Brincidofovir Search PubMed.
- M. J. Tong and S. S. Tu, Semin. Liver Dis., 2004, 24(Suppl 1), 37–44 CrossRef CAS.
- Y. Gao, F. Kong, X. Song, J. Shang, L. Yao, J. Xia, Y. Peng, W. Liu, H. Gong, M. Mu, H. Cui, T. Han, W. Chen, X. Wu, Y. Yang, X. Yan, Z. Jin, P. Wang, Q. Zhu, L. Chen, C. Zhao, D. Zhang, W. Jin, D. Wang, X. Wen, C. Liu, J. Jia, Q. Mao, Y. Ding, X. Jin, Z. Zhang, Q. Mao, G. Li and J. Niu, Clin. Infect. Dis., 2022, 74, 1925–1932 CrossRef CAS.
- K. R. Reddy, M. C. Matelich, B. G. Ugarkar, J. E. Gómez-Galeno, J. DaRe, K. Ollis, Z. Sun, W. Craigo, T. J. Colby, J. M. Fujitaki, S. H. Boyer, P. D. van Poelje and M. D. Erion, J. Med. Chem., 2008, 51, 666–676 CrossRef CAS.
- C. Wassner, N. Bradley and Y. Lee, J. Int. Assoc. Provid. AIDS Care, 2020, 19 DOI:10.1177/2325958220919231.
- J. E. Song and J. Y. Park, Expert Opin. Pharmacother., 2021, 22, 2427–2433 CrossRef CAS PubMed.
- H. Kwakwa, J. Bran, J. Ruff, S. Sharaf, H. Seung, S. Choe and J. V. Chua, Viruses, 2025, 17, 510 CrossRef CAS PubMed.
- Y. S. Lim, M. L. Yu, J. Choi, C. Y. Chen, W. M. Choi, W. Kang, G. A. Kim, H. J. Kim, Y. B. Lee, J. H. Lee, N. H. Park, S. Y. Kwon, S. Y. Park, J. H. Kim, G. H. Choi, E. S. Jang, C. H. Chen, Y. C. Hsu, M. J. Bair, P. N. Cheng, H. D. Tung, T. S. Chang, C. C. Lo, K. C. Tseng, S. S. Yang, C. Y. Peng and S. Han, Lancet Gastroenterol. Hepatol., 2025, 10, 295–305 CrossRef CAS.
- M. Gräfe, M. S. Schäfer, C. Leithner, K. Allers, T. Schneider, E. E. A. Algharably, R. Kreutz and T. G. Riemer, Pol. Arch. Med. Wewn., 2024, 134, 16861 Search PubMed.
- F.-R. Alexandre, E. Badaroux, J. P. Bilello, S. Bot, T. Bouisset, G. Brandt, S. Cappelle, C. Chapron, D. Chaves, T. Convard, C. Counor, D. Da Costa, D. Dukhan, M. Gay, G. Gosselin, J.-F. Griffon, K. Gupta, B. Hernandez-Santiago, M. La Colla, M.-P. Lioure, J. Milhau, J.-L. Paparin, J. Peyronnet, C. Parsy, C. Pierra Rouvière, H. Rahali, R. Rahali, A. Salanson, M. Seifer, I. Serra, D. Standring, D. Surleraux and C. B. Dousson, Bioorg. Med. Chem. Lett., 2017, 27, 4323–4330 CrossRef CAS PubMed.
- K. E. Squires, D. L. Mayers, G. R. Bluemling, A. A. Kolykhalov, D. B. Guthrie, P. Reddy, D. G. Mitchell, M. T. Saindane, Z. M. Sticher, V. Edpuganti and A. D. L. Rosa, Antimicrob. Agents Chemother., 2020, 64 DOI:10.1128/aac.00836-20.
- R. T. Eastman, J. S. Roth, K. R. Brimacombe, A. Simeonov, M. Shen, S. Patnaik and M. D. Hall, ACS Cent. Sci., 2020, 6, 672–683 CrossRef PubMed.
- S. S. Good, J. Westover, K. H. Jung, X. J. Zhou, A. Moussa, P. La Colla, G. Collu, B. Canard and J. P. Sommadossi, Antimicrob. Agents Chemother., 2021, 65 DOI:10.1128/aac.02479-20.
- A. M. Lam, C. Espiritu, S. Bansal, H. M. Micolochick Steuer, V. Zennou, M. J. Otto and P. A. Furman, J. Virol., 2011, 85, 12334–12342 CrossRef PubMed.
- P. G. Reddy, D. Bao, W. Chang, B. K. Chun, J. Du, D. Nagarathnam, S. Rachakonda, B. S. Ross, H. R. Zhang, S. Bansal, C. L. Espiritu, M. Keilman, A. M. Lam, C. Niu, H. M. Steuer, P. A. Furman, M. J. Otto and M. J. Sofia, Bioorg. Med. Chem. Lett., 2010, 20, 7376–7380 CrossRef PubMed.
- J. T. Witkowski, R. K. Robins, G. P. Khare and R. W. Sidwell, J. Med. Chem., 1973, 16, 935–937 CrossRef PubMed.
- R. L. Murphy, N. M. Kivel, C. Zala, C. Ochoa, P. Tharnish, J. Mathew, M. L. Pascual and R. F. Schinazi, Antiviral Ther., 2010, 15, 185–192 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Tuniyazi
Abuduani
,
Mahesh
Kasthuri
,
Zhe
Chen
,
Zahira
Tber
,
Mohammed
Loubidi
,
HongWang
Zhang
,
Longhu
Zhou
,
Shaoman
Zhou
,
Chenwei
Li
,
Amita
Kumari
,
Sijia
Tao
,
John M.
Wiseman
,
Selwyn J.
Hurwitz
,
Franck
Amblard
* and
Raymond F.
Schinazi
,
Tuniyazi
Abuduani
,
Mahesh
Kasthuri
,
Zhe
Chen
,
Zahira
Tber
,
Mohammed
Loubidi
,
HongWang
Zhang
,
Longhu
Zhou
,
Shaoman
Zhou
,
Chenwei
Li
,
Amita
Kumari
,
Sijia
Tao
,
John M.
Wiseman
,
Selwyn J.
Hurwitz
,
Franck
Amblard
* and
Raymond F.
Schinazi


 Valacyclovir (Valtrex)
Valacyclovir (Valtrex) Valganciclovir
Valganciclovir
 Famciclovir (Famvir)
Famciclovir (Famvir)
 Valopicitabine (NM-283)
Valopicitabine (NM-283)
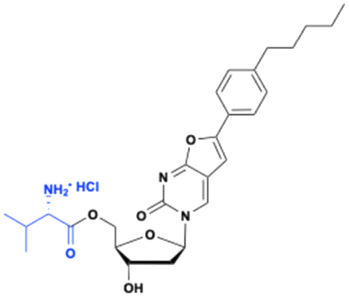 Valnivudine hydrochloride (FV-100)
Valnivudine hydrochloride (FV-100) Molnupiravir (Lagevrio)
Molnupiravir (Lagevrio)
 Lumicitabine (ALS-8176)
Lumicitabine (ALS-8176)
 Mericitabine
Mericitabine
 Balapiravir (R1626)
Balapiravir (R1626)
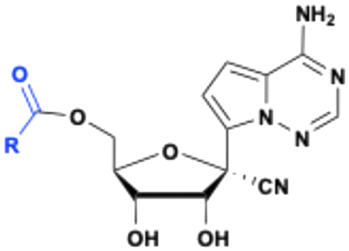 Obeldesivir (GS-5245, ATV006)
Obeldesivir (GS-5245, ATV006)
 Deuremidevir (VV116)
Deuremidevir (VV116)
 Brincidofovir (CMX001, Tembexa)
Brincidofovir (CMX001, Tembexa)
 Adefovir dipivoxil (Hepsera)
Adefovir dipivoxil (Hepsera)
 Pradefovir mesylate (MB-06886)
Pradefovir mesylate (MB-06886)
 Tenofovir disoproxil fumarate (Viread, TDF)
Tenofovir disoproxil fumarate (Viread, TDF)
 Besifovir dipivoxil maleate (LB80380)
Besifovir dipivoxil maleate (LB80380)
 Tenofovir alafenamide (Vemlidy, TAF)
Tenofovir alafenamide (Vemlidy, TAF)
 BMS-986094 (INX-08189)
BMS-986094 (INX-08189)
 IDX-184
IDX-184
 ALS-2200 (epimeric mixture)
ALS-2200 (epimeric mixture) Sofosbuvir (Sovaldi)
Sofosbuvir (Sovaldi)
 Uprifosbuvir (MK-3682)
Uprifosbuvir (MK-3682)
 ATI-2173
ATI-2173
 AL-335 (JNJ-4178, and adafosbuvir)
AL-335 (JNJ-4178, and adafosbuvir)
 Remdesivir (Veklury)
Remdesivir (Veklury)
 Bemnifosbuvir (BEM; AT-527)
Bemnifosbuvir (BEM; AT-527)
 AT-752 (Rp-diastereomer)
AT-752 (Rp-diastereomer)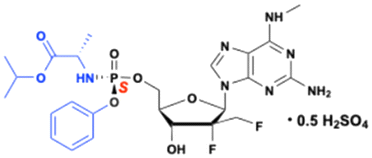 2′-α-Fluoro-2′-β-C-(fluoromethyl)guanosine
2′-α-Fluoro-2′-β-C-(fluoromethyl)guanosine
 PSI-352938 (PSI-938, GS-0938)
PSI-352938 (PSI-938, GS-0938)
 Taribavirin/hydrochloride (Viramidine)
Taribavirin/hydrochloride (Viramidine)
 Abacavir
Abacavir
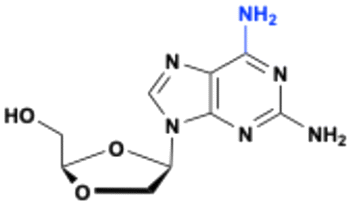 Amdoxovir/DAPD
Amdoxovir/DAPD






























