DOI:
10.1039/D6MA00182C
(Paper)
Mater. Adv., 2026,
7, 4307-4328
Computational evaluation of pressure effects on cubic ferromagnetic perovskites ACrBr3 (A = K, Rb, Cs, Fr): materials engineering perspectives for spintronics and optoelectronics via DFT
Received
8th February 2026
, Accepted 13th March 2026
First published on 13th March 2026
Abstract
In this study, the pressure-dependent properties of cubic ferromagnetic metal halide single perovskites ACrBr3 (A = K, Rb, Cs, Fr) were systematically investigated using density functional theory (DFT). The tolerance factor and octahedral factor indicate that the investigated compounds are stable in a cubic structure having the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m(221) space group. Structural analysis shows that both unit cell volume and lattice constants decrease with the increasing external hydrostatic pressure range (0–30 GPa). Mechanical stability is confirmed through Born stability criteria, and pressure enhances the elastic constants, further supporting their robustness for device integration. Spin-polarized band structure and density of states (DOS) analyses confirm the half-metallicity, where the spin-up state is metallic and the spin-down state exhibits a band gap. Our studied materials have 100% spin polarization at the Fermi level. Pressure decreases the band gap for all of our investigated materials. The total magnetic moment in all compounds remains approximately an integer value, confirming their suitability for spintronic applications. Thermodynamic parameters, including Debye temperature and melting temperature, exhibit an upward trend with increasing pressure, indicating enhanced stability. Optical properties such as dielectric constants, conductivity, absorption coefficient, reflectivity, refractive index, and loss functions of our studied materials show the highest intensity in the UV region and increase with the applied pressures. These findings highlight the multifunctional potential of ACrBr3 (A = K, Rb, Cs, Fr) perovskites in next-generation spintronic and optoelectronic devices.
m(221) space group. Structural analysis shows that both unit cell volume and lattice constants decrease with the increasing external hydrostatic pressure range (0–30 GPa). Mechanical stability is confirmed through Born stability criteria, and pressure enhances the elastic constants, further supporting their robustness for device integration. Spin-polarized band structure and density of states (DOS) analyses confirm the half-metallicity, where the spin-up state is metallic and the spin-down state exhibits a band gap. Our studied materials have 100% spin polarization at the Fermi level. Pressure decreases the band gap for all of our investigated materials. The total magnetic moment in all compounds remains approximately an integer value, confirming their suitability for spintronic applications. Thermodynamic parameters, including Debye temperature and melting temperature, exhibit an upward trend with increasing pressure, indicating enhanced stability. Optical properties such as dielectric constants, conductivity, absorption coefficient, reflectivity, refractive index, and loss functions of our studied materials show the highest intensity in the UV region and increase with the applied pressures. These findings highlight the multifunctional potential of ACrBr3 (A = K, Rb, Cs, Fr) perovskites in next-generation spintronic and optoelectronic devices.
1. Introduction
Spintronics has emerged as an important research domain in modern condensed-matter and device physics, as it offers an alternative pathway to conventional charge-based electronics by utilizing electron spin.1,2 Recently, it has been found that half metals, a novel class of materials, are intriguing options in the area of spintronics. A half-metallic material exhibits metallic characteristics for one spin state while displaying semiconductor or insulator behavior for the opposite spin state. Therefore, we can classify these materials as hybrid materials due to the distinct electronic properties associated with the two spin states. Spin-polarized currents eliminate thermal challenges that integrated circuits and other electronic equipment frequently face, since they do not lose energy as heat.3 NiMnSb and PtMnSb half-Heusler materials are the first half-metallic ferromagnets discovered by Groot et al.4 Afterwards, diverse structures of various half-metallic ferromagnetic materials were investigated, such as spinels MgNiX4,5 single perovskites PrMnO36 and BaNpO3,7 double perovskites Cs2AgMBr68 and Ba2XNbO6,9 Cr doped TiO2,10 half-Heusler alloy CoTcSn,11 ternary half-Heusler alloy GeNaZ,12 full-Heusler alloy CoNiMnSi,13 and zinc blende CrSb.14 Transition metal-based cubic perovskites show remarkable half-metallic ferromagnetic properties.
The typical formula for perovskite materials is ABX3, where A and B are cations, while X is an oxygen or a halogen. The A site is occupied by an alkali or alkaline-earth metal, whereas the B site is typically filled by transition-metal or f-block elements.15 Due to their novel chemical formulations, crystal structures, and excellent magnetic and optoelectronic features, metal halide perovskites (MHPs) have recently gained a lot of attention and have been considered for various uses, including photovoltaics, photonics, light-emitting diodes, and light-controlling magnetic devices and photonics.16–19 Lead-based perovskites are disadvantageous for massive applications due to toxicity and instability in the presence of heat, light, and moisture, although they have potential uses.20 As a result, materials based on lead-free perovskites such as A2BBiCl621 and X2YBiCl622 have become particularly desirable.
Materials' properties can be tailored through the application of various external control parameters, enabling systematic modulation of their functional behavior.23 Researchers have been showing particular interest in the pressure effect on halide perovskites24,25 as the influence of pressure on the structural, optical, mechanical, and electrical characteristics of materials is significant. Higher applied pressure compresses the crystal lattice and shrinks the unit-cell volume, which causes the electronic band gap to become smaller, as observed in various papers.26 It is possible to change the band gap from indirect to direct under hydrostatic pressure.27
One of the most effective methods for accurately conducting theoretical research on the physical and chemical properties of a significant number of condensed matter systems is via the application of first principles studies.28 The present study provides a theoretical investigation of transition-metal-based halide single perovskites ACrBr3 (A = K, Rb, Cs, Fr) over the pressure range of 0–30 GPa (0, 10, 20, and 30 GPa) for potential applications in optoelectronics and spintronics.
Our results match those previously published by Alburaih et al. for KCrBr329 and Ullah et al. for RbCrBr330 at 0 GPa. This similarity indicates that the computational method used in this work is reliable, confirming the validity of our present study. What makes our research unique is that it looks at how pressure affects the different properties of our materials for possible applications in multiple fields. B-site elements dominate the electronic behavior, as the compound retains its half-metallicity regardless of whether the A-site is occupied by K, Rb, Cs, or Fr.
2. Computational details
We utilized density functional theory (DFT) based first-principles methods implemented in the Cambridge Serial Total Energy Package (CASTEP), where the plane-wave pseudopotential approach was applied for all calculations.31 OTFG ultrasoft pseudopotentials were employed for addressing the charged particle interactions.32 Geometry optimization was performed with convergence criteria: an energy tolerance of 2 × 10−5 eV per atom, a maximum force of 0.05 eV Å−1, a maximum stress of 0.1 GPa, a maximum displacement of 0.002 Å, and maximum iterations of 1000 using the BFGS (Broyden–Fletcher–Goldfarb–Shanno) algorithm.33 The Monkhorst–Pack scheme was utilized for sampling k-points in the Brillouin zone. We used a 400 eV plane-wave cutoff and a 4 × 4 × 4 k-point mesh for all of our calculations throughout our investigations. For evaluating exchange–correlation energy, the spin-polarized GGA functional was implemented using the Perdew–Berke–Ernzerhof (PBE) approach for structural, electrical, magnetic, and optical properties.34,35 To obtain more accurate electronic and magnetic results, we use the GGA+U approximation, which introduces a Hubbard U term to account for the localized Coulomb interactions among strongly correlated electrons. Elastic and thermal properties were calculated using the GGA-PBEsol functional for refined reliability.36
3. Results and discussion
3.1 Structural features
Our investigated ACrBr3 (A = K, Rb, Cs, Fr) materials are all transition metal-based cubic perovskites having a space group Pmm (international number 221) and 5 atoms in the formula unit, in which A site atoms are positioned at K/Rb/Cs/Fr at (0,0,0), Cr is at 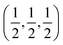 and Br is at
and Br is at 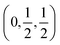 Wyckoff atomic Positions. The polyhedral view of our studied compounds is shown in Fig. 1(a). Convergence tests were performed using 4 × 4 × 4, 6 × 6 × 6, and 8 × 8 × 8 k-point meshes and cutoff energies from 300 to 600 eV, as shown in Fig. 1(b). Since the energy change was very small, and the lowest energy was obtained at 4 × 4 × 4, a cutoff energy of 400 eV with a 4 × 4 × 4 k-point mesh was used for all calculations. Table 1 demonstrates the lattice parameters, unit cell volume, and energy both under spin-polarized and non-spin-polarized conditions using the GGA functional at 0, 10, 20, and 30 GPa pressures (Table 2).
Wyckoff atomic Positions. The polyhedral view of our studied compounds is shown in Fig. 1(a). Convergence tests were performed using 4 × 4 × 4, 6 × 6 × 6, and 8 × 8 × 8 k-point meshes and cutoff energies from 300 to 600 eV, as shown in Fig. 1(b). Since the energy change was very small, and the lowest energy was obtained at 4 × 4 × 4, a cutoff energy of 400 eV with a 4 × 4 × 4 k-point mesh was used for all calculations. Table 1 demonstrates the lattice parameters, unit cell volume, and energy both under spin-polarized and non-spin-polarized conditions using the GGA functional at 0, 10, 20, and 30 GPa pressures (Table 2).
 |
| | Fig. 1 (a) Polyhedral view of the unit cell and (b) cutoff energy, k-point convergence test of the ACrBr3 (A = K, Rb, Cs, Fr) perovskites. | |
Table 1 Optimized lattice constants a (Å), equilibrium volume V0 (Å3), energy, EFM (eV), energy, EAFM (eV), energy ENM (eV) and normalized lattice parameters calculated using the GGA functional at 0, 10, 20 and 30 GPa pressures of ACrBr3 (A = K, Rb, Cs, Fr) compounds
| Compounds |
Parameters |
0 GPa |
10 GPa |
20 GPa |
30 GPa |
| KCrBr3 |
Lattice constant a (Å) |
5.31 |
4.94 |
4.76 |
4.63 |
| Non-spin polarized (Å) |
5.17, 5.1829 |
4.86 |
4.68 |
4.56 |
| Cell volume, V0 (FM) (Å3) |
149.52 |
120.37 |
107.80 |
99.24 |
| Cell volume, V0 (NM) (Å3) |
138.45 |
115.13 |
102.41 |
94.74 |
| Energy, EFM (eV) |
−4586.98 |
−4586.34 |
−4585.24 |
−4583.94 |
| Energy, EAFM (eV) |
−4586.95 |
−4586.33 |
−4585.19 |
−4583.87 |
| Energy, ENM (eV) |
−4584.60 |
−4584.27 |
−4583.15 |
−4581.97 |
| Normalized lattice constants a/a0 |
1 |
0.93 |
0.90 |
0.87 |
| Normalized volume V/V0 |
1 |
0.81 |
0.72 |
0.66 |
|
|
| RbCrBr3 |
Lattice constant a (Å) |
5.34 |
4.96 |
4.78 |
4.65 |
| Non-spin polarized a/b/c (Å) |
5.21, 5.1730 |
4.89 |
4.70 |
4.58 |
| Cell volume, V0 (FM) (Å3) |
152.52, 138.5530 |
122.38 |
109.19 |
100.74 |
| Cell volume, V0 (NM) (Å3) |
141.56 |
116.58 |
103.99 |
96.26 |
| Energy, EFM (eV) |
−4486.19 |
−4485.51 |
−4484.34 |
−4483.04 |
| Energy, EAFM (eV) |
−4486.17 |
−4485.48 |
−4484.29 |
−4482.98 |
| Energy, ENM (eV) |
−4483.79 |
−4483.38 |
−4482.25 |
−4481.06 |
| Normalized lattice constants a/a0 |
1 |
0.93 |
0.90 |
0.87 |
| Normalized volume V/V0 |
1 |
0.80 |
0.72 |
0.66 |
|
|
| CsCrBr3 |
Lattice constant a (Å) |
5.40 |
5.01 |
4.82 |
4.70 |
| Non-spin polarized a/b/c (Å) |
5.28 |
4.94 |
4.75 |
4.63 |
| Cell volume, V0 (FM) (Å3) |
157.57 |
126.02 |
112.45 |
103.80 |
| Cell volume, V0 (NM) (Å3) |
147.02 |
119.53 |
107.28 |
99.21 |
| Energy, EFM (eV) |
−4570.77 |
−4570.05 |
−4568.85 |
−4567.53 |
| Energy, EAFM (eV) |
−4570.75 |
−4570.02 |
−4568.80 |
−4567.45 |
| Energy, ENM (eV) |
−4568.31 |
−4567.76 |
−4566.72 |
−4565.47 |
| Normalized lattice constants a/a0 |
1 |
0.93 |
0.89 |
0.87 |
| Normalized volume V/V0 |
1 |
0.80 |
0.71 |
0.66 |
|
|
| FrCrBr3 |
Lattice constant a (Å) |
5.43 |
5.05 |
4.86 |
4.73 |
| Non-spin polarized a/b/c (Å) |
5.32 |
4.96 |
4.78 |
4.66 |
| Cell volume, V0 (FM) (Å3) |
160.53 |
128.43 |
114.52 |
105.68 |
| Cell volume, V0 (NM) (Å3) |
150.54 |
121.99 |
109.40 |
101.08 |
| Energy, EFM (eV) |
−4990.58 |
−4989.86 |
−4988.62 |
−4987.27 |
| Energy, EAFM (eV) |
−4990.55 |
−4989.82 |
−4988.58 |
−4987.21 |
| Energy, ENM (eV) |
−4988.09 |
−4987.52 |
−4986.46 |
−4985.19 |
| Normalized lattice constants a/a0 |
1 |
0.93 |
0.90 |
0.87 |
| Normalized volume V/V0 |
1 |
0.80 |
0.71 |
0.66 |
Table 2 Formation and cohesive energies calculated at applied pressures of 0, 10, 20, and 30 GPa for ACrBr3 (A = K, Rb, Cs, Fr) compounds
| Energy, (eV) |
Compounds |
Pressure |
| 0 GPa |
10 GPa |
20 GPa |
30 GPa |
|
E
Formation
|
KCrBr3 |
−2.75, −2.6529 |
−3.39 |
−2.42 |
−2.44 |
| RbCrBr3 |
−2.77, −2.2130 |
−3.46 |
−2.49 |
−2.48 |
| CsCrBr3 |
−2.80 |
−3.52 |
−2.45 |
−2.34 |
| FrCrBr3 |
−2.76 |
−3.47 |
−2.47 |
−2.36 |
|
|
|
E
Cohesive
|
KCrBr3 |
12.96 |
10.53 |
11.96 |
12.02 |
| RbCrBr3 |
12.64 |
11.62 |
12.02 |
12.52 |
| CsCrBr3 |
12.78 |
11.5 |
11.79 |
11.76 |
| FrCrBr3 |
12.84 |
11.14 |
11.79 |
11.82 |
All four examined perovskites reveal a ferromagnetic state with the lowest energy, which makes it energetically more favorable than both the antiferromagnetic and non-magnetic states shown in Table 1. The apparent rise in cell volume from the non-magnetic to magnetic condition represents a magnetoelastic effect that influences the material's structural characteristics.15 Pressure decreases the lattice constants and lattice volume, as seen from Table 1 and Fig. 2(a) and (b). Normalized lattice parameters and normalized volume are also presented in Fig. 2(d) and (e) to better understand the pressure effect among them. Any deviance from the optimal cubic structure can be assessed by the tolerance factor (t) that typically ranges from 0.81 to 1.11 for cubic perovskites.37 The Goldschmidt tolerance factor, t, is given as,
| | 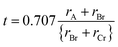 | (1) |
where
rA,
rBr, and
rCr represent the atomic radii of the A-site atom, Br atom, and Cr atom, respectively, which are calculated by using Shannon's effective ionic radii, and the values of
t are depicted in
Table 3. These values are close to unity, suggesting that the compounds are geometrically compatible with the cubic perovskite structure, although slight deviations may indicate minor structural distortions. In addition, the octahedral factor is determined using the following formula,
2 and
Table 3 shows that its values fall within the expected range.
| | 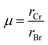 | (2) |
The formation energies of ACrBr
3 are calculated to make sure that they might be fabricated experimentally using the following expression,
38| | 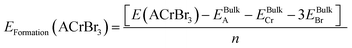 | (3) |
| | 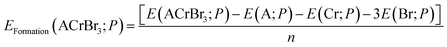 | (4) |
Here
E(ACrBr
3) denotes the total energy of our compound and
EBulkA,
EBulkCr, and
EBulkBr represent the total atomic energies of A, Cr, and Br atoms under bulk conditions, which are determined from the structural optimization calculations. The determined negative values of formation energies for all of our compounds ensure synthesizability and that our materials are energetically favorable relative to their isolated constituent elements.
39 The expression for cohesive energy is given as:
12| | 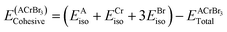 | (5) |
Here,
EAiso,
ECriso and
EBriso represent the isolated energies of atom A, Cr, and Br, respectively, and
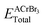
is the total energy of ACrBr
3 calculated from the structural optimization. Our calculated cohesive energy ranges from 10.53 to 12.84 eV. Such large values ensure the chemical stability of our investigated compound. The calculated Mulliken charges, bond lengths, and bond populations of ACrBr
3 (A = K, Rb, Cs, Fr) from the GGA method are presented in
Table 3 and
Fig. 3, 4.
 |
| | Fig. 2 Pressure-dependent variation of (a) lattice constants a (Å), (b) volume V0 (Å3), (c) total energies (eV), (d) normalized lattice constants, and (e) normalized volume for ACrBr3 (A = K, Rb, Cs, Fr) calculated using the GGA-PBE functional at pressures of 0, 10, 20, and 30 GPa. | |
Table 3 Tolerance factor (t), octahedral factor (μ), bond lengths (Å), bond populations and Mulliken charges (electrons) of ACrBr3 (A = K, Rb, Cs, Fr) from the GGA-PBE functional at 0 GPa
| Compounds |
Tolerance factor (t) |
Octahedral factor (μ) |
Bond |
Population |
Bond lengths (Å) |
Species |
s
|
p
|
d
|
Total |
Charge (electrons) |
| KCrBr3 |
0.99 |
0.52 |
Cr–Br |
−0.43 |
2.65 |
K |
2.29 |
6.66 |
0 |
8.94 |
0.06 |
|
|
|
|
Cr |
2.65 |
6.91 |
4.71 |
14.27 |
−0.27 |
|
|
|
|
Br |
1.53 |
5.40 |
0 |
6.93 |
0.07 |
|
|
| RbCrBr3 |
1.01 |
0.52 |
Cr–Br |
−0.47 |
2.67 |
Rb |
2.28 |
6.61 |
0 |
8.89 |
0.11 |
|
|
|
|
Cr |
2.61 |
6.91 |
4.70 |
14.22 |
−0.22 |
|
|
|
|
Br |
1.56 |
5.41 |
0 |
6.96 |
0.04 |
|
|
| CsCrBr3 |
1.05 |
0.52 |
Cr–Br |
−0.26 |
2.70 |
Cs |
2.28 |
6.49 |
0 |
8.77 |
0.23 |
|
|
|
|
Cr |
2.57 |
6.88 |
4.68 |
14.14 |
−0.14 |
|
|
|
|
Br |
1.61 |
5.42 |
0 |
7.03 |
−0.03 |
|
|
| FrCrBr3 |
1.07 |
0.52 |
Cr–Br |
−0.21 |
2.72 |
Fr |
2.26 |
6.43 |
0 |
8.69 |
0.31 |
|
|
|
|
Cr |
2.56 |
6.87 |
4.67 |
14.10 |
−0.10 |
|
|
|
|
Br |
1.65 |
5.42 |
0 |
7.07 |
−0.07 |
 |
| | Fig. 3 Variation of cohesive energy for ACrBr3 (A = K, Rb, Cs, Fr) compounds under applied pressures of 0, 10, 20, and 30 GPa. | |
 |
| | Fig. 4 Pressure-dependent variation of bond length (Å) for ACrBr3 (A = K, Rb, Cs, Fr) perovskites calculated using the GGA-PBE functional. | |
3.2 Elastic and mechanical characteristics
In materials science, elastic constants are vital factors in determining if a crystal structure is mechanically stable, along with knowing other crucial mechanical properties involving brittleness, ductility, stiffness, and anisotropy. The elastic energy in a cubic lattice can be written using the following expression, as it has only distinct elastic constants (C11, C12 and C44):40| | 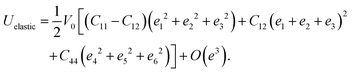 | (6) |
Here V0 is the unstrained volume and O(e3) is the strain order e3 or higher. Only when this energy becomes positive, the crystal structure has dynamical stability. For this reason, Born stability requirements, which are (C11 − C12) > 0; C11 + 2C12 > 0; C44 > 0; C11 > 0, must be followed for a cubic crystal structure to become a mechanically stable structure.41 All our studied compounds are mechanically stable, as shown in Table 4. We also analyzed these constants across various pressures, with the goal of better understanding the impact of pressure on various mechanical properties, to know the dynamical stability of our compounds, which are shown in Table 4. The pressure-induced stability condition is:42C11 − C12 − 2P > 0; C12 + 2C12 > 0; C44 − P > 0. The pressure-induced stability criteria are not satisfied for KCrBr3 at 30 GPa. However, the other compounds within the applied pressure range maintain dynamical stability.
Table 4 Elastic stiffness constants Cij (GPa), Born stability, and dynamical stability criteria of ACrBr3 (A = K, Rb, Cs, Fr) compounds using the GGA-PBEsol functional at applied pressures of 0, 10, 20, and 30 GPa
| Compounds |
Pressure, P |
Elastic stiffness constants |
Born stability |
Dynamical stability |
|
C
11
|
C
12
|
C
44
|
C
11 − C12 > 0 |
C
11 + 2C12 > 0 |
C
11 > 0 |
C
44 > 0 |
C
11 − C12 − 2P > 0 |
C
11 + 2C12 > 0 |
C
44 − P > 0 |
| KCrBr3 |
0 |
61.98, 64.2129 |
18.12, 42.3029 |
14.20, 3.9829 |
43.86 |
98.22 |
61.98 |
14.20 |
43.86 |
98.22 |
14.2 |
| 10 |
145.91 |
38.17 |
19.65 |
107.74 |
222.25 |
145.91 |
19.65 |
87.74 |
222.25 |
9.65 |
| 20 |
217.82 |
59.62 |
24.5 |
158.2 |
337.06 |
217.82 |
24.5 |
118.2 |
337.06 |
4.5 |
| 30 |
279.96 |
77.64 |
27.16 |
202.32 |
435.24 |
279.96 |
27.16 |
142.32 |
435.24 |
−2.84 |
|
|
| RbCrBr3 |
0 |
59.79 |
16.23 |
13.91 |
43.56 |
92.25 |
59.79 |
13.91 |
43.56 |
92.25 |
13.91 |
| 10 |
148.84 |
40.14 |
22.49 |
108.7 |
229.12 |
148.84 |
22.49 |
88.7 |
229.12 |
12.49 |
| 20 |
220.26 |
61.85 |
30.77 |
158.41 |
343.96 |
220.26 |
30.77 |
118.41 |
343.96 |
10.77 |
| 30 |
287.59 |
80.29 |
35.63 |
207.3 |
448.17 |
287.59 |
35.63 |
147.3 |
448.17 |
5.63 |
|
|
| CsCrBr3 |
0 |
60.04 |
17.96 |
17.51 |
42.08 |
95.96 |
60.04 |
17.51 |
42.08 |
95.96 |
17.51 |
| 10 |
145.06 |
41.85 |
28.16 |
103.21 |
228.76 |
145.06 |
28.16 |
83.21 |
228.76 |
18.16 |
| 20 |
219.61 |
63.33 |
38.79 |
156.28 |
346.27 |
219.61 |
38.79 |
116.28 |
346.27 |
18.79 |
| 30 |
287.59 |
84.17 |
47.36 |
203.42 |
455.93 |
287.59 |
47.36 |
143.42 |
455.93 |
17.36 |
| FrCrBr3 |
0 |
58.65 |
19.13 |
18.5 |
39.52 |
96.91 |
58.65 |
18.5 |
39.52 |
96.91 |
18.5 |
| 10 |
140.76 |
44.24 |
31.56 |
96.52 |
229.24 |
140.76 |
31.56 |
76.52 |
229.24 |
21.56 |
| 20 |
215.11 |
66.48 |
42.31 |
148.63 |
348.07 |
215.11 |
42.31 |
108.63 |
348.07 |
22.31 |
| 30 |
281.77 |
86.71 |
52.38 |
195.06 |
455.19 |
281.77 |
52.38 |
135.06 |
455.19 |
22.38 |
Table 4 and Fig. 5 show that all three elastic constants for our studied materials increase with increasing pressure. For technological and industrial purposes, Young's modulus is an essential factor when studying the rigidity of any material.43
| | 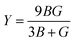 | (7) |
From
Table 5 and
Fig. 6, it is observed that as pressure increases, the value
Y also rises for all materials, meaning our materials become stiffer with the applied pressure. Among the materials we examined, Fr-based perovskites exhibit the highest stiffness as well as structural integrity in the pressure range of 0–30 GPa and can better resist the applied deformation.
| | 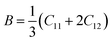 | (8) |
From our calculated values of
B (30.75–32.74 GPa) without applied pressure, it is found that our materials may be used in a thin film solar cell because of their flexibility and softness.
44 The following equations can be utilized to describe
GV and
GR from the Voigt and Reuss assumptions using the elastic constants.
| | 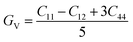 | (9) |
| | 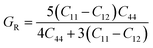 | (10) |
From the Hill
45 concept, we can determine the shear modulus
G of a crystal lattice from
GV and
GR as:
| | 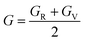 | (11) |
The ability of a material to resist shear deformation is known as the shear modulus. FrCrBr
3 shows the highest shear modulus among our other investigated compounds with and without applied pressure, as seen from
Table 5 and
Fig. 6. Without applied pressure, our materials may be used in flexible electronics. The elastic constants show that, without applied pressure and under most pressures,
Y >
B >
G except for KCrBr
3 and CsCrBr
3 above 10 GPa pressure. So our materials show a greater tendency of linear deformation over volumetric contraction. Due to the increase of
Y,
B, and
G with pressure, our materials become harder with pressure. Using
ν, we can assess the bonding nature of the compound. If
ν = 0.25, then it means that there exists an ionic bond, if
ν > 0.25, then the bond is metallic, and if
ν < 0.25, then a covalent bond is anticipated.
43 K- and Rb-based materials show metallic bonds under all our investigated pressures, whereas Cs- and Fr-based materials show ionic bonds at 0 GPa pressure, which turn into metallic bonds under pressure.
| | 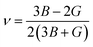 | (12) |
Ductile materials have potential applications in flexible electronics as well as photovoltaic cells, as the ability to sustain mechanical deformation without fracturing is needed.
46 Also, ductility and brittleness can be identified by this parameter. If
ν > 0.26 then the material is ductile. The pressure effect on this value can be visualized from
Table 5 and
Fig. 6, which shows that pressure can enhance the ductility of our studied compounds. The value of Pugh's ratio (
B/
G) tells us whether our studied compounds are ductile or brittle. If this value remains less than 1.75, then the materials are brittle; otherwise, they show a ductile nature. CsCrBr
3 and FrCrBr
3 at 0 GPa pressure show a value less than this value, meaning they are brittle under ambient conditions. With the application of external pressure, they start to show ductility, meaning that pressure increases this ratio significantly.
The Cauchy pressure,
Cp in
eqn (13), is another factor which is also used to differentiate between ductile and brittle materials. From
Table 5, it is found that pressure increases the ductility of our studied materials. All three parameters
ν,
B/
G, and
Cp show a similar ductile or brittle nature. The Zener anisotropy
47 index
A can be used for determining the anisotropy of cubic crystals, that is:
| | 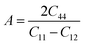 | (14) |
If
A = 1, then the structure is isotropic, meaning the material is the same in all directions, and having any value other than 1 means the material is anisotropic.
48 The value of this parameter gradually drops from unity under applied pressure, which suggests that all of our materials turn more anisotropic. Vickers hardness
HV is calculated using the following formula:
49| | 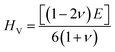 | (15) |
HV has an important role in making cutting instruments, coatings, building materials, and wear-resistant parts and in other industries. A high value of
HV means the material can withstand plastic deformation, and this parameter is increased with the applied pressure for all our investigated materials, as seen from
Fig. 6 and
Table 5. The Kleinman parameter
ζ has the following expression:
| | 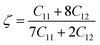 | (16) |
It falls between 0 and 1. If
ζ = 0, the bond is stretching and
ζ = 1 denotes the bending bond. The calculated values of
ζ with changes in pressure are shown in
Table 5. It has a vital role in designing semiconductors with certain optical and electrical characteristics that can be used in LEDs or photonic devices.
50 The machinability index can be calculated from the following relation:
51| | 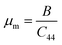 | (17) |
The value of
μm is greater than 2 except for the Cs and Fr based compounds, which remain below 2 at 0 GPa. When the pressure is applied, this value is also enhanced. Exceeding a machinability index of 2 demonstrates outstanding lubrication property and smaller friction and is suitable for industrial uses.
 |
| | Fig. 5 Variation of elastic stiffness constants C11, C12, and C44 (GPa) with pressure using the GGA-PBEsol functional for ACrBr3 (A = K, Rb, Cs, Fr) compounds. | |
Table 5 Various elastic constants calculated using the GGA-PBEsol functional for ACrBr3 (A = K, Rb, Cs, Fr) compounds under applied pressures of 0, 10, 20, and 30 GPa
| Compounds |
Pressure, P |
Young's modulus (Hill) Y |
Bulk modulus (B) |
Shear modulus (Voigt) GV |
Shear modulus (Reuss) GR |
Shear modulus (Hill) (G) |
Cauchy pressure (Cp) |
Kleinman parameter (ζ) |
Anisotropy factor (A) |
Poisson ratio (ν) |
Pugh's ratio (B/G) |
Vickers hardness, HV |
Machinability index, μm |
| KCrBr3 |
0 |
43.28 |
32.74 |
17.29 |
16.53 |
16.91 |
3.92 |
0.44, 0.7529 |
0.65, 0.3629 |
0.28, 0.4429 |
1.94, 8.1929 |
2.48 |
2.31 |
| 10 |
78.93 |
74.08 |
33.34 |
26.34 |
29.84 |
18.52 |
0.411 |
0.36 |
0.32 |
2.48 |
3.53 |
3.77 |
| 20 |
107.49 |
112.35 |
46.34 |
33.84 |
40.09 |
35.12 |
0.42 |
0.31 |
0.34 |
2.8 |
4.26 |
4.59 |
| 30 |
128.67 |
145.08 |
56.76 |
38.39 |
47.58 |
50.48 |
0.43 |
0.27 |
0.35 |
3.05 |
4.69 |
5.34 |
|
|
| RbCrBr3 |
0 |
42.33 |
30.75 |
17.06 |
16.26 |
16.66 |
2.32 |
0.42 |
0.64 |
0.27 |
1.85 |
2.55 |
2.21 |
| 10 |
84.94 |
76.37 |
35.23 |
29.38 |
32.31 |
17.65 |
0.42 |
0.41 |
0.31 |
2.36 |
3.99 |
3.40 |
| 20 |
120.41 |
114.65 |
50.14 |
40.73 |
45.44 |
31.08 |
0.43 |
0.39 |
0.32 |
2.52 |
5.30 |
3.73 |
| 30 |
148.33 |
149.39 |
62.83 |
48.31 |
55.57 |
44.66 |
0.43 |
0.34 |
0.33 |
2.69 |
6.13 |
4.19 |
|
|
| CsCrBr3 |
0 |
47.26 |
31.99 |
18.92 |
18.77 |
18.85 |
0.45 |
0.45 |
0.83 |
0.25 |
1.70 |
3.09 |
1.83 |
| 10 |
93.26 |
76.25 |
37.54 |
34.41 |
35.98 |
13.69 |
0.44 |
0.54 |
0.30 |
2.12 |
4.89 |
2.71 |
| 20 |
134.62 |
115.42 |
54.53 |
48.57 |
51.55 |
24.54 |
0.44 |
0.50 |
0.31 |
2.24 |
6.68 |
2.98 |
| 30 |
169.90 |
151.98 |
69.1 |
60.23 |
64.67 |
36.81 |
0.44 |
0.47 |
0.31 |
2.35 |
8.03 |
3.21 |
|
|
| FrCrBr3 |
0 |
47.64 |
32.30 |
19.00 |
18.98 |
18.99 |
0.63 |
0.47 |
0.94 |
0.25 |
1.70 |
3.11 |
1.75 |
| 10 |
96.54 |
76.41 |
38.24 |
36.63 |
37.44 |
12.68 |
0.46 |
0.65 |
0.29 |
2.04 |
5.26 |
2.42 |
| 20 |
138.25 |
116.02 |
55.11 |
51.11 |
53.11 |
24.17 |
0.46 |
0.57 |
0.30 |
2.18 |
7.03 |
2.74 |
| 30 |
176.03 |
151.73 |
70.44 |
64.28 |
67.36 |
34.33 |
0.45 |
0.54 |
0.31 |
2.25 |
8.68 |
2.90 |
 |
| | Fig. 6 Calculated various elastic moduli and their variation with pressure (GPa) using the GGA-PBEsol functional for ACrBr3 (A = K, Rb, Cs, Fr). | |
3.3 Electronic characteristics
Electronic property analysis plays an important role in determining the applicability of the halide perovskites in various fields. Spin-polarized band structures, density of states (DOS), and partial density of states (PDOS) have been investigated for all of our materials over a pressure range of 0 to 30 eV. Fig. 7 presents our examined band structures in both spin-up and spin-down states along the high symmetry points G–F–Q–Z–G in the Brillouin zone using both GGA and GGA+U functionals. The band gap values of d/f electron-based systems are underestimated by GGA. We have used GGA+U, where the Hubbard potential is 2.50 eV for the Cr atom.52 The band gap value is enhanced by including the U parameter, which takes it significantly closer to the experimental values.53 The Fermi level EF is set at 0 eV in the horizontal line. The conduction band is situated above this line, whereas the valence band is situated below. In spin-up states (↑), all of our studied materials show metallic nature, as there exist some bands in the Fermi level in both the GGA and GGA+U functionals. In the spin down state (↓), there exists a band gap Eg between the valence and conduction bands, and the band gap values are shown in Table 6 and Fig. 7. Our studied materials over the pressure range can be classified as half metals because of their two different nature in spin up and down states. In the spin down state, the valence band maximum (VBM) and conduction band minimum (CBM) lie at points Q and G, respectively, so our studied compounds possess an indirect band gap for all investigated pressure ranges. Pressure has a vital effect on the band structure. From Fig. 8, with the applied pressure, all of our materials show a decrease in band gap with the applied pressure. At 30 GPa, all of our materials exhibit metallic behavior in the spin-up channel and a wide-band-gap semiconducting nature in the spin-down channel, thereby maintaining half-metallicity, which is useful in electronic and optoelectronic applications for their high optical phonon energies. The half-metallic gap is defined as the minimum energy difference between the Fermi level and the nearest band edge, either the valence band maximum or the conduction band minimum.54 Mathematically, EHM = min[(EF − EVBM), (ECBM − EF)], where EF is the Fermi level, EVBM is the valence band maximum, ECBM is the conduction band minimum and EHM is the lowest quantity of energy needed to create holes in the minority spin channel and create spin excitation.15 According to Table 6 our calculated half-metallic gap is large enough for spintronic applications55 like spin field-effect transistors, magnetoresistive-based devices, and racetrack memory.56
 |
| | Fig. 7 Spin-polarized band structures of ACrBr3 (A = K, Rb, Cs, Fr) obtained from GGA-PBE and GGA+U functionals at applied pressures of 0, 10, 20, and 30 GPa. | |
Table 6 Calculated minority-spin band gaps (eV) and half-metallic gaps (eV) using GGA and GGA+U functionals at applied pressures of ACrBr3 (A = K, Rb, Cs, Fr) compounds
| Halide perovskites |
Pressure (GPa) |
Band gap, Eg (eV) |
| GGA |
GGA+U |
| Spin-down band gap |
Half-metallic gap |
Spin-down band gap |
Half-metallic gap |
| KCrBr3 |
0 |
3.46, 3.429 |
0.82, 0.329 |
4.14 |
0.103 |
| 10 |
3.02 |
0.94 |
3.93 |
1.59 |
| 20 |
2.65 |
1.16 |
3.47 |
0.48 |
| 30 |
— |
— |
2.21 |
0.09 |
|
|
| RbCrBr3 |
0 |
3.48 |
0.79 |
4.29 |
0.1 |
| 10 |
3.07 |
1 |
3.97 |
1.57 |
| 20 |
1.231 |
0.05 |
3.51 |
0.51 |
| 30 |
— |
— |
3.13 |
1 |
|
|
| CsCrBr3 |
0 |
3.5 |
0.74 |
5.14 |
0.91 |
| 10 |
3.13 |
1.09 |
4.03 |
1.54 |
| 20 |
2.79 |
1.22 |
3.61 |
0.56 |
| 30 |
— |
— |
3.24 |
1.11 |
|
|
| FrCrBr3 |
0 |
3.52 |
0.7 |
4.2 |
0.09 |
| 10 |
3.19 |
1.17 |
4.1 |
1.51 |
| 20 |
2.87 |
1.23 |
3.7 |
0.59 |
| 30 |
— |
— |
3.23 |
1.23 |
 |
| | Fig. 8 Variation of band gap (eV) with applied pressures of 0, 10, 20, and 30 GPa for ACrBr3 (A = K, Rb, Cs, Fr) compounds using the GGA+U functional. | |
Density of states (DOS) and partial density of states (PDOS) are also investigated to further evaluate the half-metallicity. ACrBr3 (A = K, Rb, Cs, Fr) exhibit half-metallic behavior, determined using both GGA and GGA+U, as the spin-up channel shows metallic states at the Fermi level while the spin-down channel shows a gap as depicted by the DOS structure over 0–30 GPa in Fig. 9 using GGA and GGA+U. From PDOS using the GGA+U functional at 0 GPa, the states around EF mainly arise from Cr-3d and Br-4p hybridization, as illustrated in Fig. 10, and this hybridization makes our studied materials a half metal. At elevated pressure, the minority-spin gap narrows as hybridization becomes stronger.
 |
| | Fig. 9 Spin-polarized density of states (DOS) (electrons per eV) of ACrBr3 (A = K, Rb, Cs, Fr) perovskites using the GGA and GGA+U functionals under applied pressures of 0, 10, 20, and 30 GPa. | |
 |
| | Fig. 10 Calculated spin-polarized partial density of states (PDOS) of ACrBr3 (A = K, Rb, Cs, Fr) perovskites using the GGA+U functional. | |
3.4 Spin polarization
The spin polarization is related to the spin-polarized DOS by the following formula:11| | 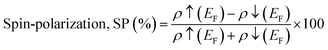 | (18) |
Here ρ↑(EF) and ρ↓(EF) denote the majority and minority spin density at the Fermi level. If ρ↑(EF) = 0 or ρ↓(EF) = 0, it means any one of the spin states is responsible for electron movement and maximum SP occurs. All of our materials have 100% SP at the Fermi level, which further illustrates the half-metallic characteristics.
3.5 Magnetic properties and Curie temperature
Magnetic properties of our compounds are investigated using GGA and GGA+U functionals. Table 7 and Fig. 11 illustrate the total and atomic magnetic moments in Bohr magnetons (μB). The total magnetic moment is calculated by using the following formula:| | | μT = A (A = K, Rb, Cs, Fr) + Cr + 3Br | (19) |
The calculated total magnetic moments range from 3.98 to 4.02 and −3.99, which are either integer or nearly integer values. Such values verify that all our investigated materials are ferromagnetic.57 Only the Cr atom significantly contributes to the magnetic moment, whereas other atoms make a negligible contribution. From the Slater–Pauling (SP) rule, the total magnetic moment (μT) per unit cell for transition-metal-based half-metallic materials is,where ZT denotes the total number of valence electrons in our material. In our compounds ACrBr3 (A = K, Rb, Cs, Fr), A-site materials (K, Rb, Cs, and Fr) have 1 valence electron, chromium has 6 valence electrons (3d5 4s1), and the bromine ([Ar] 4s2 3d10 4p5) atom has 7 valence electrons. So, ZT = 1(A) + 6(Cr) + 3 × 7(Br) = 28 electrons and μT = (28 − 24) = 4μB, consistent with our results derived from individual atomic contributions.
Table 7 Spin magnetic moment (total and atomic) in μB of ACrBr3 (A = K, Rb, Cs, Fr) at various pressures using the GGA-PBE and GGA+U functionals
| Magnetic moment |
Compounds |
0 GPa |
10 GPa |
20 GPa |
30 GPa |
| GGA |
GGA+U |
GGA |
GGA+U |
GGA |
GGA+U |
GGA |
GGA+U |
| Moment-μK |
KCrBr3 |
−0.03 |
0.03, 0.001129 |
0.04 |
0.04 |
0.05 |
0.04 |
0.06 |
0.05 |
| Moment-μCr |
−3.75 |
3.93, 3.834129 |
3.69 |
3.69 |
3.41 |
3.55 |
3.31 |
3.44 |
| Moment-μBr |
−0.07 |
0.02, −0.038329 |
0.09 |
0.09 |
0.18 |
0.14 |
0.21 |
0.17 |
| Total μ |
−3.99 |
4.02, 429 |
4 |
4 |
4 |
4.01 |
4 |
4 |
| Moment-μRb |
RbCrBr3 |
0.03 |
0.03 |
0.04 |
0.04 |
0.05 |
0.05 |
0.06 |
0.06 |
| Moment-μCr |
3.77 |
3.94 |
3.55 |
3.70 |
3.42 |
3.56 |
3.32 |
3.45 |
| Moment-μBr |
0.07 |
0.01 |
0.13 |
0.09 |
0.18 |
0.13 |
0.21 |
0.16 |
| Total μ |
4.01 |
4 |
3.98 |
4.01 |
4.01 |
4 |
4.01 |
3.99 |
| Moment-μCs |
CsCrBr3 |
0.03 |
0.03 |
0.05 |
0.05 |
0.06 |
0.06 |
0.08 |
0.07 |
| Moment-μCr |
3.79 |
3.97 |
3.58 |
3.73 |
3.44 |
3.58 |
3.34 |
3.48 |
| Moment-μBr |
0.06 |
0.00 |
0.12 |
0.08 |
0.16 |
0.12 |
0.19 |
0.15 |
| Total μ |
4 |
4 |
3.99 |
4.02 |
3.98 |
4 |
3.99 |
4 |
| Moment-μFr |
FrCrBr3 |
0.03 |
0.03 |
0.05 |
0.04 |
0.06 |
0.06 |
0.07 |
0.07 |
| Moment-μCr |
3.81 |
3.98 |
3.60 |
3.75 |
3.46 |
3.60 |
3.36 |
3.50 |
| Moment-μBr |
0.05 |
−0.00 |
0.12 |
0.07 |
0.16 |
0.11 |
0.19 |
0.14 |
| Total μ |
3.99 |
4.01 |
4.01 |
4 |
4 |
3.99 |
4 |
3.99 |
 |
| | Fig. 11 Magnetic moments in μB of (a) KCrBr3, (b) RbCrBr3, (c) CsCrBr3, and (d) FrCrBr3 single perovskites calculated using the GGA+U functional. | |
The temperature above which spontaneous magnetization of a material is lost is known as the Curie temperature. We can evaluate the Curie temperature by using the following formula,58
| |  | (21) |
Here,
kB denotes the Boltzmann constant and
EFM and
EAFM represent the total energies of the ferromagnetic and antiferromagnetic states, respectively. The calculated Curie temperature and pressure effect are shown in
Table 8 and
Fig. 12. Applied pressure leads to an increase in the estimated Curie temperature for all compounds at all pressures, except for the KCrBr
3 compound at 10 GPa, where the
TC value shows a decrease.
Table 8 Calculated Curie temperature, TC (K) at applied pressures 0, 10, 20, and 30 GPa of ACrBr3 (A = K, Rb, Cs, Fr) compounds
| Compounds |
Curie temperature, TC (K) |
| 0 GPa |
10 GPa |
20 GPa |
30 GPa |
| KCrBr3 |
194.78 |
59.93 |
423.57 |
539.55 |
| RbCrBr3 |
126.43 |
219.59 |
407.62 |
473.98 |
| CsCrBr3 |
131.28 |
220.80 |
384.42 |
580.33 |
| FrCrBr3 |
203.63 |
280.07 |
340.21 |
483.38 |
 |
| | Fig. 12 Variation of the Curie temperature, TC (K), with applied pressure (GPa) for ACrBr3 (A = K, Rb, Cs, Fr) compounds. | |
3.6 Thermal properties
The investigation of thermal properties is essential to understand how energy and heat behave, interact, and transform within solid materials. By employing the calculated elastic constants, the Debye temperature, θD, can be determined by the following expressions:| |  | (22) |
| | 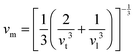 | (23) |
From Navier's equation,59| | 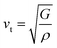 | (24) |
| | 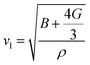 | (25) |
Here, θD denotes the Debye temperature, h stands for Planck's constant, kB is Boltzmann's constant, n is the number of atoms in the molecule, NA is Avogadro's number, M is the molecular weight, ρ is the density, and vm is the average sound velocity. With the application of pressure, the value of B and G increases, which results in an increase in average sound velocity as seen from Table 9. As a result, Debye temperature also increases with the increase of applied pressure. This high value of Debye temperature suggests that our materials are expected to have high melting points, high hardness, and large thermal expansion coefficients and might be applied to thermal resistance materials. The temperature at which a solid melt into a liquid under normal atmospheric pressure is known as the melting temperature and has the following expression:| | 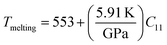 | (26) |
Applying pressure results in an increase in the melting temperature of all our investigated materials as seen from Fig. 13. The Debye frequency represented by ωD, gives information on the transmission of heat at the atomic scale and can be calculated using the following expression:60| | 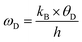 | (27) |
With the increase in pressure, the value of ωD also increases, resulting in greater heat capacity as well as thermal conductivity. The highest value is shown for Cs based compounds, illustrated in Table 9. The minimum thermal conductivity (Kmin) can be computed from the Clarke formula, which is:61| | 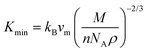 | (28) |
The application of pressure enhances Kmin, hence Rb and Cs based materials exhibit the highest values at 30 GPa pressure, as shown in Table 9.
Table 9 Effect of pressure on density ρ (kg m−3), Debye temperature θD (K), melting temperature Tm (K), Debye frequency ωD (THz) and minimum thermal conductivity Kmin (W m−1 K−1) of ACrBr3 (A = K, Rb, Cs, Fr) perovskites using the GGA-PBEsol functional
| Compounds |
Pressure (GPa) |
Density, ρ (kg m−3) |
Longitudinal sound velocity vl (m s−1) |
Transverse sound velocity vt (m s−1) |
Average sound velocity vm (m s−1) |
Debye temperature θD (K) |
Melting temperature Tm (K) |
Debye frequency ωD (THz) |
Minimum thermal conductivity Kmin (W m−1 K−1) |
| KCrBr3 |
0 |
3930.17 |
3750.64 |
2074.28 |
2311.05 |
226.93 |
919.30 |
4.73 |
0.35 |
| 10 |
4745.74 |
4898.31 |
2507.54 |
2808.96 |
293.71 |
1415.33 |
6.12 |
0.48 |
| 20 |
5295.15 |
5595.74 |
2751.56 |
3089.70 |
335.08 |
1840.32 |
6.98 |
0.56 |
| 30 |
5719.69 |
6037.92 |
2884.20 |
3243.70 |
360.94 |
2207.56 |
7.52 |
0.62 |
|
|
| RbCrBr3 |
0 |
4428.482 |
3458.28 |
1939.59 |
2158.59 |
211.12 |
906.36 |
4.40 |
0.32 |
| 10 |
5368.305 |
4717.09 |
2453.30 |
2745.41 |
286.31 |
1432.64 |
5.96 |
0.46 |
| 20 |
5962.115 |
5421.41 |
2760.70 |
3093.58 |
334.10 |
1854.74 |
6.96 |
0.56 |
| 30 |
6433.694 |
5893.76 |
2938.94 |
3297.46 |
365.27 |
2252.66 |
7.61 |
0.63 |
|
|
| CsCrBr3 |
0 |
4863.08 |
3427.29 |
1968.79 |
2186.69 |
212.11 |
907.84 |
4.42 |
0.32 |
| 10 |
5878.27 |
4597.03 |
2474.04 |
2762.09 |
285.40 |
1410.31 |
5.94 |
0.46 |
| 20 |
6531.10 |
5310.02 |
2809.45 |
3140.34 |
336.07 |
1850.90 |
7.0 |
0.56 |
| 30 |
7039.01 |
5817.30 |
3031.07 |
3391.56 |
372.13 |
2252.66 |
7.75 |
0.63 |
|
|
| FrCrBr3 |
0 |
5779.795 |
3157.41 |
1812.62 |
2013.34 |
194.01 |
899.62 |
4.04 |
0.29 |
| 10 |
7000.131 |
4248.15 |
2312.68 |
2579.77 |
264.99 |
1384.89 |
5.52 |
0.42 |
| 20 |
7774.39 |
4902.24 |
2613.70 |
2919.97 |
310.60 |
1824.3 |
6.47 |
0.51 |
| 30 |
8378.75 |
5369.18 |
2835.38 |
3169.74 |
345.69 |
2218.26 |
7.20 |
0.59 |
 |
| | Fig. 13 Variation of (a) Debye temperature θD (K), (b) melting temperature Tm (K), (c) Debye frequency ωD (THz), and (d) minimum thermal conductivity Kmin (W m−1 K−1) with applied pressure 0–30 GPa for ACrBr3 (A = K, Rb, Cs, Fr) compounds. | |
3.7 Optical properties
Investigating the optical properties of materials is essential, as it lets us know how they react to incident electromagnetic radiation. This understanding is critical for the design and selection of materials used in optoelectronic device applications. Fig. 14 shows the optical properties, absorption, energy loss, real and imaginary dielectric constants, refractive index, real optical conductivity, and optical reflectivity of our compounds ACrBr3 (A = K, Rb, Cs, Fr) in the 0–30 eV photon energy range under applied pressures of 0, 10, 20, and 30 GPa. Primarily, we can describe a material's response to electromagnetic radiation as its dielectric function. The complex dielectric function, ε(ω), can be found by the following equation:| | | ε(ω) = ε1(ω) + iε2(ω) | (29) |
Here, ε1(ω) is the real part and ε2(ω) is the imaginary part of the complex dielectric function. The degree of charge recombination is demonstrated by the dielectric function's static value ε1(0), which is related to the manner in which optoelectronic devices perform. The real static dielectric constant ε1(0) is highest when the photon energy is 0 eV, and its value is 22.55 eV, 22.78 eV, 24.09 eV, and 24.38, respectively, for K, Rb, Cs, and Fr based materials; it shows a decreasing trend with increasing applied pressure for all investigated systems as seen in Fig. 14(a). As the photon energy increases from 0 eV, the real dielectric response decreases up to approximately 1.05–2.60 eV, after which it rises and displays minor peaks in the ultraviolet range. Such a transition from the visible to the ultraviolet spectrum demonstrates the promise of the materials we examined for usage in optoelectronic applications.62 These peak intensities increase with pressure and also follow a progressive enhancement from K to Fr. Negative values of the real components of the dielectric constants are observed for a certain photon energy range for all of the materials across all pressure ranges. This indicates that our materials behave like a metal at this phonon energy, reflecting incident electromagnetic radiation rather than transmission. The imaginary part of the dielectric function is also presented in Fig. 14(b). The unreal component of the dielectric function ε2(ω) governs a substance's capacity to pass on or collect light. Energy losses in our substances are demonstrated by their peaks and the transition from the valence band maximum to the conduction band minimum, which correspond to interband electronic transitions. With increasing external pressure and substitution at the A-site from K to Fr, the magnitude of ε2(ω) also rises. Even though all our studied compounds exhibit a similar qualitative trend the peak values differ. Increasing values of ε2(ω) suggest that the material becomes less transparent, absorbs more light, and provides increased optical loss.63
 |
| | Fig. 14 Photon energy-dependent optical properties: (a) the real part of the dielectric function ε1(ω), (b) the imaginary part of the dielectric function ε2(ω), (c) optical conductivity σ(ω), (d) absorption coefficient I(ω), (e) reflectivity R(ω), (f) refractive index, η(ω), and (g) the loss function L(ω) of ACrBr3 (A = K, Rb, Cs, Fr) compounds at various pressures using the GGA-PBE functional. | |
Optical conductivity is the flow of electrons when an electromagnetic wave strikes the material. It additionally provides information on the conduction process. When a material can absorb more photons then it can conduct more electrons, meaning conductivity increases, and both of the graphs show comparable trends. The real part of the optical conductivity under pressure variation is shown in Fig. 14(c) and connected with ε2(ω) by the following relationship:64
| | 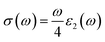 | (30) |
All four of our investigated compounds show 0 conductivity when the photon energy does not exist. Conductivity for all of our materials is expected to increase with the increase of the photon energy and reach a minor peak, and then it starts to decrease in the infrared (IR) region and becomes minimum in the visible region, as shown in
Fig. 14(c). After that, it starts to increase from the visible to the ultraviolet region and shows its highest peak in the UV region. Pressure application increases conductivity, narrowing the band gap of all of our studied materials, which is consistent with our electronic analysis results. The highest conductivity is for the Fr based compound at 30 GPa, whose value is 9.39 (1 fs
−1) at 10 eV.
The absorption coefficient can be computed using the following equation,65
| | 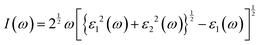 | (31) |
where
ε1(
ω) and
ε2(
ω) are the real and imaginary parts of the dielectric function, and the light frequency is represented by
ω. This parameter shows a similar trend to the conductivity graph. Under applied pressure, we observe a significant enhancement in the absorption coefficient of all the materials we investigated, and the highest peak is in the UV range, which is 11–18 eV photon energy, which is suitable for the optoelectronic applications.
66| | 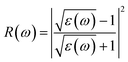 | (32) |
Reflectivity,
R(
ω), is an optical property and is defined as the ratio of the quantity of light energy falling on a material's surface to the amount of light energy reflected from that surface, and can be calculated from the above equation (
eqn (32)). The value of reflectivity without electromagnetic radiation is shown in
Fig. 14(e), which almost ranges from 43 to 44% without applied pressure and becomes 31–32% at 30 GPa pressure. Our materials show the highest reflectivity in the UV region, and reflectivity significantly increases with the increase of applied pressure. The Fr based material shows the highest reflectivity, which is 52.6% at 18.62 eV photon energy at 30 GPa pressure.
When light passes through a medium, the amount of light that bounces off from that medium is known as the refractive index. Light can travel quickly within the medium when this value is smaller, and the amount of refraction is less. The refractive index, η(ω), is determined using the following equation:67
| | 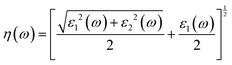 | (33) |
All four materials have a high peak at low energy that slowly drops as the energy increases. When pressure is applied, the refractive index becomes a bit higher in the low-energy region. The investigated spectrum is shown in
Fig. 14(f). The overall spectral response is almost the same for all our investigated materials.
The loss function L(ω) as a function of incident photon energy is shown in Fig. 14(g), and this function is the amount of energy an electron loses while traveling through a material. We can compute this using the following formula:68
| | 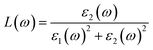 | (34) |
The highest energy loss occurs in the ultraviolet regions for all of our studied compounds. Pressure has a vital effect on this function, as pressure not only changes the peak value but also shifts the photon energy for which the highest value of the loss function can be found. The highest energy loss for the Cs-based compound is 7.9 at 25 eV photon energy for 30 GPa pressure. The Rb-based material has the highest peak at 20 GPa, and the other two materials also have the highest value at 30 GPa. This peak is generated by bulk plasmonic excitation at a specific incident light energy and the bulk plasma frequency, which is the corresponding frequency at that energy.
69 According to these results, our materials may be promising candidates for spin-polarized optoelectronic devices as well as additional domains
68,70 (
Table 10).
Table 10 Static reflectivity, R(0), at various applied pressures for ACrBr3 (A = K, Rb, Cs, Fr)
| Compounds |
Reflectivity |
Pressure (GPa) |
| 0 |
10 |
20 |
30 |
| KCrBr3 |
R(0) |
0.43 |
0.38 |
0.35 |
0.33 |
| RbCrBr3 |
0.43 |
0.38 |
0.35 |
0.33 |
| CsCrBr3 |
0.44 |
0.39 |
0.36 |
0.33 |
| FrCrBr3 |
0.45 |
0.40 |
0.38 |
0.35 |
4. Conclusions
In this work, we extensively investigated the structural, mechanical, magnetic, electronic, thermal, and optical properties of cubic ACrBr3 (A = K, Rb, Cs, Fr) perovskites under applied pressures of 0, 10, 20, and 30 GPa using GGA-PBE, GGA+U, and GGA-PBEsol functionals. The analysis reveals that Cs- and Fr-based compounds, initially brittle at ambient pressure, transform into ductile materials above 10 GPa, while K- and Rb-based perovskites remain ductile across all studied conditions. Band structure and density of states analyses confirm half-metallicity, and the integer magnetic moment demonstrates stable ferromagnetism. Increasing pressure reduces lattice constants, cell volume, and band gap, while simultaneously enhancing elastic constants, thereby improving rigidity and resilience essential for device integration. Around 10 GPa, the half-metallic gap is maximized for most materials while preserving mechanical stability. Thermodynamic parameters, including Debye temperature and melting temperature, show an upward trend with pressure, indicating enhanced thermal stability. Optical properties are also strongly pressure-dependent, with optical parameters increasing as pressure rises, and the optical conductivity exhibits the largest deviation. Collectively, these findings establish ACrBr3 (A = K, Rb, Cs, Fr) perovskites as robust multifunctional candidates, combining favorable magnetic, mechanical, thermodynamic, and optical characteristics, underscoring their suitability for next-generation spintronic, optoelectronic devices and offering a pathway toward materials capable of showing reliable performance under diverse environmental and operational conditions.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting the findings of this study, including structural parameters, electronic band structures, magnetic properties, elastic properties, and optical properties of ACrBr3 (K, Rb, Cs, Fr), are available from the corresponding author upon reasonable request. No restrictions apply to data sharing.
References
- S. Das Sarma, Spintronics: a new class of device based on electron spin, rather than on charge, may yield the next generation of microelectronics, Am. Sci., 2001, 89(6), 516–523, DOI:10.1511/2001.6.516.
- M. Y. Sofi, M. S. Khan and M. A. Khan, Spin exchange, electronic correlation, and thermoelectric transport in Rb2GeMBr6 (M = V, Mn, Ni) halide double perovskites from first principles calculations, J. Mater. Chem. C, 2026, 14(1), 444–467 Search PubMed.
- S. A. Mir, A. Q. Seh and D. C. Gupta, spintronic applications: BaMO3 (M¼ Mg and Ca), RSC Adv., 2020, 2, 36241–36252, 10.1039/d0ra06739c.
- R. A. De Groot, F. M. Mueller, P. G. Van Engen and K. H. J. Buschow, Half-metallic ferromagnets and their magneto-optical properties, J. Appl. Phys., 1984, 55(6), 2151–2154 Search PubMed.
- V. A. Online, RSC Adv., 2025, 24002–24018, 10.1039/d5ra03555d.
- M. I. Hussain, R. M. A. Khalil, F. Hussain, M. Imran, A. M. Rana and S. Kim, Investigations of structural, electronic and optical properties of YInO3 (Y = Rb, Cs, Fr) perovskite oxides using mBJ approximation for optoelectronic applications: a first principles study, Mater. Sci. Semicond. Process., 2020, 113, 105064 Search PubMed.
- S. A. Khandy and D. C. Gupta, Structural, elastic and magneto-electronic properties of half-metallic BaNpO3 perovskite, Mater. Chem. Phys., 2017, 198, 380–385 Search PubMed.
- M. Y. Sofi, M. S. Khan and M. A. Khan, Harnessing the half-metallicity and thermoelectric insights in Cs2AgMBr6 (M = V, Mn, Ni) double halide perovskites: a DFT study, Mater. Sci. Semicond. Process., 2025, 186, 109023 Search PubMed.
- S. A. Khandy and D. C. Gupta, Insight view of double perovskites Ba2XNbO6 (X = Ho, Yb) for spintronics and thermoelectric applications, Int. J. Energy Res., 2021, 45(9), 13338–13354 Search PubMed.
- G. Y. Gao, K. L. Yao, Z. L. Liu, Y. L. Li, J. L. Jiang and Y. C. Li, Half-metallic ferromagnetism of Cr-doped rutile TiO2: a first-principles pseudopotential study, Phys. B, 2006, 382(1–2), 14–16 Search PubMed.
- Y. Sefir,
et al., Structural, electronic, magnetic and thermodynamic properties of the new multifunctional half-Heusler alloy CoTcSn: Half-metallic and ferromagnetic behaviour, Pramana, 2021, 95, 95, DOI:10.1007/s12043-021-02125-w.
- L. Beldi, Y. Zaoui, K. O. Obodo, H. Bendaoud and B. Bouhafs, d0 Half-Metallic Ferromagnetism in GeNaZ (Z = Ca, Sr, and Ba) Ternary Half-Heusler Alloys: an Ab initio Investigation, J. Supercond. Novel Magn., 2020, 3121–3132 Search PubMed.
- S. Omran, Structural stability, electronic structure and magnetic properties of the new hypothetical half-metallic ferromagnetic full-Heusler alloy CoNiMnSi, Mater. Sci., 2016, 34(1), 85–93 Search PubMed.
- B.-G. Liu, Robust half-metallic ferromagnetism in zinc blende CrSb, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67(17), 172411 Search PubMed.
- A. Rahman, A. Kabir and T. Mahmud, Computational insights into transition metal-based BaCoX3 (X = Cl, Br, I) halide perovskites for spintronics, photovoltaics, and renewable energy devices, Sci. Rep., 2024, 3, 1–20 Search PubMed.
- J. Berry,
et al., Hybrid Organic–Inorganic Perovskites (HOIPs): Opportunities and Challenges, Adv. Mater., 2015, 5102–5112, DOI:10.1002/adma.201502294.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, The emergence of perovskite solar cells, Nat. Photonics, 2014, 8(7), 506–514 Search PubMed.
- F. Hao, C. C. Stoumpos, D. H. Cao, R. P. H. Chang and M. G. Kanatzidis, Lead-free solid-state organic–inorganic halide perovskite solar cells, Nat. Photonics, 2014, 8(6), 489–494 Search PubMed.
- S. Kazim, M. K. Nazeeruddin, M. Grätzel and S. Ahmad, Perovskite as light harvester: a game changer in photovoltaics, Angew. Chem., Int. Ed., 2014, 53(11), 2812–2824 Search PubMed.
- X.-G. Zhao, D. Yang, J.-C. Ren, Y. Sun, Z. Xiao and L. Zhang, Rational design of halide double perovskites for optoelectronic applications, Joule, 2018, 2(9), 1662–1673 Search PubMed.
- K. Mądra-Gackowska, M. Gackowski and Ł. Szeleszczuk, Density Functional Theory Insights into A2BBiCl6 (A = Cs, K; B = Ag, Au) Halide Double Perovskites for Next-Generation Photovoltaics, Phys. B, 2026, 418286 Search PubMed.
- Ł. Szeleszczuk, K. Mądra-Gackowska, V. B. Hacholli and M. Gackowski, Designing Efficient Energy Materials: Optoelectronic and Thermoelectric Perspectives of X2YBiCl6 (X = Cs, Na; Y = Ag, Au) Double Perovskites, J. Phys. Chem. Solids, 2025, 113293 Search PubMed.
- Ł. Szeleszczuk, K. Mądra-Gackowska and M. Gackowski, Exploring the Effects of Oxygen Replacement by Sulfur in SrTiO3: A Comprehensive DFT Investigation, J. Phys. Chem. Solids, 2026, 113575 Search PubMed.
- D. Ghosh, A. Aziz, J. A. Dawson, A. B. Walker and M. S. Islam, Putting the Squeeze on Lead Iodide Perovskites: Pressure-Induced E ff ects To Tune Their Structural and Optoelectronic Behavior, Chem. Mater., 2019, 31, 4063–4071, DOI:10.1021/acs.chemmater.9b00648.
- A. Ja, Y. Lin, C. M. Beavers, J. Voss, W. L. Mao and H. I. Karunadasa, High-Pressure Single-Crystal Structures of 3D Lead-Halide Hybrid Perovskites and Pressure Effects on their Electronic and Optical Properties, ACS Cent. Sci., 2016, 2–10, DOI:10.1021/acscentsci.6b00055.
- V. A. Online, D. Liu, Q. Li, H. Jing and K. Wu, Pressure-induced effects in the inorganic halide, RSC Adv., 2019, 3279–3284, 10.1039/c8ra10251a.
- L. Li, Y. Wang, D. Liu, C. Ma, M. G. Brik and A. Suchocki, Comparative fi rst-principles calculations of the electronic, optical, elastic and thermodynamic properties of XCaF3 (X¼ K, Rb, Cs) cubic perovskites, Mater. Chem. Phys., 2017, 188, 39–48, DOI:10.1016/j.matchemphys.2016.12.033.
- C. Li, B. Wang, R. Wang, H. Wang and X. Lu, First-principles study of structural, elastic, electronic, and optical properties of hexagonal BiAlO3, Phys. B, 2008, 403(4), 539–543 Search PubMed.
- H. A. Alburaih and S. Nazir, DFT Calculations of Half-Metallic Ferromagnetism and Transport Properties of Cubic KCrX3 (X = Cl, Br, I) Halides for Spintronics and Energy Conversion Applications DFT Calculations of Half-Metallic Ferromagnetism and Transport Properties of Cubic KCrX3 (X = Cl, Br, I) Halides for Spintronics and Energy Conversion Applications, J. Electrochem. Soc., 2023, 170(11), 114501, DOI:10.1149/1945-7111/ad0669.
- M. U. Khan, A. U. Rahman, M. Ahmad, M. Yaseen and M. A. Ahmed, Exploring room-temperature ferromagnetism and half-metallicity in new halide perovskites RbCrX3 (X: F, Cl, Br, I) using first-principles and Monte Carlo simulations, Eur. Phys. J. Plus, 2024, 139, 106, DOI:10.1140/epjp/s13360-024-04898-6.
- P. J. D. Lindan,
et al., First-principles simulation: ideas, illustrations and the CASTEP code, J. Phys.: Condens. Matter, 2002, 14(11), 2717–2744 Search PubMed.
- M. Karouchi,
et al., Investigating the structural, electronic, and optical properties of the novel double perovskite K2AgBiI6 using DFT, Front. Mater., 2024, 11, 1–11, DOI:10.3389/fmats.2024.1448400.
- T. H. Fischer and J. Almlof, General methods for geometry and wave function optimization, J. Phys. Chem., 1992, 96(24), 9768–9774 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 Search PubMed.
- M. Y. Sofi, M. S. Khan and M. A. Khan, Eco-friendly lead-free halide double perovskites A2CuMCl6 (A = K, Rb; M = Sb, Bi): stability, thermoelectric, and optoelectronic advancements through theoretical insights, J. Mater. Chem. C, 2024, 12(39), 16045–16058 Search PubMed.
- R. Hossain, S. Khanom and P. Mondal, Materials Advances (Pb): a comparative first-principles study for multifunctional device applications, Mater. Adv., 2026, 618–640, 10.1039/d5ma00961h.
- M. Y. Sofi, M. S. Khan and M. A. Khan, Materials Advances candidates for semiconductor spintronics and, Mater. Adv., 2024, 4913–4931, 10.1039/d3ma01160g.
- M. K. Shahzad,
et al., Computational investigation on physical properties of lead based perovskite RPbBr3 (R = Cs, Hg, and Ga) materials for photovoltaic applications, Sci. Rep., 2024, 1–13, DOI:10.1038/s41598-024-70586-1.
- Ł. Szeleszczuk, K. Mądra-Gackowska and M. Gackowski, Optoelectronic and Thermoelectric Performance of Cubic X2YInO6 (X = Ba, Sr; Y = Nb, V) Double Perovskites: A First-Principles Approach, Phys. B, 2025, 417958 Search PubMed.
- K. A. Persson, Lattice instabilities in metallic elements, Rev. Mod. Phys., 2012, 84, 945–986, DOI:10.1103/RevModPhys.84.945.
- M. A. Haq, Ultra-violet to visible band gap engineering of cubic halide KCaCl3 perovskite under pressure for optoelectronic applications: insights from DFT, RSC Adv., 2021, 36367–36378, 10.1039/D1RA06430D.
- J. Gao, Q.-J. Liu and B. Tang, Elastic stability criteria of seven crystal systems and their application under pressure: taking carbon as an example, J. Appl. Phys., 2023, 133(13), 135901 Search PubMed.
- A. Bakar, A. O. Alrashdi, M. M. Fadhali, A. Afaq, H. A. Yakout and M. Asif, Effect of pressure on structural, elastic and mechanical properties of cubic perovskites XCoO3 (X¼ Nd, Pr) from first-principles investigations, J. Mater. Res. Technol., 2022, 19, 4233–4241, DOI:10.1016/j.jmrt.2022.06.126.
- N. Hasan, Structural, elastic and optoelectronic properties of, RSC Adv., 2022, 7961–7972, 10.1039/d2ra00546h.
- R. Hill, The elastic behaviour of a crystalline aggregate, Proc. Phys. Soc., London, Sect. A, 1952, 65(5), 349 Search PubMed.
- M. Faghihnasiri, M. Izadifard and M. E. Ghazi, DFT study of mechanical properties and stability of cubic methylammonium lead halide perovskites (CH3NH3PbX3, X = I, Br, Cl), J. Phys. Chem. C, 2017, 121(48), 27059–27070 Search PubMed.
- S. I. Ranganathan and M. Ostoja-starzewski, Universal Elastic Anisotropy Index, Phys. Rev. Lett., 2008, 055504, 3–6, DOI:10.1103/PhysRevLett.101.055504.
- A. Hosen, R. Islam and S. Haque, Heliyon Exploring the influence of pressure-induced semiconductor-to-metal transition on the physical properties of cubic perovskites FrXCl 3 (X = Ge and Sn), Heliyon, 2024, 10(7), e27581, DOI:10.1016/j.heliyon.2024.e27581.
- M. H. Miah,
et al., First-principles study of the structural, mechanical, electronic, optical, and elastic properties of non-toxic XGeBr3 (X = K, Rb, and Cs) perovskite for optoelectronic and radiation sensing applications, Mater. Chem. Phys., 2024, 319, 129377 Search PubMed.
- S. Muramatsu and M. Kitamura, Simple expressions for elastic constants C11, C12, and C44 and internal displacements of semiconductors, J. Appl. Phys., 1993, 73(9), 4270–4272 Search PubMed.
- M. I. Naher and S. H. Naqib, An ab initio study on structural, elastic, electronic, bonding, thermal, and optical properties of topological Weyl semimetal TaX (X = P, As), Sci. Rep., 2021, 1–21, DOI:10.1038/s41598-021-85074-z.
- A. Ganguly, V. Murthy and K. Kannoorpatti, Structural and electronic properties of chromium carbides and Fe-substituted chromium carbides Structural and electronic properties of chromium carbides and Fe-substituted chromium carbides, Mater. Res. Express, 2020, 7(5), 056508 Search PubMed.
- L. B. M. Taibi and A. B. M. Abd, Dielectric relaxation, electrical conductivity and optical studies of solid – state synthesized – EuCrO3, J. Mater. Sci.: Mater. Electron., 2020, 31(1), 354–360, DOI:10.1007/s10854-019-02533-0.
- K. S. R. Kumar, Half-metallic behavior of Co2YZ (Y5V, Cr; Z5Al, Ga) under pressure: a DFT study, Appl. Phys. A, 2014, 1199–1209, DOI:10.1007/s00339-013-8211-4.
- G. Y. Gao, L. Hu, K. L. Yao, B. Luo and N. Liu, Large half-metallic gaps in the quaternary Heusler alloys CoFeCrZ (Z = Al, Si, Ga, Ge): a first-principles study, J. Alloys Compd., 2013, 551, 539–543 Search PubMed.
- S. A. Mir and D. C. Gupta, New ferromagnetic half-metallic perovskites for spintronic applications: BaMO3 (M = Mg and Ca), RSC Adv., 2020, 10(60), 36241–36252 Search PubMed.
- M. Y. Sofi,
et al., Pioneering computational insights into the structural, magnetic, and thermoelectric properties of A3XN (A = Co, Fe; X = Cu, Zn) anti-perovskites for advanced material applications, Mater. Sci. Semicond. Process., 2025, 185, 108925 Search PubMed.
- V. N. Jafarova, First-Principles Study of the Effects of Dopant Concentration on the Electronic, Magnetic,
and Structural Properties of Mn-Doped ZnO, J. Supercond. Novel Magn., 2026, 39, 31 Search PubMed.
- P. Ravindran, L. Fast, P. A. Korzhavyi, B. Johansson, J. Wills and O. Eriksson, Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2, J. Appl. Phys., 1998, 84(9), 4891–4904 Search PubMed.
- M. A. Rehman, J. ur Rehman and M. B. Tahir, Density functional theory study of structural, electronic, optical, mechanical, and thermodynamic properties of halide double perovskites Cs2AgBiX6 (X = Cl, Br, I) for photovoltaic applications, J. Phys. Chem. Solids, 2023, 181, 111443 Search PubMed.
- N. Shahadath and R. Kabir, OPEN DFT analysis of structural, electronic, optical, and thermodynamic properties of LiXI3 (where X = Ca, Sr, Ba) halide perovskites for optoelectronics, Sci. Rep., 2025, 1–23 Search PubMed.
- S. M. Ramay, Study of pressure induced physical properties of ZnZrO3 perovskite using density functional theory, Chem. Phys. Lett., 2020, 137601, DOI:10.1016/j.cplett.2020.137601.
- Y. Didi,
et al., Computational insights into spin-polarized density functional theory applied to actinide-based perovskites XBkO3 (X = Sr, Ra, Pb), Sci. Rep., 2025, 1–15 Search PubMed.
- J. M. Khoshman, P. Jakkala, D. C. Ingram and M. E. Kordesch, Optical conductivity tuning and electrical properties of a-BexZnyO thin films, J. Non.-Cryst. Solids, 2016, 440, 31–37 Search PubMed.
- X. Cr, M. M. Parvaiz, A. Khalil and B. Tahir, magnetic, optical, elastic and hydrogen storage, RSC Adv., 2024, 8385–8396, 10.1039/d4ra00204k.
- M. K. Butt and M. Yaseen, First Principle Insight into the Structural, Optoelectronic, Half Metallic, and Mechanical Properties of Cubic Perovskite NdInO3, Arabian J. Sci. Eng., 2020, 4967–4974 Search PubMed.
- A. Ramzan, M. Y. Sofi, M. S. Khan, J. Ali and M. A. Khan, Regular Article – Computational Methods First-principles investigation of Mg-based MgAl2X4 (X = S, Se) spinels for optoelectronic and energy harvesting, Eur. Phys. J. B, 2025, 98(5), 1–11, DOI:10.1140/epjb/s10051-025-00937-y.
- R. Hossain, S. Khanom, P. Mondal and F. Ahmed, Signature of half-metallicity and pressure-induced physical properties of Cubic ACrO3 (A = Si,Ge,Sn) multiferroic by DFT calculation, Mater. Adv., 2025, 6, 6550–6566, 10.1039/d5ma00169b.
- A. Hossain, M. S. I. Sarker, M. K. R. Khan and M. M. Rahman, Spin effect on electronic, magnetic and optical properties of spinel CoFe2O4: A DFT study, Mater. Sci. Eng., B, 2020, 253, 114496, DOI:10.1016/j.mseb.2020.114496.
- A. Javed, Q. Ain, A. Kumar, L. El, A. Kais and O. S. E. Shcheklein, Spin-Polarized DFT Based Investigation of Cs2LiMoBr6: A Promising Candidate for Spintronics, Optoelectronics and Sustainable Energy Systems, J. Supercond. Novel Magn., 2026, 1–12 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab and
Farid
Ahmed
a
*ab and
Farid
Ahmed
a
 and Br is at
and Br is at  Wyckoff atomic Positions. The polyhedral view of our studied compounds is shown in Fig. 1(a). Convergence tests were performed using 4 × 4 × 4, 6 × 6 × 6, and 8 × 8 × 8 k-point meshes and cutoff energies from 300 to 600 eV, as shown in Fig. 1(b). Since the energy change was very small, and the lowest energy was obtained at 4 × 4 × 4, a cutoff energy of 400 eV with a 4 × 4 × 4 k-point mesh was used for all calculations. Table 1 demonstrates the lattice parameters, unit cell volume, and energy both under spin-polarized and non-spin-polarized conditions using the GGA functional at 0, 10, 20, and 30 GPa pressures (Table 2).
Wyckoff atomic Positions. The polyhedral view of our studied compounds is shown in Fig. 1(a). Convergence tests were performed using 4 × 4 × 4, 6 × 6 × 6, and 8 × 8 × 8 k-point meshes and cutoff energies from 300 to 600 eV, as shown in Fig. 1(b). Since the energy change was very small, and the lowest energy was obtained at 4 × 4 × 4, a cutoff energy of 400 eV with a 4 × 4 × 4 k-point mesh was used for all calculations. Table 1 demonstrates the lattice parameters, unit cell volume, and energy both under spin-polarized and non-spin-polarized conditions using the GGA functional at 0, 10, 20, and 30 GPa pressures (Table 2).






 is the total energy of ACrBr3 calculated from the structural optimization. Our calculated cohesive energy ranges from 10.53 to 12.84 eV. Such large values ensure the chemical stability of our investigated compound. The calculated Mulliken charges, bond lengths, and bond populations of ACrBr3 (A = K, Rb, Cs, Fr) from the GGA method are presented in Table 3 and Fig. 3, 4.
is the total energy of ACrBr3 calculated from the structural optimization. Our calculated cohesive energy ranges from 10.53 to 12.84 eV. Such large values ensure the chemical stability of our investigated compound. The calculated Mulliken charges, bond lengths, and bond populations of ACrBr3 (A = K, Rb, Cs, Fr) from the GGA method are presented in Table 3 and Fig. 3, 4.





































