DOI:
10.1039/D5MA01466B
(Paper)
Mater. Adv., 2026,
7, 2859-2873
Unveiling a reline deep eutectic solvent (DES) for controlled synthesis of phase-pure CuFeS2 towards dielectric and electrocatalytic applications
Received
15th December 2025
, Accepted 22nd January 2026
First published on 23rd January 2026
Abstract
CuFeS2, a heavy-metal free transition metal chalcogenide (TMC), has remained unexplored despite having dielectric and electrocatalytic properties, primarily due to difficulty in controlling its phase and composition. Herein, we have developed a deep eutectic solvent (DES)-based solvothermal method for synthesizing stable and phase-pure chalcopyrite CuFeS2 with flower-like morphology consisting of triangular nanoplates. Different sulphur precursors such as thiourea, thioacetamide, sodium sulphide, and thio-reline DES were investigated, and among them, only thiourea resulted in the formation of phase-pure CuFeS2. The solvent “reline DES” serves as a reducer, solvent, and stabilizer for CuFeS2 nanocrystals and remains thermally stable up to 200 °C under solvothermal conditions, as verified by visual and spectroscopic analyses. The synthesized CuFeS2 exhibits a frequency and temperature-dependent dielectric constant and dielectric loss behavior. Furthermore, alternating current (AC) conductivity measurements indicated semiconductor-like behavior as conductivity increased with temperature, consistent with the reduction in real and imaginary parts of impedance. Also, CuFeS2 achieved an optimal oxygen evolution reaction (OER) performance with an overpotential of 360 mV at 10 mA cm−2 and a Tafel slope of 148.7 mV dec−1. This work presents a simple, eco-friendly route to synthesize phase-pure CuFeS2 with a high dielectric constant, low loss, excellent thermal stability, and good OER activity.
1. Introduction
Intermittent renewable energy has driven the demand for cost-effective energy storage and high-performance dielectric materials with high energy density and low loss.1–4 Transition metal chalcogenide (TMC) materials are of research interest for dielectric applications due to their various characteristics, including limited visible transparency, low optical attenuation, high refractive index, and strong optical nonlinearity.5,6 Due to the high polarizability of chalcogens, TMCs possess lower band gaps and higher conductivity than oxides, making them attractive for electronic and energy storage applications.7 Furthermore, electrochemical water splitting (EWS) at low overpotential, which includes the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), is a crucial process for generating green hydrogen and oxygen. However, the OER is recognized as the bottleneck in the water-splitting system due to the slow reaction kinetics associated with the four-electron transport mechanism. Noble metal electrocatalysts like Pt and Ru are highly efficient for water electrolysis, but their high cost and scarcity limit their broader applicability.8 Recently, more affordable and efficient electrocatalysts, including metal oxides, hydroxides, sulfides, selenides, carbides, and phosphides, have been investigated as benchmark electrocatalysts for water splitting.9
Ternary I–III–VI2 chalcogenides are valued for their stability, easy synthesis, and versatile properties, enabling applications in energy and electronics.10–12 Recently, nanostructured copper-based chalcogenides have garnered significant attention due to their abundance, low cost and relatively non-toxic nature compared to heavy metal-based chalcogenides.11,13 Amongst them, copper iron sulfide (CuFeS2), in which trivalent Fe atoms replace the group III atoms, is a promising candidate for replacing lead, cadmium, and other heavy metals due to its abundance and low environmental impact.14,15 CuFeS2 occurs naturally and is known as “chalcopyrite”, with its characteristics of golden luster that aids in its identification.16 The widespread occurrence of CuFeS2 in nature suggests its stability under terrestrial conditions.17 Though composed of earth-abundant elements like copper (Cu) and iron (Fe), comprehensive studies on this copper-based semiconductor's nanocrystals remain scarce in the scientific literature. Unlike other heavy metal-free TMCs, CuFeS2 possesses dielectric, thermoelectric and ferroelectric properties, making it ideal for various energy applications. Several chemical route methods, viz. wet chemical, thermal pyrolysis, microwave-assisted, hydrothermal, solvothermal, heat up, hot injection, etc., have been extensively utilized for the synthesis of CuFeS2 with a variety of morphologies, including spherical, quasi-spherical, clusters, cube, rod-like, quasi cuboctahedra, pyramidal, nanosheets, triangle, hexagonal, etc.18 The high melting points of constituent elements in the Cu–Fe–S system and the narrow thermodynamic stability window make it challenging to prepare it in a single-phase state.19 There are three main stable phases, viz. CuFeS2 (chalcopyrite), CuFe2S3 (cubanite), and Cu5FeS4 (bornite), of ternary Cu–Fe–S systems.19 Also, the intrinsic thermodynamic instability of CuFeS2 in the reaction environment, coupled with the formation of individual copper and iron sulfide phases (CuS, Cu2S, Cu2−xS, FeS, Fe7S8, etc.), poses significant challenges for controlled synthesis of pure-phase chalcopyrite.14,20 For instance, Kumar et al. employed a microwave-assisted synthesis technique for CuFeS2; however, the resulting material contains FeS and CuS impurity phases.15 Similarly, the PXRD pattern of CuFeS2 reported by Gurin et al. contains extra XRD peaks at 2θ values of 20° and 33°, suggesting the presence of an extraneous phase in the parent material.21 Pron et al. reported an Fe-deficient phase that matches the chalcopyrite phase, which warrants distinct classification as an Fe-deficient impure phase.22 Hence, developing new synthetic protocols that ensure phase selectivity during the synthesis of CuFeS2 is highly desirable. Most syntheses previously adopted for CuFeS2 require high-boiling toxic organic solvents and complicated procedures involving air-free Schlenk-line techniques for phase-controlled growth of metal sulphide nanocrystals.18 Efforts are intensifying to develop simpler, safer, and more sustainable synthetic protocols, moving away from toxic solvents and complex procedures. In this context, deep eutectic solvents (DESs) as sustainable solvents are gaining widespread attention for nanomaterial synthesis.23,24 DESs, formed from Lewis/Brønsted acids or bases, are easily prepared with high purity through simple mixing and gentle warming, minimizing waste generation and eliminating the need for post-synthesis purification steps, making them cost-effective ionic solvents.23,25 The extensive hydrogen bonding network within these DESs acts as a supramolecular template, facilitating the nucleation and growth of nanocrystals under solvothermal synthetic conditions.24,26–29 They function simultaneously as solvents, reactants, and templates, minimizing the need for multiple reagents.28 While DESs have been used for morphology-controlled synthesis of TMC nanocrystals,28,30 their use for phase-controlled synthesis under solvothermal conditions remains unexplored.
The choice of sulfur precursors and their chemical reactivity play a crucial role in determining the phase of the TMC nanocrystals during their nucleation and growth.31,32 For instance, the Macdonald group showed that sulfur precursor reactivity, governed by C–S bond strength, directs phase formation in iron sulfides, with weaker bonds yielding sulfur-rich phases.33 The Macdonald group further achieved phase-controlled cobalt sulfides by tuning thiourea reactivity, where faster decomposition favored sulfur-rich phases like CoS2, while slower decomposition rates produced sulfur-deficient phases.31 Similarly, the Hogarth group demonstrated that nickel dithiocarbamate decomposition pathways control phase selectivity between NiS and Ni3S4.34 Meng et al. proposed a thiourea-controlled model for Cu–Sb–S, where precursor concentration and temperature dictated oxidation states and phase outcomes.19 Despite these advances, the role of DESs with various sulfur precursors in phase control synthesis of TMCs remains unexplored.
Herein, we report a facile solvothermal route using a reline DES to obtain phase-pure, stable CuFeS2 with flower-like morphology, overcoming challenges of phase purity and stability. Its dielectric behavior (50–400 °C, 100 Hz–1 MHz), AC conductivity, and impedance were examined to probe charge transport, and OER performance was evaluated in 1 M KOH using a three-electrode setup.
2. Experimental section
2.1. Chemicals and materials
Choline chloride (>98%) and thiourea (>99%) were obtained from Tokyo Chemical Industry Co. Ltd (Tokyo, Japan). Cupric nitrate trihydrate (Cu(NO3)2·3H2O, >99.5%) and urea (99%) were purchased from Central Drug House (P) Ltd (New Delhi, India). Ferric nitrate nonahydrate (Fe(NO3)3·9H2O, >99%) was purchased from Merck. Thioacetamide (>99%), potassium hydroxide (KOH, 85%) and sodium sulphide flakes were purchased from Sigma-Aldrich and Sisco Research Laboratories (SRL) Pvt. Ltd – India, respectively. Carbon black (99.9%) was purchased from Otto Chemie, India, and Nafion D-521 (5 wt% in water and 1-propanol) was obtained from Alfa Aesar. Unless noted, all water used was distilled water. The chemicals mentioned above were used without further purification.
2.2. Preparation of the choline chloride–urea (ChCl-U) DES
The choline chloride–urea (reline) DES was prepared by mixing ChCl as a HBA and urea as a HBD in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio at 80 °C under continuous stirring until a homogeneous and colorless liquid was formed.
:2 molar ratio at 80 °C under continuous stirring until a homogeneous and colorless liquid was formed.
2.3. Preparation of the choline chloride-thiourea (ChCl-TU) DES
The ChCl-TU (thio-reline) DES was prepared by mixing ChCl as a HBA and TU as a HBD in a 1:2 ratio at 80 °C under continuous magnetic stirring until a homogeneous and transparent liquid was formed.
2.4. Synthesis of copper iron sulphide (CuFeS2)
In a typical procedure, 10 mL of reline DES was taken in the Teflon liner of the autoclave. Then 1 mmol of Cu(NO3)2·3H2O, 1 mmol of Fe(NO3)3·9H2O, and 4 mmol of thiourea (TU) were mixed homogeneously under magnetic stirring. Then, the above mixture was packed in a stainless-steel autoclave and heated at 200 °C for 12 h (Fig. 1). After that, the autoclave was cooled naturally to room temperature. Similar conditions were employed for other sulfur precursors [thioacetamide (TAA) and sodium sulfide (SS)] for the synthesis of Cu–Fe–S systems. With thio-reline as the DES, only the specified molar ratio of Cu and Fe precursors was added, as thio-reline inherently acts as an in situ sulfur source.
 |
| | Fig. 1 Schematic illustration of solvothermal synthesis of CuFeS2 using different sulphur precursors at 200 °C for 12 h. | |
2.5. Post-synthesis washing
The reaction mixtures were washed thrice under centrifugation with distilled water at 10000 rpm for 5 minutes. Due to the high viscosity of the reline DES, some DES remained adhered on the surface of powder particles. To free from the DES, the material was further washed five times with an ethanol–water mixture (1:3 ratio) and finally two additional washes with ethanol. The obtained material was dried at 60 °C overnight in a hot air oven. For materials synthesized in the thio-reline DES, washing with water was ineffective in removing the thio-reline DES. Therefore, ethanol washes were performed ten times under centrifugation (10000 rpm for 5 minutes), followed by drying in the same manner.
2.6. Dielectric spectroscopy measurement
The dielectric spectroscopy measurements were carried out with a conventional two-terminal sample holder using an LCR analyzer (Model PSM 1735 N4L) with variation of electric field frequency from 100Hz to 1 MHz over a 50–400 °C temperature range. A disk-shaped pellet with a diameter of 6 mm and a thickness of 1.1 mm was prepared by applying a uniaxial pressure of 5 tons. The pellet was then sintered at a temperature of 200 °C for 4 h and subsequently used for dielectric, conductivity, and impedance studies.
2.7. Electrochemical measurements
All electrochemical measurements were carried out in a three-electrode cell setup using a PGSTAT302N potentiostat/galvanostat (Metrohm Autolab). A mixture of 9 mg of the catalyst, 1 mg of carbon black, 35 µL of Nafion, and 150 µL of ethanol was evenly dispersed by ultrasonic mixing for 30 minutes. The catalyst ink (100 µL) was then applied onto a 1 cm × 1 cm of carbon cloth (CC) of 1.5 cm × 1 cm dimension using a pipette and dried at room temperature to serve as a working electrode. The drop-cast CC samples served as the working electrode, while a Pt wire was used as the counter electrode, and a Hg/HgO (1 M KOH) electrode acted as the reference electrode. The electrolyte used was a 1 M KOH solution saturated with nitrogen. RuO2 was used as a benchmark catalyst, tested under the same mass loading to evaluate catalytic activity. Before the OER tests, all samples were conditioned for 100 cycles at a scan rate of 200 mV s−1 using cyclic voltammetry (CV) to achieve a stable state. The linear sweep voltammetry (LSV) polarization curves for the sample were measured at a scan rate of 5 mV s−1 over a potential range of 0 to 1 V versus the Hg/HgO reference electrode. All reported potentials were referenced to the reversible hydrogen electrode (RHE) and converted using the Nernst equation: E(RHE) = E(Hg/HgO) + 0.0592pH + 0.098 V. Electrochemical impedance spectroscopy (EIS) measurements were performed in nitrogen-saturated 1.0 M KOH electrolyte, covering a frequency range from 104 to 0.1 Hz at an amplitude of 5 mV. The SI provides detailed insights into overpotential calculations, Tafel slope determination, and the evaluation of the electrochemical active surface area (ECSA), among other relevant aspects.
2.8. Materials characterization
Details regarding materials characterization methods are provided in the SI.
3. Results and discussion
3.1. Characterization of the reline DES
The reline DES was prepared by mixing choline chloride (ChCl) and urea (U) in a 1:2 molar ratio and heating the mixture at 80 °C until a homogeneous and transparent liquid was formed. Fig. S1 shows the FTIR spectra of ChCl, urea and the ChCl–urea (1:2) DES. The characteristic FTIR spectral pattern of the reline DES is essentially a combination of the spectral patterns of ChCl and urea. For pure urea, the bands at 3428 cm−1 and 3337 cm−1 are attributed to the anti-symmetric and symmetric N–H stretching vibrations, respectively. The band at 1675 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, while the band at 1457 cm−1 is associated with the C–N stretching vibration of urea. For ChCl, a distinct characteristic band centred at 3222 cm−1 corresponds to the O–H stretching vibration. The vibrational band at 1485 cm−1 is associated with the CH2 bending vibration of the alkyl chain in ChCl.35–37 However, the bands for anti-symmetric N–H stretching (3428 cm−1), symmetric N–H stretching (3337 cm−1), and O–H stretching vibrations (3257 cm−1) shifted to 3410, 3318, and 3202 cm−1, respectively, for the ChCl–urea DES, indicating hydrogen bonding between ChCl and urea. The hydrogen bonds in the ChCl–urea DES may exist as NH⋯NH, NH⋯OH, HO⋯HO and OH⋯NH interactions.38 Moreover, the band-related to νas CCO of ChCl at 954 cm−1 also exists in the reline DES spectrum, indicating that the structure of the choline cation (Ch+) is preserved in the reline DES. Furthermore, the proton nuclear magnetic resonance (1H-NMR) spectroscopic technique was used to investigate the interactions between ChCl as the HBA and urea as the HBD in the reline DES, as shown in Fig. S2–S4. The difference in chemical shifts between each proton in the DES and the neat components (ChCl or urea) was used effectively to get insights into the hydrogen bond interactions. This approach focuses on the chemical shift variation (Δδ) rather than the absolute chemical shift (δ) to describe and analyze these spectroscopic changes. Table S1 presents the chemical shifts of protons in the O–H and N–H groups of ChCl, urea, and the corresponding DESs, along with the associated Δδ values. In the context of an X–H⋯Y hydrogen bond, a decrease in electron density around the proton in the X–H bond leads to a downfield shift, whereas an increase in electron density causes an upfield shift.39 It was demonstrated that when ChCl and urea form the DES, the chemical shift of the proton in the O–H group of ChCl decreases. This indicates that the hydrogen bond between the Cl atom in ChCl and the H atom in the N–H of urea weakens the hydrogen bonding between ChCl molecules themselves. As a result, the electron density around the proton in the O–H group of ChCl increases, leading to an upfield shift. However, for the H proton in the N–H group of urea, the observed downfield shifts suggest that the hydrogen bond between the Cl atom of ChCl and the H atom of the N–H group in urea is stronger than the hydrogen bonds between urea molecules, which results in a decrease in electron density around the proton. These findings align with the conclusions drawn from the FT-IR analysis of the DESs. Hence, the presence of hydrogen bonding interactions and the successful formation of the reline DES are concluded.
O stretching, while the band at 1457 cm−1 is associated with the C–N stretching vibration of urea. For ChCl, a distinct characteristic band centred at 3222 cm−1 corresponds to the O–H stretching vibration. The vibrational band at 1485 cm−1 is associated with the CH2 bending vibration of the alkyl chain in ChCl.35–37 However, the bands for anti-symmetric N–H stretching (3428 cm−1), symmetric N–H stretching (3337 cm−1), and O–H stretching vibrations (3257 cm−1) shifted to 3410, 3318, and 3202 cm−1, respectively, for the ChCl–urea DES, indicating hydrogen bonding between ChCl and urea. The hydrogen bonds in the ChCl–urea DES may exist as NH⋯NH, NH⋯OH, HO⋯HO and OH⋯NH interactions.38 Moreover, the band-related to νas CCO of ChCl at 954 cm−1 also exists in the reline DES spectrum, indicating that the structure of the choline cation (Ch+) is preserved in the reline DES. Furthermore, the proton nuclear magnetic resonance (1H-NMR) spectroscopic technique was used to investigate the interactions between ChCl as the HBA and urea as the HBD in the reline DES, as shown in Fig. S2–S4. The difference in chemical shifts between each proton in the DES and the neat components (ChCl or urea) was used effectively to get insights into the hydrogen bond interactions. This approach focuses on the chemical shift variation (Δδ) rather than the absolute chemical shift (δ) to describe and analyze these spectroscopic changes. Table S1 presents the chemical shifts of protons in the O–H and N–H groups of ChCl, urea, and the corresponding DESs, along with the associated Δδ values. In the context of an X–H⋯Y hydrogen bond, a decrease in electron density around the proton in the X–H bond leads to a downfield shift, whereas an increase in electron density causes an upfield shift.39 It was demonstrated that when ChCl and urea form the DES, the chemical shift of the proton in the O–H group of ChCl decreases. This indicates that the hydrogen bond between the Cl atom in ChCl and the H atom in the N–H of urea weakens the hydrogen bonding between ChCl molecules themselves. As a result, the electron density around the proton in the O–H group of ChCl increases, leading to an upfield shift. However, for the H proton in the N–H group of urea, the observed downfield shifts suggest that the hydrogen bond between the Cl atom of ChCl and the H atom of the N–H group in urea is stronger than the hydrogen bonds between urea molecules, which results in a decrease in electron density around the proton. These findings align with the conclusions drawn from the FT-IR analysis of the DESs. Hence, the presence of hydrogen bonding interactions and the successful formation of the reline DES are concluded.
3.2. Selection of metal salt precursors and sulfur precursors and the role of the DES in the synthesis of CuFeS2
Precursor selection is key in solvothermal synthesis, enabling controlled nucleation of the desired multinary phase while suppressing competing binary phases and impurities.40 Cu(NO3)2·3H2O and Fe(NO3)3·9H2O were used as the copper and iron precursors, respectively, with the reaction at 200 °C for 12 h. Hydrated metal salts of copper and iron were chosen for the synthesis to enhance the solubility of the precursors in the DES. Additionally, even a tiny amount of water from these hydrated metal salts within the DES matrix can boost the DES's fluidity, conductivity, and polarity.41–43 This, in turn, promotes the nucleation and growth of metal chalcogenide nanocrystals in the DES solvent. Moreover, selecting Fe and Cu nitrate salts is advantageous for achieving phase-pure synthesis of ternary Cu–Fe–S systems due to their similar counter anions. This similarity facilitates a comparable decomposition rate, promoting the uniform incorporation of Fe and Cu cations into the developing nanocrystals.
In our synthetic approach, different sulfur precursors such as thiourea (TU), thioacetamide (TAA), and sodium sulfide (SS) were chosen, each differing in their chemical reactivity and the rate at which they release sulfur, keeping copper and iron precursors fixed (Fig. 1).44,45 Initially, the Cu:Fe mmol ratio was fixed at 1:1, a deliberate choice to align with the chemical formula of chalcopyrite (CuFeS2) and to minimize the formation of secondary phases. Keeping the Cu:Fe ratio at 1:1, the TU concentration (as the sulfur source) was systematically varied from 1 to 8 mmol (Fig. S5 and S6). It was observed that at lower TU concentration (less than 4 mmol), sulphur deficient phases such as Cu5FeS4 (bornite) were evolved along with CuFeS2, as evidenced from the PXRD patterns (Fig. S5). Favorably, phase pure chalcopyrite was formed when 4 mmol of TU was used (Fig. 2). Beyond 4 mmol of TU, the product contained chalcopyrite together with secondary phases as well excess sulfur (Fig. S6). Thus, appropriate control of the TU concentration is thus key for synthesizing phase-pure CuFeS2 with a Cu:Fe ratio of 1:1, preventing unwanted secondary sulphide phases that arise from sulfur imbalance. Further experiments were performed by varying the Cu:Fe mmol ratio while maintaining the thiourea (TU) concentration at 4 mmol (Fig. S7). At a Cu:Fe ratio of 2:1, a mixture of CuFeS2 and Cu5FeS4 was obtained, whereas at a Cu:Fe ratio of 1:2, CuFeS2 coexisted with Cu2Fe4S7 (Fig. S7). These findings demonstrate that any deviation from the stoichiometric (Cu:Fe:TU)1:1:4 ratio promotes the formation of secondary sulfide phases alongside chalcopyrite, highlighting the critical role of metals and sulfur precursor ratios in dictating phase purity during synthesis. Additionally, solvothermal synthesis was carried out at 200 °C for 12 h with a Cu:Fe ratio of 1:1, employing 4 mmol of either Na2S or TAA as the sulfur source in separate trials (Fig. S8). In both cases, phase-pure CuFeS2 did not form. To further assess the role of reaction duration, the synthesis time was extended to 24 h; however, even under these prolonged conditions, neither Na2S nor TAA yielded phase-pure CuFeS2 (Fig. S9 and S10). These results emphasize that the choice of sulfur precursor is a decisive factor in achieving phase-pure CuFeS2.
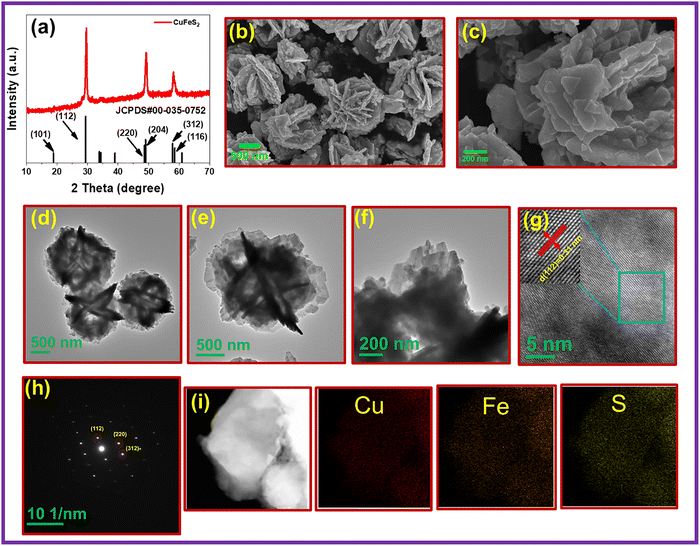 |
| | Fig. 2 (a) PXRD pattern, (b and c) FE-SEM micrographs, (d–f) TEM, (g) HRTEM and inverse FFT, (h) SAED pattern, and (i) elemental mapping of phase pure CuFeS2 synthesized in reline using TU as a sulfur precursor. | |
Na2S, being ionic, quickly dissociates to release sulfur species. It is also well known that TU is less reactive and decomposes at a higher temperature than TAA.46 Thus, TU, TAA, and Na2S decompose when the precursor solution (in DES) is heated, releasing S2− ions, as shown in eqn (1)–(3).47
| | | NH2CSNH2 + 2OH− → CH2N2 + S2− + 2H2O | (1) |
| | | CH3CSNH2 + 2OH− → CH3COOH+ S2− + NH3 | (2) |
TU and TAA both contain –CS and –NH2 functional groups. However, due to two –NH2 groups, TU forms stronger bonds with metal precursors or growing nuclei than TAA. This strong bonding can inhibit secondary crystallization and prevent nanoparticles from adsorbing onto the surface of primary nanocrystals. Therefore, we believe that the variations in phase evolution when using TU and TAA as sulfur precursors can be attributed to the influence of these functional groups. Unlike the organic sulfur precursors TU and TAA, Na2S directly releases S2− ions into the solution without undergoing reactions with hydroxide ions (OH−) in the basic reline DES (eqn (1)–(3)), resulting in an abundance of S2− ions in the solution before the solvothermal reaction starts.47 TU can rapidly decompose at temperatures above 100 °C in a basic solvent, producing ample S2− ions. When TU is used as the sulfur precursor, all the diffraction peaks of the as-synthesized material correspond to the tetragonal phase of CuFeS2 (Fig. 2a). The diffraction peaks can be indexed to the tetragonal phase of chalcopyrite with JCPDS# no 00-035-0752 (Fig. 2a). It is essential to mention that the XRD pattern does not exhibit any discernible peaks associated with CuS, Cu2S, FeS2, Fe3S4, or any oxide/hydroxides. No peaks associated with impurity phases of copper or iron are observed, indicating that the material is phase-pure. On the other hand, when TAA was used, the synthesized material contained CuFeS2 and impurity phases of CuS and CuFe2S3 (Fig. S8). Similarly, with Na2S as the sulfur precursor, the PXRD pattern of the synthesized material revealed phases including CuFeS2, CuFe2S3, CuS, and FeS (Fig. S8). Under optimized solvothermal conditions (200 °C, 12 h), the thio-reline DES (ChCl–TU) was employed as a solvent, template, and in situ sulfur source for CuFeS2 synthesis. PXRD revealed the formation of a mixed Cu–Fe–S phase comprising CuFeS2 and CuFe2S3, the latter containing both Fe2+ and Fe3+ (Fig. S11). The above discussion demonstrates that different sulfur precursors yield varying product phase selectivity under similar synthetic conditions. The Raman spectra of as-synthesized CuFeS2 exhibit three distinct peaks at 219, 279, and 393 cm−1, corresponding to S(0) lattice vibrations, Cu(I)–S stretching, and Fe(III)–S stretching, respectively (Fig. S12). Fig. 2b and c display the FE-SEM micrographs captured in the InLens mode, highlighting a micro flower-like morphology. In these, triangular nanoplates are stacked together to form larger, irregularly shaped nanostructures. This stacking occurs due to Ostwald ripening (OR), which results from the extended reaction time (12 h) in the solvothermal synthesis process. The TEM micrographs also reveal the flower-like morphology consisting of triangular nanoplates (Fig. 2d–f). The HR-TEM, SAED pattern, and inverse FFT shown in Fig. 2g and h demonstrate that the CuFeS2 nanocrystals lack noticeable dislocations or stacking faults. The presence of distinct diffraction spots in the SAED pattern in Fig. 2h indicates their single-crystalline nature, with an interplanar spacing of 0.31 nm (Fig. 2g), corresponding to the (112) plane of the tetragonal phase of CuFeS2. Furthermore, STEM-EDS mapping was employed to confirm the uniform distribution of Cu, Fe, and S throughout the nanoplates (Fig. 2i). Fig. 3a shows the full survey X-ray photoelectron spectroscopy (XPS) spectrum, revealing the presence of Cu, Fe, S, C, and O elements in the material. All binding energies were referenced to the C 1s peak at 284.8 eV. The presence of C and O in the material can be attributed to either atmospheric adsorption or the solvent used during synthesis. In the core level Cu 2p spectrum, the peaks at Cu 2p3/2 (931.9 eV) and Cu 2p1/2 (951.7 eV), with a spin–orbit splitting of 19.8 eV, correspond to the Cu+ oxidation state (Fig. 3b). The peaks at Cu 2p3/2 (933.4 eV) and Cu 2p1/2 (954.9 eV) correspond to the Cu2+ oxidation state. The satellite peak at 948.5 eV in the Cu 2p spectrum further confirms the presence of Cu2+.48 The core-level Fe 2p spectrum shows two sets of main spin–orbit doublets. The peaks at 709.1 and 720.3 eV are assigned to the Fe2+ 2p3/2 and 2p1/2 states, respectively, while the peaks at 711.4 and 725.2 eV correspond to the Fe3+ 2p3/2 and 2p1/2 states (Fig. 3c). In addition to these main features, the peak at 714.6 eV is attributed to a satellite associated with Fe2+, whereas the higher binding energy peaks at 729.4 and 731.9 eV arise from characteristic satellite features of Fe3+ species.49 Thus, deconvoluted Fe 2p peaks show the coexistence of Fe2+ and Fe3+ species, consistent with the mixed oxidation state of Fe in chalcopyrite (Fig. 3c). In the S 2p core-level spectrum, the deconvoluted peaks at 161.1 and 162.3 eV with a spin–orbit splitting of 1.2 eV are assigned to the S 2p3/2 and S 2p1/2 components of S2−species, respectively.50
 |
| | Fig. 3 Survey spectrum (a) and high-resolution XPS full spectrum of (b) Cu 2p; (c) Fe 2p; (d) S 2p; (e) C 1s; and (f) O 1s of CuFeS2. | |
In addition, the peaks located at 163.0 and 164.2 eV correspond to the S 2p3/2 and S 2p1/2 components of disulfide (S22−) species, respectively (Fig. 3d). The peak observed at 168.9 eV in the S 2p spectrum corresponds to surface-oxidized sulfur species (–SOx).28 Moreover, the C 1s spectrum exhibits a dominant peak at 284.8 eV attributed to C–C/C–H, along with components at 286.3 eV corresponding to C–O and at 288.8 eV assigned to O–CO functional groups (Fig. 3e). The O 1s spectrum shows a peak at 529.9 eV attributed to lattice Cu/Fe–O species in CuFeS2, a component at 531.2 eV assigned to Cu/Fe–OH groups, a peak at 531.8 eV corresponding to adsorbed OH−/C–O species, and a higher-binding-energy feature at 532.4 eV associated with oxidized sulfur (sulfate) species (Fig. 3f). In conclusion, Cu, Fe, and S are present in mixed valence states, with Cu existing as Cu+/Cu2+, Fe as Fe2+/Fe3+, and S as S2−/S22−, along with a minor contribution from surface-oxidized sulfur (–SOx–) species. The thermal stability of CuFeS2 was evaluated by TGA under N2, showing only ∼7% weight loss up to 800 °C (Fig. S13), indicating excellent thermal stability. Additionally, heating the nanocrystals in 1-octadecene at 300 °C for 2.5 h caused no phase transition or decomposition, as confirmed by PXRD (Fig. S14).51 To assess moisture durability, the multinary chalcogenide was stirred in water for 48 h and compared with a freshly prepared reference; PXRD revealed degradation of CuFeS2 with the formation of CuS impurities after water exposure (Fig. S15). Moreover, CuFeS2 exhibits a type IV isotherm with H3 hysteresis, a BET surface area of 19.3 m2 g−1, an average pore size of 3.4 nm, and a total pore volume of 0.343 cm3 g−1, indicating a predominantly mesoporous structure (Fig. S16a and b). The optical properties of CuFeS2 nanocrystals were examined by UV-vis-NIR diffuse reflectance spectroscopy (200–1400 nm). As shown in Fig. S16c, the reflectance decreases gradually from 1400 to 650 nm and drops sharply between 650 and 400 nm due to strong absorption by CuFeS2 nanocrystals. The estimated direct band gap for the as-synthesized CuFeS2 was found to be 2.6 eV (Fig. S16d). This optical band gap value is similar to that reported by Teranishi et al.52 and much higher than the band gap (0.6 eV) of bulk CuFeS2. The refractive index and optical bandgap are key parameters for optoelectronic applications. CuFeS2 exhibits high refractive index values (2.46–2.54, Table S3), which favor enhanced light absorption and optical confinement, desirable properties for photodetectors and solar cells. Additionally, the clear difference between dark and illuminated I–V curves indicates photocarrier generation under light illumination, highlighting the potential of CuFeS2 for optoelectronic applications (Fig. S17 and 18).
3.3. Insights into the role of DES in the synthesis of CuFeS2
To understand the role of reline DES in the synthesis of CuFeS2, additional experiments were conducted to identify the gaseous species generated during high-temperature synthesis. A moist litmus test confirmed the liberation of alkaline gases (may be NH3) by the decomposition of reline (eqn (4)).53| | | NH2CONH2 + H2O → 2NH3 + CO2 | (4) |
The generated ammonia may partially react with residual water to form NH4+ and OH− ions.
| | | NH3 + H2O → NH4+ + OH− | (5) |
This hydroxide generated in situ causes alkalinity of the reline DES by shifting the pH (eqn (5)). Also, colorimetric investigations utilizing Berthelot's reaction indicated the existence of ammonium ions (NH4+) in pure reline, likely generated during its preparation by heating the mixture of choline chloride and urea solids to 80 °C (Fig. S19). NH3, OH− and Cl− (derived from choline chloride) may react with precursor metal ions to form (Cu(NH3)x(OH)yClz) and (Fe(NH3)x(OH)yClz) species, which undergo decomposition and further reaction with in situ generated sulfur species by the decomposition of TU to form Cu–Fe–S nuclei. Also, the reline DES acts as a preorganizer and a directing agent for reactive components within the reaction medium. Thus, reline acts as a solvent, reactant, and structural directing agent for the growth of nanostructured CuFeS2. Meanwhile, the coordination of TU to metal precursors forms Cu–TU and Fe–TU complexes, which act as a reservoir for Cu and Fe ions, thereby restricting the availability of free metal precursors.54–56 At elevated temperatures, the complexes decompose, releasing ions gradually to regulate nucleation and form CuFeS2 crystallites. Moreover, TU decomposition generates a reducing environment inside the sealed autoclave under solvothermal conditions,19 which converts Cu2+ to Cu+ during synthesis, which then incorporates into the growing nanocrystal nuclei, facilitating the formation of the CuFeS2. This reduction is typically accompanied by the dissolution and oxidation of TU, resulting in the formation of formamidine disulfide and other intermediate products.19 Thus, TU functions as a sulfur source and as a coordinating and reducing agent. Initially, thiourea might undergo isomerization, and in this process, nitric acid is generated as a by-product (Scheme 1), and its accumulation can hinder the further growth of crystallites.54 We propose that the OH− produced by urea hydrolysis with residual water of metal salt precursors in the DES will neutralize the acid, inhibiting the accumulation of acids and facilitating desired reactions. TAA forms a less stable complex with metal precursors, which suggests reduced control over the release of metal ions during nanocrystal growth. Consequently, using TAA as the sulfur precursor results in lower phase selectivity in the resulting product compared to TU. In the case of Na2S, the rapid release of sulfide leads to a lack of control over nucleation and growth processes, which is primarily responsible for non-selective product formation. As a result, multiple products, including binary phases of Cu and Fe, were formed rather than phase-pure CuFeS2, as evident from PXRD analysis (Fig. S8).
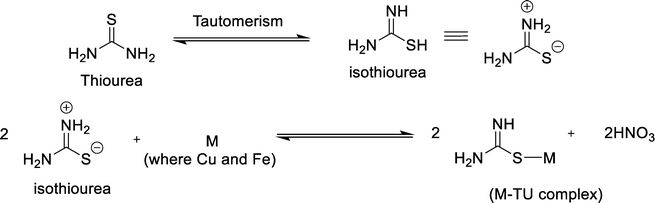 |
| | Scheme 1 Illustration of tautomerism and coordination of TU to metal ions in the precursor solution. | |
3.4. Insights into phase evolution of CuFeS2 through time-dependent ex situ PXRD
Solvothermal reactions were performed using the reline DES as the solvent and TU as the sulfur source at 200 °C, with reaction times varied from 1 to 12 h. Ex situ PXRD patterns were recorded for materials obtained after washing and drying the products from each hourly reaction to track the phase evolution of CuFeS2 (Fig. S20). After 1 hour, the PXRD pattern of the isolated powdered material indicated the concurrent presence of CuS2, Cu9S5, CuFeS2, CuS, and FeS1.7 phases. Up to 9 hours, similar mixed phases of copper and iron compounds were detected, with variations in PXRD peak intensities, suggesting the interconversion of CuS, CuS2, Cu9S5, and FeS1.7 binary phases. Notably, after 10 h, the CuS, CuS2 and Cu9S5 phases disappeared, though the FeS1.7 phase persisted, as evidenced by the XRD peak at 47.4°. This suggests that CuxSy interacted with Fe phases and became integrated into the growing Cu–Fe–S nucleus. After 11 hours of reaction, the PXRD peak at 29.5° associated with the CuFeS2 phase intensified, while a weak diffraction peak corresponding to FeS1.7 remained present in the product. Additionally, the impurity peaks disappeared as the reaction time increased, resulting in a pure tetragonal phase of CuFeS2. This shift indicates a transition from a mixed-phase binary or ternary chalcogenide to a single-phase ternary CuFeS2. Actually, in the early stages of the reaction (1–10 h), multiple intermediate sulfide phases such as CuS, CuS2, FeS1.7, and Cu9S5 coexist, reflecting incomplete reaction and metastable phase formation. As the reaction progresses, these intermediates undergo further transformation, enabling the nucleation and growth of the thermodynamically stable CuFeS2 phase. At this stage, secondary phases are gradually consumed, leading to complete conversion into the target chalcopyrite structure. Such behavior is characteristic of solvothermal synthesis, where an extended reaction time facilitates the transformation of metastable intermediates into a phase-pure final product.
3.5. Thermostability testing of the reline DES
To assess the thermostability, the reline DES was heated for different time intervals at 200 °C, and the resulting DES was evaluated for thermostability by visual and spectroscopic methods. After heating at 200 °C for one hour, there was no change in the colour or state of the DES, and no charring or particulates were observed (Table S2). The same characteristics were observed after three hours of heating. Still, after 6 hours, the DES turned yellowish, with no signs of particulate matter or char, and emitted a strong, pungent ammonia-like odor, indicating slight decomposition of the DES. After 6 hours, the DES remains liquid and remains so even after 12 hours of heating. This visual observation is complemented by time-dependent ATR-FTIR analysis of liquids. Reline exhibits absorptions at 3320, 3193, 2960, 1636, 1603, 1436, 1150, 952, and 784 cm−1 corresponding to the symmetric N–H stretch, N–H and O–H stretch of amine and hydroxyl, –CH3 stretch, CO stretch for amide, N–H scissoring band, C–N and C–O of urea, –CCO of Ch+ and N–H bending mode of urea (Fig. S1 and S21). There are no noticeable changes in the absorption frequencies of various functional groups in the ChCl–U DES after being heated at 200 °C for 12 hours. This indicates the DES's remarkable stability at 200 °C during extended heat treatment. However, as the heating duration increases from 1 to 12 hours, the stretching frequency of the N–H bending mode shifts to a lower wavenumber. This could be attributed to changes in the number of hydrogen bonds between the DES components. Additionally, the colour gradually shifts from colorless to light yellow and eventually to deep yellow, which is attributed to the decomposition of urea and the subsequent formation of biurets. This change indicates the chemical transformations within the DES during heating.
4. Dielectric response study
The dielectric properties of materials are characterized by the complex permittivity (ε) expressed in complex form as in eqn (6):where εr is the real part of the complex permittivity (also called the dielectric constant) while εi is the imaginary part of the complex permittivity. The real part of the dielectric constant reflects the energy stored in the dielectric due to polarization when subjected to an electric field. At the same time, the imaginary part assesses the energy dissipated within the dielectric as a consequence of the applied electric field. The dielectric constant (εr) is evaluated by using eqn (7):| | 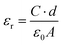 | (7) |
where d is the pellet (or sample) thickness, A is the sample area, C is the measured capacitance, and ε0 is the permittivity of the free space (or vacuum dielectric constant) (8.854 × 10−12 F m−1). Chemical reactions within the material induce modifications in its bonding characteristics, impacting its electrical polarizability. This resulted in a shift in the total energy stored and dissipated under applied electric fields, quantified by the dielectric loss parameter (tan(δ)). It is given as the ratio of the imaginary part ε″ and the real part ε′, i.e., tan(δ) = ε″/ε′.
4.1. Variation of the dielectric constant and dielectric loss with frequency at different temperatures
Fig. 4a and b show the frequency dependence of the dielectric constant and dielectric loss tangent (tanδ) of the CuFeS2 material. As the frequency increases, the εr values exhibit a declining trend and stabilize at higher frequencies (Fig. 4a). This is attributed to the reduced ability of electric dipoles to respond quickly to abrupt changes in the frequency of the applied electric field at higher frequencies.57 According to the Maxwell–Wagner theory, all the polarization mechanisms, such as space charge, dipolar, and ionic polarization, contribute to the dielectric strength at lower frequencies. As the frequency rises, the space charge polarization has insufficient time to keep pace with the applied external frequency of the electric field. Consequently, it relaxes, resulting in a decrease in the dielectric properties of the material as the frequency increases. Thus, dipolar and interfacial polarization's contribution decreases with a frequency rise. Fig. 4a also shows that the dielectric constant increases with the rise in temperature with respect to frequency. Rising temperatures result in increased mobility of charge carriers, amplifying the polarization within the material and leading to elevated dielectric constant values.
 |
| | Fig. 4 Plots of (a) dielectric constant vs. frequency, (b) tan(δ) vs. frequency, (c) dielectric constant vs. temperature, and (d) tan(δ) vs. temperature of CuFeS2. | |
The dielectric loss tangent (tanδ) represents the energy dissipation that occurs when polarization lags behind the applied electric field, primarily due to the presence of grain boundaries.58Fig. 4b illustrates the variation of dielectric loss with frequency at different temperatures. It is observed that dielectric loss (tanδ) decreases as frequency increases at all temperatures. Dielectric loss (tanδ) primarily arises from the hopping of localized electric charge carriers and the formation of defect-induced dipoles. The high dielectric loss at low frequencies is mainly due to the hopping of localized charge carriers. In the low-frequency range, resistive grain boundaries are more effective. This increases energy requirements for exchanging charge carriers through cation–anion–cation (c–a–c) interactions, increasing dielectric loss.58 At high frequencies, conductive grains dominate, requiring less energy for charge carrier exchange, which results in lower energy loss. It is evident that dielectric loss diminishes as the frequency increases and stabilizes in the higher frequency range across all temperatures. The tanδ versus frequency plot also demonstrates that the dielectric loss tangent values increase with temperature at each specific frequency. This rise is likely due to the enhanced conduction of thermally activated charge carriers.
4.2. Variation of the dielectric constant and dielectric loss with the temperature at different frequencies
Fig. 4c illustrates the temperature-dependent variation of the dielectric constant across various frequencies within the range of 1 kHz to 1 MHz. It is observed that the dielectric constant increases with an increase in temperature. With rising temperatures, thermal energy enhances the mobility or hopping rate of the charge carriers, which improves the formation of more dipoles within the material and can more easily align with the applied electric field, leading to greater dielectric polarization and, consequently, a higher dielectric constant. Interestingly, the temperature dependence of the dielectric constant varies across different frequencies. At 1 kHz, the dielectric constant increases more rapidly than other frequencies. The four polarization types, interfacial, dipolar, ionic, and electronic, contribute to the overall dielectric constant. The elevated dielectric constant at 1 kHz is attributed to interfacial and dipolar polarization, which are strongly dependent on frequency and temperature.42 At frequencies above 105 Hz, only electronic and ionic polarization contribute to the dielectric constant, and this contribution is temperature-independent. As a result, the temperature has minimal impact of high-frequency at dielectric constants, leading to low dispersion in the dielectric constant. Fig. 4d shows how dielectric loss varies with temperature across different frequencies of 1 kHz, 10 kHz, 100 kHz, 500 kHz, and 1 MHz. It can be observed that dielectric loss increases sharply with rising temperature at each frequency. The sharp increase in tanδ at higher temperatures can be attributed to lattice vibrations caused by the rising temperature.59 These vibrations create phonons, which interact with thermally activated charge carriers, leading to increased scattering and, consequently, a significant rise in tanδ. The high dielectric loss could also be attributed to crystal defects and the dominance of conductivity at elevated temperatures.60 As the temperature rises, charge carriers become more mobile, increasing dielectric loss and polarization. Furthermore, the high dielectric loss at elevated temperatures may be due to charge accumulation at the grain boundaries.61 As these carriers gather at grain boundaries, they create localized regions of high charge density. This accumulation can increase the resistance and cause more energy dissipation as heat. As the temperature rises, the tanδ versus temperature plot shows a decline after reaching a peak value at all frequencies. This behavior is characteristic of relaxation losses. As the frequency increases, charge carriers struggle to keep up with the rapidly changing alternating field, resulting in decreased polarization. The system needs additional energy to restore polarization, which can be supplied by increasing the temperature. Therefore, higher temperatures are required to restore polarization as the frequency rises. This results in a shift of the dielectric loss maxima towards higher temperatures as the frequency increases.
4.3. AC conductivity analysis
AC conductivity (σac) contributes to the material polarizability and is calculated from dielectric measurements using the following relation (eqn (8)):| | σac = ω![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) tanδεrεo tanδεrεo | (8) |
where σac is the AC conductivity, ω is the angular frequency, ε0 is the permittivity of the free space, εr is the real part of the dielectric constant, and tanδ is the dielectric loss.58Fig. 5a illustrates the behavior of the AC conductivity when subjected to varying frequencies at different temperatures. At lower frequencies, the AC conductivity increases gradually due to the ineffective impact of the electric field on the mobility of charge carriers. However, as the frequency increases, the acceleration in AC conductivity becomes more pronounced. Conductivity arises from the hopping of charges, and it increases with rising temperature. The charge carriers' hopping rate is low at lower frequencies, resulting in lower AC conductivity. At elevated frequencies, the hopping rate of charge carriers increases; consequently, there is greater AC conductivity. Fig. 5b depicts the change in AC conductivity with the inverse of temperature at different chosen frequencies. The AC conductivity increases with an increase in temperature due to increased drift mobility of the charged carriers, which subsequently increases charge hopping. Each frequency has a distinct slope in different temperature regions, as observed in Region I and Region II.
 |
| | Fig. 5 Plots of (a) AC conductivity vs. frequency at various temperatures; (b) temperature response of AC conductivity of CuFeS2 upon varying the frequency. | |
In the low-temperature region (Region II), the AC conductivity values increase gradually as the temperature rises. In contrast, in the high-temperature region (Region I), the conductivity experiences a more pronounced rise with temperature. This suggests the existence of multiple conduction mechanisms.58 At low temperatures, the mobility of charge carriers is generally reduced because the thermal energy available to move the carriers is limited. This results in lower AC conductivity. The activation energy required for charge carriers to overcome the band gap or potential barriers is higher at low temperatures, leading to lower conductivity. At high temperatures, the band gap between the valence and conduction bands seems to be narrow, facilitating easier electron transitions from the valence band to the conduction band. This results in higher conductivity values compared to lower temperatures. Dielectric relaxation processes may be slower at low temperatures, affecting the AC conductivity by limiting the rate at which charge carriers can respond to the alternating electric field. Faster dielectric relaxation processes at higher temperatures enable charge carriers to follow the alternating field more effectively, thereby enhancing AC conductivity.
4.4. Complex impedance spectroscopic analysis
To garner additional insights into the electrical transport mechanism, impedance measurements were conducted as a function of frequency across a range of temperatures. At a specific frequency, the complex impedance (Z) can be represented by its real and imaginary components as shown in eqn (9):where Z′ and Z″ are the real and imaginary parts of impedance, respectively. This is represented as Z′ = Zcosθ and Z″ = Zsinθ. The real part of impedance (Z′) describes the resistance to direct current (DC) or the resistive component of the material. It is associated with the energy dissipation in the material due to resistive losses. The imaginary part of impedance (Z″) represents the reactance, which is related to the storage and release of electrical energy in the material.
Fig. 6a and b depicts the change in Z′ and Z″ with frequency across 100 Hz to 1 MHz and over a temperature range from 50 °C to 400 °C. The Z′ values decrease with increase in both frequency and temperature. The graph shows a sigmoid trend in the low-frequency region and a plateau or saturation zone at high frequencies. The reduction in Z′ with increasing frequency is attributed to the rise in AC conductivity. Additionally, as shown in Fig. 6a, the value of Z′ remains nearly zero at 300 °C and 400 °C. As frequency and temperature increase, Z′ decreases steadily, and at higher frequencies, it levels out across all temperatures. This behavior is due to the loss of space-charge polarization at elevated frequencies. Fig. 6b illustrates how imaginary impedance varies with the frequency of the applied field. The imaginary part of impedance Z″ increases with frequency, reaches a peak value  , and decreases steadily with further increases in frequency. The peak maxima of imaginary impedance, i.e.,
, and decreases steadily with further increases in frequency. The peak maxima of imaginary impedance, i.e., 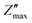 in dielectrics, typically indicate the frequency at which the material exhibits maximum dielectric loss. The magnitude of the peak represents the maximum energy dissipation due to dielectric losses at a specific frequency. Higher peaks suggest more significant energy loss in the dielectric material at that frequency. The frequency corresponding to the
in dielectrics, typically indicate the frequency at which the material exhibits maximum dielectric loss. The magnitude of the peak represents the maximum energy dissipation due to dielectric losses at a specific frequency. Higher peaks suggest more significant energy loss in the dielectric material at that frequency. The frequency corresponding to the 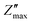 is referred to as the relaxation frequency, fmax. Here,
is referred to as the relaxation frequency, fmax. Here, 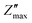 shifts toward higher frequencies as the temperature increases, suggesting the presence of non-Debye relaxation phenomena in the samples.62 This relaxation behaviour can be attributed to charge accumulation at the grain boundaries, which become polarized in response to the applied electric field. As the temperature rises, the value of
shifts toward higher frequencies as the temperature increases, suggesting the presence of non-Debye relaxation phenomena in the samples.62 This relaxation behaviour can be attributed to charge accumulation at the grain boundaries, which become polarized in response to the applied electric field. As the temperature rises, the value of 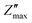 decreases, indicating a reduction in the material's resistive properties and an increase in the mobility of charge carriers within the material. This results in more efficient charge transport and, consequently, a reduction in the imaginary impedance's peak value, reflecting energy loss due to charge movement. Additionally, the broadening of the peak with increasing temperature suggests the presence of multiple relaxation processes.58
decreases, indicating a reduction in the material's resistive properties and an increase in the mobility of charge carriers within the material. This results in more efficient charge transport and, consequently, a reduction in the imaginary impedance's peak value, reflecting energy loss due to charge movement. Additionally, the broadening of the peak with increasing temperature suggests the presence of multiple relaxation processes.58
 |
| | Fig. 6 (a) Plot of (Z′) vs. frequency and (b) Z″ vs. frequency at different temperatures. | |
5. Electrochemical OER performance of CuFeS2 electrodes
The OER is key to numerous energy-related applications, including water electrolysis for H2 production, metal–air batteries, and CO2 reduction electrolyzers. To evaluate the OER performance of CuFeS2, comprehensive linear sweep voltammetry (LSV) tests were conducted at a scan rate of 5 mV s−1 in a standard three-electrode system using 1 M KOH as the alkaline electrolyte (Fig. 7a–f). The carbon cloth (CC) substrate shows no OER activity, as evident from the LSV, with the current density failing to reach the benchmark value of 10 mA cm−2 (Fig. 7a). Analysis of the OER polarization curves revealed that CuFeS2 required an overpotential of 360 mV to achieve a current density of 10 mA cm−2, as illustrated in Fig. 7a. The OER performance of the CuFeS2 electrode is at par with or surpasses that of other previously reported electrocatalysts, as summarized in Table S4. Under the same catalyst loading, the benchmark RuO2 catalyst exhibits an overpotential of 270 mV (Fig. 7a and b). To gain deeper insights into the OER kinetics, the Tafel slope (mV dec−1) was analyzed. A lower Tafel slope indicates faster reaction kinetics and, consequently, greater electrochemical activity. The Tafel slope values calculated for CuFeS2 and RuO2 are 148.7 and 90 mV dec−1, respectively (Fig. 7c).
 |
| | Fig. 7 Electrocatalytic OER performance of CuFeS2 and the benchmark catalyst RuO2: (a) LSV curves recorded at a scan rate of 5 mV s−1; (b) overpotential at 10 mA cm−2; (c) Tafel slopes; (d,e) CV curves at different scan rates in the non-faradaic region for CuFeS2 and RuO2, respectively; (f) current density versus the scan rate used to determine Cdl and the corresponding (g) ECSA; (h) Nyquist plots; and (i) long-term stability test of the CuFeS2 electrode at 10 mA cm−2 for 125 h. | |
The electrochemical surface-active sites (ECSA) of the electrode material are determined by identifying a non-faradaic CV region. Cyclic voltammetry (CV) is performed at various scan rates, ranging from 20 to 100 mV s−1, within this non-faradaic region, as depicted in Fig. 7d and e. The half-difference between the anodic and cathodic current densities (ΔJ = (Ja − Jc)/2) was plotted against the scan rates at the central potential, and a linear relationship was observed between the current densities and scan rates, as evident from Fig. 7f. From the slope of this plot, the double-layer capacitance (Cdl) was calculated to be 35.25 mF cm−2 for CuFeS2 and 42.77 mF cm−2 for RuO2. Since the ECSA is directly proportional to Cdl, a larger Cdl signifies a larger ECSA. The calculated ECSAs for CuFeS2 and RuO2 are 881 and 1069.3 cm2 respectively (Fig. 7g). EIS studies were performed to investigate the OER activities of CuFeS2 further, as shown in Fig. 7h. The Nyquist plots show semicircular curves, with the diameter of CuFeS2 representing the charge transfer resistance (Rct) between the catalyst surface and the solution. The intercept of the semicircle with the real axis in the high-frequency region corresponds to the solution resistance (Rs). The Nyquist plot is analyzed using a hypothetical equivalent circuit model (Randles circuit) consisting of three components, as shown in Fig. S22 and S23. The fitted Rs and Rct for CuFeS2 are 857 mΩ and 53.5 Ω, respectively, while for RuO2 the corresponding Rs and Rct values are 1 Ω and 3.1 Ω. Since electrocatalyst stability is a key factor in evaluating electrocatalytic performance, we investigated the OER durability of CuFeS2 using chronopotentiometry at 10 mA cm−2, as shown in Fig. 7i. The chronoamperometric data reveal that the current density of CuFeS2 remained stable for 125 hours. The LSV recorded after the long-term stability test shows a slight decrease in current density, while the overpotential remains nearly unchanged (Fig. S24). For the stability test, 2501 CV cycles were performed at a scan rate of 100 mV s−1, and the LSV recorded after completion of the cycles shows a slight decrease in current density (Fig. S25). Furthermore, turnover frequency (TOF) was calculated using eqn (10), adopting a conservative assumption of 100% faradaic efficiency and assuming that all formula units of the catalyst are actively participating in the OER.63,64
| | 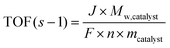 | (10) |
where
J represents the current density at the specified overpotential (10 mA cm
−2). The term
Mw,catalyst denotes the molar mass of the synthesized catalyst (g mol
−1), while
F corresponds to the Faraday constant. The parameter
n indicates the number of electrons transferred during the oxygen evolution reaction (OER), which is four (4) in this case. The variable
m refers to the mass loading of the electrocatalyst per geometric electrode area (g cm
−2). It was observed that, at identical catalyst loading and current density, CuFeS
2 exhibits a TOF of 9.8 × 10
−4 s
−1, whereas RuO
2 shows a TOF of 7.1 × 10
−4 s
−1.
Furthermore, after the stability test, the Fe concentration in the electrolyte solution was found to be 1.3 ppm, while the Cu concentration was 0.6 ppm. After the 125 hour stability test, the XPS survey spectrum of the electrocatalyst confirms the presence of Cu, Fe, S, O, C, and F (Fig. S26a). The fluorine signal originates from residual Nafion, which was used as a binder during the preparation of the catalyst ink. The Cu 2p3/2 and Cu 2p1/2 peaks at 932.5 and 952.3 eV (Δ = 19.8 eV) are assigned to Cu+, while the Cu 2p3/2 and Cu 2p1/2 peaks at 933.6 and 953.9 eV correspond to Cu2+, with the shake-up satellite at ∼943.4 eV confirming the presence of Cu2+ species (Fig. S26b). After the stability test, more pronounced Cu2+ features or satellite peaks appear, indicating surface oxidation of the electrocatalyst CuFeS2. The XPS spectrum of Cu 2p showed that all peaks shifted to higher binding energy after the OER, indicating a change in the electronic environment of copper species at the catalyst surface.65 After the stability test in 1 M KOH, the Fe 2p XPS spectrum shows peaks at 710.8 and 714.2 eV corresponding to the 2p3/2 of Fe2+ and Fe3+, while the peaks at 719.8 and 724.8 eV correspond to the 2p3/2 of Fe2+ and Fe3+. Satellite peaks are observed at 714.4, 727.5, and 733.7 eV, with enhanced intensity after the stability test, indicating surface oxidation and the formation of Fe3+ species (Fig. S26c). The S 2p spectrum shows peaks at 161.2 and 162.4 eV corresponding to S2− 2p3/2 and 2p1/2, peaks at 163.4 and 164.6 eV assigned to disulfide (S22−) 2p3/2 and 2p1/2, and peaks at 169.1 and 170.3 eV with enhanced intensity compared to that of the pristine catalyst attributed to surface-oxidized sulfur species (–SOx) (Fig. S 26d).50 After the stability test, the C 1s spectrum shows peaks at 284.8 eV (C–C), 285.4 eV (C–O), 286.5 eV (C–O–C), and 289.3 eV (O–CO), indicating surface carbon functionalities, while new peaks at 292.6 and 293.6 eV correspond to –CF2 and –CF3 groups from Nafion, and the peak at 296.3 eV arises from the K+ ion derived from the KOH electrolyte (Fig. S26e).66–68 The O 1s spectrum shows peaks at 530.1 eV (lattice Cu/Fe–O), 531.2 eV (Cu/Fe–OH), 531.8 eV (adsorbed OH−/C–O), and 532.4 eV, which are attributed to oxidized sulfur species (sulfate/–SO3H) from Nafion50,67 (Fig. S26f). Overall, the catalyst demonstrates excellent stability after 125 h of electrolysis in 1 M KOH, with XPS analysis showing surface oxidation of Cu and Fe to Cu2+ and Fe3+ species, formation of surface-oxidized sulfur (–SOx), retention of Nafion-derived –CF2 and –CF3 groups, and the presence of lattice and hydroxyl oxygen species, indicating that the structural integrity and key surface functionalities are largely preserved. After the long-term chronopotentiometry (CP) test, it was observed that the flower-like morphology of CuFeS2 was retained, as evident from the TEM micrograph in Fig. S27a and b. The HRTEM image with the inset FFT shows a lattice spacing of 0.31 nm corresponding to the (112) plane of chalcopyrite CuFeS2 (Fig. S27c). The SAED pattern further confirms the single-crystalline nature of the material (Fig. S27d). Additionally, the EDS elemental mapping demonstrates the uniform distribution of Cu, Fe, and S across the catalyst surface (Fig. S27e). Thus, the catalyst largely preserved its original composition and morphology, exhibiting only negligible degradation.
6. Conclusion
In summary, phase-pure chalcopyrite (CuFeS2) was successfully synthesized via a solvothermal approach employing the reline DES as the reaction medium. Among the sulfur precursors tested (thiourea, thioacetamide, and Na2S), only thiourea yielded CuFeS2 with high phase selectivity under comparable solvothermal conditions. Ex situ PXRD analysis indicated that, at shorter reaction times, secondary binary and ternary sulfide intermediates were formed, which were subsequently transformed into phase-pure ternary CuFeS2 chalcogenide at the optimized reaction duration. The stability of the reline DES was investigated by visual inspection and FTIR spectroscopy to evaluate its effectiveness as a solvent under solvothermal conditions. I–V measurements were conducted to assess the suitability of the material for optoelectronic applications. Dielectric measurements over broad frequency and temperature ranges showed that CuFeS2 possesses a high dielectric constant, low loss, and strong thermal stability, indicating its potential for electronic applications. Moreover, CuFeS2 demonstrated excellent performance toward the alkaline OER, with a low overpotential of 360 mV@10 mA cm−2. This work establishes the material's potential in electronic and energy applications and demonstrates the effectiveness of eco-friendly DES routes for designing advanced TMCs.
Author contributions
B. M., M. G., and H. A. conducted the experimental investigation, analyzed and validated the data, wrote the original draft, and reviewed and edited the manuscript. S. H. conceived the project, supervised the research, and reviewed and edited the manuscript. All authors have approved the final version of the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. It includes detailed characterisation methods, electrochemical and refractive index calculations, and additional plots of FTIR, NMR, PXRD, Raman, TGA, BET, UV–Vis DRS, and TEM micrographs. See DOI: https://doi.org/10.1039/d5ma01466b.
Data are also available upon request from the authors.
Acknowledgements
The authors acknowledge IIT Patna, India, for research support and fellowships.
References
- H. Jafarizadeh, E. Yamini, S. M. Zolfaghari, F. Esmaeilion, M. E. H. Assad and M. Soltani, Energy Rep., 2024, 12, 2179–2192 CrossRef.
- A. A. Balaraman and S. Dutta, J. Phys. D: Appl. Phys., 2022, 55, 183002 Search PubMed.
- F. Yan, J. Qian, S. Wang and J. Zhai, Nano Energy, 2024, 123, 109394 CrossRef CAS.
- M. Yang, W. Ren, M. Guo and Y. Shen, Small, 2022, 18, 2205247 CrossRef CAS.
- B. Gholipour, A. Karvounis, J. Yin, C. Soci, K. F. MacDonald and N. I. Zheludev, NPG Asia Mater., 2018, 10, 533–539 CrossRef CAS.
- K. O. Čajko, M. Dimitrievska, D. L. Sekulić, D. M. Petrović and S. R. Lukić-Petrović, J. Mater. Sci.: Mater. Electron., 2021, 32, 6688–6700 CrossRef.
- I. Chung and M. G. Kanatzidis, Chem. Mater., 2014, 26, 849–869 CrossRef CAS.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- G. Mayakrishnan, R. Vanaraj, J. Xiong, M. Farooq, A. Ullah, K. Zhang, S. C. Kim and I. S. Kim, Energy Environ. Mater., 2024, 7, e12788 Search PubMed.
- C. Coughlan, M. Ibáñez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef CAS PubMed.
- L. Liu, B. Bai, X. Yang, Z. Du and G. Jia, Chem. Rev., 2023, 123, 3625–3692 CrossRef CAS PubMed.
- S. Shishodia, B. Chouchene, T. Gries and R. Schneider, Nanomaterials, 2023, 13, 2889 Search PubMed.
- S. Lee, C. E. Hoyer, C. Liao, X. Li and V. C. Holmberg, Small, 2022, 18, 2104975 CrossRef CAS PubMed.
- B. Bhattacharyya, C. Balischewski, C. Pacholski, A. Pandey, I. Bald and A. Taubert, Adv. Opt. Mater., 2023, 11, 2202411 Search PubMed.
- B. Kumar, S. V. Singh, A. Chattopadhyay, S. Biring and B. N. Pal, ACS Omega, 2020, 5, 25947–25953 CrossRef CAS PubMed.
- A. Sugathan, B. Bhattacharyya, V. Kishore, A. Kumar, G. P. Rajasekar, D. Sarma and A. Pandey, J. Phys. Chem. Lett., 2018, 9, 696–701 CrossRef CAS.
- B. Bhattacharyya and A. Pandey, J. Am. Chem. Soc., 2016, 138, 10207–10213 CrossRef CAS PubMed.
- B. S. Shah, J. B. Raval, D. Kumar, S. H. Chaki and M. P. Deshpande, J. Alloys Compd., 2023, 938, 168566 CrossRef CAS.
- X. Meng, J. Xu, G. Zhao, L. Zhang, J. Cheng, Y. Pu, W. Li and L. Li, J. Alloys Compd., 2024, 992, 174590 CrossRef CAS.
- R. A. Yund and G. Kullerud, J. Petrol., 1966, 7, 454–488 CrossRef CAS.
- A. Malyarevich, K. Yumashev, N. Posnov, V. Mikhailov and V. Gurin, Appl. Phys. B: Lasers Opt., 2000, 70, 111–116 CrossRef CAS.
- G. Gabka, P. Bujak, A. Ostrowski, W. Tomaszewski, W. Lisowski, J. W. Sobczak and A. Pron, Inorg. Chem., 2016, 55, 6660–6669 CrossRef CAS.
- O. Długosz, Materials, 2023, 16, 627 CrossRef PubMed.
- O. S. Hammond and A.-V. Mudring, ChemComm, 2022, 58, 3865–3892 RSC.
- Y. Ma, Y. Yang, T. Li, S. Hussain and M. Zhu, Green Chem., 2024, 26, 3627–3669 Search PubMed.
- A. L. Siegel, L. Adhikari, S. Salik and G. A. Baker, Curr. Opin. Green Sustain. Chem., 2023, 41, 100770 CrossRef CAS.
- R. Deng, M. Gao, B. Zhang and Q. Zhang, Adv. Energy Mater., 2024, 14, 2303707 Search PubMed.
- B. Mahto, A. A. Khan, A. Barhoi and S. Hussain, ACS Appl. Nano Mater., 2023, 6, 6784–6797 CrossRef CAS.
- B. Mahto, H. Ali, A. Barhoi and S. Hussain, Coord. Chem. Rev., 2025, 527, 216406 CrossRef CAS.
- S. Kumar-Krishnan, E. Prokhorov, O. A. De Fuentes, M. Ramírez, N. Bogdanchikova, I. Sanchez, J. Mota-Morales and G. Luna-Bárcenas, J. Mater. Chem. A, 2015, 3, 15869–15875 RSC.
- P. H. Edwards, J. R. Bairan Espano and J. E. Macdonald, Chem. Mater., 2024, 36, 7186–7196 CrossRef.
- V. Mauritz and R. W. Crisp, J. Mater. Chem. C, 2024, 12, 11319–11334 Search PubMed.
- J. M. Rhodes, C. A. Jones, L. B. Thal and J. E. Macdonald, Chem. Mater., 2017, 29, 8521–8530 CrossRef CAS.
- A. Roffey, N. Hollingsworth, H.-U. Islam, M. Mercy, G. Sankar, C. R. A. Catlow, G. Hogarth and N. H. de Leeuw, Nanoscale, 2016, 8, 11067–11075 Search PubMed.
- Y. Li, Y. Li, H. Li, X. Fan, H. Yan, M. Cai, X. Xu and M. Zhu, Friction, 2023, 11, 76–92 CrossRef CAS.
- S. Azmi, A. Klimek and E. Frackowiak, Chem. Eng. J., 2022, 444, 136594 CrossRef CAS.
- D. Yue, Y. Jia, Y. Yao, J. Sun and Y. Jing, Electrochim. Acta, 2012, 65, 30–36 CrossRef CAS.
- C. Du, B. Zhao, X.-B. Chen, N. Birbilis and H. Yang, Sci. Rep., 2016, 6, 29225 CrossRef CAS.
- H. Wang, S. Liu, Y. Zhao, J. Wang and Z. Yu, ACS Sustainable Chem. Eng., 2019, 7, 7760–7767 Search PubMed.
- A. N. Roth, A. P. Porter, S. Horger, K. Ochoa-Romero, G. Guirado, A. J. Rossini and J. Vela, Chem. Mater., 2024, 36, 4542–4552 Search PubMed.
- N. M. Stephens and E. A. Smith, Langmuir, 2022, 38, 14017–14024 Search PubMed.
- T. El Achkar, S. Fourmentin and H. Greige-Gerges, J. Mol. Liq., 2019, 288, 111028 Search PubMed.
- F. Gabriele, M. Chiarini, R. Germani, M. Tiecco and N. Spreti, J. Mol. Liq., 2019, 291, 111301 CrossRef CAS.
- T. J. Huang, R. Lee Guang-Ren, X. Yin, C. Tang, G. Qi and H. Gong, Cryst. Growth Des., 2017, 17, 73–79 Search PubMed.
- J. Li, R. Hübner, M. Deconinck, A. Bora, M. Göbel, D. Schwarz, G. Chen, G. Wang, S. A. Yang, Y. Vaynzof and V. Lesnyak, ACS Appl. Nano Mater., 2023, 6, 9475–9483 Search PubMed.
- R. K. Sharma, Y. N. Chouryal, S. Chaudhari, J. Saravanakumar, S. R. Dey and P. Ghosh, ACS Appl. Mater. Interfaces, 2017, 9, 11651–11661 Search PubMed.
- Q. Ma, Y. Wang, J. Kong and H. Jia, Ceram. Int., 2016, 42, 2854–2860 Search PubMed.
- Y. Bao, K. Zhou, J. Ma, Q. Li, L. Deng, D. Jin and H. Qiu, J. Phys. Chem. Lett., 2024, 15, 10592–10601 Search PubMed.
- S. Wan, L. Bao, J. Xiao, J. He, X. Gao, H. Wang, Y. Cao and R. Ma, Phys. B, 2025, 717, 417812 Search PubMed.
- S. Huang, X. Cong, T. Ye, L. Liu, K. Peng, L. Zhang, J. Bao, P. Gao, Q. Chen and Q. He, Inorg. Chem. Front., 2023, 10, 2387–2398 Search PubMed.
- B. A. Tappan, G. Barim, J. C. Kwok and R. L. Brutchey, Chem. Mater., 2018, 30, 5704–5713 Search PubMed.
- T. Teranishi, J. Phys. Soc. Jpn., 1961, 16, 1881–1887 Search PubMed.
- S. Datta, J. Mahin, E. Liberti, I. Manasi, K. J. Edler and L. Torrente-Murciano, ACS Sustainable Chem. Eng., 2023, 11, 10242–10251 Search PubMed.
- Y. Liu, J. Hu, C. Ngo, S. Prikhodko, S. Kodambaka, J. Li and R. Richards, Appl. Catal., B, 2011, 106, 212–219 Search PubMed.
- K. Ramasamy, H. Sims, W. H. Butler and A. Gupta, Chem. Mater., 2014, 26, 2891–2899 CrossRef CAS.
- J. van Embden, K. Latham, N. W. Duffy and Y. Tachibana, J. Am. Chem. Soc., 2013, 135, 11562–11571 Search PubMed.
- J. Mallick, A. Shukla, S. K. Panda, S. K. Biswal, S. N. Rout, M. K. Yadav and M. Kar, J. Alloys Compd., 2024, 978, 173403 CrossRef CAS.
- S. K. Paswan, L. K. Pradhan, P. Kumar, S. Kumari, M. Kar and L. Kumar, Phys. Scr., 2022, 97, 095812 CrossRef CAS.
- S. Joshi, M. Kumar, S. Chhoker, G. Srivastava, M. Jewariya and V. Singh, J. Mol. Struct., 2014, 1076, 55–62 CrossRef.
- C. Behera, P. R. Das and R. Choudhary, J. Electrons. Mater., 2014, 43, 3539–3549 Search PubMed.
- C. Rayssi, S. E. Kossi, J. Dhahri and K. Khirouni, RSC Adv., 2018, 8, 17139–17150 Search PubMed.
- P. Biswas, T. Das, A. Dev, A. Shukla, S. Datta, R. K. Singh and M. Kar, Ceram. Int., 2024, 50, 30514–30532 CrossRef.
- A. T. Swesi, J. Masud and M. Nath, Energy Environ. Sci., 2016, 9, 1771–1782 RSC.
- Y. Cui, S. Yang, J. Zhu, Z. Wang, S. Chen, J. Qi and H. Wang, Molecules, 2025, 30, 2181 CrossRef.
- T. H. N. Ngo, J. D. Riches, J. Love and A. P. O’Mullane, Electrochim. Acta, 2025, 540, 147172 Search PubMed.
- M. Usselmann, J. Bansmann and A. J. C. Kuehne, Adv. Mater., 2023, 35, 2208484 Search PubMed.
- A. K. Friedman, W. Shi, Y. Losovyj, A. R. Siedle and L. A. Baker, J. Electrochem. Soc., 2018, 165, H733–H741 CrossRef.
- J. G. Gluschke, J. Seidl, R. W. Lyttleton, K. Nguyen, M. Lagier, F. Meyer, P. Krogstrup, J. Nygård, S. Lehmann, A. B. Mostert, P. Meredith and A. P. Micolich, Mater. Horiz., 2021, 8, 224–233 RSC.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Monika
Gupta
,
Haider
Ali
and
Sahid
Hussain
,
Monika
Gupta
,
Haider
Ali
and
Sahid
Hussain







 , and decreases steadily with further increases in frequency. The peak maxima of imaginary impedance, i.e.,
, and decreases steadily with further increases in frequency. The peak maxima of imaginary impedance, i.e.,  in dielectrics, typically indicate the frequency at which the material exhibits maximum dielectric loss. The magnitude of the peak represents the maximum energy dissipation due to dielectric losses at a specific frequency. Higher peaks suggest more significant energy loss in the dielectric material at that frequency. The frequency corresponding to the
in dielectrics, typically indicate the frequency at which the material exhibits maximum dielectric loss. The magnitude of the peak represents the maximum energy dissipation due to dielectric losses at a specific frequency. Higher peaks suggest more significant energy loss in the dielectric material at that frequency. The frequency corresponding to the  is referred to as the relaxation frequency, fmax. Here,
is referred to as the relaxation frequency, fmax. Here,  shifts toward higher frequencies as the temperature increases, suggesting the presence of non-Debye relaxation phenomena in the samples.62 This relaxation behaviour can be attributed to charge accumulation at the grain boundaries, which become polarized in response to the applied electric field. As the temperature rises, the value of
shifts toward higher frequencies as the temperature increases, suggesting the presence of non-Debye relaxation phenomena in the samples.62 This relaxation behaviour can be attributed to charge accumulation at the grain boundaries, which become polarized in response to the applied electric field. As the temperature rises, the value of  decreases, indicating a reduction in the material's resistive properties and an increase in the mobility of charge carriers within the material. This results in more efficient charge transport and, consequently, a reduction in the imaginary impedance's peak value, reflecting energy loss due to charge movement. Additionally, the broadening of the peak with increasing temperature suggests the presence of multiple relaxation processes.58
decreases, indicating a reduction in the material's resistive properties and an increase in the mobility of charge carriers within the material. This results in more efficient charge transport and, consequently, a reduction in the imaginary impedance's peak value, reflecting energy loss due to charge movement. Additionally, the broadening of the peak with increasing temperature suggests the presence of multiple relaxation processes.58

