DOI:
10.1039/D5MA01384D
(Paper)
Mater. Adv., 2026,
7, 2675-2689
First-principles calculation of rhombohedral perovskite MgAgF3: a DFT study of optical, electronic, elastic, thermodynamic and mechanical properties
Received
29th November 2025
, Accepted 19th January 2026
First published on 21st January 2026
Abstract
This research utilizes density functional theory (DFT) to comprehensively explore the structural, elastic, electronic, optical, thermodynamic and mechanical characteristics of the halide perovskite MgAgF3, which crystallizes in a rhombohedral lattice that belongs to the trigonal crystal system. Elastic constant evaluations confirm mechanical stability and ductility (Pugh's ratio = 2.14, Poisson's ratio = 0.28), alongside slight elastic anisotropy (AU = 0.07369). Band structure calculations reveal an indirect bandgap of 2.16 eV using GGA-PBE, with electronic states near the Fermi level dominated by hybridized Ag-4d and F-2p orbitals, indicating mixed ionic-covalent bonding. Optical analysis reveals pronounced anisotropy, a refractive index between 1.5 and 1.7, and significant ultraviolet absorption in the 8–12 eV range, with a marked energy-loss peak at 63 eV signifying strong plasmon resonance. Phonon dispersion affirms dynamic stability and thermodynamic predictions, and the consistency with Debye theory shows regular heat capacity behavior. The machinability index (2.093) and ductile behavior suggest suitability for practical processing. Despite being computational, the study lays foundational insights into MgAgF3's potential in flexible optical and electronic applications, such as polarized light detectors and UV photonic systems, emphasizing the need for future experimental validation.
1. Introduction
Understanding the physical properties of materials is crucial for their deployment in advanced technologies. Among the wide range of functional materials, perovskite-structured compounds have received significant attention due to their remarkable physical properties and broad applicability in science and technology.1–4 Perovskites are extensively studied because they not only exhibit diverse and useful characteristics but are also commonly found in the Earth's crust.5 While the archetypal perovskites are oxygen-based, with the general chemical formula ABO3, there also exist halide analogues, including fluoro-perovskites (ABF3), where A and B represent alkali or alkaline earth elements and fluorine acts as the anion. Fluorine, the most electronegative element, demonstrates a strong tendency to form chemically stable fluorides, particularly with low-electronegativity alkali and alkaline earth metals.
Fluoro-perovskites form a fascinating class of materials because of their mechanically stable crystal structures and excellent optoelectronic properties. They exhibit energy band gaps ranging from semiconducting (1–4 eV) to insulating (above 4 eV) regimes, which makes them promising for a wide spectrum of applications. In recent years, they have gained considerable attention owing to their practical uses in photolithography lenses, photo-dosimeters, flashlight materials, and semiconductor fabrication.6–8 A number of fluoro-perovskite compounds have been studied both experimentally and computationally,9 with their band gaps frequently reported as wide.10–12
Beyond electronic characteristics, fluoride perovskites also display interesting structural13 and functional properties, including piezoelectricity,14 ferromagnetism,15 photoluminescence,16,17 high-temperature superconductivity, and colossal magnetoresistance.18 The discovery of high-temperature superconducting oxides, along with the advancement of perovskite solar cells exhibiting power conversion efficiencies of around 21% and a theoretical limit of up to 31%, has significantly intensified research interest in this class of materials.19,20 Specific fluoro-perovskites such as KMgF3,21,22 NaSrF3, NaBaF3, and LiBaF323 have already shown promising results in the UV spectral regions,24 enabling their use in transparent and low-loss optical windows, lenses and prisms. Alongside these developments, halide-based perovskites have also emerged as an attractive class of low-cost and highly efficient materials for photovoltaic applications.25,26 Yet, despite their promise, early studies were hindered by limited efficiencies, structural instabilities, and challenges associated with conventional fabrication methods.25,27
Many fluoro-perovskites have been extensively studied for their structural, electronic, and optoelectronic properties,28 yet the comprehensive physical characteristics of rhombohedral MgAgF3 remain largely unexplored. In this work, we employ density functional theory (DFT) to systematically examine its structural, electronic, optical, vibrational and thermodynamic behaviors. Our analysis covers lattice parameters, band structure, density of states, charge distribution, optical responses, phonon dynamics, heat capacity, and population analysis to assess MgAgF3's potential for technological applications.
This comprehensive study contributes to a deeper understanding of MgAgF3 and its potential role in the development of next-generation optoelectronic and energy-related devices.
2. Computational methodology
First-principles density functional theory (DFT) calculations were carried out using the CASTEP module within Material Studio to investigate the structural, electronic, and optical properties of MgAgF3. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was employed due to its reliable balance among accuracy, efficiency in solid-state simulations, and computational cost.29,30 Spin polarization was disabled considering the non-magnetic nature of the material.31 Ultrasoft pseudopotentials described the ion-electron interactions and were utilized to reduce the required plane-wave cutoff energy while maintaining accuracy.32 The Koelling–Harmon scalar relativistic approximation was utilized to incorporate relativistic effects, particularly relevant due to the presence of silver (Ag) atoms.33 A plane-wave basis set cutoff energy of 500 eV was chosen following convergence tests.34 The Brillouin zone was sampled using a Monkhorst–Pack k-point mesh of 9 × 9 × 3 for accurate integration.35 Structural optimization and self-consistent field (SCF) calculations were conducted with fine energy convergence criteria to ensure computational accuracy.36 The crystal structure corresponds to a trigonal system with a rhombohedral lattice (space group R3c), characterized by lattice parameters a = b = 5.52 Å, c = 13.55 Å, and angles α = β = 90°, γ = 120° (data from Materials Project database37). To assess the anisotropic nature of optical properties, frequency-dependent dielectric functions and optical conductivities were calculated along three crystallographic polarization directions: (001), (010), and (100). This enabled a comprehensive evaluation of the directional dependence of electronic transitions and optical responses, crucial for potential photonic applications.38 Simulations included full structural optimization, electronic band structure, total and partial density of states (DOS/PDOS), reflectivity, refractivity, absorption, loss function, optical conductivity, dielectric function, phonon dispersion, charge density mapping, and Mulliken population analysis to comprehensively investigate the electronic, structural, optical, and vibrational properties of MgAgF3. The DFT method has proven to be one of the most accurate methods for the computation of the electronic structure of solids.39–45
3. Results and discussion
3.1. Structural properties
The optimized structure of MgAgF3 crystallizes in a rhombohedral lattice, which belongs to the trigonal crystal system. This phase can be regarded as a distorted derivative of the ideal cubic perovskite, stabilized by cooperative rotations and tilting of the MgF6 octahedra. Such distortion lowers the crystal symmetry from cubic to rhombohedral, resulting in subtle variations in lattice parameters and atomic coordination. The planar and three-dimensional diagram of the cell is shown in Fig. 1. Mg2+ is bonded to six equivalent F− ions to form corner-sharing MgF6 octahedra. The coordinates of Mg, Ag, and F in the cell are (0, 0, 0), (0, 0, 0.25), and (0.561576, 0.561576, 0.25), respectively. The lattice constants are a = b = 5. 52 Å, c = 13.55 Å, α = β = 90°, γ = 120°.
 |
| | Fig. 1 Crystal structure of rhombohedral MgAgF3. | |
From the Mulliken bond overlap populations, we can get bond lengths and total number of bonds for MgAgF3. The analysis of bond population reveals anti-bonding interactions between F–Mg, F–F and Mg–Ag atoms, indicated by negative overlap populations. Such overlap occurs when electron density between atoms resides in destabilizing orbitals, which weakens bond formation.46 In addition, the F–Ag bonds exhibit significantly higher positive overlap populations compared to other pairs, which suggests stronger covalent character. This aligns with the shorter bond lengths observed for F–Ag, which further confirms their stability relative to the anti-bonding or weakly bonding interactions elsewhere in the structure (Table 1).
Table 1 Bond number, bond population and bond length of MgAgF3
| Compound |
Bond |
Bond number (n) |
Bond overlap population (p) |
Bond length (d) |
| MgAgF3 |
F–Mg |
36 |
−0.84 |
2.04323 |
| F–Ag |
18 |
0.15 |
2.43764 |
| F–Ag |
36 |
0.05 |
2.86680 |
| F–F |
36 |
−0.03 |
2.86680 |
| F–F |
18 |
−0.06 |
2.91216 |
| Mg–Ag |
12 |
−0.26 |
3.46927 |
| Mg–Ag |
12 |
−0.05 |
3.48298 |
| F–F |
18 |
−0.01 |
3.49881 |
3.2. Elastic and mechanical properties
Understanding the mechanical stability of materials is crucial in materials engineering. The elasticity of a material explains how it deforms under strain before rebounding to its original nature when the load is removed.47 The mechanical properties calculated are the elastic constants, bulk and shear moduli, Cauchy pressure, anisotropy, machinability, hardness and Poisson's ratio. Elastic constants of materials are important in determining the crystals’ response to external forces and the strength of materials. The Cauchy pressure, Poisson and Pugh's ratio are extremely accurate and powerful in classifying materials as either ductile or brittle.48 The mechanical stability is confirmed from the elastic constants using the Born stability criteria.49
The trigonal structures have seven independent elastic constants and are stable if eqn (1)–(4) are obeyed.50
| | | C11 > 0, C33 > 0, C44 > 0 | (2) |
| | 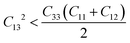 | (3) |
| | | [C44(C11 − C12) − 2C142] > 0 ≡ C44C66 | (4) |
As the elastic constants satisfy Born's stability restrictions, we can infer that MgAgF3 is mechanically stable.
Due to the lack of comparable elastic constants for MgAgF3, other functional materials (AgMgF3 and KMgF3) are compared from the previous calculation results to judge the correctness of the calculation methods used.46,51
Table 2 shows that the elastic constants of other ABX3 functional materials, as calculated using the ab initio method, closely match those reported in previous studies. This agreement confirms the reliability of the DFT approach and the overall accuracy of the calculations. Therefore, it can be confidently concluded that the elastic constants obtained for MgAgF3 are accurate and trustworthy.
Table 2 Elastic constants and Cauchy pressure comparison table (in GPa)
| Materials |
C
11
|
C
12
|
C
13
|
C
14
|
C
22
|
C
23
|
C
33
|
C
44
|
C
55
|
C
66
|
C
12–C44 |
| MgAgF3 |
132.94 |
58.22 |
51.75 |
−2.151 |
132.94 |
51.75 |
103.44 |
36.41 |
36.41 |
37.36 |
21.81 |
| AgMgF3 (cubic)46 |
129.52 |
46.39 |
— |
— |
— |
— |
— |
31.30 |
— |
— |
15.09 |
| KMgF3 (cubic)51 |
121.87 |
45.65 |
— |
— |
— |
— |
— |
37.02 |
— |
— |
8.63 |
The bulk and shear moduli can be approximated using either the Voigt52 or the Reuss53,54 theories and their averages are calculated using the Hill55 averaging scheme. From the Voigt theory,42 the bulk BV and shear GV moduli are estimated using eqn (5) and (6)
| | 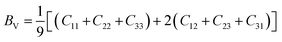 | (5) |
| | 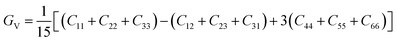 | (6) |
The Reuss scheme, the bulk
BR and shear
GR moduli are determined using
eqn (7) and (8),
| | 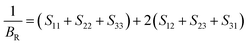 | (7) |
| | 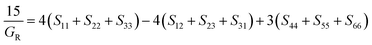 | (8) |
The averages of the bulk
BH and the shear
GH moduli from the two schemes are addressed using
eqn (9) and (10),
| | 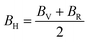 | (9) |
| | 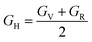 | (10) |
The Poisson's ratio
ν and the Young's modulus
EH were obtained from the bulk and shear moduli using
eqn (11) and (12),
| | 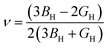 | (11) |
| | 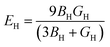 | (12) |
Key mechanical design properties are strength, hardness, stiffness, toughness and ductility. These can be tested from calculated results of shear modulus, Young's modulus, and bulk modulus (Table 3).
Table 3 Computed elastic constants (Cij), bulk modulus (B), shear modulus (G), Pugh's ratio (B/G), Young's modulus (E), Poisson's ratio (ν), and Anisotropy factor (A) of MgAgF3 and other cubic perovskites
| Materials |
C
11 (GPa) |
C
12 (GPa) |
C
44 (GPa) |
G (GPa) |
B (GPa) |
B/G |
E (GPa) |
ν
|
A
|
| MgAgF3 (rhombohedral) |
132.94 |
58.22 |
36.41 |
35.69 |
76.22 |
2.14 |
92.61 |
0.28 |
0.073 |
| AgMgF3 (cubic)46 |
129.52 |
46.39 |
31.30 |
35.07 |
74.10 |
2.11 |
90.87 |
0.30 |
0.75 |
| KMgF3 (cubic)51 |
121.87 |
45.65 |
37.02 |
37.43 |
71.03 |
1.89 |
95.53 |
0.28 |
0.97 |
The bulk, shear, and Young's modulus measure a material's response to volume, shear, and longitudinal stress, respectively. High values indicate strong resistance to compressibility and deformation.56 The Young's modulus of rhombohedral MgAgF3 is higher than that of cubic AgMgF3, but lower than that of KMgF3.46,51 So, we can say that MgAgF3 has a higher resistivity to plastic deformation than the cubic AgMgF3 material. These higher values reflect that the rhombohedral structure of MgAgF3 has superior resistance to fracture and mechanical damage compared to cubic AgMgF3. To characterize the stiffness of a material, the measurement of bulk modulus (B) is required. The material will be stiffer if the value of B is higher.57 In the present calculations, the results reveal the higher stiffness of the rhombohedral MgAgF3 compound than the other compounds. Furthermore, the Poisson's ratio ν is sometimes used to approximate bonding character, and it is fundamentally a macroscopic elastic constant reflecting the material's overall structural rigidity and packing density, not the microscopic bond type. In our study, the Poisson's ratio for rhombohedral MgAgF3 is 0.29. This value falls within the typical range for most crystalline solids (0.25 < ν < 0.35). Given the material's low elastic anisotropy index of A = 0.073, this value of ν primarily reflects the material's near-isotropic elastic response and mechanical stability.
The brittleness of the materials was checked using the Pugh criteria58 where the ratio of the bulk to the shear modulus is related to the critical value of 1.75. Materials with B/G > 1.75 are ductile and those with ratios less than the critical value are considered brittle.59 In this case, the ratios of the bulk modulus to the shear modulus are respectively 2.14 and 2.11, and the Poisson's ratios are respectively 0.28 and 0.30, indicating that the rhombohedral and cubic structures of MgAgF3 are ductile. The Poisson's ratio of 0.28 indicates that the rhombohedral MgAgF3 is a plastic material.60
The machinability index (B/C44)61 is an indicator of the ease with which a material can be processed.50 The high machinability index of MgAgF3 is 2.093, which suggests that it is suitable for easy mechanical manipulation, making it more favorable for the manufacturing of solar cells.50
3.2.1. Elastic anisotropy.
Elastic anisotropy determines many basic properties of materials and is important for predicting the fracture toughness of materials. The universal anisotropy index AU and percent anisotropy indices of compression and shear (AB and AG) are used to evaluate the elastic anisotropy of a material, and they are expressed as follows:62,63| | 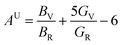 | (13) |
| | 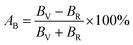 | (14) |
| | 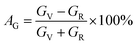 | (15) |
For AU, AB, and AG, if the value is 0, the crystal is isotropic. The greater their deviation from 0, the higher the degree of anisotropy. The AU for MgAgF3 was 0.07369, indicating that the degree of universal anisotropy of MgAgF3 is slightly low. Also, the AB and AG for MgAgF3 were 0.99 and 0.53, respectively, indicating that the compound has a high compressive anisotropy and medium shear anisotropy.
The shear anisotropic factors can be used to measure the degree of anisotropy in atomic bonding in different crystal planes. We employed the anisotropy factors A1, A2, and A3. The index of A1 represented the shear anisotropy factor between [011] and [010] crystal orientations on the (100) crystal plane, Similarly, A2 was the shear anisotropy factor between the [101] and [001] orientations on the (010) crystal plane, and A3 was that between the [110] and [010] orientations on the (001) crystal plane, for a trigonal crystal system with rhombohedral lattice system, the expressions are as follows:50
| | 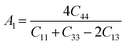 | (16) |
| | 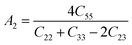 | (17) |
| | 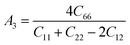 | (18) |
The greater the difference between the anisotropy factors and 1, the higher the anisotropy of the crystal. Here, MgAgF3 has a value of A1, A2 and A3 of 1.09, 1.21 and 1, respectively, indicating that MgAgF3 has a shear anisotropy along the planes [100] and [010] and has a shear isotropy along the plane [001] (Table 4).
Table 4 Calculated anisotropic index of MgAgF3
| Compound |
A
U
|
A
B
|
A
G
|
A
1
|
A
2
|
A
3
|
| MgAgF3 |
0.07369 |
0.99 |
0.53 |
1.09 |
1.21 |
1 |
The elastic anisotropy of a crystal is an important parameter in engineering science because a large value always means a high possibility of microcracks in that crystal.64 To more intuitively understand the anisotropy characteristics of MgAgF3, three-dimensional surface maps of anisotropy, Young's modulus and Shear modulus are drawn in Fig. 2.
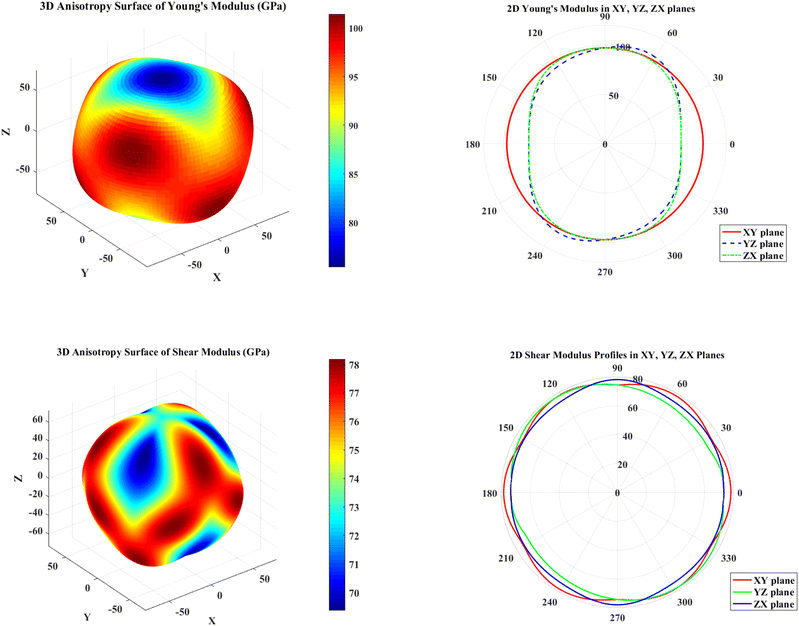 |
| | Fig. 2 3D and 2D directional Young's modulus and shear modulus. | |
For a material with A = 1 in all crystallographic directions, the resulting three-dimensional representation is a perfect sphere.65 However, the material in this paper is anisotropic, so the surface maps have some degree of depression in some directions. In general, the greater the deviation between the 3D surface structure and a sphere, the higher the anisotropic property.65 It can be observed in Fig. 2 that all the images deviate from the standard sphere, which indicates that they are all anisotropic.
3.3. Electronic properties
3.3.1. Band structure and density of states.
The electronic band structure, partial density of state (PDOS) and total density of state (TDOS) of MgAgF3 are depicted in Fig. 3. The PDOS and TDOS are defined in terms of number of states at occupied or unoccupied energy levels in statistical and solid–state physics.66 They indicate hybridization among orbital electrons as well as bonding characteristics within the compound. The Fermi level, EF, is indicated by the horizontal dashed line. In the band diagram, the purely valence and conduction bands are shown by blue lines, whereas the bands crossing the EF are indicated with red lines.
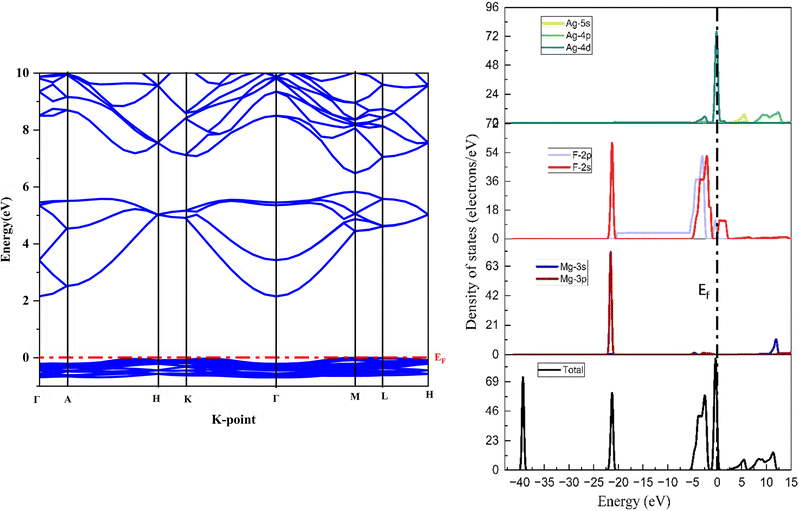 |
| | Fig. 3 Calculated band structure of MgAgF3 and total and partial density of states of MgAgF3. | |
We have calculated the band gap and band energy using the GGA-PBE pseudopotentials method in the rhombohedral MgAgF3 structure. From the band structure, it is clear that the valence band maximum (VBM) of the MgAgF3 compound is located at point M. However, the conduction band minimum (CBM) is found at the point Γ, showing that MgAgF3 has an indirect band gap at (M–Γ), similar to other reported fluoride perovskites in the literature.67–69 As seen in Table 5, our calculated band gap for MgAgF3 equals 2.16 eV. It can be said that our calculated results predict the material as a semiconductor. In contrast, the band gap for the cubic structure was found to be 1.95 eV.46
Table 5 Calculated band gaps (Eg in eV) of the MgAgF3 compounds
| Materials |
Band gap (eV) |
| MgAgF3 (rhombohedral)[our work] |
2.16 |
| AgMgF3 (cubic)[other work,46] |
1.95 |
The calculated band structure (Fig. 3) can be divided into six portions. The flat band located around −40 eV arises from Mg-3p states, while the band at −21.4 eV originates from F-2s states. The band positioned around −2.5 eV is mainly derived from F-2p states. In addition, the band located near −0.32 eV arises from Ag-4d states. The band around 10 eV originates from Ag-4p states, whereas another band associated with Mg-3s states appears at approximately 12 eV. At an indirect band gap of 2.16 eV, the symmetry point Γ–M separates the valence bands from the conduction bands. The Fermi level, EF = 0 eV, is primarily composed of Ag-4d and F-2p, F-2s states. The partial density of states (PDOS) analysis indicates that the upper valence band region is mainly composed of F-2p states with noticeable hybridization with Ag-4d orbitals. This Ag-4d and F-2p hybridization enhances the dispersion of the valence bands near the VBM. In contrast, the conduction band minimum is predominantly derived from Ag-4d states, which are relatively localized and therefore give rise to less dispersive conduction bands.
The distinct orbital character and dispersion behavior of the valence and conduction bands result in their extrema appearing at different k-points, thereby giving rise to the observed indirect band gap. The Mg-3s and Mg-3p states contribute mainly to higher-energy regions and play a minor role near the band edges. Overall, the electronic structure near the band gap of MgAgF3 is governed by Ag-4d and F-2p states, whose hybridization critically determines the band dispersion, highlighting their crucial role in determining the conductivity of MgAgF3.
3.3.2. Charge density.
The charge density distribution of a compound provides visual explanation of the nature of bonding among different atoms. The ionic character of any material can be related to the charge transfer between the cation and anion, while covalent character is related to the sharing of the charge between the cation and anion. Fig. 4 presents the valence electronic charge density (in the units of e Å−3) map of MgAgF3 perovskite along the (100) and (001) planes. Due to its anisotropic nature, it showed deviation in charge density along different planes. The adjacent scale on the right side of the plot of Fig. 4 reveals the acuity of charge (electron) density. The high and low intensity are shown by blue and red colors, respectively. The visual comparison of the corresponding charge density plots shows that the bonding nature of these compounds is partially ionic and partially covalent. The d orbital of Ag makes up most of the charge density around the Ag atom, while the p orbital of F mainly contributes to the density. In Fig. 4, it can be seen that the bond between Ag and F is covalent, while maximum charge resides closer to Ag and F sites. The variation in electronegativity of subsisting ions in the crystal lattice is favorable for such types of bonding. The Ag–F bond in MgAgF3 coincides with the hybridization between Ag-4d and F 2p orbitals evident from PDOS in Fig. 3. It is further noted that the bond between Mg and F is strongly ionic, with no charge sharing among the contours of the respective atoms. A significant amount of charge is transferred from the Mg ion to F because of the large electronegativity difference between the ions. Hence MgAgF3 has a mixed covalent and ionic bond nature with the maximum density distributed in the Ag–F bond direction.
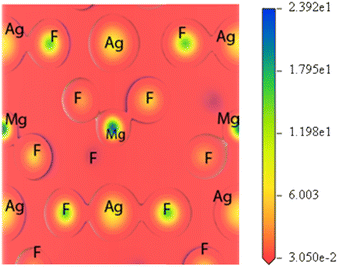 |
| | Fig. 4 Charge density of MgAgF3. | |
3.3.3. Population analysis of MgAgF3.
To analyze the electronic charge distribution in MgAgF3, we have used two complementary methods: Mulliken population analysis70 and Hirshfeld population analysis.71 The calculations were performed using the CASTEP program, which implements the approach of Sanchez-Portal et al.72 In this method, the plane wave basis set transforms to a linear combination of the atomic orbitals to calculate the Mulliken populations and the Mulliken atomic charges. While Mulliken analysis provides useful insights into charge transfer, its dependence on electron wave functions can sometimes lead to discrepancies with experimental observations.46 To address this, we also computed Hirshfeld charges, which rely on atomic electron densities for improved accuracy. A positive Mulliken or Hirshfeld charge indicates that the charge is transferred away from the atom, and if it is negative, the charge is received by the atom.73 From Table 6, we can see that, Mg and Ag are transferring an electron, while F is receiving an electron.
Table 6 Charge spilling, orbital populations (electron), atomic Mulliken charges (electron), formal charge, effective valence and Hirshfeld charge (electron) of MgAgF3
| Compound |
Charge spilling |
Species |
Mulliken atomic population |
Mulliken charge |
Formal ionic charge |
Effective valence |
Hirsfeld charge |
Effective valence |
| s |
p |
D |
f |
Total |
| MgAgF3 |
0.16 |
Mg |
0.256 |
5.950 |
0.000 |
0.000 |
6.206 |
1.794 |
2 |
0.206 |
0.31 |
1.69 |
| Ag |
0.295 |
0.382 |
9.994 |
0.000 |
10.671 |
0.329 |
1 |
0.671 |
0.39 |
0.61 |
| F |
1.987 |
5.721 |
0.000 |
0.000 |
7.708 |
−0.708 |
−1 |
0.292 |
−0.23 |
0.77 |
3.4. Optical properties
The interaction of a material with incident electromagnetic radiation is comprehensively characterized by a suite of fundamental optical parameters such as dielectric function, refractive index, optical conductivity, absorption coefficient, reflectivity, and the energy loss function. These interconnected properties serve as critical descriptors of the material's electrodynamic behavior, offering deep insights into its electronic structure, light-matter interactions (the interaction between electrons and photons), and energy dissipation mechanisms. To investigate the optical characteristics of a material, the complex dielectric function, ε(ω), serves as a foundational parameter. It is mathematically defined as:| | | ε(ω) = ε1(ω) + iε2(ω) | (19) |
where ε1(ω) denotes the real part, associated with the dispersive response of the material, and ε2(ω) represents the imaginary part, linked to energy absorption from the incident electromagnetic field.74,75 The imaginary component ε2(ω) is calculated based on the electronic band structure by evaluating the momentum matrix elements between occupied and unoccupied states.76 In contrast, the real component ε1(ω) is derived from ε2(ω) using the Kramers–Kronig transformation.77| | 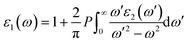 | (20) |
| | 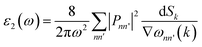 | (21) |
Pronounced features appearing at higher photon energies in the optical spectra are commonly associated with plasmon excitations, which arise from collective oscillations of the valence electron density rather than from individual interband transitions. The energy of these collective modes, referred to as the plasmon frequency, is primarily determined by the valence electron density and provides insight into the collective electronic response of the material.78 The optical constants of MgAgF3 – such as conductivity, dielectric function, absorption spectrum, refractive index, energy loss function, and reflectivity – can be evaluated using the corresponding theoretical formulations and equations,
| | 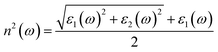 | (22) |
| | 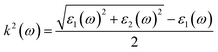 | (23) |
| | 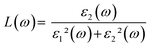 | (24) |
| | 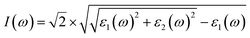 | (25) |
| | 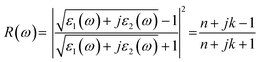 | (26) |
| | 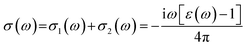 | (27) |
where
ε1(
ω) and
ε2(
ω) are the real and imaginary part of the dielectric function, respectively,
n(
ω) is the refractive index,
k(
ω) represents the extinction coefficient,
L(
ω) is the loss function,
I(
ω) is the absorption coefficient,
R(
ω) is the reflectivity, and
σ(
ω) is the optical conductivity. The optical parameters discussed above were determined using the equilibrium lattice constants optimized at the GGA level, considering the photon energy range extending up to 65 eV. The energy loss function characterizes the amount of energy dissipated as fast-moving electrons traverse a material. Its peak corresponds to the plasma resonance, and the associated frequency is referred to as the plasma frequency. The energy-loss function of MgAgF
3 displays a pronounced and well-defined peak at approximately 63 eV for all crystallographic orientations [
Fig. 5], indicative of a bulk plasmon excitation.
79,80 This feature corresponds to the plasma frequency, at which collective oscillations of the valence electrons dominate the optical response, and beyond which the material exhibits a progressively increased transparency to incident electromagnetic radiation. Additional minor peaks in the 10–25 eV range suggest weaker collective electron excitations. The sharpness of the primary peak implies minimal energy dissipation during electron traversal, which is advantageous for reducing optical losses. Such behavior enhances the efficiency of light–matter interactions, highlighting MgAgF
3's potential for ultrahigh-density optical data storage applications. The reflectivity spectrum of MgAgF
3 exhibits pronounced anisotropic behavior across the (001), (010), and (100) crystallographic directions [
Fig. 5]. In the low to mid-energy range (0–50 eV), the material maintains a low reflectivity (<0.2), indicating high transparency, strong photon penetration and minimal surface reflection, attributes which are beneficial for anti-reflective applications. A significant rise in reflectivity is detected around 63 eV, corresponding to the plasma resonance frequency. This feature arises from plasmon excitation, where the real component of the dielectric function approaches zero, resulting in increased reflectivity and a shift toward metallic-like optical behavior at elevated photon energies. The steepness of this peak suggests minimal energy dissipation, enhancing the material's suitability for high-frequency photonic devices, UV-reflective coatings, and ultrafast optical switches. The absorption coefficient spectra of MgAgF
3 [
Fig. 5] exhibit pronounced energy-dependent behavior across the (001), (010), and (100) crystallographic orientations. Notably, distinct absorption peaks are observed in the ultraviolet (UV) region, particularly between 8–12 eV and around 17 eV, with a significant rise beyond 50 eV, culminating in a sharp peak near 63 eV. While the lower-energy absorption features arise from interband electronic transitions, the high-energy peak near 63 eV is primarily associated with plasmon excitation and a collective electronic response, primarily involving the excitation of electrons from the fluorine (F) 2p orbitals in the valence band to the magnesium (Mg) 3s/3p and silver (Ag) 4d orbitals in the conduction band.
81,82 Such transitions are indicative of the material's electronic structure and are consistent with the observed optical absorption characteristics. The directional dependence of these optical responses underscores MgAgF
3's potential in polarization-sensitive applications and advanced optoelectronic systems. The real (
n) and imaginary (
k) components of the complex refractive index are extracted from the real
ε1(
ω) and imaginary
ε2(
ω) parts of the dielectric function, respectively. These parameters govern the phase propagation and attenuation behavior of electromagnetic waves within the medium, thereby providing critical insights into the material's optical density and transparency characteristics. MgAgF
3 demonstrates clear optical anisotropy along the (001), (010), and (100) direction, with noticeable directional variation in both refractive index and extinction coefficient [
Fig. 5]. The static refractive index ranges from 1.5 to 1.7, suggesting moderate photon confinement. Strong absorption peaks in the UV region (8–12 eV and ∼17 eV) reveal pronounced interband transitions,
78 while a distinct transparency window between 25–45 eV is consistent across all axes. Beyond 45 eV, enhanced optical responses imply the involvement of deeper electronic states, emphasizing MgAgF
3's potential in polarization-sensitive and energy selective optoelectronic devices. Conductivity describes how a material responds to a time-varying external electric field, specifically reflecting changes in its electrical behavior under the influence of incident light. The optical conductivity spectra of MgAgF
3 along the (001), (010), and (100) crystallographic directions reveal distinct anisotropic behavior
83 [
Fig. 5] governed by the material's direction-dependent electronic structure. In all directions, the real part of the conductivity (
σ1) features prominent peaks within the 10–20 eV energy range and a strong enhancement beyond 50 eV, peaking sharply near 63 eV. These peaks signify a bulk plasmon excitation, where incident photons promote electrons from the fluorine (F) 2p orbitals in the valence band to the unoccupied magnesium (Mg) 3s/3p and silver (Ag) 4d orbitals in the conduction band. Among the polarizations, the (001) direction exhibits the highest
σ1 magnitude, indicating stronger light–matter interaction and higher transition probability along this axis. The (010) and (100) orientations present similar spectral features but with slightly diminished intensities and minor shifts in peak positions, reflecting directional dependence in transition dynamics and charge carrier response. The imaginary part (
σ2) shows negative excursions across all directions, particularly in high-energy regions, denoting energy storage and delayed current response typical of dielectric-like behavior. This consistent interband activity and frequency-sensitive response across axes highlight MgAgF
3's potential in polarization-sensitive photonic and optoelectronic applications, especially in systems demanding tunable conductivity and controlled light absorption at specific energy ranges (
e.g. UV to soft X-ray frequencies). The dielectric function spectra of MgAgF
3 were analyzed along the (001), (010), and (100) crystallographic directions [
Fig. 5], revealing anisotropic optical behavior.
84 For all polarizations, the real part (
ε1) exhibits a high static dielectric constant (∼3.0), indicating strong low-frequency polarization. The (001) direction shows multiple pronounced peaks in
ε1 across the UV range, suggesting significant dispersion effects. Similarly, the (010) and (100) orientations display comparable peak structures but with subtle variations in peak intensity and position, reflecting directional dependence in electronic polarizability. The imaginary part (
ε2), corresponding to optical absorption, presents intense peaks between 8–20 eV and above 50 eV for all three directions, attributed to interband electronic transitions primarily from fluorine (F) 2p valence states to magnesium (Mg) 3s/3p and silver (Ag) 4d conduction states. Notably, the peak intensities differ slightly among the polarizations, with (001) showing the strongest absorption features, indicating higher photon-electron interaction along this axis. The plasmonic nature of the high-energy optical response is further corroborated by the dielectric function. As illustrated in
Fig. 5, the real part of the dielectric function,
ε1(
ω), crosses zero around 60–63 eV, while the imaginary part,
ε2(
ω), remains relatively small. This behavior fulfills the conventional plasmon resonance condition, indicating that the pronounced high-energy peak originates from a collective electronic excitation rather than from interband optical transitions. Importantly, because plasmon energies are determined by the overall valence electron density, they are not limited by the material's fundamental band gap (
Table 7).
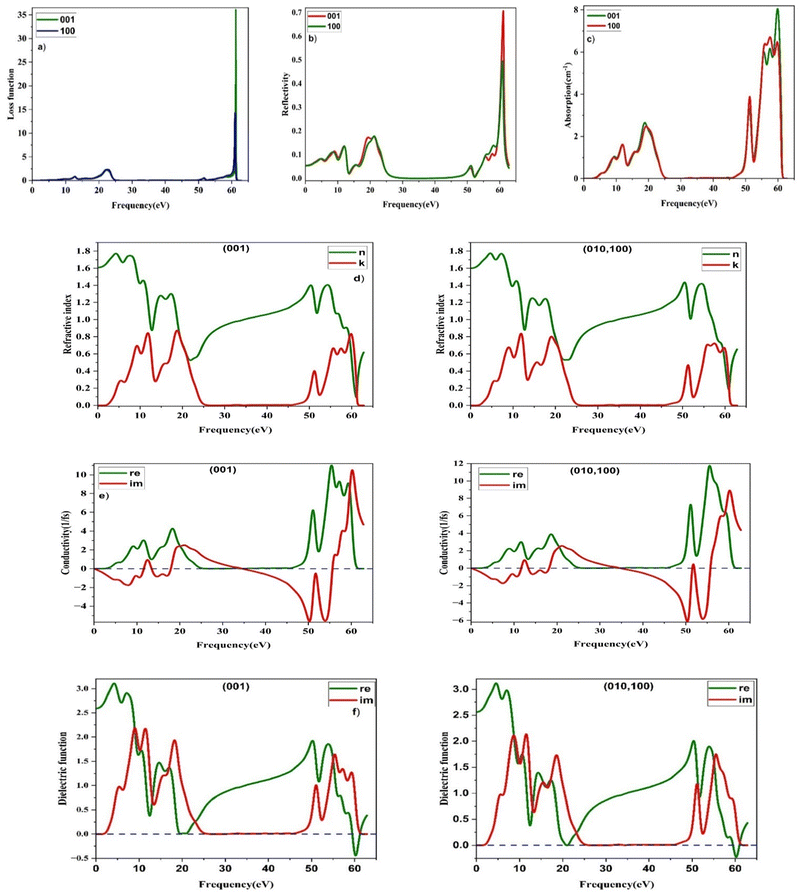 |
| | Fig. 5 Calculated optical constants of MgAgF3 – (a) energy-loss spectrum, (b) reflectivity, (c) absorption spectrum, (d) refractive index and extinction coefficient, (e) conductivity, and (f) real (ε1) and imaginary (ε2) parts of the dielectric function of MgAgF3 along the (001), (010), and (100) crystallographic directions. The bulk plasmon resonance is identified by the zero-crossing of ε1(ω) at near 63 eV, where ε2(ω) is small. | |
Table 7 Comparison of optical properties with previous work
| Optical properties |
MgAgF3 (R3c) |
AgMgF3 (Pm3m)85 |
AgMgF3 (Pm3m)81 |
AgMgF3 (Pm3m)67 |
Novelty |
| Band Gap |
2.16 eV (indirect M → Γ) |
≈5.24 eV (indirect Γ → X) |
≈0.78 eV (indirect Γ → X) |
1.95 eV (indirect M → Γ) |
Different band gap due to rhombohedral distortion, suggesting different optical transparency range |
| Refractive index |
n(0) ≈ 1.5–1.7 (anisotropic with polarization dependence) |
n(0) ≈ 1.8 (isotropic cubic) |
n(0) ≈ 1.618 (isotropic cubic) and superluminal behavior |
n(0) ≈ 1.6 (isotropic cubic) |
Anisotropic refractive index (cubic study only, isotropic) |
| Absorption coefficient |
Strong UV peaks at 8–12 eV and ∼17 eV, transparent below ∼8 eV |
No absorption below ∼7.5 eV; strong UV absorption at higher energies |
Strong UV peaks at 19.4 eV (main) and 4–25 eV range overall |
Visible-near IR absorption at 2.1 eV |
Identification of double UV absorption peaks associated with the rhombohedral symmetry |
| Reflectivity |
Very low (<0.2) up to ∼50 eV; plasmon peak at ∼63 eV |
Low (<0.1) up to ∼5 eV; rises to 0.75 (13–16 eV) |
R(0) = 5.6% and maximum R(ω) = 18.67% near 21.67 eV. |
R(0) = 5.6% and maximum R(ω) ≤ 10% at 0 to 4 eV. |
Evidence of high-energy plasmon resonance in MgAgF3, demonstrating high-energy collective excitations |
| Optical anisotropy |
Explicitly calculated; different ε1(ω), ε2(ω) along different axes |
Not studied (cubic only, isotropic) |
Not studied (cubic only, isotropic) |
Not studied (cubic only, isotropic) |
Comprehensive analysis of direction-dependent dielectric functions |
Unlike previous works,67,81,85 where the ideal cubic phase of AgMgF3 was examined and reported an isotropic refractive index, the present work provides the first prediction of the full optical response of the rhombohedral (R3c) phase of MgAgF3. Our calculations reveal a markedly smaller indirect band gap of 2.16 eV and pronounced optical anisotropy, including direction-dependent refractive indices (1.5–1.7), distinct UV absorption peaks at 8–12 eV and ∼17 eV, and a high-energy plasmon resonance near 63 eV. These findings not only complement previous cubic-phase studies but also open new avenues for anisotropic UV photonics and optoelectronic applications of MgAgF3.
3.5. Thermodynamic properties
From the graph we get three important thermodynamic properties: free energy, enthalpy, and T* entropy changing with temperature for a perovskite material. At low temperatures (near 0 K), all three values start from around zero, consistent with the third law of thermodynamics, which says entropy and thermal motion should approach zero at absolute zero temperature.86 The free energy (G) gradually decreases as the temperature increases. This is expected because more thermal vibrations make the system less stable, lowering the free energy. The downward slope reflects increasing entropy contribution (G = H − TS).87 The enthalpy (H) increases steadily with temperature, showing that the material absorbs more energy as heat increases its internal vibrations.88 The T* entropy (TS) rises sharply, especially beyond 300 K, showing how vibrational disorder increases at higher temperatures (Fig. 6). The steep growth indicates strong phonon activity at elevated temperatures, typical for complex crystals.86 These curves are derived from phonon calculations using the quasi-harmonic approximation (QHA), which estimates how lattice vibrations contribute to thermodynamic stability. From this data we can predict how materials behave under heat, aiding in the design of stable devices or materials for thermal applications.88
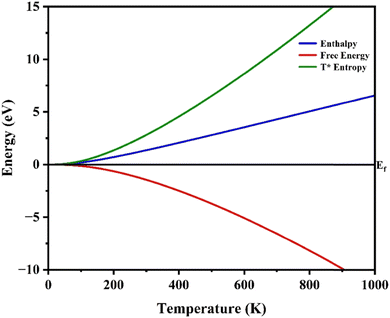 |
| | Fig. 6 Free energy, enthalpy and T* entropy of MgAgF3. | |
3.5.1. Heat capacity (Debye graph).
This graph shows how the heat capacity at constant volume (Cv) of the perovskite material changes with temperature, following the behavior predicted by the Debye model.89 At low temperatures (<100 K), the heat capacity increases sharply in a non-linear way. This is consistent with the Debye law, which states that at very low temperatures, the heat capacity of a solid grows as the cube of the temperature (Cv ∝ T3), due to only low-frequency phonons being excited.89 As temperature increases further (around 200–400 K), the slope of the curve begins to flatten. This reflects the fact that more phonon modes are getting populated with energy, and the material approaches thermal saturation.86 At high temperatures (>500 K), the heat capacity levels off and reaches a constant value, close to the Dulong–Petit limit (∼3R per atom or ∼6R per unit cell, depending on the basis) (Fig. 7).89,90 This behavior tells us that this material behaves like a typical crystalline solid with well-defined phonon modes and no anharmonic instabilities across the temperature range.
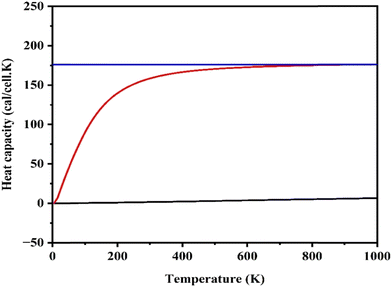 |
| | Fig. 7 Heat capacity of MgAgF3. | |
3.5.2. Phonon DOS.
The phonon density of states (DOS) graph shows the vibrational characteristics of a perovskite material, with phonon frequencies ranging from 0 to approximately 16 THz. The absence of significant imaginary (negative) frequencies suggests that the structure is dynamically stable.86 The low-frequency region (0–3 THz) is dominated by acoustic phonon modes, which are very important for thermal conductivity, and they contribute significantly to specific heat at low temperatures.87 Peaks in the phonon DOS correspond to collective vibrational modes involving specific atomic sublattices, where low-frequency peaks generally arise from heavier atoms and high-frequency regions are dominated by lighter atomic vibrations. Here, the presence of multiple sharp peaks indicates vibrational mode localization and potential phonon band degeneracy (Fig. 8).87 Phonon DOS serves as a bridge between microscopic lattice vibrations and macroscopic thermodynamic behavior. These characteristics are essential for interpreting thermal properties such as specific heat and entropy using quasi-harmonic approximations or more advanced methods like the Boltzmann transport equation.86,91
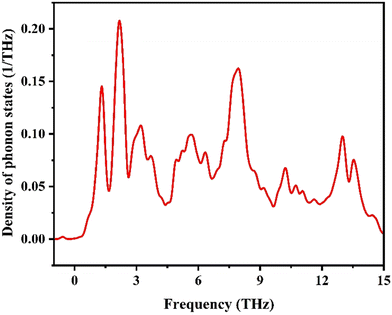 |
| | Fig. 8 Density of phonon states. | |
3.5.3. Phonon dispersion.
The phonon dispersion graph shows the vibrational modes of a perovskite material along directions in the Brillouin zone, labeled Γ–A–H–K–Γ–M–L–H. The dynamic stability of rhombohedral MgAgF3 (space group R3c) was examined through phonon dispersion calculations performed using density functional theory. The phonon spectra were calculated with a carefully converged plane wave cutoff energy of 340 eV and a Γ-centered k-point sampling to ensure numerical accuracy. As shown in Fig. 9, the resulting phonon dispersion curves exhibit no imaginary (negative) frequencies throughout the entire Brillouin zone, including the vicinity of the Γ point. The absence of soft modes confirms that the optimized rhombohedral structure corresponds to a true local minimum on the potential energy surface and is dynamically stable. The dispersion consists of three acoustic branches originating from the Γ point and a set of well separated optical branches extending up to approximately 10 THz, reflecting the vibrational contributions of Mg, Ag, and F atoms. These results conclusively establish the dynamic stability of rhombohedral MgAgF3 and support its viability as a stable fluoro perovskite phase.
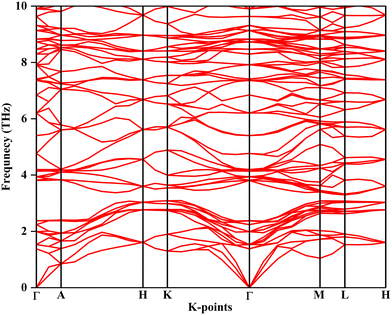 |
| | Fig. 9 Phonon dispersion. | |
4. Conclusions
In this study, we have, for the first time, carried out a comprehensive investigation of the rhombohedral (R3c) phase of MgAgF3 using density functional theory. Our structural optimization confirms a robust trigonal R3c perovskite phase, composed of corner-sharing MgF6 octahedra, with optimized lattice parameters a = b = 5.52 Å and c = 13.55 Å. Unlike earlier reports on the cubic phase, our results uncover a distinctly different electronic character, with an indirect band gap of 2.157 eV (M → Γ) and marked optical anisotropy in the ultraviolet range. Mechanical analysis reveals that MgAgF3 combines moderate stiffness with ductility (B = 78.01 GPa, G = 36.42 GPa, E = 92.61 GPa, ν = 0.2975, B/G = 2.14), alongside slight elastic anisotropy (AU = 0.07369), confirming its mechanical resilience. Optical properties further emphasize its technological promise, displaying strong UV absorption (∼8–12 eV and ∼17 eV), a wide transparency window (25–45 eV), and a pronounced plasmon resonance near ∼63 eV, which together highlight its potential in UV optoelectronics, optical coatings, and radiation shielding. The full elastic tensor confirms mechanical stability and ductility (Pugh's ratio 2.14), while the machinability index (2.09) and low universal anisotropy factor highlight the favorable processing and mechanical characteristics of MgAgF3. Moreover, phonon dispersion and heat capacity analyses establish its dynamic and thermodynamic stability, reinforcing its suitability for real-world applications.
Overall, these findings extend and enrich previous cubic-phase studies, presenting a more holistic understanding of MgAgF3. By demonstrating its stability, unique electronic and optical behavior, and promising mechanical characteristics, our work positions rhombohedral MgAgF3 as a strong candidate for next-generation ultraviolet photonic and optoelectronic devices.
Conflicts of interest
There are no conflicts of interest to declare.
Data availability
Relevant datasets generated and analyzed during the current investigation may be obtained from the corresponding author upon reasonable request.
Acknowledgements
The authors gratefully acknowledge Dr. Md. Rasadujjaman for his helpful comments and assistance in improving the clarity and presentation of this manuscript.
References
- N. Erum and M. A. Iqbal, Ab initio study of high dielectric constant oxide-perovskite: perspective for miniaturization technology, Mater. Res. Express, 2017, 4, 025904 Search PubMed.
- A. H. Reshak, M. S. Abu-Jafar and Y. Al-Douri, Two symmetric n-type interfaces SrTiO3/LaAlO3 in perovskite: Electronic properties from density functional theory, J. Appl. Phys., 2016, 119, 245303 Search PubMed.
- N. Moulay,
et al., Predictive study of structural, electronic, magnetic and thermodynamic properties of XFeO3 (X = Ag, Zr and Ru) multiferroic materials in cubic perovskite structure: first-principles calculations, Mater. Sci.-Pol., 2015, 33, 402–413 Search PubMed.
- F. Litimein, R. Khenata, A. Bouhemadou, Y. Al-Douri and S. B. Omran, First-principle calculations to investigate the elastic and thermodynamic properties of RBRh3 (R = Sc, Y and La) perovskite compounds, Mol. Phys., 2012, 110, 121–128 Search PubMed.
- R. Terki, H. Feraoun, G. Bertrand and H. Aourag, Full potential calculation of structural, elastic and electronic properties of BaZrO3 and SrZrO3, Phys. Status Solidi B, 2005, 242, 1054–1062 Search PubMed.
- T. Nishimatsu,
et al., Band Structures of Perovskite-Like Fluorides for Vacuum-Ultraviolet-Transparent Lens Materials, Jpn. J. Appl. Phys., 2002, 41, L365–L367 Search PubMed.
- M. Husain,
et al., Insight into the physical properties of the inter-metallic titanium-based binary compounds, Eur. Phys. J. Plus, 2021, 136, 624 Search PubMed.
- N. Rahman,
et al., First principle study of structural, electronic, optical and mechanical properties of cubic fluoro-perovskites: (CdXF3, X = Y, Bi), Eur. Phys. J. Plus, 2021, 136, 347 Search PubMed.
- S. Körbel, M. A. L. Marques and S. Botti, Stability and electronic properties of new inorganic perovskites from high-throughput ab initio calculations, J. Mater. Chem. C, 2016, 4, 3157–3167 Search PubMed.
- N. Chouit,
et al., First-principles study of the structural, electronic and thermal properties of CaLiF3, Phys. Scr., 2013, 88, 035702 Search PubMed.
- T. Seddik,
et al., Elastic, electronic and thermodynamic properties of fluoro-perovskite KZnF3 via first-principles calculations, Appl. Phys. A: Mater. Sci. Process., 2012, 106, 645–653 Search PubMed.
- H. Benmhidi, H. Rached, D. Rached and M. Benkabou, Ab Initio Study of Electronic Structure, Elastic and Transport Properties of Fluoroperovskite LiBeF3, J. Electron. Mater., 2017, 46, 2205–2210 Search PubMed.
- P. Parhi, J. Kramer and V. Manivannan, Microwave initiated hydrothermal synthesis of nano-sized complex fluorides, KMF3 (K = Zn, Mn, Co, and Fe), J. Mater. Sci., 2008, 43, 5540–5545 Search PubMed.
- M. Eibschütz, H. J. Guggenheim, S. H. Wemple, I. Camlibel and M. DiDomenico, Ferroelectricity in BaM2 + F4, Phys. Lett. A, 1969, 29, 409–410 Search PubMed.
- S. Margadonna and G. Karotsis, Cooperative Jahn-Teller Distortion, Phase Transitions, and Weak Ferromagnetism in the KCrF3 Perovskite, J. Am. Chem. Soc., 2006, 128, 16436–16437 Search PubMed.
- F. Zhang, Y. Mao, T. J. Park and S. S. Wong, Inside Front Cover: Green Synthesis and Property Characterization of Single-Crystalline Perovskite Fluoride Nanorods, Adv. Funct. Mater., 2008, 18, adfm.200890007 Search PubMed.
- A. V. Gektin, I. M. Krasovitskaya and N. V. Shiran, High-temperature thermoluminescence of KMgF3-based crystals, J. Lumin., 1997, 72–74, 664–666 Search PubMed.
- C. N. R. Rao, Novel materials, materials design and synthetic strategies: recent advances and new directions, J. Mater. Chem., 1999, 9, 1–14 Search PubMed.
- A. S. Bhalla, R. Guo and R. Roy, The perovskite structure—a review of its role in ceramic science and technology, Mater. Res. Innov., 2000, 4, 3–26 CrossRef CAS.
-
M. R. Levy, Crystal Structure and Defect Property Predictions in Ceramic Materials, Imperial College of Science, Technology and Medicine, 2005, ch. 3, pp. 79–144 Search PubMed.
- M. Sahnoun,
et al., Full potential calculation of structural, electronic and optical properties of KMgF3, Mater. Chem. Phys., 2005, 91, 185–191 Search PubMed.
- M. Yanagihara,
et al., Vacuum ultraviolet field emission lamp utilizing KMgF3 thin film phosphor, APL Mater., 2014, 2, 046110 Search PubMed.
-
Y. Oyama, Optical Member for Vacuum Ultraviolet, and Aligner and Device Manufacture Method Using Same, ( 2004).
- Z. Wang,
et al., A new 12L-hexagonal perovskite Cs4 Mg3 CaF12: structural transition derived from the partial substitution of Mg2+ with Ca2+, RSC Adv., 2014, 4, 54194–54198 Search PubMed.
- P. J. Edwardson,
et al., Ferroelectricity in perovskitelike NaCaF 3 predicted ab initio, Phys. Rev. B:Condens. Matter Mater. Phys., 1989, 39, 9738–9741 Search PubMed.
- N. T. Mahmoud, R. Shaltaf, M. Alyami, M. Alshaaer and R. Habib, Ab initio stability to efficiency study of SrGeO3 perovskite, MRS Energy Sustain., 2024, 11, 647–658 Search PubMed.
- L. L. Boyer,
et al., Predicted properties of NaCaF3, AIP Conf. Proc., 2000, 535, 364–371 Search PubMed.
- I. Bourachid,
et al., Insight into the structural, electronic, mechanical and optical properties of inorganic lead bromide perovskite APbBr3 (A = Li, Na, K, Rb, and Cs), Comput. Condens. Matter, 2020, 24, e00478 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- Z. Wu and R. E. Cohen, More accurate generalized gradient approximation for solids, Phys. Rev. B:Condens. Matter Mater. Phys., 2006, 73, 235116 Search PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 Search PubMed.
- D. Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism, Phys. Rev. B:Condens. Matter Mater. Phys., 1990, 41, 7892–7895 Search PubMed.
- D. D. Koelling and B. N. Harmon, A technique for relativistic spin-polarised calculations, J. Phys. C-Solid State Phys., 1977, 10, 3107–3114 Search PubMed.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients, Rev. Mod. Phys., 1992, 64, 1045–1097 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B:Condens. Matter Mater. Phys., 1976, 13, 5188–5192 Search PubMed.
- M. D. Segall,
et al., First-principles simulation: ideas, illustrations and the CASTEP code, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 Search PubMed.
- MgAgF3 parameters.
- M. Gajdoš, K. Hummer, G. Kresse, J. Furthmüller and F. Bechstedt, Linear optical properties in the projector-augmented wave methodology, Phys. Rev. B:Condens. Matter Mater. Phys., 2006, 73, 045112 Search PubMed.
- A. Hassan,
et al., Effect of heteroatoms on structural, electronic and spectroscopic properties of polyfuran, polythiophene and polypyrrole: A hybrid DFT approach, J. Mol. Struct., 2023, 1274, 134484 Search PubMed.
- H. Yu,
et al., Coupling ferroelectric polarization and anisotropic charge migration for enhanced CO2 photoreduction, Appl. Catal., B, 2021, 284, 119709 Search PubMed.
- R. Ullah,
et al., Pressure-dependent elasto-mechanical stability and thermoelectric properties of MYbF3 (M = Rb, Cs) materials for renewable energy, Int. J. Energy Res., 2021, 45, 8711–8723 Search PubMed.
- S. Tabassam,
et al., Co2YZ (Y = Cr, Nb, Ta, V and Z = Al, Ga) Heusler alloys under the effect of pressure and strain, J. Mol. Graph. Model., 2021, 104, 107841 Search PubMed.
- R. Singla, S. Kumar, T. A. Hackett, A. H. Reshak and M. K. Kashyap, Genesis of magnetism in graphene/MoS2 van der Waals heterostructures via interface engineering using Cr-adsorption, J. Alloys Compd., 2021, 859, 157776 Search PubMed.
- D. M. Hoat,
et al., Strain effect on the electronic and optical properties of 2D Tetrahexcarbon: a DFT-based study, Indian J. Phys., 2021, 95, 2365–2373 Search PubMed.
- M. Husain,
et al., Insight into the physical properties of the inter-metallic titanium-based binary compounds, Eur. Phys. J. Plus, 2021, 136, 624 Search PubMed.
- G. Surucu, A. Gencer, O. Surucu and Md. A. Ali, DFT Insights into Noble Gold-Based Compound Li5 AuP2: Effect of Pressure on Physical Properties, ACS Omega, 2023, 8, 15673–15683 CrossRef CAS PubMed.
- Md. S. Alam,
et al., Tuning band gap and enhancing optical functions of AGeF3 (A = K, Rb) under pressure for improved optoelectronic applications, Sci. Rep., 2022, 12, 8663 Search PubMed.
- M. R. Molla, M. Saiduzzaman, T. I. Asif, W. A. Dujana and K. M. Hossain, Electronic phase transition from semiconducting to metallic in cubic halide CsYbCl3 perovskite under hydrostatic pressure, Phys. Rev. B:Condens. Matter Mater. Phys., 2022, 630, 413650 Search PubMed.
- F. Mouhat and F.-X. Coudert, Necessary and sufficient elastic stability conditions in various crystal systems, Phys. Rev. B:Condens. Matter Mater. Phys., 2014, 90, 224104 Search PubMed.
- M. M. Namisi, R. J. Musembi, W. M. Mulwa and B. O. Aduda, DFT study of cubic, tetragonal and trigonal structures of KGeCl3 perovskites for photovoltaic applications, Comput. Condens. Matter, 2023, 34, e00772 Search PubMed.
- S. S. Essaoud, S. M. Al Azar, A. A. Mousa and R. S. Masharfe, Characterization of structural, dynamic, optoelectronic, thermodynamic, mechanical and thermoelectric properties of AMgF3 (A = K or Ag) fluoro-perovskites compounds, Phys. Scr., 2023, 98(3), 035820 Search PubMed.
- Q.-J. Liu, H. Qin, Z. Jiao, F.-S. Liu and Z.-T. Liu, First-principles calculations of structural, elastic, and electronic properties of trigonal ZnSnO3 under pressure, Mater. Chem. Phys., 2016, 180, 75–81 Search PubMed.
- S. Kuma and M. M. Woldemariam, Structural, Electronic, Lattice Dynamic, and Elastic Properties of SnTiO3 and PbTiO3 Using Density Functional Theory, Adv. Condens. Matter Phys., 2019, 1–12 Search PubMed.
- A. Reuss, Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle, Z. Angew. Math. Mech., 1929, 9, 49–58 Search PubMed.
- R. Hill, The Elastic Behaviour of a Crystalline Aggregate, Proc. Phys. Soc. Sect. A, 1952, 65, 349–354 CrossRef.
- H. Bochaoui and M. El Bouabdellati, Feasibility of cubic and trigonal KGeBr3 perovskite material in photovoltaic applications: Structural, optical, electronic, and mechanical properties via a DFT study, Phys. Rev. B:Condens. Matter Mater. Phys., 2024, 695, 416516 Search PubMed.
- S. A. Dar, V. Srivastava, U. K. Sakalle, A. Rashid and G. Pagare, First-principles investigation on electronic structure, magnetic, mechanical and thermodynamic properties of SrPuO3 perovskite oxide, Mater. Res. Express, 2018, 5, 026106 Search PubMed.
- H. Dong, C. Chen, S. Wang, W. Duan and J. Li, Elastic properties of tetragonal BiFeO3 from first-principles calculations, Appl. Phys. Lett., 2013, 102, 182905 Search PubMed.
- M. Mbilo and R. Musembi, First-Principle Calculations to Investigate Structural, Electronic, Elastic, Mechanical, and Optical Properties of K2CuX (X = As, Sb) Ternary Compounds, Adv. Mater. Sci. Eng., 2022, 1–10 Search PubMed.
- Y. Benkaddour,
et al., First-Principle Calculations of Structural, Elastic, and Electronic Properties of Intermetallic Rare Earth R2Ni2Pb (R = Ho, Lu, and Sm) Compounds, J. Supercond. Nov. Magn., 2018, 31, 395–403 CrossRef CAS.
- M. H. K. Rubel,
et al., Newly synthesized A-site ordered cubic-perovskite superconductor (Ba0.54K0.46)4Bi4O12: A DFT investigation, Phys. C, 2020, 574, 1353669 Search PubMed.
- S. I. Ranganathan and M. Ostoja-Starzewski, Universal Elastic Anisotropy Index, Phys. Rev. Lett., 2008, 101, 055504 Search PubMed.
- S. J. Edrees, M. M. Shukur and M. M. Obeid, First-principle analysis of the structural, mechanical, optical and electronic properties of wollastonite monoclinic polymorph, Comput. Condens. Matter, 2018, 14, 20–26 Search PubMed.
- V. Tvergaard and J. W. Hutchinson, Microcracking in Ceramics Induced by Thermal Expansion or Elastic Anisotropy, J. Am. Ceram. Soc., 1988, 71, 157–166 CrossRef CAS.
- H. Yan, Z. Wei, M. Zhang and Q. Wei, Exploration of stable stoichiometries, ground-state structures, and mechanical properties of the W–Si system, Ceram. Int., 2020, 46, 17034–17043 Search PubMed.
- W.-C. Hu, Y. Liu, D.-J. Li, X.-Q. Zeng and C.-S. Xu, First-principles study of structural and electronic properties of C14-type Laves phase Al2Zr and Al2Hf, Comput. Mater. Sci., 2014, 83, 27–34 Search PubMed.
- Y. Selmani and L. Bahmad, Structural, mechanical, optoelectronic properties of AgMgX3 (X = F, Cl, and Br) perovskites: DFT calculations, Mod. Phys. Lett. B, 2025, 39, 2550166 CrossRef CAS.
- M. Harmel,
et al., Full-Potential Calculation of Structural, Electronic, and Thermodynamic Properties of Fluoroperovskite (M = Be and Mg), Int. J. Thermophys., 2012, 33, 2339–2350 Search PubMed.
- S. Khan, N. Mehmood, R. Ahmad, A. Kalsoom and K. Hameed, Analysis of structural, elastic and optoelectronic properties of indium-based halide perovskites InACl3 (A = Ge, Sn, Pb) using density functional theory, Mater. Sci. Semicond. Process., 2022, 150, 106973 Search PubMed.
- R. S. Mulliken, Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I, J. Chem. Phys., 1955, 23, 1833–1840 Search PubMed.
- F. L. Hirshfeld, Bonded-atom fragments for describing molecular charge densities, Theor. Chim. Acta, 1977, 44, 129–138 Search PubMed.
- D. Sanchez-Portal, E. Artacho and J. M. Soler, Projection of plane-wave calculations into atomic orbitals, Solid State Commun., 1995, 95, 685–690 Search PubMed.
- M. A. Hadi,
et al., A comparative DFT exploration on M- and A-site double transition metal MAX phase, Ti3ZnC2, Open Ceram., 2022, 12, 100308 CrossRef CAS.
-
M. Fox, Optical Properties of Solids, Oxford Univ. Press, Oxford, 2008 Search PubMed.
- R. Saniz, L.-H. Ye, T. Shishidou and A. J. Freeman, Structural, electronic, and optical properties of NiAl 3: First-principles calculations, Phys. Rev. B:Condens. Matter Mater. Phys., 2006, 74, 014209 CrossRef.
- A. S. Jbara, J. Munir, B. Ul Haq and M. A. Saeed, Density functional theory study of mixed halide influence on structures and optoelectronic attributes of CsPb(I/Br)3, Appl. Opt., 2020, 59, 3751 Search PubMed.
- H. Tributsch, Solar Energy-Assisted Electrochemical Splitting of Water. Some Energetical, Kinetical and Catalytical Considerations Verified on MoS2 Layer Crystal Surfaces, Z. Naturforsch., A:Phys. Sci., 1977, 32, 972–985 Search PubMed.
- X.-H. Zhu, X.-B. Xiao, X.-R. Chen and B.-G. Liu, Electronic structure, magnetism and optical properties of orthorhombic GdFeO3 from first principles, RSC Adv., 2017, 7, 4054–4061 Search PubMed.
-
R. F. Egerton, Electron Energy-Loss Spectroscopy in the Electron Microscope, Springer, US, Boston, MA, 2011 DOI:10.1007/978-1-4419-9583-4.
- A. Mock,
et al., Band-to-band transitions, selection rules, effective mass, and excitonic contributions in monoclinic β-Ga2O3, Phys. Rev. B, 2017, 96, 245205 CrossRef.
- G. Murtaza,
et al., First principle study of cubic perovskites: AgTF3 (T = Mg, Zn), Phys. Rev. B:Condens. Matter Mater. Phys., 2011, 406, 4584–4589 Search PubMed.
- M. Hilfiker,
et al., Anisotropic dielectric functions, band-to-band transitions, and critical points in α-Ga2O3, Appl. Phys. Lett., 2021, 118, 062103 Search PubMed.
- H. Khan,
et al., Computational Study of Elastic, Structural, Electronic, and Optical Properties of GaMF3 (M = Be and Ge) Fluoroperovskites, Based on Density Functional Theory, Molecules, 2022, 27, 5264 Search PubMed.
- F. A. Rodríguez Ortiz,
et al., The Anisotropic Complex Dielectric Function of CsPbBr3 Perovskite Nanorods Obtained via an Iterative Matrix Inversion Method, J. Phys. Chem. C, 2023, 127, 14812–14821 Search PubMed.
- S. Sâad Essaoud, S. M. Al Azar, A. A. Mousa and R. S. Masharfe, Characterization of structural, dynamic, optoelectronic, thermodynamic, mechanical and thermoelectric properties of AMgF3 (A = K or Ag) fluoro-perovskites compounds, Phys. Scr., 2023, 98, 035820 Search PubMed.
- A. Togo and I. Tanaka, First principles phonon calculations in materials science, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- S. Baroni, S. De Gironcoli, A. Dal Corso and P. Giannozzi, Phonons and related crystal properties from density-functional perturbation theory, Rev. Mod. Phys., 2001, 73, 515–562 CrossRef CAS.
- N. Mounet and N. Marzari, First-principles determination of the structural, vibrational and thermodynamic properties of diamond, graphite, and derivatives, Phys. Rev. B:Condens. Matter Mater. Phys., 2005, 71, 205214 Search PubMed.
-
C. Kittel, Introduction to Solid State Physics, Wiley, Hoboken, NJ, 2005 Search PubMed.
-
D. C. Wallace, Thermodynamics of Crystals, Dover publication, Mineola (N.Y.), 1998 Search PubMed.
- A. Castellanos-Gomez,
et al., Periodic spatial variation of the electron-phonon interaction
in epitaxial graphene on Ru(0001), Appl. Phys. Lett., 2013, 102, 063114 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Jannat Ara
Jui
,
Jannat Ara
Jui































