DOI:
10.1039/D5MA01325A
(Paper)
Mater. Adv., 2026,
7, 5183-5193
Unlocking high-energy and long-life supercapacitors via Zn–MnO2/MoS2 heterostructure engineering
Received
14th November 2025
, Accepted 9th April 2026
First published on 11th April 2026
Abstract
Materials engineering plays a pivotal role in determining the energy storage efficiency of supercapacitors. In this work, a Zn–MnO2/MoS2 heterostructure was synthesized via a hydrothermal route, where the synergistic coupling of Zn-doped MnO2 with conductive MoS2 nanosheets significantly enhanced redox activity, electronic conductivity, surface area and structural stability. Zn doping not only expanded the MnO2 lattice to facilitate faster ion diffusion but also induced oxygen vacancies, providing additional active sites for charge storage. Meanwhile, MoS2 offered a conductive 2D framework that buffered volume changes and accelerated electron transport. As a result, the Zn–MnO2/MoS2 electrode delivered a high capacitance of 1440 F g−1 at 2.85 A g−1, outperforming individual Zn–MnO2 (1250 F g−1) and MnO2/MoS2 (1370 F g−1) as well as previously reported electrodes in 1 M KOH. Furthermore, the assembled Zn–MnO2/MoS2//AC device exhibited a specific capacitance of 147 F g−1 at 2.85 A g−1, an excellent energy density of 59 Wh kg−1 at a power density of 1145 W kg−1 and outstanding cycling stability with ∼91% retention over 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. These experimental and theoretical insights highlight the strong potential of Zn–MnO2/MoS2 heterostructures for next-generation practical supercapacitor applications.
000 cycles. These experimental and theoretical insights highlight the strong potential of Zn–MnO2/MoS2 heterostructures for next-generation practical supercapacitor applications.
1. Introduction
In the past decade, the extensive use of fossil fuels has resulted in serious environmental damage, leading to increased interest in renewable and sustainable energy sources. Among these, electrochemical energy storage systems particularly lithium-ion batteries and supercapacitors (SCs) have become prominent portable and environmentally friendly power sources.1 However, each system has inherent limitations. For instance, batteries often suffer from gradual material degradation over time and low power density.2 In contrast, SCs are emerging as promising alternatives particularly for applications such as uninterrupted power supplies, energy storage systems and hybrid vehicles. They offer significant benefits, including exceptionally high theoretical energy density, superior power density, rapid charge–discharge capability and broad operational temperature tolerance.3 Despite these benefits, SCs face critical challenges most notably their limited practical specific capacitance and cycling stability, which constrain their broader applicability. One of the most important factors influencing the performance of SCs is selection of electrode materials.4 So far, researchers have investigated numerous materials involving various transition metal-based compounds, graphene, conducting polymers, carbon nanotubes and metal–organic frameworks, for instance, hydroxides, phosphides and sulfides.5–9 Despite their excellent electrochemical performance, these materials remain impractical for widespread use due to their scarcity and high production costs. In contrast, MnO2 has garnered considerable attention due to the multivalence of Mn, its low cost, abundance, high theoretical capacitance and environmental friendliness.10 However, despite its high theoretical capacitance, the practical deployment of pristine MnO2 is significantly limited by its intrinsically low electrical conductivity. This results in inferior rate capability, sluggish charge-transfer kinetics and restricted utilization of electroactive sites. Consequently, the experimentally observed capacitance rapidly deteriorates at high current densities and remains well below the theoretical value.11 Therefore, to resolve these issues, various MnO2-based composites including MnO2–NiO, MnO2/ZnO, Mn2O3–SnO2, MnO2/C, Fe2O3/MnO2/rGO, MnO2/Graphene/CNT, CNTs@MnO2@Polypyrrole and carbon nanosheets/MnO2/NiCo2O4 have been examined as electrode materials to mitigate these constraints for SC applications.12–19 These materials have improved the surface area and volume expansion of MnO2-based electrodes. However, the trade-off between electrical conductivity, long-term cycling stability and energy density in the development of practical SCs remains a major bottleneck. These limitations require rational materials engineering to improve the electronic structure and transport pathways of MnO2.
Doping engineering has proven to be an effective strategy for tailoring the electronic properties and lattice structure of MnO2. In particular, Zn2+ doping enhances electrical conductivity and ion-transport kinetics by introducing defect states and oxygen vacancies and inducing lattice distortion.20 However, doping alone often provides limited improvement, as long-range electron transport remains a critical challenge. To mitigate this limitation, constructing heterostructures with conductive frameworks offers a complementary and efficient approach.
Two-dimensional transition-metal dichalcogenides (MoS2) have attracted significant attention as conductive frameworks owing to their layered structure and favorable electrical properties.21 Integrating MnO2 with a 2D MoS2 framework creates abundant heterointerfaces, shortens ion-diffusion pathways and facilitates rapid electron transport.22 Moreover, the synergistic coupling between doped MnO2 and MoS2 is expected to further improve structural stability during electrochemical cycling. For example, Md. Roxy Islam et al. designed MoS2/MnO2 nanocomposites exhibiting 95% retention after 10000 cycles with a capacitance of 199 F g−1 at 0.04 A g−1.23 Jing Ran et al. developed a hollow MnO2@MoS2/RGO structure delivering 743 F g−1 and retaining 88.5% capacity over 5000 cycles.24
In the present work, the Zn–MnO2/MoS2 heterostructure was successfully synthesized using a hydrothermal technique and assessed for SC applications. The fabricated Zn–MnO2/MoS2 electrode exhibited a specific capacitance of 1440 F g−1 at 2.85 A g−1. Moreover, the assembled asymmetric coin cell device provided a significant energy density of 59 Wh kg−1 at a power density of 1145 W kg−1. Additionally, the device retained 91% of its capacity after 14000 cycles, validating remarkable electrochemical stability and significant potential for next-generation energy storage devices.
2. Experimental section
2.1. Chemicals and reagents
Manganese chloride tetrahydrate (MnCl2·4H2O), potassium hydroxide (KOH), zinc acetate dihydrate (Zn(CH3COO)2·2H2O), ethanol and acetone were acquired from Shanghai Aladdin Biochemical Technology Co., Ltd. Potassium permanganate (KMnO4) was procured from Shanghai Macklin Biochemical Co., Ltd. Sodium molybdate dihydrate (Na2MoO4·2H2O), manganese sulfate monohydrate (MnSO4·H2O) and thiourea (CH4N2S) were supplied by Nanjing Wanking Chemical Glassware and Instrument Co., Ltd.
2.2. Synthesis of Zn–MnO2/MoS2
The synthesis of the Zn–MnO2/MoS2 heterostructure was carried out as follows. In the first step, Zn–MnO2 nanorods were synthesized according to a previously reported work via a hydrothermal method.25 Initially, 0.5 M manganese chloride tetrahydrate (MnCl2·4H2O) and 0.025 M zinc acetate dihydrate (Zn(CH3COO)2·2H2O) were dissolved in 50 mL of deionised (DI) water, subsequently undergoing sonication for 30 minutes using a probe sonicator. The prepared solution was placed into a 50 ml stainless steel Teflon-lined autoclave and the reaction was carried out at 140 °C for 12 hours.
Secondly, 0.3 M thiourea (CH4N2S) and 0.2 M sodium molybdate (Na2MoO4) were prepared in DI water and added to the already prepared Zn–MnO2 aqueous solution. The mixture was agitated for 1 hour in order to produce a homogenous solution. The final solution was poured into a stainless-steel autoclave lined with Teflon and hydrothermally treated at 200 °C for 24 hours. After natural cooling to room temperature, the resultant brown precipitate was collected by filtering and thoroughly washed with acetone and ethanol. Finally, the product was dried at 70 °C for 5 hours. For comparison, a similar process was adopted to synthesize the pristine MnO2, MoS2 and MnO2/MoS2 nanostructures without the addition of zinc precursors.
2.3. Microstructural characterization
X-ray diffraction (XRD, Bruker D8 Advance) was utilized to analyse the structural properties of the synthesised materials. High-resolution transmission electron microscopy (HRTEM, JEOL JEM-201, 200 kV), energy-dispersive spectroscopy (EDS), and field emission scanning electron microscopy (FE-SEM, TESCAN MIRA-3) were carried out to evaluate the surface morphology and elemental composition. Raman spectroscopy was performed utilising a Horiba Xplora system with an excitation laser to study the vibrational modes of the synthesized material. X-ray photoelectron spectroscopy (XPS) was employed to assess the surface chemistry and elemental oxidation states of the samples using the Super ESCA beamline of the ELETTRA synchrotron facility in Trieste, Italy. To determine the Brunauer–Emmett–Teller (BET) specific surface area, N2 adsorption–desorption isotherms were analysed. To further clarify the reaction kinetics, density functional theory (DFT) calculations were performed.
2.4. Electrochemical measurements
Electrochemical measurements of the fabricated MnO2, MoS2, Zn–MnO2, MnO2/MoS2 and Zn–MnO2/MoS2 electrodes (mass loading ∼2 mg cm−2) were performed using a CHI660E electrochemical workstation in 1 M KOH aqueous electrolyte. A three-electrode configuration was employed, with the synthesized MnO2-based heterostructure as the working electrode, platinum as the counter electrode and Ag/AgCl as the reference electrode. Cyclic voltammetry (CV) was conducted at scan rates of 10–80 mV s−1, and galvanostatic charge–discharge (GCD) at current densities in the range of 2.85–11.47 A g−1. The electrochemical impedance spectroscopy (EIS) was performed across the frequency range of 0.1 Hz–140 kHz.
The specific capacitance (Cs) was determined from the GCD curves using the following equation:
| | 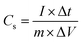 | (1) |
where
m represents the active mass of materials, Δ
t denotes the discharge period,
I signifies the discharge current, and Δ
V indicates the voltage window width including the IR drop.
2.5. Fabrication of an asymmetric supercapacitor (Zn–MnO2/MoS2//AC) device
An asymmetric supercapacitor device (Zn–MnO2/MoS2//AC) was fabricated using Zn–MnO2/MoS2 as the positive electrode, and activated carbon (AC) as the negative electrode. The cell employed a cellulose membrane separator (Whatman GF/D, thickness ∼0.2 mm) electrolyte to provide ionic conductivity while avoiding electrode contact, and an aqueous solution of 1 M KOH. The electrochemical performance of the assembled device was evaluated using a two-electrode configuration. The active mass of material on the positive (m+) and negative (m−) electrodes was calculated based on the charge balance condition (q+ = q−), expressed by the following relationship:| | 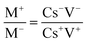 | (2) |
Moreover, the power (P) and energy (E) density of the device were measured using the following equations:| | 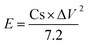 | (3) |
| | 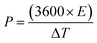 | (4) |
2.6. Computational features
DFT computations were conducted using the plane-wave pseudopotential scheme of the VASP algorithm.26 This research employed the Perdew–Burke–Ernzerhof (PBE) functional to model exchange–correlation interactions inside the generalized gradient approximation (GGA) framework.27 To model the α-MnO2 phase, a primitive tetragonal unit cell was validated by experimental data, using the optimal lattice parameters c = 2.925 Å and a = b = 9.907 Å. One Zn atom was substituted for one Mn atom, resulting in a doping concentration of around 6%. For defect computations, interactions between periodic pictures of defects were minimized using a √2 × √2 × 1 supercell of 48 atoms. The rotationally invariant Hubbard U correction was used to handle electronic correlation effects, yielding effective U values of 3.9 eV for Mn and 4.7 eV for Zn, as adopted from prior computational studies.28–31 A ferromagnetic spin configuration was applied to α-MnO2 in alignment with previous theoretical reports.32 The execution of a 500 eV kinetic energy cutoff resulted in the truncation of the plane-wave basis set. The convergence criteria were established at 0.025 eV Å−1 for ionic relaxation forces and 1 × 10−5 eV for total energy. A 3 × 3 × 12 Monkhorst–Pack k-point mesh was utilized for the primitive cell in order to execute Brillouin zone integrations:| | 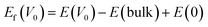 | (5) |
Herein, E(V0) and E(bulk) are the total energies of the defective and pristine supercells, respectively, and E(0) is the chemical potential of a single oxygen atom. The value of E(0) was taken to be −4.52 eV, consistent with high-throughput computational databases.
3. Results and discussion
3.1. Morphological, structural and compositional analysis
The phase composition and crystalline structure of the MnO2, MoS2, MnO2/MoS2 and Zn–MnO2/MoS2 heterostructures were determined using XRD as depicted in Fig. 1(a). The characteristic diffraction peaks identified at 12.71° (110), 18.01° (200), 28.62° (310), 36.49° (400), 37.52° (211), 41.20° (420), 49.63° (411) and 60.13° (521) revealed the tetragonal phase (space group, I4/m) of α-MnO2 as supported by JCPDS No. 00-044-0141.32 The exclusive peaks at 2θ = 14.41°, 32.98°, 38.33° and 58.33°, were precisely correlated with the (002), (100), (103), and (110) diffraction planes of the hexagonal 2H-MoS2 (molybdenite phase, JCPDS No. 03-065-0160), respectively.33 Furthermore, the XRD pattern of the MnO2/MoS2 composite exhibits characteristic diffraction peaks of both α-MnO2 (marked with *) and MoS2 (denoted by ♦), confirming the successful formation of the MnO2/MoS2 heterojunction. Similarly, the Zn–MnO2/MoS2 heterostructure retains the prominent diffraction peaks of α-MnO2 including the (200), (310), (400), (211), and (521) planes, together with the characteristic peaks of MoS2. No additional impurity peaks are detected, verifying the effective synthesis of the Zn–MnO2/MoS2 heterostructure without the formation of secondary phases. The inset of Fig. 1(b) presents a comparison of Zn-doped and pristine MnO2 in the 2θ range of 10–30° corresponding to the (110), (200) and (310) peaks. A careful examination reveals a slight shift of these diffraction peaks toward lower angles upon Zn doping. This peak shift can be attributed to lattice expansion induced by charge imbalance and lattice distortion arising from the incorporation of Zn2+ ions into the MnO2 crystal lattice. Such lattice distortion is indicative of successful Zn doping and may generate local strain and defect sites, which are expected to influence the structural and electrochemical properties of the material.34
 |
| | Fig. 1 (a) Typical XRD spectra of the MnO2, MoS2, MnO2/MoS2 and Zn–MnO2/MoS2 heterostructures. (b) Comparison XRD patterns of the Zn-doped and pristine MnO2 samples. | |
The structural vibration modes of molecules of the Zn–MnO2/MoS2 heterostructure were examined by employing Raman spectroscopy. Fig. 2 presents the Raman spectra of MnO2, Zn–MnO2, MoS2 and Zn–MnO2/MoS2 heterostructures. As observed, the spectra exhibit modes at 382, 405 and 635 cm−1. The distinctive peak at 635 cm−1 correlates to the A1g mode of MnO2, arising from the breathing vibrations of the [MnO6] octahedral.35 The prominent peaks noted at 405 and 382 cm−1 can be attributed to the A1g and E12 g phonon modes of 2H-MoS2, respectively.36 Moreover, the presence of characteristic vibrational modes of both MnO2 and MoS2, verify the successful formation of the heterostructure. Additionally, the Zn doped MnO2 shows a shift towards higher wavenumbers as clearly indicated by the green dotted line, consistent with previously reported studies.37 This shift can be attributed to changes in the local bonding environment and lattice strain caused by Zn incorporation, as well as interactions at the MoS2 interface.
 |
| | Fig. 2 Raman shift of the MnO2, MoS2, Zn–MnO2, and Zn–MnO2/MoS2 heterostructures. | |
Fig. 3(a)–(c) present the N2 adsorption–desorption isotherms and pore size distribution of MnO2, MoS2, and the Zn–MnO2/MoS2 heterostructure measured at 77 K. All samples exhibited type IV adsorption–desorption isotherms with H3-type hysteresis loops, confirming the predominance of mesoporous channels. Notably, the Zn–MnO2/MoS2 heterostructure shows a markedly higher adsorption volume across the entire relative pressure (p/p0) range compared with pristine MnO2 and MoS2. The specific surface area of the heterostructure was calculated to be 215 m2 g−1, substantially exceeding that of MnO2 (85 m2 g−1) and MoS2 (45 m2 g−1). Morever, Fig. 3(b) and (c) present the BJH pore size distribution curves derived from the N2 desorption isotherms. The Zn–MnO2/MoS2 composite exhibits a relatively uniform mesoporous structure, with a narrow pore size distribution centered in a narrow pore size distribution predominantly located within the mesoporous region (≈3–10 nm). In contrast, pristine MnO2 and MoS2 display broader pore size distributions extending towards larger mesopores, indicating a higher degree of structural heterogeneity. The pronounced peak observed in the pore size distribution confirms the formation of well-defined mesopores, which is advantageous for electrochemical energy storage applications. Moreover, the combination of high surface area and accessible mesopores facilitates enhanced electrode–electrolyte contact, thereby improving ion transport kinetics and charge storage efficiency. This enhancement can be ascribed to the synergistic effects of MoS2 integration and Zn doping, which presumably create structural defects and promote the formation of additional accessible mesopores. The mesopores enhance rapid ion transport consequently augmenting the material's power output and rate capability.38 Such an engineered architecture facilitates abundant active sites, thereby fulfilling the structural requirements for high-performance energy storage devices.39–41
 |
| | Fig. 3 (a) N2 absorption–desorption isotherms, and the pore size distribution of (b) the Zn–MnO2/MoS2 heterostructure and (c) MnO2 and MoS2. | |
Fig. 4(a) displays the morphology of the MnO2 which consists of high-quality nanorods with lengths in microns and uniform diameters in the range of nanometers. Fig. 4(b) presents the surface morphology of the MoS2 structure. The microstructure consists of a loosely packed, sheet-like structure composed of thin, layered nanosheets with irregular edges. These nanosheets are interconnected, forming a porous, flower-like architecture with abundant voids and interlayer spacing. Fig. 4(c) displays the morphology of the Zn–MnO2/MoS2 heterostructure. The developed material is composed of irregularly shaped, agglomerated nanosheets with a rough texture, along with distinct, rod-like or needle-shaped nanostructures distributed throughout the matrix. The composite structure clearly shows the successful integration of MoS2 nanosheets with Zn-doped MnO2 nanorods. The presence of 1D Zn-doped MnO2 nanorods is very suitable for electrochemical energy storage due to greater surface accessibility and rapid ion diffusion.
 |
| | Fig. 4 (a)–(c) SEM images of the Zn–MnO2/MoS2 heterostructure network at different magnifications, (d)–(f) TEM images, (g) and (h) HRTEM images, and (i) EDS spectrum of the Zn–MnO2/MoS2 heterostructure. | |
The detailed morphological and structural characteristics were analyzed using TEM. Fig. 4(d) contains a low-magnification TEM image of pristine MnO2 indicating randomly dispersed nanorods. These nanorods exhibit a uniform diameter of approximately 75 nm and extend to micrometre-scale lengths. Fig. 4(e) describes the TEM image of MoS2, revealing well-dispersed and directionally aligned nanosheets. In Fig. 4(f), the Zn–MnO2/MoS2 heterostructure displays MnO2 nanorods anchored onto MoS2 nanosheets, signifying the successful formation of a hybrid heterostructure. HRTEM analysis, as shown in Fig. 4(g), validates the polycrystalline nature of the Zn–MnO2/MoS2 composite. Fig. 4(h), extracted from the rectangular region marked in Fig. 2(g), shows clear lattice fringes with measured d-spacings of ∼0.31 nm and ∼0.27 nm, which correspond to the (110) plane of MnO2 and the (100) plane of layered hexagonal MoS2, respectively. Furthermore, the EDS spectrum in Fig. 4(i) reveals prominent peaks for Mo, Mn, S, O and Zn, thereby confirming the successful formation of the composite. In comparison, Fig. S1(a) and (b) present EDS spectra of pristine MnO2 and MoS2, where the elemental compositions closely match their respective theoretical stoichiometry.
XPS analysis was employed to examine the surface valence states of the Zn–MnO2/MoS2 heterostructures. Fig. 5(a) presents the XPS spectra of the Mn 2p region. The coexistence of Mn3+ and Mn4+ states is evidenced by the observation of two distinct peaks in the Mn 2p3/2 region at 644.1 eV (Mn4+) and 642.6 eV (Mn3+). Similarly, the peaks at 655.5 eV (Mn4+) and 653.9 eV (Mn3+) in the Mn 2p1/2 region further validate the existence of both oxidation states.42,43 The energy separation between Mn 2p3/2 and Mn 2p1/2 is ∼11.86 eV, consistent with the characteristic spin–orbit splitting of Mn in mixed oxidation states.41 It is noteworthy that oxygen vacancies can induce the formation of trivalent Mn3+ indicating the reduction of M4+ to M3+ by accelerating the kinetics of surface redox reactions.44 The Mn 2p XPS spectra were analyzed using quantitative peak deconvolution to evaluate the Mn3+/Mn4+ ratio. The Mn3+/Mn4+ ratio increased from 1.4 (MnO2) to 3.1 (Zn–MnO2/MoS2 heterostructure), indicating an enhanced proportion of Mn3+ species upon Zn doping. This increase suggests a higher concentration of oxygen vacancies in the Zn-doped sample. The observed shift in Mn valence states can be attributed to charge compensation effects associated with Zn incorporation, which modifies the local electronic structure and promotes the formation of Mn3+ species.45Fig. 5(b) presents the spectra of Zn 2p, thereby corroborating the successful incorporation of Zn into the host MnO2. Two separate peaks at 1023.5 eV and 1046.6 eV correlate to the Zn2+ 2p3/2 and Zn2+ 2p1/2 states respectively, with a spin–orbit energy of 23.1 eV.46Fig. 5(c) and (d) display the O 1s spectra emphasizing two prominent peaks at 529.4 eV and 530.7 eV, representing lattice oxygen (Olatt) and surface oxygen vacancies (Ov) respectively.47 Significantly, the enhanced oxygen vacancies in the heterostructure are evidenced by the increase in intensity ratio (OV/Olatt) ∼ 0.56 compared with that of pristine MnO2 (0.44). As shown in Fig. 3(e), the Mo 3d spectrum was deconvoluted into two discrete peaks located at 228.2 eV and 231.6 eV corresponding to Mo4+ 3d5/2 and 3d3/2 respectively. Fig. 5(f) illustrates the S 2p spectra, which exhibit discrete peaks at 161.1 eV and 162.3 eV associated with S2− 2p3/2 and S2− 2p1/2 of MoS2, respectively. All of these XPS findings support the effective synthesis of the Zn–MnO2/MoS2 heterostructure and are in good agreement with the literature.48–50
 |
| | Fig. 5 XPS analysis of Zn–MnO2/MoS2: high resolution spectra of (a) Mn 2p, (b) Zn 2p, (c) and (d) O 1s, (e) Mo 3d, and (f) S−2. | |
3.2. Electrochemical measurements
3.2.1. Cyclic voltammetry.
Fig. 6(a) reveals the comparative CV profiles of the MnO2, MoS2, Zn–MnO2, MnO2/MoS2 and Zn–MnO2/MoS2 electrodes measured at a scan rate of 10 mV s−1 within a stable potential window of −0.4 to 0.6 V. All electrodes show an approximately rectangular CV curve and demonstrate pseudo-capacitance behaviour. In comparison, the area under the CV curve of the Zn–MnO2/MoS2 electrode is higher than its individual counterpart (Zn–MnO2 and MnO2/MoS2) electrodes. The significantly enhanced enclosed area of the curve is considered due to the increase in active sites and surface area. Fig. 6(b) depicts the CV response of the Zn–MnO2/MoS2 electrode at different sweep rates in the range of ∼10–80 mV s−1. It is found that the enclosed area of the CV loop correspondingly increases with the increase in scan rate, reflecting an increase in capacitive current. The shape of the CV curves remains constant across various scan rates indicating excellent electrochemical reversibility and symmetrical redox kinetics. However, it is found that, at higher scan rates, slight distortions in loop closure are observed, likely due to kinetic limitations associated with multi-electron redox transitions Mn4+/Mn3+ and Mo4+/Mo3+. These processes demand sufficient time for ion migration and interfacial charge redistribution, which becomes rate-limiting under rapid scanning conditions.38 In comparison, the CV responses of MnO2, MoS2, Zn–MnO2 and MnO2/MoS2 electrodes at various sweep rates are presented in Fig. S3(a)–(d). Fig. 6(c) elucidates the relationship between scan rate (v) and peak current (i) obtained from the CV curves shown in Fig. 6(b) offering valuable insights into its underlying charge storage mechanism. The relationship between the scan rate and peak current is described by the power law.Typically, the b-value is found using the log(i) against log(v) plot, in which b is the slope of the linear fit. A b-value close to 0.5 and 1.0 corresponds to diffusion and surface-driven capacitive controlled mechanisms, respectively, thereby highlighting the dominant kinetic process governing the electrode behaviour.51,52 The computed b-values from Fig. 6(c), 0.65 and 0.67, confirm the coexistence of both diffusion controlled and capacitive charge storage mechanisms in the Zn–MnO2/MoS2 electrode. Additionally, based on the CV curves, the reaction kinetics were further investigated using Dunn's equations to distinguish between capacitive and diffusion-controlled contributions.53The equation has been modified to,| | 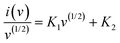 | (8) |
where, i and v are the current and scan rate, respectively, and K1v and K1v(1/2) are the capacitive and diffusion currents, respectively. Fig. 6(d) reveals the charge contribution of the Zn–MnO2/MoS2 electrode at different sweep rates. At a low sweep rate of 10 mV s−1, a significant portion of the charge arises from diffusion-controlled processes, as the ions have enough time to permeate into the bulk of the electrode and participate in faradaic redox reactions, while the capacitive contribution accounts for nearly 75%. With increasing scan rate (20–40 mV s−1), the capacitive contribution progressively dominates, since the reduced diffusion time limits ion insertion into the inner active sites, thereby favouring surface-controlled storage. At higher scan rates (60–80 mV s−1), the capacitive mechanism overwhelmingly prevails, contributing over 90% of the total charge. Even at a high scan rate, the composite's extensive mesopores allow for efficient charge storage by reducing ion diffusion pathways and accelerating electrolyte penetration. A wide interfacial area and continuous electron transport channels are also provided by the 2D conductive MoS2 framework, which greatly enhances the surface-controlled capacitive process. Zn–MnO2 and MoS2 have strong covalent interfacial contact that facilitates rapid redox reactions and lowers charge transfer resistance. This trend demonstrates that the electrode exhibits a rapid charge storage response, with capacitive behaviour dominating at fast sweep rates, underscoring its potential and excellent rate capability for high-power energy storage applications.
 |
| | Fig. 6 (a) Comparative CV response at sweep rate of 10 mV s−1 of all electrodes. (b) CV curves of the Zn–MnO2/MoS2 electrode at different sweep rates. (c) Relationship between logi(A) and logV(mV s−1). (d) Diffusion and capacitive contribution ratios. (e) Comparative analysis of the GCD curve of electrodes at 2.85 A g−1. (f) GCD curves of the Zn–MnO2/MoS2 electrode at different current densities. (g) The specific capacitance variation of all electrodes. (h) Nyquist plot of MnO2, MoS2, Zn–MnO2, MnO2/MoS2 and Zn–MnO2/MoS2 electrodes; the inset shows the kinetic parameters of the electrodes. (i) Cyclic performance of the Zn–MnO2/MoS2 for 14000 cycles at 2.85 A g−1. | |
3.2.2. Galvanostatic charge–discharge (GCD).
Fig. 6(e) represents the comparative GCD profiles of the MnO2, MoS2, Zn–MnO2, MnO2/MoS2 and Zn–MnO2/MoS2 electrodes, measured within a stable potential window of −0.2 to 0.6 V at a current density of 2.85 A g−1. The nearly symmetrical triangular-shaped GCD curves confirm the characteristic capacitive behavior exhibited by all electrodes. Notably, the Zn–MnO2/MoS2 heterostructure electrode exhibits a prolonged discharge time relative to the other three electrodes, suggesting enhanced charge storage capabilities. Fig. 6(f) reveals the GCD curves of the Zn–MnO2/MoS2 electrode at different current densities ranging from 2.85 to 11.43 A g−1. The specific capacitance of the electrodes was determined from the GCD measurements based on eqn (1), The calculated specific capacitance values at 2.85, 5.71, 8.57 and 11.43 A g−1 are 1440, 1184, 980 and 870 F g−1, respectively. The electrode achieved a maximum specific capacitance of 1440 F g−1 at a current density of 2.85 A g−1. At higher current density of 11.43 A g−1 the electrode maintains a specific capacitance of 870 F g−1 with a retention of around 60%, showing outstanding rate capability. For comparison, the GCD curves of the MnO2, MoS2, Zn–MnO2 and MnO2/MoS2 electrodes were also measured at different current densities, as represented in Fig. S4(a)–(d). Furthermore, the specific capacitance values of all five electrodes across the same current density range (2.85 to 11.43 A g−1) are shown in Fig. 6(g). These findings provide clear evidence that the heterostructure electrode offers improved capacitance. The heterostructure electrode achieved much better capacitance than the pristine MnO2, MoS2, Zn–MnO2 and MnO2/MoS2 electrodes, as well as previously reported MnO2 based electrodes, as shown in Table 1.
Table 1 Performance comparison of the designed Zn–MnO2/MoS2 electrode with previously reported MnO2-based electrodes
| Material |
Electrolyte |
Current density (A g−1) |
Specific capacitance (F g−1) |
Cycling stability (%) |
Charge transfer resistance (Rct) |
Cycle number |
Ref. |
| MoS2/MnO2 |
Na2SO4 |
0.04 |
199.12 |
95 |
2.5 |
10000 |
23
|
| MoS2/Mn3O4 |
Na2SO4 |
1 |
172 |
69.3 |
1.41 |
2000 |
62
|
| MoS2/MnO2 |
PVA/H3PO4 |
0.8 |
212 |
84.1 |
700 |
5000 |
63
|
| MnO2@MoS2 |
KOH |
1 |
352 |
72 |
2 |
2000 |
39
|
| POAP/MoS2/MnO2 |
HClO4 |
1 |
529 |
93 |
2.85 |
4000 |
64
|
| ZnMnO2/CC |
Na2SO4 |
1 |
667 |
92 |
105.4 |
8000 |
31
|
| ZnMnO/HPC |
KOH |
0.5 |
400 |
99.5 |
15 |
10000 |
65
|
| MnO2/graphene/PANI |
Na2SO4 |
1 |
870 |
93 |
— |
5000 |
66
|
| ZnS/MnO2 |
KOH |
1 |
1002 |
95 |
0.48 |
5000 |
67
|
| CQDs/ε-MnO2 |
Na2SO4 |
1 |
334 |
90 |
3 |
6000 |
68
|
| MnO2@Co2NiO4 |
KOH |
1 |
1025 |
73 |
0.356 |
5000 |
69
|
| MnO2−xFy |
Mg (NO3)2 |
0.5 |
491.5 |
84 |
0.71 |
10000 |
70
|
| Zn–MnO2/MoS2 |
KOH |
2.85 |
1440 |
95 |
44.07 |
14000 |
This work |
3.2.3. Electrochemical impedance spectroscopy (EIS).
EIS was performed to determine the charge transfer, ion diffusion mechanism and kinetics of electrons within the electrode materials in 1 M KOH electrolyte. Fig. 6(h) presents the Nyquist plots illustrating the imaginary and real components of impedance for the MnO2, MoS2, Zn–MnO2, and Zn–MnO2/MoS2 electrodes. The Nyquist plots exhibit semicircular and linear slopes in the high-frequency region. The semicircle diameter at the electrode–electrolyte interface is directly related to the charge transfer resistance (Rct). The semi-circle of the bare MnO2 electrode is greater than that of MoS2, Zn–MnO2 and Zn–MnO2/MoS2 electrodes. In contrast, the Zn–MnO2/MoS2 heterostructure exhibits the smallest semicircle diameter, demonstrating the lowest Rct among all samples. An AC equivalent circuit model as illustrated in Fig. 4(h) reveals that the measured Rct value for the MnO2, MoS2, Zn–MnO2, MnO2/MoS2 and Zn–MnO2/MoS2 electrodes are 204, 128.9, 78.56, 65.03 and 44.07 Ω, respectively. The fitting parameters of synthesized electrodes are shown in Table 2. The notably low Rct value of the Zn–MnO2/MoS2 electrode indicates the high intrinsic conductivity of MoS2 and the generation of oxygen vacancies induced by Zn reflects excellent electrical conductivity within the electrochemical system. The synergistic interaction between Zn–MnO2 and MoS2 significantly improved the reaction kinetics, resulting in exceptional electrochemical energy storage capability.
Table 2 The fitted parameters of all electrodes
| Material |
R
s (Ω) |
R
ct (Ω) |
| MnO2 |
8.05 |
204 |
| MoS2 |
1.19 |
128.9 |
| Zn–MnO2 |
6.65 |
78.56 |
| MnO2/MoS2 |
5.38 |
65.03 |
| Zn–MnO2/MoS2 |
4.42 |
44.07 |
Fig. S6 presents the Bode phase plots for the MnO2, MoS2, Zn–MnO2, MnO2/MoS2, and Zn–MnO2/MoS2 heterostructures, elucidating the frequency-dependent electrochemical kinetics. A 0° phase angle signifies a perfect resistor, +90° indicates an ideal inductor, and −90° denotes an ideal capacitor. At high frequencies, it is crucial to observe that the phase angle decreased for all electrodes owing to the increasing effects of ohmic resistance and diffusion constraints. That being said, Zn–MnO2/MoS2 retains a relatively greater phase angle, which is indicative of lower internal resistance and more effective electron–ion transport. In the middle frequency range (102–103 Hz), the phase angle attains a maximum that corresponds to the characteristic relaxation frequency related to interfacial charge transfer. The Zn–MnO2/MoS2 electrode exhibits this peak at a higher frequency and a less negative phase angle compared to the other samples, signifying accelerated charge-transfer kinetics and a reduced time constant, attributable to the synergistic interaction between Zn-doped MnO2 and conductive MoS2. At low frequency ranges (10–102 Hz), all electrodes display significant negative phase angles, signifying the prevalence of capacitive and pseudocapacitive processes resulting from surface redox reactions and ion adsorption. The Zn–MnO2/MoS2 electrode exhibited a smaller negative phase angle, indicating a more optimal capacitive response, contributed to better electrolyte accessibility and an increase in electroactive sites within the hybrid structure. This reaction highlights limitations in mass transport, such as the diffusion of ions through the electrolyte or within a porous electrode structure.
3.3. DFT calculations
DFT calculations were employed to support and validate the main experimental results, with an emphasis on the contribution of Zn doping in α-MnO2 toward improving the electrode's electrochemical performance. Fig. 7(a) and (b) present the partial density of states (PDOS) for both α-MnO2 and Zn–MnO2. For MnO2, a noticeable band gap of approximately 1.0 eV is observed, indicative of its semiconducting nature. Upon Zn incorporation, the band gap closes, leading to a metallic ground state. This transition implies a significant enhancement in the electronic conductivity of MnO2 due to Zn doping, predicted to enhance the kinetics of the redox process at the electrode. In addition to the electronic structure, the impact of Zn doping on defect formation was also investigated by calculating the oxygen vacancy formation energy for a nearest-neighbor O atom. The vacancy formation energy for MnO2 is found to be 1.55 eV, whereas for the O atom adjacent to the Zn dopant, the energy decreases significantly to 0.30 eV. This substantial reduction indicates that Zn doping promotes the formation of oxygen vacancies. The close observation shows that oxygen vacancies increase the electrode material specific capacitance by introducing more electrochemically active sites. Furthermore, the formation of a heterostructure between Zn–MnO2 and MoS2 is expected to introduce even more active sites benefiting not only from the high surface-to-volume ratio of MoS2 but also from defect generation during heterostructure formation. Overall, these theoretical insights confirm that Zn doping in MnO2 enhances both conductivity and defect-induced activity, thereby improving redox kinetics and capacitance. To further corroborate experimental findings, we performed Bader charge analysis to estimate the net atomic charges on Mn in MnO2, calculated as the difference between the electronic population of pristine Mn and that in α-MnO2. Overall, a net charge of ∼ +1.80 e is found on the Mn atom in α-MnO2, suggesting a net electron transfer of ∼1.80 e to oxygen atoms, which is in good agreement with previous DFT calculations.54 Interestingly, there is no significant change in Mn Bader charges due to Zn doping. In contrast, a noticeable increase in electronic population is observed due to a single oxygen vacancy such that the number of electrons increases by ∼0.25 e for the Mn atom adjacent to the oxygen vacancy. Since the Zn dopant promotes the oxygen vacancy formation process, this means that the oxidation state of Mn will reduce due to Zn doping, which is qualitatively consistent with the experimental results, where an increase in Mn3+ population was observed due to oxygen vacancies. These findings are in strong agreement with the experimental observations, validating the synergistic role of Zn doping and heterostructure engineering in optimizing electrochemical performance. Finally, we point out that a study of the MnO2/MoS2 heterostructure is currently not feasible owing to the two materials having different symmetries, thus requiring a prohibitively large supercell. We nevertheless discuss the possible beneficial effects of such heterostructures.
 |
| | Fig. 7 Partial density of states of (a) α-MnO2 and (b) Zn doped α-MnO2. The dashed vertical line represents the Fermi level. | |
3.4. Performance of the asymmetric supercapacitor (Zn–MnO2/MoS2//AC) device
To assess the practical applicability of the synthesized heterostructure, an asymmetric coin cell supercapacitor (Zn–MnO2/MoS2//AC) was assembled with activated carbon (AC) as the anode and Zn–MnO2/MoS2 as the cathode utilizing 1 M KOH/PVA as the electrolyte. Fig. 8(a) is a schematic of the fabricated Zn–MnO2/MoS2//AC device. As shown in Fig. 8(b), the CV curves exhibit a quasi-rectangular shape at scan rates of 10 to 60 mV s−1, indicating efficient ion transport and optimal capacitive behavior. As represented in Fig. 8(c), the GCD curves exhibit approximately symmetrical triangular shapes at current densities ranging from 2.85 to 14.29 A g−1, indicating higher columbic efficiency and superior reversibility. The specific capacitances computed from the GCD data are exhibited in Fig. 8(d). The device possesses outstanding rate capability, offering a high specific capacitance of 147 F g−1 at a current density of 2.85 A g−1 and retaining 91 F g−1 even at 14.29 A g−1. Fig. S7(a) illustrates the energy density and power density of the device, as calculated using eqn (3) and (4). Remarkably, with a power density of 1145 W kg−1, the device attained an exceptional energy density of around 59 Wh kg−1 within a potential window of 0.0–1.7 V, surpassing previously reported MnO2-based devices.38,55–61Fig. 8(e) presents a radar plot comparing the energy density, power density, operating potential window and specific capacitance achieved in this work with those reported in previous studies, with detailed comparative data provided in Table S2. Fig. 8(f) shows the Nyquist plot, demonstrating low internal resistance and enhanced charge-transfer kinetics, which signify the device's effective electrochemical performance. As shown in Fig. S7(b) the cycling stability demonstrates 91% capacitance retention over 14000 cycles, confirming the superior long-term electrochemical stability of the Zn–MnO2/MoS2//AC device. Additionally, the self-discharge behaviour of the Zn–MnO2/MoS2//AC device demonstrated an extensive voltage drop during the initial phase, with the voltage diminishing from 2.4 volts to around 1.2 volts within the first 10 hours as shown in Fig. S7(c). Subsequently, the voltage stabilized at approximately 1.0 volts, preserving around 42% of its initial value after 140 hours. Moreover, the average self-discharge rate of the device was approximately 0.42% per hour over 140 hours, however, this rate markedly dropped to around 0.05% per hour during the constant voltage phase. In addition, the areal capacitance of the device, shown in Fig. S9, was calculated from the linear GCD curves (Fig. S8). The device exhibits a high areal capacitance of 205 mF cm−2 at a current density of 0.5 mA cm−2, demonstrating excellent capacitive performance. This value is comparable to those of recently reported devices listed in Table S1. The improved performance can be ascribed to several synergistic effects: (i) the heterostructure introduces structural defects and abundant electroactive interfaces; (ii) the nanorod–nanosheet morphology promotes rapid ion diffusion and mass transport and (iii) Zn doping generates oxygen vacancies, enhancing electrical conductivity and reaction kinetics.
 |
| | Fig. 8 The electrochemical performance of the Zn–MnO2/MoS2//AC device. (a) Structural illustration of the fabricated device. (b) CV response at different scanning rates between 10 and 60 mV s−1. (c) GCD curves at a current density of 2.85 to 11.47 A g−1. (d) Variation of capacitance at different current densities. (e) Radar graphs of the operating potential window, specific capacitance, energy density and power density of different devices. (f) Nyquist plot of Zn–MnO2/MoS2//AC. | |
4. Conclusion
The Zn–MnO2/MoS2 heterostructure was successfully synthesized using a hydrothermal strategy. The combined experimental and DFT analyses confirmed that Zn doping introduces oxygen vacancies and enhances electronic conductivity, leading to superior charge storage capability. Unlike pristine or binary electrodes, the Zn–MnO2/MoS2 heterostructure uniquely integrates the high redox activity of MnO2 with the layered conductivity of MoS2, while Zn incorporation introduces defect sites that accelerate charge transport, thereby achieving an exceptional specific capacitance of 1440 F g−1 at 2.85 A g−1. The asymmetric coin cell (Zn–MnO2/MoS2//AC) further demonstrated a high energy density of 59 Wh kg−1 at 1145 W kg−1 with excellent cycling stability (91% retention after 14000 cycles), underscoring its robustness for long-term operation. The improved performance is a result of the synergistic effect of Zn doping and the heterostructure interface, which is rarely achieved in conventional electrode designs. Looking ahead, scalable synthesis, device integration, and hybridization with other 2D systems could advance the Zn–MnO2/MoS2 heterostructure toward next-generation supercapacitors for portable electronics, electric vehicles and grid storage.
Author contributions
M. S. B. performed the experimental work, and was primarily responsible for data collection, analysis, and drafting of the manuscript. M. A. assisted with methodology development, formal analysis, and the preparation of figures. S. J. performed the DFT study. S. K. provided supervision and technical resources throughout the study. N. A. contributed to experimental investigations and data curation. A. G. contributed to project administration and supervision. A. N. provided guidance, supervision, and critical revisions of the manuscript. M. A. conceived the overall research idea, supervised the project, and contributed significantly to manuscript writing, review, and editing.
Conflicts of interest
No financial conflicts of interest are declared by the authors.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary information (SI). See DOI: https://doi.org/10.1039/d5ma01325a.
Acknowledgements
This work was supported by the Pakistan Atomic Energy Commission (PAEC). The authors are thankful to COMSTECH, Pakistan and SESAME, Jordan, for supporting the experiments performed at the MS/XPD beamline under the SESAME users’ program. Help and support from ICTP in performing the experiments at Elettra – Synchrotron under the ICTP-Elettra users program is also acknowledged.
References
- H. Shen, X. Kong, P. Zhang, X. Song, H. Wang and Y. Zhang, J. Alloys Compd., 2021, 853, 157357 CrossRef CAS.
- N. S. Padalkar, J. A. Shingade and J. P. Park, Mater. Today Phys., 2025, 53, 101711 CrossRef CAS.
- X. Ren, H. Wang, J. Chen, W. Xu, Q. He, H. Wang, F. Zhan, S. Chen and L. Chen, Small, 2023, 19, 2204121 CrossRef CAS PubMed.
- N. Zahra, M. Shahbaz, M. Saleem, M. Z. Khan, M. Irshad, S. Sharif, J. H. Koh, M. A. Marwat, G. Lee and M. Irfan, Mater. Today Sustainability, 2025, 30, 101099 CrossRef.
- N. Guo, R. Ma, P. Feng, D. Wang, B. Zhang, L. Wang, D. Jia and M. Li, Int. J. Biol. Macromol., 2024, 262, 130254 CrossRef CAS PubMed.
- R. Liang, Y. Du, P. Xiao, J. Cheng, S. Yuan, Y. Chen, J. Yuan and J. Chen, Nanomaterials, 2021, 11, 1248 CrossRef CAS PubMed.
-
R. Fatima, S. Naseer, M. R. H. S. Gilani, M. Aamir and J. Akhtar, Sustainable Materials for Electrochemical Capacitors, 2023, pp. 33–64.
- C. M. Han and K. S. Lee, Appl. Chem. Eng., 2025, 36, 253–259 Search PubMed.
- Z. Huang, W. Zhou, D. Li and J. Xu, Chem. Rec., 2025, 25, e202400233 CrossRef CAS PubMed.
- M. Nepal, G. S. Gudavalli and T. P. Dhakal, ACS Omega, 2025, 10, 3439–3448 CrossRef CAS.
- X. Cao, X. Liu, Q. Zou and W. Sun, RSC Adv., 2025, 15, 45480–45499 RSC.
- V. Vinisha, S. Narayanasamy, A. Matharasi, G. Jaffrin and J. M. Linet, Electrochim. Acta, 2025, 518, 145785 CrossRef.
- B. L. Vijayan, S. G. Krishnan, N. K. M. Zain, M. Harilal, A. Yar, I. I. Misnon, J. O. Dennis, M. M. Yusoff and R. Jose, Chem. Eng. J., 2017, 327, 962–972 CrossRef CAS.
- A. Sayah, N. Boumaza, F. Habelhames, A. Bahloul, H. Bencherif, A. Tounsi, L. Lamiri and B. Nessark, J. Mater. Sci.: Mater. Electron., 2024, 35, 62 CrossRef CAS.
- J. Jablonskiene, D. Simkunaite, J. Vaiciuniene, G. Stalnionis, A. Drabavicius, V. Jasulaitiene, V. Pakstas, L. Tamasauskaite-Tamasiunaite and E. Norkus, Crystals, 2021, 11, 784 CrossRef CAS.
- M. Geerthana, S. Prabhu and R. Ramesh, J. Energy Storage, 2022, 47, 103529 CrossRef.
- Y. Liu, D. He, J. Duan, Y. Wang and S. Li, Mater. Chem. Phys., 2014, 147, 141–146 CrossRef CAS.
- A. H. P. de Oliveira, M. L. F. Nascimento and H. P. de Oliveira, Mater. Res., 2016, 19, 1080–1087 CrossRef CAS.
- X. Hong, C. Deng, X. Wang, W. Dong and B. Liang, J. Energy Storage, 2022, 53, 105086 CrossRef.
- S. Li, Q. He, F. Gao, H. Liu, H. Gao, Y. Xue and L. Li, J. Alloys Compd., 2025, 1010, 177895 CrossRef CAS.
- P. Patel, A. Pandey, V. Bonu, K. K. Madapu, O. P. Khatri and H. C. Barshilia, Phys. B, 2025, 705, 417087 CrossRef CAS.
- X. Liao, Y. Zhao, J. Wang, W. Yang, L. Xu, X. Tian, Y. Shuang, K. A. Owusu, M. Yan and L. Mai, Nano Res., 2018, 11, 2083–2092 CrossRef CAS.
- M. R. Islam, M. Rahaman, M. M. Billah and M. R. Islam, Mater. Adv., 2024, 5, 5307–5321 RSC.
- J. Ran, Y. Liu, T. Yang, H. Feng, H. Zhan and H. Shi, J. Energy Storage, 2023, 64, 107216 CrossRef.
- K. Ashokkumar, S. Dhanapandian, S. Suthakaran and N. Krishnakumar, Mater. Today: Proc., 2022, 62, 425–428 CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. Humphreys and A. P. Sutton, Phys. Rev. B, 1998, 57, 1505 CrossRef CAS.
- D. A. Kitchaev, H. Peng, Y. Liu, J. Sun, J. P. Perdew and G. Ceder, Phys. Rev. B, 2016, 93, 045132 CrossRef.
- H. V. Thang and G. Pacchioni, J. Phys. Chem. C, 2018, 122, 20880–20887 CrossRef CAS.
- S. Abbas, T. H. Bokhari, A. Zafar, S. Javed, S. Karim, H. Sun, S. Hussain, A. Khalid, Y. Yu and R. T. A. Khan, J. Energy Storage, 2024, 87, 111455 CrossRef CAS.
- D. A. Tompsett, S. C. Parker and M. S. Islam, J. Mater. Chem. A, 2014, 2, 15509–15518 RSC.
- A. Yadav, A. K. Sharma, J. Yadav, S. Bhasker, G. Mishra, H. P. Bhasker, S. P. Patel, P. K. Dhawan and D. K. Chaudhary, Z. Naturforsch., A: Phys. Sci., 2025, 80, 345–362 CrossRef CAS.
- D. İskenderoğlu, K. Ç. Demir, H. Güney and M. E. Güldüren, Adv. Eng. Mater., 2025, e202501856 Search PubMed.
- E. D. Blinov, E. V. Kulchakovskaya, N. A. Sokovikov, V. A. Svetlichnyi, S. A. Kulinich and O. V. Vodyankina, Nanomaterials, 2025, 15, 166 CrossRef CAS PubMed.
- P. Yao, X. Gao, F. Xie, G. Lv, H. Yang, R. Snyders, C. Bittencourt and W. Li, J. Alloys Compd., 2025, 1014, 178678 CrossRef CAS.
- I. Elhamdi, H. Souissi, O. Taktak, J. Elghoul, S. Kammoun, E. Dhahri and B. F. Costa, RSC Adv., 2022, 12, 13074–13086 RSC.
- M. M. A. Siddiqui, S. Abbas, F. Faiz, A. Zafar, S. Karim, H. Sun, A. Khalid, Y. Faiz, Y. Yu and H. Sultan, New J. Chem., 2025, 49, 8888–8897 RSC.
- N. Kanaujiya, N. Kumar, A. Srivastava, Y. Sharma and G. D. Varma, J. Electroanal. Chem., 2018, 824, 226–237 CrossRef CAS.
- R. Kumar and R. Thangappan, Energy Fuels, 2024, 38, 11216–11232 CrossRef.
- M. M. H. Raza, A. S. Alzahrani, M. Al-Rasheidi, M. Y. Bhat and F. Khan, J. Alloys Compd., 2025, 182265 CrossRef CAS.
- G. Wang, Y. Wang, B. Guan, J. Liu, Y. Zhang, X. Shi, C. Tang, G. Li, Y. Li and X. Wang, Small, 2021, 17, 2104557 CrossRef CAS PubMed.
- Y. Jiang, D. Ba, Y. Li and J. Liu, Adv. Sci., 2020, 7, 1902795 CrossRef CAS PubMed.
- L. Kang, C. Huang, N. Zhang, J. Zhang, C. Luo, C. Wang, X. Zhou and X. Wu, J. Alloys Compd., 2019, 809, 151790 CrossRef CAS.
- G. A. Ribeiro, S. L. de Lima, K. E. Santos, J. P. Mendonça, P. Macena, E. C. Pessanha, T. C. Cordeiro, J. Gardener, G. Solórzano and J. E. Fonsaca, Discover NANO, 2023, 18, 147 CrossRef CAS PubMed.
- J. Wu, W. Raza, P. Wang, A. Hussain, Y. Ding, J. Yu, Y. Wu and J. Zhao, Electrochim. Acta, 2022, 418, 140339 CrossRef CAS.
- Z.-H. Huang, Y. Song, D.-Y. Feng, Z. Sun, X. Sun and X.-X. Liu, ACS Nano, 2018, 12, 3557–3567 CrossRef CAS PubMed.
- N. P. Kondekar, M. G. Boebinger, E. V. Woods and M. T. McDowell, ACS Appl. Mater. Interfaces, 2017, 9, 32394–32404 CrossRef CAS PubMed.
- H. Dong, C. Gong, R. Addou, S. McDonnell, A. Azcatl, X. Qin, W. Wang, W. Wang, C. L. Hinkle and R. M. Wallace, ACS Appl. Mater. Interfaces, 2017, 9, 38977–38983 CrossRef CAS PubMed.
- M. Baker, R. Gilmore, C. Lenardi and W. Gissler, Appl. Surf. Sci., 1999, 150, 255–262 CrossRef CAS.
- P. Matheswaran, P. Karuppiah and P. Thangavelu, Ionics, 2021, 27, 1769–1780 CrossRef CAS.
- D. R. Patil, B. Koteswararao, K. Begari, A. Yogi, M. Moussa and D. P. Dubal, ACS Appl. Energy Mater., 2019, 2, 2972–2981 CrossRef CAS.
- Q. Li, J. Zhou, R. Liu and L. Han, Dalton Trans., 2019, 48, 17163–17168 RSC.
- M. Rittiruam, P. Buapin, T. Saelee, P. Khajondetchairit, S. Kheawhom, B. Alling, S. Praserthdam, A. Ektarawong and P. Praserthdam, J. Alloys Compd., 2022, 926, 166929 CrossRef CAS.
- Z. Pan, L. Jin, C. Yang, X. Ji and M. Liu, Chem. Eng. J., 2023, 470, 144084 CrossRef CAS.
- A. Zhang, N. Mao, Y. Zhong, W. Zheng, L. Cui, C. Barrow, J. Razal, W. Yang and J. Liu, Composites, Part B, 2021, 215, 108756 CrossRef CAS.
- X. Cheng, L. Zhang, L. Li, H. Wu, J. Zheng, J. Yao and G. Li, Colloids Surf., A, 2025, 709, 136163 CrossRef CAS.
- Y. Zhang, K. Li, X. Wang, S. Xiong, F. Chen and Y. Li, Sci. Rep., 2026, 16, 301 CrossRef CAS PubMed.
- M. Yu, R. Liu, J. Liu, S. Li and Y. Ma, Small, 2017, 13, 1702616 CrossRef PubMed.
- A. Philip and A. R. Kumar, J. Alloys Compd., 2025, 1010, 177249 CrossRef CAS.
- W. Tao, H. Quan, Z. Tu, Z. Zhang and D. Chen, J. Colloid Interface Sci., 2025, 683, 1–13 CrossRef CAS PubMed.
- M. Wang, H. Fei, P. Zhang and L. Yin, Electrochim. Acta, 2016, 209, 389–398 CrossRef CAS.
- D. Sahoo, J. Shakya, S. Choudhury, S. S. Roy, L. Devi, B. Singh, S. Ghosh and B. Kaviraj, ACS Omega, 2022, 7, 16895–16905 CrossRef CAS PubMed.
- H. Heydari, M. Abdouss, S. Mazinani, J. S. Shayeh and A. M. Bazargan, J. Energy Storage, 2022, 48, 103905 CrossRef.
- Y. Luo, J. Li, C. Chen and W. Liu, Sci. Rep., 2025, 15, 6393 CrossRef CAS PubMed.
- A. Xu, S. Zhang, L. Yin, W. Cao, Z. Zhao and Y. Qin, Electrochim. Acta, 2025, 524, 146048 CrossRef CAS.
- M. Abdullah, P. John, K. Jabbour, M. I. Ahmad, S. Khan, M. S. Waheed, M. D. Albaqami, M. Sheikh, M. F. Ehsan and M. N. Ashiq, J. Energy Storage, 2024, 78, 110034 CrossRef CAS.
- H. Quan, W. Zeng, W. Chen, Y. Wang, W. Tao and D. Chen, J. Alloys Compd., 2023, 938, 168524 CrossRef CAS.
- S. Ren, S. Zhang, X. Zhang, Y. Jiang, C. Xu, Z. Lei, M. Wei, G. Zhu, Y. Zhao and H. Xu, J. Energy Storage, 2025, 128, 117207 CrossRef CAS.
- X. Zhang, Y. Wei, X. Yang, C. Hou, Y. Chen and D. Wang, J. Energy Storage, 2025, 122, 116677 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence h,
Abdul
Ghaffar
*b,
Amjad
Nisar
h,
Abdul
Ghaffar
*b,
Amjad
Nisar













