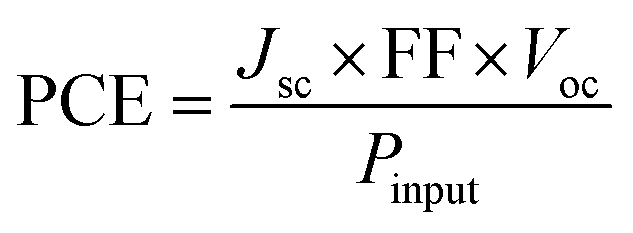DOI:
10.1039/D5MA00925A
(Paper)
Mater. Adv., 2026,
7, 351-365
Computational assessment of the photovoltaic potential in efficient donor–acceptor non-fullerene molecules
Received
18th August 2025
, Accepted 2nd November 2025
First published on 4th November 2025
Abstract
For the advancement of organic solar cell (OSC) technology, it is essential to develop photovoltaic (PV) materials with enhanced conduction band properties, optimized electrochemical behavior, and higher power conversion efficiency (PCE). Quantum chemical modeling is a key component of the design of high-performance organic photovoltaics (OPVs) and optoelectronics. The aim of this work is to determine the most promising materials for OSCs by modeling and characterizing several unique non-fullerene (NF) donor–acceptor complexes (AMF1–AMF6) formed from the R [Ph(T-IDIC)2] phenylene core with two T-IDIC arm molecules. The MPW1PW91/6-31G(d,p) basis set is used in this research to assess the electrical properties, charge transport features, and molecular structures of these compounds using density functional theory (DFT) and time-dependent DFT (TD-DFT). Important analyses include studies of charge transfer, open-circuit voltage (Voc), density of states (DOS), transition density matrix (TDM), molecular electrostatic potential (MEP), and natural bond orbital (NBO). Compared to the reference molecule AMF (R), the results demonstrate a better λmax value of 689.22 nm in the solvent phase (DCM), a lowered band gap (Eg) of 1.015 eV, and a binding energy (Eb) of −0.251 eV. Additionally, in comparison to the reference molecule, the developed molecules exhibit a high open-circuit voltage (Voc) of 1.262 V and a fill factor (FF) of 90.20%. Furthermore, due to the higher exciton dissociation rate, AMF6 exhibits the highest potential for solar energy applications among the studied systems. The results from TDM and MEP analyses show that the tailored AMF6-based non-fullerene molecule outperforms traditional fullerene-based systems, paving the way for more efficient and environmentally friendly organic electronic devices. The results provide insightful guidance for improving the stability, performance, and design of opto-electronics and solar energy conversion devices.
1. Introduction
Because of their tunable optoelectronic features, mechanical flexibility, light weight, and solution processability, organic solar cells (OSCs) have become a viable next-generation photovoltaic technology.1–3 Through logical molecular design and donor–acceptor (D–A) interface optimization, non-fullerene acceptors (NFAs) have recently revolutionized OSC efficiency, achieving power conversion efficiencies (PCEs) above 19%.4–6 Because of their planar conjugated backbones, which support strong π–π stacking, high charge mobility, and improved light absorption, indacenodithiophene (IDT) and indacenodithienothiophene (IDTT)-based structures have garnered a lot of interest among NFAs.7,8
Exciton dissociation and charge carrier mobility are improved when T-IDIC arms are added to the Ph(T-IDIC)2 molecule because they stabilize the LUMO energy level and improve the donor–acceptor interface. Furthermore, the planar π-extended system facilitates efficient molecule packing and intermolecular interactions, which are essential for enhancing the active layer shape and charge transport properties in OSC devices.9–11
Before synthesis, novel NFAs' electrical, optical, and photovoltaic characteristics can be reliably predicted and correlated using computational modeling utilizing density functional theory (DFT).12,13 Ph(T-IDIC)2's electronic delocalization, border orbital alignment, open-circuit voltage (Voc), and reorganization energy can all be thoroughly understood through theoretical research, which also offers important information about the materials’ suitability for photovoltaic applications. In this study, we concentrate on a recently developed molecule called Ph(T-IDIC)2, which is made up of two T-IDIC arms (4,4,9,9-tetramethyl-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene-IDIC units) connected symmetrically to a para-phenylene (Ph) linker. To fine-tune the border molecular orbitals and optical bandgap, this structural configuration combines an extended π-conjugated backbone with terminal 2-(3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (IC) moieties, which function as potent electron-withdrawing groups. In order to facilitate intramolecular charge transfer (ICT) and preserve balanced charge transport paths between donor and acceptor segments, the phenylene core provides exceptional planarity and stiffness. All things considered, this work offers a thorough theoretical investigation of Ph(T-IDIC)2 as a conjugated, planar, non-fullerene acceptor, highlighting its structure–property correlations and promise as a high-performance part of organic solar cells.
A similar class based on infrastructure of atomic clusters known as carboranes are made up of boron and carbon atoms that have their hydrogen atoms replaced. These atoms are bound together by delocalized electrons in a three-center, two-electron pattern known as “3-D aromaticity”.14,15 It has also been shown on multiple occasions that emissive carborane species are the photoactive element in organic light-emitting diodes.16–21 Numerous conjugated polymers, whose emissive qualities depend on the choice of carborane isomer and substituent site, as well as a certain tuneability based on the choice of conjugated co-monomers, are other examples of emissive species containing carborane.14,22,23 A variety of strategies have been investigated for incorporating carborane into polymeric species. These strategies include utilizing carborane in the backbone of the polymer, fusing it into conjugated monomers, and connecting it to side chains that solubilize the polymer.14,24–28 In order to investigate further the possible uses of carborane-containing conjugated materials, Anies et al. previously demonstrated that they could be employed as the semiconductor layer in organic field-effect transistors: polymers containing fused benzocarane and benzocaranodithiophene monomers, as well as polythiophenes with pendant carboranes, demonstrated p-type behavior, whereas P(NDI2OD-T2)-inspired polymers containing carborane in the backbone displayed n-type behavior.26,27,29,30 A phthalocyanine unit connected to a carborane moiety has also been used to create devices similar to this one.31
Additionally, the polythiophene and NDI based polymers mentioned represent the sole instances, to our current understanding, where carborane-containing compounds have been incorporated into organic photovoltaic (OPV) devices. Carborane-containing polythiophenes have demonstrated a peak PCE of 2.0% when paired with PCBM as the acceptor material, while carborane-containing NDI polymers achieved 1.8% PCE using PM6 as the donor material.26,30 Other noteworthy materials utilized in photovoltaic applications include o-carborane derivatives serving as electron and hole transport layers in perovskite solar cells, bis(dicarbollide) species acting as redox shuttles in dye-sensitized solar cells (DSSCs), and o-carborane linked phthalocyanines serving as photosensitizers in DSSCs.32–34 Anies et al. (2023) delved deeper into the design approaches for incorporating carborane-containing materials into organic photovoltaics (OPVs). Introducing a set of four carborane-based conjugated small molecules, each featuring one of the three icosahedral isomers alongside a fluorinated variant, Anies et al., introduced the inaugural instances of carborane-containing non-fullerene acceptors (NFAs). Analysis of the device data validates this approach as promising for carborane-containing OPV materials, notably with the fluorinated variant demonstrating PCE values surpassing 5% – a significant improvement, more than doubling the previous record for such materials. A comparative assessment of the various NFAs underscores the substantial influence of the carborane isomer selection on device performance, with o-carborane emerging as the optimal performer.14
In this research, we utilize the molecule [Ph(T-IDIC)2] as a reference (R)14 as Anies et al. disclosed all its properties and PCE from all effective perspectives. In response to this reference molecule (R), we designed six novel molecules (AMF1–AMF6) by substituting the end-capped groups for newly designed acceptor molecules. All the developed compounds (AMF1–AMF6) and the R molecule have geometrical analyses like reorganization energy, DOS, and Voc theoretically computed to examine the molecule's functioning potential. By altering the end-capped groups on the new compounds, we suggested that they are completely more effective than the R molecule at producing better PCE. Through these analyses, we can determine that different types of NFAs need effective side-chain groups in order to display outstanding morphology and a more effective device. Electron acceptors with various chemical configurations would thereby improve the PV and optoelectronic characteristics of the solar cells. Six novel Ph(T-IDIC)2-based non-fullerene acceptors (AMF1–AMF6) were designed by modifying the end groups. Theoretical analyses show improved charge transport and photovoltaic properties, demonstrating that end-group engineering effectively enhances device performance.
2. Computational methodology
All of the geometrical values were computed using the Gaussian 0935 program. Novel molecules were optimized using the Gauss view program. The optimal values of the reference molecule have been determined using the functionals B3LYP,36 MPW1PW91,37 CAM-B3LYP,38,39 and ωB97XD40 in conjunction with the basic set 6-31G(d,p).36 We also calculated UV absorption values for the reference molecule for each functional and compared them in the form of a graph to determine which function has a value that is near to the experimental absorption value and has been utilized for additional calculations. The UV values in the graph showed a level where significant absorption has been taken into account out of all the levels. The conductor-like polarizable continuum model (CPCM) with dichloromethane (DCM) as the solvent has been employed to account for solvation effects. Each molecule's value has been tuned at the appropriate DFT functional. The optical, band gap energy, natural bond order (NBO) analysis, fill factor (FF), FMO analysis, DOS, Voc, TDM, and MEP investigation, as well as the analysis of binding energy (Eb) of reference R and newly developed molecules have been examined using the MPW1PW91/6-31G(d,p) level of DFT.
3. Results and discussion
3.1. Selection of the DFT functional and molecular design
The para-substituted carbazole-based compound incorporating indacenodithienothiophene acceptor units, [p-Cb(T-IDIC)2], was synthesized experimentally. To minimize computational cost, it was modified to [Ph(T-IDIC)2], which served as the reference (R) molecule in this study.14 Six novel compounds (AMF1–AMF6) have been created by modifying the molecule's end-capped groups. We have modified these six molecules to enhance the end-capped R molecule's photophysical and optoelectronic properties. Firstly, we estimated the λmax in the gas phase for the R (AMF) molecule with four different functionals that include B3LYP, CAM–B3LYP, MPW1PW91, and B97XD using the 6-31G(d,p) basic set implemented in DFT. 785.21, 518.69, 712.48, and 496.80 nm were the results of λmax values that were attained theoretically in the gas phase. The literature states that the experimental λmax value for R is 640 nm. The R molecule optimization at different DFT functionals has been displayed in Fig. 1. Among these, the experimental value of R and the λmax values derived from MPW1PW91/6-31G(d,p) are rather close to it. Because of this, we have chosen the MPW1PW91/6-31G(d,p) level of DFT to characterize this planned series (AMF1–AMF6) in more detail. Furthermore, the IDIC units in the reference system are conjugated, which require a functional that can accurately describe the electronic delocalization. The MPW1PW91 functional performs well for conjugated systems.22 So, the MPW1PW91 functional is computationally more efficient than the pure post-Hartree–Fock methods,41,42 making it suitable for larger systems like p-Cb(T-IDIC) and Ph(T-IDIC). The R (AMF) molecule and the newly proposed structures of AMF1–AMF6 have been displayed in Fig. 2.
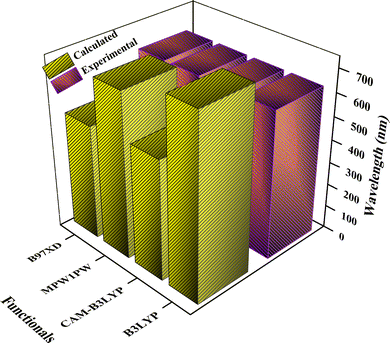 |
| | Fig. 1 UV-visible bar chart of the R molecule calculated using four functionals at the 6-31G(d,p) DFT level. | |
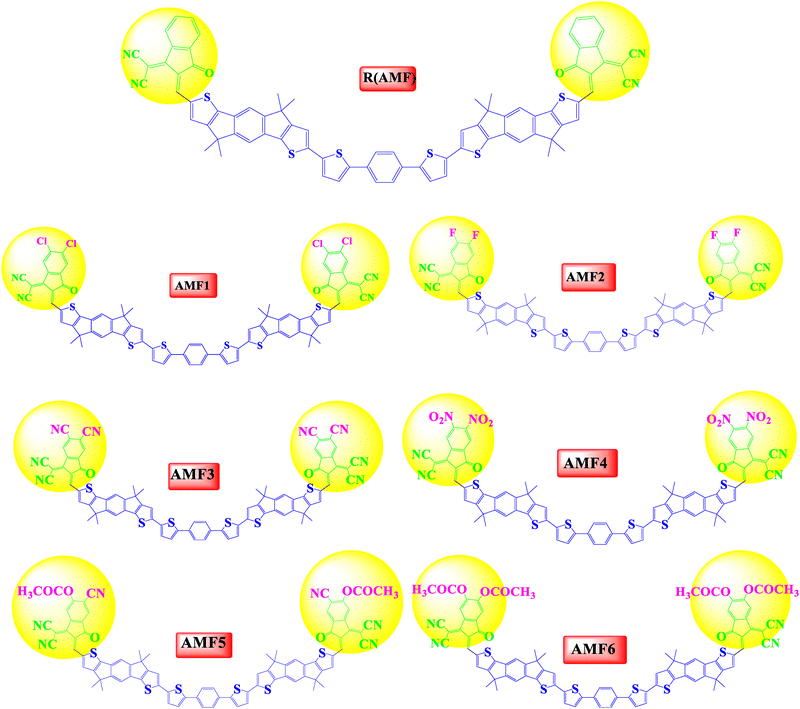 |
| | Fig. 2 Optimized chemical structures of AMF1–AMF6 and the reference molecule R. | |
The current study describes how end-capped moieties were used to modify the end-capping of a reference molecule like 2-(2-methylene-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (R), 2-(5,6-dichloro-2-methylene-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (AMF1), 2-(5,6-difluoro-2-methylene-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (AMF2), 1-(dicyanomethylene)-2-methylene-3-oxo-2,3-dihydro-1H-indene-5,6-dicarbonitrile (AMF3), 2-(2-methylene-5,6-dinitro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (AMF4), 6-cyano-1-(dicyanomethylene)-2-methylene-3-oxo-2,3-dihydro-1H-inden-5-yl acetate compound with methane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (AMF5), and 1-(dicyanomethylene)-2-methylene-3-oxo-2,3-dihydro-1H-indene-5,6-diyl diacetate (AMF6), In this way, we have designed six new molecules (AMF1–AMF6).
:1) (AMF5), and 1-(dicyanomethylene)-2-methylene-3-oxo-2,3-dihydro-1H-indene-5,6-diyl diacetate (AMF6), In this way, we have designed six new molecules (AMF1–AMF6).
3.2. Frontier molecular orbital (FMO) analysis
Each molecule's chemical structure (R and AMF1–AMF6) has been examined in this study as shown in Fig. 2. For R and customized molecules (AMF1–AMF6), we have employed the DFT level of theory by using MPW1PW91/6-31G(d,p) and the optimized structure has been given in Fig. 3. Frontier molecular orbitals (FMOs), which have an impact on the charge transfer shown in Fig. 3, display the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). The energy difference between the molecule's LUMO and HOMO, which function as valence and conduction bands, respectively, indicates a bandgap.43,44 A molecule's efficiency is indicated by its bandgap; a lower bandgap indicates higher performance, and vice versa as shown in Fig. 4. Additionally, Table 1 displays the outcomes of the FMO of R and the customized molecules (AMF1–AMF6). The theoretical values of the reference molecule's EHOMO, ELUMO and Eg, (−7.670 eV, −6.209 eV, and 1.461 eV) have been shown in this table. The customized molecules (AMF1–AMF6) have the following EHOMO values: −7.539, −7.530, −7.543, −7.542 to 7.408, and −7.255, in that order. Individually, these molecules have the following ELUMO values: −6.174, −6.175, −6.2925, −6.237, −6.234, and −6.240 eV. The customized molecules' bandgaps have been measured and found to be in that order (1.364, 1.355, 1.251, 1.304, 1.173, and 1.015 eV). These results led to the conclusion that all the modified molecules have smaller bandgaps than the reference molecule (R).
 |
| | Fig. 3 HOMO–LUMO distribution behavior of R and the designed molecules (AMF1–AMF6). | |
 |
| | Fig. 4 HOMO–LUMO energies of R and AMF1–AMF6 calculated at the MPW1PW91/6-31G(d,p) level. | |
Table 1 The EHOMO, ELUMO, and band gap energy (Eg) values of the reference and designed molecules
| Molecules |
E
HOMO (eV) |
E
LUMO (eV) |
E
g (eV) |
| R |
−7.670 |
−6.209 |
1.461 |
|
AMF1
|
−7.539 |
−6.174 |
1.364 |
|
AMF2
|
−7.530 |
−6.175 |
1.355 |
|
AMF3
|
−7.543 |
−6.292 |
1.251 |
|
AMF4
|
−7.542 |
−6.237 |
1.304 |
|
AMF5
|
−7.408 |
−6.234 |
1.173 |
|
AMF6
|
−7.255 |
−6.240 |
1.015 |
The 1.46 eV difference seen in the R molecule indicates that the acceptor's endcap is essential for enhancing the band gap reduction from R. The suggested molecule AMF1 has an Eg value of 1.364 eV because its end-capped units contain the electron-withdrawing group Cl. The 1.355 eV band gap in the proposed molecule AMF2 is demonstrated by the effect of F groups at the acceptor's end. The electronic gap of the molecule AMF3 shows a sufficient and significant difference of 1.251 eV since the acceptor's end-capped molecules have CN groups on both arms. The molecule AMF6's smallest electronic gap (Eg), 1.015 eV, showed that COOCH3 linked to the terminal ends of both arms of the acceptor molecule has a larger band gap-closing capacity than the R molecule and all other generated molecules. By improving the molecule's ability to remove electrons, these methyl ester functions stabilize the LUMO level and make it easier for the LUMO and HOMO energy levels to separate more clearly. The generated molecules AMF5–AMF6 have methyl ester groups on their end-capped acceptor. In order to maximize the electrical characteristics of NFA in OPVs, the electronic gap must be significantly reduced as a result of the stabilization. Thus, of all the modified molecules, AMF6 exhibits the lowest bandgap (1.015 eV), suggesting that it has effective photovoltaic properties. Because AMF6 has an appropriate end-capped acceptor like –COOCH3, its gap is lower than that of the reference molecule. This is because by lowering the LUMO energy more significantly than the HOMO energy, a narrower gap increases the light absorption range, enabling the absorption of lower-energy photons and enhancing photocurrent generation. The energy-gap (Eg) has been computed using eqn (1).35,45
where
Eg is the molecules' energy gap,
ELUMO is the LUMO energy, and
EHOMO is the HOMO energy in
eqn (1). The HOMO and LUMO energies at the MPW1PW91 functional level with the 6-31G(d,p) basis-set of all molecules are given in
Table 1. All the molecules, including the reference one, have the following descending order of
Eg: R >
AMF1 >
AMF2 >
AMF4 >
AMF3 >
AMF5 >
AMF6. Additionally,
AMF6 has the highest PCE due to its lowest energy gap. Using the same computational method MPW1PW91 and the 6-31G(d,p) basic set of DFT, we have drawn the results of FMO and DOS.
46
3.3. Density of state (DOS) analysis
The DOS analysis for the R molecule and the newly designed AMF1–AMF6 has been estimated, and it gives details on the energy level increments per unit energy level (Fig. 5). At a particular energy level, the DOS mainly shows the amount of energy that is not being used for occupation. This is also used to look at how chemicals are distributed. There is no room for any quantity of energy when the state's density is zero. Three different color types are available in the DOS graphs that are red, green, and black. In this graph, the green lines reflect the EHOMO (donor), whereas the red lines represent the ELUMO (acceptor). In the DOS graphical representation, the distance between the colored lines indicates the energy differential. The functional level MPW1PW91/6-31G(d,p) was utilized to compute the DOS plots for the reference R and acceptor compounds (AMF1–AMF6). According to quantum physics, a molecule's DOS is a system of energy distribution between its energy levels. Examining DOS revealed the function of FMO. Subsequent FMO and electron charge density supply calculations revealed that the charge dispersion of electrons on molecular orbitals (MOs) has been changed by acceptor parts with different degrees of withdrawing groups.47 Crucially, the DOS occupied MOs of all the newly formed compounds (AMF1–AMF6) were found to be comparable, suggesting that occupied MOs are spreading via the acceptor component of the molecule.48 Moreover, the DOS plots show that the HOMOs originate in the donor region of the molecules and exist throughout as the behavior of the electron was analyzed as a result of excitations at different energy levels. The DOS of unoccupied orbitals in novel compounds (AMF1–AMF6) has been compared, and it has been found that they all follow the same pattern. The DOS graphs, which show the energy levels surrounding the donor component, are shown in Fig. 5 where the green line on the graph indicates that the electrons occupy the energy levels surrounded by the donor section, while the red line represents the acceptor's electron density. The amount of energy required to activate the electrons is represented by DOS space between the acceptor and donor.49 In all the newly formed molecules, the smallest amount of energy is needed to raise the electrons between the donor and acceptor sections.
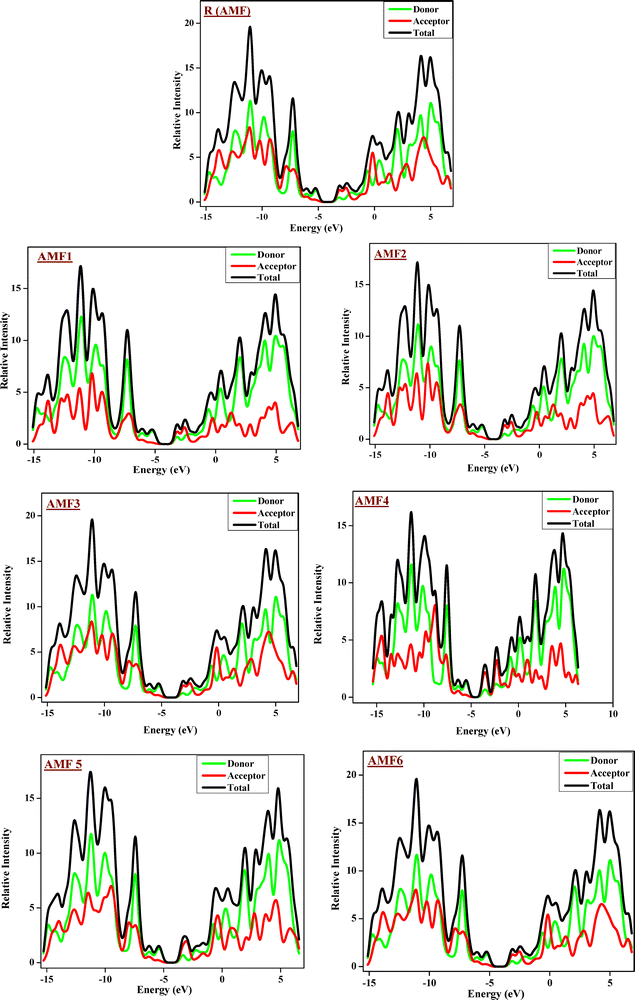 |
| | Fig. 5 DOS plots of R and AMF1–AMF6 molecules computed at the MPW1PW91/6-31G(d,p) DFT level. | |
3.4. Optical properties (UV-visible analysis)
Another important component in estimating the operational efficiency of OSCs is a molecule's optical characteristics.50–52 At the MPW1PW91/6-31G(d,p) level of computation, the optical properties of the reference and new molecules (AMF1–AMF6) in the solvent (DCM) phase were examined. Table 2 presents the results of the measurement and analysis of various optical properties in the solvent (dichloromethane) phase, including oscillator strength, the excitation-energy (Ex) of the newly designed and R molecule, and the experimentally calculated absorption maxima (λmax). The redshift happens in the case of designed molecules with an effective end-capped acceptor group, which increases the light harvesting capability. The conjugation plays a crucial part in the red-shifting (λmax) value. In the solvent (DCM), the λmax values of the reference (R) and tailored molecules (AMF1, AMF2, AMF3, AMF4, AMF5, and AMF6) are 708.31 nm, 714.99 nm, 732.07 nm, 805.92 nm, 796.83 nm, 715.68 nm and 689.22 nm respectively. As compared to other developed and reference molecules, AMF6 displayed a bathochromic shift, with a maximum λmax value moving toward the red, and this is due to the end-capped acceptor molecules.53,54 All the maximum absorption (λmax) values in the solvent (DCM) phase are listed in descending order: AMF3 >AMF4 > AMF2 > AMF5 > AMF1 > R > AMF6. Fig. 6 displays the UV-Vis absorption spectra in a solvent (DCM) phase. The Ex-values in the solvent phase of the reference (R) and tailored molecules (AMF1–AMF6) are as follows: 1.7504 eV, 1.7341 eV, 1.6936 eV, 1.5384 eV, 1.5560 eV, 1.7324 eV and 1.7989 eV respectively. Based on these results, each designed molecule exhibits performance comparable to the reference compound. Furthermore, among the designed molecules AMF6 is a suitable molecule for better solar cell performance due to least energy of excitation.
Table 2 The absorption maxima (λmax), excitation energy (Ex), oscillator strength (fos) and molecular orbital (MO) assignments were analyzed to illustrate the transition nature of the designed (AMF1–AMF6) and reference (R) molecules in the solvent (DCM) phase
| Molecules |
λ
max
(nm) |
λ
max (nm) calculated |
E
x
(eV) |
f
os
|
Major MO assignment |
|
Experimental value was taken from ref. 14.
|
| R |
640 |
708.31 |
1.7504 |
3.5080 |
HOMO → LUMO (87.22%) |
|
AMF1
|
714.99 |
1.7341 |
3.2242 |
HOMO → LUMO (86.97%) |
|
AMF2
|
732.07 |
1.6936 |
3.1417 |
HOMO → LUMO (86.73%) |
|
AMF3
|
805.92 |
1.5384 |
2.3616 |
HOMO → LUMO (85.04%) |
|
AMF4
|
796.83 |
1.5560 |
1.7913 |
HOMO → LUMO (87.53%) |
|
AMF5
|
715.68 |
1.7324 |
2.9979 |
HOMO → LUMO (90.80%) |
|
AMF6
|
689.22 |
1.7989 |
2.9374 |
HOMO → LUMO (93.23%) |
 |
| | Fig. 6 UV absorption spectrum in the solvent (DCM) phase of R and the designed molecules (AMF1–AMF6). | |
3.5. Natural bond order analysis
The D–A molecule's charge carrier affinity has been measured using the natural bond orbital (NBO)55 charge analysis. The computed electronic charges (Q) carried by the molecules are shown in Table 3. Differences in the electrical behavior of the molecules have a major effect on their reactivity.56 Therefore, the quantum mechanical reactivity descriptors,57 which comprise chemical potential (μ), softness (σ), and hardness (η), have been used to analyze the chemical reactivity of the compounds. The results have been displayed in Table 3. Reactivity and η, or resistance to deformation, are inversely correlated.58 A molecule's chemical reactivity decreases as its hardness increases.59,60 Compounds with a significant electron, such as R, AMF1, and AMF2, have η values of 0.730, 0.682, and 0.677 eV, and for other designed molecules (AMF3–AMF6), the values fall between 0.507 to 0.652. In contrast to other compounds, the AMF6 molecule exhibits the lowest chemical potentials, suggesting that it is more chemically reactive than R and other designed molecules. Both positive and negative NBO charge levels can be maintained via the acceptor's end caps. As a result, the molecules' electrical behavior and chemical reactivity are in good agreement. Therefore, Table 3 also displayed the electronic properties derived from NBO analysis for the R molecule and its derivative molecules (AMF1–AMF6).
Table 3 Ionization potential (IP), electron affinity (EA), chemical hardness (η), chemical softness (σ), chemical potential (μ), electronegativity (χ), and electrophilicity index (ω) of the designed (AMF1–AMF6) and reference (R) molecules
| Molecules |
IP |
EA |
Chemical hardness (η) |
Chemical softness (σ) |
Chemical potential (μ) |
Electronegativity χ = −µ |
Electrophilicity index (ω) |
| R |
7.670 |
6.209 |
0.730 |
1.368 |
−6.940 |
6.940 |
1.461 |
|
AMF1
|
7.539 |
6.174 |
0.682 |
1.465 |
−6.856 |
6.856 |
1.364 |
|
AMF2
|
7.530 |
6.175 |
0.677 |
1.475 |
−6.852 |
6.852 |
1.355 |
|
AMF3
|
7.543 |
6.292 |
0.625 |
1.598 |
−6.918 |
6.918 |
1.251 |
|
AMF4
|
7.542 |
6.237 |
0.652 |
1.532 |
−6.889 |
6.889 |
1.304 |
|
AMF5
|
7.408 |
6.234 |
0.586 |
1.704 |
−6.821 |
6.821 |
1.173 |
|
AMF6
|
7.255 |
6.240 |
0.507 |
1.968 |
−6.747 |
6.747 |
1.015 |
The properties, which include electron affinity (EA), ionization potential (IP), and related indices, reveal details about the electrical structure and reactivity of the molecules. Small variations in EA and IP indicate the differences in the electron-donating and electron-withdrawing capabilities of the molecules. Two essential parameters in electronic characteristics that characterize the affinities of charge transfer are IP and EA.61 High EA values have been seen in compounds composed of electron-accepting molecules.62AMF3 and AMF6, for instance, have the greatest EA value of any designed molecule. This is because the AMF3 and AMF6 molecules stabilize the HOMO concentrations and higher negative energies, which facilitates electron transport, in contrast to other electron-deficient molecules. R and the developed molecules have EA values lower than the AMF3 and AMF6 molecules, ranging from 6.174 to 6.240 eV.63 Similarly, electron addition has been made easier in molecules with methyl ester end-capped acceptor molecules due to the reduced computed ionization energy and electron affinity. Recombination rates and mobility are higher in AMF6 due to softness (σ), which affects how well charge carriers can separate and move. The ability of a molecule to receive electrons is measured by its electrophilicity (ω).64 In order to maximize charge transfer and reduce energy losses, electrophilicity can be used to forecast when the energy levels of D–A materials will align. AMF6 has the ability to generate more photocurrent since it can absorb more photons and accept more electrons. Band gap alignment, mobility of charge carriers, and charge transport efficiency all are significantly influenced by electronegativity (χ).65 When combined, these factors help to maximize PV cells' performance, efficiency, and design. Due to AMF6 having the lowest chemical hardness (η = 0.507) and maximum EA, which indicates the enhanced capacity to attract electrons, AMF6 stands out as a highly active molecule among the series. The electrophilicity index (ω) and chemical softness (σ) further emphasize AMF6's unique electrical properties. With its high softness (1.968) and electrophilicity index (ω) value (1.015), AMF6's high electronegativity (χ = 6.747) and very negative chemical potential (μ = −6.747) are compatible. With these characteristics, AMF6 is the most electrophilic and electron-deficient molecule, which makes it ideal for applications in organic electronics that demand high electron-accepting capacity. These descriptors' recurrent patterns show how substituent changes alter a molecule's electrical characteristics and reactivity.
3.6. Open circuit voltage (Voc) analysis
V
oc (open circuit voltage), which is crucial to comprehending how OSCs work, is used to quantify the PCE of these devices. It is the total current extracted from any given device. It is the maximum voltage that can be extracted, particularly from solar devices when they are in the null current state.66 Both (i) photogenerated current and (ii) saturation voltage, which are crucial in solar devices, are the two main current types that influence Voc. The Voc calculation pattern at the MPW1PW91/6-31G(d,p) basis set for R and the AMF1–AMF6 molecules has been displayed in Fig. 7. The Voc reading has been used to gauge OSC performance evaluation. Outstanding new molecules are typically created under the best conditions, which have large Voc values and a minimized HOMO–LUMO (Eg) gap. Here, the value of Voc has been determined using the Scharber equation.67| | | Voc = [EDHOMO] − [EAHOMO] – 0.3 | (2) |
where EDHOMO is the HOMO energy of the donor, EAHOMO is the LUMO energy of the acceptor molecule and 0.3 eV accounts for the empirical energy loss in eqn (2). Table 4 depicts the computed values of Voc.
 |
| | Fig. 7 Values of Voc for AMF1–AMF6 and R molecules with the standard PBDB-T donor. | |
Table 4 Energy of the HOMO (donor), energy of the LUMO (acceptor) and open circuit voltage (Voc) of R and all the proposed molecules
| S. no. |
E
DHOMO (eV) |
E
AHOMO (eV) |
V
oc (V) |
| R |
−5.33 |
−6.209 |
1.179 |
|
AMF01
|
−5.33 |
−6.174 |
1.144 |
|
AMF02
|
−5.33 |
−6.175 |
1.145 |
|
AMF03
|
−5.33 |
−6.292 |
1.262 |
|
AMF04
|
−5.33 |
−6.237 |
1.207 |
|
AMF05
|
−5.33 |
−6.234 |
1.204 |
|
AMF06
|
−5.33 |
−6.240 |
1.210 |
In this work, the donor polymer PBDB-T (LUMO = −3.44 eV, HOMO = −5.33 eV)68 has been used as a standard to satisfy the requirements of this eqn (3). The HOMO of the polymer has been compared to the LUMO levels of the acceptor molecules (R) and (AMF1–AMF6). Eqn (2)'s results have been depicted in Fig. 7. The Voc values of the suggested molecules (AMF1–AMF6) are 1.144, 1.145, 1.262, 1.207, 1.204, and 1.210 V, respectively, while the reference (R) molecule has a Voc value of 1.179 V. Ultimately, with a Voc value of 1.262 V, AMF3 had the highest Voc of all the compounds examined. This is because, in contrast to other compounds under investigation, the AMF3 molecule's extended conjugation and effective end-capped group raise the Voc value.69 Additionally, the typical polymer PBDB-T has a higher HOMO level than the molecules R and AMF1–AMF6 under investigation, which leads to increased conduction power between the donor–acceptor units. Altogether, the designed AMF3 has the highest Voc value. Because of its superior photophysical, electrical, and photovoltaic properties, AMF3 can be considered for solar cell applications.
3.7. Fill factor (FF) and power conversion efficiency (PCE)
The FF is essential for evaluating the performance of OSCs. The efficiency of converting light into unstable current is indicated by the FF measurement. A higher FF results in a higher electric output relative to the theoretical value. Voc, FF, and short-circuit current density (Jsc) in photovoltaics (solar cells) are the three primary factors that a photovoltaic device employs to calculate its output potential. In the current work, the FF has been calculated by using eqn (3).39| | 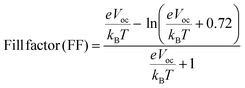 | (3) |
The variables in eqn (3) are the temperature (T), which is equal to 298 K, the elementary charge “e” which is equal to 1, and the Boltzmann constant, kB, for which the experimental value is 8.617 × 10−5 eV per kelvin. For AMF3, the calculated FF is 0.9021. Because its Voc value is higher than those of the R molecule and other created molecules, the AMF3 molecule has a higher FF than other designed and R molecules. Eqn (4) has been used to find the PCE for each of the proposed compounds (AMF1–AMF6).
| | 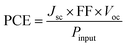 | (4) |
Pinput provides the external light source power. The designed molecules (
AMF1,
AMF2,
AMF4,
AMF5, and
AMF6) and R molecule have a lower PCE than the
AMF3 molecule, according to all theoretical calculations.
Table 5 represents the computed results for
Voc, FF, and FF%.
Table 5 Computed values of Voc, fill factor (FF), and FF % of all the studied molecules
| S. no. |
V
oc (V) |
FF |
FF% |
| R |
1.179 |
0.8967 |
89.67 |
|
AMF1
|
1.144 |
0.8943 |
89.43 |
|
AMF2
|
1.145 |
0.8944 |
89.44 |
|
AMF3
|
1.262 |
0.9021 |
90.20 |
|
AMF4
|
1.207 |
0.8986 |
89.86 |
|
AMF5
|
1.204 |
0.8984 |
89.84 |
|
AMF6
|
1.210 |
0.8988 |
89.88 |
3.8. MEP analysis
Molecular electrostatic potential (MEP) investigation provides an important rationale for the presence of charge separation and dynamic charging site fluctuations inside molecules.70 Since the combination of charging sites and charge positions varies from molecule to molecule, each molecule has a unique set of data that represents each charge site. The molecular efficiency is significantly influenced by its constituent parts.71 MEP analysis has been performed on R and the proposed compounds (AMF1–AMF6). Fig. 8 displays the findings from the MEP study. The molecule's neutral region is represented by the light green portion, its electropositive region by the blue portion, and its negative portions by the red portion. According to the MEP analysis, all the suggested molecules (AMF1–AMF6) have shown the exact location of the charges. Compared to the R molecule, the MEP study indicates that the suggested compounds (AMF1–AMF6) have the potential to improve photophysical qualities.
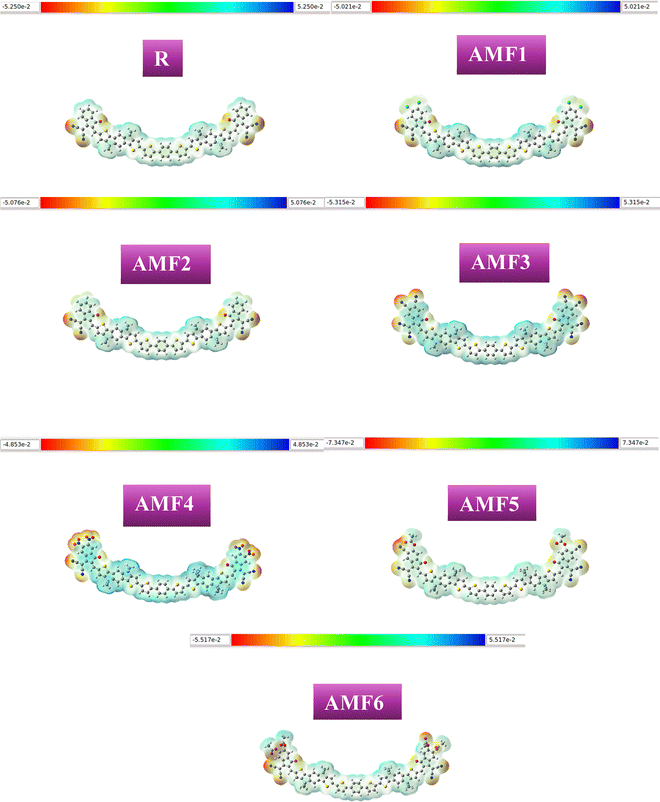 |
| | Fig. 8 MEP plots of R and each of the customized (AMF1–AMF6) molecules. | |
3.9. Transition density matrix (TDM)
The main purpose of the TDM analysis is to measure the degree of transition between NFAs and their original state. TDM analysis has been used to examine and investigate the electron transition process in OSCs. Three-dimensional maps known as TDMs are useful for identifying the donor and acceptor regions of a molecule while it is in an excited state. Together with the R molecule, TDM transitions were estimated and computed for every molecule.72 The examination of the customized molecules (AMF1–AMF6) with the reference one was calculated with Multiwfn software, the MPW1PW91/6-31G(d,p) basis set, and n states = 6. In Fig. 9, the 3D TDM spectrum of AFM6 has been displayed. Other molecules have a similar TDM spectrum of electronic transition from donor to acceptor. The low relevance of hydrogen atoms means that they are not automatically included in TDM displays. All the studied molecules are made up of three parts: acceptor, donor, and acceptor (A–D–A). The TDM is used to estimate electron excitation, the delocalization and localization of electron and hole pairs, and the interaction between the donor and acceptor sections of a molecule. As shown in Fig. 9, the AMF 6 molecule has a high degree of electron coherence on its acceptor side and a low degree on its donor side. Another beneficial factor that is crucial for determining the materials' working potential in a solar cell is binding energy (Eb), which is a noteworthy and well established fact.73 When assessing the electrical and optical characteristics of OSCs as well as their exciting separation potential, the Eb remains a helpful factor.
 |
| | Fig. 9 TDM plot of the tailored AMF6 molecule. | |
3.10. Exciton binding energy (Eb) analysis
A further important factor in determining the operational performance of PV materials is the computation of binding energy (Eb). The current density (Jsc) of the materials used to make devices was found to have an inverse connection with Eb, that is, the greater the Jsc, the smaller the Eb. Furthermore, by facilitating a higher hole–electron overlaying phenomenon to speed up the charge conversion process on the interface region, the lower Eb also has a major impact. As a result, as indicated in Table 6, the calculated Eb and Eopt for the R and the proposed series (AMF1–AMF6) have been presented.
Table 6 Calculated results of Eb of R and the designed compounds (AMF1–AMF6)
| Molecules |
E
H–L (eV) |
E
opt (eV) |
E
b (eV) |
| R (AMF) |
1.461 |
1.750 |
−0.289 |
|
AMF1
|
1.364 |
1.734 |
−0.369 |
|
AMF2
|
1.355 |
1.693 |
−0.338 |
|
AMF3
|
1.251 |
1.538 |
−0.287 |
|
AMF4
|
1.304 |
1.556 |
−0.251 |
|
AMF5
|
1.173 |
1.732 |
−0.559 |
|
AMF6
|
1.0157493 |
1.7989 |
−0.773 |
Specifically, eqn (5) has been employed, where Eb is the exciton binding energy, the energy required to separate an electron–hole pair (exciton) into free charge carriers, which reflects how strongly the electron and hole are bound together after photoexcitation. EH–L is the electronic band gap obtained from ground-state DFT calculations and Eopt is the optical band gap, the energy corresponding to the first electronic excitation. In this way, the Eb for R is −0.289 eV, but the Eb for the newly constructed series (AMF1–AMF6) are, in that order, −0.369, −0.338, −0.287, −0.251, −0.559, and −0.773 eV, respectively, as depicted in Table 6 and Fig. 10. A negative binding energy for solar cell materials means that a stable configuration is produced by an energetically beneficial interaction between components, such as a donor–acceptor pair or molecule-adsorbent combination. This implies that the molecules work well together to create a complex that facilitates charge transfer mechanisms that are essential for the operation of the device. Stronger contact and improved electronic coupling are generally implied by larger magnitude (more negative) binding energy. These factors improve charge transport efficiency and exciton dissociation, two processes that are critical to increasing solar cell performance. An ideal equilibrium is necessary because too strong binding impedes charge separation. So, the Eb value of AMF6 is significantly lower than that of R and other developed materials. This appealing value of Eb, as opposed to R, demonstrated an effective design strategy for creating a robust PV material with the required properties. Consequently, the molecule under study with an Eb value below R also shows more dissociation, which raises the density of charge. Additionally, all the molecules (R and AMF1–AMF6) exhibit reduced Eb, which improves the efficiency of exciton dissociation in the excited state by more successfully illuminating the charge separation. Therefore, all developed compounds can be used as acceptor molecules in solar cell applications because their Eb values are lower and comparable to reference molecule values. By lowering the Eg, changing terminal end caps of acceptors can effectively modify PV properties, according to the present discussion. The obtained computational results provide useful insights for developing high-performance Ph(T-IDIC)2 derivatives analogous to p-Cb(T-IDIC)2, featuring improved optoelectronic characteristics.
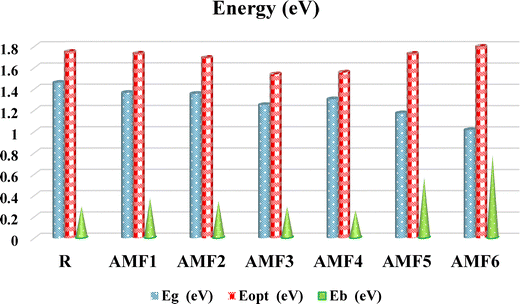 |
| | Fig. 10 Comparison between Eb, Eopt, and Eg of R and the designed molecules (AMF1–AMF6). | |
4. Conclusion
In conclusion, the application of quantum chemical modeling to non-fullerene donor and acceptor systems has played a crucial role in the rational design of high-performance OPVs and advanced optoelectronic materials. Computational methods that can predict energy levels and molecular interactions have made it possible to create compounds with better light absorption and effective charge transport properties. In this work, six non-fullerene small molecule acceptors (NFSMAs) have been designated as AMF1–AMF6 by altering the end cap of the acceptor unit of the Ph(T-IDIC)2 molecule. Their electronic structures, geometries, and photophysical properties have been investigated using density functional theory (DFT) and time-dependent DFT (TD-DFT) methods utilizing the MPW1PW91/6-31G(d,p) basis set. Compared to the R molecule's band gap (Eg = 1.461 eV), AMF1–AMF6 exhibited narrower band gaps (1.015–1.364 eV) with red-shifted absorption spectra that enhance their light-harvesting capabilities. These molecules demonstrated excellent optoelectronic properties and high exciton dissociation efficiency with AMF6 emerging as the most promising candidate due to its low band gap (1.015 eV) and strong electron-withdrawing characteristics. Furthermore, the designed molecule (AMF3) exhibited a high open-circuit voltage (Voc = 1.262 V) and better MEP and TDM properties as compared to the R molecule, highlighting its potential for OSC applications. These results demonstrate the successful incorporation of end-capped electron-accepting units into high-performance OSC designs and offer a valuable approach for the design and optimization of future photoactive materials.
Conflicts of interest
The authors declare that they have no conflict of interest
Data availability
The datasets generated during the current study are available from the authors on reasonable request.
Acknowledgements
The authors are grateful to the Department of Chemistry, Government College University Faisalabad (GCUF), Faisalabad 38000, Pakistan, and acknowledge the support provided by the President’s International Fellowship Initiative (PIFI) Project No. 2024VEA0015 of the Chinese Academy of Sciences (CAS).
References
- D. Luo,
et al., Recent progress in organic solar cells based on non-fullerene acceptors: materials to devices, J. Mater. Chem. A, 2022, 10, 3255–3295 RSC.
- J. Shi,
et al., Designing high performance all-small-molecule solar cells with non-fullerene acceptors: comprehensive studies on photoexcitation dynamics and charge separation kinetics, Energy Environ. Sci., 2018, 11, 211–220 RSC.
- A. Rani,
et al., Molecular engineering of benzothiadiazole core based non-fullerene acceptors to tune the optoelectronic properties of perovskite solar cells, Comput. Theor. Chem., 2024, 1237, 114637 CrossRef CAS.
- C. Gu,
et al., Recent advances in small molecular design for high performance non-fullerene organic solar cells, Mol. Syst. Des. Eng., 2022, 7, 832–855 RSC.
- M. Mainville,
et al., Theoretical Insights into Optoelectronic Properties of Non-Fullerene Acceptors for the Design of Organic Photovoltaics, ACS Appl. Energy Mater., 2021, 4(10), 11090–11100 CrossRef CAS.
- M. U. Saeed,
et al., Impact of end-group modifications and planarity on BDP-based non-fullerene acceptors for high-performance organic solar cells by using DFT approach, J. Mol. Model., 2022, 28(12), 397 CrossRef CAS PubMed.
- M. U. Khan,
et al., Theoretical design and prediction of novel fluorene-based non-fullerene acceptors for environmentally friendly organic solar cell, Arabian J. Chem., 2023, 1(16), 104374 CrossRef.
- Y. Li,
et al., Indacenodithiophene: a promising building block for high performance polymer solar cells, J. Mater. Chem. A, 2017, 5, 10798–10814 RSC.
- U. Q. Zheng,
et al., Rational molecular engineering towards efficient non-fullerene small molecule acceptors for inverted bulk heterojunction organic solar cells, Chem. Commun., 2014, 50, 1591–1594 RSC.
- Z. Xie,
et al., High Mobility Emissive Organic Semiconductors for Optoelectronic Devices, J. Am. Chem. Soc., 2025, 147(3), 2239–2256 CrossRef CAS PubMed.
- S. Y. Kim,
et al., Influence of π-conjugation structural changes on intramolecular charge transfer and photoinduced electron transfer in donor–π–acceptor dyads, Phys. Chem. Chem. Phys., 2017, 19, 426–435 RSC.
- J. L. Bredas, Mind the gap!, Mater. Horiz., 2014, 1, 17–19 RSC.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- F. Aniés,
et al., A comparison of para, meta, and ortho-carborane centred non-fullerene acceptors for organic solar cells, J. Mater. Chem. C, 2023, 11(12), 3989–3996 RSC.
- X. Wu,
et al., Mechanically triggered reversible stepwise tricolor switching and thermochromism of anthracene-o-carborane dyad, Chem. Sci., 2018, 9(23), 5270–5277 RSC.
- L. Böhling,
et al., Substituent Effects on the Fluorescence Properties of ortho-Carboranes: Unusual Emission Behaviour in C-(2′-Pyridyl)-ortho-carboranes, Eur. J. Inorg. Chem., 2016,(3), 403–412 CrossRef.
- K.-R. Wee,
et al., Carborane-based optoelectronically active organic molecules: wide band gap host materials for blue phosphorescence, J. Am. Chem. Soc., 2012, 134(43), 17982–17990 CrossRef CAS PubMed.
- R. Furue,
et al., Aggregation-Induced Delayed Fluorescence Based on Donor/Acceptor-Tethered Janus Carborane Triads: Unique Photophysical Properties of Nondoped OLEDs, Angew. Chem., Int. Ed., 2016, 55(25), 7171–7175 CrossRef CAS.
- A. R. Davis, J. J. Peterson and K. R. Carter, Effect of o-Carborane on the Optoelectronic and Device-Level Properties of Poly (fluorene) s, ACS Macro Lett., 2012, 1(4), 469–472 CrossRef CAS.
- Y. Kim,
et al., Homoleptic tris-cyclometalated iridium complexes with substituted o-carboranes: green phosphorescent emitters for highly efficient solution-processed organic light-emitting diodes, Inorg. Chem., 2016, 55(2), 909–917 CrossRef CAS PubMed.
- S. Kim,
et al., Insights into the effects of substitution position on the photophysics of mono-o-carborane-substituted pyrenes, Inorg. Chem. Front., 2020, 7(16), 2949–2959 RSC.
- M. Tominaga,
et al., Synthesis and characterization of an alternating copolymer with 1, 2-disubstituted and 9, 12-disubstituted o-carborane units, Polym. J., 2014, 46(10), 740–744 CrossRef CAS.
- J. Ochi, K. Tanaka and Y. Chujo, Experimental proof for emission annihilation through bond elongation at the carbon–carbon bond in o-carborane with fused biphenyl-substituted compounds, Dalton Trans., 2021, 50(3), 1025–1033 RSC.
- X. Zhang, L. M. Rendina and M. Müllner, Carborane-Containing Polymers: Synthesis, Properties, and Applications, ACS Polym. Au, 2023, 4(1), 7–33 CrossRef CAS PubMed.
- A. Marfavi, P. Kavianpour and L. M. Rendina, Carboranes in drug discovery, chemical biology and molecular imaging, Nat. Rev. Chem., 2022, 6(7), 486–504 CrossRef.
- J. Marshall,
et al., Polythiophenes with vinylene linked ortho, meta and para-carborane sidechains, Polym. Chem., 2014, 5(21), 6190–6199 RSC.
- J. Marshall,
et al., Incorporation of benzocarborane into conjugated polymer systems: synthesis, characterisation and optoelectronic properties, J. Mater. Chem. C, 2014, 2(2), 232–239 RSC.
- J. J. Peterson,
et al., Carborane-containing poly (fluorene): response to solvent vapors and amines, ACS Appl. Mater. Interfaces, 2011, 3(6), 1796–1799 CrossRef CAS.
- J. Marshall,
et al., Benzocarborano [2, 1-b: 3, 4-b′] dithiophene containing conjugated polymers: Synthesis, characterization, and optoelectronic properties, Macromolecules, 2014, 47(1), 89–96 CrossRef CAS.
- F. Aniés,
et al., N-type polymer semiconductors incorporating para, meta, and ortho-carborane in the conjugated backbone, Polymer, 2022, 240, 124481 CrossRef.
- I. Nar, A. Atsay, A. Altındal and E. Hamuryudan, Inorg. Chem., 2018, 57, 2199–2208 CrossRef CAS.
- S. Şener, Ball-Type Dioxy-o-Carborane Bridged Cobaltphthalocyanine: Synthesis, Characterization and DFT Studies For Dye-Sensitized Solar Cells as Photosensitizer, Heterocycl. Commun., 2020, 26(1), 37–45 CrossRef.
- J. Ordóñez-Hernández, J. G. Planas and R. Núñez, Carborane-based BODIPY dyes: synthesis, structural analysis, photophysics and applications, Front. Chem., 2024, 12, 1485301 CrossRef.
- V. M. Alpatova,
et al., Multicomponent molecular systems based on porphyrins, 1, 3, 5-triazine and carboranes: Synthesis and characterization, Molecules, 2022, 27(19), 6200 CrossRef CAS.
- M. Adnan,
et al., In silico designing of efficient C-shape non-fullerene acceptor molecules having quinoid structure with remarkable photovoltaic properties for high-performance organic solar cells, Optik, 2021, 241, 166839 CrossRef CAS.
- B. Civalleri,
et al., B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals, CrystEngComm, 2008, 10(4), 405–410 RSC.
- Z. Yang,
et al., Designing triphenylamine derivative dyes for highly effective dye-sensitized solar cells with near-infrared light harvesting up to 1100 nm, RSC Adv., 2014, 4(89), 48750–48757 RSC.
- Z.-P. Yu,
et al., Simple non-fused electron acceptors for efficient and stable organic solar cells, Nat. Commun., 2019, 10(1), 2152 CrossRef.
- M. Waqas,
et al., Impact of end-capped modification of MO-IDT based non-fullerene small molecule acceptors to improve the photovoltaic properties of organic solar cells, J. Mol. Graphics Model., 2022, 116, 108255 CrossRef CAS PubMed.
- J. Yuan,
et al., Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core, Joule, 2019, 3(4), 1140–1151 CrossRef CAS.
- A. R. Epstein,
et al., Assessing the Accuracy of Density Functional Approximations for Predicting Hydrolysis Reaction Kinetics, J. Chem. Theory Comput., 2023, 19(11), 3159–3171 CrossRef CAS PubMed.
- M. U. Rahman,
et al., Computational chemistry unveiled: a critical analysis of theoretical coordination chemistry and nanostructured materials, Chem. Prod. Process Model., 2024, 19(4), 473–515 CrossRef CAS.
- M. Adnan, Z. Irshad and J. K. Lee, Facile all-dip-coating deposition of highly efficient (CH 3) 3 NPbI 3− x Cl x perovskite materials from aqueous non-halide lead precursor, RSC Adv., 2020, 10(48), 29010–29017 RSC.
- R. Hussain,
et al., Enhancement in photovoltaic properties of N, N-diethylaniline based donor materials by bridging core modifications for efficient solar cells, ChemistrySelect, 2020, 5(17), 5022–5034 CrossRef CAS.
- M. Adnan,
et al., Banana-shaped nonfullerene acceptor molecules for highly stable and efficient organic solar cells, Energy Fuels, 2021, 35(14), 11496–11506 CrossRef CAS.
- M. U. Khan,
et al., In silico modeling of new “Y-Series”-based near-infrared sensitive non-fullerene acceptors for efficient organic solar cells, ACS Omega, 2020, 5(37), 24125–24137 CrossRef CAS.
- M. U. Khan,
et al., Designing spirobifullerene core based three-dimensional cross shape acceptor materials with promising photovoltaic properties for high-efficiency organic solar cells, Int. J. Quantum Chem., 2020, 120(22), e26377 CrossRef CAS.
- M. Adnan and J. K. Lee, Highly efficient planar
heterojunction perovskite solar cells with sequentially dip-coated deposited perovskite layers from a non-halide aqueous lead precursor, RSC Adv., 2020, 10(9), 5454–5461 RSC.
- R. Hussain,
et al., Efficient designing of triphenylamine-based hole transport materials with outstanding photovoltaic characteristics for organic solar cells, J. Mater. Sci., 2021, 56, 5113–5131 CrossRef CAS.
- E. U. Rashid,
et al., Engineering of W-shaped benzodithiophenedione-based small molecular acceptors with improved optoelectronic properties for high efficiency organic solar cells, RSC Adv., 2022, 12(34), 21801–21820 RSC.
- J. C. Garcia-Alvarez and S. Gozem, Absorption Intensities of Organic Molecules from Electronic Structure Calculations versus Experiments: the Effect of Solvation, Method, Basis Set, and Transition Moment Gauge, J. Chem. Theory Comput., 2024, 20(16), 7227–7243 CAS.
- M. Yahya, Insights into structural, solvent effect, molecular properties and NLO behavior of hemithioindigo-photoisomerization: A DFT study, J. Mol. Liq., 2021, 342, 116944 CrossRef CAS.
- Z. Irshad, M. Adnan and J. K. Lee, Simple preparation of highly efficient MA x FA1− x PbI3 perovskite films from an aqueous halide-free lead precursor by all dip-coating approach and application in high-performance perovskite solar cells, J. Mater. Sci., 2022, 57(3), 1936–1946 CrossRef CAS.
- Z. Irshad, M. Adnan and J. K. Lee, Efficient planar heterojunction inverted perovskite solar cells with perovskite materials deposited using an aqueous non-halide lead precursor, Bull. Korean Chem. Soc., 2020, 41(9), 937–942 CrossRef CAS.
- F. Weinhold, C. Landis and E. Glendening, What is NBO analysis and how is it useful?, Int. Rev. Phys. Chem., 2016, 35(3), 399–440 Search PubMed.
- L. Lauberte,
et al., Lignin modification supported by DFT-based theoretical study as a way to produce competitive natural antioxidants, Molecules, 2019, 24(9), 1794 Search PubMed.
- C. H. Suresh, G. S. Remya and P. K. Anjalikrishna, Molecular electrostatic potential analysis: A powerful tool to interpret and predict chemical reactivity, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12(5), e1601 Search PubMed.
- D. Labonte, A.-K. Lenz and M. L. Oyen, On the relationship between indentation hardness and modulus, and the damage resistance of biological materials, Acta Biomater., 2017, 57, 373–383 CrossRef CAS.
- H. Sajid,
et al., Modification of the N-methyl-4, 5-diazacarbazole D-π-A with a π-conjugated bridge to enhance excited-state properties for superior utilization in green energy applications: A theoretical approach, Colloids Surf., A, 2024, 134508 CrossRef CAS.
- A. A. Sangolkar,
et al., Electronic and photophysical properties of an atomically thin bowl-shaped beryllene encapsulated inside the cavity of [6] cycloparaphenylene (Be n@[6] CPP), Phys. Chem. Chem. Phys., 2023, 25(34), 23262–23276 RSC.
- M. Umar,
et al., A first-principles study on the multilayer graphene nanosheets anode performance for boron-ion battery, Nanomaterials, 2022, 12(8), 1280 CrossRef CAS PubMed.
- C. Song,
et al., Extension of π-conjugation and enhancement of electron-withdrawing ability at terminal indenedione for A-π-D-π-A small molecules for application in organic solar cells, Org. Electron., 2020, 81, 105679 CrossRef CAS.
- S. Ahmed, R. Dutta and D. J. Kalita, Strategical designing of diketopyrrolopyrrole-thiophene based donor-acceptor type organic oligomers and study their transport properties: A DFT/TD-DFT perspective, Chem. Phys. Lett., 2019, 730, 14–25 CrossRef CAS.
- S. Sarı and N. Karakuş, Effect of electron-donating and-withdrawing substitutions in naphthoquinone sensitizers: The structure engineering of dyes for DSSCs in Quantum Chemical Study, Chem. Phys., 2024, 587, 112439 CrossRef.
- P. Jaiswal,
et al., Electrical and optical properties of CNTs additives chalcogenide glasses, Micro Nanosyst., 2021, 13(2), 146–157 CrossRef CAS.
- M. Y. Mehboob,
et al., Quantum chemical design of near-infrared sensitive fused ring electron acceptors containing selenophene as π-bridge for high-performance organic solar cells, J. Phys. Org. Chem., 2021, 34(8), e4204 CrossRef CAS.
- M. Y. Mehboob,
et al., Designing of benzodithiophene core-based small molecular acceptors for efficient non-fullerene organic solar cells, Spectrochim. Acta, Part A, 2021, 244, 118873 CrossRef CAS.
- M. Muzammil,
et al., Boosting the performance of PBDB-T/ITIC based organic solar cell: a theoretical analysis utilizing SCAPS-1D, Chem. Phys. Imp., 2024, 8, 100407 CrossRef.
- F. Zafar,
et al., End-capped engineering of truxene core based acceptor materials for high performance organic solar cells: theoretical understanding and prediction, Opt. Quantum Electron., 2021, 53, 1–24 CrossRef.
- A. Farooq,
et al., Predicting high performance optoelectronic attributes containing iso-indacenodithiophene-based photovoltaic materials for future solar cell technology, Comput. Theor. Chem., 2024, 1234, 114549 CrossRef CAS.
- G. Marchetti,
et al., Syk inhibitors: New computational insights into their intraerythrocytic action in plasmodium falciparum malaria, Int. J. Mol. Sci., 2020, 21(19), 7009 CrossRef CAS.
- S. Riaz,
et al., Ab initio study of two-dimensional cross-shaped non-fullerene acceptors for efficient organic solar cells, ACS Omega, 2022, 7(12), 10638–10648 CrossRef CAS.
- M. Y. Mehboob,
et al., Impact of π-linker modifications on the photovoltaic performance of rainbow-shaped acceptor molecules for high performance organic solar cell applications, Phys. B, 2022, 625, 413465 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab