DOI:
10.1039/D5MA00779H
(Paper)
Mater. Adv., 2026,
7, 4646-4658
Revealing the anodic performance of Janus Mxenes (ABN, A = Sc; B = Zr, Nb, Mo) for magnesium-ion batteries: a first-principles study
Received
20th July 2025
, Accepted 30th March 2026
First published on 16th April 2026
Abstract
Rechargeable magnesium-ion batteries (MIBs) have attracted attention as alternatives to lithium-ion batteries for next-generation energy storage systems due to their low cost and the abundance of magnesium. However, the absence of suitable electrode components for long-term use has prevented the exploration of MIBs. In this work, three computationally predicted Janus MXenes, namely ScZrN, ScNbN, and ScMoN, are investigated as anode materials using density functional theory (DFT). Various parameters, including adsorption energy, charge density difference (CDD), band structure, density of states (DOS), and charge transfer, were analyzed. All the nanosheets exhibit metallic behavior, confirmed by the band structures from HSE06 hybrid functional calculations. These MXenes demonstrate good structural stability, evidenced by negative cohesive energy and Gibbs free energy values. In the adsorption energy calculations, Mg atoms are adsorbed on the three nanosheets with favorable adsorption energies of −1.66 eV to −2.44 eV. Low diffusion barriers of 0.49, 0.26, and 0.37 eV are observed for the ScZrN, ScNbN, and ScMoN, respectively. Moderately high specific capacities of 555.2, 666.4, and 384.5 mAh g−1 are calculated for ScNbN, ScZrN, and ScMoN, respectively. Therefore, these materials can be considered as potential anode candidates for MIBs, although they require further experimental validation.
1. Introduction
At present, fossil fuels are the most widely used sources of energy. In the future, environmental pollution and fuel shortages will be our main challenges. Using fossil fuels for energy generation has led to unprecedented environmental harm and climate change. Scientists are trying to reduce the dependence on fossil fuels by developing efficient energy conversion techniques. Rechargeable batteries are important in an energy-based society because they are cost-effective and effective energy storage devices that permit the transfer of energy from green sources into the power system while minimizing demand for fossil fuels. LIBs are one of the most significant energy storage technologies that power electric vehicles and exhibit significant popularity in portable electronics. However, issues with lithium supply, high production costs, and safety issues make using LIBs for massive energy storage challenging.1 There appears to be an increasing demand for alternatives to lithium. Rechargeable Na-ion, K-ion, and MIBs have received much interest because of their possibility of outstanding efficiency in energy storage applications. The ions of sodium (1.02 Å) and potassium (1.38 Å) are larger than lithium ions (0.76 Å), making quicker movement possible.2,3 The use of rechargeable MIBs has been introduced as an appealing candidate for power storage since it is more abundant in the Earth's crust (Mg abundance = 104 × Li abundance),4 less expensive (24 times less than lithium),5 more environmentally benign, and has a higher melting point compared with lithium. The basic properties of Mg (narrow ion radius: 0.72 Å, divalent nature, higher electron density donor ability, favorable reduction potential, and mostly no dendrite growth) render it an ideal material.6 Mg metal volumetrically (3833 mAh cm−3) has a larger capacity than Li (2046 mAh cm−3), and this allows the construction of much higher energy batteries.7 Two-dimensional (2D) materials have generated much focus regarding their special qualities and potential applications. The synthetic process of graphene nanosheets has established a foundation for the ensuing identification of further 2D materials, like several new 2D materials, including phosphorene (865 mAh g−1),8 borophene (1240 mAh g−1),9 defective graphene (762 mAh g−1),10 transition metal dichalcogenides (389 mAh g−1),11 transition metal carbides (263 mAh g−1)12etc. Graphene, which has a specific capacity of 372 mAh g−1, is mostly used as an anode material in rechargeable batteries.13 MXenes are carbides, nitrides, or carbonitrides of early transition metals. Transition metal nitrides (TMNs) are commonly derived from MAX phases, where M represents a transition metal, A is an A-group element, and X is nitrogen.14 These materials have attracted considerable attention due to their high electrical conductivity, excellent chemical stability, mechanical robustness, and high melting temperatures. These qualities make them ideal for electrocatalysis, supercapacitors, and rechargeable batteries.15 TMNs have been considered for application as high-performance electrodes for MIBs because of their excellent characteristics.
Several research studies have explored the synthesis and characterization of various substances for utilization as ion storage battery electrodes. In 2008, Zhang et al. conducted a study on conductive paper with added MXene for lithium applications in batteries. According to their research, MXene's special features might reduce the effect of shuttling in room-temperature sodium sulfur (RT-Na–S) cells.16 Hongli et al. observed that the V2N MXene monolayer is both metallic and stable. It enables Li, Na, and Mg ions to move smoothly (barriers <0.06 eV). It is capable of holding 925 mAh g−1 of lithium, 463 mAh g−1 of sodium, and 1850 mAh g−1 of magnesium. This makes it excellent for batteries, especially for Na and Mg ions, which store more charge than Li.17 Zhang et al. observed that the MoN2 monolayer is highly effective as a rechargeable anode. It can hold 432 mAh g−1 as a cathode for lithium-ion batteries and 864 mAh g−1 as an anode for NIBs. MoN2 and other TMNs are ideal for large-capacity batteries.18 Wu et al. studied the TiNbS4 TMD with two dimensions as an anode material through DFT. They observed that TiNbS4 had a low average open-circuit voltage (0.63 V) and a high theoretical capacity (990 mAh g−1), which exceeds that of graphite (372 mAh g−1).19 The metallic properties, low diffusion barriers, and significant specific capacities (1034, 561, and 707 mAh g−1, respectively) of Zr2N, Hf2N, and ZrHfN MXenes make them promising anode materials for calcium-ion batteries, as reported by Tanvir et al. They are ideal for ion storage due to these qualities.20 Ling et al. used DFT calculations to study pure and boron-doped graphene as NIB anode materials. They found that boron-doped graphene, with a sodiation voltage of 0.44 V and a specific capacity of ∼762 mAh g−1, is efficient for Na adsorption.10
The structural and electronic features, absorption behavior, diffusion barriers and electrochemical properties of Janus Mxene, such as ScZrN, ScNbN, and ScMoN, nanosheets, including specific capacity and open-circuit voltage, have all been studied using DFT study with GGA–PBE. The investigated Janus nitride MXenes (ScZrN, ScNbN, and ScMoN) are designed as hypothetical structures motivated by recent experimental and theoretical advances that demonstrate the feasibility of Janus MXenes. Experimentally, Janus transition-metal carbides and nitrides such as Mo2TiC2Tx, V4C3Tx, and Ti2N have been synthesized through selective etching and surface substitution processes, which confirms that chemical asymmetry and multilayer transition-metal configurations can be stabilized.21,22 Theoretically, several studies have also predicted the structural stability and feasible synthesis of hetero-metallic and nitride Janus MXenes, including ScMoN, TiCrCT2, and Sc2C using similar methodologies.23,24
In our case, the stability of ScZrN, ScNbN, and ScMoN monolayers is validated through negative cohesive energies, Gibbs free energy calculations, and vibrational frequency analyses, which demonstrate both thermodynamic and dynamical stability. While these structures have not yet been experimentally realized, their formation is thermodynamically plausible based on the close lattice matching and chemical compatibility of Sc–N and (Zr/Nb/Mo)–N bonds, as observed in experimentally known ternary nitrides and MAX phases. This study has demonstrated these materials’ potential for use as MIB electrodes, encouraging optimism and providing a way for further investigation and development in the field of metal ion batteries.
2. Computational details
The material studio simulation tool was utilized to perform all the calculations, using DFT with the DMol3 method. The Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) was used for electron exchange–correlation due to its combined accuracy and computational efficiency, particularly for investigating the geometry and surface reactivity of periodic systems.25 GGA provides a reliable description of total energy and energy barriers. To further calculate the electronic structure results, HSE06 hybrid functional calculations were performed for the band structure and density of states (DOS), which supports the reliability of the PBE–GGA results. We used the Grimme dispersion correction method (DFT-D2) to accurately calculate the van der Waals interactions. The DFT semi-core pseudopotential was paired with double numerical plus polarization (DNP) basis sets to accommodate for relativistic effects in the core electrons.26 The MIB anode was taken from a 3 × 3 × 1 supercell and contained 27 atoms of 2D TMN nanosheets. We performed convergence tests for the global orbital cut-off, and k-point mesh. 5.6 Å was selected as the global orbital cut-off to improve quality. A 4 × 4 × 1 k-point mesh was used to optimize the geometry. A fixed vacuum of 20 Å was maintained to avoid interactions between adjacent layers.27 This study employs the following mathematical formulas to calculate the cohesive energy, open circuit voltage, and work function. The Janus nanosheet (ScZrN, ScMoN and ScNbN) cohesive energies were evaluated for structural stability by utilizing eqn (1).20 This indicates the energy required to reduce a crystal into each of its neutral atoms.| |  | (1) |
where EABN denotes the total energy of the ternary transition metal nitride, EA and EB represent the energies of elements A (Sc) and B (Zr/Nb/Mo), and EN represents the energy of nitrogen (N) atoms, respectively. The variables a, b, and c indicate the number of A, B, and N atoms within the nanosheet, and n = a + b + c is the total atom count.
For calculating the adsorption energy per unit atom, we employed the formula as follows:28
| | 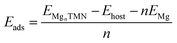 | (2) |
where adsorption energy is denoted by
Eads,
EMgnTMN is the total energy of the TMN nanosheets with n number of Mg atoms adsorbed,
Ehost denotes the energy of the considered MXene,
EMg is the energy of a single Mg of a bulk Mg cluster, and
n represents the number of Mg atoms adsorbed.
The average OCV, which measures the performance of ion storage batteries, is calculated by comparing the total energy before and after a change. N represents the overall atom number within the nanosheets. The charging and discharging operations of TMN as an anode material can be considered as29
| | | Mgx1TMN + (x2 − x1)Mg2+ +2(x2 − x1)e ↔ Mgx2TMN | (3) |
where
x1 and
x2 represent two different magnesium concentrations within the TMN nanosheets during battery operation. The value
x1 corresponds to the Mg content at a certain state of charge, while
x2 represents the Mg content at another state of charge after additional Mg ions have been inserted.
The average OCV in the concentration range x1 < x < x2 is determined using:30
| |  | (4) |
where

and

are the total energies of the TMN having
x1 and
x2 Mg atoms.
The work function variation due to Mg adsorption was computed utilizing the following equation.20
| |  | (5) |
where
φi and
φj represent the work function of the TMN nanosheets before and after Mg adsorption, respectively. Comparing these values allows us to evaluate how Mg adsorption affects the electronic surface properties of the nanosheets.
3. Results and discussion
3.1. Geometric structure
In this study, we have chosen a 3 × 3 × 1 supercell of three Janus ScZrN, ScMoN, and ScNbN nanosheets as an anode material for Mg-ion batteries. Each nanosheet contains a total of 27 atoms, with nine atoms of each element. In each structure, the Sc atoms are sandwiched between a Zr, Mo, or Nb layer and an N layer, resulting in a stable and well-ordered configuration. The optimized ground-state structures and calculated cohesive energies confirm the energetic stability of the nanosheets. These nanosheets are optimized in the ground state and illustrated in Fig. 1. Previously, Pan et al. investigated two-dimensional transition-metal nitride monolayers as an anode material for Li-ion batteries.31 Using eqn (1), we determined the cohesive energies of ScZrN, ScNbN, and ScMoN to be –5.98 eV, –5.95 eV, and –5.44 eV, respectively. Our results are consistent with a previous study by Dong et al., who calculated a cohesive energy of −5.39 eV for monolayer VN2.32 The negative cohesive energy values confirm that all the structures are energetically stable, with more negative values indicating higher structural stability.33 Moreover, positive vibrational modes are found for ScNbN, and ScMoN, except ScZrN, where only one negative vibrational mode is found at −0.9 cm−1. Negative values of Gibbs free energy have been found for all the nanosheets, which are −1.0, −1.15, and −1.1 kcal mol−1 for ScZrN, ScNbN, and ScMoN, respectively, indicating the thermodynamic stability. To better understand the electrical features, we analyzed the band structure and density of states (DOS) of our proposed structures using the HSE06 hybrid functional. The results are shown in Fig. 1. The band structure calculations were performed along the high-symmetry path Γ–M–K–Γ in reciprocal space, with the corresponding coordinates defined as Γ (0, 0, 0) → M (0, 0.5, 0) → K (−0.333, 0.667, 0) → Γ (0, 0, 0). In our investigation, the conduction band crosses the Fermi level, indicating that the nanosheets are metallic, which is consistent with previous findings.34 The DOS spectra of the studied systems (ScZrN, ScNbN, and ScMoN) showed consistent non-zero DOS at the Fermi level, which confirms that each material retains its metallic nature. As a result, these nanosheets represent a promising option for anode materials in rechargeable battery applications.
 |
| | Fig. 1 Optimized structures, band structures, and density of states (DOS) of (a) ScZrN, (b) ScNbN, and (c) ScMoN nanosheets. | |
3.2. Single Mg adsorbed on ScZrN, ScNbN, and ScMoN
Adsorption energy is required to determine the most suitable nanosheet adsorption positions and study how chemicals interact. To investigate the most favorable adsorption site on the nanosheets, we have calculated the adsorption energy by adsorbing a single Mg by choosing different adsorption sites on the nanosheets. In this study, we have chosen six adsorption sites for each nanosheet. For ScZrN, six possible adsorption sites were examined, located on the top and back of the Sc, Zr, and N atoms, as illustrated in Fig. 2. The corresponding adsorption energies, charge transfer values, and Mg–surface distances are summarized in Table 1. Among all the sites, A6 (–2.05 eV) exhibits the most favorable adsorption, as indicated by the highest negative adsorption energy, whereas A1 (–1.66 eV) corresponds to the weakest adsorption. These trends are consistent with the Mg–surface distances listed in Table 1, where stronger adsorption is associated with shorter interaction distances.
 |
| | Fig. 2 Top, back, and side views of a single Mg atom adsorbed on the ScZrN nanosheet at six different adsorption sites (A1–A6). | |
Table 1 The calculated adsorption energy (Eads in eV), charge transfer of the Mg atom (Q in e), and distance from Mg to the nanosheets (d in Å). All charge transfer values indicate electron transfer from Mg to the nanosheet
| Structure |
Atom |
Adsorption position |
Adsorption site |
E
ads (eV) |
Charge transfer |
d (Å) |
| Hirshfeld |
Mulliken |
| ScZrN |
Sc |
Top |
A1 |
−1.66 |
0.228 |
0.429 |
2.662 |
| Back |
A2 |
−2.11 |
0.259 |
0.399 |
3.055 |
| Zr |
Top |
A3 |
−1.78 |
0.249 |
0.331 |
3.044 |
| Back |
A4 |
−1.88 |
0.251 |
0.487 |
2.686 |
| N |
Top |
A5 |
−1.81 |
0.245 |
0.334 |
3.029 |
| Back |
A6 |
−2.05 |
0.262 |
0.397 |
3.073 |
| ScNbN |
Sc |
Top |
B1 |
−2.30 |
0.225 |
0.522 |
2.960 |
| Back |
B2 |
−2.30 |
0.225 |
0.519 |
2.963 |
| Nb |
Top |
B3 |
−1.77 |
0.250 |
0.344 |
3.040 |
| Back |
B4 |
−1.92 |
0.234 |
0.614 |
2.685 |
| N |
Top |
B5 |
−1.82 |
0.245 |
0.346 |
3.020 |
| Back |
B6 |
−2.26 |
0.224 |
0.514 |
2.967 |
| ScMoN |
Sc |
Top |
C1 |
3.77 |
0.139 |
0.077 |
1.701 |
| Back |
C2 |
−2.53 |
0.231 |
0.134 |
2.882 |
| Mo |
Top |
C3 |
−1.70 |
0.254 |
0.341 |
3.005 |
| Back |
C4 |
−2.08 |
0.262 |
0.381 |
2.531 |
| N |
Top |
C5 |
−1.80 |
0.252 |
0.354 |
2.881 |
| Back |
C6 |
−2.59 |
0.217 |
0.149 |
3.001 |
For ScNbN, six adsorption sites were examined, corresponding to the top and back positions of Sc (B1 and B2), Nb (B3 and B4), and N (B5 and B6), as illustrated in Fig. 3. The adsorption energies were calculated to be –2.30, –2.30, –1.77, –1.92, –1.82, and –2.26 eV for Mg adsorption on the B1, B2, B3, B4, B5, and B6 sites, respectively. Consistent with the findings of Bolen et al., who found that the most stable adsorption of Mg2+ on 2-octyl acrylate occurred at energies around −1.90 eV,35 our results confirm that Mg adsorption on ScNbN is energetically favorable. Among all examined sites, B1 and B2 (both –2.30 eV) exhibit the most favorable adsorption, while the weakest adsorption occurs at the B3 site (–1.77 eV). These observations are consistent with the Mg–surface distances, where the more favorable sites (B1 and B2) show shorter separations compared to the weaker B3 site. For ScMoN, six adsorption sites were examined corresponding to the top and back positions of Sc (C1, C2), Mo (C3, C4), and N (C5, C6), as shown in Fig. 4. Consistent with Yang et al., who reported Mg adsorption energies of −0.58 eV and −0.61 eV at the C and V sites on M-VS2, and −2.69 eV at the C site on the B-VS2 nanosheet,36 our results show that Mg adsorption on ScMoN is exothermic at most sites. Among all sites, C6 (–2.59 eV) is the most favorable, whereas C1 exhibits a positive adsorption energy (3.77 eV), indicating a repulsive interaction and making it unsuitable for Mg adsorption.
 |
| | Fig. 3 Top, back, and side views of a single Mg atom adsorbed on the ScNbN nanosheet at six different adsorption sites (B1–B6). | |
 |
| | Fig. 4 Top, back, and side views of a single Mg atom adsorbed on the ScMoN nanosheet at six different adsorption sites (C1–C6). | |
To obtain a deeper understanding of the adsorption characteristics, we have calculated the Mulliken and Hirshfeld charge analysis to illustrate the charge transfer nature between Mg and the nanosheets. From Table 1, we can see that all the charge transfer values are positive, which means the charge is transferred from the Mg atom to the nanosheets. In Mulliken charge analysis, we observe a significant charge transfer of 0.397e (A1), 0.522e (B1), and 0.134e (C1) from the Mg atom to the ScZrN, ScNbN, and ScMoN nanosheets, respectively. Similarly, in Hirshfeld charge analysis, the charge transfers are 0.262e, 0.225e, and 0.231e for the same systems. Zhang et al. found that, on g-C3N4, each Mg ion carries a charge of 0.280 e, which is consistent with our findings.37
Charge density difference (CDD) analysis helps to visualize the spatial distribution of charge transfer upon adsorption. We have also calculated the CDD map using the following formula.38
| | | Δρ = ρMg+sheet − ρsheet − ρMg | (6) |
where
ρMg+sheet is the total charge (or electron) density of the whole system, and
ρsheet and
ρMg represent the electronic charge distributions of nitride MXenes and Mg atoms, respectively. In the CDD map illustration (
Fig. 5), the purple color represents electron accumulation, and electron depletion is represented by red color. The iso-surface value was set to 0.01 e Å
−3. The CDD map shows that when a single Mg is adsorbed on ScZrN, ScNbN, and ScMoN, a significant amount of charge is transferred from Mg to the nanosheets.
 |
| | Fig. 5 Charge density difference (CDD) maps of a single Mg adsorbed on (a) ScZrN, (b) ScNbN, and (c) ScMoN. | |
3.3. Multiple Mg adsorb on ScZrN, ScNbN and ScMoN
To examine the storage mechanism, the number of Mg atoms was gradually increased on the favorable adsorption site of the ScZrN, ScNbN, and ScMoN nanosheets (Fig. 6). Initially, Mg atoms were adsorbed on the favorable sites of ScZrN, ScNbN, and ScMoN, respectively, as these sites exhibited the highest adsorption energies. Then, Mg adatoms were distributed on both sides (a maximum of 10 atoms on each side) of the nanosheets to reduce repulsive interactions. This work analyzes the adsorption characteristics and charge transfer, with the number of adatoms ranging from 1 to 20. During this process, the Mg adatoms exhibited slight displacements from their initial positions. When a maximum of 20 atoms were distributed symmetrically (10 per side) across the nanosheets, they align along different lines due to mutual repulsion. However, no structural deformation is observed. The optimized configurations for the Mg concentrations (4, 8, 15, and 20 atoms) are presented in Fig. 6.
 |
| | Fig. 6 Front and side views of optimized nanosheets adsorbed with different numbers of Mg atoms. Rows 1 and 2: ScZrN, rows 3 and 4: ScNbN, and rows 5 and 6: ScMoN. Panels are labeled as (a) 4 Mg, (b) 8 Mg, (c) 15 Mg, and (d) 20 Mg, indicating the number of adsorbed Mg atoms. | |
In the case of ScZrN, the adsorption energy for a single Mg atom concentration was −2.05 eV. Then, the adsorption energy increased steadily, reaching a maximum of −2.31 eV for 15 Mg and then decreasing to −2.11 eV for 20 Mg atoms. For ScNbN, initially, the adsorption energy was −2.30 eV; when 2 Mg were adsorbed the adsorption energy reduced to −2.02 eV, and then it gradually increased, and for 20 Mg adsorption, it became −2.43 eV. Similarly, during the single atom adsorption on ScNbN the adsorption energy was −2.30 eV and then it decreases to −2.10 eV for 2 Mg adsorption. Then, it increases with slight variation observed for 4 and 6 Mg adsorption, and for 20 Mg adsorption, it became −2.51 eV. Negative adsorption energy indicates an exothermic and energetically favorable adsorption process. With the increasing number of ion concentrations, the adsorption energy becomes more negative, which indicates that the adsorption process is more favorable. This pattern shows that the Mg atom and our suggested Mxenes are interacting more strongly, which improves the capability of these materials for effective ion storage applications. A similar behavior was reported by Saharan et al., who observed an increase in adsorption energy with ion concentration in Zn-ion battery systems.39
From Fig. 7, it is evident that the Hirshfeld and Mulliken charge transfers decrease as adsorbed Mg atoms increase due to the increasing electrostatic repulsion among the positively charged Mg ions. Specifically, for the ScZrN nanosheet, the Hirshfeld charges decreased steadily from 0.262e (1 Mg) to 0.041e (20 Mg), while the Mulliken charges dropped from 0.397e to negative values (reaching −0.021e). Similar trends were observed for ScNbN and ScMoN. In ScNbN, the Hirshfeld charge declined from 0.225e (1 Mg) to 0.019e (20 Mg), while the Mulliken charge decreased from 0.522e to −0.015e for the same Mg adsorption range. For ScMoN, the Hirshfeld charges decreased from 0.231e to 0.016e, while the Mulliken charges decreased from 0.134e to −0.099e, corresponding to 1 Mg to 20 Mg adsorption, respectively. From our Mulliken charge analysis, we observed that the charge transfer decreases as the Mg ion concentration increases. However, the charge transfer becomes negative for up to 15 Mg adatoms (or 20 Mg for ScNbN), indicating cluster formation. For this reason, we calculated a maximum of 20 Mg adsorption on these nanosheets.
 |
| | Fig. 7 Illustration of the variation of charge transfer from the Mg to the nanosheets (a–c) and work function (d) with respect to increasing Mg atom adsorption on the nanosheets. | |
The differences in electrical conductivity before and after Mg ion adsorption were evaluated using the combined total and partial density of states with spin-up and down orientation (Fig. 8). The ScZrN, ScNbN, and ScMoN optimized structures demonstrated metallic properties, exhibited by a non-zero DOS at the Fermi level. As seen in the DOS, additional states are available at the Fermi level after Mg adsorption, which makes our proposed nanosheets even more conducting. According to the increased DOS, Mg atoms intercalate to form Mg cations by providing electrons to the nanosheets. This electron donation supports increased Mg-ion storage capacity, which is becoming essential to improving battery performance and conductivity. This increase implies that more Mg atoms can be stored efficiently, and the enhancement in DOS also supports a higher concentration of magnesium atoms, which boosts battery performance. These results support the possibility of using ScZrN, ScNbN, and ScMoN as efficient MIB electrode materials. The work function represents the energy needed to free an electron from one material into a vacuum.40 However, we utilized work function analysis in this study to confirm electron transport to the nanosheets. The initial work functions of ScZrN, ScNbN, and ScMoN were 5.14 eV, 5.17 eV, and 5.15 eV, respectively. All three nanosheets showed significant reductions upon Mg adsorption. Upon 10 Mg adsorption, the work function significantly decreases to 3.46 eV (about 32.68%) for ScZrN, 3.48 eV (about 32.69%) for ScNbN, and 3.51 eV (around 31.84%) for ScMoN. This sharp drop points to higher surface electron activity, which improves the ion storage efficiency.
 |
| | Fig. 8 Density of states of (a) ScZrN, (b) ScNbN and (c) ScMoN. Fermi levels are at zero energy, which is denoted by the dotted line. | |
3.4. Diffusion barrier
A promising electrode material should have high mobility and a low diffusion barrier. In ion storage batteries, the diffusion barrier impacts the charge and discharge rates, which are affected by how quickly Mg ions move from one location to another throughout the electrode. In the present study, we have investigated the diffusion behavior of Mg ions on the surface of ScZrN, ScNbN, and ScMoN by using the LST/QST method.
According to the Arrhenius equation, metallic atom motion is measured by the diffusion barrier at a constant temperature, following the equation41
| |  | (7) |
where
Eb,
kB and
T are the migration barrier, Boltzmann constant and absolute temperature, respectively.
Fig. 9 illustrates the four paths chosen to calculate the energy barrier for each. In the case of ScZrN, the calculated energy barriers are about 0.31 eV and 0.35 eV when Mg moves along the N → Zr and Zr → N paths, respectively. For the N → N path and its reverse direction, the energy barrier is 0.22 eV, indicating identical values in both directions. When the distance is doubled (N → N), the energy barrier increases to 0.917 eV. Therefore, the average energy barrier for ScZrN is 0.49 eV. Moreover, for ScNbN, the first two migration paths are from the N site to the transition metal sites (Nb and Sc), with energy barriers of 0.09 eV and 0.299 eV, respectively. The reverse paths from Nb to N and Sc to N exhibit energy barriers of 0.03 eV and 0.16 eV, respectively. Compared to the shortest migration path, the N → Nb pathway shows an energy barrier of about 0.521 eV. Therefore, the average diffusion barrier for ScNbN is 0.26 eV.
 |
| | Fig. 9 Mg ion diffusion through the different paths on the (a)–(c) ScZrN, (d)–(f) ScNbN, and (g)–(i) ScMoN nanosheets. | |
On the other hand, in ScMoN, Mg migration along the Mo to Mo path shows a low energy barrier of 0.15 eV, whereas the reverse direction (Mo to Mo) presents a slightly higher barrier of 0.17 eV. Migration along the Mo to N and N to Mo paths involves energy barriers of 0.27 eV and 0.36 eV, respectively. When the migration path distance is doubled along the Mo → Mo direction, the energy barrier increases and the obtained values are 0.618 eV and 0.640 eV. The average diffusion barrier is 0.37 eV. The smallest diffusion barriers were observed on ScNbN and ScMoN, compared to ScZrN. Accordingly, the Mg ion mobility across these three nanosheets follows the order of ScZrN > ScMoN > ScNbN, indicating better diffusion performance with lower energy barriers. Notably, the diffusion barriers for all three materials fall within a range of 0.26–0.49 eV, which is significantly lower than those predicted by Chaney et al., who reported Mg diffusion barriers on MXenes (M2CS2, where M = Mo, Nb, Ti, V, Zr) in the range of 0.4–0.65 eV.42
3.5. Specific capacity and OCV
The specific capacity (SC) and the open circuit voltage (OCV) are the two important factors in examining the overall efficiency of an electrode material. The maximum capacity of the anode material was determined using the following equation43| |  | (8) |
where F is the Faraday constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 801 mAh mol−1), n and z are the valence electrons of Mg ions and the total number of ions in the electrochemical system.44 The maximum specific capacity depends on the highest number of alkali atoms that can be adsorbed onto the anode material during charging, as this determines the amount of electricity released during discharge. To explore this, we progressively increase the concentration of Mg atoms on Nitride MXene nanosheets by introducing varying amounts of Mg (from 2 to 20 atoms), continuing until noticeable structural deformation occurs in the material's geometry. The calculated specific capacities are 555.2, 666.4, and 384.5 mAh g−1 for ScNbN, ScZrN, and ScMoN, respectively. According to Siriwardane et al., the gravimetric capacities of Janus MXenes Ti2CSO and Ti2CSSe as Mg-ion battery anodes were reported to be 524.54 mAh g−1 and 345.22 mAh g−1, respectively, which is consistent with our findings.45 Panigrahi et al. investigated Si2BN nanosheets as anode materials for magnesium-ion batteries and reported a specific capacity of 647.90 mAh g−1.46
801 mAh mol−1), n and z are the valence electrons of Mg ions and the total number of ions in the electrochemical system.44 The maximum specific capacity depends on the highest number of alkali atoms that can be adsorbed onto the anode material during charging, as this determines the amount of electricity released during discharge. To explore this, we progressively increase the concentration of Mg atoms on Nitride MXene nanosheets by introducing varying amounts of Mg (from 2 to 20 atoms), continuing until noticeable structural deformation occurs in the material's geometry. The calculated specific capacities are 555.2, 666.4, and 384.5 mAh g−1 for ScNbN, ScZrN, and ScMoN, respectively. According to Siriwardane et al., the gravimetric capacities of Janus MXenes Ti2CSO and Ti2CSSe as Mg-ion battery anodes were reported to be 524.54 mAh g−1 and 345.22 mAh g−1, respectively, which is consistent with our findings.45 Panigrahi et al. investigated Si2BN nanosheets as anode materials for magnesium-ion batteries and reported a specific capacity of 647.90 mAh g−1.46
OCV is an essential factor for evaluating the efficiency of MIBs and is obtained using eqn (4). It implies the motivation for Mg-ion migration within the anode and cathode during the charging/discharging periods. The calculated OCV values for varying numbers of Mg ions on ScNbN, ScZrN, and ScMoN are presented in Fig. 10. Calculations were performed up to a maximum of 15 Mg ions adsorbed, as beyond this point, the materials tend to form clusters. The OCV for ScZrN steadily improves with the concentration of adsorbed Mg ions. It starts at 1.12 V, shows a slight decrease to 0.97 V upon adsorption of the second Mg ion, and then continuously increases to 1.26 V with the adsorption of up to 10 Mg ions. However, an unexpected drop to 0.76 V is noted at the 15th Mg-ion adsorption. Similarly, for ScNbN, the OCV was measured at several key stages of Mg-ion intercalation. It started at 0.86 V with a single adsorbed Mg ion and increased to 1.25 V upon adding three ions. A slight dip to 1.18 V occurred at the adsorption of the 4th ion, followed by a steady rise up to the 10th Mg ion. The OCV then decreased to 1.19 V on adsorption of the 15th ion. For ScMoN, the computed OCV is 0.83 V for the first Mg ion and rises to 1.40 V for the second. Upon adding the third Mg ion, it drops suddenly to 1.03 V. From the fourth to the eighth ion, the voltage gradually increases, then slowly decreases from 1.34 V to 1.24 V beyond the eighth adsorption. This significant decrease suggests that Mg-ion adsorption beyond this point may cause electrochemical instability or reduce the charge transfer efficiency. However, no structural distortions were observed in the optimized Janus TMN nanosheets; the material remains electrochemically active even at higher Mg-ion concentrations. Our predicted OCV trend strongly agrees with previous studies, which reported similar OCV behavior during Zn-ion adsorption on the m-Ti3C2S2 and b-Ti3C2S2 MXenes.39 The average OCV values were approximately 1.08 V, 1.67 V, and 1.21 V for Mg-ion intercalation on ScZrN, ScNbN, and ScMoN, respectively. Among these, ScNbN exhibited the greatest average OCV, which can be related to the more significant energy transfer between the Mg ions and the host material. Consequently, ScNbN demonstrated a higher Mg storage capacity than ScZrN and ScMoN. Overall, all three materials exhibit average OCVs within a suitable range, highlighting their potential as anode materials. Our predicted average OCV values are generally consistent with those reported by Xu et al., who observed an OCV of approximately 1.45 V for C@PVI-(TPC)Co in Zn-ion batteries.47
 |
| | Fig. 10 OCV curves of (a) ScNbN, (b) ScZrN, and (c) ScMoN, respectively. | |
4. Conclusions
First-principles calculations were performed to evaluate the potential of transition metal nitrides as anode materials for MIBs. In this work, ScZrN, ScNbN, and ScMoN are considered as computationally predicted monolayers. Although the results may depend on the chosen exchange–correlation method, they help to identify possible candidates through a screening approach. The study systematically investigated the structural, electronic, adsorption, and electrochemical properties of TMN nanosheets, focusing on specific charge capacity and open-circuit voltage. The excellent structural stability of ScZrN, ScNbN, and ScMoN is confirmed by their calculated cohesive energies of –5.98 eV, –5.95 eV, and –5.44 eV per atom, respectively. All three nanosheets exhibit metallic behavior, which is favorable for ion transport. The favorable adsorption energies of −2.05 eV (ScZrN), −2.30 eV (ScNbN), and −2.53 eV (ScMoN) were calculated. The ScZrN, ScNbN, and ScMoN nanosheets exhibit relatively low diffusion barriers for Mg atoms, which suggests optimal conditions for ion mobility and efficient battery performance. The diffusion barriers are 0.49, 0.26, and 0.37 eV, and the specific capacities are 555.2, 666.4, and 384.5 mAh g−1 for ScZrN, ScNbN, and ScMoN, respectively. These selected materials have not been experimentally verified yet. However, the outcomes of this investigation demonstrate that each of the materials is a promising candidate at the theoretical level for MIB anodes, which could be explored in future experimental studies and applied in practical systems.
Conflicts of interest
There are no conflicts to declare.
Data availability
All data supporting the findings of this study, including optimized structures, adsorption energy values, charge transfer, work function, etc., are available within the article. Additional data can be provided by the corresponding author upon reasonable request.
Acknowledgements
We gratefully acknowledge the Bangladesh Research and Education Network (BdREN) for providing computational access.
References
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li and N. Tavajohi, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS.
- C. Lin, Y. Wang, F. Zhong, H. Yu, Y. Yan and S. Wu, Chem. Eng. J., 2021, 407, 126991 CrossRef CAS.
- X. He, N. Iqbal, U. Ghani and T. Li, J. Alloys Compd., 2024, 981, 173680 CrossRef CAS.
- F. Bella, S. De Luca, L. Fagiolari, D. Versaci, J. Amici, C. Francia and S. Bodoardo, Nanomaterials, 2021, 11, 810 CrossRef CAS PubMed.
- P. Novák, R. Imhof and O. Haas, Electrochim. Acta, 1999, 45, 351–367 CrossRef.
- L. Song, H. Li, P. Wang, Y. Shang, Y. Yang and Z. Wu, Chin. J. Chem. Eng., 2024, 69, 199–211 CrossRef CAS.
- R. Xu, X. Gao, Y. Chen, C. Peng, Z. Zhang, C. Wang, H. Sun, X. Chen and L. Cui, Mater. Today Phys., 2023, 36, 101186 CrossRef CAS.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 13921–13928 RSC.
- X. Zhang, J. Hu, Y. Cheng, H. Y. Yang, Y. Yao and S. A. Yang, Nanoscale, 2016, 8, 15340–15347 RSC.
- C. Ling and F. Mizuno, Phys. Chem. Chem. Phys., 2014, 16, 10419–10424 RSC.
- E. Yang, H. Ji and Y. Jung, J. Phys. Chem. C, 2015, 119, 26374–26380 CrossRef CAS.
- D. Çakır, C. Sevik, O. Gülseren and F. M. Peeters, J. Mater. Chem. A, 2016, 4, 6029–6035 RSC.
- G. Wang, X. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049–2053 CrossRef CAS.
- X. Chen, Z. Kong, N. Li, X. Zhao and C. Sun, Phys. Chem. Chem. Phys., 2016, 18, 32937–32943 RSC.
- Z. G. Yang, H. M. Xu, T. Y. Shuai, Q. N. Zhan, Z. J. Zhang, K. Huang, C. Dai and G. R. Li, Nanoscale, 2023, 15, 11777–11800 RSC.
- J. Zhang, Z. Xie, W. Li, S. Dong and M. Qu, Carbon, 2014, 74, 153–162 CrossRef CAS.
- H. Liu, Y. Cai, Z. Guo and J. Zhou, ACS Omega, 2022, 7, 17756–17764 CrossRef CAS PubMed.
- X. Zhang, Z. Yu, S.-S. Wang, S. Guan, H. Y. Yang, Y. Yao and S. A. Yang, J. Mater. Chem. A, 2016, 4, 15224–15231 RSC.
- Y. Wu, C. Ren and Q. Wei, Appl. Surf. Sci., 2023, 615, 156322 CrossRef CAS.
- T. Ahmed, A. A. Piya and S. U. Daula Shamim, Nanoscale Adv., 2024, 6, 3441–3449 RSC.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- C. Caddeo, M. I. Saba, S. Meloni, A. Filippetti and A. Mattoni, ACS Nano, 2017, 11, 9183–9190 CrossRef CAS PubMed.
- S. Ram, N. A. Koshi, S. C. Lee and S. Bhattacharjee, J. Phys. Chem. C, 2024, 128, 7323–7333 CrossRef CAS.
- W. Jin, S. Wu and Z. Wang, Physica E, 2018, 103, 307–313 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- K. C. Wasalathilake, G. A. Ayoko and C. Yan, Carbon, 2018, 140, 276–285 CrossRef CAS.
- X. Wang, Y. Cai, S. Wu and B. Li, Appl. Surf. Sci., 2020, 525, 146501 CrossRef CAS.
- X. Wang, Z. Zeng, H. Ahn and G. Wang, Appl. Phys. Lett., 2009, 95, 183103 CrossRef.
- H. R. Jiang, Z. Lu, M. C. Wu, F. Ciucci and T. S. Zhao, Nano Energy, 2016, 23, 97–104 CrossRef CAS.
- C. Ling and F. Mizuno, Chem. Mater., 2012, 24, 3943–3951 CrossRef CAS.
- H. Pan, J. Mater. Chem. A, 2015, 3, 21486–21493 RSC.
- Y. Dong, Z. Tang, P. Liang, H. Wan, H. Wang, L. Wang, H. Shu and D. Chao, J. Colloid Interface Sci., 2021, 593, 51–58 CrossRef CAS PubMed.
- Y. Ding, Y. Wang, J. Ni, L. Shi, S. Shi and W. Tang, Phys. B, 2011, 406, 2254–2260 CrossRef CAS.
- T. Jing, D. Liang, J. Hao, M. Deng and S. Cai, Phys. Chem. Chem. Phys., 2019, 21, 5394–5401 RSC.
- E. Bolen, J. S. Dolado and A. Ayuela, Polymers, 2025, 17, 799 CrossRef CAS PubMed.
- J. Yang, J. Wang, X. Dong, L. Zhu, D. Hou, W. Zeng and J. Wang, Appl. Surf. Sci., 2021, 544, 148775 CrossRef CAS.
- J. Zhang, G. Liu, H. Hu, L. Wu, Q. Wang, X. Xin, S. Li and P. Lu, Appl. Surf. Sci., 2019, 487, 1026–1032 CrossRef CAS.
- Z.-K. Tang, Y.-N. Zhang, D.-Y. Zhang, W.-M. Lau and L.-M. Liu, Sci. Rep., 2014, 4, 7007 CrossRef CAS PubMed.
- S. Saharan, U. Ghanekar and S. Meena, Nanoscale, 2025, 17, 7898–7907 RSC.
- L. Lin, R. Jacobs, T. Ma, D. Chen, J. Booske, D. Morgan, L. Lin, R. Jacobs, T. Ma, D. Chen, J. Booske and D. Morgan, PhRvP, 2023, 19, 37001 CAS.
- S. U. D. Shamim, M. K. Hossain, S. M. Hasan, A. A. Piya, M. S. Rahman, M. A. Hossain and F. Ahmed, Appl. Surf. Sci., 2022, 579, 152147 CrossRef CAS.
- G. Chaney, D. Çakır, F. M. Peeters and C. Ataca, Phys. Chem. Chem. Phys., 2021, 23, 25424–25433 RSC.
- L. Xiong, J. Hu, S. Yu, M. Wu, B. Xu and C. Ouyang, Phys. Chem. Chem. Phys., 2019, 21, 7053–7060 RSC.
- B. Tian, T. Huang, J. Guo, H. Shu, Y. Wang and J. Dai, Mater. Sci. Semicond. Process., 2020, 109, 104946 CrossRef CAS.
- E. M. D. Siriwardane and J. Hu, J. Phys. Chem. C, 2021, 125, 12469–12477 CrossRef CAS.
- P. Panigrahi, S. B. Mishra, T. Hussain, B. R. K. Nanda and R. Ahuja, ACS Appl. Nano Mater., 2020, 3, 9055–9063 CrossRef CAS.
- C. Xu, P. Guo, K. Yang, C. Lu, P. Wei and J. Liu, ChemCatChem, 2023, 15, e202300404 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Obaidullah
,
Obaidullah



 and
and  are the total energies of the TMN having x1 and x2 Mg atoms.
are the total energies of the TMN having x1 and x2 Mg atoms.












