DOI:
10.1039/D6LP00040A
(Paper)
RSC Appl. Polym., 2026, Advance Article
An attapulgite–polysulfide composite for highly selective and scalable recovery of precious metals from gold ore residues and wastewater
Received
2nd February 2026
, Accepted 9th April 2026
First published on 13th April 2026
Abstract
Precious metals play a crucial role in a multitude of sectors, owing to their superior physicochemical attributes, yet their limited natural abundance leads to supply shortages. Consequently, it is imperative to devise an affordable adsorbent capable of effectively extracting gold from dilute gold ore tailings and industrial effluents. This study delineates an economical approach for fabricating a composite sorbent, integrating attapulgite with a sulfur-rich polymer. The polymer (named Poly(S-AM) or IV polymer) was synthesized through inverse vulcanization using elemental sulfur (S8) and acrylamide (AM), both of which are cost-effective and readily accessible. This Poly(S-AM) was then merged with acid-modified attapulgite (HAPT) via a mechanochemical process, giving the Poly(S-AM)/HAPT composite. With Au(III) serving as a representative ion, the composite achieved a peak adsorption capacity of 964.15 mg g−1, demonstrating a selectivity greater than 98% for Au(III) and Ag(I) in gold ore tailings and wastewater. Notably, the material maintained over 80.14% of its Au(III) adsorption capability after seven regeneration cycles. A scaled-up continuous adsorption cyclic trial was performed using 1 kg of gold mine wastewater, with the results indicating that after one cycle, the adsorption rates reached as high as 78.12% for Au(III) and 97.69% for Ag(I). The integration of HAPT not only curtails the overall cost but also enhances the material's dispersibility, acid resistance and recyclability, without diminishing its adsorption efficacy. These findings highlight the composite's strong economic feasibility and application potential, offering a promising route for efficient precious-metal recovery and value-added utilization of IV polymers.
1. Introduction
It is well known that conventional polymers (e.g., polyethylene and polypropylene) typically feature carbon–carbon (C–C) covalent bonds as their primary backbone skeleton. To enhance properties such as mechanical strength, thermal stability, and solvent resistance, crosslinking agents–such as divinylbenzene–are commonly introduced to create three-dimensional networks. A classic example is vulcanization, discovered by Charles Goodyear in 1839, in which sulfur serves as a crosslinking agent to reinforce natural rubber by forming covalent bridges between polymer chains. Although elemental sulfur (S8) is capable of self-polymerization, the resulting linear polysulfide chains are thermally unstable and prone to rapid degradation.1 Consequently, traditional rubber vulcanization typically incorporates only a small amount of sulfur (usually <5 wt%) into organic polymers to enhance mechanical strength, abrasion resistance, and heat tolerance.2 This process laid the foundation for modern polymer crosslinking technology.
In contrast, Pyun and co-workers pioneered the inverse vulcanization (IV) strategy in 2013. Subsequently, researchers have developed various improved methods, such as photoinduced3–5 and TBAF-catalyzed6 approaches, to achieve synthesis under mild conditions, enabling the synthesis of inverse vulcanized polymers (IV polymers) with fundamentally different architectures.1,7,8 Through thermal ring-opening polymerization, a polymer backbone with sulfur–sulfur (S–S) bonds as the main chain is formed, while small molecule olefins (e.g., diisopropenylbenzene, DIB) are embedded between sulfur chains as crosslinkers. In IV, the polymer structure is transformed from a “carbon backbone” to a “sulfur backbone”, resulting in sulfur-rich (typically >50 wt%) polymers.1 By harnessing sulfur – an abundant byproduct of the petroleum and natural gas industry – this approach enables value-added resource utilization and aligns closely with green chemistry principles.9 Sulfur-rich polymers generated via IV exhibit unique properties, including a high refractive index and notable infrared transparency.10 More importantly, their high sulfur content provides exceptional heavy metal adsorption capability (particularly for ions such as Hg2+ and Au3+),11 as sulfur atoms readily form strong coordination bonds with these metals.12,13 This feature allows IV-derived materials to overcome many of the limitations associated with traditional adsorbents. However, many sulfur-rich polymers suffer from low porosity, limited dispersibility, and a scarcity of accessible active sites, necessitating further structural optimization. For example, Yang et al. fabricated a porous high-sulfur polymer using edible oil as a crosslinker and a NaCl/urea mixture as a porogen, achieving an Au(III) adsorption capacity of 20.98 mg g−1 at pH 1.0.14 Subsequent studies have investigated various approaches such as supercritical CO2 foaming,15 electrospinning,16 salt templating,17 porogen-assisted fabrication,18 and functional-group modification19,20 to further enhance adsorbent performance. Notably, Chalker reported a substantial decrease in adsorption efficiency (from 95% to 26%) over three cycles, highlighting the challenge of regeneration and the need for more economical and durable polysulfide supports.21 These efforts have yielded ionic liquid-modified sulfur-rich copolymers capable of selectively adsorbing gold, silver, palladium, and platinum, achieving an Au(III) adsorption capacity of up to 636.5 mg g−1.20 However, these materials still exhibit lower adsorption capacities than many advanced adsorbents. Moreover, they often require additional processing to improve performance and continue to face challenges related to dispersibility and reusability (typically remaining effective only for three cycles). Some variants also present economic disadvantages. Notably, polysulfides prepared via inverse vulcanization and related polymerization reactions have previously been employed for gold binding and integrated into complete leaching processes for gold mining and electronic waste recycling. Studies from the Hasell, Chalker, and Jenkins laboratories have demonstrated that such sulfur-rich materials exhibit excellent gold-binding capabilities in practical leaching systems, providing important precedents for the application of inverse vulcanized polymers in precious metal recovery.22–27
Attapulgite (APT), a natural magnesium aluminosilicate clay (Mg5(H2O)4[Si4O10]2(OH)2) with a chain-layer structure possesses a high specific surface area, permanent negative surface charge, and intrinsic metal-adsorption capability without causing secondary pollution.28,29 Integrating APT into sulfur-rich polymers offers a promising strategy for creating cost-effective materials for metal recovery.
Noble metals, particularly gold, are highly valued for their unique physicochemical properties and are extensively employed across diverse modern industries.30 However, as non-renewable resources with limited natural reserves and increasing annual demand, the efficient recovery of these metals from secondary sources has become increasingly critical. The retrieval of noble metals from industrial waste streams not only mitigates resource scarcity but also contributes to reducing environmental pollution.31,32 Current recovery methods include solvent extraction, chemical precipitation, membrane filtration, adsorption, and ion exchange.33–39 Among these, adsorption is particularly notable for its operational simplicity, cost-effectiveness, and high selectivity.40,41 Nevertheless, conventional adsorbents—such as activated carbon, functionalized resins, and metal–organic frameworks—often suffer from drawbacks including complex synthesis, lengthy processing, high cost, limited adsorption capacity, and inadequate selectivity under highly acidic or multi-ion conditions, restricting their large scale application.7,42,43
Herein, we present a novel composite (Poly(S-AM)/HAPT) fabricated by scaffolding a sulfur-rich polymer onto acid-purified attapulgite (HAPT). The polymer Poly(S-AM) was synthesized via inverse vulcanization of low-cost, commercially available elemental sulfur (S8) and acrylamide (AM) (Scheme 1). The resulting Poly(S-AM)/HAPT composite reduces overall material cost while significantly improving dispersibility and recyclability, all without sacrificing the material's core adsorption functionality and performance. The composite demonstrates a maximum Au(III) adsorption capacity of 964.2 mg g−1, selective adsorption rates exceeding 98% for both Au(III) and Ag(I) in simulated tailings and wastewater, and retains over 80% of its initial adsorption efficiency after seven cycles. In a continuous adsorption scale-up test using 1 kg of gold mine wastewater, the adsorption rate for Au(III) was 78.12% and for Ag(I) 97.69% in the first cycle. From the sixth cycle onward, both metals reached 100% adsorption. This strategy offers an economically feasible and practical solution for the efficient recovery of precious metals, particularly gold and silver, while also enabling the high-value utilization of both sulfur-rich polymers and attapulgite resources.
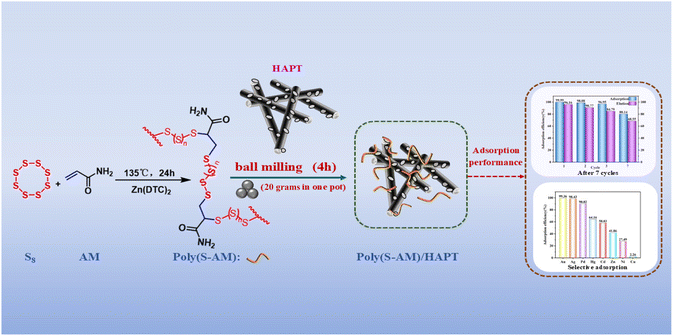 |
| | Scheme 1 Preparation of the Poly(S-AM)/HAPT composite material. | |
2. Experimental
2.1. Chemicals and materials
Sulfur (S8, ≥99.5%) was purchased from Shanghai Chemical Reagent Co., Ltd, acrylamide (AM, ≥98.5%) from Linhai Chemical Factory, zinc diethyldithiocarbamate (Zn(DTC)2, 99%) from Shanghai Titan Scientific Co., Ltd, deuterated DMSO-D6 (C2D6OS, 99.8%) + TMS (0.03%) from Adamas, attapulgite (APT) from Gansu Western Attapulgite Application Research Institute, sulfuric acid (H2SO4, AR grade) from Shanghai Titan Scientific Co., Ltd, and gold ion standard solution (GSB 04-1715-2004) from China Array Research Technology Group Co., Ltd. All chemicals were used as received, without any additional purification/treatment.
2.2. Synthesis of the Poly(S5-AM5)/HAPT composite material
The Poly(S5-AM5)/HAPT composite was prepared through a mechanochemical method as follows: a precisely weighed mixture of Poly(S5-AM5) (500 mg, the synthesis of Poly(S5-AM5) is detailed in the SI (section 1.1) and acid-purified HAPT (500 mg) was loaded into a 100 mL stainless steel grinding jar containing 15 stainless steel grinding balls (5 mm diameter). Mechanical grinding was conducted at room temperature for 4 hours at a rotational speed of 300 revolutions per minute. Upon completion of the grinding process, the system was allowed to cool naturally to room temperature before opening the jar. The resulting black solid product, designated as Poly(S5-AM5)/HAPT, was collected for subsequent characterization. Furthermore, a series of composite materials with varying compositions (denoted as Poly(S5-AM5)x/HAPTy) were systematically prepared by adjusting the mass ratios of the starting materials, where x represents the mass fraction of Poly(S5-AM5) and y represents the mass fraction of HAPT.
2.3. Adsorption experiment of Au(III)
2.5 mg of Poly(S5-AM5)/HAPT was added to a 20 mL centrifuge tube containing 5 mL of metal ion solution and stirred at room temperature for 12 hours. The adsorbent was then separated by centrifugation and filtration. The metal ion concentrations before and after adsorption were measured using inductively coupled plasma optical emission spectrometry/mass spectrometry (ICP-OES/MS, Agilent 5110).
The adsorption performance was evaluated using adsorption efficiency (eqn (1)) and adsorption capacity (eqn (2)):
| |
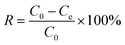 | (1) |
| |
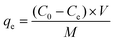 | (2) |
where qe denotes the adsorption capacity (mg g−1), C0 represents the initial concentration (mg L−1), Ce indicates the equilibrium concentration (mg L−1), M stands for the adsorbent mass (mg), and V refers to the solution volume (mL).20
3. Results and discussion
3.1. Synthesis of the Poly(S5-AM5)/HAPT composite
During the planetary ball milling process of the Poly(S5-AM5) and HAPT mixture, the applied mechanical forces reduce the particle size of the Poly(S5-AM5) component, increase its contact area with HAPT, and promote physical adsorption and diffusion between the two components. As a result, part of the Poly(S5-AM5) components becomes uniformly coated on the HAPT surface, while another portion penetrates the pores and interlayer spaces of HAPT, effectively immobilizing the Poly(S5-AM5) polymer powder onto the HAPT framework.
3.2. Characterization
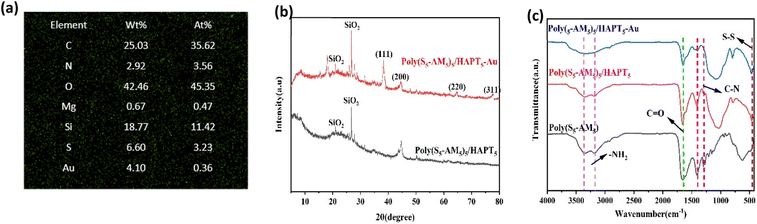
The characterization of Poly(S5-AM5) (Fig. S1) and Poly(S5-AM5)x/HAPTy (Fig. 1(a)) was first performed by using Fourier Transform Infrared Spectroscopy (FT-IR). The FT-IR spectra results showed that the peaks at 3355, 3186 and 1663 cm−1 were attributed to the stretching vibrations of two N–H bonds and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in AM, respectively. Additionally, the characteristic stretching vibration peaks of CC–H and CC bonds (3029 cm−1 and 1623 cm−1) in Poly(S5-AM5) and Poly(S5-AM5)x/HAPTy completely disappeared.44 New peaks at 620 and 456 cm−1 were assigned to the absorption peaks of C–S and S–S bonds.45 A new characteristic peak emerged near 1033 cm−1 in Poly(S5-AM5)x/HAPTy, corresponding to the stretching vibration of the Si–O–Si bond.46 These results confirm the successful polymerization of AM with S8 and the formation of the composite between Poly(S5-AM5) and HAPT. Moreover, the characteristic chemical bonds of each component remained intact without cleavage or reconstruction during the composite formation, and the Si–O–Si characteristic peak showed no significant shift or splitting, indicating a preserved original crystal framework of HAPT. This suggests that the two materials were successfully combined through physical loading. X-ray diffraction (XRD) patterns (Fig. 1(b)) revealed that no characteristic diffraction peaks of S8 were observed in Poly(S5-AM5)5/HAPT5, indicating the complete absence of residual elemental sulfur. The Poly(S5-AM5) component in Poly(S5-AM5)5/HAPT5 formed an amorphous copolymer,47 while the characteristic diffraction peak of SiO2 appeared at 26.7,48 which further confirmed the successful formation of the composite. Thermogravimetric analysis (TGA) (Fig. 1(c)) revealed significant weight loss peaks for the composite of Poly(S5-AM5)5/HAPT5 at 160–355 °C and 355–550 °C, which were absent in HAPT. These peaks correspond to the thermal decomposition of amide groups49 and sulfur chains in Poly(S5-AM5)5, respectively, confirming their successful incorporation onto the HAPT surface. Differential scanning calorimetry (DSC) results (Fig. S4) showed that, as HAPT's melting point is well above 1500 °C,50 the DSC measurements primarily reflected the characteristics of the Poly(S5-AM5) matrix. No distinct glass transition temperature (Tg) was observed for Poly(S5-AM5), and no melting peak corresponding to elemental sulfur was detected at 119 °C, indicating that crystalline sulfur was completely converted into a polysulfide copolymer. Fig. 2(a–c) show the scanning electron microscopy (SEM) images of natural APT, acidified APT (HAPT), and the Poly(S5-AM5)5/HAPT5 composite material, respectively. The unacidified APT exhibits typical rod-like or fibrous aggregated morphology with significant agglomeration between particles, which is attributed to its strong native interlayer interactions and pore blockage by impurities. After acid treatment, the agglomerated structure of APT is effectively dispersed, resulting in significantly improved dispersion of the rod-like crystals, a roughened surface, and the appearance of numerous micropores. This transformation is ascribed to the dissolution of impurities and the exfoliation of the interlayer structure during acidification. Upon compositing HAPT with Poly(S5-AM5), the surface of HAPT becomes uniformly coated with the Poly(S5-AM5) material. The rod-like morphology is partially covered, and the pore structures are further filled, leading to a more regular overall morphology. Notably, no exposed HAPT particles are observed, indicating a tight integration between the two components. Energy-dispersive X-ray spectroscopy (EDS) analysis of HAPT confirms the presence of C, S, N, O, Mg, and Si elements in the material (Fig. S8(b)–(h)). The TEM images of Poly(S5-AM5)5/HAPT5 (Fig. 2(d–f)) further illustrate that nanoscale particles are uniformly attached to the surface and layers of HAPT fibers. No obvious large-scale agglomerated particles are observed, and a small amount of particles fills the pores on the fiber surface. EDS analysis (Fig. S9(b)–(h)) showed increased S and N contents compared to those of pure HAPT, further confirming the successful composite formation and uniform loading of Poly(S5-AM5) on HAPT surfaces. N2 adsorption–desorption experiments using an automated surface area and porosity analyzer (BET) (Fig. 3(a)–(d)) demonstrated that Poly(S5-AM5)5/HAPT5 exhibited a significant decrease in the BET surface area (from 207.746 m2 g−1 to 0.513 m2 g−1) and the total pore volume (from 0.352 cm3 g−1 to 0.001 cm3 g−1) compared to HAPT. This indicates the infiltration of Poly(S5-AM5) into HAPT pores and the coverage of original pore walls, further evidencing the physical loading mechanism for the material combination.51
O bond in AM, respectively. Additionally, the characteristic stretching vibration peaks of CC–H and CC bonds (3029 cm−1 and 1623 cm−1) in Poly(S5-AM5) and Poly(S5-AM5)x/HAPTy completely disappeared.44 New peaks at 620 and 456 cm−1 were assigned to the absorption peaks of C–S and S–S bonds.45 A new characteristic peak emerged near 1033 cm−1 in Poly(S5-AM5)x/HAPTy, corresponding to the stretching vibration of the Si–O–Si bond.46 These results confirm the successful polymerization of AM with S8 and the formation of the composite between Poly(S5-AM5) and HAPT. Moreover, the characteristic chemical bonds of each component remained intact without cleavage or reconstruction during the composite formation, and the Si–O–Si characteristic peak showed no significant shift or splitting, indicating a preserved original crystal framework of HAPT. This suggests that the two materials were successfully combined through physical loading. X-ray diffraction (XRD) patterns (Fig. 1(b)) revealed that no characteristic diffraction peaks of S8 were observed in Poly(S5-AM5)5/HAPT5, indicating the complete absence of residual elemental sulfur. The Poly(S5-AM5) component in Poly(S5-AM5)5/HAPT5 formed an amorphous copolymer,47 while the characteristic diffraction peak of SiO2 appeared at 26.7,48 which further confirmed the successful formation of the composite. Thermogravimetric analysis (TGA) (Fig. 1(c)) revealed significant weight loss peaks for the composite of Poly(S5-AM5)5/HAPT5 at 160–355 °C and 355–550 °C, which were absent in HAPT. These peaks correspond to the thermal decomposition of amide groups49 and sulfur chains in Poly(S5-AM5)5, respectively, confirming their successful incorporation onto the HAPT surface. Differential scanning calorimetry (DSC) results (Fig. S4) showed that, as HAPT's melting point is well above 1500 °C,50 the DSC measurements primarily reflected the characteristics of the Poly(S5-AM5) matrix. No distinct glass transition temperature (Tg) was observed for Poly(S5-AM5), and no melting peak corresponding to elemental sulfur was detected at 119 °C, indicating that crystalline sulfur was completely converted into a polysulfide copolymer. Fig. 2(a–c) show the scanning electron microscopy (SEM) images of natural APT, acidified APT (HAPT), and the Poly(S5-AM5)5/HAPT5 composite material, respectively. The unacidified APT exhibits typical rod-like or fibrous aggregated morphology with significant agglomeration between particles, which is attributed to its strong native interlayer interactions and pore blockage by impurities. After acid treatment, the agglomerated structure of APT is effectively dispersed, resulting in significantly improved dispersion of the rod-like crystals, a roughened surface, and the appearance of numerous micropores. This transformation is ascribed to the dissolution of impurities and the exfoliation of the interlayer structure during acidification. Upon compositing HAPT with Poly(S5-AM5), the surface of HAPT becomes uniformly coated with the Poly(S5-AM5) material. The rod-like morphology is partially covered, and the pore structures are further filled, leading to a more regular overall morphology. Notably, no exposed HAPT particles are observed, indicating a tight integration between the two components. Energy-dispersive X-ray spectroscopy (EDS) analysis of HAPT confirms the presence of C, S, N, O, Mg, and Si elements in the material (Fig. S8(b)–(h)). The TEM images of Poly(S5-AM5)5/HAPT5 (Fig. 2(d–f)) further illustrate that nanoscale particles are uniformly attached to the surface and layers of HAPT fibers. No obvious large-scale agglomerated particles are observed, and a small amount of particles fills the pores on the fiber surface. EDS analysis (Fig. S9(b)–(h)) showed increased S and N contents compared to those of pure HAPT, further confirming the successful composite formation and uniform loading of Poly(S5-AM5) on HAPT surfaces. N2 adsorption–desorption experiments using an automated surface area and porosity analyzer (BET) (Fig. 3(a)–(d)) demonstrated that Poly(S5-AM5)5/HAPT5 exhibited a significant decrease in the BET surface area (from 207.746 m2 g−1 to 0.513 m2 g−1) and the total pore volume (from 0.352 cm3 g−1 to 0.001 cm3 g−1) compared to HAPT. This indicates the infiltration of Poly(S5-AM5) into HAPT pores and the coverage of original pore walls, further evidencing the physical loading mechanism for the material combination.51
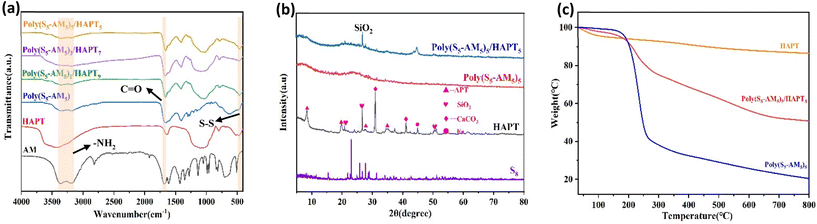 |
| | Fig. 1 (a) FT-IR spectra of Poly(S5-AM5) and Poly(S5-AM5)x/HAPTy. (b) XRD patterns of S8, HAPT, and composite Poly(S5-AM5)5/HAPT5. (c) TGA curves of HAPT and Poly(S5-AM5)5/HAPT5. | |
 |
| | Fig. 2 Scanning electron microscopy (SEM) images of (a) natural APT; (b) acidified APT (HAPT); (c) Poly(S5-AM5)5/HAPT5 composite material. TEM images of Poly(S5-AM5)5/HAPT5 of different sizes: (d) 100 nm, (e) 50 nm and (f) 20 nm. | |
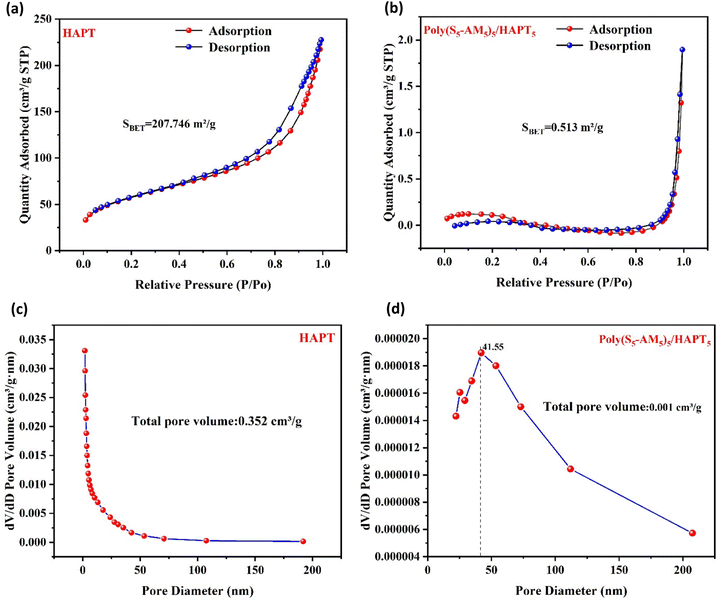 |
| | Fig. 3 (a) N2 adsorption–desorption isotherm of HAPT at 77 K; (b) N2 adsorption–desorption isotherm of Poly(S5-AM5)5/HAPT5 at 77 K; (c) pore size distribution of HAPT from BJH analysis; (d) pore size distribution of Poly(S5-AM5)5/HAPT5 from BJH analysis. | |
4. Adsorption experiments
4.1. The effects of S8 content, Poly(S5-AM5) loading and initial solution pH on Au(III) adsorption performance
Poly(Sx-AMy) with varying sulfur contents was prepared by adjusting the Poly(Sx-AMy) ratio to investigate the effect of sulfur content on Au(III) adsorption capacity (Fig. S6). The results demonstrated a significant increase in adsorption capacity as the sulfur content rose from 30 wt% to 50 wt%, followed by a decrease with a further increase to 70 wt%. Poly(S5-AM5) exhibited optimal adsorption performance, achieving a maximum capacity of 1085.03 mg g−1, and was therefore selected for subsequent composite preparation. Further studies on Poly(S5-AM5)x/HAPTy composites with varying Poly(S5-AM5) loading (Fig. S7) revealed a marked improvement in adsorption capacity as the Poly(S5-AM5) content increased from 10 wt% to 50 wt%, with Poly(S5-AM5)5/HAPT5 showing the best performance (964.15 mg g−1). As shown in Fig. 4(a), in the absence of HAPT, Poly(S5-AM5) exhibited a high adsorption capacity, which can be primarily attributed to its abundant polysulfide active sites that selectively coordinate with Au(III), significantly enhancing the adsorption performance of the material. After the incorporation of HAPT, the content of Poly(S5-AM5) in the composite was reduced to half of its original amount. However, the adsorption capacity did not decrease proportionally. It is speculated that HAPT, serving as a support, improved the dispersion of the active components, allowing the active sites to be more fully exposed and utilized, thereby compensating to some extent for the reduction in the active component content. The solution pH had a significant effect on the adsorption capability of Poly(S5-AM5)5/HAPT5 (Fig. 4(b)). Under acidic conditions (pH = 3), the adsorption efficiency reached 99%, while a considerable decrease in adsorption capacity was observed as the pH increased from 3 to 10. This phenomenon can be attributed to the enhanced protonation of amino groups in acidic media, which suppress proton competition with Au(III) and maintain the activity of carbonyl/sulfhydryl adsorption sites, thereby facilitating Au(III) coordination.52 At higher pH values, Au(III) forms various complexes that compete for adsorption sites.20 Therefore, the adsorbent demonstrated excellent acid resistance and retained its superior Au(III) adsorption capacity across a wide pH range, making it highly suitable for precious metal recovery, particularly gold, in complex environments.
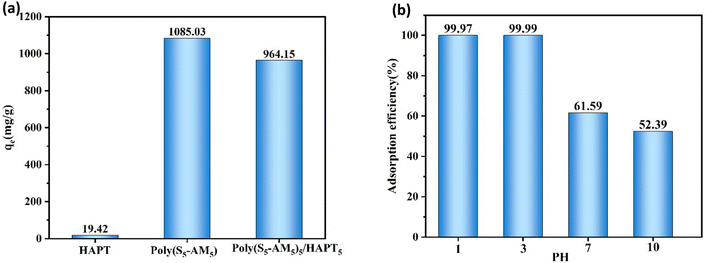 |
| | Fig. 4 (a) Comparison diagram of the maximum saturated adsorption capacities among HAPT, Poly(S5-AM5), and Poly(S5-AM5)5/HAPT5. (b) Effect of solution pH on the adsorption of Au(III) by Poly(S5-AM5)5/HAPT5. | |
4.2. Adsorption isotherm
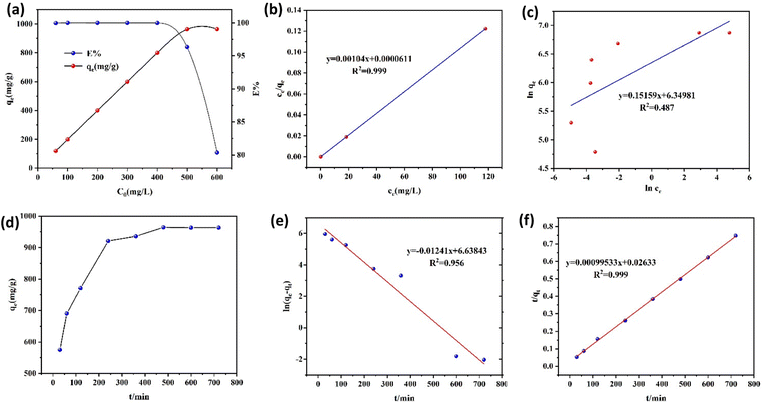
Fig. 5(a) illustrates the impact of the initial Au(III) concentration on the adsorption efficiency of Poly(S5-AM5)5/HAPT5. To investigate the adsorption mechanism, Langmuir (eqn (3)) and Freundlich (eqn (4)) isotherm models were employed to fit the experimental data, as shown in the following expressions:| |
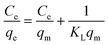 | (3) |
| |
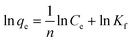 | (4) |
where qe (mg g−1) represents the monolayer saturation adsorption capacity; KL (L mg−1) is the Langmuir adsorption equilibrium constant; Kf and n are Freundlich constants. Fig. 5(b) and (c) present the fitting curves of Langmuir and Freundlich models for Au(III) adsorption by Poly(S5-AM5)5/HAPT5, respectively. The specific parameters of both models are listed in Table S2. The correlation coefficient of the Langmuir model (0.999) was significantly higher than that of the Freundlich model (0.487), indicating monolayer adsorption. The theoretical saturation adsorption capacity (961.54 mg g−1) closely matched the experimentally measured value (964.15 mg g−1).
 |
| | Fig. 5 (a) Effect of initial concentration on Au(III) adsorption by Poly(S5-AM5)5/HAPT5; (b) Langmuir adsorption model of Poly(S5-AM5)5/HAPT5; (c) Freundlich adsorption model of Poly(S5-AM5)5/HAPT5; (d) time-dependent Au(III) adsorption by Poly(S5-AM5)5/HAPT5; (e) pseudo-first-order kinetic model of Poly(S5-AM5)5/HAPT5; (f) pseudo-second-order kinetic model of Poly(S5-AM5)5/HAPT5. | |
4.3. Adsorption kinetics
Fig. 5(d) shows the effect of adsorption time on the adsorption performance. In the first 240 minutes, Poly(S5-AM5)5/HAPT5 exhibited rapid Au(III) adsorption, followed by a gradual decrease in the adsorption rate between 240 and 480 min, ultimately reaching equilibrium at approximately 480 minutes. After this point, the adsorption rate stabilized between 480 and 720 min as the available adsorption sites became saturated. As a result, 480 min was selected as the optimal stirring time for subsequent experiments. The adsorption process was further analyzed using pseudo-first-order (eqn (5)) and pseudo-second-order (eqn (6)) kinetic models:| |
ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − K1t qe − K1t
| (5) |
| |
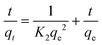 | (6) |
where qe (mg g−1) is the equilibrium adsorption capacity; qt (mg g−1) is the adsorption amount at time t; K1 (min−1) and K2 (g mg−1 min−1) are the rate constants for pseudo-first-order and pseudo-second-order models, respectively. As shown in Fig. 5(e) and (f), the pseudo-second-order model showed a higher correlation coefficient (0.999) compared to the pseudo-first-order model (0.956). Additionally, the theoretical saturation adsorption capacity calculated from the pseudo-second-order model (1004.69 mg g−1) closely aligned with the experimental value (964.15 mg g−1), with detailed parameters listed in Table S3. These results suggest that the pseudo-second-order model more accurately describes the adsorption process, indicating chemisorption as the dominant mechanism.53
4.4. Competitive adsorption and recycling performance
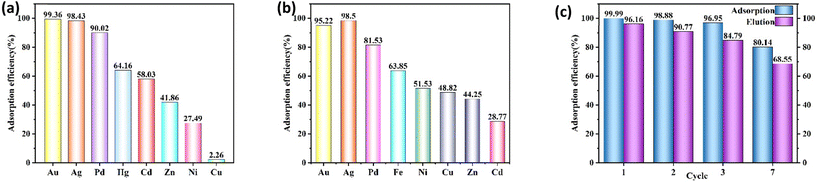
To evaluate its practical application performance, Poly(S5-AM5)5/HAPT5 was tested for competitive ion adsorption using real gold ore tailings. First, the gold ore tailings sample was microwave-digested and diluted, with the initial metal ion concentrations shown in Table S4. The digested solution was then mixed with Poly(S5-AM5)5/HAPT5 at pH = 1 and stirred at room temperature for 8 hours. Notably, in the presence of multiple coexisting ions, Poly(S5-AM5)5/HAPT5 exhibited excellent selective adsorption for precious metals such as Au(III), Ag(I), and Pd(II), with removal efficiencies exceeding 90%. In contrast, the material showed only relatively weak affinity for other metals (Fig. 6a). When gold mine wastewater was treated with 2.5 mg of Poly(S5-AM5)5/HAPT5 under the same conditions, the removal rates for the precious metals Au(III), Ag(I), and Pd(II) reached over 81%, while only relatively weak affinity was observed for other metals (Fig. 6b), with the initial metal concentrations listed in Table S5. To assess the recyclability, 20 mg Poly(S5-AM5)5/HAPT5 was added to 20 mL of 100 mg L−1 Au(III) solution and stirred for 8 h at room temperature. After the adsorption process, the adsorbent was first eluted using a desorption agent (8 wt% HCl + 10 wt% thiourea) and then rinsed 4–5 times with distilled water to ensure complete Au removal. The regenerated adsorbent was then ready for the next cycle. As shown in Fig. 6c, the adsorption efficiency remained high at 80.14% after 7 cycles. As shown in Table S6, compared with some existing polysulfide adsorbents, this material demonstrates relatively superior adsorption performance. To further evaluate the practical engineering application potential of Poly(S5-AM5)5/HAPT5, this study constructed a dynamic cyclic scale-up adsorption setup (Fig. 7(a and b)). Using 1 kg of gold mine wastewater as the treatment target, continuous adsorption cycle scale-up tests were conducted. After preparing 80 g of the adsorbent into small pellets using a circular pelletizer, the pellets were packed into an adsorption column. A peristaltic pump was employed to drive the wastewater circulation, with the pump speed set at 30 rpm and the adsorption temperature maintained at room temperature. The time required for one complete cycle was measured to be 40 minutes. During the experiment, wastewater samples from the 1st, 3rd, 6th and 12th cycles were collected for adsorption performance testing of Au(III) and Ag(I). The results are shown in Fig. 7(c). In the first cycle, the adsorption rates reached 78.12% for Au(III) and 97.69% for Ag(I). By the third cycle, the adsorption rate for Au(III) increased to 81.85%, while that for Ag(I) reached 99.02%. With increasing cycle numbers, the adsorption rates for both Au(III) and Ag(I) achieved 100% as early as the 6th cycle. In summary, these results further confirm that the material exhibits good reusability and excellent adsorption performance. In practical applications, even a single cycle can effectively adsorb and recover precious metals from gold mine wastewater, demonstrating its potential for scalable implementation.
 |
| | Fig. 6 (a) Competitive ion adsorption experiment of Poly(S5-AM5)5/HAPT5 in gold ore residue. (b) Competitive ion adsorption experiment of Poly(S5-AM5)5/HAPT5 in gold mine wastewater. (c) Recyclability test results. | |
 |
| | Fig. 7 (a) Schematic illustration of the cyclic adsorption of Au(III) and Ag(I) from 1 kg of gold mine wastewater by Poly(S5-AM5)5/HAPT5. (b) Practical operation schematic of the cyclic adsorption of Au(III) and Ag(I) from 1 kg of gold mine wastewater by Poly(S5-AM5)5/HAPT5. (c) Adsorption performance diagram of Poly(S5-AM5)5/HAPT5 for Au(III) and Ag(I) in 1 kg of gold mine wastewater through cyclic experiments. | |
4.5. Adsorption mechanism
Poly(S5-AM5)5/HAPT5 exhibited an adsorption capacity of 964.15 mg g−1 for Au(III), showing outstanding gold adsorption performance. This outstanding performance originates from its unique material composite and synthesis strategy. The enhancement in adsorption performance may be attributed to the following reasons: introducing mechanical activation processes during adsorbent preparation may help create a richer pore structure and more active sites, thereby enhancing its adsorption performance; precise chemical modification of the material surface by introducing specific functional groups (such as amide groups) can significantly enhance its selective adsorption capacity for target metal ions. Introducing amide as hydrophilic groups can also effectively improve the material's dispersibility and accessibility in aqueous phases, which is crucial for adsorption performance.
In order to gain a deeper understanding of the adsorption mechanism, the characterization of the adsorption process of Au(III) by Poly(S5-AM5)5/HAPT5 was further investigated using SEM, PXRD, FT-IR and XPS analyses. SEM images of Poly(S5-AM5)5/HAPT5-Au (Fig. S10(a)) revealed Au distributed across the material's surface and around the pores. EDS mapping (Fig. S10(b)–(h)) and (Fig. 8a) confirmed Au adsorption. XRD patterns (Fig. 8b) of Poly(S5-AM5)5/HAPT5 before and after Au(III) adsorption showed four new diffraction peaks at 2θ = 38.3°, 44.5°, 64.7°, and 77.6°, corresponding to the Au(111), (200), (220), and (311) crystal planes, respectively. These peaks indicate the reduction of Au(III) to metallic Au.20 The FT-IR spectrum of Poly(S5-AM5)5/HAPT5-Au shows shifts in the stretching vibration peaks of N–H, CO, C–N, C–S, and S–S to 3345, 3181, 1656, 1404, 616, and 453 cm−1, respectively (Fig. 8c), compared with those of Poly(S5-AM5)5 and Poly(S5-AM5)5/HAPT5, which appear at 3355, 3186, 1663, 1410, 620, and 456 cm−1). Notably, the C–N stretching vibration peak weakens significantly. These changes further indicate that the adsorption of Au(III) by Poly(S5-AM5)5/HAPT5 may involve electrostatic interaction or coordination adsorption. XPS analysis of Poly(S5-AM5)5/HAPT5-Au (Fig. 9(a)) showed a peak at 83.4 eV binding energy for Au 4f. High-resolution Au 4f spectra (Fig. 9(d)) revealed peaks at 84.32 eV (Au(0) 4f7/2) and 88.01 eV (Au(0) 4f5/2), with minor contributions at 86.42 eV (Au(III) 4f7/2) and 89.87 eV (Au(III) 4f5/2). The significantly higher concentration of Au(0) versus Au(III) confirmed the reduction of Au(III) to Au(0) during adsorption, likely mediated by polysulfide24 species in the material. These findings are consistent with XRD results, further validating the reduction process. In the deconvoluted C 1s spectrum (Fig. 9(b)), characteristic peaks at 284.80 eV, 285.37 eV, 286.56 eV and 288.29 eV correspond to C–C, C–S, C–N and CO bonds, respectively. After adsorption (Fig. 9(e)), these peaks shifted to 284.87 eV, 285.77 eV, 288.77 eV and 292.42 eV. For the S 2p spectrum (Fig. 9(c)), the doublet at 163.50 eV (S 2p3/2) and 164.62 eV (S 2p1/2) is attributed to S–S bonds, while peaks at 161.94 eV (S 2p3/2) and 162.87 eV (S 2p1/2) correspond to C–S bonds. After adsorption (Fig. 9(f)), the S–S bond peaks shifted to 163.67 eV and 164.81 eV, and the C–S bond peaks to 162.17 eV and 163.41 eV. The overall increase in binding energies of C 1s and S 2p indicates coordination between sulfur atoms and Au(III).54,55 The lower binding energy of Au(0) compared to Au(III) suggests higher electron density at the Au centers adsorbed on Poly(S5-AM5)5/HAPT5, likely due to electron donation from sulfur atoms in Poly(S5-AM5)5/HAPT5 to Au(III).56 These results demonstrate that coordination interactions dominate the adsorption mechanism of Au(III) by Poly(S5-AM5)5/HAPT5, with sulfur-containing functional groups playing a critical role in the process.
 |
| | Fig. 8 (a) EDS mapping image of Poly(S5-AM5)5/HAPT5 after adsorbing Au, (b) XRD patterns of Poly(S5-AM5)5/HAPT5 before and after adsorbing Au, and (c) FT-IR spectra of Poly(S5-AM5)5, Poly(S5-AM5)5/HAPT5, and Poly(S5-AM5)5/HAPT5 after Au adsorption. | |
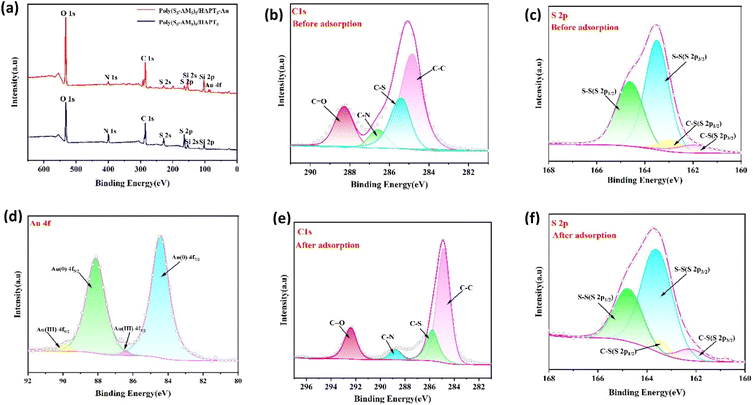 |
| | Fig. 9 (a) XPS spectra of Poly(S5-AM5)5/HAPT5 and Poly(S5-AM5)5/HAPT5-Au, (b) C 1s spectrum before adsorption, (c) S 2p spectrum before adsorption, (d) Au 4f spectrum after adsorption, (e) C 1s spectrum after adsorption, and (f) S 2p spectrum after adsorption. | |
Based on the above characterization and analysis, the adsorption mechanism is illustrated in Fig. 10. Poly(S5-AM5)5/HAPT5 provides selective adsorption sites where N, O, and S atoms coordinate with Au(III). Simultaneously, electrostatic adsorptions occur between the protonated amino groups in Poly(S5-AM5)5/HAPT5 and the anionic complex AuCl4−. The Poly(S5-AM5)5/HAPT5 material also facilitates the gradual reduction of Au(III) to Au(0), with sulfur-containing functional groups playing a crucial role in this process. In addition, the porous structure and high specific surface area of HAPT provide adsorption channels and sufficient dispersion templates for the adsorbent throughout the adsorption process, preventing polymer agglomeration. By confining and strengthening adsorption forces through its pore channels and layered architecture, the material effectively suppresses desorption. This stabilizes the adsorbate during the adsorption process, enhancing overall adsorption performance by using pore confinement to reinforce adsorption forces and reduce desorption. Furthermore, surface groups on HAPT, such as Si–OH and Al–OH, can form coordination bonds or complexes with metal ions like Ag+ and Au3+ thereby further enhancing the adsorption performance.57
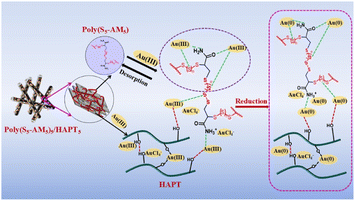 |
| | Fig. 10 Adsorption mechanism of Poly(S5-AM5)5/HAPT5. | |
Conclusions
This study successfully achieves synergistic optimization between cost control and performance stability in precious metal adsorption materials by constructing a polysulfide–APT composite system. It shows that the composite maintains excellent structural stability in complex wastewater environments. As shown in Table S6, compared with some existing polysulfide adsorbents, this material demonstrates relatively superior adsorption performance. Notably, its sustained stability during multi-cycle dynamic adsorption provides critical support for large-scale continuous operation. This work not only expands the application scope of attapulgite in environmental functional materials but also offers an innovative technological pathway for high-value utilization of IV polymers.
Author contributions
Z. J. Q. conceived the project. S. J. W., Y. J. M., X. J. L. and X. C. W. carried out the experimental work and performed the characterization studies. X. F. W. and Z. J. Q. discussed the results and thoroughly revised the manuscript. All authors contributed to the preparation of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6lp00040a.
Acknowledgements
The work was supported by the National Nature Science Foundation of China (No. 22571258), the Gansu Province Major Science and Technology Project (No. 25ZDWA008), the Natural Science Foundation of Gansu Province (25JRRA025) and the Gansu Province University Industry Support Plan (No. 2024CYZC-08).
References
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y. E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5(6), 518–524 CrossRef CAS PubMed.
- N. Payungwong, H. Cheng, K. Nakajima, C. C. Ho and J. Sakdapipanich, Chin. J. Polym. Sci., 2025, 43(1), 70–82 CrossRef CAS.
- J. Jia, J. Liu, Z. Q. Wang, T. Liu, P. Yan, X. Q. Gong, C. Zhao, L. Chen, C. Miao, W. Zhao, S. Cai, X. C. Wang, A. I. Cooper, X. Wu, T. Hasell and Z. J. Quan, Nat. Chem., 2022, 14(11), 1249–1257 CrossRef CAS PubMed.
- J. Jia, P. Yan, S. D. Cai, Y. Cui, X. Xun, J. Liu, H. Wang, L. Dodd, X. Hu, D. Lester, X. C. Wang, X. Wu, T. Hasell and Z. J. Quan, Eur. Polym. J., 2024, 207, 112815 CrossRef CAS.
- J. Jia, Y. Chai, X. Xun, Y. Gao, T. Qiao, X. Wang, X. C. Wang, T. Hasell, X. Wu and Z. J. Quan, Macromol. Rapid Commun., 2025, 46(6), 2400998 CrossRef CAS PubMed.
- X. R. Cao, X. J. Liu, W. P. Li, D. P. Chen, T. Hasell, X. Wu, X. C. Wang and Z. J. Quan, Green Chem., 2025, 27(7), 1974–1983 RSC.
- A. Batnasan, K. Haga, H.-H. Huang and A. Shibayama, Metals, 2019, 9(3), 363 CrossRef CAS.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55(50), 15486–15502 CrossRef CAS PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19(12), 2748–2761 RSC.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K. J. Kim, J. van der Laan, N. A. Nguyen, E. L. Dereniak, M. E. Mackay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26(19), 3014–3018 CrossRef CAS PubMed.
- J. M. Chalker, M. Mann, M. J. H. Worthington and L. J. Esdaile, Org. Mater., 2021, 03(02), 362–373 CrossRef CAS.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55(5), 1714–1718 CrossRef CAS PubMed.
- F. G. Müller, L. S. Lisboa and J. M. Chalker, Adv. Sustainable Syst., 2023, 7(5), 2300010 CrossRef.
- Z. Ren, X. Jiang, L. Liu, C. Yin, S. Wang and X. Yang, J. Mol. Liq., 2021, 328, 115437 CrossRef CAS.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllister and S. M. Howdle, Chem. Commun., 2016, 52(31), 5383–5386 RSC.
- M. W. Thielke, L. A. Bultema, D. D. Brauer, B. Richter, M. Fischer and P. Theato, Polymers, 2016, 8(7), 266 CrossRef PubMed.
- V. Diniz, J. C. Bear, S. Rath and C. R. Crick, Sci. Rep., 2024, 14(1), 8144 CrossRef CAS PubMed.
- S. Petcher, B. Zhang and T. Hasell, Chem. Commun., 2021, 57(41), 5059–5062 RSC.
- M. L. Eder, C. B. Call and C. L. Jenkins, ACS Appl. Polym. Mater., 2022, 4(2), 1110–1116 CrossRef CAS.
- X. Zhou, Y. Cui, X. Xun, J. Jia, X. C. Wang and Z. J. Quan, Sep. Purif. Technol., 2025, 365, 132679 CrossRef CAS.
- N. A. Lundquist, M. J. H. Worthington, N. Adamson, C. T. Gibson, M. R. Johnston, A. V. Ellis and J. M. Chalker, RSC Adv., 2018, 8(3), 1232–1236 RSC.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10(1), 647 CrossRef PubMed.
- J. M. M. Pople, T. P. Nicholls, L. N. Pham, W. M. Bloch, L. S. Lisboa, M. V. Perkins, C. T. Gibson, M. L. Coote, Z. Jia and J. M. Chalker, J. Am. Chem. Soc., 2023, 145(21), 11798–11810 CrossRef CAS PubMed.
- M. Mann, T. P. Nicholls, H. D. Patel, L. S. Lisboa, J. M. M. Pople, L. N. Pham, M. J. H. Worthington, M. R. Smith, Y. Yin, G. G. Andersson, C. T. Gibson, L. J. Esdaile, C. E. Lenehan, M. L. Coote, Z. Jia and J. M. Chalker, Nat. Sustain., 2025, 8(8), 947–956 CrossRef.
- J. Rollins, C. B. Call, D. Herrera and C. L. Jenkins, ACS Appl. Polym. Mater., 2025, 7(13), 8529–8537 CrossRef CAS.
- W. Cao, F. Dai, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2020, 142(2), 978–986 CrossRef CAS PubMed.
- M. Mann, T. Nicholls, H. Patel, L. Lisboa, J. Pople, L. N. Pham, M. Worthington, M. Smith, Y. Yin, G. Andersson, C. Gibson, L. Esdaile, C. Lenehan, M. Coote, Z. Jia and J. Chalker, ChemRxiv, 2024, DOI:10.26434/chemrxiv-2024-1xd84.
- Y. Wang, Y. Feng, J. Jiang and J. Yao, ACS Sustainable Chem. Eng., 2019, 7(2), 1855–1869 CrossRef CAS.
- J. Liu, J. Zhong, Z. Chen, J. Mao, J. Liu, Z. Zhang, X. Li and S. Ren, Appl. Surf. Sci., 2021, 565, 150398 CrossRef CAS.
- Y. Wei, W. Zhang and J. Gao, Green Chem., 2024, 26(10), 5684–5707 RSC.
- A. Islam, A. M. Swaraz, S. H. Teo, Y. H. Taufiq-Yap, D. V. N. Vo, M. L. Ibrahim, G. Abdulkreem-Alsultan, U. Rashid and M. R. Awual, J. Cleaner Prod., 2021, 323, 129015 CrossRef CAS.
- C. Wang, G. Lin, J. Zhao, S. Wang, L. Zhang, Y. Xi, X. Li and Y. Ying, Chem. Eng. J., 2020, 380, 122511 CrossRef CAS.
- M. John, S. Heuss-Aßbichler, K. Tandon and A. Ullrich, J. Water Process Eng., 2019, 30, 100532 CrossRef.
- S. Rascón-Leon, M. M. Castillo-Ortega, I. Santos-Sauceda, G. T. Munive, D. E. Rodriguez-Felix, T. D. Castillo-Castro, J. C. Encinas, J. L. Valenzuela-García, J. M. Quiroz-Castillo, B. García-Gaitan, P. J. Herrera-Franco, J. Alvarez-Sanchez, J. Z. Ramírez and L. S. Quiroz-Castillo, Polym. Bull., 2018, 75(7), 3241–3265 CrossRef.
- Q. Meng, X. Yan and G. Li, J. Cleaner Prod., 2021, 323, 129115 CrossRef CAS.
- J. W. Choi, M. H. Song, J. K. Bediako and Y. S. Yun, Environ. Pollut., 2020, 266, 115167 CrossRef CAS PubMed.
- Z. Feng, Y. Ma, V. Natarajan, Q. Zhao, X. Ma and J. Zhan, Sens. Actuators, B, 2018, 255, 884–890 CrossRef CAS.
- H. Murakami, S. Nishihama and K. Yoshizuka, Hydrometallurgy, 2015, 157, 194–198 CrossRef CAS.
- X. Xun, J. Jia, X. C. Wang, X. Wu and Z. J. Quan, Polym. Chem., 2025, 16(2), 149–155 RSC.
- Y. Chen, Q. Qiao, J. Cao, H. Li and Z. Bian, Joule, 2021, 5(12), 3097–3115 CrossRef CAS.
- M. Soleimani and T. Kaghazchi, Bioresour. Technol., 2008, 99(13), 5374–5383 CrossRef CAS PubMed.
- L. Fu, L. Zhang, S. Wang, B. Zhang and J. Peng, J. Taiwan Inst. Chem. Eng., 2017, 80, 342–348 CrossRef CAS.
- X. Liu, R. Liu, Y. Lu, Q. Sun, W. Xue, M. Cheng and Y. Yang, Sep. Purif. Technol., 2024, 328, 125049 CrossRef CAS.
- A. Siddiqua, N. M. Ranjha, S. Rehman, H. Shoukat, N. Ramzan and H. Sultana, Polym. Bull., 2022, 79(9), 7655–7677 CrossRef CAS.
- A. S. M. Ghumman, R. Shamsuddin, Z. A. Alothman, A. Waheed, A. M. Aljuwayid, R. Sabir, A. Abbasi and A. Sami, ACS Omega, 2024, 9(4), 4831–4840 CrossRef CAS PubMed.
- A. K. Antosik, E. Makuch and K. Gziut, J. Polym. Res., 2022, 29(4), 135 CrossRef CAS.
- S. Zeng, L. Li, D. Zhao, J. Liu, W. Niu, N. Wang and S. Chen, J. Phys. Chem. C, 2017, 121(5), 2495–2503 CrossRef CAS.
- W. Tian, L. Wu, R. Huang, A. Wang, Y. Lu, N. Tang and L. Gao, AIP Adv., 2024, 14(2), 025017 CrossRef CAS.
- F. Zamani-Babgohari, A. Irannejad, G. R. Khayati and M. Kalantari, Thermochim. Acta, 2023, 725, 179532 CrossRef CAS.
- Z. Peng and D. Chen, J. Polym. Sci., Part B: Polym. Phys., 2006, 44(3), 534–540 CrossRef CAS.
- J. Ge, M. Wang, T. Sun, L. Dong and H. Liu, J. Appl. Polym. Sci., 2025, 142(33), e57304 CrossRef CAS.
- Z. Wang, Z. Lu, Y. Xia, Y. Wei, H. Liu, H. Tang, X. Liu, J. Shi, J. Zhang and C. Liu, Water Res., 2025, 282, 123624 CrossRef CAS PubMed.
- Y. Sun, C. Yang, Y. Fu, T. Guo, G. Yan and J. Hu, J. Environ. Chem. Eng., 2023, 11(3), 109806 CrossRef CAS.
- Y. Liao, M. Liu, J. Liu, J. h. Zhu, J. j. Liu, X. C. Wang and Z. J. Quan, New J. Chem., 2021, 45(35), 16205–16212 RSC.
- R. A. Dop, D. R. Neill and T. Hasell, ACS Appl. Mater. Interfaces, 2023, 15(17), 20822–20832 CrossRef CAS PubMed.
- T. M. Ivanova, R. V. Linko, A. V. Petrov, M. I. Bazanov and K. M. Dyumaev, Russ. J. Inorg. Chem., 2008, 53(11), 1784–1787 CrossRef.
- J. Zhang, H. Chen and A. Wang, Polym. Compos., 2007, 28(2), 208–213 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xiao-Feng Wu
a,
Xiao-Feng Wu














