
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual end-functionalisation of poly(beta-amino ester) gene delivery vectors using multicomponent chemistry
Lewis
O'Shaughnessy
 a,
Rahman
Khosravi
a,
James
Robins
a,
Akosua
Anane-Adjei
b,
Mariarosa
Mazza
b,
Naoto
Hori
a,
Pratik
Gurnani
*c and
Cameron
Alexander
*a
a,
Rahman
Khosravi
a,
James
Robins
a,
Akosua
Anane-Adjei
b,
Mariarosa
Mazza
b,
Naoto
Hori
a,
Pratik
Gurnani
*c and
Cameron
Alexander
*a
aDivision of Molecular Therapeutics and Formulation, Boots Science Building, School of Pharmacy, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: cameron.alexander@nottingham.ac.uk
bAdvanced Drug Delivery, Pharmaceutical Sciences, Biopharmaceuticals R&D, AstraZeneca, Cambridge, UK
cUCL School of Pharmacy, University College London, 29-39 Brunswick Square, Bloomsbury, London, WC1N 1AX, UK. E-mail: p.gurnani@ucl.ac.uk
First published on 16th December 2025
Abstract
The use of RNA therapeutics provides a potent tool to enhance patient outcomes, but successful RNA delivery requires efficient and safe vectors. Cationic polymers provide one technology platform for this delivery and among these materials, poly(beta-amino esters) (PBAEs) have emerged as efficient and well tolerated vectors. Changing the end group of these materials can have a profound impact on their physical and biological properties, and the development of new pathways for end-group functionalisation can provide access to untapped material libraries for further development. We therefore developed a synthetic pathway that exploits the Passerini 3-component reaction as a means to incorporate aldehyde and isocyanide materials into the end-groups of an acid terminated PBAE. Polyplexes were then prepared and studied for encapsulation efficiencies, formulation properties and gene transfectability in vitro. Select polymers demonstrated high mRNA transfection efficiency in HEK293T cells. Our findings indicate that this synthetic pathway provides a versatile and adaptable pathway for the further modification of PBAEs and that this modification serves to provide new materials with enhanced nucleic acid delivery properties.
Introduction
Despite rapid and continuous evolution of healthcare technologies, many conditions still remain beyond our capacity to treat via conventional methods; we lack high quality treatment for many cancers and inherited conditions, and do not have effective vaccines for many communicable diseases.1 In particular, diseases caused by aberrant proteins can be difficult to treat with small molecule pharmaceutics, as the vast majority of these proteins remain conventionally “undruggable”, typically owing to flat functional interfaces, lacking binding sites for drug interactions.2,3 Researchers are now turning to biologics as a potential solution, with ribonucleic acid (RNA) therapeutics offering an alternative avenue of treatment, functioning by modulating protein levels within cells to effect changes in disease progression and patient outcomes.1,4One way to utilise this technology, is the delivery of messenger RNA (mRNA) to support the expression otherwise absent proteins, which has been used most significantly for vaccination purposes when mRNA coding for the SARS-CoV-2 spike antigen was delivered to humans, resulting in protein expression and subsequently triggering an immune response.1,5
However, RNA is a large, charged molecule, which can not effectively cross cell membranes, and is rapidly cleared from the circulation by endogenous nuclease enzymes.6,7 Consequently, RNA therapeutics require safe and effective vectors to allow the payload to enter the cell cytoplasm where it is active, and protect them from this prior degradation. Commonly investigated vector technologies include viral vectors, lipid nanoparticles and polyelectrolyte complexes (polyplexes).8,9 Formed by electrostatic attractions between negatively charged RNA and cationic polymers, polyplexes are self-assembled nanoparticles that provide a means for RNA to enter into cells.10 Among the many polymers studied, poly(beta-amino ester)s (PBAE)s provide a robust, well established and well researched medium for RNA delivery. PBAEs are synthesised by aza-Michael addition reaction between a diacrylate and either a primary amine or a bis-secondary amine, providing a high yielding, efficient polymerisation route. The resultant polymers are easily biodegradable via hydrolysis of the ester groups within the polymer backbone, exhibit low cytotoxicity, and can effectively release RNA within the cell interior.11,12 Efficient and simple synthesis from a wide range of available starting materials allows for the development of extensive polymer libraries, and has been behind the development and study of thousands of different PBAEs.13
Since the initial development of PBAEs, research has highlighted the importance of the PBAE end group identity, with small changes in end group functionality leading to outsized impacts on polymer and polyplex properties.14,15 The aza-Michael addition process detailed above is typically conducted with a slight excess of acrylate monomer, resulting in acrylate terminated polymers, the end-groups of which can then be further modified. In particular, the introduction of amine end-capping groups has become well established and widely studied, owing to the ease with which amines can be joined to acrylate terminated PBAEs via an additional aza-Michael addition. Prior research has found that incorporating amine end-capping units onto acrylate terminated PBAEs can increase DNA delivery five-fold, and simultaneously reduce cytotoxicity.16 Subsequent work demonstrated that the identity of the end-capping unit plays a more significant role in determining transfection efficiency than any physical characteristics of the polyplex.17 As a result, researchers continue to explore the introduction of new end-capping units to enhance RNA delivery.18,19
Further research into end-cap modification includes the use of thiol containing small molecules, which can also be incorporated into PBAEs via thiol-Michael addition reactions, with several reports that these modifications also enhance RNA delivery beyond that of the acrylate terminated polymer.20,21
Michael addition techniques have also been utilised to introduce other polymers as end-capping units, creating co-polymers. This has included reactions between acrylate terminated PBAEs and thiol terminated poly(ethylene glycol) (PEG) to improve particle stability by generating PEG-ylated copolymers,22 and also the synthesis of a PBAE-polysarcosine copolymer, achieved by generating thiol terminated polysarcosine in situ, which could then undergo Michael addition reactions with acrylate terminated PBAEs.23 Synthesis of these copolymers generally reduced the cytotoxicity and serum protein interaction of the polymer relative to unfunctionalised PBAE.23
However, current end-capping technologies are restricted to the introduction of just one functional group per polymer end-group, severely limiting the chemical space available for modification. Introducing additional groups onto the polymer termini could further enhance gene delivery applications, beyond what has been achieved with single-group modifications. Consequently, utilising alternative chemistries could not only further broaden the scope of accessible starting materials for end-capping of PBAEs, but could also increase the number of groups introduced simultaneously to the polymer, to enhance gene delivery. Tapping into new chemical space and synthesising novel biomaterials via the incorporation of novel end-capping agents would provide a route towards unique polymer properties and characteristics. In turn, this could lead to materials with further optimised transfection capabilities.
The use of multi-component reactions (MCRs) is one possible way to achieve this goal. Typically highly efficient and versatile procedures, MCRs combine 3 or more reagents in a single step, providing direct pathways to highly functionalised materials, attaining high yields under mild conditions.
Among the wide range of existing MCRs, each combines a unique combination of substrates into a different product, allowing researchers to utilise different MCRs with a wide range of different functional groups. For example, the A3 reaction involves the reaction of aldehyde, an alkyne and an amine to form a propargyl-amine,24 while the Mannich reaction uses a non-enolizable aldehyde with a primary or secondary amine and an enolizable carbonyl group to form β-amino-carbonyl compounds.25 Two of the most widely studied MCRs are the isocyanide based Passerini 3-component reaction (P3CR) and Ugi 4-component reaction (U4CR), which both progress in a similar manner. The P3CR combines an aldehyde with a carboxylic acid and an isocyanide to form an α-acyloxy amide,26 whereas the U4CR combines these same reagents with an additional amine component to form a bis-amide.
An appropriately chosen MCR could be used to introduce a wide range of new materials to PBAEs as end-capping units, and importantly could provide a means to simultaneously introduce two or more end-capping motifs simultaneously. Whilst there exist reports of MCRs being used to post-modify different polymers, to the best of our knowledge there have been no reports of the use of MCRs to modify PBAE termini.
In this work we demonstrated the successful use of an MCR as a means to introduce novel end-capping units onto a PBAE chain. By synthesising first an acid terminated polymer, we were able to use the P3CR to modify these termini simultaneously with both aldehyde- and isocyanide-derived motifs. Neither of these functionalities have previously been incorporated into PBAE end-caps, and so this chemistry opens up two new untapped libraries of substrates for research. We then tested the capability of these polymers to complex with RNA, before testing their in vitro cytocompatibility and their capacity to deliver model mRNA.
Materials and methods
Materials
The following chemicals were used as received:1,6-Hexanediol diacrylate, 4-amino-1-butanol (Aldrich), 3-mercaptopropanoic acid (Aldrich), tert-butyl isocyanide, n-butyl isocyanide, cyclohexyl isocyanide, benzaldehyde, p-anisaldehyde, 2-nitrobenzaldehyde, 4-pyridine carboxaldehyde, 4-ethynyl benzaldehyde, 4-(trifluoromethyl)benzaldehyde, 4-nitro benzaldehyde, 2,4-dinitrobenzaldehyde, 2-imidazole carboxaldehyde, dodecanal, sodium chloride, hydrochloric acid and sodium hydroxide were obtained from Sigma-Aldrich. 2-Pyridine carboxaldehyde was obtained from Acros Organics.
For NMR, chloroform-d (99.8% D atom) and DMSO-d6 were obtained from Sigma-Aldrich and used as received.
All solvents were obtained from Fisher Scientific and were of HPLC grade and used without purification.
Carbon coated copper TEM grids were obtained from Agar Scientific.
HEK 293T cells and MDA-MB-231 cells were both obtained from ATCC.
Permanently fLuc expressing MDA-MB-231 cells were established and gifted by Prof. Anna Grabowska, School of Medicine, University of Nottingham.
Permanently mCherry-Gal9 expressing HEK293T cells were established and provided by Dr Sal Jones, School of Medicine, University of Nottingham, utilising mCherry-GAL9 plasmid, as a gift from Prof. Alan Sabirsh (Addgene plasmid #166689; https://n2t.net/addgene:166689; RRID:Addgene_166689) and pCMV-ZFN-AAVS1, from Dr Dmitriy Mazurov (Addgene plasmid # 89707; https://n2t.net/addgene:89707; RRID:Addgene_89707).
Dulbecco's Modified Eagle's Medium (DMEM), OptiMEM reduced serum medium, Phosphate buffered saline (PBS) and Trypsin-EDTA were obtained from Gibco, ThermoFisher.
Foetal bovine serum, L-glutamine, penicillin streptomycin, Quant-iT RiboGreen assay, 3 M sodium acetate buffer solution, nuclease free water, Lipofectamine MessengerMAX, Lipofectamine 3000, Triton X-100, Hoechst 33342 and RNase Zap were all obtained from Fisher Scientific. PrestoBlue reagent and ONE-Glo Luciferase assay system were purchased from Promega, UK. RiboGreen quantitation kit was purchased from ThermoFisher. CleanCap fLuc mRNA was obtained from Trilink Biotechnologies and ARCA EGFP mRNA (5-moUTP) was obtained from ApexBio. PolyA was purchased from Sigma-Aldrich.
Methods
NMR spectroscopy. 1H and 13C NMR spectra were recorded on a Bruker DPX-400 spectrometer using deuterated solvent (materials section). DOSY NMR spectra were recorded on a Bruker AV(III)500 spectrometer, using deuterated solvent (materials section). The spectra produced were analysed with MestReNova 14.2.0 (Mestrelab Research S.L.).
Size exclusion chromatography (SEC). A Polymer Laboratories PL-50 instrument equipped with differential refractive index (DRI) was used for SEC analysis. The system was fitted with 2× PLgel Mixed D columns (300 × 7.5 mm) and a PLgel 5 µm guard column. The eluent used was DMF with LiBr 0.01 mol dm−3 (equivalent to 0.088% w/v). Samples were run at 1 mL min−1 at 50 °C. Poly(methyl methacrylate) standards (Agilent EasyVials) were used for calibration between 955
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500–550 g mol−1. Analyte samples were filtered through a membrane with 0.22 μm pore size before injection. Experimental molar mass (Mn, SEC) and dispersity (Đ) values of synthesised polymers were determined by conventional calibration using Cirrus Gel Permeation Chromatography (GPC) software.
500–550 g mol−1. Analyte samples were filtered through a membrane with 0.22 μm pore size before injection. Experimental molar mass (Mn, SEC) and dispersity (Đ) values of synthesised polymers were determined by conventional calibration using Cirrus Gel Permeation Chromatography (GPC) software.
Fourier transform infrared (FTIR) spectrometry. FTIR spectrometry was performed on an Agilent Cary630 FTIR with an attenuated total reflection (ATR) module between the wavenumber range of 4000–650 cm−1.
DLS and zeta potential. A dynamic light scattering (DLS), Zetasizer nano-ZS90 (Malvern, Inc.), instrument was used to characterise the particles in terms of the size, PDI and zeta potential. The instrument was used at 25 °C to determine the zeta potential, average hydrodynamic diameters, and polydispersity index (PDI) of polyelectrolyte complexes. Polyplexes were prepared at an RNA concentration of 20 μg mL−1 at a range of NP ratios (32, 64 and 128).
Transmission electron microscopy. Polyplexes were prepared at an RNA concentration of 20 μg mL−1 at NP ratio 64 in 25 mM sodium acetate buffer.
For imaging, glow discharged Formvar/carbon coated TEM grids were used, on which were placed the formulation samples (10 µL), which were left for 3 minutes, then the excess solution was removed and the samples washed of excess buffer with ultrapure water. Excess water was removed and the samples were allowed to dry at room temperature. Following this, 10 µL of 2% aqueous uranyl acetate was applied to each grid and left for 30 seconds. After air drying, imaging was performed using a Tecnai G2 Spirit TEM with BioTwin lens configuration (Thermo Fisher Scientific, Eindhoven, The Netherlands) at an accelerating voltage of 100 kV.
Polymer buffering capacity. The buffering capacity of the PBAEs was evaluated by acid–base titration over the pH range of 11.0–3.0. 2 mg of polymer was dissolved in 30 mL of 0.1 M NaCl aqueous solution, and the solution was adjusted to pH 11.0 using 0.1 M NaOH. Precise titres (between 2–20 µL) of 0.1 M HCl were added until a pH of 3 was achieved. pH after each addition of HCl was recorded. 0.1 M NaCl was set as negative control. The proton buffering capacity of polymers when pH is changed from 7.4 to 5.5 was calculated using eqn (2) and (3).
Preparation of polyplexes. Polymers were stored as stock solutions (100 mg mL−1 in DMSO) and used to prepare polyplexes, which were formed by electrostatic interactions between positively charged polymers and negatively charged RNA. Polyplexes at the desired NP ratio were made by preparing the appropriate working dilutions of polymer and RNA in 25 mM NaOAc buffer (pH: 5.5). Equal volumes of these polymer and RNA solutions were gently mixed using a pipette and incubated at room temperature for 10 minutes.
Encapsulation efficiency determination using Ribogreen assay. Fluorescence-based Quant-iT RiboGreen assay was utilised to detect free mRNA in solution after polymer encapsulation. Polyplexes were prepared at an RNA concentration of 2.5 μg mL−1 in 25 mM NaOAc buffer at a range of NP ratios (32, 64 and 128) and subsequently diluted by a factor of 10 with 1× TE buffer and the assay was completed according to the manufacturer's protocol. Samples were then loaded on a black, 96-well plate, and analysed for fluorescence on a Tecan Spark microplate reader at an excitation of 480 nm and emission at 525 nm.
The readout was used to calculate the amount of free mRNA in solution, which in turn was used to calculate the amount of mRNA encapsulated in the polyplex.
Cell culture. HEK 293T cells and MDA-MB-231 cells were both cultured in Dulbecco's Modified Eagle's Medium (DMEM) for cell culture supplemented with foetal bovine serum (10%), L-glutamine (1%) and penicillin streptomycin (1%). Cells were grown to 90% confluence in a humidified incubator at 37 °C, 5% CO2 and detached with 1× trypsin/EDTA. Viability was assessed using Trypan blue staining.
In vitro transfection with fLuc mRNA. In vitro transfection experiments were performed in both HEK293T and MDA-MB-231 cell lines, with the commercially available transfection reagent Lipofectamine Messenger MAX™ used as a positive control. 24 hours prior to treatment, cells were seeded in a clear 96-well plate at a density of 25 × 103 cells per well (HEK293T) or 10 × 103 cells per well (MDA-MB-231) in DMEM. At time of treatment, the growth media was removed and cells were transfected with 200 μL of OptiMEM containing fLuc mRNA polyplexes, unless stated otherwise, polyplexes were utilised at a concentration of 250 ng mL−1 RNA (prepared at 2.5 μg mL−1 in 25 mM NaOAc and diluted 1
:10 in OptiMEM). At 24 hours post-treatment, 150 μL of media was removed and 50 μL of ONE-Glo D-luciferin substrate was placed into each well and mixed well by pipette. The plate was then incubated at 37 °C for 10 minutes before the total 100 μL was placed in a white 96-well plate and a Tecan Spark plate reader was used to determine the luminescence.
Cell viability determined by PrestoBlue metabolic activity assay:. To investigate the cytotoxicity of the formulations, a cell viability assay was performed in both HEK293T and MDA-MB-231 cell lines, with untreated cells and cells treated with 1% Triton X-100 used as negative and positive controls. 24 hours prior to treatment, cells were seeded in a clear 96-well plate at a density of 25 × 103 cells per well (HEK293T) or 10 × 103 cells per well (MDA-MB-231) in DMEM. At time of treatment, the growth media was removed and cells were transfected with 200 μL of OptiMEM containing fLuc mRNA polyplexes, unless stated otherwise, polyplexes were utilised at a concentration of 250 ng mL−1 mRNA (prepared at 2.5 μg mL−1 in 25 mM NaOAc and diluted 1
:10 in OptiMEM). At 24 hours post-treatment, all media was removed from the cells and replaced with 100 μL of 10% PrestoBlue reagent (diluted in PBS) and incubated for 1 hour at 37 °C. The total volume was transferred to a black 96-well plate and a Tecan Spark plate reader was used to measure the fluorescence intensity (Excitation: 535 nm Emission: 615 nm) of each well. Metabolic activity of the negative control was accepted as 100% and activity of the positive control was accepted as 0%. Metabolic activity (%) of the samples was calculated from eqn (1).
Calculation of relative metabolic activity:
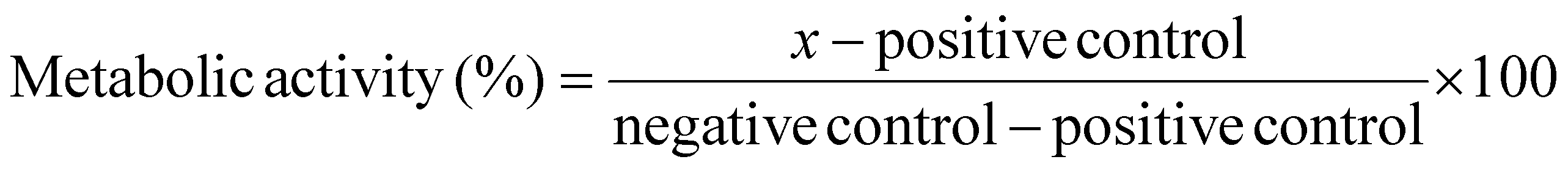 | (1) |
Confocal microscopy study. 24 hours prior to treatment, mCherry-Gal9 expressing HEK293T cells were seeded at a density of 25 × 103 cells per well in a glass bottomed, black walled 96-well plate in DMEM. At time of treatment, the growth media was removed and cells were transfected with 200 μL of OptiMEM containing polyplexes prepared from PBAEs and EGFP mRNA at NP 64, at a concentration of 500 ng mL−1 mRNA, along with 0.01% v/v Hoechst 33342 stain. 90 minutes after treatment, the cells were transferred to a ZEISS Celldiscoverer 7 automated confocal microscope, and imaged under brightfield illumination and at three different sets of excitation/emission wavelengths for fluorescence imaging: 348/455 nm (Hoechst 33342); 488/509 nm (GFP); and 587/610 nm (mCherry). Repeat images were collected at regular intervals until 23 hours after treatment.
Procedure for COOH-(pHDD-4AB)-COOH synthesis. 4-Amino-1-butanol (802.3 mg, 9 mmol) was dissolved in 2.7 ml of DMSO along with 1,6-hexanediol diacrylate (2240.1 mg, 9.9 mmol). The reaction mix was stirred at 90 °C for 24 hours and then cooled to room temperature before the addition of 3-mercaptopropanoic acid (477.6 mg, 4.5 mmol), followed by a further 24 hours of stirring at room temperature. The reaction mix was diluted with 3 mL of THF and precipitated from ice-cold diethyl ether, the polymer was then resuspended in THF and precipitated from THF a further 3 times.
General procedure for P3CR end-capping of COOH terminated PBAEs. A sample (200 mg) of COOH-(pHDD-4AB)-COOH was dissolved in 2 mL of DCM along with the chosen aldehyde (2 mmol) and stirred at room temperature for 30 minutes. At this point, the appropriate isocyanide (2 mmol) was added and the reaction mix was stirred for a further 1 day. Purification was achieved by precipitating the polymer from ice-cold diethyl ether, the polymer was then redissolved in 4 mL of DCM and precipitation repeated twice more.
Results and discussion
Synthetic design
As a procedure for effective end-capping of PBAEs, the P3CR was selected as the MCR of choice, as it would allow for the effective introduction of two different functional groups simultaneously. The P3CR reaction combines an aldehyde with a carboxylic acid and an isocyanide into an α-acyloxy amide within a single step,27–29 and has been widely used in polymer science for post-modification of polymer chains and the introduction of end capping units into different polymer families. It remains a popular methodology as it provides a clean and efficient synthetic pathway that proceeds under mild conditions, without the need for a catalyst. Moreover, the P3CR tolerates a wide variety of functional groups and solvents, and so can be applied successfully to a large library of substrates.29,30 Previous uses of the P3CR for polymeric modifications include work the introduction of PEG chains onto the termini of a star shaped polymer,31 and its use to modify the termini of an aldehyde terminated PEG chain.32 However, there have been no prior reports of its application to PBAEs. Within this work, it was established as an ideal reaction to introduce wide functional diversity into a polymer library, via a rapid, easy, efficient and catalyst-free procedure. By developing an acid terminated PBAE it should prove possible to modify those same PBAE end groups with both aldehydes and isocyanides, opening up two libraries of chemical space to be explored. Neither class of molecule has previously been used for PBAE modification.Synthesis and characterisation of an acid terminated PBAE
To allow for subsequent functionalisation, a PBAE was first synthesised with carboxylic acid terminal groups, which would be amenable to functionalisation via the P3CR. A polymer backbone derived from 1,6-hexanedioldiacrylate (HDD) and 4-amino-1-butanol (4AB) was chosen as it has previously been shown to have effective RNA transfection capabilities.33–35 Initially, an acrylate terminated polymer was synthesised by established aza-Michael addition protocols,33,35 before subsequent end-capping with 3-mercaptopropanoic acid. The polycation was synthesised by dissolving 4AB (1 equiv.) and HDD (1.1 equiv.) in DMSO (0.3 mL per mmol of 4AB), which was heated to 90 °C for 24 hours. The 1:1.1 ratio of monomers was chosen to ensure that the polymer was terminated with acrylate groups for further functionalisation, and the high concentration was to ensure a high degree of polymerisation to maximise polymer length. To incorporate the desired terminal carboxylic acid functionalities, a subsequent thiol-Michael addition was conducted with 3-mercaptopropanoic acid. The reaction mix was cooled to room temperature, and 0.5 equivalents of 3-mercaptopropanoic acid was added before the reaction was stirred for a further 24 hours. Reaction completion was determined by NMR spectroscopy, which was used to ascertain the complete elimination of the acrylate peaks at 6.4–5.8 ppm.
In addition, two further polymers were synthesised for comparison purposes. An acrylate terminated polymer (pHDD-4AB) was synthesised as the simplest possible HDD/4AB derived PBAE and used as a comparator for an unfunctionalised polymer. pHDD-4AB was synthesised by the same protocol as outlined above, except once the initial reaction was cooled to room temperature, the product was immediately purified by precipitation from ice cold diethyl ether, as described above. Additionally, an amine terminated PBAE, end-capped with 4-amino-1-butanol (OH-(pHDD-4AB)-OH) was also synthesised following the above procedure, but in this instance, the 3-mercaptopropanoic acid was instead replaced with 0.2 equivalents of 4-amino-1-butanol. In this case 4-aminobutanol was chosen as the simplest possible amine end-cap to use with this PBAE backbone; as amine terminated PBAEs are generally found to outperform acrylate terminated polymers with regards to transfection capability and cytotoxicity, and the 4AB terminated polymer was deemed a useful comparison for biological testing.16 To facilitate direct and accurate comparison between the acrylate, amine and acid terminated polymers, all three polymers were obtained from the same reaction mix. Following 24 hours of stirring at 90 °C, the reaction mix was divided into three equal aliquots and each material was synthesised according to the procedures outlined above.
Characterisation of the polymers was achieved via NMR spectroscopy and SEC, with the results outlined in Fig. 1 below. Integration of signals for terminal groups within the NMR spectrum, compared against integration for the repeat unit was used to establish an average degree of polymerisation (DP) of 15 for each polymer, resulting in an Mn,NMR of 5000–5200 Da for each polymer (dependent on mass of the end groups). The mass obtained by SEC was significantly higher at over 10000 Da, but again was similar in each case, the divergence between NMR spectroscopy derived polymer mass and SEC derived polymer masses is likely to result from the use of PMMA standards for SEC calibration, these standards may not accurately match the behaviour of PBAEs within the SEC columns. The dispersity measured by SEC was lower than that expected for step growth polymerisation (2), but it is anticipated that this is a result of the purification by precipitation, which would have removed low mass oligomers and narrowed the overall mass distribution of the polymer. Results from the NMR spectroscopy and SEC analyses for each polymer were compared, showing that in each case, incorporating the acid group into the structure via this procedure did not detrimentally impact polymer length, or structure. As desired, the polymer structures differed only in the identity of the end groups. The other change of note was an unexpected peak within the 1H NMR spectrum for COOH-(pHDD-4AB)-COOH at 4.5 ppm. We attribute this to protonation of amines within the polymer chain, as a result of the large amount of acid used in the reaction.
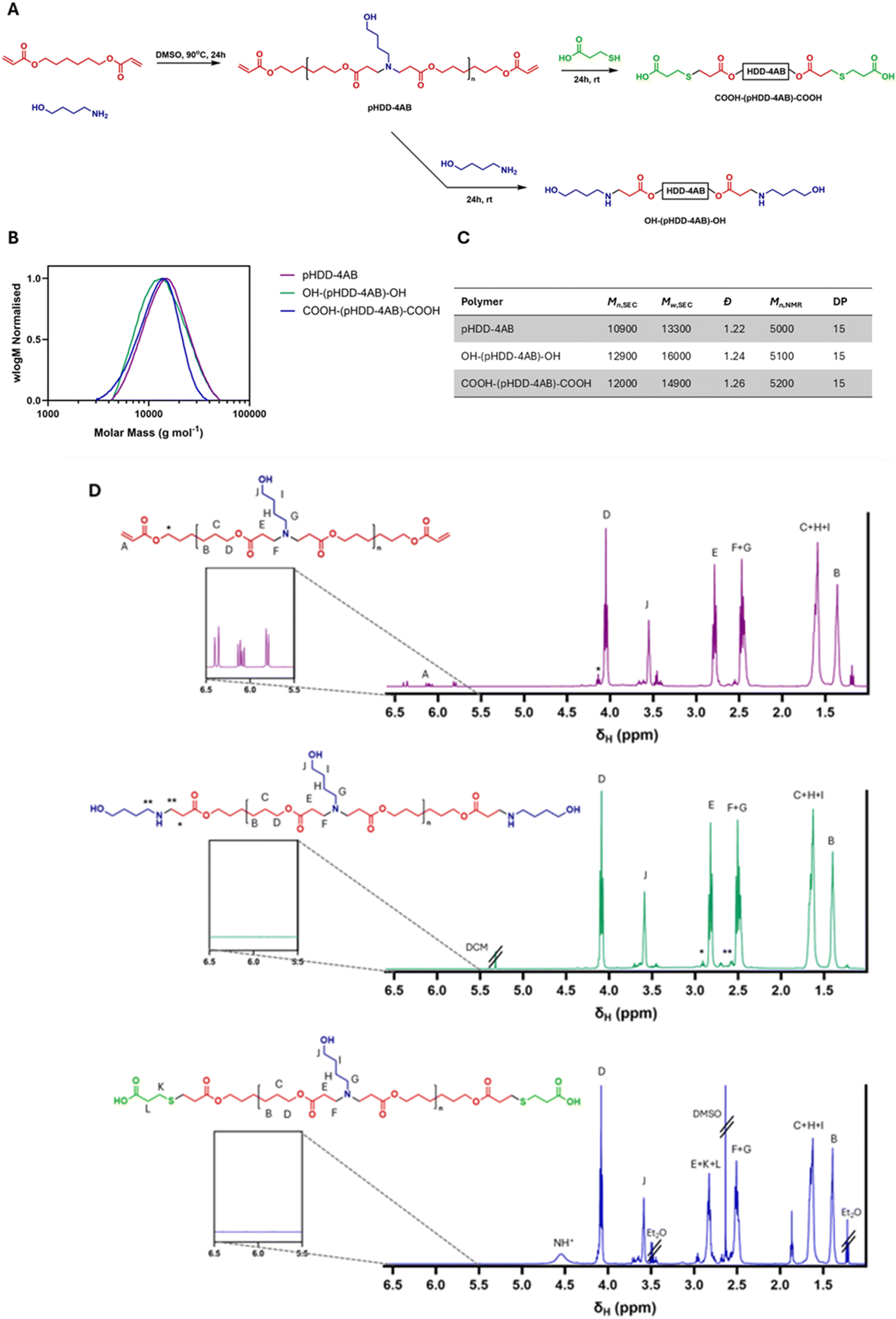 | ||
| Fig. 1 Synthesis and characterisation of PBAEs. (A) Synthetic scheme for pHDD-4AB and the derivatives OH-(pHDD-4AB)-OH and COOH-(pHDD-4AB)-COOH. (B) Molecular weight distributions of the three polymers, as measured by SEC established using 0.01 M LiBr in DMF at 50 °C as the eluent. (C) Molecular weights and dispersities of the polymers as calculated from DMF-SEC, alongside Mn and DP calculated by NMR. (D) 1H NMR (400 MHz, CDCl3) spectra for the three PBAEs. Cut-outs show an enhancement of the 5.5–6.5 ppm region to highlight the presence or absence of acrylate peaks. | ||
Modification of an acid terminated PBAE via the P3CR
Once the synthesis of an acid terminated PBAE was established, this polymer was then modified by the P3CR to introduce new end-capping structures. For an initial reaction, the aldehyde chosen was benzaldehyde (deemed Al1), as it would be easy to identify via1H NMR spectroscopy, with the peaks from the benzene ring being located far from any peaks from the pHDD-4AB backbone. The isocyanide of choice was tert-butyl isocyanide (termed I1), so chosen for its long-established use in successful P3CR syntheses.36Polymer COOH-(pHDD-4AB)-COOH was dissolved in DCM along with an excess of benzaldehyde and stirred at room temperature for 30 minutes. This premixing step was to allow full complexation of the acid and aldehyde components before addition of the isocyanide, to reduce competition with other reactions, minimising side products and maximising yield.37–39 After this, tert-butyl isocyanide was added and the reaction mix was allowed to stir for 24 hours. After this time the reaction was purified by repeated precipitation from diethyl ether, to isolate the polymer Al1/I1-(pHDD-4AB)-Al1/I1. Following this procedure, successful functionalisation of the polymer end groups was determined by both 1D 1H NMR spectroscopy and diffusion ordered NMR spectroscopy (DOSY). 1D 1H NMR spectroscopy showed the presence of peaks between 7.35 and 7.40 ppm, indicative of the aromatic group introduced from benzaldehyde, as well as a sharp singlet at 1.35 ppm, consistent with the tertiary butyl group introduced from the isocyanide. A small but important peak was also identified at 5.97 ppm, this peak is highly distinctive and is indicative of the CH group between the ester and amide groups formed by the P3CR. Further, the use of DOSY confirmed that the peaks associated with the newly introduced phenyl and tert-butyl groups correlated with the same diffusion coefficient as peaks on the main polymer chain, strongly suggesting that these motifs became covalently bound to the polymer as desired, rather than remaining free as small molecules in solution.
The resultant polymer Al1/I1-(pHDD-4AB)-Al1/I1 was also analysed by SEC, and compared against the starting sample of COOH-(pHDD-4AB)-COOH, the respective SEC derived molecular weight distributions are shown in Fig. 2 below. A slight shift in the traces can be observed, with the SEC calculated Mn increasing from 7700 g mol−1 for COOH-(pHDD-4AB)-COOH to 10200 g mol−1 for Al1/I1-(pHDD-4AB)-Al1/I1. A significant increase in chain length was not anticipated from the P3CR, as this reaction does not significantly impact chain length; the new groups incorporated into the polymer are only introduced onto the polymer termini and account for an increase in mass of just 380 Da. This disparity in SEC calculated Mn may result from differences in behaviour in the SEC columns between these two polymers. Alternatively, the increase may result from the purification process. Both COOH-(pHDD-4AB)-COOH and Al1/I1-(pHDD-4AB)-Al1/I1 were purified by precipitation, but as Al1/I1-(pHDD-4AB)-Al1/I1 was derived from COOH-(pHDD-4AB)-COOH it was subject to more rounds of precipitation overall. This could lead to more elimination of short oligomers from the polymer mix, leading to a slightly narrower molecular weight distribution and a greater average mass, which is reflected in the SEC curve.
 | ||
| Fig. 2 (A) 1H NMR spectroscopy (500 MHz, CDCl3-d1) of Al1/I1-(pHDD-4AB)-A1/I1. (B) DOSY (500 MHz, CDCl3-d1) for Al1/I1-(pHDD-4AB)-A1/I1. (C) Molecular weight distributions of Al1/I1-(pHDD-4AB)-A1/I1 and COOH-(pHDD-4AB)-COOH as measured via SEC, established using 0.01 M LiBr in DMF at 50 °C as the eluent. | ||
Following the successful synthesis and characterisation of Al1/I1-(pHDD-4AB)-Al1/I1, a number of other aldehydes were incorporated into the end-capping protocol to develop a small polymer library. In total a further 10 aldehydes (Al2-Al11, shown in Fig. 3 below) successfully reacted in the manner desired. In all cases, the reaction protocol was unchanged, with the exception of Al10 (2-imidazole carboxaldehyde), which was insoluble in DCM, and so necessitated the use of DMF as a solvent instead. In the synthesis of Al10/I1-(pHDD-4AB)-Al10/I1, purification was incomplete following precipitation from ice cold diethyl ether, and the polymer was further purified by dialysis against ultrapure water for 24 hours, with a 3.5 kDa SnakeSkin™ dialysis membrane. The aldehydes used were chosen to incorporate a wide range of functionalities and are generally reflective of the range of materials that are available commercially. Other functionalities could be possible, but may require additional synthetic preparation. In each case, polymers were characterised by NMR spectroscopy and SEC (full characterisation in the SI) (Table 1).
 | ||
| Fig. 3 General synthetic scheme for the incorporation of novel P3CR end-capping units into a PBAE and scope of materials used. | ||
| Polymer | M n (Da) | Đ | Mass recovery after P3CR | % P3CR conversionb |
|---|---|---|---|---|
| a Calculated by SEC using 0.01 M LiBr in DMF at 50 °C as the eluent. b Calculated by NMR. | ||||
| pHDD-4AB | 10900 |
1.22 | N/A | N/A |
| OH-(pHDD-4AB)-OH | 12900 |
1.24 | N/A | N/A |
| COOH-(pHDD-4AB)-COOH | 12000 |
1.26 | N/A | N/A |
| Al1/I1-(pHDD-4AB)-Al1/I1 | 10200 |
1.23 | 25% | 38% |
| Al2/I1-(pHDD-4AB)-Al2/I1 | 11800 |
1.27 | 59% | 48% |
| Al3/I1-(pHDD-4AB)-Al3/I1 | 11200 |
1.37 | 67% | 36% |
| Al4/I1-(pHDD-4AB)-Al4/I1 | 12700 |
1.45 | 64% | 80% |
| Al5/I1-(pHDD-4AB)-Al5/I1 | 8900 | 1.48 | 68% | 61% |
| Al6/I1-(pHDD-4AB)-Al6/I1 | 11100 |
1.23 | 53% | 35% |
| Al7/I1-(pHDD-4AB)-Al7/I1 | 12100 |
1.17 | 41% | 71% |
| Al8/I1-(pHDD-4AB)-Al8/I1 | 9000 | 1.65 | 73% | 95% |
| Al9/I1-(pHDD-4AB)-Al9/I1 | 7500 | 1.84 | 82% | 52% |
| Al10/I1-(pHDD-4AB)-Al10/I1 | 11600 |
1.32 | 25% | 24% |
| Al11/I1-(pHDD-4AB)-Al11/I1 | 16300 |
1.13 | 24% | 54% |
| Al3/I2-(pHDD-4AB)-Al3/I2 | 4700 | 1.43 | 26% | 55% |
| Al5/I2-(pHDD-4AB)-Al5/I2 | 6100 | 1.94 | 76% | 94% |
| Al6/I2-(pHDD-4AB)-Al6/I2 | 7900 | 1.76 | 70% | 88% |
| Al3/I3-(pHDD-4AB)-Al3/I3 | 3700 | 1.53 | 46% | 21% |
| Al5/I3-(pHDD-4AB)-Al5/I3 | 5200 | 1.80 | 78% | 97% |
| Al6/I3-(pHDD-4AB)-Al6/I3 | 5200 | 1.68 | 74% | 93% |
In addition to these materials, reactions were also attempted with a further six aldehydes: 2,4-dihydroxybenzaldehyde, 2,4-dimethoxybenzaldehyde, indole-3-carboxaldehyde, 4-(dimethylamino)benzaldehyde, 4-hydroxybenzaldehyde and pyrrole-2-carboxaldehyde. However, under the conditions used in this work, the attempted syntheses were unsuccessful, and following work up of the reaction mixes, the NMR spectra showed no deviation from the spectrum of the starting material COOH-(pHDD-4AB)-COOH, with no signs of any peaks for the aldehyde functional groups, or for the tert-butyl group.
Whilst the incorporation of aldehydes into the PBAE end-cap was the primary motivation for this work, the P3CR protocol also introduces an isocyanide into the end-cap at the same time. The isocyanide forms a significant proportion of the new end-cap and like the aldehyde, could have profound impacts on the polymer properties and transfection capabilities. With this in mind, two further isocyanides were introduced to the reaction to begin testing the possible scope of the synthetic procedure, and to investigate the relative importance of changing the aldehyde and isocyanide groups. In this instance, the isocyanides chosen were cyclohexyl isocyanide (I2) and n-butyl isocyanide (I3), both are relatively unfunctionalised, but are commercially available and have previously been shown to react successfully via a P3CR process. Each of these two isocyanides was incorporated into the same reaction protocol developed before, with the established COOH-(pHDD-4AB)-COOH backbone along with aldehydes Al3, Al5 and Al6, chosen to cover a range of the functionalities previously studied. Each reaction proceeded successfully, delivering 6 more polymers for the library of end-capped PBAEs. In future, a wider range of isocyanides could be utilised, providing an opportunity to incorporate further functionality into the polymer end group.
Formulation of the end-capped PBAEs with mRNA
There are many reports within the literature of PBAEs being used for RNA transfection purposes. For this purpose, the PBAE is complexed with RNA to form polyplex nanoparticles to act as vectors for the RNA payload, protecting them from degradation in the blood and facilitating transport across cell membranes.6,40,41 The physical properties of polyplex nanoparticles have a significant impact on their transfection capability, and so it was important to study whether changing the PBAE end group led to any significant changes in these properties. To study this, the polymers were formulated with PolyA as a model mRNA at three different nitrogen to phosphate (NP) ratios: 32, 64 and 128. On preparation of these nanoparticles, hydrodynamic diameter and zeta potential were then measured by dynamic light scattering (DLS). Each polymer was diluted to the desired concentration in 25 mM sodium acetate buffer (pH 5.5), chosen to ensure protonation of the amine groups; the polymer solution was then mixed with an equal volume of PolyA dissolved in the same buffer, and mixed gently by repeated pipetting. DLS measurements were conducted immediately following polyplex preparation. The results of the studies are shown below in Fig. 4. | ||
| Fig. 4 (A) Hydrodynamic diameters of polymer/PolyA polyplexes as measured by dynamic light scattering. (B) Zeta potential of polymer/PolyA polyplexes as measured by dynamic light scattering. Each bar is the mean of three readings, points indicate individual readings. Grey outline: PBAE with no P3CR end-cap. Solid outline: P3CR end-cap with I1, I2 or I3. (C) TEM images collected from a Tecnai T12 BioTWIN Transmission Electron Microscope, showing polyplexes formed at NP 64 between PolyA and pHDD-4AB derived PBAEs. Polyplexes are stained with uranyl acetate and appear black. (D) Percentage mRNA encapsulation efficiency of each polymer as quantified by RiboGreen assays. Each bar is the mean of three independent replicates, points indicate individual measurements. | ||
The acrylate and amine terminated polymers pHDD-4AB and OH-(pHDD-4AB)-OH were considered to be benchmarks for formulation testing of the end-capped PBAEs, as their physicochemical and transfection properties are well established. Under the conditions studied, both of these control polymers formed polyplexes with diameters of approximately 200 nm or below, and with high, positive zeta potentials. Polyplex size polydispersity (PDI, shown in Fig. S2) was generally low for these polymers, below 0.2 under most conditions, although notably this rose substantially at higher NP ratios for the alcohol terminated polymer. As anticipated, the introduction of terminal carboxylic acid end groups to this polymer structure both increased the hydrodynamic radius of the polyplexes, and reduced the zeta potential. This is likely to result from the negative charge of the carboxylate groups at pH 5.5, reducing the overall charge density on the polymer, in turn reducing the attraction to negatively charged PolyA, and reducing the overall charge within the resultant polyplexes.
Functionalisation of the end group via P3CR had even more profound impacts on polyplex size and charge, with significant differences driven by both the aldehyde group and the isocyanide. In general, the incorporation of benzaldehyde, 2-pyridine carboxaldehyde, 4-ethynylbenzaldehyde and 2-imidazole carboxaldehyde (Al1, Al2, Al6 and Al10) with tert-butyl isocyanide (I1) led to substantial increases in polyplex diameter across the NP ratios studied. The PBAEs terminated with 2,4-dinitrobenzaldehyde and dodecanal (Al9 and Al11) had diameters comparable to the acid capped polymer at NP 32 and 64, although smaller at NP 128. The remaining aldehydes (p-anisaldehyde, 2-nitrobenzaldehyde, 4-pyridine carboxaldehyde, 4-(trifluoromethyl)benzaldehyde and 4-nitrobenzaldehyde, Al3-Al5, Al7 and Al8), when introduced alongside tert-butyl isocyanide, led to a reduction in diameter compared to the acid capped PBAE, all producing polyplexes with diameters of approximately 200 nm or below at the NP ratios studied. These polyplexes were generally larger than those formed by OH-(pHDD-4AB)-OH at NP 32 and 64, although at NP 128, these modified polymers formed slightly smaller polyplexes than their 4-amino-1-butanol terminated counterpart. For the majority of these polyplexes, PDI remained relatively low (approximately 0.2 or below) although higher PDIs (around 0.25) were observed for the Al6 terminated polymer as well as the Al10 terminated polymer at NP 128.
Changing the isocyanide used had an even more significant impact on polyplex size. The polymers modified with cyclohexyl isocyanide and n-butyl isocyanide (I2 and I3) formed significantly larger polyplexes at all NP ratios than their tert-butyl modified counterparts. The p-anisaldehyde and 4-ethynylbenzaldehyde modified polymers formed exceptionally large polyplexes, which had diameters greater than 1000 nm at all NP ratios. Notably the p-anisaldehyde terminated polymers also exhibited a much higher PDI than the rest of the polyplex library. Whilst polyplexes from the tert-butyl/4-ethynylbenzaldehyde (Al6/I1) terminated PBAE were also relatively large, the tert-butyl/p-anisaldehyde (Al3/I1) capped PBAE had formed relatively small polyplexes, suggesting that the isocyanide structure plays an equally large role in determining polyplex properties. The polymers capped with 4-pyridine carboxaldehyde (Al5) formed smaller polyplexes than these others, but these were all substantially larger than the amine capped PBAE, the acid capped PBAE and also the tert-butyl counterparts. Only the cyclohexyl isocyanide/4-pyridine carboxaldehyde (Al5/I2) terminated polymer formed any polyplexes below 200 nm in diameter, and from the formulation conditions studied, this only occurred at NP 32. All three of the isocyanides used in the end-capping process introduced only unfunctionalised alkane groups into the amide component of the end-cap, and so it is likely that the increase in polyplex size on changing from I1 to I2 and I3 was driven by steric effects, rather than dipole interactions or hydrogen bonding. As all of the isocyanide groups are large, these end groups are all likely to impact how the polymers pack with both RNA and other polymers within the polyplexes.
Changes to zeta potential did not correlate with changes to diameter. Most polymers prepared with tert-butyl isocyanide had positive zeta potentials at all NP ratios, which were generally of a similar magnitude to the zeta potentials of pHDD-4AB, OH-(pHDD-4AB)-OH and COOH-(pHDD-4AB)-COOH polyplexes. Although notably the 2-nitrobenzaldehyde and dodecanal (Al4 and Al11) derived polymers resulted in polyplexes with the highest zeta potentials, exceeding those of OH-(pHDD-4AB)-OH at most NP ratios, while 4-(trifluoromethyl)benzaldehyde (Al7) terminated polymers led to slightly lower zeta-potentials, and the p-anisaldehyde and 4-nitrobenzaldehyde (Al3 and Al8) derived polymers resulted in the lowest values of all, with near zero values for most formulations.
Once again, changing the isocyanide component from tert-butyl isocyanide to cyclohexyl or n-butyl isocyanide had an even greater effect than any change in aldehyde. All six polymers capped with these isocyanides formed polyplexes with strongly negative zeta potentials at all NP ratios. It is possible that these polyplexes had different conformations to those formed from tert-butyl derived polymers, with negatively charged PolyA presented on the surface of the polyplexes. Like the changes in diameter, this could be driven by differences in sterics or solubility of the isocyanide group.
It is anticipated that the polymers forming the smallest polyplexes, and those with non-zero zeta potentials will have the greatest transfection capabilities.42–45 The measurements made here would predict the greatest transfection capability would be obtained from polymers end-capped with I1 and Al4, Al5 and Al8.
The images obtained by TEM were in accord with the data obtained by DLS measurements. All polyplexes appeared to be roughly circular in cross section, which indicates an overall spherical structure to the particles. TEM imaging of OH-(pHDD-4AB)-OH indicated that this polymer formed the smallest polyplexes, with diameters of typically 100 nm, which was in line with measurements by DLS.
Similarly, as measured by DLS, the polyplexes from the five end-capped polymers were all observed via TEM to be generally larger than those from the amine capped polymer. However, between these five polymers there were some discrepancies between the results obtained from DLS and those obtained from TEM. While DLS measured the smallest polyplexes as being formed by Al11/I1-(pHDD-4AB)-Al11/I1, under TEM imaging, the smallest particles observed were formed by Al2/I1-(pHDD-4AB)-Al2/I1. Al2/I1 terminated polymers generally formed polyplexes that appeared under 200 nm, whereas most polyplexes from the other four polymers were above this threshold. However, the throughput capacity of TEM is too low to study the entire sample and so these observations may not be representative of the complete set of polyplexes present within the formulation.
Encapsulation efficiency
A RiboGreen assay was used to assess quantitatively the capabilities of the end-capped polymers to encapsulate RNA, relative to their acrylate and amine terminated counterparts.46 Polyplexes were formulated as before at NP ratios of 32, 64 and 128, by mixing equal volumes of polymer solution and PolyA solution, before free PolyA was quantified by the RiboGreen reagent, and this value used to calculate the percentage of RNA encapsulated within the polyplex at each NP ratio.23 The results of this study are shown in Fig. 4D.The encapsulation efficiencies of pHDD-4AB and OH-(pHDD-4AB)-OH were high at over 95% across the NP ratios studied, which is broadly in line with expectations for PBAEs. On replacing the acrylate and 4-amino-1-butanol end-cap with a carboxylic acid group there was a significant fall in encapsulation efficiency; a reduction of approximately 40% at NP 32, and a reduction of 10% at the higher NP 128. This drop can be justified by an overall decrease in polymer charge density, as these carboxylate groups are likely to remain deprotonated, and so will introduce negative charges into the polymer structure.
The results of reacting this carboxylate group via the P3CR to introduce new end groups were much more varied and were strongly dependent on the identities of the structures introduced via this reaction.
For example, introduction of I1 (tertiary butyl isocyanide) along with either pyridine motif (Al2 and Al5), or the different nitro-benzaldehydes (Al4, Al8 and Al9) resulted in an encapsulation efficiency higher than that of the carboxylate terminated polymer at all NP ratios. At NP 64 and 128 the encapsulation efficiency for all these polymers was in excess of 90%, and was above 95% for many of the formulations. Incorporation of these end groups converted the carboxylate group to an ester, restoring the overall polymer charge of the original pHDD-4AB chain.
However, the same was not observed for all new end groups; introduction of Al1 (benzaldehyde), Al6 (4-ethynylbenzaldehyde) and Al11 (dodecanal) led to high encapsulation (over 90%) at NP 128, but at NP 32, the encapsulation efficiency observed was comparable or lower than that of COOH-(pHDD-4AB)-COOH (ranging between 13 and 55%). Introduction of Al3 (p-anisaldehyde), Al7 (4-(trifluoromethyl)benzaldehyde) and Al10 (2-imidazole carboxaldehyde) led to an even greater reduction in encapsulation efficiency, with limited encapsulation achieved across the range of NP ratios studied, in almost every case encapsulation was measured to be at least 10–20% lower than that of COOH-(pHDD-4AB)-COOH at the same NP ratio. An even more pronounced effect was observed with the polymers derived from different isocyanides; the I2 (cyclohexyl) and I3 (n-butyl) derived polymers led to a low encapsulation efficiency (below 50%) under all formulation conditions.
Polymer pKa titrations
To understand the impact of end-capping on buffering capacity, an acid–base titration was performed for each polymer between a pH range of 11.0 to 3.0.23 0.1 M NaCl was set as negative control, and PEI was used as a positive control.Buffering capacity was then calculated within the pH range 7.4 to 5.5, so chosen because it relates to endosomal acidification, and so is considered to be important for endo/lysosomal escape of polymeric RNA vectors.23,47,48 The buffering capacity was calculated relative to both the mass of polymer used and the moles of protonatable amine according to the equations below.
Calculation of buffering capacity of polymer between pH 7.4 to 5.5 normalised to mass of polymer:
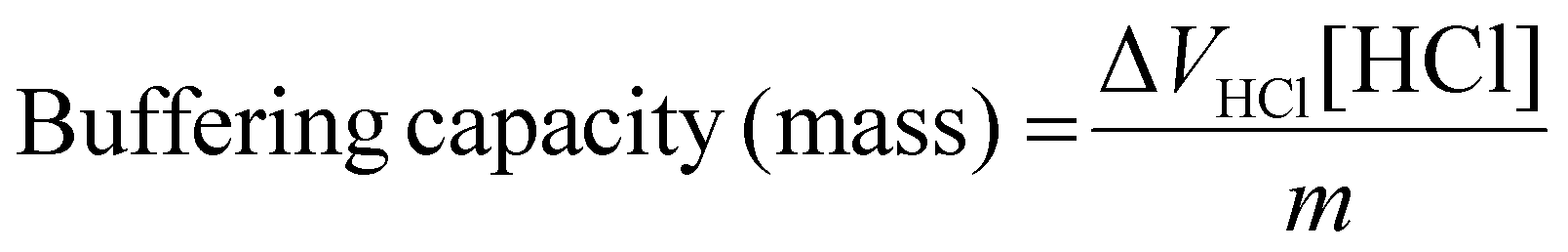 | (2) |
Calculation of buffering capacity of polymer between pH 7.4 to 5.5 normalised to moles of ionisable amines present:
 | (3) |
The results of these calculations are shown in Fig. 5 below. Full data are shown in Table S1 in the SI.
 | ||
| Fig. 5 Buffering capacities of the polymers calculated by titration against 0.1 M HCl, and normalised against both mass of polymer, and moles of protonatable amine. PEI is used as a control. For all P3CR terminated polymers, the nomenclature has been shortened to just the identity of the end-capping unit to ease readability. (A) Buffering capacity normalised against mass of polymer. (B) Buffering capacity normalised against moles of protonatable amine. | ||
Most of the P3CR end-capped polymers had broadly similar buffering capacities to the unfunctionalised, acrylate terminated pHDD-4AB, with capacities between 0.0025 and 0.0031 moles of H+ buffered per gram of polymer (equivalent to buffering 0.80 and 0.98 moles of H+ per mole of ionisable amine). This was anticipated as the amines in each polymer are primarily contained in the pHDD-4AB core that is shared by each polymer. When normalised by polymer mass, this value was substantially lower than that for PEI, owing to the far greater amine density that PEI has compared to PBAEs, however, when normalised by number of protonatable amines, many of the PBAEs showed a far greater buffering capacity within this range than PEI. This is likely the result of the neighbouring group effect granted by adjacent amine groups in the structure of PEI, which imparts a particularly broad buffering capacity relative to PBAEs.49
However, there were a number of polymers that deviated significantly from this trend. The Al9/I1 (2,4-dinitrobenzaldehyde/tert-butyl isocyanide) capped and Al11/I1 (dodecanal/tert-butyl isocyanide capped) polymers showed slightly lower buffering capacities (approximately 55% that of pHDD-4AB). Whereas Al1/I1-(pHDD-4AB)-Al1/I1 (benzaldehyde/tert-butyl isocyanide terminated), had a slightly higher buffering capacity. Additionally, all of the polymers capped using cyclohexyl isocyanide had significantly lower buffering capacities (approximately 1 third of the capacity of pHDD-4AB), and the n-butyl isocyanide capped polymers had generally lower values, but were more varied depending on the aldehyde component, with the p-anisaldehyde and 4-pyridine carboxaldehyde derived polymers only having slightly lower buffering capacities, whereas the 4-ethynyl benzaldehyde variant had a substantially lower capacity.
The cause of these difference is unknown, but as the polymers only differ in the identity of the end groups, they may be driven by end group solubility. Both the C12 tail from dodecanal (Al11) and the cyclohexyl ring from cyclohexyl isocyanide (I2) reduce the aqueous solubility of the polymer, which may in turn impact polymer conformation or aggregation within solution. A number of behaviours could then impact the local environment of a number of the amine groups, this could include effects such as steric crowding by large hydrophobic domains, or self-assembly of small clusters with hydrophobic core regions. These different local environments could hinder the protonation of any amines contained within these regions.
mRNA transfection
The toxicity and transfection capabilities of the PBAEs were then studied in HEK293T cell lines, chosen as they are well studied in pilot transfections with many RNAs, and subsequently in MDA-MB-231 cells, which are Triple Negative Breast Cancer (TNBC) cells and a candidate for RNA therapies. A firefly luciferase (fLuc) encoding mRNA was chosen as a model RNA for transfection, and was complexed with the Passerini end-capped PBAEs at NP 64, along with OH-(pHDD-4AB)-OH as a model amine end-capped PBAE control, as well as the commercial transfection agent Lipofectamine MessengerMAX™ as a positive control and free mRNA in solution as a negative control.Transfection efficiency was assessed via a One-Glo luciferase assay, and standardised against luciferase expression from transfection by Lipofectamine MessengerMAX™. At the same time, cell metabolic activity following transfection was used as a measure of overall cell viability, measured via a PrestoBlue assay, which was standardised against cells treated with buffer and cells killed with Triton X-100 as positive and negative controls (Fig. 6).
 | ||
| Fig. 6 Metabolic activity and transfection efficiency of PBAE/mRNA polyplexes in HEK293T cells (A and B) and in MDA-MB-231 cells (C and D). Effect of PBAE polyplexes formulated with firefly luciferase encoding mRNA at polymer/mRNA NP ratio of 64 on transfection efficiency (A and C) and metabolic activity (B and D). Cells were treated with polyplexes containing mRNA at a concentration of 250 ng mL−1 (50 ng per well) in serum-free OptiMEM. In vitro transfection efficiency was analysed after 24 h post-transfection using the Promega ONE-GLO luciferase assay and compared against Lipofectamine Messenger MAX™ (positive control) and naked mRNA (negative control). Transfection efficiency is expressed as a proportion of the luminescence achieved by transfection with the positive control. Metabolic activity as calculated from PrestoBlue assay was compared against Triton X (positive control) and untreated cells (negative control), and calculated by normalising metabolic activity to untreated cells. Bars represent the mean ± SD of three biologically independent replicates, points represent the average of three technical repeats. *p < 0.05, **p < 0.01 etc., ***p < 0.001, ****p < 0.0001. | ||
In HEK 293T cells, luciferase expression obtained by the control polymer OH-(pHDD-4AB)-OH was approximately 50 times lower than that achieved by Lipofectamine, but introducing a Passerini derived end-cap onto the same polymer backbone had profound effects on polymer transfection capability. Many of the novel Passerini end-capped polymers showed a substantial increase in luciferase expression relative to the amine terminated control, with five end-capped polymers showing a statistically significant increase. Protein expression from each of these polymers was comparable to the level obtained by treatment with Lipofectamine. However, four of the modified polymers also demonstrated a significant reduction in transfection capability relative to the amine terminated comparison.
The changes in transfection capability were driven by the identity of both the aldehyde and the isocyanide involved in the end-capping reaction. A particularly large increase in transfection capacity (approximately 70 times greater than OH-(pHDD-4AB)-OH) was obtained by the introduction of an imidazole motif (Al10), this was in spite of the large particle size for polyplexes with this PBAE. Previous studies have observed that the presence of imidazoles appears to be associated with increased endosomal escape of nucleic acid delivery systems, owing to the buffering capacity and pKa range of the imidazole motif.50,51 High transfection efficiency was also observed for the pyridine containing Al5/I1 and Al2/I1 end groups (respectively approximately 80 and 50 times greater than the OH-(pHDD-4AB)-OH control polymer). The expected pKa of these end groups is also low (approximately 5.25), and so it is possible that they may behave similarly to imidazole, which is expected to protonate as the endosome acidifies. However, we were unable to validate fully this assumption experimentally.
The incorporation of dodecanal via the Al11/I1 cap also led to a significant increase in transfection capability, with protein expression over 50 times greater than achieved by the amine capped control; high transfection efficiency has previously been reported for PBAEs that incorporate a mixture of alcohol groups and long alkyl side chains.52 A similar effect may also drive the high transfection capability observed for the benzyl and 4-ethynylbenzyl incorporating end groups (Al1/I1 and Al6/I1), with the increased hydrophobicity from these groups stabilising the polyplexes until they reach the cytoplasm.52 However, many of the polymers with hydrophobic end groups failed to increase transfection capability beyond that observed for the controls. This may result from the differing physical properties of the polyplex nanoparticles. Alternatively, these differences in transfection capability could be driven by differences in membrane interaction of the polymer end groups, with certain end-caps driving nanoparticle internalisation owing to interactions with membrane functionalities and receptors.
Notably however, on changing the isocyanide group from a tertiary butyl motif to either a cyclohexyl or n-butyl group, many of these trends changed. Whereas the p-anisaldehyde group led to nearly 10-fold lower transfection than the amine capped polymer when paired with a tertiary butyl group, much higher transfection capability was achieved with both I2 and I3. Transfection with both the I2 and I3 polymers was 50 times greater than the amine capped control. Whilst this is somewhat at odds with expectations given the large size of the nanoparticles, it should be noted that both the Al3/I2 and Al3/I3 capped polymers resulted in particles with a particularly high size polydispersity. Therefore, despite the high average size, there may be large numbers of small particles present that are able to effectively transfect the cells. By contrast, both the 4-pyridine and 4-ethynylbenzyl motifs saw a significant decrease in transfection capability when the isocyanide group was changed. Given the numerous barriers to successful intracellular RNA delivery, there may be different, competing effects driven by different combinations of the aldehyde and isocyanide groups incorporated by the P3CR, for example, enhanced endosomal escape could be mitigated by reduced internalisation of the polyplex, or poor release of the RNA.
Across the range of polymers studied, toxicity was generally low, with metabolic activity for most of the end-capped polymers comparable to both OH-(pHDD-4AB)-OH and Lipofectamine. This is in line with previous studies of PBAEs and they have remained popular for transfection because of this low cytotoxicity.11 However, both the Al1/I1 and Al11/I1 caps led to a slight increase in toxicity, with average cell survival dropping by 10–15%, however these differences were non-significant. Both of these end-caps are relatively hydrophobic, but that is not unique to these motifs within this study. Six polymers with the I2 and I3 derived end-caps were also unusual, as incorporation of these motifs generally led to very high metabolic activity, which in several cases exceeded 100%. This may be a sign that the polymers were inducing metabolic stress in the cells, leading to a spike in metabolic activity in an attempt to survive.53,54
Within MDA-MB-231 cells, overall transfection capability of all PBAEs relative to Lipofectamine showed approximately a 10–20 fold reduction compared to transfection in HEK293T cells. This was not entirely unexpected given that MDA-MB-231 cells are known to be harder to transfect. However, the general trends amongst the polymers were preserved over the change in cell line, and most of the end-capped polymers still outperformed OH-(pHDD-4AB)-OH. There were some differences in transfection in the MDA-MB-231 cells, with the Al5/I1, Al7/I1 and Al11/I1 capped polymers all showing a greater reduction in transfection capability than the other PBAEs. Whereas in HEK293T cells, these three polymers all induced comparable luciferase expression to Lipofectamine, in MDA-MB-231 cells, relative expression was approximately 100 times lower. By contrast, the Al3/I1, Al9/I1, Al10/I1 and Al5/I3 capped polymers were among the least impacted by the change in cell line, showing just a 10-fold reduction in relative luciferase expression between the two cell lines. Although overall the levels of protein expression induced by the Al3/I1 and Al9/I1 terminated polymers were very low.
When studying changes in isocyanide groups, the overall trends in transfection efficiency were similar across both cell lines. On changing from HEK293T to MDA-MB-231 cells, all the I2 and I3 capped polymers displayed a roughly 10- to 20-fold reduction in relative protein expression, which was in line with the average for the I1 terminated polymers. Expression induced by Al3/I2 and Al3/I3 capped polymers was still significantly greater than that induced by Al3/I1 capped polymers (roughly 100 times greater). Among the Al5 caps, expression from the I1 terminated polymer was 5–20 times higher than from the I2 and I3 variants, and with Al6, the protein expression associated with the I1 terminated polymer was over 2000 times greater than with the I2 or I3 counterparts. The Al6/I2 and Al6/I3 capped polymers induced a very low level of transfection, it appears that the combination of the ethynylbenzene structure and the n-butyl and cyclohexyl side groups into the PBAE end-cap severely inhibits transfection.
Metabolic activity studies also led to similar results in MDA-MB-231 cells as had been observed in HEK293T cells, and none of the polymers studied led to a significant reduction in metabolic activity relative to either OH-(pHDD-4AB)-OH, or lipofectamine. Generally, most PBAEs were non-toxic, as is typically reported in previous studies. However, in this case both the Al4/I1 and Al11/I1 terminated polymers resulted in a metabolic activity below 80%. Although this was also observed in HEK293T cells for the Al11/I1 polymer, this was not the case for Al4/I1which was observed as being non-toxic in HEK293T cells. There is some evidence that MDA-MB-231 cells exhibit increased membrane fluidity relative to non-cancerous cell lines55 and so any polymers that interact with the cellular membrane may show different behaviours across the different cell-lines. If the 2-nitrobenzene motif is involved in any membrane interactions, then this difference in membrane behaviour may explain the toxicity difference between the cell lines. A large number of polymers also caused a very high metabolic activity, in excess of 100%, which again may result from inducing a stressed state in the cells, leading to upregulated metabolism.
Microscopy studies of cellular transfection
Having observed the differences in transfection driven by changes in end groups, further investigations were undertaken to better understand the mechanisms behind these differences. To gather information on the kinetics of transfection cells were treated with a green fluorescent protein (GFP) encoding mRNA and imaged at regular intervals. For this work, a modified HEK293T cell line that expresses mCherry-tagged galectin-9 (Gal-9) was used for the transfection procedure. Previous work has shown Gal-9 to be a marker for endosomal damage during transfection by LNPs, manifest by recruitment of Gal-9 to endosomal membranes. In this cell line, localised endosomal damage can be visualised by fluorescence microscopy, which can indicate the accumulation of the mCherry-tagged Gal-9 molecules.56Al6/I1-(pHDD-4AB)-Al6/I1, Al10/I1-(pHDD-4AB)-Al10/I1 and Al11/I1-(pHDD-4AB)-Al11/I1 were selected for the study, along with OH-(pHDD-4AB)-OH as a model PBAE for comparison. The polymers were chosen for their high transfection capability with fLuc mRNA, and were formulated with an mRNA coding for green fluorescent protein (GFP), at NP ratio 64. Cells were treated following the same protocol as before, and then imaged at regular time points to study endosomal damage and GFP expression. The outcome is shown in Fig. 7 below.
 | ||
| Fig. 7 Timelapse imaging of cells expressing mCherry-Gal9 when treated with differing polyplexes of GFP encoding mRNA, with additional nuclear staining achieved with Hoechst 33342. Cells imaged between 2 and 23 hours after dosing. Lipofectamine MessengerMax™ used as a positive control. Scale bar is 100 μm. Brightness has been increased by 20% to enhance visualisation. | ||
Following treatment with Lipofectamine, GFP expression was observed across a much greater number of cells than with treatment by any of the polymers trialled, a result that was somewhat at odds with observations from fLuc mRNA transfections, which showed comparable outcomes between treatment with Lipofectamine and the three end-capped polymers Al6/I1-(pHDD-4AB)-Al6/I1, Al10/I1-(pHDD-4AB)-Al10/I1 and Al11/I1-(pHDD-4AB)-Al11/I1. Some differences in expression are to be expected, as the GFP and fLuc RNAs may not be translated with equal efficiency, and additionally, the fLuc assay measures protein expression at the end of the experiment, whereas microscopy studies image GFP expression continuously.
Additionally, it was also observed that GFP expression occurred much earlier in Lipofectamine treated cells than in cells treated with the various PBAEs. Whereas Lipofectamine treated cells expressed GFP just two hours after treatment, Al10/I1-(pHDD-4AB)-Al10/I1 and Al11/I1-(pHDD-4AB)-Al11/I1 treated cells first displayed clear GFP expression after approximately 4 hours, and OH-(pHDD-4AB)-OH and Al6/I1-(pHDD-4AB)-Al6/I1 treated cells did not display significant GFP expression until 6–10 hours after treatment. All three Passerini capped polymers had outperformed OH-(pHDD-4AB)-OH in tests of fLuc mRNA transfection, so it was again unexpected that the Al6/I1 terminated polymer only performed comparably to OH-(pHDD-4AB)-OH with GFP mRNA.
However, it was immediately apparent that as well as the diffuse expression of GFP observed throughout the cells, a second source of emission was present in images of the Passerini terminated polymers. On studying the images, bright puncta can be observed in the GFP channel for cells treated by all of the polymers capped with the P3CR, but not by OH-(pHDD-4AB)-OH or Lipofectamine.
We have previously reported observations of non-traditional intrinsic luminescence resulting from the cellular internalisation of polymers derived directly from Passerini 3-component polymerisation, which resulted in similar bright puncta.57 On closer inspection, colocalised puncta can be identified across the different channels used here (shown in the SI, Fig. S5), suggesting a similar broad spectrum luminescence. The previously reported behaviour was postulated to be linked to a form of aggregation induced emission caused by macromolecular crowding within the polyplexes and exacerbated by cellular internalisation. The behaviour of these PBAEs is somewhat similar, and is likely linked to the ester/amide motif introduced by the P3CR, which has some structural similarity to common motifs in other polymer systems that have led to non-traditional intrinsic luminescence.58,59 Notably the luminescence is not limited to those polymers with aromatic end-capping units (Al6 and Al10) as it was also observed in cells treated with the Al11 (dodecanal) capped polymer.
Whilst this innate luminescence property of the polymers could be useful for tracking intracellular trafficking of polyplexes, in this instance, meaningful conclusions were limited by the use of the mCherry-Gal9 expressing cell line, making it difficult to distinguish puncta caused by Gal9 accumulation from those linked to intrinsic polymeric luminescence.
Conclusions
In this work, we have developed a novel method that utilises the P3CR to introduce end-capping units onto PBAEs. 11 aldehydes and three isocyanides have been successfully incorporated onto a pHDD-4AB backbone, and the development of this methodology opens up a wide untapped library of materials that can be used to modify PBAEs, to introduce new diversity and resultant changes in physical and biological properties. The nature of the methodology is such that it should prove compatible with a with range of different PBAE architectures, as well as a wide variety of end-capping substrates. This provides a route for the synthesis and investigation of a wide range of new materials, systematically varying the polymer chain, the aldehyde and the isocyanide used via an approach similar to combinatorial chemistry could lead to significant discoveries on the relative impacts of each component. As part of such investigations, it would be useful to study hyper-branched PBAEs, as well as linear variants. A similar technology could also be used to link aldehyde terminated polymers to acid terminated PBAEs to develop a range of copolymers that may not be accessible via other chemistries. Further work could also exploit the same acid-terminated PBAEs with the U4CR as a means to introduce three-functional groups simultaneously into each end-group.Changing the terminal group of the PBAE had a significant impact on nanoparticle properties and RNA complexation capability, and the majority of the resultant polyplexes were well tolerated in cells, and proved potent transfection agents relative to the amine terminated PBAE. Many polymers were found to delivery mRNA with a similar efficiency to Lipofectamine. Future investigations of biodistribution and transfection capabilities in vivo would be useful for the further development of this technology.
Initial microscopy studies suggest that these Passerini capped polymers may possess intrinsic non-traditional luminescence properties on aggregation. This property could provide a useful tool in the future to track nanoparticle delivery within cells without the need to introduce additional fluorescent motifs.
Author contributions
Lewis O'Shaughnessy: conceptualisation, methodology, validation, formal analysis, investigation, visualisation and writing – original draft. Rahman Khosravi: investigation and methodology. James Robins: investigation, methodology and visualisation. Mariarosa Mazza: conceptualisation, supervision; writing – review& editing; funding acquisition and resources. Akosua Anane-Adjei: conceptualisation, supervision, writing – review & editing, funding acquisition and resources. Naoto Hori: conceptualisation, supervision and writing – review & editing. Pratik Gurnani: conceptualisation, supervision and writing – review & editing. Cameron Alexander: conceptualisation, supervision; writing – review& editing; funding acquisition and resources.Conflicts of interest
The authors declare no conflict of interest.Data availability
All data in this paper are included in the Supplementary information (SI), covering polymer characterisation, TEM, Dynamic Light Scattering, polymer pH titration curves, calculated buffering capacity, and microscopy studies of cellular transfection. See DOI: https://doi.org/10.1039/d5lp00251f.Other relevant data are available on request via the University of Nottingham, cameron.alexander@nottingham.ac.uk.
Acknowledgements
We thank UKRI/EPSRC for funding [grant EP/S023054/1]. We also thank Esme Ireson and Paul Cooling for expert technical support. The Nanoscale & Macroscale Research Centre (NMRC) is acknowledged for providing the facilities for TEM and related analysis, and we thank the School of Life Sciences imaging facility (SLIM) and staff for their contribution to this publication.References
- J. Witten, Y. Hu, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2307798120 CrossRef CAS PubMed.
- X. Xie, T. Yu, X. Li, N. Zhang, L. J. Foster, C. Peng, W. Huang and G. He, Signal Transduction Targeted Ther., 2023, 8, 335 CrossRef PubMed.
- C. M. Crews, Chem. Biol., 2010, 17, 551–555 CrossRef CAS PubMed.
- S. Chatterjee, E. Kon, P. Sharma and D. Peer, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2307800120 CrossRef CAS PubMed.
- S. Berger, U. Lächelt and E. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2307799120 CrossRef CAS PubMed.
- S. F. Dowdy, Nat. Biotechnol., 2017, 35, 222–229 CrossRef CAS PubMed.
- P. J. White, F. Anastasopoulos, C. W. Pouton and B. J. Boyd, Expert Rev. Mol. Med., 2009, 11, e10 CrossRef PubMed.
- D. Li, C. Liu, Y. Li, R. Tenchov, J. M. Sasso, D. Zhang, D. Li, L. Zou, X. Wang and Q. Zhou, ACS Pharmacol. Transl. Sci., 2023, 6, 943–969 CrossRef CAS PubMed.
- S. Taranejoo, J. Liu, P. Verma and K. Hourigan, J. Appl. Polym. Sci., 2015, 132, 42096 CrossRef.
- A. K. Blakney, P. F. McKay, K. Hu, K. Samnuan, N. Jain, A. Brown, A. Thomas, P. Rogers, K. Polra, H. Sallah, J. Yeow, Y. Zhu, M. M. Stevens, A. Geall and R. J. Shattock, J. Controlled Release, 2021, 338, 201–210 CrossRef CAS PubMed.
- S. Iqbal, Y. Qu, Z. Dong, J. Zhao, A. R. Khan, S. Rehman and Z. Zhao, Eur. Polym. J., 2020, 141, 110097 CrossRef CAS.
- H. L. Kim, G. Saravanakumar, S. Lee, S. Jang, S. Kang, M. Park, S. Sobha, S.-H. Park, S.-M. Kim, J.-A. Lee, E. Shin, Y.-j. Kim, H.-S. Jeong, D. Kim and W. J. Kim, Biomaterials, 2025, 314, 122896 CrossRef CAS PubMed.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS.
- D. G. Anderson, A. Akinc, N. Hossain and R. Langer, Mol. Ther., 2005, 11, 426–434 CrossRef CAS PubMed.
- H. Yong, L. Lin, Z. Li, R. Guo, C. Wang, S. Liu and D. Zhou, Nano Lett., 2024, 24, 9368–9376 CrossRef CAS PubMed.
- J. J. Green, G. T. Zugates, N. C. Tedford, Y. H. Huang, L. G. Griffith, D. A. Lauffenburger, J. A. Sawicki, R. Langer and D. G. Anderson, Adv. Mater., 2007, 19, 2836–2842 CrossRef CAS.
- J. C. Sunshine, D. Y. Peng and J. J. Green, Mol. Pharm., 2012, 9, 3375–3383 CrossRef CAS PubMed.
- C. Li, S. Y. Tzeng, L. E. Tellier and J. J. Green, ACS Appl. Mater. Interfaces, 2013, 5, 5947–5953 CrossRef CAS PubMed.
- B. Chen, Q. Ren, P. Jiang, Q. Wu, Q. Shuai and Y. Yan, Macromol. Biosci., 2024, 2400168 CrossRef CAS PubMed.
- P. Dosta, N. Segovia, A. Cascante, V. Ramos and S. Borrós, Acta Biomater., 2015, 20, 82–93 CrossRef CAS PubMed.
- N. Segovia, P. Dosta, A. Cascante, V. Ramos and S. Borrós, Acta Biomater., 2014, 10, 2147–2158 CrossRef CAS PubMed.
- J. Kim, Y. Kang, S. Y. Tzeng and J. J. Green, Acta Biomater., 2016, 41, 293–301 CrossRef CAS PubMed.
- H. Bayraktutan, R. J. Kopiasz, A. Elsherbeny, M. Martinez Espuga, N. Gumus, U. C. Oz, K. Polra, P. F. McKay, R. J. Shattock, P. Ordóñez-Morán, A. Mata, C. Alexander and P. Gurnani, Polym. Chem., 2024, 15, 1862–1876 RSC.
- I. Jesin and G. C. Nandi, Eur. J. Org. Chem., 2019, 2019, 2704–2720 CrossRef CAS.
- M. Arend, B. Westermann and N. Risch, Angew. Chem., Int. Ed., 1998, 37, 1044–1070 CrossRef PubMed.
- S. Kumar, A. Arora, S. Kumar, R. Kumar, J. Maity and B. K. Singh, Eur. Polym. J., 2023, 190, 112004 CrossRef CAS.
- R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46–48 CrossRef CAS PubMed.
- M. Passerini, Gazz. Chim. Ital., 1921, 51, 181–189 CAS.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- R. Kakuchi and Y. Okura, Polym. J., 2020, 52, 1057–1066 CrossRef CAS.
- S. Oelmann, A. Travanut, D. Barther, M. Romero, S. M. Howdle, C. Alexander and M. A. R. Meier, Biomacromolecules, 2019, 20, 90–101 CrossRef CAS PubMed.
- L. Li, X.-W. Kan, X.-X. Deng, C.-C. Song, F.-S. Du and Z.-C. Li, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 865–873 CrossRef CAS.
- N. K. Dastgerdi, N. Gumus, H. Bayraktutan, D. Jackson, K. Polra, P. F. McKay, F. Atyabi, R. Dinarvand, R. J. Shattock, L. Martinez-Pomares, P. Gurnani and C. Alexander, Nanoscale Adv., 2024, 6, 1409–1422 RSC.
- H. Lopez-Bertoni, K. L. Kozielski, Y. Rui, B. Lal, H. Vaughan, D. R. Wilson, N. Mihelson, C. G. Eberhart, J. Laterra and J. J. Green, Nano Lett., 2018, 18, 4086–4094 CrossRef CAS PubMed.
- N. Karimi, K. Mansouri, M. Soleiman-Beigi and A. Fattahi, Adv. Pharm. Bull., 2020, 10, 221–232 CrossRef CAS PubMed.
- M. Zhao, N. Liu, R.-H. Zhao, P.-F. Zhang, S.-N. Li, Y. Yue and K.-L. Deng, ACS Appl. Bio Mater., 2019, 2, 1714–1723 CrossRef CAS PubMed.
- B. A. D. Neto, M. N. Eberlin and J. Sherwood, Eur. J. Org. Chem., 2022, 2022, e202200172 CrossRef CAS.
- S. Maeda, S. Komagawa, M. Uchiyama and K. Morokuma, Angew. Chem., Int. Ed., 2011, 50, 644–649 CrossRef CAS PubMed.
- R. Ramozzi and K. Morokuma, J. Org. Chem., 2015, 80, 5652–5657 CrossRef CAS PubMed.
- R. Bholakant, H. Qian, J. Zhang, X. Huang, D. Huang, J. Feijen, Y. Zhong and W. Chen, Biomacromolecules, 2020, 21, 2966–2982 CrossRef CAS PubMed.
- X. Wang, Z. Zhang and N. Hadjichristidis, Prog. Polym. Sci., 2023, 136, 101634 CrossRef CAS.
- N. Bono, B. Coloma Smith, F. Moreschi, A. Redaelli, A. Gautieri and G. Candiani, Nanoscale, 2021, 13, 8333–8342 RSC.
- D. Pezzoli and G. Candiani, J. Nanopart. Res., 2013, 15, 1523 CrossRef.
- G. Candiani, D. Pezzoli, M. Cabras, S. Ristori, C. Pellegrini, A. Kajaste-Rudnitski, E. Vicenzi, C. Sala and M. Zanda, J. Gene Med., 2008, 10, 637–645 CrossRef CAS PubMed.
- F. Sakurai, R. Inoue, Y. Nishino, A. Okuda, O. Matsumoto, T. Taga, F. Yamashita, Y. Takakura and M. Hashida, J. Controlled Release, 2000, 66, 255–269 CrossRef CAS PubMed.
- L. J. Jones, S. T. Yue, C.-Y. Cheung and V. L. Singer, Anal. Biochem., 1998, 265, 368–374 CrossRef CAS PubMed.
- A. Alazzo, N. Gumus, P. Gurnani, S. Stolnik, R. Rahman, K. Spriggs and C. Alexander, J. Mater. Chem. B, 2022, 10, 236–246 RSC.
- Y. B. Hu, E. B. Dammer, R. J. Ren and G. Wang, Transl. Neurodegener., 2015, 4, 18 CrossRef PubMed.
- C. E. Gallops, C. Yu, J. D. Ziebarth and Y. Wang, ACS Omega, 2019, 4, 7255–7264 CrossRef CAS.
- N. P. Truong, W. Gu, I. Prasadam, Z. Jia, R. Crawford, Y. Xiao and M. J. Monteiro, Nat. Commun., 2013, 4, 1902 CrossRef PubMed.
- M. Gillard, Z. Jia, J. J. C. Hou, M. Song, P. P. Gray, T. P. Munro and M. J. Monteiro, Biomacromolecules, 2014, 15, 3569–3576 CrossRef CAS PubMed.
- A. A. Eltoukhy, D. Chen, C. A. Alabi, R. Langer and D. G. Anderson, Adv. Mater., 2013, 25, 1487–1493 CrossRef CAS PubMed.
- S. Fulda, A. M. Gorman, O. Hori and A. Samali, Int. J. Cell Biol., 2010, 2010, 214074 Search PubMed.
- R. J. Cavanagh, P. A. Smith and S. Stolnik, Mol. Pharm., 2019, 16, 618–631 CrossRef CAS PubMed.
- C. Angelucci, G. Maulucci, G. Lama, G. Proietti, A. Colabianchi, M. Papi, A. Maiorana, M. De Spirito, A. Micera, O. B. Balzamino, A. Di Leone, R. Masetti and G. Sica, PLoS One, 2012, 7, e50804 CrossRef CAS PubMed.
- M. J. Munson, G. O'Driscoll, A. M. Silva, E. Lázaro-Ibáñez, A. Gallud, J. T. Wilson, A. Collén, E. K. Esbjörner and A. Sabirsh, Commun. Biol., 2021, 4, 211 CrossRef CAS PubMed.
- L. O'Shaughnessy, A. Anane-Adjei, M. Mazza, N. Hori, P. Gurnani and C. Alexander, Polym. Chem., 2025, 16, 538–548 RSC.
- A. Zia, J. R. Finnegan, J. P. Morrow, W. Yin, J. J. Jasieniak, E. Pentzer, S. Thickett, T. P. Davis and K. Kempe, Biomacromolecules, 2021, 22, 4794–4804 CrossRef CAS PubMed.
- A. Monti, C. Bruckmann, F. Blasi, M. Ruvo, L. Vitagliano and N. Doti, Chem. Commun., 2021, 57, 3720–3723 RSC.
| This journal is © The Royal Society of Chemistry 2026 |