Pd@Ce metal organic framework for dual reduction of nitrophenols and chromium(VI)
Received
18th March 2026
, Accepted 13th April 2026
First published on 15th April 2026
Abstract
In view of the promising photocatalytic activities of Ce-based materials, herein a cerium terephthalate metal–organic framework (Ce–BDC) has been decorated with Pd NPs via a double-solvent method and the characteristic features established through PXRD, FTIR, FESEM, and BET surface area analysis. The excellent redox activity, electron transfer and unique photo-harvesting ability of the material highlighted it as an active member in the arena of photocatalyst materials. Interestingly these features were demonstrated through application in the degradation of environmental pollutants, such as potassium dichromate ions (Cr(VI)) and 4-nitrophenol (4-NP). In particular, its photo-responsive catalytic activity was evaluated in aqueous medium at room temperature for Cr(VI) and 4-NP removal. Pd@Ce–BDC achieved ∼94% reduction of 4-NP to 4-aminophenol within 10 min at room temperature (k = 0.13 min−1), while ∼95% of Cr(VI) was reduced to Cr(III) within 20 min under sunlight irradiation (k = 0.206 min−1). These findings underscore its promise as a highly efficient photocatalyst for environmental remediation, where the interplay of framework design and redox-active centers governs performance.
Introduction
Water, the foundation of life on Earth, is essential for all living beings. However, rapid population growth and industrialization have led to increasing problems of water scarcity and contamination worldwide. Noxious contaminants like heavy metals and industrial chemicals are becoming an increasing concern due to their persistence and toxicity. Among heavy metals, chromium is recognized as the second most hazardous pollutant after lead and is commonly found in wastewater discharged from textile, electroplating, and leather industries. Chromium exists mainly in two oxidation states: hexavalent chromium (Cr(VI)) and trivalent chromium (Cr(III)). Cr(VI) is highly toxic, a group A carcinogen, water-soluble, and a proven mutagen, posing serious threats to both environmental and human health. Therefore, the reduction of Cr(VI) to the less toxic Cr(III) form is of great importance. Unlike Cr(VI), Cr(III) is an essential trace element for biological systems and tends to form insoluble hydroxides under controlled pH conditions, making it easier to remove from aqueous systems and safer for environmental applications.1 Similarly, 4-nitrophenol (4-NP) is considered as a highly toxic pollutant found in pharmaceutical industries due to high stability and solubility in aqueous media. The presence of the nitro group (–NO2) notably alters the electron density of the aromatic ring through strong inductive (–I) and resonance (−M) effects. As a result, the ring becomes more reactive and unstable, leading to the formation of harmful intermediates such as nitro radicals and nitroso compounds, which can disrupt biological functions by inducing methemoglobinemia through the oxidation of ferrous iron to ferric iron in haemoglobin.2,3
These two pollutants exhibit high biological and chemical stability in the environment. Consequently, the degradation of these pollutants using functional materials remains a key focus in environmental research.4 Traditionally, Pd-based complexes have been widely employed for catalytic applications due to their superior activity and selectivity compared to other noble metal NPs. They also exhibit strong interactions with hydrogen molecules, resulting in efficient hydrogen activation for reduction of organic pollutants like nitroaromatic compounds.5 Moreover, a well-known plasma effect of Pd NPs enables effective light harvesting, thereby promoting photocatalytic degradation of pollutants like Cr(VI).6 Despite these advantages, the practical application of Pd-based catalysts is constrained by separation and recovery, leading to poor recyclability and increased consumption of expensive Pd.7,8 In addition, single Pd NPs are easily aggregated during catalysis, resulting in deactivation and rapid loss of activity of Pd NPs. To overcome these drawbacks, significant efforts have been directed towards engineering suitable catalyst supports to enhance Pd dispersion and reduce metal loading. A variety of support materials, including zeolites, silica, metal oxides, carbonaceous materials and organic polymers, have been explored to improve catalytic efficiency and reusability, but these systems remain suboptimal due to NP agglomeration and metal leaching during catalytic cycles, which lead to activity loss.9 In this context metal–organic frameworks (MOFs) have emerged as highly promising supports for noble metal catalysts owing to their exceptionally high surface area, adjustable pore structures and robust thermal stability. Also, the unique functional groups on the organic linkers within MOFs can easily immobilise catalytically active metal ions, avoiding the loss of expensive metals and increasing stability.10–12 MOFs, particularly lanthanide-based MOFs, have attracted significant attention for catalysis.13 Among them, the cerium-based MOF Ce–BDC stands out due to the presence of Ce4+ ions with low-lying empty 4f orbitals, which contribute to enhanced photocatalytic performance. These orbitals allow efficient ligand-to-metal charge transfer (LMCT), resulting in effective charge separation and reduced electron–hole recombination rate as compared to other metal centers like Zr, Hf, Th, Ti, or U.14 In addition, the Ce4+/Ce3+ redox metal centre within Ce–BDC facilitates rapid electron transfer during redox processes, further enhancing its photocatalytic efficiency. Additionally, its tunable structure, water stability, high porosity, and large surface area can be engineered to optimize adsorptive and catalytic properties for efficient pollutant removal.15 In this context, the interface formed between Pd NPs and Ce–BDC can modulate the electronic structure and promote efficient charge transfer, thereby significantly improving catalytic activity, stability and recyclability. Furthermore, Pd@Ce–BDC typically exhibits a smaller band gap (2.1 eV), enabling effective absorption of visible light and, thus, significantly improving photocatalytic efficiency on light irradiation. Zhao et al. used CeO2@Ni-MOF for the reduction of 4-NP to 4-aminophenol (4-AP); CeO2 was chosen for its ability to provide abundant active sites, facilitate electron transfer, and synergize with Ni-MOF's adsorption capabilities, resulting in a highly efficient, selective, and stable catalyst.16 Similarly, Kohantorabi et al. chose CeO2 nanorods as the support for CuNi nanoparticles in their study on the catalytic reduction of 4-NP due to CeO2 nanorods exhibiting superior surface oxygen release and storage capability, which is crucial for redox reactions involved in catalytic processes.17 In another study, Anandkumar et al. highlighted that the interaction at the interface between gold and cerium oxide, mainly redox cycling between Ce3+ and Ce4+, is directly involved in the efficient hydrogenation of 4-NP to 4-AP.18 Ce-MOFs like Ce-MOF-808 with Pd/Au support are efficient for 4-NP reduction via enhanced electron transfer. Collectively, these studies highlight the versatility of Ce in improving catalytic performance by facilitating electron transfer and offering structural adaptability.19 They also emphasize the critical role of surface area and redox-active centers in the rational design of efficient catalytic systems, as illustrated by selected examples in Table S1.
Building on these insights we have synthesized Pd@Ce–BDC via a double-solvent method, enabling immobilization of Pd NPs on the Ce–BDC framework. The mixed-valence Ce3+/Ce4+ electronic structure of Ce–BDC provides strong anchoring sites for Pd through electron donation, thereby lowering Pd adsorption energy and stabilizing Pd clusters against agglomeration.20 The resulting Pd and Ce–BDC interface promotes efficient electron transfer, band gap narrowing, and accessible active sites. These synergistic effects collectively enhance catalytic activity, structural stability and recyclability making Pd@Ce–BDC an effective catalyst for both 4-NP and Cr(VI) reduction. The photocatalytic reduction using Pd@Ce–BDC was evaluated by optimising parameters such as catalytic dosage, pH of the solution, etc. 40 ppm of toxic Cr(VI) was reduced to Cr(III) at neutral pH (pH = 7) and 0.5 mM of 4-NP was reduced to 4-AP (pH = 7) using 5 mg of Pd@Ce–BDC catalyst at room temperature using visible light. Notably the catalyst retained its framework integrity up to the 8th cycle in 4-NP reduction and the 7th cycle in Cr(VI) reduction underscoring robust stability and reusability. In contrast, pristine Ce–BDC exhibited significantly lower activity, reducing 40 ppm Cr(VI) and 0.5 mM 4-NP under acidic conditions, emphasizing the role of Pd in modulating the electronic structure and catalytic efficiency of the Ce–BDC framework. The ability of Pd@Ce–BDC to operate efficiently at neutral pH and room temperature represents a substantial advantage for practical wastewater remediation, minimizing secondary chemical inputs while maintaining high catalytic efficiency.
Experimental
Synthesis of the Pd@Ce-MOF
Ce–BDC was synthesized employing a reported procedure.21 Pd NPs-loaded Ce–BDC was prepared by using a double-solvent method. In a typical experiment, 200 mg of Ce–BDC was dispersed in 40 mL of acetonitrile under continuous stirring to ensure uniform suspension. Separately, palladium acetate solution (0.1 M) prepared in methanol was dropwise added to the Ce–BDC and the mixture was continuously stirred for 4 h to facilitate the adsorption of Pd2+ within the Ce–BDC framework. The resulting solid was then filtered, washed with DI water and dried at 80 °C. Subsequently, after dehydration, freshly prepared NaBH4 (0.05 mM) was added under stirring leading to the formation of a gray suspension, indicating the in situ reduction of Pd2+ to Pd NPs as shown in Scheme 1. The final product was recovered by centrifugation and dried at 100 °C to obtain Pd NPs decorated on Ce–BDC (Pd@Ce–BDC), which was used for subsequent catalytic studies.22
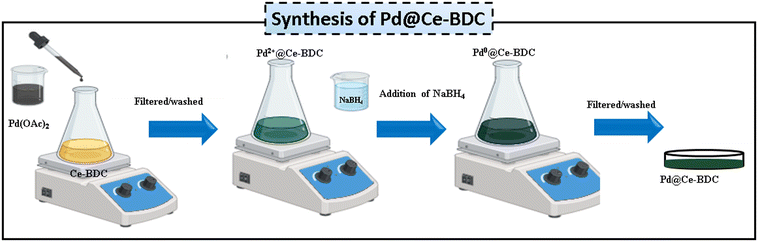 |
| | Scheme 1 Synthesis procedure of Pd@Ce-BDC. | |
Catalytic test for reduction of Cr(VI)
To evaluate the photocatalytic performance of the synthesized catalysts, Cr(VI) was selected as a representative toxic heavy metal pollutant. A 40 ppm aqueous solution of potassium dichromate (K2Cr2O7) was prepared according to a standard method. Prior to the experiment, the catalysts were activated for 10 hours and ground into fine powders. For the photocatalytic test, 5 mg of catalyst was dispersed in 30 mL of Cr(VI) solution and kept in the dark for 15 minutes to establish adsorption desorption equilibrium, allowing adsorption of Cr(VI) on surface of the catalyst before light exposure. Following dark reaction, the suspension was exposed to light, and the Cr(VI) concentration was tracked using a JASCO V-770 UV-visible spectrophotometer within the wavelength range of 300–450 nm. The absorbance of the Cr(VI) solution decreased over time and markedly diminished within 20 minutes, indicating efficient reduction of Cr(VI) to Cr(III) in neutral pH as shown in Fig. 3a. To further investigate the reaction mechanism, the formation of H2O2 as a byproduct of the photocatalytic Cr(VI) reduction was confirmed by titrating the post-reaction K2Cr2O7 solution with a standardized KMnO4 solution (>99%, Sigma-Aldrich, Germany). This additional analysis provided insight into the redox processes involved during the photocatalytic degradation of Cr(VI) under sunlight.23,24
Catalytic test for 4-NP reduction
The reduction of 4-NP using NaBH4 was selected as another model reaction to evaluate the catalytic performance of Pd@Ce–BDC, using the same spectrophotometer employed for Cr(VI) analysis. The activation of the catalyst was performed as in the Cr(VI) reduction procedure, the catalyst being used for the reduction of 4-NP. In a standard quartz cuvette having path length 1 cm, 1 mL of an aqueous 4-NP solution (0.5 mM) was mixed with 0.6 mL of freshly prepared aqueous NaBH4 solution (0.05 mM). After thorough mixing, 1.5 mL of dispersed catalyst solution was introduced and tested at varying concentrations of 3, 5, and 10 mg at neutral pH.12,25
Results and discussion
The Ce–BDC MOF was synthesized via a hydrothermal method, wherein Ce metal nodes were coordinated with terephthalic acid (BDC) linkers to construct a crystalline porous framework. The synthesized powder material was characterized with various techniques. PXRD, XPS, Raman, TGA, FTIR etc. confirmed the coordination of Ce and BDC linkage and generation of photo-responsive ‘Ce–O’ cluster. In this regard, the reported crystallographic information file was in accord with the PXRD pattern of synthesized powder compound. Therefore, the phase purity of the material is very similar to that of the well-known Uio-66 framework. The insight of structural architecture reveals that the central metal ion displays an eight-coordinated geometry, wherein Ce atoms are coordinated with BDC by two distinct types of oxygen environments.
As depicted in Fig. 1, the Ce(IV) ions display an eight-coordinated geometry, with each cerium atom participating in a hexanuclear cluster formed from six Ce6O4(OH)4 secondary building units (SBUs). The triangular faces of each Ce(IV) cluster are alternately coordinated by μ3-O and μ3-OH groups, leading to the formation of a hexanuclear cluster denoted as Ce6(μ3-O)4(μ3-OH)42+.26 The BDC ligand contains two carboxylate groups (–COO−), each with two oxygen atoms. These carboxylate groups act as bidentate ligands, enabling them to bind simultaneously to a single cerium ion through both oxygen atoms. This interaction creates a stable coordination environment around the cerium ion, enhancing the stability and functionality of the Ce–BDC complex.27 This structure provides accessible cerium sites for reactions and exhibits reversible Ce4+/Ce3+ redox transitions due to the stabilization of mixed oxidation states within the framework, attributed to delocalized π-electrons in terephthalate ligands that facilitate electron transfer.28
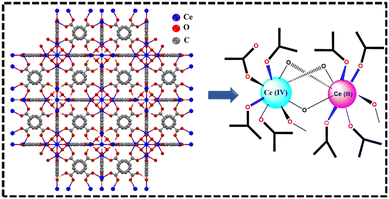 |
| | Fig. 1 Schematic representation and molecular structure of the synthesized Ce–BDC. | |
The crystallinity and phase purity of Ce–BDC and Pd@Ce–BDC were analyzed using powder X-ray diffraction (PXRD) within the 2θ range of 5–50°. The PXRD pattern of Ce–BDC MOF exhibits sharp reflections at 2θ values of 7.2°, 8.3°, 11.7°, 13.8°, and 14.4°, corresponding to the (111), (200), (220), (311), and (222) planes, respectively. These peaks closely correspond to those reported in the literature, confirming the well-crystallized nature, high structural integrity, and successful synthesis of Ce–BDC as validated by PXRD analysis.28,29 The diffraction pattern of Pd@Ce–BDC closely matches with that of Ce–BDC, indicating that Pd NP loading does not alter the crystalline phase or disrupt the framework integrity of the MOF support as shown in Fig. 2a.
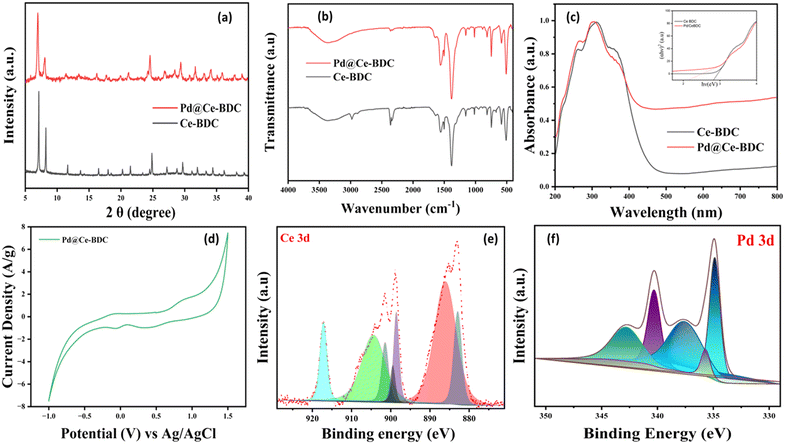 |
| | Fig. 2 (a) PXRD patterns, (b) FTIR spectra and (c) UV-DRS spectra of Ce–BDC and Pd@Ce–BDC. (d) Cyclic voltammetry and XPS peaks of (e) Ce 3d and (f) Pd 3d of Pd@Ce–BDC. | |
FTIR analysis confirmed the presence of functional groups in the organic linkers and revealed the coordination of metal ions, indicating successful framework formation. In the spectrum of Ce–BDC, a distinct peak at 511 cm−1 corresponds to Ce–O vibrations, while bands between 1000 and 1200 cm−1 and at 737 cm−1 are assigned to C–O stretching and O–H bending of the H2BDC ligand. These collectively confirm the coordination of BDC to cerium centers and the presence of key structural units essential for the Ce–BDC framework as shown in Fig. 2b.30,31 The absence of a new peak after Pd NPs loading indicates a stable coordination environment which is attributed to the low Pd loading and high dispersion within the framework. FESEM analysis of Ce–BDC revealed the presence of irregularly shaped, intergrown nanoparticles with a tendency to form aggregated clusters. The particles displayed rough surfaces and heterogeneous morphology, matching with the literature. After the deposition the overall framework remain intact,32 indicating no significant structural damages, leading to the formation of a Pd NPs and Ce–BDC interface. Furthermore, energy dispersive X-ray (EDX) analysis confirms the presence of Pd, with loading of approximately 2.0 wt%, indicating successful incorporation of Pd in Ce–BDC MOF (Fig. S1–S4). Furthermore, nanoparticle deposition on Ce–BDC can be confirmed through MOF. Upon Pd NP deposition, the BET surface area decreased from 544 m2 g−1 to 61 m2 g−1 accompanied by a reduction in pore volume from 0.24 to 0.08 cm3 g−1. This confirms Pd NP incorporation on the porous Ce–BDC support (Fig. S5).
The semiconducting nature of Ce–BDC, a key factor for photocatalysis, was evaluated using UV-visible DRS, which revealed distinct optical absorption characteristics. The UV-visible diffuse reflectance spectrum of the Ce–BDC MOF exhibits strong absorption in the ultraviolet region (∼200–350 nm),33,34 which originates from π–π* electronic transitions of the BDC organic linker along with LMCT from carboxylate 2p orbitals to the Ce 4f/5d states.15,35 A gradual absorption tail extending into the visible region is also observed, which can be ascribed to the coexistence of Ce3+/Ce4+ redox couples that promote enhanced light absorption. Upon loading of Pd nanoparticles, the absorption edge shows a slight blue shift, indicating electronic interaction between the Pd species and the Ce–BDC framework. Correspondingly, the optical band gap decreases from 2.7 eV for pristine Ce–BDC to 2.1 eV for Pd@Ce–BDC, demonstrating that Pd incorporation effectively narrows the band gap and facilitates electron transfer during reduction of 4-NP as depicted in Fig. 2c. XPS analysis was performed to gain detailed insights into the elemental composition, chemical states, and surface interactions of the material. Furthermore, to study the elemental composition and surface chemical state of Pd NPs immobilised on Ce–BDC, XPS was used as shown in Fig. 2e. The survey spectrum verifies the existence of Pd in addition to the framework constituents, namely C, N, O and Ce, indicating the successful incorporation of Pd on the Ce–BDC support. The high-resolution C 1s spectrum can be deconvoluted into multiple components with a peak corresponding to sp2 hybridized carbon at 284.5 eV and oxygen-containing carbon species appearing at higher binding energies between 285 and 289 eV reflecting the functional group of Ce–BDC (Fig. S7a and b). This Ce 3d spectrum indicates the coexistence of both Ce4+ and Ce3+ oxidation states. Specifically, Ce4+ is characterized by peaks of 3d3/2 (918.4, 901.1, 899.1 eV) and 3d5/2 (882.4 eV). The peaks at 904.4 and 885.9 eV correspond to Ce3+ states in 3d3/2 and 3d5/2, respectively. These findings indicate that the Ce–BDC framework exhibits a mixed-valence state involving both Ce3+ and Ce4+, responsible for its redox activity.36–38 The Pd 3d core-level spectrum exhibits characteristic doublets corresponding to different Pd chemical states. The peaks appearing at approximately 336 and 341 eV are attributed to Pd 3d5/2 and Pd 3d3/2 levels of metallic Pd(0), respectively. In contrast, the signals observed at around 338 and 343 eV correspond to the Pd 3d5/2 and Pd 3d3/2 components of Pd2+species as shown in Fig. 2f. The partial oxidation of Pd2+ is ascribed to strong interactions between Pd and surface-absorbed oxygen which improve the adherence of Pd on the Ce–BDC support, providing stable anchoring sites and structural stability.9 The specific binding energies and the relative proportion of Ce3+ and Ce4+ are responsible for redox properties of the framework.
Cyclic voltammetry (CV) experiments were conducted to investigate the redox behavior of the prepared MOF. It exhibits distinct and reversible redox peaks, supporting fast electron transfer and reversible Ce3+ and Ce4+ cycling under applied potentials. The electrode performance was evaluated at various scan rates, 100 mV s−1, using a three-electrode configuration in a mixed electrolyte solution of 3 M Na2SO4. The resulting CV curves displayed well-defined redox peaks within the range of 1.0 to 1.5 V. Redox pair for Ce(IV)/Ce(III) was around 0.3–0.5 V (anodic) and −0.1 to 0.0 V (cathodic) vs. Ag/AgCl, reflecting reversible mixed-valence cerium states enhanced by the MOF's porosity and ligand coordination. Pd incorporation shifts or amplifies these peaks (e.g., anodic ∼0.4 V, cathodic ≃0.05 V), with additional Pd(0)/Pd(II) oxidation near 0.8–0.9 V, matching the observed 0.89 V peak as shown in Fig. 2d.9,38
Photocatalytic reduction of Cr(VI) using Pd@Ce–BDC
The photocatalytic reduction of Cr(VI) using Pd@Ce–BDC as a catalyst was attributed to its enhanced electron transfer and visible-light-harvesting ability. The results demonstrated complete removal of 40 ppm Cr(VI) within 20 minutes using only 5 mg of the catalyst, highlighting its high efficiency as shown in Fig. 3a. The degradation efficiency was calculated using the following equation:22,34| | 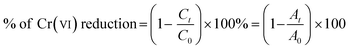 | (1) |
where Ct and C0 represent the concentration of Cr(VI) at different times t and initial concentration, whereas At and A0 represent the absorbance at 370 nm at time t and at the initial time. The photocatalytic reduction followed pseudo-first-order kinetics, expressed by the following equation (eqn (2)):39where k and t represent the observed rate constant and reaction time, respectively. As shown in Fig. 3b, a plot of ln(C0/Ct) vs. time results in a straight line from which the rate constant (k) was determined to be 0.11 min−1 with correlation coefficient (R2) > 0.99.
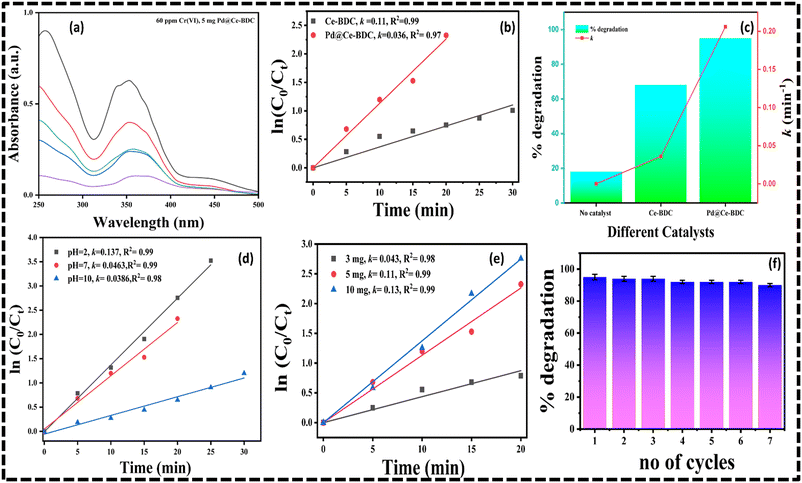 |
| | Fig. 3 (a) UV-visible spectra. (b) Pseudo-first-order kinetic models of Cr(VI) reduction using Ce–BDC and Pd@Ce–BDC. (c) Histogram of k value with % efficiency vs. catalytic dosage for Cr(VI) reduction. (d) Ln(Ct/C0) vs. time for different pH. (e) Ln(Ct/C0) vs. time for different dosages. (f) Histogram showing reusability after Cr(VI) reduction. | |
The catalyst dosage was optimized as 5 mg by maintaining a constant concentration of 40 ppm Cr(VI) solution. By increasing the catalyst dosage from 3 mg to 10 mg, both the rate of reaction and catalytic efficiency improved significantly. The rate constant for 3 mg of Pd@Ce–BDC catalyst was found to be 0.043 min−1 with a reduction efficiency of 68.3%. Upon increasing the dosage to 10 mg, the rate of reaction and efficiency drastically increased to 0.13 min−1 and 93.9% (Fig. 3c) respectively as plotted in Fig. 3e. This improvement is attributed to the greater number of available active sites, enabling enhanced interaction between the catalyst and Cr(VI) species. However, the slight increase in rate constant and efficiency when increasing the dosage from 5 mg to 10 mg is likely due to the saturation of active sites and possible mass transfer limitations, resulting in only marginal performance gains. The reduction rate of hexavalent chromium is strongly influenced by pH. The pseudo-first-order rate constant decreased from 0.13 min−1 to 0.038 min−1 by increasing the pH from 2 to 10, as shown in Fig. 3d. This may be due to the fact that, at acidic and neutral pH, Cr(VI) (HCrO4− or Cr2O72−) carries a negative charge, while the Pd@Ce–BDC surface may become positively charged due to protonation of surface sites. This electrostatic attraction between the positively charged Pd@Ce–BDC and the anionic Cr(VI) species facilitates closer interaction and accelerates electron transfer, enhancing the reduction rate. Plots of the relation between rate constant and % degradation efficiency were obtained for different catalytic dosages with constant concentration of Cr(VI) solution as shown in Fig. 3f. The photocatalytic reduction of Cr(VI) generally occurs through multiple steps, starting with photoexcitation and the formation of reactive species. Among these, two active species, superoxide radicals (O2−˙) and photogenerated electrons, play an important role in the process. To gain insight into the detailed role of active species, a scavenger experiment was conducted using EDTA, isopropanol (IPA), silver nitrate (AgNO3) and benzoquinone (BQ) to selectively quench h+, OH˙, electrons and O2−˙, respectively. As shown in Fig. S5, adding EDTA had little effect on the reduction efficiency, suggesting that h+ plays a minor role. However, the presence of AgNO3 and BQ significantly decreased Cr(VI) reduction, indicating that electrons and O2−˙ are the main species driving the photocatalytic process.40,41
Catalytic reduction of 4-NP using Pd@Ce–BDC
The catalytic performance of Pd@Ce–BDC was evaluated by capturing UV-visible spectra at regular time intervals, specifically tracking the decrease in absorbance at 400 nm, corresponding to the reduction of 4-NP. Fig. 4a shows the time-dependent UV-visible absorption spectra for the reduction of 4-NP using the catalyst. The catalytic performance of Ce–BDC in the reduction of 4-NP to 4-AP using NaBH4 was assessed based on key parameters such as pH, catalyst dosage, and overall reduction efficiency. In this reduction process, 4-NP exhibits a prominent absorption peak at 317 nm in the UV spectrum, which shifts to 400 nm after the addition of NaBH4. This shift corresponds to the formation of 4-nitrophenolate ions.
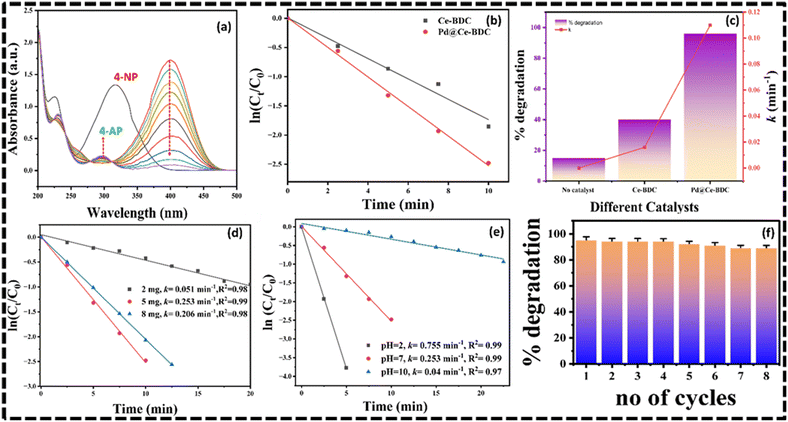 |
| | Fig. 4 (a) UV-visible spectra. (b) Pseudo-first-order kinetic models of 4-NP reduction using Ce–BDC and Pd@Ce–BDC. (c) Histogram of k value with % efficiency vs. catalytic dosage for 4-NP reduction. (d) Ln(Ct/C0) vs. time at different dosages for 4-NP reduction. (e) Ln(Ct/C0) vs. time for different pH. (f) Histogram showing reusability after 4-NP reduction. | |
However, in the absence of a catalyst, no conversion of 4-NP was observed, even after 40 minutes, despite the reaction being thermodynamically feasible. This is because the reduction of 4-NP to 4-AP is kinetically hindered by a high energy barrier. The absorption spectrum of the mixture of 4-NP and NaBH4 remains unchanged even after two hours, indicating no reaction progression without a catalyst42 as depicted in Fig. 4a. Upon the addition of the catalyst, a blue shift in absorption peak towards 298 nm was observed, confirming the formation of 4-AP. Additionally, the isosbestic point at 313 nm suggests that no side reactions occurred, and 4-AP was the sole product.43 In the presence of excess NaBH4, the reduction of 4-NP followed pseudo-first-order kinetics, described by the following equation (eqn (3)):
where
Ct is the concentration at time
t,
k is the pseudo-first-order rate constant, and
C0 is the initial concentration. Pd@Ce–BDC exhibits catalytic activity and follows pseudo-first-order kinetics
44 with a rate constant 0.023 min
−1 as shown in
Fig. 4d. The efficiency is attributed to the redox properties of cerium, particularly its facile Ce
4+/Ce
3+ transition, which enhances electron transfer and accelerates reaction kinetics. Moreover, the increase in pH caused by excess NaBH
4 helps stabilize BH
4− and suppress hydrolysis, ensuring that the catalytic effect is primarily governed by the metal centers in Pd@Ce–BDC. The conversion rate of 4-NP was calculated using the same equation as applied for Cr(
VI) reduction, as described in
eqn (1).
The catalyst dosages were optimized by maintaining a constant concentration of 4-NP. Increasing the catalyst dosage from 2 mg to 8 mg significantly improved both the rate of reaction and reduction efficiency. The rate constant for 2 mg of Pd@Ce–BDC catalyst was found to be 0.051 min−1, whereas at 5 mg, the rate of reaction and efficiency drastically increased to 0.253 min−1 and 95% respectively (Fig. 4c and d). This increase in reaction rate with higher catalyst dosage is attributed to the increased availability of active sites, enabling greater interaction with 4-NP molecules. On further increasing the catalyst dose to 8 mg, the rate constant decreased to 0.206 min−1. This is because at higher catalyst loadings, particle aggregation reduces the effective active surface area.
The reduction rate of 4-NP is highly dependent on pH, as reflected in the present study, where the pseudo-first-order rate constant decreased when the pH increased from 2 to 7 and 11. By increasing the pH of solution from 2 to 11 the rate constant decreases from 0.755 min−1 to 0.04 min−1 as depicted in Fig. 4e. Acidic conditions provide more protons (H+), which are necessary for the reduction of the nitro group (–NO2) to the corresponding amine (–NH2). The reduction process typically involves the transfer of electrons and protons to the nitro group, so higher proton concentration speeds up the reaction. Plots of the relation between rate constant and % degradation efficiency were obtained for different catalytic dosages with constant concentration of 4-NP solution as shown in Fig. 4f.
Catalytic reduction mechanism
The plausible mechanism of photocatalytic reduction of Cr(VI) is explained below and illustrated in Fig. 5. Upon exposure to visible light, electrons in the valence band (VB) are excited to the conduction band (CB) generating excitons, also known as photogenerated free electrons and holes in the CB and VB respectively. In Ce–BDC, LMCT is strongly favoured especially for Ce4+ nodes, which have low-lying empty 4f orbitals in the presence of light. This LMCT facilitates efficient separation of electrons and holes, reducing their rapid recombination and increasing carrier lifetime, thereby enhancing the photocatalytic activity.
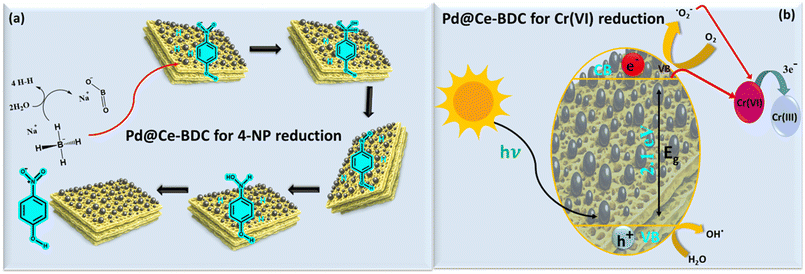 |
| | Fig. 5 Plausible mechanistic pathways showing reduction of (a) 4-NP and (b) Cr(VI). | |
The proposed mechanism of Cr(VI) reduction includes the following steps:
| | | Pd@Ce–BDC + hν → Pd@Ce–BDC (e− + h+) | (4) |
| | | O2−˙ + Cr(IV) → Cr(III) + O2 | (6) |
| | | h+ + 2H2O → 2H+ + H2O2 | (8) |
| | | O2−˙/OH˙/ + Cr(III) → Cr(VI) | (10) |
| | | Cr(VI) + 3e−CB → Cr(III) | (11) |
The photogenerated electrons and superoxide radicals are mainly responsible for photocatalytic reduction of Cr(
VI) which is evident from the scavenger experiment. When BQ was added to the reaction system, the photocatalytic reduction efficiency was decreased from 95% to 61%, whereas the addition of AgNO
3 suppressed the photocatalytic activity up to 40% confirming their significant involvement in the reduction process. Photogenerated holes are responsible for production of hydroxyl radicals (OH˙) from water and H
2O
2 indicating the conversion of Cr(
III) to Cr(
VI)
via re-oxidation as shown in Fig. S5. The combined effects of charge carrier separation and the presence of reactive species drive the overall photocatalytic process, aligning with previously reported studies.
45 The plausible mechanism of reduction of 4-NP can be discussed in terms of both electron transfer and redox property of Pd@Ce–BDC. Initially, BH
4− ions and 4-NP molecules adsorb onto the Pd@Ce–BDC surface, facilitated by π–π interactions and electrostatic attractions between negatively charged 4-NP and Pd@Ce–BDC. Pd(0) nanoparticles adsorb BH
4− and nitrophenol on their electron-rich surfaces. The small size and high dispersion of Pd(0) in supports like Ce–BDC maximize active sites and prevent agglomeration for sustained activity. This cerium nano-MOF with appended Pd NPs stabilizes the hydride ions on its surface, hence smoothen the reduction. In contrast the redox property of Ce–BDC is further promoted by the intrinsic redox property of cerium centres (Ce
3+/Ce
4+), which further transfer electrons in the catalytic cycle. During catalysis, partial oxidation of Ce
3+ to Ce
4+ occurs upon electron donation, followed by regeneration of Ce
3+ through interaction with BH
4− for continuous electron flow. Overall, the superior catalytic activity arises from the synergistic interaction of Pd(0) nanoparticles and the redox-active Ce–BDC framework, where Pd/Ce interfacial charge transfer and reversible Ce
3+/Ce
4+ redox cycling collectively promote efficient and sustained reduction of 4-NP.
46 The reusability of the Ce–BDC catalyst was also evaluated for both 4-NP and Cr(
VI) reduction. Following each catalytic run, the material was recovered
via centrifugation, rinsed thoroughly with methanol and water, and dried at 70 °C before reuse. Under optimized conditions, the efficiency for 4-NP reduction declined slightly from 95% to 89% after four successive cycles as shown in
Fig. 4f, whereas for Cr(
VI) reduction it dropped from 95% to 90% after three successive cycles as shown in
Fig. 3f. This gradual loss in activity can be ascribed to: (i) minimal leaching of Ce ions into the reaction medium, reducing the number of available active sites; (ii) slight mass loss during recovery and washing; and (iii) partial pore blockage by adsorbed intermediates or products. Despite this minor reduction in catalytic performance, PXRD patterns confirmed that Ce–BDC retained its crystallinity and structural framework even after repeated use
41 as shown in Fig. S8.
Conclusions
In summary, a Ce-oxo SBU-based MOF has been synthesized and Pd NPs deposited creating an interface, well characterized with standard methods. The study reveals the formation of a stable, redox as well as photoactive molecular system to achieve structural morphology corresponding to the ideal UiO-66. The well-defined porosity and molecular construction can be utilised in catalytic reactions. Here, taking advantage of chemical stability, photo-harvesting property, and redox properties, it has been utilised for water purification through solar light, which is itself a green technology and a new way towards an alternative source in view of the energy crisis. In particular, at room temperature and neutral pH, 4-NP was completely reduced within just 10 minutes, while Cr(VI) reduction required around 20 minutes. Remarkably, Pd@Ce–BDC exhibited ∼95% efficiency for 4-NP reduction and ∼94% efficiency for Cr(VI) reduction under solar irradiation. The catalyst exhibited excellent reusability for eight consecutive cycles for reduction of 4-NP, and seven consecutive cycles for reduction of Cr(IV), making it a sustainable photocatalyst for effective environmental remediation.
Conflicts of interest
There are no conflicts of interest to declare.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author upon reasonable request.
Supplementary information (SI): the SI includes experimental details, characterization techniques, FESEM, BET, scavenger experiment, reusability study and comparison table. See DOI: https://doi.org/10.1039/d6lf00091f.
Acknowledgements
We acknowledge the Central Research Facility, KIIT for providing characterization facilities and DRDO-DRDE project (LSRB/01/15001/LSRB-429/BTB/2024) for funding.
References
- H. Kaur, S. Sinha, V. Krishnan and R. R. Koner, Ind. Eng. Chem. Res., 2020, 59, 8538–8550 CrossRef CAS.
- A. O. Cardoso Juarez, E. I. O. Lopez, M. K. Kesarla and N. K. R. Bogireddy, ACS Omega, 2024, 9, 33335–33350 CrossRef CAS PubMed.
- T. J. Martin, A. K. Goodhead, K. Acharya, I. M. Head, J. R. Snape and R. J. Davenport, Environ. Sci. Technol., 2017, 51, 7236–7244 CrossRef CAS PubMed.
- M. Lei, P. Wang, X. Ke, J. Xie, M. Yue, M. Zhao, K. Zhang, Y. Dong, Q. Xu and C. Zou, Electron, 2025, 3, e43 CrossRef CAS.
- X. Zhao, Y. Chang, W.-J. Chen, Q. Wu, X. Pan, K. Chen and B. Weng, ACS Omega, 2021, 7, 17–31 CrossRef PubMed.
- M. L. de Souza, D. P. Dos Santos and P. Corio, RSC Adv., 2018, 8, 28753–28762 RSC.
- M. Yamada, M. Rajiv Gandhi and A. Shibayama, Sci. Rep., 2018, 8, 16909 CrossRef PubMed.
- X.-Z. Wei, H. Wang, J. Liu and L. Ma, Inorg. Chem. Commun., 2023, 154, 110956 CrossRef CAS.
- A. Lin, A. A. Ibrahim, P. Arab, H. M. El-Kaderi and M. S. El-Shall, ACS Appl. Mater. Interfaces, 2017, 9, 17961–17968 CrossRef CAS PubMed.
- B. Dutta, S. Datta and M. H. Mir, Chem. Commun., 2024, 60, 9149–9162 RSC.
- F. Zheng, S. Cao, Z. Yang, Y. Sun, Z. Shen, Y. Wang and H. Pang, Energy Fuels, 2024, 38, 11494–11520 CrossRef CAS.
- J. Panda, S. P. Biswal, H. S. Jena, A. Mitra, R. Samantray and R. Sahu, Catalysts, 2022, 12, 494 CrossRef CAS.
- Y. Zhang, S. Liu, Z.-S. Zhao, Z. Wang, R. Zhang, L. Liu and Z.-B. Han, Inorg. Chem. Front., 2021, 8, 590–619 RSC.
- X.-P. Wu, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2018, 140, 7904–7912 CrossRef CAS PubMed.
- M. D. Goudarzi, N. Khosroshahi and V. Safarifard, RSC Adv., 2022, 12, 32237–32248 RSC.
- H. Zhao, X. Pang, Y. Huang, C. Ma, H. Bai and W. Fan, Inorg. Chem., 2022, 61, 19806–19816 CrossRef CAS PubMed.
- M. Kohantorabi and M. R. Gholami, Ind. Eng. Chem. Res., 2017, 56, 1159–1167 CrossRef CAS.
- M. Anandkumar, G. Vinothkumar and K. S. Babu, New J. Chem., 2017, 41, 6720–6729 RSC.
- H. M. Hassan, H. A. Alruwaili, M. S. Alhumaimess, A. H. Alanazi, M. El-Aassar, M. S. Alshammari, M. F. Hussein and I. H. Alsohaimi, Environ. Res., 2025, 264, 120340 CrossRef CAS PubMed.
- X.-L. Xu, N.-N. Wang, Y.-H. Zou, X. Qin, P. Wang, X.-Y. Lu, X.-Y. Zhang, W.-Y. Sun and Y. Lu, Nat. Commun., 2024, 15, 7273 CrossRef CAS PubMed.
- D. Grebenyuk, M. Shaulskaya, A. Shevchenko, M. Zobel, M. Tedeeva, A. Kustov, I. Sadykov and D. Tsymbarenko, ACS Omega, 2023, 8, 48394–48404 CrossRef CAS PubMed.
- G.-F. Liu, X.-X. Qiao, Y.-L. Cai, J.-Y. Xu, Y. Yan, B. Karadeniz, J. Lu and R. Cao, ACS Applied Nano Mater., 2020, 3, 11426–11433 CrossRef CAS.
- C. Hu, Y.-C. Huang, A.-L. Chang and M. Nomura, J. Colloid Interface Sci., 2019, 553, 372–381 CrossRef CAS PubMed.
- K. Wang, D. Huang, X. Li, K. Feng, M. Shao, J. Yi, W. He and L. Qiao, Electron, 2023, 1, e4 CrossRef.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CrossRef CAS.
- M. Lammert, C. Glißmann, H. Reinsch and N. Stock, Cryst. Growth Des., 2017, 17, 1125–1131 CrossRef CAS.
- Z. Hu, Y. Wang and D. Zhao, Chem. Soc. Rev., 2021, 50, 4629–4683 RSC.
- N. Nagarjun, P. Concepcion and A. Dhakshinamoorthy, Appl. Organomet. Chem., 2020, 34, e5578 CrossRef CAS.
- D. González, C. Pazo-Carballo, E. Camú, Y. Hidalgo-Rosa, X. Zarate, N. Escalona and E. Schott, Dalton Trans., 2024, 53, 10486–10498 RSC.
- S. Sangeetha and G. Krishnamurthy, Bull. Mater. Sci., 2020, 43, 269 CrossRef CAS.
- Q. Zhang, B. Yang, Y. Tian, X. Yang, R. Yu, J. Wang, T. Deng and Y. Zhang, Green Processes Synth., 2022, 11, 184–194 CrossRef CAS.
- J. He, C. Pei, Y. Yang, B. Lai, Y. Sun and L. Yang, J. Cleaner Prod., 2021, 321, 128778 CrossRef CAS.
- E. E. Ghadim, M. Walker and R. I. Walton, Dalton Trans., 2023, 52, 11143–11157 RSC.
- M. Bagheri, A. Melillo, B. Ferrer, M. Y. Masoomi and H. Garcia, ACS Appl. Mater. Interfaces, 2021, 14, 978–989 CrossRef PubMed.
- S. Karmakar, S. Barman, F. A. Rahimi, S. Biswas, S. Nath and T. K. Maji, Energy Environ. Sci., 2023, 16, 2187–2198 RSC.
- Y. Liu, G. Chen, J. Chen and H. Niu, Catalysts, 2022, 12, 775 CrossRef CAS.
- X. Hu, J. Qian, J. Yang, X. Hu, Y. Zou and N. Yang, J. Electroanal. Chem., 2023, 947, 117756 CrossRef CAS.
- S. Rojas-Buzo, P. Concepción, J. L. Olloqui-Sariego, M. Moliner and A. Corma, ACS Appl. Mater. Interfaces, 2021, 13, 31021–31030 CrossRef CAS PubMed.
- Z. Kiani, R. Zhiani, S. Khosroyar, A. Motavalizadehkakhky and M. Hosseiny, Inorg. Chem. Commun., 2021, 124, 108382 CrossRef CAS.
- S. K. Sahoo and G. Hota, ACS Applied Nano Mater., 2019, 2, 983–996 CrossRef CAS.
- J. Swain, A. Priyadarshini, S. Hajra, S. Panda, J. Panda, R. Samantaray, Y. Yamauchi, M. Han, H. J. Kim and R. Sahu, J. Alloys Compd., 2023, 965, 171438 CrossRef CAS.
- Z. Valizadeh, S. M. Hosseini, R. Cheraghi and V. Safarifard, Mater. Res. Bull., 2025, 185, 113304 CrossRef CAS.
- S. Gu, Y. Lu, J. Kaiser, M. Albrecht and M. Ballauff, Phys. Chem. Chem. Phys., 2015, 17, 28137–28143 RSC.
- M. Mansour, H. Kahri, M. Guergueb, H. Barhoumi, E. G. Puebla, B. Ayed and U. B. Demirci, React. Chem. Eng., 2022, 7, 908–916 RSC.
- L. Shi, T. Wang, H. Zhang, K. Chang, X. Meng, H. Liu and J. Ye, Adv. Sci., 2015, 2, 1500006 CrossRef PubMed.
- X. Kong, H. Zhu, C. Chen, G. Huang and Q. Chen, Chem. Phys. Lett., 2017, 684, 148–152 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Dipankar
Singha
c,
Bikash Chandra
Dhal
a,
Subrat
Swain
a,
Jagannath
Panda
c,
Dipankar
Singha
c,
Bikash Chandra
Dhal
a,
Subrat
Swain
a,
Jagannath
Panda






