
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel microfluidic multichannel electrochemical cell for multiplexed monitoring of water pollutants
Tong
Liu
 ,
Ivo
Tichý
,
Jiří
Homola
* and
Amir M.
Ashrafi
*
,
Ivo
Tichý
,
Jiří
Homola
* and
Amir M.
Ashrafi
*
Institute of Photonics and Electronics, Czech Academy of Sciences, Chaberská 1014/57, Prague, 182 51, Czech Republic. E-mail: liu@ufe.cz; tichy@ufe.cz; homola@ufe.cz; ashrafi@ufe.cz
First published on 22nd December 2025
Abstract
A novel microfluidic multichannel (4×) electrochemical cell (MMEC) was developed and used for multiplexed determination of compounds related to water quality. These include heavy metals (lead and mercury ions), catechol and hydrogen peroxide. No crosstalk between the channels of the MMEC was observed. This enabled the specific and independent modification of each MMEC channel with respect to the targeted analyte. Namely, the mercury ions were determined at the bare gold (Au) electrode, lead ions were determined at the Au electrode coated with a thin mercury film (MF), H2O2 was determined at the Au electrode electrodeposited with gold nanostructures (AuNS), and catechol at the Au electrode modified with polyurea (PU) and AuNS. The limits of detection (LODs) were determined and found to be 0.9 ppb, 0.1 ppb, 0.4 μM, and 1.6 μM for lead and mercury ions, catechol, and hydrogen peroxide, respectively. The MMEC was applied for the detection of the analytes in river water samples and in industrial wastewater and good recovery rates were obtained: from 91.8% to 109% in river water samples and 81.8% to 111.6% in industrial wastewater. In addition, comparison with the reference method (ICP-OES) was performed for the determination of Pb2+ ions and the relative error was found to be smaller than 5%. This allows the MMEC to be used for the multiplexed detection of analytes at concentrations relevant to the monitoring of the quality of water resources.
1. Introduction
Urbanization and industrialization endanger clean water, which is a fundamental requirement for human welfare and environmental sustainability. Hence, water resources must be protected, and preventive measures must be taken to avoid their pollution. This makes the development of methods and technologies for water quality monitoring an important research goal. Water pollution is mainly caused by either direct or indirect sources of contamination.1 The former results from the direct release of industrial wastewater or toxic (by-)products into water.2 Indirect pollution refers to contaminants through secondary means, such as the washing of contaminated soil or rainfall following air pollution.3 Therefore, water may contain a broad range of pollutants, including organic and inorganic compounds, pathogens, and agricultural contaminants.4 The primary water contaminants, either naturally occurring or introduced into water resources through human activities, include phenolic compounds from chemical plants, oil refineries, and paper and pulp mills,5 heavy metals from mining, and pesticides and fertilizers used in agriculture.6Effective monitoring necessitates an analytical method characterized by high sensitivity, selectivity, minimal sample volume requirements, short response time, and ability to detect a wide range of analytes, including inorganic and organic compounds as well as biomolecules. Additionally, the method should allow for miniaturization to facilitate on-site analysis.7–9 Furthermore, implementing multiplexed analysis of various analytes can improve the efficiency of the monitoring.10 So far, optical and electrochemical techniques exhibit some of the aforementioned features to varying extents.11 The development and advancement of nanomaterials and their applications in electrochemical sensors and biosensors led to improved sensitivity and selectivity.12–15 Additionally, progress in the development of miniature microfluidic devices and their introduction in electrochemical sensing significantly reduced sample volume, simplified user input, and enabled automation and on-site analysis.16,17 However, the electrochemical sensing devices allowing for multiplexed determination of analytes remain rare. In multiplexed determination using electrochemical sensors, platforms with two or more working electrodes (that share a common reference and counter electrode) are widely employed.18–21 However, this configuration may limit the range of electrochemical techniques and parameters that can be independently applied to each working electrode. Other studies reported the use of multiple screen-printed electrodes (SPEs) integrated into a microfluidic system, forming an electrode array for multiplexed determination.22 This setup allows for the use of different electrochemical techniques to quantify various analytes. However, SPEs are known to suffer from poor reproducibility and limited reusability, which negatively influence the reliability of the results and limit potential applications. The poor reproducibility of SPEs stems from the manufacturing process and the formulation of printing inks.23 Furthermore, their sensitivity and reproducibility deteriorate with their repeated use. Therefore, SPEs are preferably used as disposable electrodes24 and it is rather seldom to use SPEs in applications requiring electrochemical modifications.
In this paper, we present a novel microfluidic multichannel electrochemical cell (MMEC) for the multiplexed analysis of water pollutants. The four channels of the MMEC can be separately and specifically modified to provide an enhanced sensitivity for the corresponding analyte. As a result, four distinct electrochemical cells can be operated simultaneously for the multiplexed detection of various analytes, each utilizing different electrochemical techniques with optimized parameters. The targeted analytes include lead and mercury ions that belong to the most toxic heavy metals and the most dangerous noxious waste in water,25 catechol, a highly carcinogenic phenolic compound present in industrial wastewaters,26 and hydrogen peroxide (H2O2). Monitoring the levels of H2O2 is important because (I) it generates reactive oxygen species (ROS), causing oxidative stress in aquatic organisms;27 (II) elevated H2O2 levels can indicate industrial pollution and inefficient wastewater treatment;28 and (III) H2O2 facilitates the natural oxidative degradation of organic pollutants in water systems.29,30
Monitoring the selected analytes provides valuable information about sources of pollution, imperfections in wastewater treatment, and the oxidative degradation of organic pollutants.
2. Materials and methods
2.1 Chemicals
All the materials and reagents were used without further purification and purchased from Merck (Saint Louis, MO, USA) unless otherwise stated. Sulfuric acid (98%), nitric acid (65%), hydrochloric acid (35%), potassium chloride, sodium chloride, hydrogen peroxide solution (35% in water), magnesium chloride, calcium chloride, L-ascorbic acid, urea, D-(+)-glucose, sodium hydroxide, and standards for atomic absorption spectroscopy (AAS) of As3+, Hg2+, Pb2+, Cd2+, and Zn2+ (1000 ppm) were purchased from penta Chemicals Unlimited (Prague, Czech Republic). Catechol was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Deionized (DI) water purified with a Millipore system was used during the whole experiment (resistivity ≥18 MΩ cm). Phosphate-buffered saline (PBS) powder was used for the preparation of PBS buffer solution (after dilution in 1.0 L of water the concentrations of the components were 10 mM PBS, 140 mM NaCl, and 2.7 mM KCl, pH 7.4). Sodium acetate (SA) buffer solution (100 mM, pH 4.5 and pH 5.7) was prepared by dilution of stock solution of SA (3.0 M, pH 5.2) with water, and further adjustment of pH was accomplished with hydrochloric acid and sodium hydroxide. The prepared buffer solutions were stored at 4 °C.A solution of 1.0 mM [Ru (NH)3]2+/3+ in 100 mM KCl solution was used in the study of the scan rate dependence of the peak current and a solution of 4 mM [Fe (CN)6]3−/4− was used to determine the electroactive area using cyclic voltammetry (CV) at different scan rates.
2.2 Development of the MMEC
Four working electrodes were prepared by vacuum evaporation of gold (layer thickness: 50 nm) on the surface of soda lime glass slides (SCHOTT UK Ltd., Wolverhampton, UK) through a mask with a titanium adhesion interlayer (2 nm thick). Prior to use, the glass substrate with gold electrodes (GGE) was rinsed with DI water and ethanol, placed in ozone cleaner for 10 min, and rinsed again with ethanol and DI water and dried with a nitrogen gas stream.A printed circuit board (PCB) was designed by our group and manufactured by PragoBoard (Prague, Czech Republic). The PCB is composed of four layers: layer I, counter and reference electrodes along with a conductive connection for the counter and working electrodes; layers II and III consist of one shielding layer for the working and counter electrodes and a separate shielding layer for the reference electrodes; and layer IV, conductive connection for the reference electrode. The PCB is fabricated from ISOLA IS400 epoxy laminate with an 18 μm thick copper layer. The counter and reference electrodes are terminated with copper. The counter electrodes are coated with a layer of carbon paste by screen printing (Fig. 1aI). The conductive connections on the PCB are terminated with a connector, enabling connection to the potentiostat. Prior to use, the PCB was immersed in ELCHEMCo AG-1 silvering bath (GM Electronic, Prague, Czech Republic) and put in an Elmasonic P sonicator (P-LAB a.s., Prague, Czech Republic) with a frequency set to 80 kHz and the power adjusted to 80% for 5 min at 30 °C to replace copper with silver (Fig. 1aII). The PCB was then rinsed with DI water and ethanol to remove any impurities.
 | ||
| Fig. 1 (a) (I–III) Schematic of the main steps in the fabrication of PCB. (b) Configuration of the experimental setup. | ||
The reference electrodes were covered with a layer of Ag/AgCl (60/40) from Merck (Saint Louis, MO, USA) and a layer of graphite ink was applied to the surface of the counter electrodes by dragging a cotton bud dipped in graphite ink (Redoxme AB, Norrköping, Sweden) across them (Fig. 1aIII). The prepared PCBs were placed in a heater control box at 80 °C for 10 min to cure the Ag/AgCl paste. Later, the PCBs were rinsed with ethanol and DI water to clean the surface. Four separated electrochemical cells were formed by using a spacer. The spacer was fabricated from a 0.7 mm thick PETG sheet (NUDEC, Barcelona, Spain) and covered on both sides with a 50 μm thick layer of 7952MP laminating adhesive (3M, Prague, Czech Republic), which was precisely laser-cut to the desired shape. The adhesive layers on the spacer connect the spacer to the PCB on one side and to the GGE on the other side. The dimensions of the electrochemical compartments on the spacer were length 22.5 mm, width 1.7 mm, and depth 0.7 mm. The electrical connection between the gold electrodes and the PCB was established using silver conductive grease (Chemtronics, Kennesaw, GA, USA). The prepared multichannel electrochemical cell was secured between two poly(methyl methacrylate) (PMMA) plastic guards using steel bolts. To prevent leakage, a pressure unit with two rods was inserted into prefabricated holes in the PMMA plastic guards. Finally, the pressure unit was tightened against the multichannel electrochemical cell using a steel bolt. Solutions were introduced into the multichannel electrochemical cell via the inlet ports using a multichannel micro peristaltic pump (ISM 931 C, Ismatec) and discharged through the outlet ports. Finally, the assembled MMEC was connected to the multipotentiostat workstation consisting of four potentiostats (CompactStat.h, IVIUM Technologies, Eindhoven, the Netherlands) via a connector on the PCB. The potentiostats were programmed and operated either independently or simultaneously, as needed, using IviumSoft software (IVIUM Technologies, Eindhoven, the Netherlands). The experimental setup is shown in Fig. 1b.
2.3 Methods
The electrochemical methods used for electrode surface cleaning, characterization, and electrochemical measurements for the concentration determination of different analytes are described in the SI (sections 2.3.1–2.3.7). Additionally, the methods used for evaluation of limit of detection (LOD) and limit of quantification (LOQ), repeatability and reproducibility, interference study, recovery percentage and real sample analysis are explained in the SI (sections 2.3.8–2.3.10). Furthermore, the instrumentation used for scanning electron microscopy (SEM), energy dispersive X-ray (EDX) analysis, and atomic force microscopy (AFM) are described in the SI (section 2.3.11). The AFM images were processed with Gwyddion software.313. Results and discussion
3.1 Electrochemical characterization of MMEC
As shown in the fast Fourier-transport (FFT) spectra of chronopotentiograms recorded at channels 2–4 (Fig. 2a and b), three distinct peaks are observed at frequencies (0.3 Hz, 0.9 Hz, and 1.6 Hz) that closely match those identified in the FFT of the time-dependent potential (CV) recorded at channel 1, albeit with significantly lower magnitudes. Notably, the highest peak is observed in channel 4, which is located farther from channel 1. This suggests that the crosstalk between channels is minimal and can be considered negligible under experimental conditions. The peaks observed in the FFT spectra around 0.2 Hz are due to the peristaltic pump, and the initial peaks (0.04 Hz) are most likely related to the fitting of the obtained results and subsequent subtraction of calculated data from the experimental ones.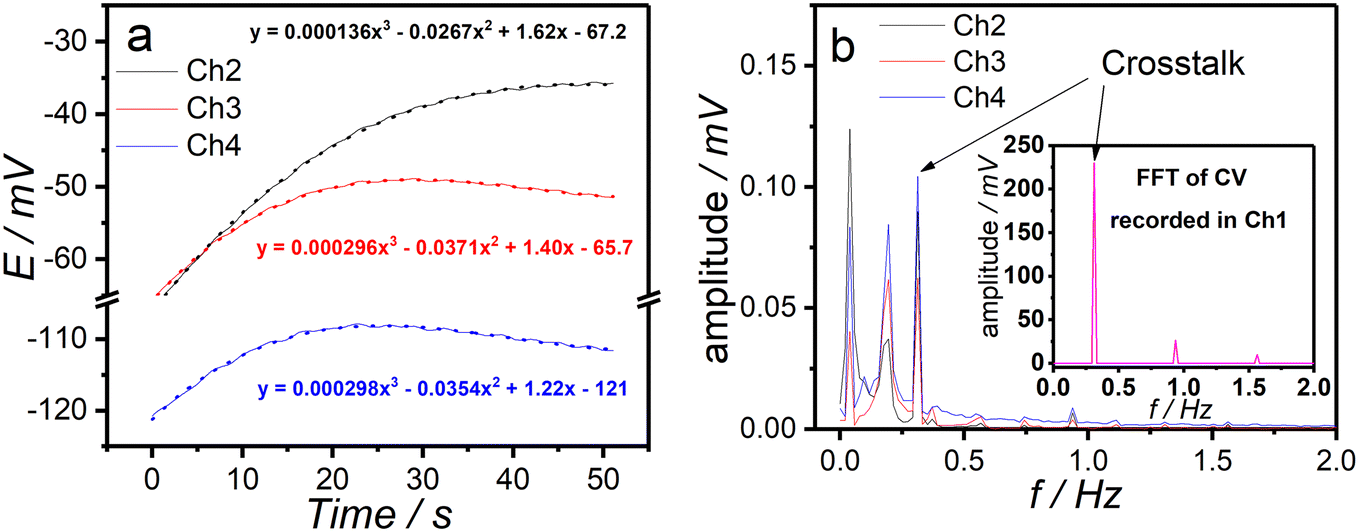 | ||
| Fig. 2 (a) The chronopotentiograms recorded at channels 2–4 in 100 mM KCl and at applied DC current of 0.5 μA along with the fitted data. (b) The FFT of residuals between the experimental and fitted data along with the FFT of the CVs run in channel 1 (inset) in 100 mM KCl during recording of chronopotentiograms in the other three channels. | ||
As follows from Fig. S1a, the peak currents in the recorded CVs exhibit a linear dependence on the square root of the scan rate in all four channels. This dependence confirms diffusion-controlled mass transport, which is characteristic of a reversible system.32 The good channel-to-channel reproducibility of the electrochemical measurements is evidenced by low RSD% values of anodic and cathodic peak currents at various scan rates (Table S2). The evaluation of reusability of the MMEC through CVs recorded every half an hour over an extended period of time (15 h) (Fig. S1b) revealed only minor changes in anodic and cathodic peak currents (Table S3).
The effect of flow rate on electrochemical measurements was evaluated by recording differential pulse voltammetry (DPV) and square wave voltammetry (SWV) responses at various flow rates while 0.1 mM [Fe(CN)6]4− was injected into the MMEC. The obtained results showed no significant differences in peak shape, peak current, or baseline current (Fig. S2a and b). Additionally, chronoamperometry measurements were performed at an applied potential of +0.3 V, corresponding to the oxidation of Fe2+ at different flow rates (Fig. S2c). Herein, two parameters were found to be sensitive to flow rate: the current response upon injection of 0.1 mM [Fe(CN)6]4− solution (S) and the baseline noise (N). We calculated the signal-to-noise (S/N) ratio at different flow rates (Fig. S2d) and found that a flow rate of 20 μL min−1 provides the optimal condition for the experiments.
3.2 Determination of mercury ions at the Au electrode by anodic stripping differential pulse voltammetry (ASDPV)
The bare gold surface appears smooth with a fine grainy texture and lacks distinct surface features, as observed in the SEM image (Fig. 3a). This grainy morphology, characterized by rounded nanometer-sized grains, is also evident in the AFM image (Fig. 3c). Furthermore, the low root mean square (RMS) roughness value calculated from the AFM data (Table S4) is consistent with the expected surface smoothness of bare gold. The electroactive area of Au was calculated following the Randles–Sevcik equation33 (Fig. S3a). The electroactive area of the bare Au electrode (11.5 mm2) differs from the geometric surface area (12.1 mm2) only slightly, which suggests that the Au electrode exhibits only minor defects. The analysis of substrates with bare Au indicates the presence of Si and O and small amounts of metal oxides due to the soda lime glass composition (Fig. 3b). | ||
| Fig. 3 (a) SEM image of bare gold electrode. (b) EDX surface mapping along with elemental analysis of bare gold electrode. (c) AFM image of bare gold electrode. (d) The DPVs recorded at the Au electrode with successive increase in Hg2+ ion concentration (1–100 ppb) in 100 mM HCl and the corresponding calibration curve (inset). | ||
The results of anodic stripping differential pulse voltammetry (ASDPV) show that in the presence of Hg2+ ions in solution, an anodic peak can be observed at the Au electrode, corresponding to the reoxidation of metallic mercury (Hg0), which had been deposited on the electrode surface during the accumulation step (Fig. 3d). Therefore, the parameters of ASDPV were optimized to achieve a high sensitivity for the determination of Hg2+ ions. It must be mentioned that usually an Au rotating electrode is utilized for the determination of Hg2+ ions to avoid hydrogen evolution during the cathodic accumulation of species at the electrode surface. Therefore, the accumulation potential is a crucial parameter in the process. Although the use of a more negative potential is expected to improve the deposition rate of the species and consequently the sensitivity, it resulted in an overlap of the peaks at +0.2 V and that at +0.4 V that corresponds to mercury reoxidation. Moreover, at more negative potentials, hydrogen evolution occurs, which can deteriorate the electrode surface and compromise reproducibility. To mitigate this issue, a deposition potential with an amplitude of +0.3 V was selected. It allows for the application of a stationary electrode (instead of a rotating electrode), as hydrogen evolution does not take place at this potential. The effect of pH on the reoxidation peak current of mercury was investigated in different electrolytes. To ensure the solubility of Hg2+ ions and prevent the formation of insoluble Hg(OH)2 species, acidic media were preferred. A well-defined peak was observed in 100 mM HCl.
The effect of the deposition time on the peak current was studied as well. The peak current increased with time till 360 s (Fig. S8f). As Hg2+ ions need to be detected at very low concentrations, the deposition time was prolonged to 360 s.
To minimize the memory effect and achieve high reproducibility, electrochemical cleaning of the electrode surface was employed by applying a potential greater than the peak potential for a specified duration. Both the cleaning potential and the cleaning time were optimized to be +0.6 V, and 60 s, respectively. The influence of pulse amplitude (Fig. S8a) and pulse time in DPV measurements on the peak current was investigated. The optimum values of these parameters were found to be 70 mV and 5 ms, respectively. Fig. 3d presents the voltammograms recorded at different concentrations of Hg2+ ions at optimum conditions along with the corresponding calibration curve. The main performance characteristics of the MMEC for the determination of Hg2+ ions (together with Pb2+ ions, H2O2, and catechol) are collected in Table 1. A linear concentration dependence of the peak current is observed within the concentration range of 1 to 100 ppb. The LOD and LOQ values were 0.2 and 0.5 ppb, respectively. Low RSD% values of reproducibility (1.2%) and repeatability (4.5%) show the high precision of the method (Table 1). Moreover, the calculated recovery rate of 99.2% (Fig. S7c and d) for buffer samples spiked with a known concentration of mercury ions further demonstrates the high accuracy of the developed sensor in detecting Hg2+ ions. The electrochemical determination of metal ions exhibits inherent selectivity to some extent, as the redox potentials of metal ions typically vary. Additionally, the strong affinity of mercury for the gold surface and formation of gold mercury amalgam34 facilitates the accumulation of mercury, enabling preconcentration and enhancing both selectivity and sensitivity.
| Pb2+ ions | Hg2+ ions | H2O2 | Catechol | |
|---|---|---|---|---|
| a The values 3 s/m and 10 s/m were used for the calculation of LOD and LOQ, respectively, where s represents the standard deviation of 3 repetitive measurements of the lowest concentration in the dynamic range, and m is the slope of the calibration curve.40 ICP-OES: inductively coupled plasma-optical emission spectrometry. | ||||
| Concentration range | 5.0–100.0 ppb | 1.0–100.0 ppb | 10.0–1000.0 μM | 5.0–50.0 μM |
| Sensitivity | 0.14 μA ppb−1 | 0.57 μA ppb−1 | 0.02 μA μM−1 | 0.27 μA μM−1 |
| LODa | 0.9 ppb | 0.2 ppb | 1.6 μM | 0.4 μM |
| LOQa | 2.9 ppb | 0.5 ppb | 5.5 μM | 1.4 μM |
| Reproducibility | 3.0% | 1.2% | 3.5% | 1.7% |
| Repeatability | 1.1% | 4.5% | 3.7% | 3.2% |
| Added (in buffer) | 25.0 ppb | 20.0 ppb | 25.0 μM | 25.0 μM |
| Found | 24.0 ppb | 19.8 ppb | 24.7 μM | 25.1 μM |
| Recovery percentage | 96.2% | 99.2% | 98.8% | 100.3% |
| Added (river water) | 20.0 ppb | 30.0 ppb | 100 μM | 20.0 μM |
| Found | 21.9 ppb | 29.4 ppb | 96.9 μM | 18.4 μM |
| Recovery percentage | 109% | 97.9% | 96.9% | 91.8% |
| Added (wastewater) | — | 20 ppb | 100 μM | 20 μM |
| Found | With MMEC: 1.55 ppm | 22.31 ppb | 95.6 μM | 16.3 μM |
| ICP-OES: 1.63 ppm | ||||
| aRecovery percentage, brelative error | b4.9 | 111.6 | 95.6 | 81.5 |
3.3 Determination of Pb2+ ions at the Au/MF electrode by anodic stripping square wave voltammetry (ASSWV)
The ex situ determination of Pb2+ was carried out at the prepared Au/MF electrode. Fig. 4d shows the ASSWVs of Pb2+ ions at the Au/MF electrode. A well-defined peak is visible at −0.5 V that is not seen in the blank. This peak belongs to the dissolution of pre-concentrated lead. Mercury film is known as an attractive substrate for the sensitive determination of heavy metals due to its capability to form an amalgam with metals.35 Furthermore, coating the Au surface with a layer of mercury film enables applying more negative potentials because it moves the onset of hydrogen evolution to more negative potentials (−1.3 V) compared to bare Au electrode (−0.7 V). | ||
| Fig. 4 (a) SEM image of the Au/MF electrode. (b) EDX surface mapping along with elemental analysis of the Au/MF electrode. (c) AFM image of the Au/MF electrode. (d) The SWVs recorded at the Au/MF electrode with successive increases in Pb2+ ion concentration (5–100 ppb) in acetate buffer pH 4.5, and the corresponding calibration curve (inset). | ||
The parameters that influence the peak's current were optimized. Moderate acidic conditions were utilized to prevent the dissolution of the mercury film at lower pH values and to avoid the precipitation of metal ions as hydroxides at natural and alkaline pHs. An acetate buffer at pH 4.5 was found to be optimal in terms of sensitivity and achieving a well-defined peak. The process of coating bare Au electrode with mercury film was studied. It was found that stepwise increasing of the deposition potential in the coating step results in a uniform film and better reproducibility of the ASSWV results. The mercury film formed on the gold substrate includes irregularly shaped particles with a characteristic size of a few micrometers, as shown in Fig. 4a. The formation of mercury film on the Au substrate was confirmed by elemental analysis as well (Fig. 4b). The AFM image of the prepared Au/MF (Fig. 4c) indicates that its structure differs significantly from that of the bare Au substrate. The increase in both surface area and roughness (RMS) after electrodeposition of the mercury film is remarkable (Table S4).
Studying the effect of accumulation potential on the lead reoxidation peak shows the optimum value to be −1.3 V, where the hydrogen evolution is uninitiated but effective accumulation of Pb2+ ions at electrode surface occurs. To reduce measurement time while maintaining sufficient sensitivity, an accumulation time of 180 s was selected for subsequent analyses. The pulse amplitude and frequency of the SWV were also optimized to be 50 mV and 80 Hz, respectively (Fig. S8c and e). Fig. 4d shows the ASSWVs obtained for various concentrations of Pb2+ ions under optimum conditions and related calibration curve. A linear dependence of the peak current on the concentration is observed within a concentration range of 5 to100 ppb (R2 = 0.990). The LOD and LOQ were determined to be 0.9 and 2.9 ppb, respectively. The low values of repeatability (1.1%) and reproducibility (3.0%) obtained from repeated measurements (Table 1) indicate good precision. The calculated recovery percentage (96.2%) (Fig. S7a and b) for buffer spiked with a known concentration of Pb2+ further shows the accuracy of the developed sensor.
3.4 Determination of H2O2 at the Au/AuNS electrode
The CV of H2O2 at the bare Au electrode (Fig. 5d) shows a broad reduction peak starting from −0.4 V. In contrast, at the Au/AuNS electrode, the onset potential of the H2O2 reduction peak is less negative (−0.2 V), and there is also a significant increase in current. The catalytic effect on H2O2 reduction can be attributed to the peroxidase-like activity of AuNS, as previously reported,12,36 along with the increased electroactive area of the electrode resulting from the deposition of AuNS. The AuNSs are evident in the AFM image (Fig. 5c), which shows an increased dimension in the z-axis following AuNS deposition when compared to the bare Au surface (Fig. 3c). When compared to the bare Au electrode, the electroactive area of the Au/AuNS electrode calculated using the Randles–Sevcik equation was larger by 41%. This is confirmed by AFM analysis that shows that after the modification with AuNS, the surface area increased by approximately 25%, and surface roughness increased nearly 25-fold (Table S4). The SEM image reveals a uniform distribution of AuNSs (Fig. 5a) which cannot be observed at the bare gold surface (Fig. 3a). These AuNSs consist of particles that have irregular shapes with characteristic dimensions of approximately 100 nm. The deposition of AuNS on the Au substrate is evidenced by EDX elemental analysis as well. After the formation of a thick layer of AuNS on the Au surface, the signal from Au showed a remarkable increase in intensity, emerging as the most distinctive feature in the analysis (Fig. 5b). | ||
| Fig. 5 (a) SEM image of the Au/AuNS electrode. (b) EDX surface mapping along with elemental analysis of the Au/AuNS electrode. (c) AFM image of the Au/AuNS electrode. (d) CVs with and without H2O2 at the Au and Au/AuNS electrodes (0.1 M KCl at 50 mV s−1). (e) Chronoamperogram recorded for different concentrations (10–1000 μM) of H2O2 at the Au/AuNS electrode and corresponding calibration curve (inset); applied potential −0.3 V, supporting electrolyte 0.1 M KCl. | ||
Nyquist plots of EIS results (Fig. 6) show a semicircle at high frequencies and a straight line at low frequencies, representing the equivalent circuit in Fig. 6 (inset). The semicircle corresponds to charge transfer resistance (Rct) and double-layer capacitance (Cdl), indicating charge transfer control at the electrode surface. At lower frequencies, the straight line represents Warburg impedance, showing diffusion-limited transport of redox active species. At high frequencies, where no electrochemical reaction occurs, the impedance corresponds to the solution resistance (Rs) and can be estimated from its intersection with the Re(Z) axis. At lower frequencies, where the double-layer capacitance is high, the impedance corresponds to (Rs+ Rct), which can be estimated from the other end of the semicircle. Thus, the Rct value correlates with the diameter of the semicircle. A constant phase element (Q) was used instead of a capacitor in the equivalent circuit fitting (solid lines) to account for non-ideal behavior at the electrode interface. Evidently, the Rct value decreased at the Au/AuNS compared to the bare Au electrode, which can be due to increased surface area and nanostructure-facilitated electron exchange.
 | ||
| Fig. 6 EIS of different electrode configurations recorded in 4 mM [Fe (CN)6]3−/4− in 100 mM KCl, at open circuit potential (OCP) superimposed with 5 mV AC voltage. The points are the experimental data which are fitted to the circuit shown in the inset (solid lines). Rs is the solution resistance, Q is the constant phase element, Rct is the charge transfer resistance, and W is the Warburg diffusion element. | ||
The effect of pH on the electrochemical redox reaction of H2O2 at the Au/AuNS electrode was evaluated over a pH range of 3 to 8 using Britton–Robinson (BR) buffer (Fig. S4). In acidic buffers (pH 3 to 5), the reduction of H2O2 initiates at +0.2 V and no significant change in the reduction current of H2O2 was observed. At pH 6, a shift in the reduction potential of H2O2 toward more negative values (−0.4 V) was observed. This was accompanied by the appearance of an oxidation peak near 0.0 V, attributed to the electrocatalytic oxidation of H2O2 at higher pH values (7 and 8). Adsorption of hydroxide species on the gold surface at higher pH values can catalyze the oxidation of H2O2.37,38 pH 5 was selected for further experiment because it is close to the pH of the water samples, while the electrochemical reduction of H2O2 generates a noticeable current response even at lower potentials. The effect of supporting electrolyte was studied by comparison of the H2O2 reduction in BR buffer and 0.1 M KCl at Au/AuNS. As shown in Fig. S5a, higher cathodic current response, even at lower potential, was obtained in 0.1 KCl compared to BR buffer. Furthermore, the impact of dissolved oxygen on the reduction of H2O2 at the Au/AuNS electrode was assessed by recording the CV of H2O2 in both deaerated and non-deaerated 0.1 M KCl. As follows from Fig. S5b, the shape of the CV remains the same even without the deaeration, and only small changes in the current were observed, which implies that dissolved oxygen does not cause any interfering effect on the reduction of H2O2 at the Au/AuNS. In fact, the dissolved oxygen reduction at the gold electrode proceeds with a higher rate in alkaline media, where the adsorption of oxygen, that is the rate-determining step, is favored. In 0.1 M KCl (pH around 5), a high overpotential is required for the absorption of oxygen which shifts the onset of dissolved oxygen reduction to more negative potentials.39 Thus, an application of a potential close to 0.0 V in chronoamperometry can help to eliminate even this minor effect of dissolved oxygen. Therefore, chronoamperometry at an applied potential of −0.3 V, corresponding to the peak current potential in CV, was employed for the determination of H2O2 (Fig. 5e). The calibration curve (Fig. 5e, inset) shows a linear relationship between concentration and current within a range of 10.0 to 1000 μM. As given in Table 1, the low RSD% values suggest high reproducibility (3.5%) and repeatability (3.7%). The LOD and LOQ were calculated to be 1.6 μM and 5.5 μM, respectively. Recovery percentage values close to 100% suggest high accuracy (Fig. S7g and h).40 As proposed by Garehbaghi et al., using a lower applied potential magnitude enhances the selectivity of H2O2 detection, as fewer redox reactions are likely to occur at less negative potentials.41
3.5 Determination of catechol at the Au/PU/AuNS
In the presence of catechol, there is an oxidation peak and related reduction peak in the CVs (Fig. 7c) around +0.1 V, which are not observed in the blank sample. These redox peaks can be attributed to the catechol redox reaction involving two electrons.42 | ||
| Fig. 7 (a) SEM image of the Au/PU/AuNS electrode. (b) AFM image of the Au/PU/AuNS electrode. (c) The CVs of 1 mM catechol at different electrode configurations; acetate buffer pH 5.7, scan rate 50 mV s−1. (d) The SWVs of catechol (5 to 50 μM) at Au/PU/AuNS along with the corresponding concentration dependence (inset); acetate buffer pH 5.7. | ||
Upon deposition of PU, a shift to more negative potential in oxidation potential was noticed. With the electrodeposition of AuNS, the current substantially increased and further shift to more negative potentials was observed in peak potentials, indicating that the PU/AuNS nanocomposite provides a transducer with promising electrical conductivity and electrochemical catalytic properties. Although the oxidation peak current at the Au/AuNS is only slightly less than that at the Au/PU/AuNs, it shows a shoulder around +0.3 V that overlaps with the catechol oxidation peak at +0.1 V in SWVs, particularly at lower concentrations (not shown). This shoulder can be due to the oxidation of adsorbed catechol at the Au/AuNs. As shown in the EIS results (Fig. 6), a decrease in charge transfer resistance (Rct) was observed by electrodeposition of PU film on the Au electrode that can be due to the larger surface area and porous structure of the PU film.43 A further decrease in charge transfer resistance was observed after deposition of AuNS. The electroactive area of Au/PU/AuNS (19.7 mm2) calculated by the Randles–Sevcik equation shows 71% increase compared to bare Au. Comparing the active surface area of Au/PU/AuNS, which was prepared using only 2 min of electrodeposition, with that of Au/AuNS (16.2 mm2), electrodeposited for 10 min, highlights the significant effect of PU modification on the active surface area of the Au/PU/AuNS electrode. The dependence of catechol redox peak currents on scan rate at the bare Au and Au/PU/AuNS electrodes (Fig. S6a and b) shows that diffusion is the dominant mechanism of mass transport, as the slope of the plot of logarithm of peak current (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ip) versus logarithm of scan rate (logν) is close to the theoretical value of 0.5 at both electrode configurations. However, the higher slope observed for Au/PU/AuNS (0.55) compared to bare Au (0.49) suggests that adsorption influences the overall electrode process in this electrode configuration, as the theoretical slope for an adsorption-controlled process is closer to 1.0.44 As follows from the SEM image (Fig. 7a), the PU layer is porous. This porosity is also evident in the AFM image (Fig. 7b), where a substantial increase in roughness (RMS) and surface area is observed by modification of the electrode with PU and AuNS (Table S4). The surface porosity facilitates the accumulation of the analyte on the electrode. Additionally, the decoration of PU with gold nanostructures (AuNS) enhances its electrical conductivity, as evidenced by a decrease in the real part of the impedance (Fig. 6). It should also be noted that the formation of a layer of PU on the Au surface protects it from deterioration. Therefore, the presence of PU/AuNS nanocomposite components can improve both sensitivity and reproducibility.
Ip) versus logarithm of scan rate (logν) is close to the theoretical value of 0.5 at both electrode configurations. However, the higher slope observed for Au/PU/AuNS (0.55) compared to bare Au (0.49) suggests that adsorption influences the overall electrode process in this electrode configuration, as the theoretical slope for an adsorption-controlled process is closer to 1.0.44 As follows from the SEM image (Fig. 7a), the PU layer is porous. This porosity is also evident in the AFM image (Fig. 7b), where a substantial increase in roughness (RMS) and surface area is observed by modification of the electrode with PU and AuNS (Table S4). The surface porosity facilitates the accumulation of the analyte on the electrode. Additionally, the decoration of PU with gold nanostructures (AuNS) enhances its electrical conductivity, as evidenced by a decrease in the real part of the impedance (Fig. 6). It should also be noted that the formation of a layer of PU on the Au surface protects it from deterioration. Therefore, the presence of PU/AuNS nanocomposite components can improve both sensitivity and reproducibility.
To assess the sensitivity and efficiency, the Au/PU/AuNS was used in the electrochemical determination of catechol. The resulting SWVs (Fig. 7d) and calibration curve (Fig. 7d, inset) demonstrate high sensitivity. The concentration dependence of oxidation peak current was found to be linear within the concentration range of 5.0–50.0 μM and LOD and LOQ were calculated to be 0.4 μM and 1.4 μM, respectively. The precision and accuracy were expressed in terms of reproducibility (1.7%), repeatability (3.2%), and recovery percentage (100%) in buffer (Fig. S7e and f and Table 1). The occurrence of the oxidation peak at a small potential amplitude of +0.1 V avoids interfering effect from other redox active substances, leading to a high selectivity.
3.6 Multiplexed determination
Based on the data presented in Table 2, the limits of detection (LODs) achieved in this study are superior to or comparable with those reported in other electrochemical sensing studies, except for a few that employed highly sophisticated electrode modification strategies. This makes the MMEC suitable for sensitive and multiplexed analysis of water pollutants. The instrumentation and its use in multiplexed monitoring are illustrated in the Appendix video. The developed MMEC enables the multiplexed detection of water pollutants, including both organic and inorganic substances, by employing highly sensitive electrochemical techniques with distinct parameters tailored for each specific analyte. Additionally, the measurement solution for each analyte can be selected independently, as each measurement is conducted in a spatially separated channel. In this study, acidic pH solutions were preferred for Hg2+ and Pb2+ ions to prevent the precipitation of metals ions in the form of metal hydroxide, and for the measurement of catechol and H2O2 different buffers with pH values of ∼5.0 and 5.7 were employed, respectively. Due to its compact size, high reproducibility and reusability, and the feasibility of a variety of surface modifications, the developed MMEC provides a straightforward platform for both online and laboratory analysis.| Analyte | Maximum allowable concentration | LOD | Linear range | Ref. |
|---|---|---|---|---|
| a World Health Organization. b Public Health England. c United States Environmental Protection Agency. | ||||
| Hg2+ | 1.0 ppba | 0.8 ppb | 2.0–100 ppb | 58 |
| 1.8 ppb | 2.0–20 ppb | 59 | ||
| 0.2 ppb | 10–120 ppb | 60 | ||
| 0.2 ppb | 1.0–100 ppb | This work | ||
| Pb2+ | 50.0 ppba | 2.6 ppb | 8.0–124 ppb | 61 |
| 1.1 ppb | 30–180 ppb | 62 | ||
| 0.8 ppb | 2.0–50 ppb | 63 | ||
| 0.9 ppb | 5.0–100 ppb | This work | ||
| H2O2 | 22.0 μMb | 3.0 μM | 500–4500 μM | 41 |
| 16 μM | 100–800 μM | 64 | ||
| 7.5 μM | 100–800 μM | 65 | ||
| 1.6 μM | 10–1000.0 μM | This work | ||
| Catechol | 30.0 μMc | 0.6 μM | 5.8–103 μM | 66 |
| 0.5 μM | 2.5–80 μM | 67 | ||
| 1.2 μM | 3.9–14.5 μM | 68 | ||
| 0.4 μM | 0.5–50 μM | This work | ||
Moreover, Table S5 lists selected electrochemical platforms enabling multiplexed sensing. For example, multiple SPEs have been combined into an electrode array connected to a multichannel potentiostat.45–47 However, reusability or integration with microfluidics was not demonstrated. Another approach utilizes electrode arrays composed of multiple working electrodes patterned on a substrate (typically glass or plastic), which share a single reference and counter electrode.48–54 This configuration is advantageous for performing identical electrochemical techniques with the same operational parameters across multiple working electrodes. However, it is not compatible with the parallel use of different electrochemical techniques. The developed MMEC represents a promising platform for multiplexed electrochemical analysis, as it comprises four independent electrochemical cells, each equipped with its own reference and counter electrodes. This configuration enables the parallel use of different electrochemical techniques under distinct operating parameters within each cell. Furthermore, its integration into a microfluidic system provides the MMEC with the advantage of precise sample handling and automation. Finally, the reusability of the MMEC offers a significant improvement over the previously reported platforms.
3.7 Interference study
Since the developed MMEC was aimed at monitoring water pollutants, an interference study was conducted to assess the selectivity of each specific channel toward its target analyte. For each analyte, the effects of other analytes and potential interfering substances were examined. As follows from Fig. S9a, no major interferences on catechol peak current upon the addition of NaCl, CaCl2, MgCl2, glucose, and Pb2+ (see section 2.3.9 in the SI) were observed. The addition of Hg2+ and H2O2 increased the peak current by almost 20%. The H2O2 current response was unaffected by the addition of the interfering species except for ascorbic acid, which resulted in a decrease of the current response by 82% (Fig. S9b), probably due to its reducing properties that convert H2O2 to H2O.55 No significant interference was observed in the Hg2+ reoxidation peak in the presence of the interfering species, except for a slight decrease (by about 18%) when catechol was added (Fig. S9c). Catechol and As3+ were found to have the most significant effect on the Pb2+ reoxidation peak current, reducing it by about 20% and 25%, respectively (Fig. S9d). The interference arises either from the chemical reaction between the analyte and the interfering species, alteration of the electrode's surface by the interfering species, or overlapping redox peaks of the interfering species and analyte.Overall, the results show that apart from the significant effect of ascorbic acid on H2O2 current response, no substantial interference was observed. As detailed in sections 3.2–3.5, several factors contribute to the selectivity of the developed methods. The selectivity arises from the specific affinity between the electrode surface and the analyte. For example, in the determination of Pb2+, selectivity is achieved through the formation of an amalgam, which facilitates the preconcentration of mercury at the Au/MF surface. Additionally, the inherent interaction between gold and mercury further promotes the accumulation of mercury on the gold electrode. Furthermore, the oxidation or reduction of each metal ion occurs at a distinct potential, minimizing interference from other metal ions present in the solution. Additionally, the selectivity stems from the potential magnitude applied in the analysis. Applying a potential close to 0.0 V (e.g. −0.3 V for the reduction of H2O2 and +0.1 V for catechol) to monitor the oxidation/reduction of the analyte for quantification in both chronoamperometry and voltammetry enhances selectivity by minimizing the oxidation or reduction of potential interfering species in the solution.
3.8 Real sample analysis
The accuracy of the determination of four analytes in Vltava River water (Prague, Czech Republic) by the MMEC approach was found to be high, as evident from the achieved recovery percentages (within the range of 91.8% to 109% for different analytes), as shown in Table 1 and Fig. S10. The results of the analysis of industrial wastewater with the developed MMEC (Fig. S11) show good recovery percentages for samples spiked with a known concentration of H2O2, catechol, or Hg2+, which were not initially present in the wastewater. The analysis of Pb2+ ions that were present in the wastewater sample shows a good agreement with the results obtained by the reference method (inductively coupled plasma-optical emission spectrometry). Although the approach requires only minimal sample pretreatment (filtering and dilution), it provides a highly efficient platform for multiplexed monitoring of pollutants in river water and industrial wastewater with complicated matrices. Although various metal ions and anions are present in the wastewater sample at high concentrations, accurate determination is still achieved due to the method's selectivity. The selectivity arises from the affinity between the electrode surface and the target analyte. Therefore, with only minimal sample pretreatment (filtration), the developed MMEC provides a highly efficient platform for multiplexed monitoring of pollutants in both river water and industrial wastewater with complex matrices.4. Conclusion
We report on the development of a novel microfluidic multichannel electrochemical cell (MMEC) and its successful implementation for water quality monitoring. It allows for a separate modification of each channel to support sensitive detection of the selected analyte. Moreover, even when different electrochemical techniques with varying parameters were applied simultaneously in each channel, no interference or crosstalk between channels was observed. The MMEC is a compact device that can provide response in a very quick manner (typically less than 4 min) making it a suitable candidate for the online monitoring of water pollutants. In order to illustrate the potential of MMEC for water quality monitoring, the system was applied for the multiplexed detection of lead ions, mercury ions, catechol, and hydrogen peroxide, and the limits of detection were determined to be 0.9 ppb, 0.6 ppb, 0.4 μM and, 4.2 μM, respectively. The system was also tested for analysis of river water and industrial wastewater samples, and good recovery percentages (ranging from 91.8% to 109% for river water and from 81.8% to 111.6% for industrial wastewater) were achieved. The experiments also demonstrated excellent reproducibility and reusability of the MMEC. Furthermore, the proposed MMEC approach demonstrates the potential of electrochemical methods for miniaturization. This may, in the future, facilitate the development of on-site automated analytical devices for water quality monitoring. The commercial SPEs are inexpensive and convenient; however, their performance is limited by poor reproducibility, and they are typically constrained to single use.56,57 Although the MMEC may appear more complicated than commercial SPEs, the assembly is rather straightforward and quick (typically takes less than 5 min). The developed MMEC remains cost-effective (although slightly more expensive than SPEs), but in contrast to SPEs, it exhibits good reproducibility and reusability (see data in Fig. S1). Moreover, we have demonstrated that the MMEC approach enables desirable surface modifications, particularly electrochemical. Furthermore, the MMEC is well suited for multiplexed analysis, as demonstrated by our experiments, which successfully detected four different types of analytes. In contrast, multiplexed analysis based on individual SPEs, each requiring its own specific surface modification and distinct electrochemical parameters, requires a considerably more complex setting and experimental procedure. The promising performance of the developed MMEC convincingly demonstrates its potential for use in food safety and clinical analysis, particularly for the multiplexed determination of biomarkers for early diagnosis.Author contributions
Tong Liu: formal analysis, investigation, methodology, validation, writing – original draft. Amir M. Ashrafi: conceptualization, data curation, formal analysis, investigation, methodology, supervision, validation, writing – original draft, and writing – review & editing. Ivo Tichý: technical support, writing – original draft. Jiří Homola: writing – review & editing, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Data availability data
Data for this article are available at Zenodo at https://doi.org/10.5281/zenodo.15230280. Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5lc00825e.Acknowledgements
This research was supported by the Czech Science Foundation (contract 20–23787X). This work has been funded by a grant from the Programme Johannes Amos Comenius under the Ministry of Education, Youth and Sports of the Czech Republic CZ.02.01.01/00/22_008/0004558 Advanced MUltiscaLe materials for key Enabling Technologies. We acknowledge Jan Vaniš for providing SEM and EDX measurements, Petr Dvorak for AFM imaging, and Karel Chadt for visualization. We also appreciate the kind collaboration of Martin Pešek and Ing. Hana Palková, Ph.D., the laboratory manager at Kovohutě (Příbram, Czech Republic).References
- J. Singh, P. Yadav, A. K. Pal and V. Mishra, Water Pollutants: Origin and Status, in Sensors in Water Pollutants Monitoring: Role of Material, ed. D. Pooja, P. Kumar, P. Singh and S. Patil, Springer Singapore, Singapore, 2020, pp. 5–20, DOI:10.1007/978-981-15-0671-0_2.
- M. A. D. Silveira, D. L. Ribeiro, G. M. Vieira, N. R. Demarco and L. P. G. d'Arce, Direct and Indirect Anthropogenic Contamination in Water Sources: Evaluation of Chromosomal Stability and Cytotoxicity Using the Allium cepa Test, Bull. Environ. Contam. Toxicol., 2018, 100(2), 216–220, DOI:10.1007/s00128-017-2232-1.
- R. K. Mishra, S. S. Mentha, Y. Misra and N. Dwivedi, Emerging pollutants of severe environmental concern in water and wastewater: A comprehensive review on current developments and future research, Water-Energy Nexus, 2023, 6, 74–95, DOI:10.1016/j.wen.2023.08.002.
- R. P. Schwarzenbach, T. Egli, T. B. Hofstetter, U. Von Gunten and B. Wehrli, Global water pollution and human health, Annu. Rev. Environ. Resour., 2010, 35(1), 109–136, DOI:10.1146/annurev-environ-100809-125342.
- W. Raza, J. Lee, N. Raza, Y. Luo, K.-H. Kim and J. Yang, Removal of phenolic compounds from industrial waste water based on membrane-based technologies, J. Ind. Eng. Chem., 2019, 71, 1–18, DOI:10.1016/j.jiec.2018.11.024.
- E. Singovszka, M. Balintova and N. Junakova, The impact of heavy metals in water from abandoned mine on human health. SN, Appl. Sci., 2020, 2(5), 934, DOI:10.1007/s42452-020-2731-2.
- A. D. da Silva, W. J. Paschoalino, R. C. Neto and L. T. Kubota, Electrochemical point-of-care devices for monitoring waterborne pathogens: Protozoa, bacteria, and viruses – An overview, Case Stud. Chem. Environ. Eng., 2022, 5, 100182, DOI:10.1016/j.cscee.2022.100182.
- M. Khodaparast, D. Sharley, S. Marshall and T. Beddoe, Advances in point-of-care and molecular techniques to detect waterborne pathogens, npj Clean Water, 2024, 7(1), 74, DOI:10.1038/s41545-024-00368-9.
- N. Mandal, S. Mitra and D. Bandyopadhyay, Paper-Sensors for Point-of-Care Monitoring of Drinking Water Quality, IEEE Sens. J., 2019, 19(18), 7936–7941, DOI:10.1109/JSEN.2019.2919269.
- A. T. Demetillo, M. V. Japitana and E. B. Taboada, A system for monitoring water quality in a large aquatic area using wireless sensor network technology, Sustainable Environ. Res., 2019, 29(1), 12, DOI:10.1186/s42834-019-0009-4.
- E. C. Okpara, T. O. Ajiboye, D. C. Onwudiwe and O. B. Wojuola, Optical and electrochemical techniques for Point-of-Care water quality monitoring: A review. Results, Chemistry, 2023, 5, 100710, DOI:10.1016/j.rechem.2022.100710.
- A. M. Ashrafi, Z. Bytesnikova, J. Barek, L. Richtera and V. Adam, A critical comparison of natural enzymes and nanozymes in biosensing and bioassays, Biosens. Bioelectron., 2021, 192, 113494, DOI:10.1016/j.bios.2021.113494.
- A. M. Ashrafi, A. Mukherjee, A. Saadati, F.-M. Matysik, L. Richtera and V. Adam, Enhancing the substrate selectivity of enzyme mimetics in biosensing and bioassay: Novel approaches, Adv. Colloid Interface Sci., 2024, 331, 103233, DOI:10.1016/j.cis.2024.103233.
- A. Mukherjee, L. Richtera, V. Adam and A. Ashrafi, Chapter 11 - Role of magnetic nanoparticles in development of biosensors for viral infection diagnostics, in Advanced Biosensors for Virus Detection, ed. R. Khan, A. Parihar, A. Kaushik and A. Kumar, Academic Press, 2022, pp. 189–202, DOI:10.1016/B978-0-12-824494-4.00002-3.
- S. Mukherjee, A. Mukherjee, Z. Bytesnikova, A. M. Ashrafi, L. Richtera and V. Adam, 2D graphene-based advanced nanoarchitectonics for electrochemical biosensors: Applications in cancer biomarker detection, Biosens. Bioelectron., 2024, 250, 116050, DOI:10.1016/j.bios.2024.116050.
- H. Adam, S. C. B. Gopinath, M. K. Md Arshad, T. Adam, U. Hashim and Z. Sauli, et al.,Integration of microfluidic channel on electrochemical-based nanobiosensors for monoplex and multiplex analyses: An overview, J. Taiwan Inst. Chem. Eng., 2023, 146, 104814, DOI:10.1016/j.jtice.2023.104814.
- Y. Huang and A. J. Mason, Lab-on-CMOS integration of microfluidics and electrochemical sensors, Lab Chip, 2013, 13(19), 3929–3934, 10.1039/C3LC50437A.
- B. S. Ferguson, S. F. Buchsbaum, T.-T. Wu, K. Hsieh, Y. Xiao, R. Sun and H. T. Soh, Genetic Analysis of H1N1 Influenza Virus from Throat Swab Samples in a Microfluidic System for Point-of-Care Diagnostics, J. Am. Chem. Soc., 2011, 133(23), 9129–9135, DOI:10.1021/ja203981w.
- J. Luo, X. Fang, D. Ye, H. Li, H. Chen, S. Zhang and J. Kong, A real-time microfluidic multiplex electrochemical loop-mediated isothermal amplification chip for differentiating bacteria, Biosens. Bioelectron., 2014, 60, 84–91, DOI:10.1016/j.bios.2014.03.073.
- C. K. Tang, A. Vaze, M. Shen and J. F. Rusling, High-Throughput Electrochemical Microfluidic Immunoarray for Multiplexed Detection of Cancer Biomarker Proteins, ACS Sens., 2016, 1(8), 1036–1043, DOI:10.1021/acssensors.6b00256.
- D. P. Wasalathanthri, V. Mani, C. K. Tang and J. F. Rusling, Microfluidic Electrochemical Array for Detection of Reactive Metabolites Formed by Cytochrome P450 Enzymes, Anal. Chem., 2011, 83(24), 9499–9506, DOI:10.1021/ac202269t.
- L. Zhu, X. Liu, J. Yang, Y. He and Y. Li, Application of Multiplex Microfluidic Electrochemical Sensors in Monitoring Hematological Tumor Biomarkers, Anal. Chem., 2020, 92(17), 11981–11986, DOI:10.1021/acs.analchem.0c02430.
- Y.-H. Chang, C.-L. Hsu, C.-J. Yuan, S.-F. Tang, H.-J. Chiang, H.-D. Jang and K.-S. Chang, Improvement of the inter-electrode reproducibility of screen-printed carbon electrodes by oxygen plasma etching and an image color level method for quality control, Mater. Sci. Eng., C, 2011, 31(7), 1265–1270, DOI:10.1016/j.msec.2011.01.001.
- R. Zeitoun, V. Adamchuk, J. Warland and A. Biswas, Paper-polished carbon screen-printed electrodes increase reusability and enhance performance in phosphomolybdate electrochemical detection, J. Electroanal. Chem., 2021, 890, 115229, DOI:10.1016/j.jelechem.2021.115229.
- S. Anubhav, S. Anuj, K. V. Rohit, L. C. Rushikesh, P. P. Pritam and N. Varad, et al., Heavy Metal Contamination of Water and Their Toxic Effect on Living Organisms, in The Toxicity of Environmental Pollutants, ed. D. Daniel Junqueira and D. Palma de O, IntechOpen, Rijeka, 2022, ch. 2, DOI:10.5772/intechopen.105075.
- A. A. Aghapour, G. Moussavi and K. Yaghmaeian, Biological degradation of catechol in wastewater using the sequencing continuous-inflow reactor (SCR), J. Environ. Health Sci. Eng., 2013, 11(1), 3, DOI:10.1186/2052-336X-11-3.
- J. J. Morris, A. L. Rose and Z. Lu, Reactive oxygen species in the world ocean and their impacts on marine ecosystems, Redox Biol., 2022, 52, 102285, DOI:10.1016/j.redox.2022.102285.
- J. J. Rueda Márquez, I. Levchuk and M. Sillanpää, Application of Catalytic Wet Peroxide Oxidation for Industrial and Urban Wastewater Treatment: A Review, Catalysts, 2018, 8(12), 673, DOI:10.3390/catal8120673.
- Y. Huang, M. Kong, S. Coffin, K. H. Cochran, D. C. Westerman and D. Schlenk, et al., Degradation of contaminants of emerging concern by UV/H2O2 for water reuse: Kinetics, mechanisms, and cytotoxicity analysis, Water Res., 2020, 174, 115587, DOI:10.1016/j.watres.2020.115587.
- G. S. Wang, C. H. Liao, H. W. Chen and H. C. Yang, Characteristics of Natural Organic Matter Degradation in Water by UV/H2O2 Treatment, Environ. Technol., 2006, 27(3), 277–287, DOI:10.1080/09593332708618638.
- D. Nečas and P. Klapetek, Gwyddion: an open-source software for SPM data analysis, Cent. Eur. J. Phys., 2012, 10(1), 181–188, DOI:10.2478/s11534-011-0096-2.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- J. E. B. Randles, Kinetics of rapid electrode reactions, Discuss. Faraday Soc., 1947, 1, 11–19, 10.1039/DF9470100011.
- M. Fiałkowski, P. Grzeszczak, R. Nowakowski and R. Hołyst, Absorption of Mercury in Gold Films and Its Further Desorption: Quantitative Morphological Study of the Surface Patterns, J. Phys. Chem. B, 2004, 108(16), 5026–5030, DOI:10.1021/jp0365413.
- A. Economou and P. R. Fielden, Mercury film electrodes: developments, trends and potentialities for electroanalysis, Analyst, 2003, 128(3), 205–213, 10.1039/B201130C.
- S. Garehbaghi, A. M. Ashrafi, V. Adam and L. Richtera, Surface modification strategies and the functional mechanisms of gold nanozyme in biosensing and bioassay, Mater. Today Bio, 2023, 20, 100656, DOI:10.1016/j.mtbio.2023.100656.
- K. Liu, S. He, L. Li, Y. Liu, Z. Huang and T. Liu, et al., Spectroscopically clean Au nanoparticles for catalytic decomposition of hydrogen peroxide, Sci. Rep., 2021, 11(1), 9709, DOI:10.1038/s41598-021-89235-y.
- B. J. Plowman, N. Thompson and A. P. O'Mullane, Probing the surface oxidation of chemically synthesised gold nanospheres and nanorods, Gold Bull., 2014, 47(3), 177–183, DOI:10.1007/s13404-014-0141-1.
- P. Rodriguez and M. T. M. Koper, Electrocatalysis on gold, Phys. Chem. Chem. Phys., 2014, 16(27), 13583–13594, 10.1039/C4CP00394B.
- H. Y. Aboul-Enein and A. Sibel, Ozkan: Electroanalytical Methods in Pharmaceutical Analysis and Their Validation, Chromatographia, 2012, 75(13), 811, DOI:10.1007/s10337-012-2268-7.
- S. Garehbaghi, V. Adam, J. Přibyl, L. Richtera and A. M. Ashrafi, An enzyme cascade biosensor based on multiwalled carbon nanotube-RuO2 nanocomposite for selective amperometric determination of lactose in milk samples, Microchem. J., 2024, 204, 111138, DOI:10.1016/j.microc.2024.111138.
- F. Jalali, A. M. Ashrafi and D. Nematollahi, Measurement of Dissolved Oxygen in Biological Fluids by Using a Modified Carbon Paste Electrode, Electroanalysis, 2009, 21(2), 201–205, DOI:10.1002/elan.200804410.
- M. Gashu, B. A. Aragaw and M. Tefera, Voltammetric determination of oxytetracycline in milk and pharmaceuticals samples using polyurea modified glassy carbon electrode, J. Food Compos. Anal., 2023, 117, 105128, DOI:10.1016/j.jfca.2023.105128.
- E. Laviron, A multilayer model for the study of space distributed redox modified electrodes: Part I. Description and discussion of the model, J. Electroanal. Chem. Interfacial Electrochem., 1980, 112(1), 1–9, DOI:10.1016/S0022-0728(80)80002-7.
- I. N. Hanitra, F. Criscuolo, N. Pankratova, S. Carrara and G. D. Micheli, Multichannel Front-End for Electrochemical Sensing of Metabolites, Drugs, and Electrolytes, IEEE Sens. J., 2020, 20(7), 3636–3645, DOI:10.1109/JSEN.2019.2959885.
- X. Liu and P. B. Lillehoj, Embroidered electrochemical sensors for biomolecular detection, Lab Chip, 2016, 16(11), 2093–2098, 10.1039/C6LC00307A.
- Y. Wan, W. Deng, Y. Su, X. Zhu, C. Peng and H. Hu, et al., Carbon nanotube-based ultrasensitive multiplexing electrochemical immunosensor for cancer biomarkers, Biosens. Bioelectron., 2011, 30(1), 93–99, DOI:10.1016/j.bios.2011.08.033.
- C. Asci, A. Sharma, R. Del-Rio-Ruiz and S. Sonkusale, A multiplexed sensing system with multimodal readout electronics and thread-based electrochemical sensors for high throughput screening, Electrochim. Acta, 2024, 501, 144798, DOI:10.1016/j.electacta.2024.144798.
- Q. Bao, G. Li, Z. Yang, J. Wei, W. Cheng, Z. Qu and L. Lin, A Time-Division Multiplexing Multi-Channel Micro-Electrochemical Workstation with Carbon-Based Material Electrodes for Online L-Trosine Detection, Sensors, 2023, 23(14), 6252, DOI:10.3390/s23146252.
- J. Gao, L. Jeffries, K. E. Mach, D. W. Craft, N. J. Thomas and V. Gau, et al., A Multiplex Electrochemical Biosensor for Bloodstream Infection Diagnosis, SLAS Technol., 2017, 22(4), 466–474, DOI:10.1177/2211068216651232.
- E. A. Lima, R. M. Snider, R. S. Reiserer, J. R. McKenzie, D. W. Kimmel and S. E. Eklund, et al., Multichamber multipotentiostat system for cellular microphysiometry, Sens. Actuators, B, 2014, 204, 536–543, DOI:10.1016/j.snb.2014.07.126.
- D. Quinton, A. Girard, L. T. Thi Kim, V. Raimbault, L. Griscom and F. Razan, et al., On-chip multi-electrochemical sensor array platform for simultaneous screening of nitric oxide and peroxynitrite, Lab Chip, 2011, 11(7), 1342–1350, 10.1039/C0LC00585A.
- J. Wu, F. Yan, J. Tang, C. Zhai and H. Ju, A disposable multianalyte electrochemical immunosensor array for automated simultaneous determination of tumor markers, Clin. Chem., 2007, 53(8), 1495–1502, DOI:10.1373/clinchem.2007.086975.
- Y. Wu, P. Xue, K. M. Hui and Y. Kang, A paper-based microfluidic electrochemical immunodevice integrated with amplification-by-polymerization for the ultrasensitive multiplexed detection of cancer biomarkers, Biosens. Bioelectron., 2014, 52, 180–187, DOI:10.1016/j.bios.2013.08.039.
- G.-Y. Chen, Y.-H. Lin, C.-H. Fu, C.-H. Lin, B. Muthiah and W. V. Espulgar, et al., Quantification of peracetic acid (PAA) in the H2O2 + acetic acid reaction by the wavelength shift analysis in near-UV/visible absorption region, Anal. Sci., 2024, 40(3), 489–499, DOI:10.1007/s44211-023-00481-8.
- G. Paimard, E. Ghasali and M. Baeza, Screen-Printed Electrodes: Fabrication, Modification, and Biosensing Applications, Chemosensors, 2023, 11(2), 113, DOI:10.3390/chemosensors11020113.
- B. Repič, K. Radan, G. Marolt, A. B. Golob and D. Kuscer, Effect of processing temperature on performance of screen-printed graphite electrodes, Mater. Chem. Phys., 2025, 337, 130455, DOI:10.1016/j.matchemphys.2025.130455.
- J. Xia, Q. Wang, M. Yang and H. Wu, Reliable electroanalysis of Hg(II) in water via flower-like porous MnCo2O4: Excellent multilayer adsorption and (Mn, Co)(II)/(Mn, Co)(III) cycles, Sens. Actuators, B, 2021, 326, 129008, DOI:10.1016/j.snb.2020.129008.
- Z. Yang, Voltammetry for quantitative determination of trace mercury ions in water via acetylene black modified carbon paste electrode, Alexandria Eng. J., 2024, 87, 107–113, DOI:10.1016/j.aej.2023.12.007.
- Q.-X. Bao, Y. Liu, Y.-Q. Liang, R. Weerasooriya, H. Li, Y.-C. Wu and X. Chen, Tea polyphenols mediated Zero-valent Iron/Reduced graphene oxide nanocomposites for electrochemical determination of Hg2+, J. Electroanal. Chem., 2022, 917, 116428, DOI:10.1016/j.jelechem.2022.116428.
- H. Han, D. Pan, Y. Li, J. Wang and C. Wang, Stripping Voltammetric Determination of Lead in Coastal Waters With a Functional Micro-Needle Electrode, Front. Mar. Sci., 2020, 7, 196, DOI:10.3389/fmars.2020.00196.
- S. Chen, Y. Wang, B. Zhang, M. Li, J. Zhang and Q. Hu, et al., Electrochemical Detection of Cd2+ and Pb2+ in Wastewater by Amino C-dot-MOF/GCE, ECS J. Solid State Sci. Technol., 2024, 13(10), 107001, DOI:10.1149/2162-8777/ad7b73.
- S. Hu, S. Zhang, J. Qin, K. Cai, C. Peng and L. Luo, et al., Simultaneous determination of lead and cadmium in water by metal oxide framework complex-modified glassy carbon electrodes, Microchem. J., 2024, 205, 111154, DOI:10.1016/j.microc.2024.111154.
- A. Mukherjee, A. M. Ashrafi, Z. Bytesnikova, P. Svec, L. Richtera and V. Adam, An investigation on the multiple roles of CeO2 nanoparticle in electrochemical sensing: Biomimetic activity and electron acceptor, J. Electroanal. Chem., 2023, 935, 117301, DOI:10.1016/j.jelechem.2023.117301.
- A. M. Ashrafi, M. Sýs, E. Sedláčková, A. Shaaban Farag, V. Adam, J. Přibyl and L. Richtera, Application of the Enzymatic Electrochemical Biosensors for Monitoring Non-Competitive Inhibition of Enzyme Activity by Heavy Metals, Sensors, 2019, 19(13), 2939, DOI:10.3390/s19132939.
- B. Huang, C. Yao, E. Wang, S. Du, J. Yang and X. Lu, Simultaneous Determination of Catechol and Hydroquinone on Nano-Co/L-Cysteine Modified Glassy Carbon Electrode, Electroanalysis, 2022, 34(2), 405–414, DOI:10.1002/elan.202100047.
- M. Stoytcheva, Z. Velkova, V. Gochev, B. Valdez and M. Curiel, An Affordable and Efficient Graphite-Coated Electrode for the Voltammetric Determination of Catechol, ChemistrySelect, 2024, 9(20), e202401418, DOI:10.1002/slct.202401418.
- P. Nochit, S.-u. Paitoon and S. Teepoo, Multiwalled Carbon Nanotube (MWCNT) Based Electrochemical Paper-Based Analytical Device (ePAD) for the Determination of Catechol in Wastewater, Anal. Lett., 2021, 54(15), 2484–2497, DOI:10.1080/00032719.2021.1872591.
| This journal is © The Royal Society of Chemistry 2026 |