
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual kinetic effect from confined iron nanoparticles in zeolite modulates high-temperature catalytic NO reduction and NH3 oxidation
Xinlin Xie,
Jibin Yuan,
Lei Liu,
Hanzi Liu* and
Zhiqiang Sun *
*
Hunan Engineering Research Center of Clean and Low-Carbon Energy Technology, School of Energy Science and Engineering, Central South University, Changsha 410083, China. E-mail: liuhz@csu.edu.cn; zqsun@csu.edu.cn
First published on 15th December 2025
Abstract
The selective catalytic reduction of ammonia (NH3-SCR) is a promising technology for abating nitrogen oxides (NOx), yet its application at high temperatures is severely hampered by the over-oxidation of ammonia, leading to a trade-off between NOx conversion and N2 selectivity. Herein, we construct a series of Fe-exchanged ZSM-5 catalysts with controlled Fe loadings (0.05–0.5 wt%) to decouple the competing reaction pathways. The optimized 0.1Fe@ZSM-5 catalyst achieves 83.0% NOx conversion at 700 °C and maintains exceptional stability for over 120 h under harsh conditions, representing a significant performance enhancement. Mechanistic investigations combining kinetic modeling and in situ spectroscopy reveal a dual kinetic regime, governed by the size of the Fe species. Catalysts with low Fe loadings favor the standard SCR pathway via stable NH4+ and  intermediates, whereas catalysts with higher loadings and larger Fe nanoparticles promote the undesirable oxidation of ammonia to NOx. The result identifies that the optimal catalytic sites for high-temperature SCR rely on a delicate balance, activating ammonia for the desired reaction while suppressing its subsequent over-oxidation. These findings provide new implications for advanced catalyst design by tuning the active site structure to navigate competing reaction kinetics.
intermediates, whereas catalysts with higher loadings and larger Fe nanoparticles promote the undesirable oxidation of ammonia to NOx. The result identifies that the optimal catalytic sites for high-temperature SCR rely on a delicate balance, activating ammonia for the desired reaction while suppressing its subsequent over-oxidation. These findings provide new implications for advanced catalyst design by tuning the active site structure to navigate competing reaction kinetics.
Keywords: Selective catalytic reduction; NH3 oxidation; Kinetics modeling; Brønsted acid site; N2 selectivity.
1 Introduction
The abatement of nitrogen oxides (NOx) from high-temperature exhaust streams is a critical challenge in emerging industrial scenarios, including the after-treatment of diesel engine emissions, end-of-pipe control for ammonia–hydrogen co-firing, and the management of emissions from coal-fired boilers under rapid load changes.1,2 The selective catalytic reduction of NOx with ammonia (NH3-SCR) over Fe-based zeolites represents a promising technology due to its exceptional efficacy in promoting NH3 dehydrogenation and N–N bond formation.3 However, the catalysts are susceptible to deactivation by sulfur poisoning and hydrothermal aging.4 More critically, the over-oxidation of ammonia at elevated temperatures severely suppresses the overall NOx reduction activity.5–8 Our previous work has shown that incorporating transition metals can enhance thermal-sulfur stability through synergistic effects;9 however, a fundamental understanding of the NH3 oxidation mechanism and its kinetics at high temperatures remains elusive. Specifically, suppressing ammonia over-oxidation above 700 °C with sufficient deNOx efficiency shows a significant yet challenging task.The catalytic oxidation of ammonia over metal-based catalysts has been studied extensively, both for its abatement as a toxic pollutant and for its potential in chemical synthesis, yielding products such as N2, NO, and N2O.10–12 The ammonia activation is facilitated by metal–oxygen sites via abstraction of a H+ proton, where the stronger oxidizability of the metal–oxygen bonds plays a crucial role. For instance, iron oxides possess a relatively low Fe–O bond energy, enabling the activation of NH3. In the case of NH3-SCR, rational ammonia oxidation is beneficial, as it accelerates the initial dehydrogenation to form the  intermediate, which couples with NO to produce N2. However, under excessively oxidizing conditions, lattice oxygen can drive the progressive dehydrogenation of adsorbed
intermediate, which couples with NO to produce N2. However, under excessively oxidizing conditions, lattice oxygen can drive the progressive dehydrogenation of adsorbed  . This process leads to the formation of highly reactive NH* or N* radicals, which are subsequently oxidized to NOx, thereby lowering the N2 selectivity and suppressing the catalytic performance for the desired NOx reduction.13,14 This inherent trade-off between NH3 conversion and N2 selectivity poses a challenge for NH3-SCR reactions on activated metal oxide surfaces.
. This process leads to the formation of highly reactive NH* or N* radicals, which are subsequently oxidized to NOx, thereby lowering the N2 selectivity and suppressing the catalytic performance for the desired NOx reduction.13,14 This inherent trade-off between NH3 conversion and N2 selectivity poses a challenge for NH3-SCR reactions on activated metal oxide surfaces.
Fe-based zeolites are energetically favored for stable NO reduction and high N2 selectivity, owing to their inherent spatial confinement and shape-selective properties.7 Previous studies have focused on the kinetics and mechanisms of the interaction between NOx and NH3.15,16 According to acid–base principles, the exchange of Fe ions into the zeolite framework generates Brønsted acid sites associated with Fe–OH moieties, which are recognized as critical for the initial adsorption and activation of NH3, thereby facilitating N–N bond coupling.17,18 However, an excessive concentration of these exposed acid sites can trigger the over-oxidation of ammonia. Current research often investigates how the catalyst's lattice structure, oxidation state, and acid site distribution influence NO conversion and N2 selectivity, with the reaction pathway of NH2NO* being a key intermediate.19–22 Recent work has demonstrated the local coordination structure of the metal sites in dictating the diffusion pathways of dehydrogenated species, where van der Waals forces can create a barrier effect that suppresses the egress of intermediates leading to side reactions.23 The disparity between Fe–O and Si–O bond lengths makes the Fe catalytic center particularly sensitive to local defects, which in turn alters the Fe–Fe and Fe–O coordination environments and the structure of the Fe–OH acid sites. By tailoring the oxidative properties and coordination environment of the catalytic Fe centers, one can modulate the product selectivity and reaction network through synergistic kinetics. Therefore, establishing a quantitative descriptor that correlates Fe–Fe/O coordination numbers and acid properties helps to fundamentally decouple the competing pathways of NO reduction and NH3 oxidation.
In this study, we report the synthesis and robust catalytic performance of a series of Fe-exchanged ZSM-5 catalysts. By developing a comprehensive kinetic model that couples the primary SCR reaction with parasitic NH3 oxidation, we demonstrate that the ion-exchanged Fe loading directly affects the high-temperature NH3–NO–O2 reaction system. Among the catalysts studied, 0.1Fe@ZSM-5 achieved a NO conversion of 83.0% and maintained exceptional stability for over 120 h at 700 °C in the presence of SO2 and water vapor. This performance represents a 15.2% enhancement in high-temperature de-NOx activity at 850 °C compared to our previous work. Mechanistically, we reveal that the exposed Fe nanoparticles facilitate the consumption of NH3-coordinated Lewis acid sites, promoting NH3 adsorption and activation on the Brønsted acid sites for the SCR reaction. Simultaneously, these nanoparticles catalyze further dehydrogenation of  intermediates at elevated temperatures, which leads to the undesired over-oxidation of ammonia to NO.
intermediates at elevated temperatures, which leads to the undesired over-oxidation of ammonia to NO.
2 Results and discussion
2.1 Characterization of Fe@ZSM-5
The Fe@ZSM-5 catalysts were synthesized via a facile ion-exchange method, schematically depicted in Fig. 1a. A series of catalysts with different iron mass loadings were prepared and are denoted as 0.05Fe@ZSM-5, 0.08Fe@ZSM-5, 0.1Fe@ZSM-5, and 0.5Fe@ZSM-5, based on the calculated Fe content during synthesis. The loading of Fe was determined by ICP-OES and the results are summarized in Table S1. Detailed synthesis procedures are provided in the Experimental and computational section. Unless otherwise specified, HZSM-5 in this work refers to the zeolite with a Si/Al ratio of 27. X-ray diffraction (XRD) patterns confirmed the retention of the MFI framework, showing characteristic reflections at 8.1° (101) and 23.2° (332) (Fig. 1b). Notably, no diffraction peaks corresponding to Fe2O3 (e.g., at 33.2°, 35.7°, or 53.6°) or other crystalline iron-containing phases were detected,24,25 indicating the dispersed state of iron species on the ZSM-5 lattice. | ||
| Fig. 1 Synthesis and structural characterization of Fe@ZSM-5 catalysts. (a) Schematic illustration of the synthesis of Fe@ZSM-5; (b) XRD patterns of the as-prepared Fe@ZSM-5 catalysts with different iron loadings; (c) TEM image of 0.5Fe@ZSM-5; (d–f) high-resolution TEM images of 0.5Fe@ZSM-5, showing Fe2O3 nanoparticles with resolved lattice fringes corresponding to the (110) and (104) planes; (g) HAADF-STEM image and the EDS elemental maps for Fe, Al, Si, and O in 0.5Fe@ZSM-5; (h–j) aberration-corrected STEM images of 0.1Fe@ZSM-5. Isolated single Fe atoms are marked by yellow circles, and Fe nanoclusters are highlighted by white squares; (k) Fe L-edge EELS spectra of 0.1Fe@ZSM-5. | ||
Transmission electron microscopy (TEM) revealed the microstructural features of 0.5Fe@ZSM-5 (Fig. 1c, S1 and S2). The large-scale image displays the characteristic morphology of the MFI framework. High-resolution TEM (HR-TEM) reveals distinct Fe2O3 nanoparticles anchored on the zeolite framework, evidenced by clear lattice fringes corresponding to the (110) and (104) planes (Fig. 1e and f). Furthermore, energy-dispersive X-ray spectroscopy (EDS) elemental maps (Fig. 1g) show a homogeneous distribution of the framework elements (Si, Al, and O), whereas Fe is concentrated in discrete locations, consistent with the presence of nanoparticles.
Atomic-resolution scanning transmission electron microscopy (AC-STEM) was used to probe the iron species in 0.1Fe@ZSM-5 at the atomic scale. The images (Fig. 1h–j and S3) revealed numerous distinct bright dots and ∼1.5 nm particles, demonstrating that 0.1Fe@ZSM-5 is composed of both dispersed Fe atoms and nanoparticles (NPs). Electron energy loss spectroscopy (EELS) was subsequently conducted to analyze the oxidation state of the Fe species. The Fe L3 and L2 edges from three EELS line scans exhibited identical positions at 708.7 and 721.5 eV, respectively, consistent with the Fe3+ reference (Fig. 1k).26 The combined results confirm the presence of small nanoparticles containing Fe3+ species on 0.1Fe@ZSM-5.
2.2 Coordination environment of Fe@ZSM-5
The physicochemical properties of the Fe@ZSM-5 catalysts were characterized by X-ray photoelectron spectroscopy (XPS), where the Fe 2p spectra varied significantly with iron loading (Fig. 2a). For samples with low iron content (0.05Fe@ZSM-5 and 0.08Fe@ZSM-5), distinct iron peaks were not resolved. In contrast, the 0.1Fe@ZSM-5 and 0.5Fe@ZSM-5 catalysts displayed well-defined peaks for Fe2+ (2p3/2 at 710.8 eV) and Fe3+ (2p3/2 and 2p1/2).27–31 The presence of Fe3+ was further confirmed by satellite peaks at 718.8 eV and 733.0 eV.32,33 Therefore, iron exists as a mixture of Fe2+ and Fe3+ in the higher-loading samples, with Fe3+ being the predominant species. The larger Fe3+ peak area for 0.5Fe@ZSM-5 is consistent with its higher total iron content. | ||
| Fig. 2 Electronic state and coordination structure characterization on FeZSM-5. (a) Fe 2p spectra; (b) O 1s XPS spectra; (c) NH3-TPD spectra; (d) H2-TPR spectra; (e) K-edge XANES spectra; (f) K-edge EXAFS spectra; (g) Fe wavelet transforms. All FT and WT signals in this study are non-phase corrected. | ||
The O 1s XPS spectra (Fig. 2b) were deconvoluted into three distinct components, corresponding to lattice oxygen (Olattice) at a binding energy of 530.2 eV, surface-adsorbed oxygen (Osurface-adsorbed) at 531.7 eV, and adsorbed hydroxyl groups (Ohydro) at 532.6 eV.34,35 It is worth noting that the parent HZSM-5 exhibits a large proportion of Osurface-adsorbed which is associated with –Al/Si bonds.36 While surface-adsorbed oxygen was the predominant species across all samples (49–54%) from parent HZSM-5 or metal oxides,36–38 the concentration of lattice oxygen exhibited a notable, non-monotonic trend with increasing Fe content. For 0.1Fe@ZSM-5, it has the highest value of lattice oxygen of about 33.1%, and concurrently displayed the lowest concentration of adsorbed hydroxyl groups (14.9%). However, 0.5Fe@ZSM-5 shared the equivalent amount of lattice oxygen and adsorbed hydroxyl groups (both 20.7%). The variation suggests that moderate Fe incorporation promotes the generation of lattice oxygen while excessive Fe loading may bring hydroxyl enrichment, which enables the catalyst to obtain higher N2 selectivity.
The surface acidity of the Fe@ZSM-5 catalysts was further investigated using NH3-TPD. As shown in Fig. 2c, the low-temperature peaks at 100–300 °C are ascribed to weak acid sites, corresponding to NH3 release from the weaker Lewis acid sites associated with exchange metal ions.39 Meanwhile, the high-temperature peaks at 400–500 °C correspond to strong acid sites exhibiting thermal stability.39,40 Compared to HZSM-5, increasing the Fe loading shifts the high-temperature NH3 desorption peak from 447 °C to approximately 410 °C, indicating a reconstruction in the acid site structure. The total quantified acidity for each Fe@ZSM-5 sample is about 3 mmol g−1, comparable to that of HZSM-5. The surface acidity was detected by pyridine IR and the spectra (Fig. S4) reveal bands at 1450, 1545, 1625 and 1490 cm−1, which are assigned to Lewis sites, Brønsted sites, and a combination of Lewis and Brønsted sites (BNH4 + LNH3 acid), respectively.41 The quantitative analysis (Table S2) of Brønsted vs. Lewis (B/L) through pyridine-IR shows a marked decrease from 41.0 for HZSM-5 to 14.4–22.6 for Fe@ZSM-5, indicating a substantial reduction in Brønsted acidity and a relative increase in Lewis acidity upon Fe incorporation. Furthermore, the H2-TPR profiles in Fig. 2d reveal the reducibility of Fe@ZSM-5. With increasing Fe loading, the initial peak corresponding to the reduction of Fe2O3 to Fe3O4 was intensified,42 while the high-temperature region associated with the reduction of FeO particles to metallic Fe (Fe2+ to Fe0) decreased in intensity.43 This suggests that the Fe2+ observed in the XPS spectra may originate from the interaction between iron and the surrounding atoms.44 The results also imply that Fe3+ predominates as the stable species at higher loadings.
X-ray absorption fine structure (XAFS) was employed to probe the coordination environment of iron within the synthesized Fe@ZSM-5 catalysts. The normalized Fe K-edge X-ray absorption near-edge structure (XANES) spectra are presented in Fig. 2e. The absorption threshold (E0) for all samples is shifted to a higher energy relative to that of Fe foil, indicating that iron predominantly exists in an oxidized state.45 As the iron loading decreases, the XANES spectra exhibit a further shift toward higher energy and a broadening of the normalized peak. These features are characteristic of the oxidation of Fe to Fe3+, suggesting a higher average oxidation state in catalysts with lower iron content,46 aligning with the fitting results in the XANES spectra.
The Fourier-transformed (FT) EXAFS spectra of the Fe@ZSM-5 catalysts (Fig. 2f) exhibit a prominent peak at ∼2.5 Å, corresponding to the Fe–Fe scattering path. The intensity of this peak strengthens with increasing iron loading, while the intensity of the Fe–O scattering path at 1.8 Å concurrently diminishes. These trends indicate that higher iron loadings promote the formation of Fe nanoclusters. In contrast, lower loadings favor a coexistence of atomically dispersed Fe species and small nanoclusters. This structural model is corroborated by wavelet transform (WT) analysis (Fig. 2g), which clearly resolves the distinct scattering contributions from Fe–O and Fe–Fe paths. Quantitative EXAFS fitting provides further validation (Table S3). As the iron loading increases from 0.05 to 0.5 wt%, the Fe–Fe coordination number rises from 0.9 to 5.4, while the Fe–O coordination number decreases from 4.1 to 1.8. These results are in agreement with the structural evolution inferred from our XANES and WT analyses.
2.3 Dual kinetic effect of NH3-SCR over Fe@ZSM-5
To identify the optimal support, we first evaluated the catalytic performance of the parent HZSM-5 zeolites with various Si/Al ratios (10, 27, 85, 130, and 240), as shown in Fig. 3a. The zeolite with a Si/Al ratio of 27 exhibited the highest activity. The catalytic performance of the Fe@ZSM-5 series in the SCR of NO with NH3 was evaluated (Fig. 3b). By varying the Fe loading amount during ion exchange from 0.05 to 0.5 wt%, two distinct kinetic regimes were observed in catalytic tests over the 400–850 °C temperature range. In the low-temperature regime (400–700 °C), the NO conversion increased with both temperature and iron loading. The 0.5Fe@ZSM-5 catalyst achieved the highest performance, with an average NO conversion of 95.1%. In contrast, the parent HZSM-5 support showed poor activity (9.3–65.3% conversion), confirming that the iron species are the primary sites for activating NH3-SCR, which improved the redox properties of the catalyst thereby accelerating the reaction rates and catalytic performance.47 However, at temperatures above 700 °C, this trend reversed. The NO conversion began to decrease with increasing temperature and iron loading, indicating a loss of activity and N2 selectivity (Fig. S5). | ||
| Fig. 3 NO catalytic reduction and NH3 oxidation. NO conversion over (a) HSZM-5 with various Si/Al ratios and (b) Fe@ZSM-5 at different reaction temperatures; (c) effect of NOx concentration on catalytic NH3 oxidation at 700–850 °C; (d) effect of gas hourly space velocity on NO conversion; (e) time-on-line NO conversion and outlet concentration of N2O and NO2 over 0.1Fe@ZSM-5 at 700 °C. Conditions: 1000 ppm NH3, 6.0 vol% O2, 1000 ppm NO, and balance N2. | ||
To investigate this high-temperature inactivation, we performed separate catalytic oxidation of NH3 in the presence of O2. The concentration of NOx produced from NH3 oxidation increased with both temperature and iron loading (Fig. 3c). This result shows that at high temperatures, NH3 oxidation becomes a dominant competing side reaction, consuming the reducing agent and thereby lowering the efficiency of NO reduction. Moreover, the lower NO concentration of pristine HZSM-5 further confirms this side reaction to the iron species. Combining the TEM and XAS results, we preliminary conclude that the iron nanoparticle size is an important factor in modulating the trade-off between NH3-SCR activity and NH3 oxidation.
The effect of gas hourly space velocity on the catalytic performance was explored (Fig. 3d). Increasing the space velocity led to a decrease in NO conversion. This was accompanied by a rise in unconverted NH3 at the outlet, while the concentrations of NO2 and N2O by-products did not change significantly. These results indicate that the drop in NO conversion at higher space velocities is due to the reduced NH3 conversion rather than the enhanced NH3 over-oxidation.
In addition, the long-term stability of the 0.1Fe@ZSM-5 catalyst was tested at 700 °C (Fig. 3e). The catalyst demonstrated robust performance over a 50 hour run, with NO conversion showing only a slight decrease from 79.5% to 77.0%. During this period, the outlet NO2 concentration increased from 4.1 to 7.5 ppm, while the N2O concentration remained stable. This suggests that the minor drop in NO conversion is linked to a slow side reaction where some NO is oxidized to NO2, which does not efficiently enter the fast SCR pathway.
Regarding practical application, we evaluated the long-term catalytic performance of synthesized 0.1Fe@ZSM-5 and HZSM-5 under exposure to 300 ppm SO2 and 8.3 vol% H2O at 700 °C. Under 300 ppm SO2, 0.1Fe@ZSM-5 deactivated gradually, with NO conversion decreasing from 83.0% to 78.5% over 50 h as shown in Fig. 4a. Introducing 8.3 vol% H2O accelerated deactivation and the conversion dropped to 60.1%. After both poisons were removed at 110 h, the catalyst partially recovered to 71.5%. For comparison, HZSM-5 showed a similar slight decline under SO2 alone, from 77.3% to 72.1% NO conversion. With both SO2 and H2O, the conversion decreased to 60.8% and did not recover after poison removal, indicating irreversible deactivation.
 | ||
Fig. 4 The long-term stability test on synthesized (a) 0.1Fe@ZSM-5 and (b) HZSM-5 catalysts in the presence of SO2 and H2O; (c) magnified HAADF-STEM image and corresponding EDS maps of the reacted 0.1Fe@ZSM-5; (d) 27Al MAS NMR spectra; (e) pyridine FTIR spectra; (f) O2-TPD patterns of the as-prepared and reacted 0.1Fe@ZSM-5. Reaction conditions: 700 °C, 1000 ppm NH3, 6.0 vol.% O2, 1000 ppm NO, 300 ppm SO2, 8.3 vol% H2O and balance N2 at a space velocity of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 533 ml gcat.−1 h−1. 533 ml gcat.−1 h−1. | ||
Further physicochemical analyses were performed after 120 h continuous reaction to probe the deactivation mechanism of 0.1Fe@ZSM-5. HAADF-STEM and EDS reveal dispersed Fe nanoparticles. Sulfur deposits distribute broadly on ZSM-5 with partial co-localization on Fe nanoparticles, while remaining overall dispersed. The 27Al MAS NMR spectra were used to track the evolution of the framework Al throughout the long-term hydrothermal ageing. As can be seen in Fig. 4d, the broad peak signal of dehydrated 0.1Fe@ZSM-5 is 4-coordinated corresponding to the Brønsted acidic Al(IV) site at 58 ppm. After SO2 + H2O ageing for 120 h, the signal intensity of the framework Al decreases, suggesting a dealumination process.48 The significant decline of the LNH3 peak from Py-IR clearly indicates the loss of Lewis acid sites after long-term poisoning (Fig. 4e). The O2-TPD patterns (Fig. 4f) of the as-prepared catalyst and after 120 h in SO2 and H2O confirm a severe loss of lattice oxygen (over 400 °C)49,50 and diminished oxygen mobility after exposure to SO2 and H2O. These observations indicate that sulfur deposition blocks active oxygen sites and suppresses lattice oxygen participation in the long-term test.
2.4 Kinetics of NO reduction and NH3 oxidation
Catalytic experiments revealed that NH3 oxidation significantly influences the overall reaction kinetics at high temperatures, with its rate being dependent on the Fe loading. Consequently, a conventional kinetic model for standard SCR fails to accurately describe the system at 700 °C (Fig. 5a). We therefore developed a dual-pathway kinetic model that considers both the NH3 oxidation and NO reduction reactions. The model parameters, governing equations, and diffusion criteria are detailed in the SI. | ||
| Fig. 5 Identification of the dual kinetic effect on 0.1Fe@ZSM-5. (a) Comparison of the conventional SCR kinetic model with experimental data under a NO + NH3 + O2 atmosphere; effect of reaction temperature on (b) NH3 concentration and (c) NOx selectivity under NH3 + O2 oxidation conditions; (d) comparison of the dual kinetic model and experimental data; in situ DRIFT spectra for (e) NO + NH3 + O2 reduction and (f) NH3 + O2 oxidation; (g) evolution of DRIFT spectra on intermediates; (h) schematic illustrating the proposed dual kinetic pathway for high-temperature SCR. | ||
Under NH3 + O2 atmospheric conditions, NH3 oxidation products on the catalyst surface can be primarily classified as N2 and NO, with selectivity varying as the temperature increases. Higher temperatures favor higher NO selectivity.
| 4NH3 + 3O2 → 2N2 + 6H2O | (1) |
| 4NH3 + 5O2 → 4NO + 6H2O | (2) |
 | (3) |
 | (4) |
 | (5) |
Experimental measurement data under NH3 + O2 conditions were analyzed by selecting data points where NH3 conversion remained below 15% to minimize diffusion effects on fitting accuracy. Here,  represents the NH3 surface coverage. After optimization, the rate constants for NH3 oxidation reactions at various temperatures yielded an activation energy of 98.3 kJ mol−1. The pre-exponential factor A and activation energy Ea for the NH3 oxidation process were subsequently determined by fitting the lnkx vs. 1/T relationship according to the Arrhenius equation. We compare the experimental measurements and model calculations for NH3 conversion and NH3-to-NO oxidation selectivity under NH3 + O2 atmospheric conditions. The calculated results demonstrate good agreement with experimental data over the 500–850 °C temperature range (Fig. 5b and c).
represents the NH3 surface coverage. After optimization, the rate constants for NH3 oxidation reactions at various temperatures yielded an activation energy of 98.3 kJ mol−1. The pre-exponential factor A and activation energy Ea for the NH3 oxidation process were subsequently determined by fitting the lnkx vs. 1/T relationship according to the Arrhenius equation. We compare the experimental measurements and model calculations for NH3 conversion and NH3-to-NO oxidation selectivity under NH3 + O2 atmospheric conditions. The calculated results demonstrate good agreement with experimental data over the 500–850 °C temperature range (Fig. 5b and c).
Under NH3 + NO + O2 atmospheric conditions, the reaction rate of NO is governed by the synergistic interplay between the catalytic reduction of NO by NH3 and the concurrent catalytic oxidation of NH3 to form NO. This dual mechanism significantly influences the overall reaction kinetics, as demonstrated below:
| 4NO + 4NH3 + O2 → 4N2 + 6H2O | (6) |
| 4NH3 + 3O2 → 2N2 + 6H2O | (7) |
| 4NH3 + 5O2 → 4NO + 6H2O | (8) |
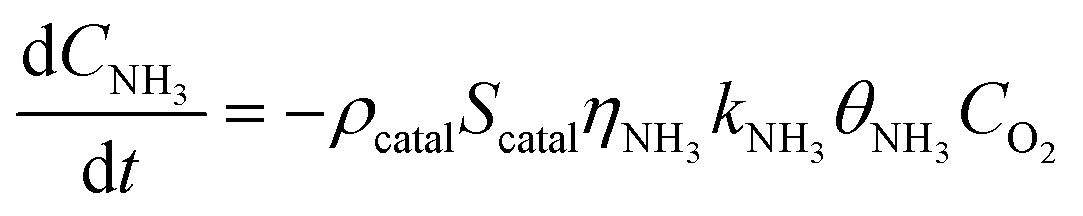 | (9) |
 | (10) |
Subsequently, the experimental data obtained under NH3 + NO + O2 conditions were systematically analyzed. Given the relatively low SNCR efficiency observed below 850 °C, gas-phase NH3 + NO + O2 reactions were excluded from consideration in this temperature range. To minimize the influence of mass transfer limitations on fitting accuracy, experimental data points with NO conversion rates below 15% were specifically selected for kinetic analysis. Through optimization procedures, the rate constants for NO catalytic reduction reactions were determined at various temperatures. The pre-exponential factor (A) and activation energy (Ea) for the NH3 oxidation process were subsequently derived by fitting the ln(kx) versus 1/T relationship according to the Arrhenius equation, as presented in Table S4.
Compared to conventional SCR kinetic models, this work introduces a dual kinetic framework that explicitly incorporates NH3 surface oxidation as a competing reaction under high-temperature conditions. A key feature of this framework is the inclusion of the selectivity of NH3 toward NO formation as a temperature-dependent parameter, which allows quantitative evaluation of how the NH3 over-oxidation pathway influences the overall SCR performance. This integration provides a more accurate description of the experimental results (Fig. 5d). As the temperature rises, the model successfully captures the transition where the NO formation rate surpasses the N2 formation rate, revealing the increasing contribution of NH3 oxidation in determining overall selectivity. By incorporating this temperature-dependent selectivity, the dual kinetic model addresses a gap in prior SCR kinetic frameworks, which typically treat NH3 oxidation as a parallel but independent side reaction.
To explore the effect of NH3 catalytic oxidation on SCR-related intermediates, in situ DRIFT measurements combined with TPSR were performed on 0.1Fe@ZSM-5 from 400 to 850 °C (Fig. 5e). Following NH3 adsorption, broad N–H vibration peaks at 3259 and 2936 cm−1 are ascribed to the asymmetric stretching as vas(NH4+) on Brønsted acid sites, where three hydrogen atoms bond to oxygen ions in the AlO4 tetrahedra.51,52 The sharp peaks at 3581 and 3660 cm−1 are attributed to Si–OH–Al and Si–OH, respectively. In the lower wavenumber region, the peak at 1287 cm−1 corresponds to  vibrations, while features at 1186 and 1621 cm−1 are assigned to vas(NH3) on Lewis acid sites, and those at 1465 and 1762 cm−1 to NH4+ vibrations on Brønsted acid sites.53 Upon heating in an NH3–NO–O2 atmosphere to 600 °C, the intensities of
vibrations, while features at 1186 and 1621 cm−1 are assigned to vas(NH3) on Lewis acid sites, and those at 1465 and 1762 cm−1 to NH4+ vibrations on Brønsted acid sites.53 Upon heating in an NH3–NO–O2 atmosphere to 600 °C, the intensities of  and NH4+ intermediates at 3259 and 1621 cm−1 decay rapidly, whereas
and NH4+ intermediates at 3259 and 1621 cm−1 decay rapidly, whereas  remains relatively stable, indicating that certain
remains relatively stable, indicating that certain  and NH4+ species serve as the initial active sites in the SCR redox cycle during activation. Notably, as the temperature further increased from 600 to 850 °C, only the absorbance of NH4+ intermediates at 3259 and 2936 cm−1 decreased significantly, whereas the band at 1465 cm−1 remained relatively stable. This observation indicates that framework Brønsted acid sites participate more actively in the synergistic conversion of NH3 and NO at high temperatures. Moreover, the time-resolved DRIFT spectra of 0.1Fe@ZSM-5 during NH3 adsorption, O2 oxidation, and NO reduction at 800 °C (Fig. S6–S8) confirm the dynamic transformation of NH4+ species during the redox cycle.
and NH4+ species serve as the initial active sites in the SCR redox cycle during activation. Notably, as the temperature further increased from 600 to 850 °C, only the absorbance of NH4+ intermediates at 3259 and 2936 cm−1 decreased significantly, whereas the band at 1465 cm−1 remained relatively stable. This observation indicates that framework Brønsted acid sites participate more actively in the synergistic conversion of NH3 and NO at high temperatures. Moreover, the time-resolved DRIFT spectra of 0.1Fe@ZSM-5 during NH3 adsorption, O2 oxidation, and NO reduction at 800 °C (Fig. S6–S8) confirm the dynamic transformation of NH4+ species during the redox cycle.
For comparison, variations in intermediates during exposure to an NH3 + O2 atmosphere at 400–850 °C were also examined under NH3 catalytic oxidation conditions (Fig. 5f). At 400 °C, the  intermediate at 1287 cm−1 is markedly lower than that in NH3 + NO + O2, but its intensity reverses and increases upon heating to 800 °C, confirming that dehydrogenation products of NH3 or NH4+ proceed via the
intermediate at 1287 cm−1 is markedly lower than that in NH3 + NO + O2, but its intensity reverses and increases upon heating to 800 °C, confirming that dehydrogenation products of NH3 or NH4+ proceed via the  pathway in high-temperature oxidizing environments, while
pathway in high-temperature oxidizing environments, while  remains relatively inert. For quantitative comparison, normalized absorbance changes of characteristic peaks with temperature are shown in Fig. 5g, suggesting that NH4+ likely participates in surface species consumption during NH3 oxidation. Drawing from these observations, we propose a molecular-level pathway governed by a dual kinetic regime (Fig. 5h). Under moderate temperature conditions, a favorable activation barrier promotes the dehydrogenation of NH3 to form
remains relatively inert. For quantitative comparison, normalized absorbance changes of characteristic peaks with temperature are shown in Fig. 5g, suggesting that NH4+ likely participates in surface species consumption during NH3 oxidation. Drawing from these observations, we propose a molecular-level pathway governed by a dual kinetic regime (Fig. 5h). Under moderate temperature conditions, a favorable activation barrier promotes the dehydrogenation of NH3 to form  intermediates. These species subsequently undergo N–N coupling to yield NH2NO*, which decomposes into N2 following the conventional SCR pathway. Conversely, at temperatures exceeding 700 °C, the higher thermal energy shifts the reaction toward an undesirable oxidation route. In this regime, lattice oxygen induces the progressive dehydrogenation of adsorbed NH3 at acid sites through sequential N–H bond scission. This process generates highly reactive NH* or N* radicals that are ultimately oxidized to NOx.
intermediates. These species subsequently undergo N–N coupling to yield NH2NO*, which decomposes into N2 following the conventional SCR pathway. Conversely, at temperatures exceeding 700 °C, the higher thermal energy shifts the reaction toward an undesirable oxidation route. In this regime, lattice oxygen induces the progressive dehydrogenation of adsorbed NH3 at acid sites through sequential N–H bond scission. This process generates highly reactive NH* or N* radicals that are ultimately oxidized to NOx.
To probe the influence of the iron nanoparticle size on the SCR mechanism, we performed in situ DRIFT experiments on the 0.05Fe@ZSM-5, 0.08Fe@ZSM-5, and 0.5Fe@ZSM-5 catalysts. Following NH3 adsorption at 200 °C (Fig. 6a–c), distinct changes in the DRIFT spectra were observed as the Fe loading increased from 0.05 to 0.5 wt%. Correlating with the XANES fitting results, which indicated increasing metallic Fe content at higher loadings, these spectral changes suggest that metallic Fe influences the NH3 adsorption behavior. Specifically, the intensity of the band corresponding to NH3 coordinated to Lewis acid sites decreased, while the band assigned to NH4+ ions at 1471 cm−1 grew substantially. This shift indicates that NH3 preferentially adsorbs at Brønsted acid sites in the presence of metallic Fe species, where it forms more stable NH4+ species. The enhanced stability at Brønsted sites suppresses the excessive oxidation of NH3 at high temperatures, thereby improving the selectivity toward N2 formation.54 Furthermore, the band at 3259 cm−1, attributed to NH4+ ions interacting with Brønsted acid sites of the zeolite, was attenuated, indicating a weaker interaction between the agglomerated iron species and the zeolite framework.
 | ||
| Fig. 6 Effect of iron nanoparticles on the high-temperature SCR mechanism. In situ DRIFT spectra of NH3 adsorption at 200 °C on (a) 0.05Fe@ZSM-5, (b) 0.08Fe@ZSM-5, and (c) 0.5Fe@ZSM-5; TPSR on (d) 0.05Fe@ZSM-5, (e) 0.08Fe@ZSM-5, and (f) 0.5Fe@ZSM-5 at 400 to 850 °C. | ||
As the temperature was increased (Fig. 6d–f), the intensity of all ammonia-related bands decreased, indicating the progressive desorption of NH3 from the acid sites. Notably, the evolution of the surface species differed significantly with iron loading. For the low-loading 0.05Fe@ZSM-5 and 0.08Fe@ZSM-5 catalysts, the bands for NH3 adsorbed on Lewis acid sites were almost completely consumed by 700–850 °C, whereas the signals for NH4+ and  species persisted. This suggests that the latter are more thermally stable and likely serve as key intermediates in the redox reaction. In contrast, for the 0.5Fe@ZSM-5 catalyst, the bands corresponding to nitrosyl species at 1846 cm−1 were significantly more intense.55 Concurrently, the consumption of the NH4+ band at 3259 cm−1 was more pronounced, indicating that the larger iron nanoparticles promote the undesirable over-oxidation of ammonia to NO at high temperatures, which is in agreement with the observed SCR performance.
species persisted. This suggests that the latter are more thermally stable and likely serve as key intermediates in the redox reaction. In contrast, for the 0.5Fe@ZSM-5 catalyst, the bands corresponding to nitrosyl species at 1846 cm−1 were significantly more intense.55 Concurrently, the consumption of the NH4+ band at 3259 cm−1 was more pronounced, indicating that the larger iron nanoparticles promote the undesirable over-oxidation of ammonia to NO at high temperatures, which is in agreement with the observed SCR performance.
3 Conclusions
In summary, we have developed a sinter-resistant 0.1Fe@ZSM-5 catalyst whose exceptional stability in high-temperature NH3-SCR is attributed to the confinement of ultrasmall Fe nanoclusters within the zeolite framework. Its structural integrity was confirmed, with minor long-term deactivation resulting from sulfur deposition rather than framework collapse or Fe sintering. Through a combination of in situ DRIFTS and kinetic analysis, we have established a dual-pathway mechanism that accurately captures the transition from the conventional NO-reduction route to the high-temperature NH3-oxidation-dominated regime. Unlike traditional SCR kinetic models, which assume independent or single-pathway behavior, the dual-pathway model explicitly incorporates the competing oxidation of NH3 by treating the selectivity of NH3 toward NO formation as a temperature-dependent parameter. This approach reveals how the interplay between NH3 consumption by oxidation and its availability for SCR modifies apparent activation energies and selectivity trends, providing improved predictive capability across the entire temperature range. These studies identified the distinct roles of key surface intermediates, providing direct evidence for as a critical species in the NO reduction pathway, which competes with the NH3 oxidation route. These findings offer a new strategy for the rational design of robust catalysts for challenging industrial applications. The development of a stable catalyst structure, coupled with a more accurate kinetic model, paves the way for advancing high-performance deNOx technologies to meet stringent emission regulations.
as a critical species in the NO reduction pathway, which competes with the NH3 oxidation route. These findings offer a new strategy for the rational design of robust catalysts for challenging industrial applications. The development of a stable catalyst structure, coupled with a more accurate kinetic model, paves the way for advancing high-performance deNOx technologies to meet stringent emission regulations.
4 Experimental and computational section
4.1 Catalyst synthesis and activity measurement
The HZSM-5 zeolite was prepared via a hydrothermal synthesis route. A precursor gel was formed by dissolving ethylenediamine (EDA, 1.20 g), tetraethyl orthosilicate (TEOS, 8.32 g), NaOH (0.3 g), and NaAlO2 (1.02 g) in an aqueous solution containing distilled water (15 g) and the structure-directing agent (SDA), tetramethylammonium hydroxide (TMADa+, 13 g). After stirring for 6 h at room temperature, the gel was transferred to a 100 mL Teflon-lined autoclave and crystallized at 170 °C for 3 days. The resulting solid product was calcined at 800 °C for 5 h (with a heating ramp of 5 °C min−1) to remove the SDA. To produce the final parent material, the calcined powder was ion-exchanged by washing three times with a 0.2 M NH4NO3 solution at 70 °C, followed by drying at 110 °C for 12 h.For the fabrication of Fe@ZSM-5, a suspension was created by mixing 10 g of HZSM-5 in 100 ml of distilled water, under intense stirring. The necessary quantity of iron acetylacetonate (Fe(C5H7O2)3) was dissolved in this mixture. Ensuing continuous magnetic agitation at 80 °C for 3 h facilitated the homogenization of the solution, which was then subjected to filtration and thrice rinsed with distilled water. The filtrate was air-dried at 110 °C for 12 h, followed by calcination in a muffle furnace at 800 °C for 5 h under ambient air conditions. The resultant catalyst particles were sieved to obtain a granule size range between 0.18 and 0.25 mm.
The catalytic performance for the NH3-SCR of NOx was evaluated in a continuous-flow quartz fixed-bed reactor over a temperature range of 400–850 °C. The standard feed gas consisted of 1000 ppm NH3, 1000 ppm NO, and 6 vol% O2, with N2 as the balance gas. The total flow rate was maintained at 2 L min−1 using mass flow controllers, corresponding to a gas hourly space velocity (GHSV) of 30000 h−1. The effluent gas concentrations (NO, NH3, N2O, NO2, and O2) were continuously monitored using an online infrared (IR) gas analyzer. To comprehensively assess the catalyst, several experiments were conducted: (i) the effect of GHSV on NO reduction was systematically investigated; (ii) long-term stability tests were performed under standard reaction conditions; (iii) the catalyst's resistance to poisoning was evaluated in the presence of 300 ppm SO2 and/or 8.3 vol% H2O. Prior to data acquisition for each point, the system was allowed to reach a steady state, and the reported values represent the average of measurements taken over a 20 minute period. The NO reduction efficiency, N2 selectivity, and apparent reaction kinetics were calculated using the following equations:
 | (11) |
 | (12) |
 | (13) |
4.2 Characterization and computational methods
The loading of Fe was determined by inductively coupled plasma (ICP, Agilent ICP-OES 725 ES). The crystallinity was assessed by X-ray diffraction (XRD) using a D8 Advance instrument with a scanning angle range (2-theta) from 5° to 90°. X-ray photoelectron spectroscopy (XPS) was conducted on a Thermo Scientific Nexsa. The obtained binding energy was externally calibrated by the carbonaceous C 1s of 284.6 eV. Surface areas and pore size distributions were quantified via the Brunauer–Emmett–Teller (BET) analysis and Barrett–Joyner–Halenda (BJH) method, determined by N2 physisorption performed on an ASAP 2460 (Micromeritics) by degassing at 150 °C for 8 h. The morphologies were investigated using scanning electron microscopy (SEM) and scanning-transmission electron microscopy (STEM) via an FEI, Tecnai F20. Electron energy loss spectroscopy (EELS) was conducted with a Gatan Model 977 Enfinium ER EELS spectrometer. In situ infrared spectroscopy and diffuse reflectance UV-vis spectroscopy (UV-3600i Plus, Shimadzu) were employed to explore the dynamics of the catalytic processes. Nuclear magnetic resonance (NMR, Bruker 400 M) and K-edge X-ray absorption fine structure (XAFS) analyses were performed with Si (111) crystal monochromators at the BL14W beam line at the Shanghai Synchrotron Radiation Facility (SSRF) (Shanghai, China). Before the analysis at the beamline, samples were placed into aluminum sample holders and sealed using Kapton tape film. The XAFS spectra were recorded at room temperature using a 4-channel silicon drift detector (SDD) Bruker 5040. The obtained spectra were refined and analyzed using Athena. Athena and Artemis software programs were employed to perform the EXAFS fitting.56 The software developed by Funke and Chukalina was applied to execute wavelet transformation using the Morlet wavelet with κ = 5 and σ = 1.57Hydrogen temperature-programmed reduction (H2-TPR) was conducted on an AutoChem II 2920 instrument to evaluate the reducibility of the catalysts. In a typical measurement, 100 mg of the sample was loaded into a U-shaped quartz tube. The catalyst was pretreated at 300 °C for 1 h under a flowing He atmosphere (heated at 10 °C min−1) to remove adsorbed water. After the sample was cooled to 50 °C, the gas flow was switched to a 10 vol% H2/He mixture (30 mL min−1). The temperature was then ramped to 800 °C at a rate of 10 °C min−1, and H2 consumption was continuously monitored with a thermal conductivity detector (TCD).
The acidic properties of the catalysts were characterized by ammonia temperature-programmed desorption (NH3-TPD) and oxygen storage and mobility were evaluated by oxygen temperature-programmed desorption (O2-TPD) using the same instrument. Prior to the analysis, 100 mg of the sample was pretreated in a He flow at 300 °C for 1 h (10 °C min−1 ramp) and cooled to 50 °C. For NH3-TPD, the sample was saturated with ammonia by exposure to a 10% NH3/Ar stream (25 mL min−1) for 1 h, followed by Ar purging for 1 h to remove physisorbed ammonia, and then heated to 800 °C at a rate of 10 °C min−1, monitoring the amount of desorbed NH3 using the TCD. In O2-TPD, oxygen saturation was achieved by exposing the pretreated sample to 10% O2/He (25 mL min−1) for 1 h, followed by Ar purging for 1 h and subsequent heating to 800 °C at 10 °C min−1.
4.3 Reaction kinetics modeling
For the fixed bed reactor, the following assumptions were made: 1. negligible volumetric flow changes along the catalyst bed, maintaining consistency throughout the reaction section; 2. negligible pressure variations before and after the catalyst bed, i.e., no pressure drop; 3. negligible radial and axial diffusion, maintaining uniform gas concentration at the interface. These assumptions simplified the model to a one-dimensional plug flow reaction process, establishing the conservation equation for gas component x:
 | (14) |
 | (15) |
The catalyst bed thickness is h, Cx represents the component concentration of reactants/products (mol m−3), primarily CNH3, CNO, and CO2 in this study, and kx (mol m−2 s−1) represents the reaction rate constant for each component. Subsequently, the rate equation was established:
| rx = kxf(Cx) | (16) |
Specifically, for the NH3 + O2 catalytic oxidation process on 0.1Fe@ZSM-5, NH3 adsorption based on the Langmuir model and O2 effects were primarily considered.58 For the comprehensive NH3 + NO + O2 reaction process, referring to standard SCR kinetic models, the reaction orders for NH3, NO, and O2 are 0, 1, and 0.5, respectively.59 The O2 content in coal combustion flue gas is approximately 6 vol%, with concentrations being orders of magnitude higher than other reaction gases, allowing neglect of concentration changes due to reaction during calculations. Additionally, according to the Mear criterion, since the external diffusion rate of catalyst particles is much lower than the reaction rate, diffusion effects on the gas–solid reaction process in this study can be neglected.60
Considering the influence of reactant gas internal diffusion within catalyst particles, an effectiveness factor η was introduced:
 | (17) |
The Thiele modulus expression is given as  , where R represents the catalyst particle diameter (m). For molecular sieve catalysts with porous structure characteristics, the comprehensive diffusion coefficient Dx,eff (m2 s−1) of gas component x is not only influenced by molecular diffusion and Knudsen diffusion (assuming a zeolite pore diameter of 10 nm) but also requires porosity correction. For the catalytic NOx reduction reaction, the effectiveness factor ϕn approaches 1, indicating that the entire process is under surface reaction control. Other model-related parameter values are shown in Table S5.
, where R represents the catalyst particle diameter (m). For molecular sieve catalysts with porous structure characteristics, the comprehensive diffusion coefficient Dx,eff (m2 s−1) of gas component x is not only influenced by molecular diffusion and Knudsen diffusion (assuming a zeolite pore diameter of 10 nm) but also requires porosity correction. For the catalytic NOx reduction reaction, the effectiveness factor ϕn approaches 1, indicating that the entire process is under surface reaction control. Other model-related parameter values are shown in Table S5.
| Dx,eff = ε2Dx | (18) |
Here, Dx,gas represents the diffusion coefficient of reaction gases in air. For NH3, the Fuller equation yields a calculated value of 0.198 cm2 s−1 under standard conditions, while the diffusion coefficient of NO molecules in air is 0.168 cm2 s−1 (with temperature corrections applied as needed). In actual reaction processes, gas–solid reactions compete with intraparticle diffusion  . The combined effects of intraparticle diffusion and the chemical reaction on the overall reaction rate can be determined through the effectiveness factor and Thiele modulus.
. The combined effects of intraparticle diffusion and the chemical reaction on the overall reaction rate can be determined through the effectiveness factor and Thiele modulus.
4.4 In situ DRIFT measurement
In situ DRIFT spectroscopic analysis was conducted using a NICOLET iS50 FTIR spectrometer. Diffuse reflectance infrared Fourier transform spectra (DRIFTS) were acquired with a Harrick Praying Mantis attachment (Model DRK-4, featuring a water-cooling circulation system). Ambient diffuse reflectance UV-vis spectroscopy was performed on a Cary 400 spectrometer (Varian) outfitted with a diffuse reflectance unit. These UV-vis and DRIFTS examinations are pivotal for elucidating the nature of transient species in the in situ SCR process, thereby enhancing our comprehension of the reaction dynamics. To optimize detector performance, specifically the mercury cadmium telluride (MCT/A) detector, liquid nitrogen was utilized to regulate its operational temperature. The temperature within the experimental chamber was managed using a Harrick temperature control unit (Model ATC-024-2), complemented with a water-cooling system to mitigate potential overheating. Prior to initiating measurements, background spectra were obtained using the prepared samples. Subsequently, the catalyst was evenly distributed within the Harrick chamber. A mixture of reaction gases (NH3/NO/O2/He) was precisely regulated using mass flow controllers (MFCs) and warmed before being introduced into the chamber, with an overall gas flow rate set at 50 mL min−1.The procedure for conducting in situ UV-vis and DRIFTS analyses comprised the following steps: (1) NH3 adsorption: the chamber was purged with He at 500 °C for 1 h and then brought back to ambient temperature. Subsequently, a flow of 1000 ppm NH3/He was introduced, gradually elevating the temperature from 100 to 800 °C at 10 °C min−1. (2) NH3 oxidation: upon reaching adsorption equilibrium (typically within 1 h) at a set temperature, 6 vol% O2 was infused into the chamber for 30 min. (3) NH3–NO–O2 co-presence and pulse: following equilibrium establishment (usually within 30 min) at the required temperature, a mixture of 1000 ppm NO, 6 vol% O2, and 1000 ppm NH3 in He was simultaneously fed into the chamber at 800 °C. For pulse tests, specific gas components were selectively discontinued. For the temperature programmed surface reaction (TPSR), the reaction chamber was heated from 400 °C to 850 °C in 1000 ppm NO and 6 vol% O2 after exposure to 1000 ppm NH3/He for 1 h. In situ DRIFT spectra were recorded every 8 seconds, and the IR spectrum presented subsequently represents the average interference pattern across eight successive scans.
The acidity of the samples was determined by pyridine infrared spectroscopy (Py-IR) on a Bruker Vector 22 spectrophotometer. Prior to the tests, the samples were shaped into tablets and degassed under vacuum at 400 °C for 1 h. After cooling to room temperature (RT), the samples were saturated with pyridine and then desorbed at RT, 200, and 450 °C under vacuum for 30 min. The IR spectra were registered in the 1400–1700 cm−1 range.
Author contributions
Xinlin Xie: investigation, software, writing – original draft. Jibin Yuan: data curation, methodology, writing – original draft. Lei Liu: methodology, writing – review & editing. Hanzi Liu: investigation, methodology, conceptualization, data curation, formal analysis, writing – original draft. Zhiqiang Sun: supervision, project administration, funding acquisition, writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Supplementary information (SI): containing additional performance and structural data, as well as in situ DRIFTS. See DOI: https://doi.org/10.1039/d5im00245a.
Acknowledgements
We acknowledge the National Natural Science Foundation of China (52306179) and the Provincial Natural Science Foundation of Hunan (2024JJ6507).References
- L. Han, S. Cai, M. Gao, J.-Y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Selective catalytic reduction of NOx with NH3 by using novel catalysts: State of the art and future prospects, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed.
- Y. Qu, G. Xu, C. Chen, J. Guo, D. Liu, H. Jia, H. Guo, S. Jia, J. Jia, Y. Zhang and L. Yan, A guideline to optimizing the performance of V2O5-MoO3/TiO2 catalysts for low-temperature SCR denitrification in industrial application, Ind. Chem. Mater., 2025 10.1039/D5IM00055F.
- S. Y. Joshi, A. Kumar, J. Luo, K. Kamasamudram, N. W. Currier and A. Yezerets, New insights into the mechanism of NH3-SCR over Cu- and Fe-zeolite catalyst: Apparent negative activation energy at high temperature and catalyst unit design consequences, Appl. Catal., A, 2018, 226, 565–574 CrossRef CAS.
- M. Liu, C. Miao and Z. Wu, Recent advances in the synthesis, characterization, and catalytic consequence of metal species confined within zeolite for hydrogen-related reactions, Ind. Chem. Mater., 2024, 2, 57–84 RSC.
- S. J. Schmieg, S. H. Oh, C. H. Kim, D. B. Brown, J. H. Lee, C. H. F. Peden and D. H. Kim, Thermal durability of Cu-CHA NH3-SCR catalysts for diesel NOx reduction, Catal. Today, 2012, 184, 252–261 CrossRef CAS.
- S. Brandenberger, O. Kröcher, M. Casapu, A. Tissler and R. Althoff, Hydrothermal deactivation of Fe-ZSM-5 catalysts for the selective catalytic reduction of NO with NH3, Appl. Catal., B, 2011, 101, 649–659 CrossRef CAS.
- X. Shi, F. Liu, L. Xie, W. Shan and H. He, NH3-SCR performance of fresh and hydrothermally aged Fe-ZSM-5 in standard and fast selective catalytic reduction reactions, Environ. Sci. Technol., 2013, 47, 3293–3298 CrossRef CAS PubMed.
- C. U. I. Odenbrand, CaSO4 deactivated V2O5-WO3/TiO2 SCR catalyst for a diesel power plant. Characterization and simulation of the kinetics of the SCR reactions, Appl. Catal., B, 2018, 234, 365–377 CrossRef CAS.
- H. Liu, C. You and H. Wang, Experimental and density functional theory studies on the zeolite-based Fe-Ni-W trimetallic catalyst for high-temperature NOx selective catalytic reduction: Identification of active sites suppressing ammonia over-oxidation, ACS Catal., 2021, 11, 1189–1201 CrossRef CAS.
- L. Gang, B. Anderson, J. Van Grondelle and R. Van Santen, Low temperature selective oxidation of ammonia to nitrogen on silver-based catalysts, Appl. Catal., B, 2003, 40, 101–110 CrossRef CAS.
- B. Bahrami, V. G. Komvokis, M. S. Ziebarth, O. S. Alexeev and M. D. Amiridis, NH3 decomposition and oxidation over noble metal-based FCC CO combustion promoters, Appl. Catal., B, 2013, 130, 25–35 CrossRef.
- Y. Tian, Z. Han, Z. Zhou, H. Zhao, Q. Zeng, Y. Li and D. Ma, Synthesis of Cu/CeTiOx tandem catalyst with dual-function sites for selective catalytic oxidation of ammonia, Chem. Eng. J., 2025, 503, 158212 CrossRef CAS.
- X. Yang, B. Zhao, Y. Zhuo, Y. Gao, C. Chen and X. Xu, DRIFTS study of ammonia activation over CaO and sulfated CaO for NO reduction by NH3, Environ. Sci. Technol., 2011, 45, 1147–1151 CrossRef CAS PubMed.
- G. Novell-Leruth, J. M. Ricart and J. Pérez-Ramírez, Pt(100)-catalyzed ammonia oxidation studied by DFT: Mechanism and microkinetics, J. Phys. Chem. C, 2008, 112, 13554–13562 CrossRef CAS.
- Y. Yang, J. Liu, F. Liu, Z. Wang, J. Ding and H. Huang, Reaction mechanism for NH3-SCR of NOx over CuMn2O4 catalyst, Chem. Eng. J., 2019, 361, 578–587 CrossRef CAS.
- X. Li, K. Li, Y. Peng, X. Li, Y. Zhang, D. Wang, J. Chen and J. Li, Interaction of phosphorus with a FeTiOx catalyst for selective catalytic reduction of NOx with NH3: Influence on surface acidity and SCR mechanism, Chem. Eng. J., 2018, 347, 173–183 CrossRef CAS.
- Y. J. Kim, H. J. Kwon, I. Heo, I.-S. Nam, B. K. Cho, J. W. Choung, M.-S. Cha and G. K. Yeo, Mn-Fe/ZSM5 as a low-temperature SCR catalyst to remove NOx from diesel engine exhaust, Appl. Catal., B, 2012, 126, 9–21 CrossRef CAS.
- M. Zhu, J.-K. Lai, U. Tumuluri, Z. Wu and I. E. Wachs, Nature of active sites and surface intermediates during SCR of NO with NH3 by supported V2O5-WO3/TiO2 catalysts, J. Am. Chem. Soc., 2017, 139, 15624–15627 CrossRef CAS PubMed.
- P. Gong, J. Xie, D. Fang, D. Han, F. He, F. Li and K. Qi, Effects of surface physicochemical properties on NH3-SCR activity of MnO2 catalysts with different crystal structures, Chin. J. Catal., 2017, 38, 1925–1934 CrossRef CAS.
- P. S. Hammershøi, P. N. R. Vennestrøm, H. Falsig, A. D. Jensen and T. V. W. Janssens, Importance of the Cu oxidation state for the SO2-poisoning of a Cu-SAPO-34 catalyst in the NH3-SCR reaction, Appl. Catal., B, 2018, 236, 377–383 CrossRef.
- Q. Zhang, T. Zhang, F. Xia, Y. Zhang, H. Wang and P. Ning, Promoting effects of acid enhancing on N2 selectivity for selectivity catalytic oxidation of NH3 over RuOx/TiO2: The mechanism study, Appl. Surf. Sci., 2020, 500, 144044 CrossRef CAS.
- Q. Zhao, B. Chen, J. Li, X. Wang, M. Crocker and C. Shi, Insights into the structure-activity relationships of highly efficient CoMn oxides for the low temperature NH3-SCR of NOx, Appl. Catal., B, 2020, 277, 119215 CrossRef CAS.
- B. E. R. Snyder, M. L. Bols, H. M. Rhoda, D. Plessers, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Cage effects control the mechanism of methane hydroxylation in zeolites, Science, 2021, 373, 327–331 CrossRef CAS PubMed.
- L.-C. Wang, Y. Zhang, J. Xu, W. Diao, S. Karakalos, B. Liu, X. Song, W. Wu, T. He and D. Ding, Non-oxidative dehydrogenation of ethane to ethylene over ZSM-5 zeolite supported iron catalysts, Appl. Catal., B, 2019, 256, 117816 CrossRef.
- S. Takenaka, Formation of filamentous carbons over supported Fe catalysts through methane decomposition, J. Catal., 2004, 222, 520–531 CrossRef CAS.
- T. P. Almeida, T. Kasama, A. R. Muxworthy, W. Williams, L. Nagy, T. W. Hansen, P. D. Brown and R. E. Dunin-Borkowski, Visualized effect of oxidation on magnetic recording fidelity in pseudo-single-domain magnetite particles, Nat. Commun., 2014, 5, 5154 CrossRef CAS.
- F. Jin, B. Wang, Y. Ning, Z. Zhang, J. Yang, H. Zhang, D. Wang and Y. Zhou, Graphene-modified mesoporous iron phosphate as superior binary sulfur host for lithium-sulfur batteries, Energy Technol., 2020, 8, 1901462 CrossRef CAS.
- Z. Xu, L. Zhang, M. Pan, Q. Jiang, Y. Huang, F. Wang and X. Liu, A bionanozyme with ultrahigh activity enables spatiotemporally controlled reactive oxygen species generation for cancer therapy, Adv. Funct. Mater., 2021, 31, 2104100 CrossRef CAS.
- F. O. Boakye, M. Fan, F. Zhang, H. Tang, R. Zhang and H. Zhang, Growth of branched heterostructure of nickel and iron phosphides on carbon cloth as electrode for hydrogen evolution reaction under wide pH ranges, J. Solid State Electrochem., 2022, 26, 875–885 CrossRef CAS.
- S. Bi, Z. Geng, Y. Wang, Z. Gao, L. Jin, M. Xue and C. Zhang, Multi-stage porous nickel-iron oxide electrode for high current alkaline water electrolysis, Adv. Funct. Mater., 2023, 33, 2214792 CrossRef CAS.
- R. Kumar, M. Mooste, Z. Ahmed, S. Akula, I. Zekker, M. Marandi, M. Käärik, J. Leis, A. Kikas, A. Treshchalov, M. Otsus, J. Aruväli, V. Kisand, A. Tamm and K. Tammeveski, Highly active ZIF-8@CNT composite catalysts as cathode materials for anion exchange membrane fuel cells, Ind. Chem. Mater., 2023, 1, 526–541 RSC.
- Y. Shaharyar, J. Y. Cheng, E. Han, A. Maron, J. Weaver, J. Marcial, J. S. McCloy, A. Goel and L. Pinckney, Elucidating the effect of iron speciation (Fe2+/Fe3+) on crystallization kinetics of sodium aluminosilicate glasses, J. Am. Ceram. Soc., 2016, 99, 2306–2315 CrossRef CAS.
- X. Zuo, X. Wang, G. Si, D. Zhang, X. Yu, Z. Guo and N. Gu, Size-dependent oxygen vacancy of iron oxide nanoparticles, Small Methods, 2025, 9, e2400685 CrossRef PubMed.
- Y. Zhou, D. Wang, Y. Li, L. Jing, S. Li, X. Chen, B. Zhang, W. Shuai, R. Tao, X. Lu and J. Liu, Critical effect of oxygen pressure in pulsed laser deposition for room temperature and high performance amorphous In-Ga-Zn-O thin film transistors, Nanomaterials, 2022, 12, 4358 CrossRef CAS PubMed.
- Y. Wu, X. Wu, J. Fan, H. Wang and Z. Wu, Insights into the roles of different iron species on zeolites for N2O selective catalytic reduction by CO, Environ. Sci. Technol., 2024, 58, 22583–22593 CrossRef CAS PubMed.
- Q. Deng, R. Zhou, Y. C. Zhang, X. Li, J. Li, S. Tu, G. Sheng, J. Wang, Z. Zeng and T. Yoskamtorn, H+-H− pairs in partially oxidized MAX phases for bifunctional catalytic conversion of furfurals into linear ketones, Angew. Chem., 2023, 135, e202211461 CrossRef.
- F. Qin, X. Fan and W. Ma, Selective oxidation of triethylamine catalyzed by Mn-Ce/ZSM-5, Langmuir, 2023, 39, 7820–7830 CrossRef CAS PubMed.
- X.-Y. Peng, L.-J. Liu, B.-X. Shen, Y. Bian and L.-C. Su, Insight into the catalytic oxidation of toluene over M/ZSM-5 (M = Cu, Mn, Fe, Ce, Ti) catalysts, J. Fuel Chem. Technol., 2023, 51, 841–851 CrossRef CAS.
- J. Luo, K. Kamasamudram, N. Currier and A. Yezerets, NH3-TPD methodology for quantifying hydrothermal aging of Cu/SSZ-13 SCR catalysts, Chem. Eng. Sci., 2018, 190, 60–67 CrossRef CAS.
- Y. Yu, X. Yi, J. Zhang, Z. Tong, C. Chen, M. Ma, C. He, J. Wang, J. Chen and B. Chen, Application of ReOx/TiO2 catalysts with excellent SO2 tolerance for the selective catalytic reduction of NOx by NH3, Catal. Sci. Technol., 2021, 11, 5125–5134 RSC.
- X. Zhong, J. Liu, L. Gao, J. Chen, X. Wang, Y. Zhang, Y. A. Wu, M. Shakeri, X. Zhang and B. Zhang, Constructing the Al deficiency in Si-O(H)-Al units based on Pt/ZSM-5 for enhanced hydrocracking of polyethylene into high-quality liquid fuel, Nano Res., 2024, 17, 10088–10098 CrossRef CAS.
- K. Krishna, G. Seijger, C. Van Den Bleek, M. Makkee, G. Mul and H. Calis, Selective catalytic reduction of NO with NH3 over Fe-ZSM-5 catalysts prepared by sublimation of FeCl3 at different temperatures, Catal. Lett., 2003, 86, 121–132 CrossRef CAS.
- J. Zeng, S. Chen, Z. Fan, C. Wang, H. Chang and J. Li, Simultaneous selective catalytic reduction of NO and N2O by NH3 over Fe-Zeolite catalysts, Ind. Eng. Chem. Res., 2020, 59, 19500–19509 CrossRef CAS.
- H. Chang, T. Zhang, H. Dang, X. Chen, Y. You, J. W. Schwank and J. Li, Fe2O3@SiTi core-shell catalyst for the selective catalytic reduction of NOx with NH3: Activity improvement and HCl tolerance, Catal. Sci. Technol., 2018, 8, 3313–3320 RSC.
- W. Kwiatek, A. Hanson, C. Paluszkiewicz, M. Gałka, M. Gajda and T. Cichocki, Application of SRIXE and XANES to the determination of the oxidation state of iron in prostate tissue sections, J. Alloys Compd., 2004, 362, 83–87 CrossRef CAS.
- S. Pongha, B. Seekoaon, W. Limphirat, P. Kidkhunthod, S. Srilomsak, Y.-M. Chiang and N. Meethong, XANES investigation of dynamic phase transition in olivine cathode for Li-ion batteries, Adv. Energy Mater., 2015, 5, 1500663 CrossRef.
- J. Zhu, C. Jin, Y. Duan, L. Zhu, J. Liu, K. Cheng, H. Cheng, F. Liu, H. Li and J. Liu, Fe-triggered Mn-mullite oxides for efficient low-temperature reduction of nitrogen oxides: Insights into the structure-activity relationship, Appl. Catal., B, 2025, 366, 125028 CrossRef CAS.
- Y. Wang, J. Han, M. Chen, W. Lv, P. Meng, W. Gao, X. Meng, W. Fan, J. Xu, W. Yan and J. Yu, Low-silica Cu-CHA zeolite enriched with Al pairs transcribed from silicoaluminophosphate seed: Synthesis and Ammonia performance, Angew. Chem., Int. Ed., 2023, 62, e202306174 CrossRef CAS PubMed.
- Q. Liang, X. Wu, D. Weng and H. Xu, Oxygen activation on Cu/Mn-Ce mixed oxides and the role in diesel soot oxidation, Catal. Today, 2008, 139, 113–118 CrossRef CAS.
- X. Ding, J. Qiu, Y. Liang, M. Zhao, J. Wang and Y. Chen, New insights into excellent catalytic performance of the Ce-modified catalyst for NO oxidation, Ind. Eng. Chem. Res., 2019, 58, 7876–7885 CrossRef CAS.
- H. Kubota, C. Liu, T. Toyao, Z. Maeno, M. Ogura, N. Nakazawa, S. Inagaki, Y. Kubota and K.-I. Shimizu, Formation and reactions of NH4NO3 during transient and steady-state NH3-SCR of NOx over H-AFX zeolites: Spectroscopic and theoretical studies, ACS Catal., 2020, 10, 2334–2344 CrossRef CAS.
- P. E. Fanning and M. A. Vannice, A DRIFTS study of Cu-ZSM-5 prior to and during its use for N2O decomposition, J. Catal., 2002, 207, 166–182 CrossRef CAS.
- L. Jia, L. Zhang, B. Liu, H. Cheng, H. Li, Z. Zhao, W. Zhu, W. Song, J. Liu and J. Liu, Interface induced by hydrothermal aging boosts the low-temperature activity of Cu-SSZ-13 for selective catalytic reduction of NOx, Environ. Sci. Technol., 2024, 58, 15038–15051 CAS.
- J. Liu, Y. Pan, Y. Duan, S. Liang, H. Cheng, M. Hua, X. Dai, F. Liu, H. Li and J. Liu, Fe1-yMnyZr4Ox solid solution for efficient low-temperature selective catalytic reduction of NOx, Chem. Eng. Sci., 2025, 313, 121750 CrossRef CAS.
- J. Cheng, D. Zheng, G. Yu, R. Xu, C. Dai, N. Liu, N. Wang and B. Chen, N2O catalytic decomposition and NH3-SCR coupling reactions over Fe-SSZ-13 catalyst: Mechanisms and interactions unraveling via experiments and DFT calculations, ACS Catal., 2023, 13, 934–947 CrossRef CAS.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- H. Funke, A. Scheinost and M. Chukalina, Wavelet analysis of extended X-ray absorption fine structure data, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 232–234 CrossRef.
- W. Eijima, G. Shibata, N. Shibayama, Y. Kobashi, H. Ogawa and K.-I. Shimizu, Kinetic modeling of steady-state NH3-SCR over a monolithic Cu-CHA catalyst, Catal. Today, 2020, 352, 237–242 CrossRef CAS.
- P. S. Metkar, M. P. Harold and V. Balakotaiah, Experimental and kinetic modeling study of NH3-SCR of NOx on Fe-ZSM-5, Cu-chabazite and combined Fe- and Cu-zeolite monolithic catalysts, Chem. Eng. Sci., 2013, 87, 51–66 CrossRef CAS.
- S. Kiil, S. K. Bhatia and K. Dam-Johansen, Modelling of catalytic oxidation of NH3 and reduction of NO on limestone during sulphur capture, Chem. Eng. Sci., 1996, 51, 587–601 CrossRef CAS.
| This journal is © Institute of Process Engineering of CAS 2026 |