
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProcess-intensified enzymatic decarboxylation using immobilized arylmalonate decarboxylase for sustainable asymmetric synthesis of α-arylpropionic acids
Jan Gerstenberger a,
Timm Werbiloa,
Marvin Haasa,
Simona Serbanb,
Alessandra Bassob,
Pablo Domínguez de Maríac and
Selin Kara*ad
a,
Timm Werbiloa,
Marvin Haasa,
Simona Serbanb,
Alessandra Bassob,
Pablo Domínguez de Maríac and
Selin Kara*ad
aInstitute of Technical Chemistry, Leibniz University Hannover, Callinstraße 5, 30167 Hannover, Germany. E-mail: selin.kara@iftc.uni-hannover.de
bSunresin New Materials, Sunresin Park, Xi'an Hi-tech Industrial Development Zone Xian, Shaanxi, P. R. China
cSustainable Momentum SL, 35011 Las Palmas de Gran Canaria, Canary Islands, Spain
dBiocatalysis and Bioprocessing Group, Department of Biological and Chemical Engineering, Aarhus University, 8000 Aarhus C, Denmark. E-mail: selin.kara@bce.au.dk
First published on 8th June 2026
Abstract
Enantiopure α-arylpropionic acids are essential pharmaceuticals, yet their biocatalytic synthesis via arylmalonate decarboxylases (AMDases) is hindered by the instability of malonic acid intermediates. Standard saponification requires an acidic workup that triggers spontaneous, non-selective malonate decarboxylation, while alternative hydrogenolysis reactions rely on potentially pyrophoric palladium catalysts. This study presents an integrated, intensified process combining alkaline hydrolysis of dimethyl malonates with enzymatic decarboxylation using an immobilized AMDase. To suppress racemic side-product formation, a reaction setup using a rotating bed reactor with in situ pH-stat control was established, ensuring substrate stability via anionic stabilization. For enzyme immobilization, C2-amino polyacrylate carriers resulted in superior stability, enabling efficient preparative-scale biotransformations. Naproxen and flurbiprofen were obtained with isolated yields up to 96% and high enantiopurity (ee > 99%), leading to a Process Mass Intensity (PMI) of, e.g., 158 kg kg naproxen−1, representing a significant improvement over the current benchmark. Likewise, CO2 production, as a Global Warming Potential (GWP) indicator, was assessed considering the three main contributors, the energy of the reaction, the decarboxylation process, and the wastewater treatment, with an impact in the range of 32 kg CO2 kg naproxen−1. Overall, integrating a safe saponification with automated process control and decarboxylases provides a scalable, green(er) framework for producing enantiopure carboxylic acids.
Green foundation1. α-Aryl-propionic acids are key pharmaceuticals. This work presents a biocatalytic platform replacing palladium catalyzed hydrogenolysis with alkaline saponification of dimethyl malonates, improving safety by avoiding pyrophoric catalysts and hydrogen, while enhancing atom economy and reducing waste via smaller ester protecting groups.2. Spontaneous malonate decarboxylation is suppressed via anionic stabilization until enzymatic decarboxylation introduces chirality in an automated pH-stat rotating bed reactor. Estimated PMI is 158 kg kg−1 naproxen, outperforming current benchmarks. With ∼90% solvent recovery and enzyme recycling, PMI may reach 73. In situ naproxen precipitation simplifies downstream processing and improves sustainability. 3. Global warming potential analysis (kg CO2 kg naproxen−1) identified wastewater treatment as the main environmental hotspot, beyond (petrochemical) malonate decarboxylation and energy invested. Future mitigation actions should focus on water- and enzyme recycling to reduce the waste. |
Introduction
Enantiopure α-arylpropionic acids are key building blocks in the pharmaceutical industry and form the core structure of many non-steroidal anti-inflammatory drugs (NSAIDs), including naproxen and ibuprofen. In profen derivatives, pharmacological activity resides in the (S)-enantiomer, whereas the (R)-enantiomer is largely inactive and may cause undesired side effects.1–3 Consequently, considerable efforts have been devoted to the enantioselective synthesis of α-arylpropionic acids.4–8 These strategies include catalytic asymmetric hydrogenation of corresponding acrylic acid precursors,9,10 the use of chiral auxiliaries,11 biocatalysis,12–16 and resolution methods.17,18Biocatalysis represents a powerful and potentially sustainable strategy to access these industrially relevant chiral pharma intermediates. Compared to traditional chemical approaches, biocatalytic methods typically proceed under milder reaction conditions and may provide significant environmental benefits.19–24 Among the available biocatalytic tools for the synthesis of enantiopure α-arylpropionic acids, arylmalonate decarboxylases (AMDases) offer an elegant route via decarboxylation of prochiral malonates (Scheme 1). First discovered by Miyamoto and Ohta in 1990, the arylmalonate decarboxylase (AMDase) was initially isolated from Bordetella bronchiseptica (Bb) in an enzyme screening campaign.25
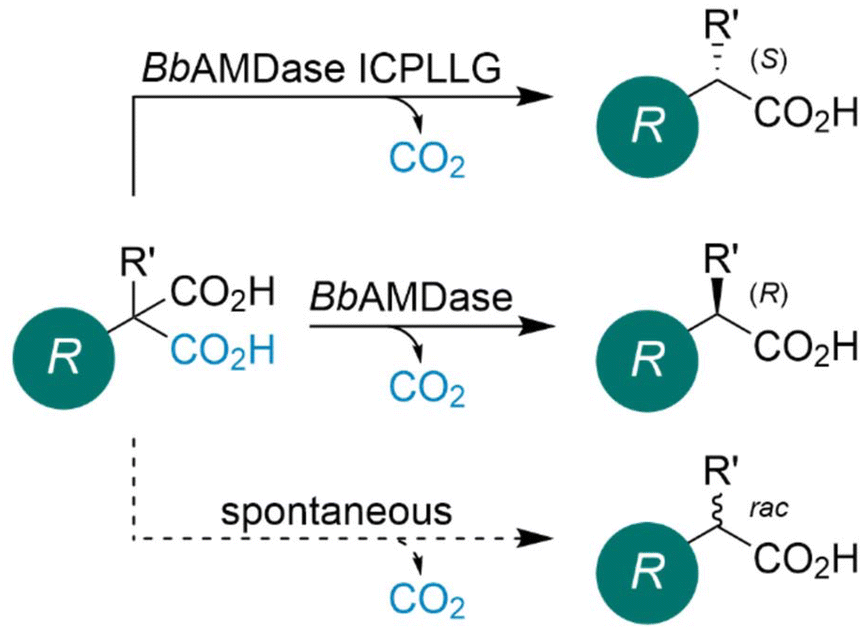 | ||
| Scheme 1 Asymmetric decarboxylation of prochiral malonic acids by AMDase for the synthesis of α-substituted carboxylic acids. Malonic acid derivatives can be prone to spontaneous decarboxylation leading to the racemic mixture of desired product. R = vinyl or conjugated systems. R′ = H, D, Me, OH, NH2, and F. | ||
Since its discovery, the natural substrate of AMDase has remained unknown.27 The enzyme is expressed recombinantly in Escherichia coli, with recent reports of efficient, scalable cultivation.28 The cofactor-independent AMDase catalyzes the asymmetric decarboxylation of prochiral α-substituted malonic acids, yielding optically pure propionic acids. All nature-derived AMDases showed predominantly (R)-selectivity (Scheme 1).29,30 Interestingly, inversion of stereoselectivity can be introduced by the double substitution G74C/C188S in BbAMDase. Further protein engineering strategies yielded the (S)-selective variant ICPLLG (V43I/G74C/A125P/V156L/M159L/C188G),31 which was used in benchtop production of flurbiprofen16 and naproxen.15 The industrial application of AMDase has been limited by several factors, most notably the free enzyme's insufficient thermal and process stability. Enzyme immobilization offers an effective strategy to enhance robustness under non-natural conditions and extend catalyst lifetime. A variety of immobilization approaches have been reported, including adsorptive, covalent, entrapment, encapsulation, and cross-linking strategies.32 Multiple immobilization strategies for AMDase have been reported in the literature.33–35 Notably, Aßmann et al. achieved a 158-fold increase in operational stability through covalent immobilization on an amino-C2 acrylate carrier.35 Beyond immobilization, Kourist and Kara groups demonstrated that ancestral sequence reconstruction can substantially improve AMDase stability, work that was conducted in parallel with the present study. Furthermore, inversion of stereoselectivity was introduced in the reconstructed ancestral variants.36
In addition to the enzyme's limited stability, the spontaneous decarboxylation of malonic acid substrates poses a major challenge for the synthesis of enantiopure products, as it leads to the formation of racemic propionic acids (Scheme 1). This side reaction is particularly pronounced under acidic conditions and at elevated temperatures. Overall, such decarboxylation reactions are widely used in organic synthesis to prepare ketones, alkenes, and aromatic derivatives from their corresponding carboxylic acid precursors.37,38 An overview of how this ‘racemization challenge’ was addressed by different research groups is provided in Scheme 2.
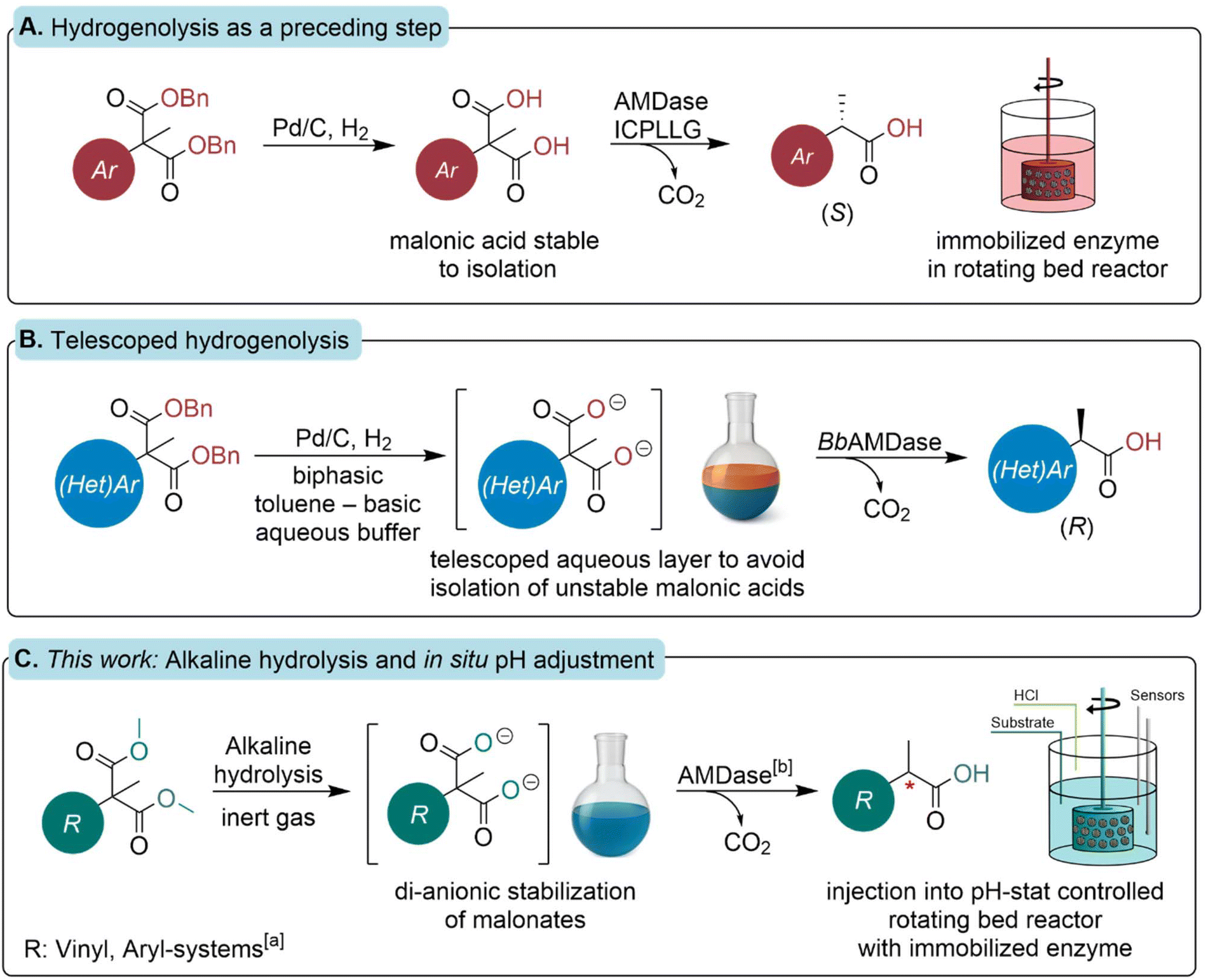 | ||
| Scheme 2 Overview of different approaches of preparative enzymatic decarboxylation utilizing AMDase. A. Hydrogenolysis of benzyl esters was reported by Gaßmeyer et al. to avoid the acidification of the reaction mixture, thereby avoiding spontaneous decarboxylation. The reaction was intensified to a bench scale in a rotating bed reactor system.16 B. This concept was further developed by Blakemore et al. to a telescoped bi-phasic process for the synthesis of heteroarylpropionic acids in preparative scale.26 C. This work shows the control of traditional saponification in preparative scale with subsequent enzymatic decarboxylation. Spontaneous decarboxylation is avoided by in situ pH adjustment in the rotating bed reactor under optimized process conditions. [a] Substrates utilized in this study: naproxen malonate (1b), flurbiprofen malonate (2b), 2-methyl-2-phenyl malonate (3b), 2-methyl-2-vinyl malonate (4b) and 2-ethyl-2-vinyl malonate (5b). [b] BbAMDase variants used: (R)-selective IPLL variant and (S)-selective ICPLLG variant. | ||
Among the different strategies, Gaßmeyer et al. reported the synthesis and isolation of malonic acids by hydrogenolysis of dibenzylmalonate precursors (Scheme 2A).16 This approach does not require acidification to process the reaction mixture, unlike traditional saponification. Furthermore, hydrogenolysis cleaves the benzylester in most cases with quantitative yield, without further purification. Thus, the received malonic acids were obtained in high purity, free of racemic product from spontaneous decarboxylation. Blakemore et al. further developed a biphasic approach that uses hydrogenolysis in a toluene phase, with a telescoped phase of a basic aqueous buffer for the solubilization of deprotected malonates.26 This enables subsequent enzymatic decarboxylation without the need to isolate the substrate (Scheme 2B). They further expanded the substrate spectrum of promiscuous AMDase to a variety of prochiral heteroaryl malonic acids. In the work presented here (Scheme 2C), we demonstrate a fundamentally different approach aimed at improving overall sustainability and handling of the route. To this end, the traditional saponification step is combined with the concept of direct conversion of deprotected malonates, without compromising the purity of either the intermediate malonates or the final products. By employing a methyl ester protection strategy rather than bulky dibenzyl esters, an improved atom economy is achieved (SI, section 1.1), while overcoming the use of palladium on carbon (Pd/C). The saponification process requires only the safe handling of acids and bases, and avoids potential explosion risks associated with laboratory-scale hydrogenations.39
Results and discussion
Experimental set-up and saponification
One of the key challenges in the enzymatic synthesis of optically pure propionic acids is the spontaneous decarboxylation of the malonic acid starting material, which yields a racemic product. To address this, we aimed at developing an intensified reaction setup aligned with Green Chemistry principles.40 We identified two process stages as most critical for inducing spontaneous decarboxylation: (i) the acidification step typically performed after saponification, and (ii) the pH of the reaction buffer itself. During a conventional acidic workup, the deprotected malonate is protonated while the exothermic neutralization simultaneously supplies the activation energy for decarboxylation. This proceeds through a six-membered intermediate (Scheme 3A), yielding an enediolate whose tautomeric equilibrium favors the undesired racemic propionic acid.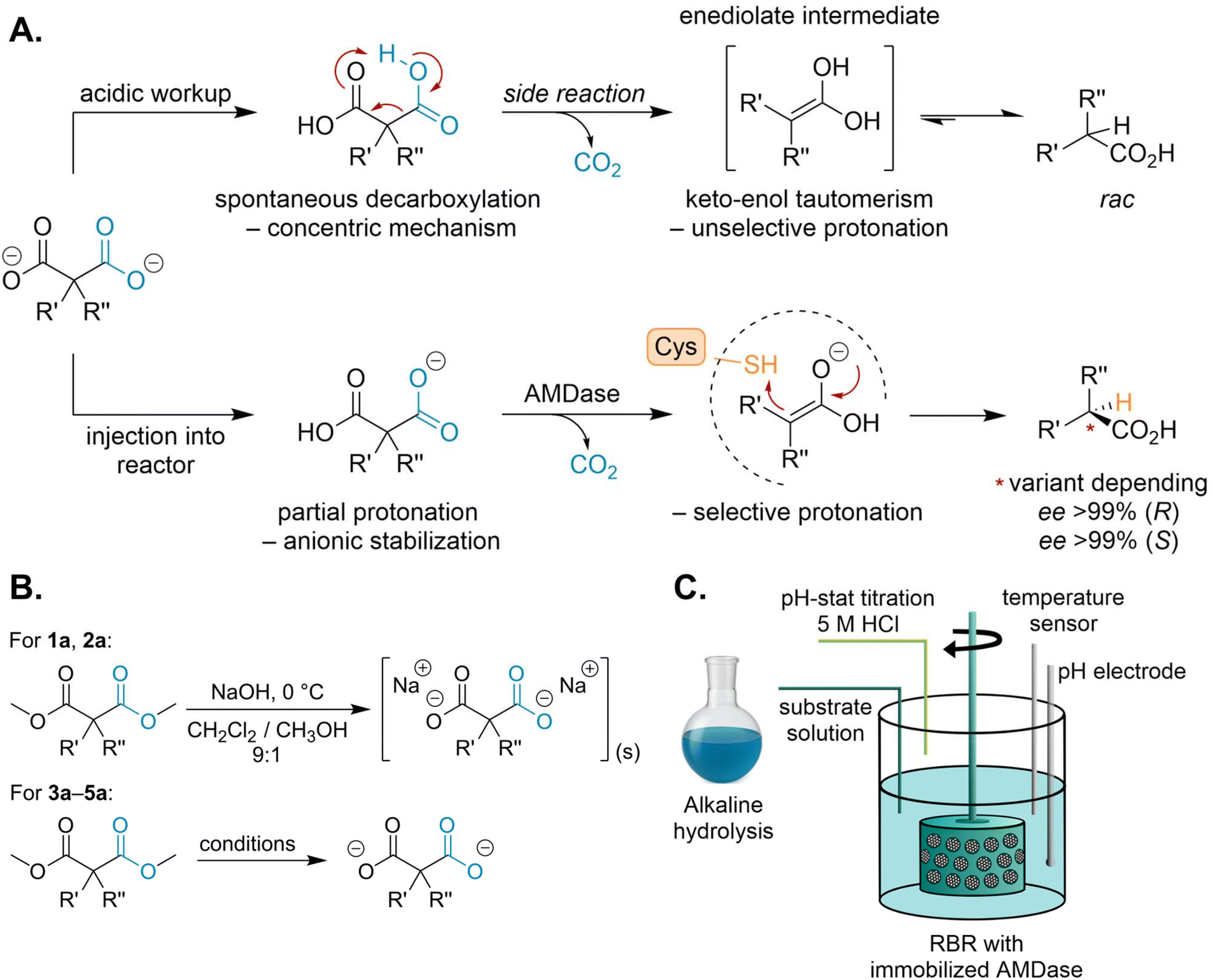 | ||
Scheme 3 A. Comparison of spontaneous decarboxylation to the asymmetric enzymatic decarboxylation of AMDase. R′ = vinyl or conjugated systems. R″ = H, D, Me, OH, NH2, F. B. Saponification reaction scheme of dimethyl malonate derivatives used in this study. Saponification conditions: 3a: LiOH, 0 °C, MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF:H2O (2:2:1). 4a and 5a: 40% (w/v) NaOH (aq.), EtOH, 0 °C (SI, section 4). C. Schematic overview of process set-up of the rotating bed reactor (RBR) utilizing a SpinChem® vessel V2 with a corresponding basket filled with immobilized enzyme on Seplife® EMC7225S amino polyacrylate resin. The alkaline substrate solution is injected by a syringe pump, and the pH controlled by a volumetric titrator in pH stat mode. :THF:H2O (2:2:1). 4a and 5a: 40% (w/v) NaOH (aq.), EtOH, 0 °C (SI, section 4). C. Schematic overview of process set-up of the rotating bed reactor (RBR) utilizing a SpinChem® vessel V2 with a corresponding basket filled with immobilized enzyme on Seplife® EMC7225S amino polyacrylate resin. The alkaline substrate solution is injected by a syringe pump, and the pH controlled by a volumetric titrator in pH stat mode. | ||
Previous approaches avoided the acidic workup by applying the clean hydrogenolysis of the benzylesters and dissolving the free malonic acid derivative in the slight alkaline enzymatic buffer. Our approach circumvents this issue by directly integrating the saponification with the biocatalytic step. It builds on the dimethyl malonate precursor saponification and avoids the full protonation itself, before enzymatic decarboxylation leads to the stable product. This design enhances atom economy and eliminates the need for hydrogenolysis, which in industrial settings may entail complex ATEX compliance and significant safety management.41
While optimizing the saponification of these dimethyl malonate derivatives, we noted that strict adherence to Schlenk conditions prevents side product formation and results in quantitative yields. This allows the resulting alkaline reaction mixture to be injected directly into the buffered solution containing the immobilized biocatalyst. The basicity of the injected substrate solution is actively compensated by a volumetric titrator, maintaining a stable pH optimized for enzyme activity and substrate stability (Scheme 3C). However, this integrated strategy is dependent on a quantitative and rapid saponification step. The choice of solvent system proved to be highly substrate dependent. The smaller precursors dimethyl 2-methyl-2-phenylmalonate (3a) and dimethyl-2-alkyl-2-vinylmalonates (4a, 5a) were readily deprotected in an aqueous sodium hydroxide solution. In contrast, larger, more non-polar substrates such as dimethyl naproxen malonate (1a) and dimethyl flurbiprofen malonate (2a) exhibited poor solubility in this reaction system. This resulted in incomplete conversions and an increased rate of spontaneous decarboxylation during the prolonged saponification process. To achieve quantitative deprotection of these substrates, we initially investigated a mixed solvent system of tetrahydrofuran, methanol, and water (2:2:1) using LiOH as the base. Although these conditions represent a common protocol for saponification, they failed to resolve the solubility limitations and still resulted in significant decarboxylation.
As conventional methods proved insufficient, we therefore explored a non-aqueous saponification strategy. The non-polar precursors were fully dissolved in a 9:1 mixture of dichloromethane (CH2Cl2) and methanol and were quantitatively deprotected at 0 °C using NaOH (Scheme 3B).42 The use of CH2Cl2 requires careful justification within a Green Chemistry framework, and the future quest for other solvent alternatives is fundamental.43 In any case, this drawback can be partly mitigated by the reaction dynamics: as the reaction proceeds, the target disodium salt of the malonate derivative precipitates from the organic solvent mixture. This precipitation enables efficient recovery and recycling of the solvent (SI, Fig. S20), which has a significant impact on the Green Metrics.44
Thus, the isolated precipitate can be freed from residual organic solvents and directly transferred to the reactor. As the aim of this work was to establish the entire novel route under high substrate loadings, and for the practicality of our bench-scale setup, in our experiments, we opted for the injection of the dissolved substrate, with pH adjustment as the flow-rate-limiting step of the whole procedure. To this end, water was added to the completed saponification mixture, and residual organic solvents were removed under reduced pressure for eventual recycling in scaled-up conditions. The resulting alkaline substrate solution was then injected into the rotating bed reactor (RBR) for the subsequent biocatalytic decarboxylation.
Immobilization of biocatalysts
Implementation of the intensified process in the RBR required heterogenization of the biocatalyst. To this end, the metal-affinity and covalent immobilization strategies reported by Aßmann et al.35 were reproduced and further optimized (SI, detailed specification of carriers used, section 3.3). The AMDase variants employed in the intensified process share a common structural fold (Fig. 1), enabling two distinct immobilization approaches: (i) affinity immobilization via the C-terminal His-tag or (ii) covalent attachment through surface-exposed lysine residues.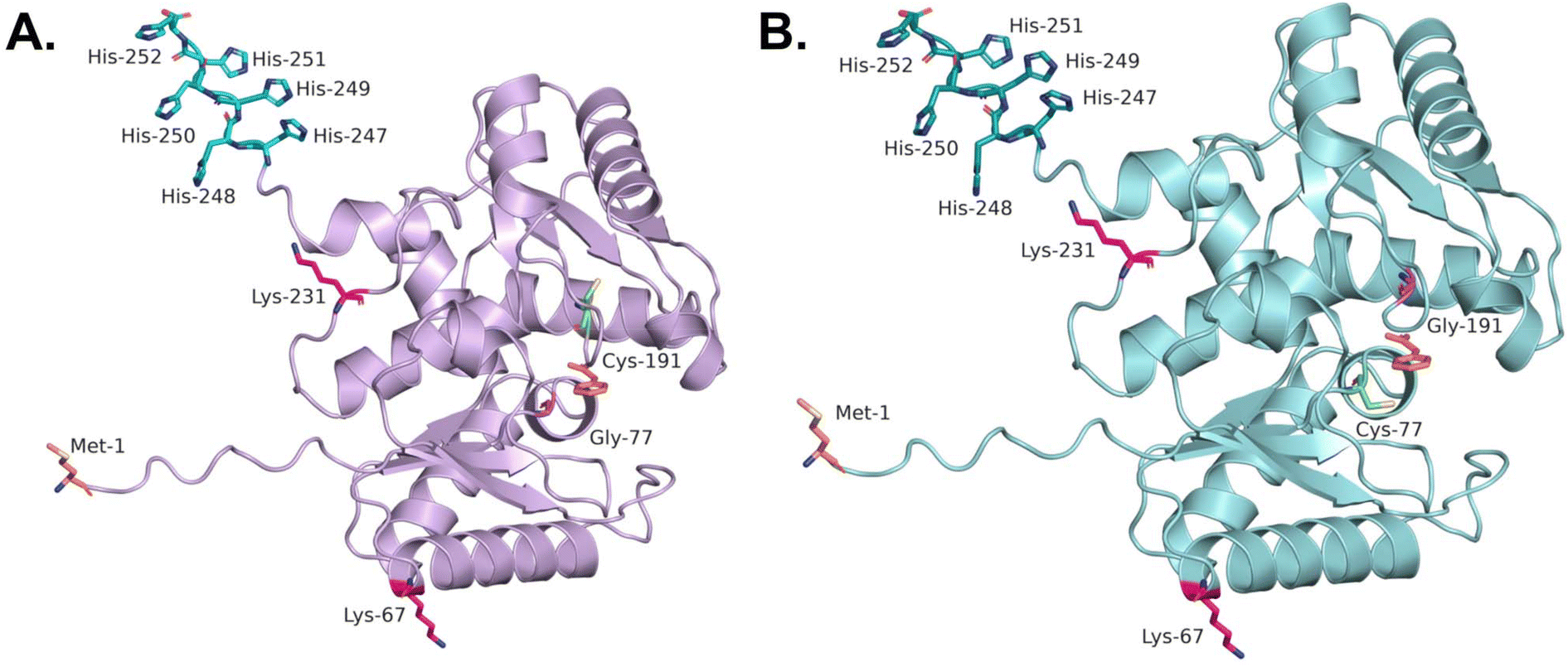 | ||
| Fig. 1 Structural comparison of AMDase IPLL (A) and AMDase ICPLLG (B) Residues of interest are marked in color: lysines in pink, His6-Tag at the C-terminus in blue/green, N-terminus in orange and active site with the reaction product phenylacetic acid based on the crystal data (3IXL) of AMDase ICPLLG.46 | ||
Carrier screening for metal affinity (IMA) immobilization
First, metal affinity (IMA) carriers Seplife® Chelex 7350S based on iminodiacetic acrylate and loaded with different divalent metal ions, were evaluated. While immobilization on Fe2+- and Cu2+-charged carriers resulted in a substantial loss of enzymatic activity, carriers loaded with Co2+, Ni2+, or Zn2+ showed no significant differences in specific activity per carrier mass. Analysis of the immobilization yield revealed the highest degree of specific binding for the Co2+-charged carrier (Fig. 2A). High binding specificity is advantageous, as reduced levels of nonspecifically adsorbed protein decrease the risk of enzyme leaching and thereby simplify downstream processing. Consequently, the Co2+ IMA carrier Seplife® Chelex 7350S/Co2+ was selected as the representative affinity immobilization system for the further assessments.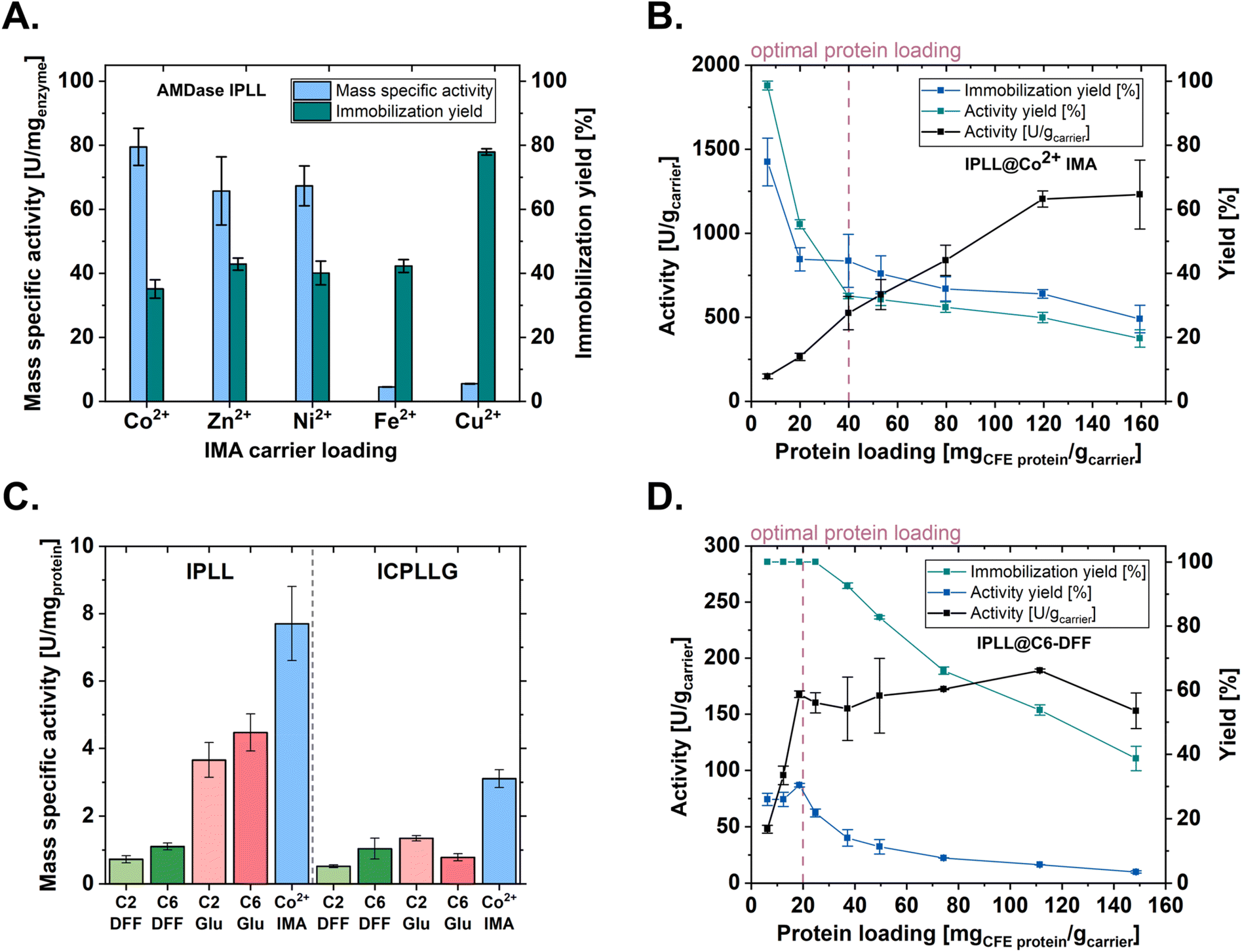 | ||
| Fig. 2 Investigation of activity characteristics of enzyme carriers for AMDase immobilization. Immobilizations were done in triplicate. All mass-specific activities were determined towards phenylmalonate (1 mL, 20 mM phenylmalonate, 50 mM HEPES buffer, pH 7.8, 30 °C). All experimental preparations involving active enzyme were done at 4 °C. Enzymatic reactions were incubated for 5 minutes at 30 °C before conducting the activity assay (SI section 3.4). Protein quantification was done by BCA-assay. A. Comparison of mass-specific activity [U mgenzyme−1] and immobilization yield of different divalent metal loading of immobilized metal affinity carrier with immobilization of AMDase IPLL. Quantification of AMDase was done by densitometric SDS-PAGE. B. Protein loading study of Co2+ metal affinity immobilization of AMDase IPLL. The protein loading of the carrier was achieved by step-wise dilution of a CFE stock solution with the reaction buffer (50 mM HEPES buffer, pH 7.8). C. Comparison of mass specific activity [U mgprotein−1] of amino polyacrylate carriers with C2 and C6 linkers and 2,5 diformylfuran (DFF) and glutaraldehyde crosslinking agent for the immobilization of AMDase IPLL and AMDase ICPLLG. D. Protein loading study of AMDase IPLL immobilized onto the C6 amino polyacrylate using glutaraldehyde as crosslinking agent. The protein loading of the carrier was achieved by step-wise dilution of a CFE stock solution with the immobilization buffer (50 mM HEPES buffer, 50 mM NaCl, pH 6.8). | ||
To gain deeper insight into AMDase affinity immobilization, the amount of enzyme applied to the carrier surface was increased stepwise using the cell-free extract (CFE). This approach enables the identification of potential multilayer formation and non-linear loading effects.45 For each carrier preparation, (i) the mass-specific activity, (ii) activity yield, and (iii) immobilization yield were determined (Fig. 2B). The carrier's specific activity increased linearly with increasing enzyme provision up to a protein loading of 120 mgCFE gcarrier−1. In contrast, both immobilization and activity yields, key parameters for the sustainable and efficient use of the biocatalyst, decreased markedly already at low protein loadings. This indicates that enzyme is not fully accessible for its catalytic activity. Balancing the carrier's specific activity with efficient enzyme utilization, an optimal loading of 40 mgCFE gcarrier−1 was selected for this system.
Carrier screening for covalent immobilization
The previous study by Aßmann et al.35 identified covalent immobilization on a C2-amino polyacrylate carrier using a glutaraldehyde linker as the most suitable option for an intensified process, particularly with respect to biocatalyst half-life, which gave a remarkable 158-fold higher operational stability compared to the free enzyme. For the present study, a very similar type of resin was evaluated, based on polyacrylate and with a porosity suitable for AMDase immobilization. Four carrier–linker combinations were evaluated: carriers functionalized with either a C2 (Seplife® EMC7225S) or C6 (Seplife® EMC7120S) spacer, each combined with a conventional glutaraldehyde linker or with 2,5-diformylfuran (DFF), a bio-based alternative for covalent enzyme immobilization.47In all cases, covalent immobilization resulted in significantly lower mass-specific activity of the total protein on the carrier compared to affinity immobilization (Fig. 2C vs. Fig. 2A). Comparing linker systems and enzyme variants revealed variant-dependent effects. The (R)-selective IPLL variant exhibited higher activity with the conventional glutaraldehyde linker (C6-Glu: 4.5 ± 0.5 U mg−1) than with the bio-based alternative DFF (C6-DFF: 1.1 ± 0.1 U mg−1). In contrast, the (S)-selective ICPLLG variant showed no clear dependence on the crosslinking reagent, and differences in mass-specific activity among the analyzed covalent immobilization strategies were not significant.
To obtain deeper insight into covalent immobilization, the effect of protein loading was investigated using the AMDase IPLL variant immobilized on a C6-amino polyacrylate carrier with the bio-based linker 2,5-diformylfuran (DFF) (Fig. 2D). In contrast to the affinity-based system (Fig. 2B), all proteins present in the CFE were covalently immobilized up to a loading of 25 mgCFE gcarrier−1. However, the immobilization yield decreased from 26% to 4% at the highest protein loading. The carrier activity increased with increasing protein loading up to the point of 19 mgCFE gcarrier−1, after which a plateau was observed. This plateau, together with the declining activity yield, defines the practical loading limit for process intensification, as additional enzyme input provides diminishing benefits. As the underlying covalent binding mechanism is expected to remain comparable across the analyzed carrier–linker configurations, the influence of different spacers or crosslinking agents on the optimal loading capacity was considered negligible. Consequently, a protein loading of 20 mgCFE gcarrier−1 was selected as the optimal condition for immobilization of AMDase on amino polyacrylate carriers.
It is interesting to note that despite the available amino groups (0.81 mmol gcarrier−1, SI, Table S12) and available surface area (≥100 m2 g−1), the carrier should be able to achieve higher loadings. However, results are in line with previously reported observations where enzyme immobilization occurs on the external section of the carrier. This is due to the Gaussian distribution of the porosity profile, which includes both micro- and macro-porosity. Consequently, the enzyme is able to effectively access 20–30% of the carrier beads.48 This is a particularly interesting observation, as the affinity enzyme carrier shows approximately 10-fold higher activity at the highest protein loading, with a quarter of the surface area (≥25 m2 g−1) and comparable particle sizes of the two carriers utilized. A similar finding was also obtained during immobilization of a ketoreductase enzyme when comparing different immobilization strategies.49
The (internal) mass transfer limitations can be attributed to the specified pore size of the C6-amino polyacrylate carrier, which is considerably smaller, with 200–400 Å compared to 800–1000 Å. Nevertheless, the primary cause of reduced catalytic activity appears to be the more stringent conditions inherent to covalent immobilization compared to the milder, reversible chelation on the IMA carrier. This is evidenced by the negligible differences in specific activity observed between identical crosslinking agents across different linker spacers. Notably, the amino polyacrylate carrier with the C2 linker (Seplife® EMC7225S) possesses a specified pore size of 500–700 Å (SI, Table S12), which theoretically should alleviate mass transfer limitations and enhance diffusion relative to the C6 alternative (Seplife® EMC7120S); however, this potential advantage did not translate into improved performance. The advantage of the larger pore size could be outweighed by the shorter distance to the carrier due to the C2 linker. The shorter distance leads to less mobility of the immobilized enzyme and possible blockage of the active center if oriented unfavorably towards the carrier. While covalent immobilization represents a more robust but harsher process, a comparison of immobilization yields (Yimmo) underscores the superior specificity of the affinity methodology (Fig. 3A).
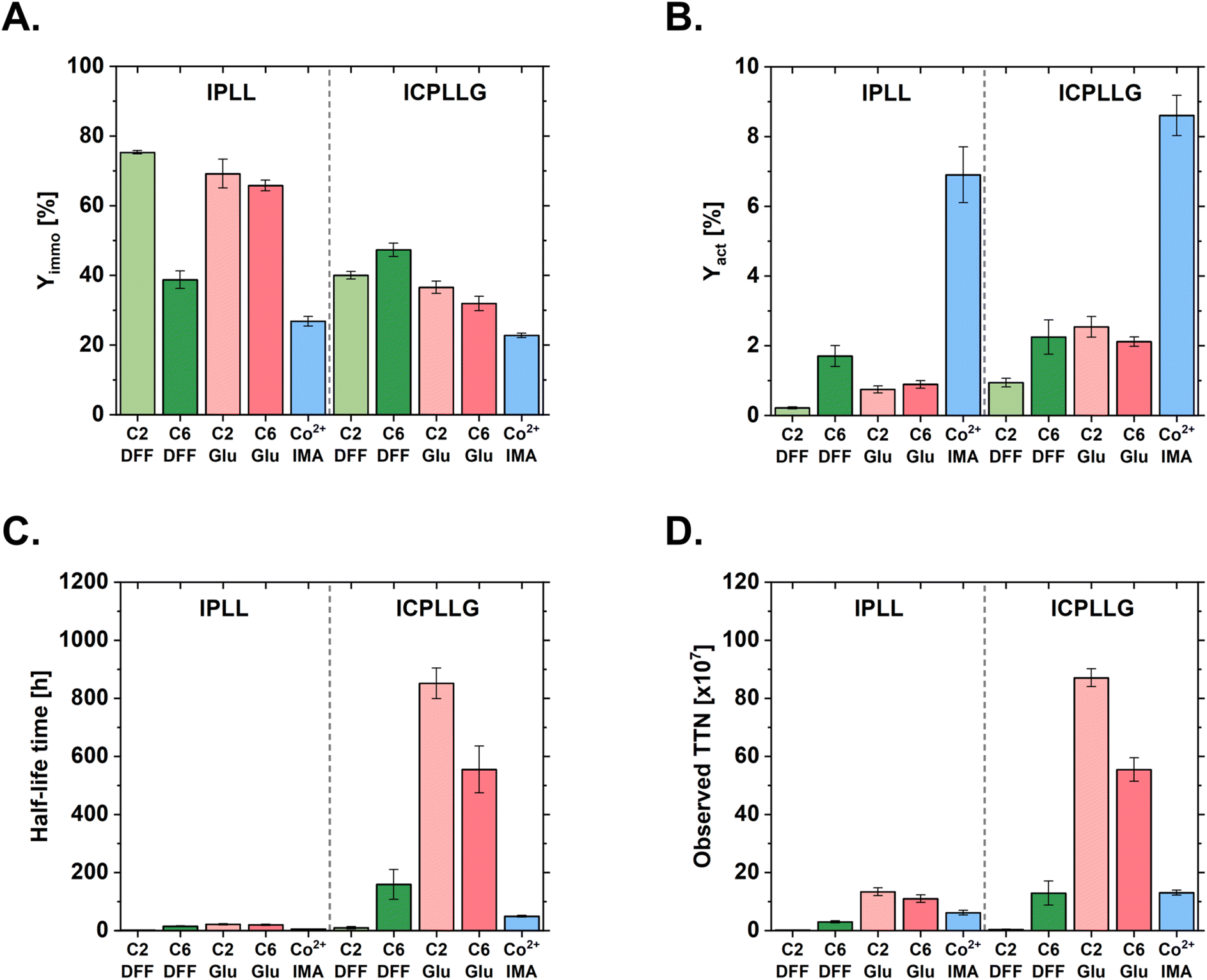 | ||
| Fig. 3 Investigation of activity and stability characteristics of enzyme carrier preparations with immobilized AMDase IPLL and AMDase ICPLLG. Immobilizations were done in triplicates. Immobilization was carried out onto amino polyacrylate carriers with C2 or C6 linkers and DFF or glutaraldehyde crosslinking agent or onto a Co2+ IMA carrier. All mass-specific activities were determined towards phenylmalonate (1 mL, 20 mM phenylmalonate, 50 mM HEPES buffer, pH 7.8, 30 °C). Protein quantification was done by BCA-assay. All experimental steps involving active enzyme were done at 4 °C. Enzymatic reactions were incubated for 5 minutes at 30 °C before conducting the activity assay (SI, section 3.4). A. Comparison of the mass specific activity [U gcarrier−1]. B. Comparison of the activity yield [%]. C. Comparison of the half-life of the immobilized biocatalyst (SI, section 3.5). D. Comparison of the observed total turnover numbers [TTN]50 resulted from the observed catalytic constant kcat value of initial activity and observed deactivation constant kdes of storage condition during storage at 30 °C (SI, section 3.5). | ||
The selectivity of the affinity approach is further elucidated by comparing Yimmo to the activity yield (Yact, Fig. 3B). The considerably lower Yimmo of the IMA immobilization is offset by a significantly higher Yact, indicating a higher proportion of correctly oriented and active enzyme. This trend was also observed, however in a mitigated form, during the covalent immobilization of (R)-selective AMDase variant IPLL onto the C6-DFF carrier and across all covalent immobilizations of the (S)-selective AMDase variant ICPLLG, especially when compared to IPLL@C2-DFF and both IPLL glutaraldehyde-crosslinked combinations.
To identify the most robust biocatalyst for an intensified process and maximize enzyme utilization, the stability of all carrier preparations was evaluated. The total turnover number (TTN) was estimated by determining the biocatalyst half-life under process-relevant conditions. Preparations were incubated at 30 °C in reaction buffer in the absence of substrate, and residual activities were measured at defined time intervals. Although this static incubation does not fully reproduce the dynamic stresses encountered under intensified process conditions, it enables the identification of critical differences in carrier stability.
Moreover, conducting the assays at the mL-scale allowed the parallel evaluation of multiple preparations, thereby reducing experimental workload and resource consumption.
Given the structural similarity between the two AMDase variants (Fig. 1), the disparity in the stability of their respective carrier preparations is remarkable, as AMDase IPLL exhibited a significantly lower half-life. For both variants, the highest stability was achieved via immobilization on C2-Glu; however, the (S)-selective ICPLLG variant demonstrated a 39.2-fold higher half-life compared to IPLL (851.2 h vs. 21.7 h, respectively; Fig. 3C). Preparations utilizing the bio-based crosslinking agent DFF resulted in considerably lower half-lives than those using traditional glutaraldehyde. Interestingly, a profound loss of initial activity was observed across all carriers during the first hours of incubation. This trend is best characterized by a biexponential decay function.51,52 This initial loss may suggest the rapid deactivation of enzymes immobilized on the outer surface of the carrier, while enzymes localized within the pores remain shielded and stabilized by the protected microenvironment. To accurately represent long-term stability, the deactivation constant (kdes) was derived from the second exponential decay phase, which describes the kinetics of the more stable enzyme fraction. This approach focuses the assessment on the enzyme fraction suitable for prolonged process times or biocatalyst recycling in an intensified process (SI, section 3.5).
A second key parameter influencing the total turnover number (TTN) of the biocatalysts is the catalytic constant (kcat), which exhibits a trend inverse to that of the variant stability profiles. Preparations of the (R)-selective IPLL variant were significantly more active than their counterparts. A direct comparison of the cell-free extracts revealed a 3.8-fold higher mass-specific activity toward the model substrate phenylmalonic acid (62.5 U mg−1), for IPLL versus 16.1 U mg−1 for ICPLLG. While the 39.2-fold disparity in stability for the C2-Glu preparations was striking, the observed TTN values were less divergent because the higher kcat values in the less stable variant acted as a counterbalancing factor. Consequently, the (S)-selective ICPLLG variant showed a 6.5-fold higher TTN compared to the more active IPLL variant (Fig. 3D).
For both AMDase variants, the highest TTN was obtained with the amino polyacrylate carrier functionalized with a C2 spacer and glutaraldehyde. This configuration was therefore selected for the intensified asymmetric synthesis of α-chiral carboxylic acids. In this context, overall productivity, reflected by the TTN of the enzyme formulation, served as the primary selection criterion, consistent with the focus on industrial performance metrics and process intensification.
Bayesian optimization (BO) for the decarboxylation in the rotating bed reactor (RBR)
To improve optical purity and enzymatic activity, the reaction pH and temperature were selected as key optimization parameters, as both strongly influence spontaneous decarboxylation. The pH range was defined according to physiologically relevant buffer systems between pH 5.5 and 9.7 (SI, section 1.9.3). The temperature was screened from 10 to 45 °C, considering the thermostability range of AMDase.35Bayesian optimization (BO), a Machine Learning model, was performed directly in the established RBR system to ensure that the identified optima were intrinsically linked to the process setup, including in situ pH adjustment. This approach enabled the identification of operating conditions that balance high enzymatic activity, required for economically viable product formation, with minimized spontaneous decarboxylation.
The iterative nature of the reactor setup required an efficient optimization strategy to minimize experimental effort. Accordingly, BO was employed instead of a traditional Design of Experiments (DoE) approach. BO is particularly well-suited for this task, as it models the response surface from an initial experimental set and iteratively refines the predicted optimum. The optimization was implemented using a validated, in-house MATLAB script (SI, section 1.9), which includes full details and validation data.
In this study, four initial conditions generated via Latin Hypercube Sampling (LHS) were selected by the script based on the defined factor ranges. Subsequent experimental points were then chosen to strategically balance exploitation of known optima with exploration of less-certain regions of the parameter space. This targeted approach markedly reduces the number of experiments required for optimization, albeit with a slight compromise in predictive accuracy. By minimizing resource, energy, and time consumption, the application of BO for process design also aligns with the principles of Green Chemistry.
Following the initial four conditions and the first optimization iteration, the effects of pH and temperature on both enantiomeric excess (ee) and initial activity of the biocatalyst preparation were visualized (Fig. 4). Spontaneous decarboxylation was found to be strongly pH-dependent, with higher pH values favoring excellent optical purity. This behavior arises from partial deprotonation of the substrate, which inhibits the six-membered transition-state mechanism responsible for spontaneous decarboxylation of malonic acids (Scheme 3A). Enhanced thermal decomposition via spontaneous decarboxylation has previously been reported for heteroaryl malonic acids.26 It should be noted, however, that the effect is more pronounced for these substrates due to their increased electron deficiency. In our experiments, temperature had little impact on the optical purity of the product but led to a notable increase in enzymatic activity, likely offsetting any accelerated decarboxylation and promoting efficient conversion to the stable propionic acid.
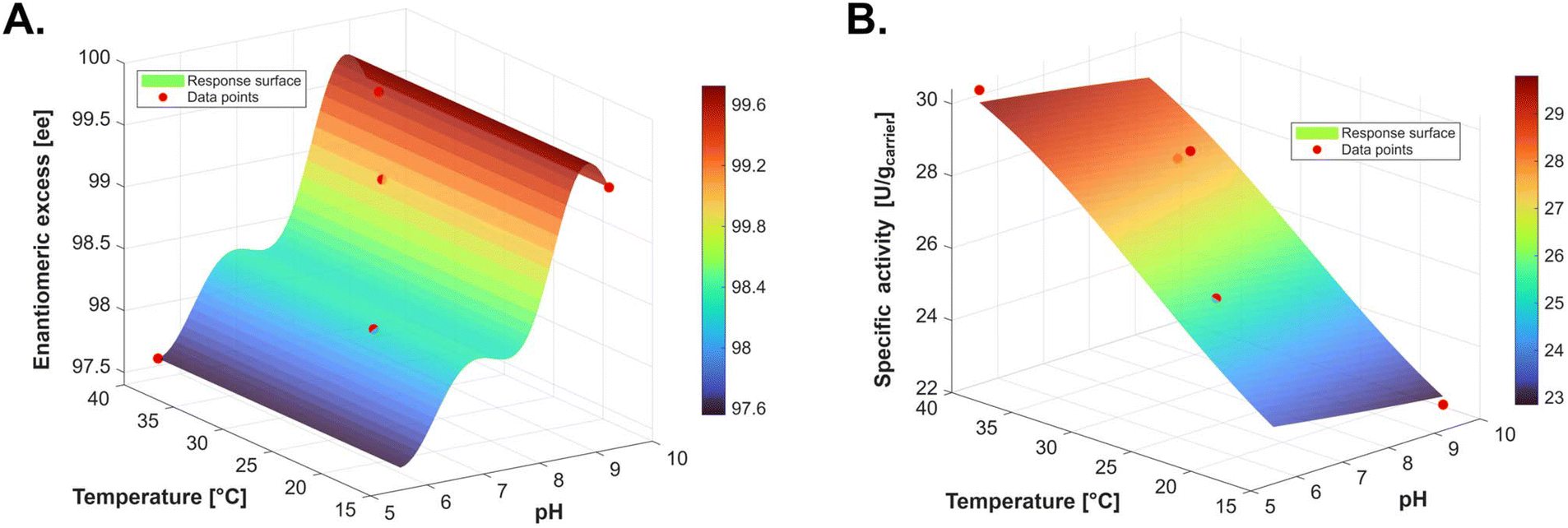 | ||
| Fig. 4 Mean functions (µ) of the Gaussian process (GP) after the initial set of four experiments and the first iteration of optimized experimental conditions for enhanced enantiomeric excess and activity of the biocatalyst (SI, section 1.9, for full details and validation data). The experimental data points were collected in a rotating bed reactor system (SpinChem® V2). The corresponding basket was filled with AMDase ICPLLG immobilized on amino polyacrylate carriers with C2 linkers and glutaraldehyde as a crosslinking agent. A. Response surface, represented by the GP mean function of the enantiomeric excess (ee) of (S)-flurbiprofen after derivatization with TMS–CH2–N2 and chiral GC measurement. B. Response surface, represented by the GP mean function of the initial activity after complete substrate injection into the rotating bed reactor system. The activity was calculated on the carrier mass utilized for biocatalytic conversion. Samples of 100 µL were terminated in ACN/H2O; 9:1, and measured by HPLC-DAD (SI, section 2.1). | ||
Preparative scale experiments
The highest enantiomeric excess was observed under slightly alkaline conditions (8.3–9.7) using AMPSO buffer. Although these BO-derived findings are mechanistically informative, their application at larger scale is limited by economic considerations.AMPSO is approximately ten times more expensive than MES, MOPS, or HEPES, which were used to access the high-pH range (8.3–9.7) in the BO study (SI, section 1.9.3). TRIS-HCl buffer serves as a cost-effective and widely utilized alternative, offering a broad buffering range from pH 7.0 to 9.0.53 However, neither AMPSO nor TRIS-HCl is classified as Good's buffers, and both have been reported to engage in metal-ion complexation.53,54 Such interactions could potentially interfere if the carrier preparation is transitioned again to affinity immobilization at a later stage. Most importantly, the choice of buffer system was found to significantly influence the AMDase activity. At a buffer molarity of 50 mM at pH 7.9, the enzymatic activity was significantly lower in TRIS-HCl compared to HEPES and MOPS buffer (SI, section 3.2). Consequently, preparative scale reactions were conducted at pH 8.1 in HEPES buffer (buffering range pH 6.8 to 8.2). This selection ensures a stable pH during the process, which can tolerate minor adjustments without drastic fluctuations. We further decided to set the reaction temperature to 30 °C for all preparative-scale reactions. This serves as a triple compromise of increased enzymatic activity, previously published higher rates of spontaneous decarboxylation,26 and the potential deactivation of the biocatalysts during longer reaction times.
To demonstrate the synthetic utility of the developed methodology for producing enantiopure arylpropionic acids, 1c–5c were synthesized in both enantiomeric forms on a preparative scale (Table 1). Upscaling these biotransformations required biocatalyst quantities exceeding the volumetric productivity achievable in standard shake-flask cultivations. Accordingly, the AMDase variants IPLL and ICPLLG were produced via high-density cultivation in a DASGIP multi-bioreactor system.28
| Product | Abbr. | Molarity [mM] | Reaction time [h] | ee [%] | Isolated yield [%] | PMIc [kg kg] | |
|---|---|---|---|---|---|---|---|
| a The conversion was determined by HPLC-DAD with 92.8 ± 0.9%. Full conversion to (R)-ethyl-3-butenoic acid could not be reached in three repetitions.b Full conversion was reached based on HPLC analysis.c Process mass intensity (PMI; the total mass of materials used for the mass of the final product) is used to benchmark the “greenness” of a process by focusing on the total mass of all materials used to produce a given mass of product. | |||||||
| (R)-Naproxen |  |
(R)-1c | 58.1 | 47 | 99.0 ± 0.1 | 94.2 | 160 |
| (S)-Naproxen | (S)-1c | 58.1 | 48 | 99.4 ± 0.3 | 96.3 | 156 | |
| (R)-Flurbiprofen | 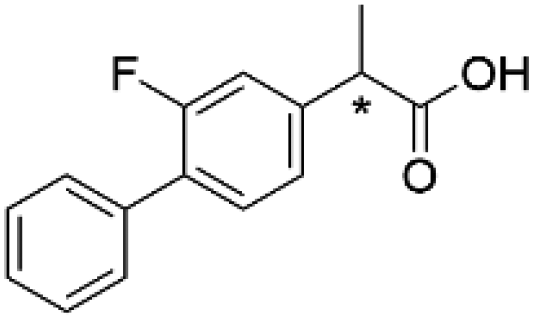 |
(R)-2c | 36.7 | 23 | 99.0 ± 0.2 | 90.2 | 204 |
| (S)-Flurbiprofen | (S)-2c | 36.7 | 17 | 99.1 ± 0.3 | 90.3 | 203 | |
| (R)-2-Phenylpropionic acid | 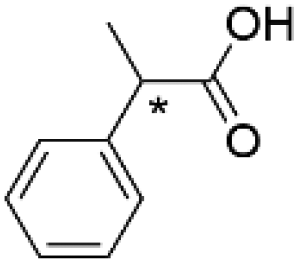 |
(R)-3c | 45.6 | 1.7 | 99.0 ± 0.0 | 87.7 | 228 |
| (S)-2-Phenylpropionic acid | (S)-3c | 45.6 | 21 | >99 | 90.2 | 250 | |
| (R)-2-Methyl-3-butenoic acid | 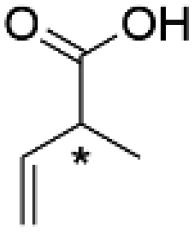 |
(R)-4c | 3.2 | 24 | 99.4 ± 0.0 | 93.6 | 4467 |
| (S)-2-Methyl-3-butenoic acid | (S)-4c | 3.2 | 24 | 96.4 ± 1.7 | 89.3 | 4686 | |
| (R)-2-Ethyl-3-butenoic acida | 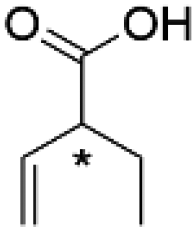 |
(R)-5c | 3.2 | 24 | 93.1 ± 0.7 | 82.9 | 4626 |
| (S)-2-Ethyl-3-butenoic acidb | (S)-5c | 3.2 | 24 | >99 | 79.8 | 4807 | |
Following biomass harvest, cells were processed into CFE by microfluidization in the immobilization buffer. The resulting CFE was used directly for immobilization without further purification. This approach, which allows partial nonspecific immobilization of other proteins, was previously shown to stabilize AMDase in repeated-batch studies by Aßmann et al.35 Among the tested carriers, polyacrylate carriers functionalized with C2 spacers and crosslinked with glutaraldehyde yielded the highest TTNs (Fig. 3) and were therefore selected for preparative-scale reactions.
All preparative-scale reactions were performed in a SpinChem® S2 RBR system. To streamline operation, the RBRs were run by using only the outer mesh, allowing rapid and homogeneous packing of enzyme carriers by hydrodynamic flow. Carrier beads were directly added to the reaction vessel and captured by convective flow driven by centrifugal suction of the basket, enabling a self-packing mechanism that greatly simplified reactor loading. All preparative reactions achieved >99% conversion. Product isolation was adapted to solubility: the NSAIDs naproxen (1c) and flurbiprofen (2c) were recovered via acid-induced precipitation, whereas the more water-soluble derivatives 3c–5c required liquid–liquid extraction. Isopropyl acetate was chosen as the extraction solvent due to its lower water solubility55 and greater resistance to acid-catalyzed hydrolysis compared with ethyl acetate, minimizing solvent loss and preventing acetic acid formation during prolonged workup or uncontrolled pH adjustment. This approach streamlined the isolation process and reduced the extent of drying required for the organic phase. All final products were dried under reduced pressure at 22 °C before characterization (SI, section 3.8).
Sustainability assessment and process mass intensity (PMI)
To evaluate the environmental footprint of this intensified process, we conducted a comparative sustainability analysis against previously reported enzymatic decarboxylation methodologies utilizing AMDase (Fig. 5). We utilized Process Mass Intensity (PMI) as the primary Green Metric.56 The PMI provides a comprehensive assessment by accounting for the total mass of all materials, including water and solvents for the production of a unit mass of product (SI, eqn (S15)). This can offer a more holistic perspective than the traditional Environmental-factor (E-factor, kgwaste kgproduct)57,58 for active pharmaceutical ingredient (API) production, as it incorporates the grade and quantity of water, a critical factor in biocatalytic systems.56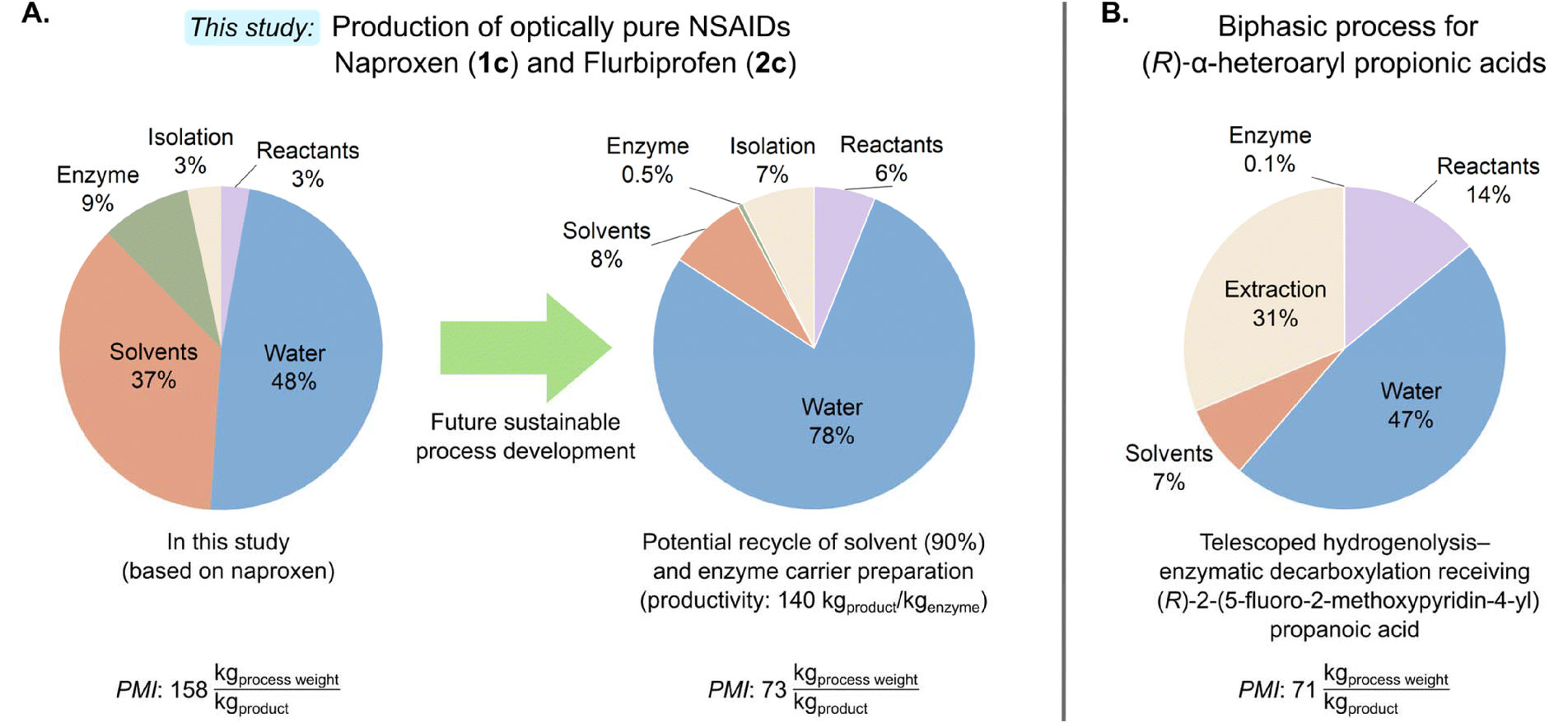 | ||
| Fig. 5 A. Visualization of the process mass distribution of the naproxen-based process of the current state-of-the-art, compared to the distribution after potential future sustainable process development. This accounts for the reuse of 90% of organic solvent after the precipitation of disodium naproxenmalonate and the recycle of enzyme carrier preparation according to the reported value of 140 kgproduct kgenzyme−1 for (S)-naproxen.15 The determined PMI values for each process are seen below the respective pie chart. B. Process mass distribution and PMI value of the biphasic process of dibenzylester hydrogenolysis and subsequent enzymatic decarboxylation for the synthesis of (R)-α-heteroaryl propionic acids (Scheme 2B). The data is determined based on the available information in the publication.26 | ||
The determined PMI values of the preparative scale reactions of this work are summarized in Table 1. The PMI assessment encompassed the entire sequence: alkaline hydrolysis, integrated enzymatic decarboxylation with in situ pH control, and final product isolation. The determined PMI values directly reflect the influence of the utilized isolation procedure (downstream), where the acid-induced precipitation of naproxen (1c) and flurbiprofen (2c) resulted in mean values of 158 kg kgproduct−1 and 203 kg kgproduct−1, respectively (with differences directly related to their substrate loadings of 58 and 37 mM). In contrast, 2-phenylpropionic acid (3c) required organic extraction due to its higher water solubility, which increased the process mass intensity (239 kg kgproduct−1). The higher PMI values observed for the 2-alkyl-3-butenoic acids 4c and 5c (∼4600 kg kgproduct−1) highlight the necessity for further optimization of the saponification step and enzyme engineering to support intensified substrate loading leading to a more proper use of resources, water and solvents.
As preparative-scale reactions were conducted in single batches, the reusability of the immobilized carrier was not included in the initial assessment. Consequently, the impact of the immobilized enzyme on the PMI is substantial compared to the use of CFE. Previous reports of optically pure naproxen production reported an E-factor of 84 kgwaste kgproduct−1,15 which translates to a PMI of 343 kg kgproduct−1 when all materials and solvents are accounted for (or 309 kg kgproduct−1 assuming biocatalyst reuse, which would be comparable to the complete E-factor). For comparison, the reported synthesis of optically pure flurbiprofen was determined to have a PMI of 580 kg kgproduct−1.16 Besides the optically pure arylpropionic acids, the telescoped biphasic process for the production of (R)-α-heteroaryl propionic acids (Scheme 2B) was determined to have a PMI of 71 kg kgproduct−1 on a scale of 50 g protected substrate.26 However, it should be noted that this early stage of optimized industrial application of AMDase relies on the organic extraction of the product.
In contrast, the precipitation option for the derivatives of naproxen (1b–c) and flurbiprofen (2b–c) shows a distinctive process advantage: besides the product precipitation of 1c and 2c, the process benefits from the precipitation of disodium naproxenmalonate (1b) and disodium flurbiprofenmalonate (2b) during the alkaline hydrolysis (Scheme 3B). This allows the implementation of organic solvent recovery (estimated at 90%), which would result in significant sustainability gains. Furthermore, the potential for enzyme carrier reusability was estimated based on literature values of 140 kgproduct kgenzyme−1 for (S)-naproxen.15
The potential of future process development is visualized in Fig. 5, illustrating the process mass distribution for the naproxen-based process. Implementing the described optimizations, the PMI could be further reduced by 54% for naproxen to 73 kg kg−1, and by 46% for flurbiprofen to 110 kg kg−1.
Apart from assessing the amount of waste generated in the processes (PMI) and allocating the main impact contributors, it is also relevant to consider the fate of such wastes when treated, as well as the impact of the energy necessary to conduct the reaction. Overall, the ultimate environmental burden of waste treatment and energy invested can be translated to CO2 formation, which acts as permanent (and comparable) waste.59 Thus, to complement ‘mass-based’ Green Metrics, we further assessed the environmental profile of the biocatalytic production of (S)-naproxen through a Global Warming Potential (GWP) analysis (Fig. 6).60,61 We evaluated the three main contributors for GWP: (i) the cumulative CO2 contributions from upstream energy demand (assuming the current impact of the European grid as 0.25 kg CO2 kWh−1);62 (ii) the stoichiometric reaction release, since malonates are often petrochemical, and a decarboxylation is conducted; (iii) and the impact of the wastewater treatment, which can be variable in CO2 production depending on the recalcitrance of the wastewater (from mild treatment to incineration).
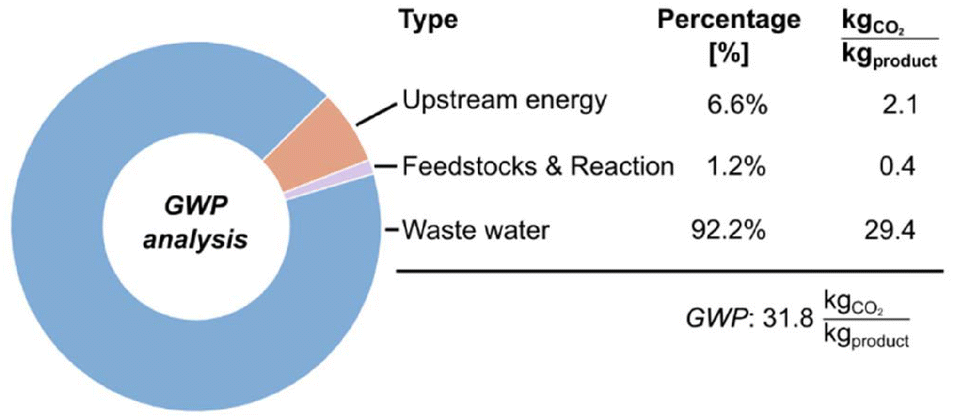 | ||
| Fig. 6 Visualization of the global warming potential (GWP) analysis of the established biocatalytic process of naproxen production. The distribution of impact factors highlights wastewater treatment as the main contributor to CO2 release. | ||
Since water is acidified during the downstream, an intermediate impact scenario between mild treatment and incineration was considered.60 Overall, we identified the wastewater treatment as the main environmental hotspot, contributing 29.4 kg CO2 kgproduct−1. This value results from the acid-induced precipitation of naproxen, which would likely necessitate a pre-treatment for the acidic wastewater before discharge into a standard treatment plant.
Furthermore, as naproxen precursors originate from petrochemical feedstocks, we accounted for the stoichiometric release of CO2 for the availability of the substrate and the enzymatic decarboxylation itself, adding 0.4 kg CO2 kgproduct−1. Combined with the required upstream energy investment of 2.1 kg CO2 kgproduct−1, the biocatalytic process established a total GWP of 31.8 kg CO2 kgproduct−1. This quantitative assessment reveals that while the enzymatic step is highly efficient, the overall carbon footprint remains intrinsically linked to the downstream neutralization requirements and subsequent wastewater treatment.
Conclusions
This work establishes an intensified, potentially sustainable biocatalytic route for the asymmetric synthesis of α-arylpropionic acids through the integration of alkaline hydrolysis and enzymatic decarboxylation. By employing dimethyl malonate precursors, the process avoids both the safety risks associated with hydrogenolysis and the low atom economy of bulky dibenzyl esters. The critical challenge of spontaneous decarboxylation was mitigated using an automated in situ pH-stat–controlled RBR, which stabilized the malonate intermediates while maintaining an optimal environment for the biocatalyst.Process intensification relied on robust immobilization of AMDase variants. C2-amino polyacrylate carriers (Seplife® EMC7225S) provided the necessary operational stability and heterogeneity for preparative-scale applications. This configuration enabled the efficient production of high-value nonsteroidal anti-inflammatory APIs, including naproxen and flurbiprofen, achieving isolated yields of up to 96% and enantiomeric excess exceeding 99%.
Quantitative sustainability metrics highlight the advantages of this integrated approach. The PMI for naproxen synthesis was 158 kg kgproduct−1, representing a significant improvement over previously reported biocatalytic processes. Moreover, the precipitation of disodium malonate salts during saponification offers a unique opportunity for organic solvent recovery and hence efficient recycling. When combined with biocatalyst recycling, this process architecture could reduce the PMI further to 73 kg kgproduct−1. Subsequently, the production of CO2 during the reaction (GWP) was assessed based on the main contributors, the energy invested, the decarboxylation step, and the wastewater treatment. Overall, actions devoted to generating less (and better treatable) wastewater appear to be relevant for future improvement actions, as that fraction is the main hotspot in GWP. Increasing substrate loadings, leading to more optimized substrate-to-water ratios, may also be envisioned for future works, en route to scale-up and potential industrial implementation.
Likewise, we have found that the solvent composition for the alkaline hydrolysis of dimethyl malonate precursors 1a and 2a is critical and requires further development. Replacing dichloromethane with other bio-based alternatives appears a necessary future action, whereby a trade-off between the solvent impact and the spontaneous decarboxylation and enantiomeric purity of the product needs to be considered.
Ultimately, these findings demonstrate that integrating process engineering with biocatalysis can deliver safer, more efficient routes to enantiopure pharmaceutical building blocks while adhering to Green Chemistry principles and identifying hotspots for future improvement.
Author contributions
J. G.: methodology, investigation, validation, formal analysis, visualization, software, writing – original draft & editing. T. W.: methodology, investigation, formal analysis, software. M. H.: investigation, formal analysis. S. S.: methodology, resources. A. B.: methodology, resources. P. D. d. M.: writing – review & editing. S. K.: conceptualization, resources, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: Bayesian optimization, analytical methods, experimental procedures, NMR spectroscopy, and substrate synthesis. See DOI: https://doi.org/10.1039/d6gc02400a.CCDC 2546401 and 2546399 contain the supplementary crystallographic data for this paper.63,64
The authors have cited additional references within the SI.65–77
Acknowledgements
The authors thank Prof. Robert Kourist (Institute of Molecular Biotechnology, Graz University of Technology, Austria) and his team for the recombinant plasmids containing the AMDase gene. The authors also thank Dr Patrick Linder and Dr Dörte Solle from the Institute of Technical Chemistry at Leibniz University Hannover for the fruitful discussions on Bayesian Optimization. The authors also thank Dr Gerald Dräger for the determination of the crystal structure of 1a and 2a. SK thanks the Ministry for Science and Culture of Lower Saxony for the Holen & Halten starting grant (grant no. 12.5-76251-17-9/20) and Aarhus Universitets Forskningsfond (AUFF, grant no. AUFF-T-2018-7-11) for financial support.References
- A. K. Schweiger, K. Miyamoto and R. Kourist, Front. Catal., 2021, 1, 1–19 CrossRef.
- A. Bonabello, M. R. Galmozzi, R. Canaparo, G. C. Isaia, L. Serpe, E. Muntoni and G. P. Zara, Anesth. Analg., 2003, 97, 402–408 CrossRef CAS PubMed.
- S. Bindu, S. Mazumder and U. Bandyopadhyay, Biochem. Pharmacol., 2020, 180, 114147 CrossRef CAS PubMed.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- H. R. Sonawane, N. S. Bellur, J. R. Ahuja and D. G. Kulkarni, Tetrahedron: Asymmetry, 1992, 3, 163–192 CrossRef CAS.
- P. J. Harrington and E. Lodewijk, Org. Process Res. Dev., 1997, 1, 72–76 CrossRef CAS.
- C. R. Smith and T. V. RajanBabu, J. Org. Chem., 2009, 74, 4896 CrossRef CAS.
- R. D. Larsen, E. G. Corley, P. Davis, P. J. Reider and E. J. J. Grabowski, J. Am. Chem. Soc., 1989, 111, 7650–7651 CrossRef CAS.
- T. Ohta, H. Takaya, M. Kitamura, K. Nagai and R. Noyori, J. Org. Chem., 1987, 52, 3174–3176 CrossRef CAS.
- H. Zhong, M. Shevlin and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 5272–5281 CrossRef CAS PubMed.
- A. Fadel, Synlett, 1992, 48–50 CrossRef CAS.
- R. Kourist, P. Domínguez de María and K. Miyamoto, Green Chem., 2011, 13, 2607–2618 RSC.
- E. Tassano, K. Faber and M. Hall, Adv. Synth. Catal., 2018, 360, 2742–2751 CrossRef CAS PubMed.
- Z. Li, Z. Wang, G. Meng, H. Lu, Z. Huang and F. Chen, Asian J. Org. Chem., 2018, 7, 763–769 CrossRef CAS.
- M. Aßmann, A. Stöbener, C. Mügge, S. K. Gaßmeyer, L. Hilterhaus, R. Kourist, A. Liese and S. Kara, React. Chem. Eng., 2017, 2, 531–540 RSC.
- S. K. Gaßmeyer, J. Wetzig, C. Mügge and R. Kourist, ChemCatChem, 2016, 8, 916–921 CrossRef.
- C. José, M. V. Toledo and L. E. Briand, Crit. Rev. Biotechnol., 2016, 36, 891–903 CrossRef PubMed.
- A. Bhattacharya and D. Murphy, Org. Process Res. Dev., 2003, 7, 717–722 CrossRef CAS.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- S. P. France, R. D. Lewis and C. A. Martinez, JACS Au, 2023, 3, 715–735 CrossRef CAS PubMed.
- T. Bayer, S. Wu, R. Snajdrova, K. Baldenius and U. Bornscheuer, Angew. Chem., Int. Ed., 2025, 64, e202505976 CrossRef CAS PubMed.
- S. Simić, E. Zukić, L. Schmermund, K. Faber, C. K. Winkler and W. Kroutil, Chem. Rev., 2022, 122, 1052–1126 CrossRef PubMed.
- T. W. Thorpe, J. R. Marshall and N. J. Turner, J. Am. Chem. Soc., 2024, 146, 7876–7884 CrossRef CAS PubMed.
- K. Miyamoto and H. Ohta, J. Am. Chem. Soc., 1990, 112, 4077–4078 CrossRef CAS.
- C. A. Blakemore, S. P. France, L. Samp, D. M. Nason, E. Yang, R. M. Howard, K. J. Coffman, Q. Yang, A. C. Smith, E. Evrard, W. Li, L. Dai, L. Yang, Z. Chen, Q. Zhang, F. He and J. Zhang, Org. Process Res. Dev., 2021, 25, 421–426 CrossRef CAS.
- K. Miyamoto and R. Kourist, Appl. Microbiol. Biotechnol., 2016, 100, 8621–8631 CrossRef CAS PubMed.
- J. Gerstenberger, T. Werbilo, R. Kourist and S. Kara, ChemBioChem, 2026, 27, e70339 CrossRef CAS PubMed.
- J. Maimanakos, J. Chow, S. K. Gaßmeyer, S. Güllert, F. Busch, R. Kourist and W. R. Streit, Front. Microbiol., 2016, 7, 1–14 Search PubMed.
- K. Okrasa, C. Levy, M. Wilding, M. Goodall, N. Baudendistel, B. Hauer, D. Leys and J. Micklefield, Angew. Chem., Int. Ed., 2009, 48, 7691–7694 CrossRef CAS PubMed.
- S. Yoshida, J. Enoki, R. Kourist and K. Miyamoto, Biosci. Biotechnol. Biochem., 2015, 79, 1965–1971 CrossRef CAS PubMed.
- P. De Santis, L. E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184 RSC.
- L. S. Wong, K. Okrasa and J. MicKlefield, Org. Biomol. Chem., 2010, 8, 782–787 RSC.
- K. Markošová, J. Husarčíková, M. Halásová, R. Kourist, M. Rosenberg, R. Stloukal, L. Zajoncová and M. Rebroš, Catalysts, 2018, 8, 603 CrossRef.
- M. Aßmann, C. Mügge, S. K. Gaßmeyer, J. Enoki, L. Hilterhaus, R. Kourist, A. Liese and S. Kara, Front. Microbiol., 2017, 8, 1–8 Search PubMed.
- E. van der Pol, J. Gerstenberger, X. Georgiadou, K. Schliep, C. Schür, S. Kara and R. Kourist, bioRxiv, 2026, preprint, DOI:10.64898/2026.01.14.699310.
- S. O. C. Mundle, G. Lacrampe-Couloume, B. S. Lollar and R. Kluger, J. Am. Chem. Soc., 2010, 132, 2430–2436 CrossRef CAS PubMed.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford university press, Oxford, 2012 Search PubMed.
- T. Chandra and J. P. Zebrowski, J. Chem. Health Saf., 2016, 23, 16–25 CrossRef.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- T. Jespen, ATEX—Explosive Atmospheres. Risk Assessment, Control and Compliance, Springer Cham, 1st edn, 2016 Search PubMed.
- V. Theodorou, K. Skobridis, A. G. Tzakos and V. Ragoussis, Tetrahedron Lett., 2007, 48, 8230–8233 CrossRef CAS.
- P. Richardson, M. C. Bryan, L. Diorazio, F. Gallou, S. Keily, I. Martinez, S. Plummer, A. B. Reed, R. Sheppard, J. Shields, G. Wrigley, C. Yeung and Á. Enríquez-García, J. Med. Chem., 2025, 68, 25625–25664 CrossRef CAS PubMed.
- P. Petermeier, P. Domínguez de María, E. Byström and S. Kara, ACS Sustainable Chem. Eng., 2024, 12, 12869–12878 CrossRef CAS PubMed.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- R. Obata and M. Nakasako, Biochemistry, 2010, 49, 1963–1969 CrossRef CAS PubMed.
- C. Danielli, L. van Langen, D. Boes, F. Asaro, S. Anselmi, F. Provenza, M. Renzi and L. Gardossi, RSC Adv., 2022, 12, 35676–35684 RSC.
- O. Pauli, A. Ecker, A. Cruz-Izquierdo, A. Basso and S. Serban, Catalysts, 2022, 12, 1–15 CrossRef.
- A. Basso, M. S. Brown, A. Cruz-Izquierdo, C. A. Martinez and S. Serban, Org. Process Res. Dev., 2022, 26, 2075–2084 CrossRef CAS.
- T. A. Rogers and A. S. Bommarius, Chem. Eng. Sci., 2010, 65, 2118–2124 CrossRef CAS PubMed.
- J. P. Henley and A. Sadana, Biotechnol. Bioeng., 1986, 28, 1277–1285 CrossRef CAS PubMed.
- C. Aymard and A. Belarbi, Enzyme Microb. Technol., 2000, 27, 612–618 CrossRef CAS PubMed.
- C. Ferreira, I. Pinto, E. Soares and H. Soares, RSC Adv., 2015, 5, 30989 RSC.
- N. I. Gumerova and A. Rompel, Angew. Chem., Int. Ed., 2026, 1–10 Search PubMed.
- L. Delhaye, A. Ceccato, P. Jacobs and C. Ko, Org. Process Res. Dev., 2007, 11, 160–164 CrossRef CAS.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- R. Sheldon, Chem. Ind., 1992, 23, 903–906 Search PubMed.
- F. Tieves, F. Tonin, E. Fernández-Fueyo, J. M. Robbins, B. Bommarius, A. S. Bommarius, M. Alcalde and F. Hollmann, Tetrahedron, 2019, 75, 1311–1314 CrossRef CAS.
- P. Domínguez de María, Curr. Opin. Green Sustainable Chem., 2025, 52, 101003 CrossRef.
- P. Domínguez de María, ChemSusChem, 2025, 202501831 CrossRef PubMed.
- P. Domínguez de María, S. Kara and F. Gallou, Molecules, 2023, 28, 6452 CrossRef PubMed.
- European Enviroment Agency, Greenhouse gas emission intensity of electricity generation, https://www.eea.europa.eu/en/analysis/maps-and-charts/co2-emission-intensity-15?activeTab=8a280073-bf94-4717-b3e2-1374b57ca99d (accessed 25 March 2026).
- CCDC 2546401: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2rgqzy.
- CCDC 2546399: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2rgqxw.
- C. E. Rasmussen and C. K. I. Williams, Gaussian Processes for Machine Learning, MIT press, Cambridge, 2006 Search PubMed.
- G. Derringer and R. Suich, J. Qual. Technol., 2018, 12, 214–219 CrossRef.
- NIST SEMATECH, e-Handbook of Statistical Methods, https://www.itl.nist.gov/div898/handbook/pri/section5/pri5322.htm, (accessed 11 February 2026).
- M. Siska, E. Pajak, K. Rosenthal, A. del Rio Chanona, E. von Lieres and L. M. Helleckes, Biotechnol. Bioeng., 2026, 805–830 CrossRef CAS PubMed.
- D. H. Ackley, A Connectionist Machine for Genetic Hillclimbing, Kluwer Academic Publishers, Boston, MA, 1987 Search PubMed.
- J. G. Proakis and D. G. Monolakis, Digital signal processing: principles, algorithms, and applications, Prentice-Hall, Inc., Upper Saddle River, New Jersey, 3rd edn, 1996 Search PubMed.
- F. Hoffmeister and T. Bäck, in Parallel Problem Solving from Nature, ed. H.-P. Schwefel and R. Männer, Springer Berlin Heidelberg, Berlin, Heidelberg, 1991, pp. 455–469 Search PubMed.
- H. H. Rosenbrock, Comput. J., 1960, 3, 175–184 CrossRef.
- E. Van Der Pol, T. Schlatzer, G. Hoffka, B. Di Geronimo, J. Eder, A. K. Schweiger, M. Karava, D. Gross, R. C. Fischer, D. Kracher, R. Kazlauskas, K. Miyamoto, S. Caroline, L. Kamerlin, R. Breinbauer and R. Kourist, J. Am. Chem. Soc., 2025, 147, 39271–39283 CrossRef CAS PubMed.
- E. Wiedenbeck, M. Kovermann, D. Gebauer and H. Cölfen, Angew. Chem., Int. Ed., 2019, 58, 19103–19109 CrossRef CAS PubMed.
- S. Dohrn, C. Luebbert, K. Lehmkemper, S. O. Kyeremateng, M. Degenhardt and G. Sadowski, Fluid Phase Equilib., 2021, 548, 113200 CrossRef CAS.
- Y. Terao, Y. Ijima, H. Kakidani and H. Ohta, Bull. Chem. Soc. Jpn., 2003, 76, 2395–2397 CrossRef CAS.
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2431–2434 CAS.
| This journal is © The Royal Society of Chemistry 2026 |