 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface-induced symmetry breaking leads to unexpected vibrational activity of melem on Cu(111)
Matthew Oakes ,
Sam Haq,
Matthew S. Dyer and
Rasmita Raval*
,
Sam Haq,
Matthew S. Dyer and
Rasmita Raval*
Surface Science Research Centre, University of Liverpool, Liverpool L69 3BX, UK. E-mail: raval@liverpool.ac.uk
First published on 2nd March 2026
Abstract
The adsorption of melem on Cu(111) at 80 K has been studied by Scanning Tunnelling Microscopy (STM), Reflection Absorption Infrared Spectroscopy (RAIRS) and Density Functional Theory (DFT) calculations. STM images and DFT calculations confirm a planar adsorption geometry and a triangular footprint at the surface, with the nitrogen atoms positioned above atop surface sites and the carbon atoms in 3-fold fcc sites. DFT calculations show that melem is adsorbed at a height of 3 Å above the surface, consistent with molecule–metal distances observed for physisorbed aromatic molecules. A small charge transfer of 0.27e− and a small tilt of the NH2 groups towards the surface are observed in the DFT calculations, endorsing the general physisorption model, albeit with a small perturbation. Simulated RAIRS spectra generated from the DFT model clearly indicate the reduction of symmetry from D3h in the gas phase to C3v in the adsorbed state, which causes specific gas phase modes to be switched on or off. The simulated RAIR spectrum is dominated by out-of-plane vibrations as expected for a planar physisorbed molecule. In contrast, the experimental RAIRS data show unexpectedly intense and broad, shifted bands arising from the in-plane NH2 scissors + ring stretching vibrational modes, suggesting the presence of significant additional intermolecular and molecule–metal interactions in the adsorbed state that lead to bond softening and increased anharmonicity in the vibrational modes.
Introduction
Molecular systems adsorbed on metallic surfaces are increasingly being studied using an array of surface sensitive techniques providing both spatial information and chemical information. When combined, detailed understanding of a molecule’s chemical nature, orientation, interactions with the surface and neighbouring molecules can be obtained. For many years, vibrational spectroscopy was at the vanguard of interrogating molecular behaviour at surfaces, due to its prowess in signposting chemical, geometric and site-symmetry information from the normal modes of vibration of the adsorbate. Increasingly, over the past two decades, scanning probe microscopies have moved to the fore in this area of investigation, with resolution down to the sub-molecular level enabling individual molecules and atoms to be imaged by Scanning Tunnelling Microscopy (STM), and even intramolecular bond distances to be measured by noncontact atomic force microscopy (nc-AFM). These approaches are often augmented by Density Functional Theory (DFT) calculations that provide detailed information on molecule adsorption, orientation and geometry. One can then pose the question: what does vibrational spectroscopy really add to this rich information base?We examine this here by investigating the adsorption of melem (2,5,8-triamino-1,3,4,6,7,9,9b-heptaazaphenalene) on Cu(111) at 80 K. Melem is an aromatic molecule containing a heptazine unit. It possesses interesting semiconducting properties and has gained recent attention for its applications in photocatalysis.1–5 Infrared spectroscopy has been used to characterize melem in its crystalline form but no detailed experimental vibrational assignment, to our knowledge, currently exists on the adsorbed state. The self-assembly of melem on Au(111) and Ag(111) surfaces has been studied by STM and is described in terms of physisorbed molecules oriented flat on the surface, with extended self-assembled networks driven by hydrogen bonding.6–10
Aromatic systems at surfaces are important in many fields11–16 and provide an interesting exemplar system for this study. Their inherent electronic properties offer a span of potential interactions with metallic surfaces, from physisorption to direct chemisorption. Their delocalised π system provides one avenue for interaction and, furthermore, the p orbitals can be ideally positioned for direct bonding with the metal. The nature of adsorption is often gauged by the molecule–surface separation, the magnitude of the adsorption energy, or the level of charge transfer.17 However, these are often difficult to establish for larger molecules. Benzene on reactive surfaces such as Pt(111), Pd(111), Rh(111), Ni(111) and Cu(110) shows molecule–surface distances ranging from 2–2.3 Å,18–21 close to what is expected for the Cu–C bond and consistent with a chemisorbed state. On more inert surfaces such as Cu(111), Au and Ag the separation is larger at ∼3 Å,22–24 and assigned to a physisorbed state. Other aromatic molecules show similar delineation of separations for the chemisorbed and physisorbed state.25–28 Additionally, a change in molecular conformation often accompanies chemisorption, deforming from the gas phase geometry. For example, porphyrin units on Cu(111) display strong interactions with molecule–surface bond distances close to those expected for Cu–C and Cu–N bonds.29–31 The stabilisation energy provided through the formation of such surface bonds can outweigh the stabilisation energy provided by aromaticity and distort molecular conformations.32
Here, the adsorption of melem on Cu(111) at 80 K was studied using STM, DFT calculations and Reflection–Absorption Infrared Spectroscopy (RAIRS) and we discuss the results from the different methods, and the insights they provide on the molecular state at the surface.
Methods
STM measurements
Low-temperature STM experiments were performed using a Createc LT-STM system within an ultrahigh vacuum (UHV) chamber. The STM was calibrated using atomic distances of the known Cu(110)-(2 × 1)O surface reconstruction.33 All measurements were taken in constant current mode using a tungsten tip and the chamber was at a base pressure of around 2 × 10−10 mbar. STM images were then processed using WSxM software.34 Processing consisted of image flattening and brightness/contrast adjustments. The Cu(111) surface was prepared and cleaned through argon ion sputtering at pressures of 2.5 × 10−5 mbar and annealing cycles to around 550 °C. The flatness and cleanliness of the sample were checked via STM and Low-Energy Electron Diffraction (LEED) prior to melem exposure. Melem was dosed from an electrically heated glass capillary tube onto a Cu(111) surface cooled to 80 K. Low temperature STM (LT-STM) measurements were conducted with the Cu crystal temperature at 80 K.RAIRS measurements
RAIRS experiments were performed in a different UHV chamber with a base pressure of 2 × 10−10 mbar and optically coupled to a Nicolet FTIR spectrum fitted with a liquid nitrogen cooled mercury cadmium telluride (MCT) detector, accessing a spectral range of 650–4000 cm−1. RAIR spectra were collected at 4 cm−1 resolution with 256 scans collected per spectrum. Where required, the resulting spectra were baseline corrected to account for reflectivity changes with changing temperature. The Cu(111) surface was prepared using the same protocols as the STM experiments, and the cleanliness of the crystal was checked via LEED.DFT
DFT calculations were performed using the plane-wave based code VASP35 with the PAW method36 to treat core electrons. All calculations were performed using the rev-vdW-DF2 functional,37 a plane-wave cutoff energy of 520 eV, and a corrective dipole moment within the vacuum region to reduce the effect of periodic dipole moments. Adsorption structures were generated on four layer Cu slabs in 6 × 6 supercells of the surface unit cell with 4 × 4 × 1 k-point grids, and with 50 Å in height leaving a vacuum layer of about 35 Å vertically between the adsorbed melem molecules and the bottom of the Cu slab in the periodic image. The isolated melem molecule with D3h symmetry was computed within a cubic cell of length 20 Å at the Gamma point only. Structures were optimised in fixed cells until forces fell below 0.01 Å. Adsorption energies are calculated relative to the clean copper surface, gas phase melem molecules and adsorbed atomic hydrogen. Normal mode frequencies and infrared intensities were calculated in the harmonic approximation by finite differences using the Atomic Simulation Environment.38 RAIRS intensities of the adsorbed melem molecule were simulated by calculating only the component of dipole moments perpendicular to the plane of the surface.Results and discussion
Scanning tunnelling microscopy of melem adsorption on Cu(111) at 80 K
Melem is a planar molecule possessing three-fold symmetry with its molecular structure broadly resembling a triangular shape with edges of ∼6.9 Å. Fig. 1 shows a series of STM images from the adsorption of melem on Cu(111) at 80 K. Across the surface, triangular features with edge lengths of ∼7 Å were observed, consistent with intact melem adsorbed in a flat orientation. Fig. 1a depicts melem’s scaled structure overlaid onto an imaged triangular unit. Fig. 1c and d show that these molecular features group together in small aggregates rather than being dispersed as single units across the surface. Examination of the small assemblies shows that neighbouring molecules mimic the most stable hydrogen bonding motif of melem, where the amine group of one melem acts as a hydrogen bond donor to the ring nitrogen at the exterior edge of its neighbour, resulting in a slight stagger. This interaction is also observed in the crystalline structure of melem and in the highly organised surface assemblies it forms on Ag(111) and Au(111).2,6–10,39–41 However, on Cu(111) such highly ordered self-assemblies are not observed at 80 K; even when the coverage is increased, the system is restricted to varying levels of localised ordering of the triangular units. | ||
| Fig. 1 (a) 4.5 nm × 4.5 nm scanning tunnelling microscopy image of individual melem molecules adsorbed on Cu(111) at 80 K with Vt = −0.15 V and It = 250 pA and imaged at 80 K. An overlay of the chemical structure of melem can be seen imposed on a single triangular feature confirming their match in size. (b) Chemical structure of melem with its dimensions. (c and d) Scanning tunnelling microscopy images taken at 80 K after melem deposition on Cu(111) at 80 K with a low surface coverage, where: (c) 50 nm × 50 nm, Vt = −0.15 V and It = 250 pA, (d) 20 nm × 20 nm, Vt = −0.15 V and It = 250 pA. The Cu(111) close-packed directions are depicted in (d). | ||
Density functional theory studies of melem adsorption on Cu(111)
Density Functional Theory (DFT) calculations of melem adsorption on Cu(111) show that the intact melem molecule preferentially binds to Cu(111) in a flat-lying orientation, in agreement with the STM observations described above (Fig. 2). In its optimised adsorption geometry, the dimensional matching of an individual melem with respect to the underlying Cu(111) surface results in a C3v adsorption site symmetry, which enables the nitrogen atoms to be positioned above atop surface sites, and the carbon atoms in 3-fold fcc sites. Additionally, melem is positioned 3 Å above the metallic surface which, in comparison to other aromatic systems on Cu(111) and other reactive surfaces, suggests a physisorbed state.18–20,22,28,29,42 Overall, the melem molecular structure remains fairly unperturbed compared to the gas-phase, with its heptazine ring remaining planar and a slight distortion of the NH2 groups, which bend 7° towards the surface. This configuration has an adsorption energy of −1.4 eV per molecule, with a small electron exchange of 0.27 e− observed from the molecule to the surface. | ||
| Fig. 2 Density Functional Theory (DFT) optimised adsorption geometry of melem on Cu(111). The calculation was carried out in a 6 × 6 supercell. (a) A top–down view of this state, (b) a side profile view of the same structure. The inset in (b) depicts the resulting NH2 tilt towards the surface upon adsorption. | ||
RAIRS of melem on Cu(111) at 80 K
The RAIR spectra obtained for low and high coverage of melem on Cu(111) at 80 K in the monolayer regime are shown in Fig. 3a and b. Three broad bands are observed at 1650 cm−1, 1610 cm−1 and 1475 cm−1, alongside sharper bands at 820 and 800 cm−1, with the latter attenuating at higher coverage. To aid vibrational assignment, the IR vibrational modes of gas-phase melem and the RAIR spectrum of melem adsorbed on Cu(111) were also calculated and are shown as Fig. 3d and c, respectively. | ||
| Fig. 3 (a and b) Reflection-Absorption Infrared Spectra (RAIRS) of melem on Cu(111) at 80 K. From top to bottom, each spectrum represents an increase in melem coverage on the surface, within the first monolayer regime. Spectra were taken with 256 scans and baseline corrected accordingly. (c) DFT calculated RAIR spectrum of melem on Cu(111) with the binding motif depicted in Fig. 2, possessing C3v symmetry. (d) DFT calculated IR spectrum of a single melem molecule in its most stable D3h confirmation. In both (c) and (d), the symmetry species of each calculated normal mode can be seen labelled. | ||
There are a number of points of interest: first, both the calculated and experimental RAIRS spectra differ significantly from the gas phase spectrum; second, the simulated RAIR spectrum is in good agreement with the experimental data in terms of the number of bands observed, albeit with a small offset in vibration wavenumbers, which is often seen for calculations that work within the simple harmonic approximation; and, third: there is, however, a difference between the experimental and simulated RAIRS data in terms of the relative intensities of the vibrational modes in the 1440–1700 cm−1 region vs. the 800–820 cm−1 region, with experimental data showing strong integrated intensity in the former region compared to the latter. It is noted that the calculations do not extract information on intrinsic bandwidths so the data are simply depicted assuming a Gaussian profile and a FWHM of 10 cm−1. Each of these points is discussed separately below.
Melem adsorption site symmetry and vibrational mode activity
The vibrational modes observed in the calculated RAIR spectrum are depicted for the adsorbed species in Fig. 4, showing the atomic displacement vectors. All three vibrations within the 1300–1700 cm−1 region (1591 cm−1, 1509 cm−1 and 1345 cm−1) are in-plane and consist mainly of NH2 scissoring modes mixed with contributions from heptazine ring stretches. The weak vibration at 1125 cm−1 is also a mixed mode, with the majority of displacement in the C–N stretches within the heptazine ring and small contributions from the NH2 scissoring modes. The out-of-plane heptazine ring-buckling vibration is calculated to be at 761 cm−1. For the gas phase molecule, calculations show the in-plane vibrations, consisting of mainly NH2/heptazine coupled motions, possess the largest intensities. Previously reported theoretical calculations of the single molecule and small melem clusters also reveal strong mixing of the heptazine ring stretching modes and the NH2 deformation modes.40 We note that these calculations agree with our description of modes and do not align with the previous vibrational mode assignments of melem in the powder form which, without the aid of theoretical calculations, assign the highest frequency mode in this region solely to the NH2 bending vibration and the remaining modes to the heptazine ring modes.2,3,39,43–49 | ||
| Fig. 4 DFT models depicting the vibrational vector displacements of melem on Cu(111) corresponding to the RAIRS active modes seen in Fig. 3c. | ||
The observation of both in- and out-of-plane modes in the RAIR spectrum is at odds with both the STM and DFT data, which show that the most stable adsorption configuration is associated with the molecular plane parallel to metal surface. Therefore, the observation of strong in-plane vibrational modes in RAIRS for this adsorption geometry is unexpected, where the metal surface selection rule dictates that a change in dipole moment perpendicular to the surface is necessary for RAIRS activity. Therefore, for physisorbed flat-lying molecules, in principle, the expectation would be for the spectrum to be dominated by modes corresponding to out-of-plane vibrations, i.e. the bands observed around 820 cm−1, while the in-plane modes in the 1300–1700 cm−1 region should be screened by the metal surface and hence absent or significantly attenuated in the RAIR spectrum.
To account for the significant change between the gas phase and RAIR spectra and the observation of the strong in-plane modes in RAIRS, we consider the adsorption symmetry alongside the more rigorous application of the metal surface selection rule, namely that only those vibrational modes that transform as the most symmetric irreducible representation are formally allowed. The isolated gas phase melem molecule belongs to the D3h point group where the molecule is completely planar and its amine group nitrogens are sp2 hybridised, resulting in the N–H bonds being aligned in the molecular plane. The normal modes of vibration corresponding to this gas phase geometry were calculated and are illustrated in Fig. 5 and listed in Table 1.
 | ||
| Fig. 5 Calculated normal modes of vibration for gas phase melem with corresponding vibrational frequency. | ||
| Gas phase melem (D3h) | Melem/Cu(111) 80 K (C3v) | ||||||
|---|---|---|---|---|---|---|---|
| Calculated | Calculated | Experimental | |||||
| Wavenumber (cm−1) | Symmetry species | Normalised % intensity | Wavenumber (cm−1) | Symmetry species | Normalised % intensity | Wavenumber (cm−1) | |
| ν59–60 | 3642 | E′ | 5.6 | ||||
| ν58 | 3641 | A2′ | 0 | ||||
| ν57 | 3505 | A1′ | 0 | 3457 | A1 | 1 | |
| ν55–56 | 3504 | E′ | 13.2 | ||||
| ν53–54 | 1633 | E′ | 44.6 | ||||
| ν52 | 1611 | A1′ | 0 | 1591 | A1 | 4 | 1650 |
| ν50–51 | 1576 | E′ | 100 | ||||
| ν49 | 1515 | A1′ | 0 | 1509 | A1 | 11.9 | 1610 |
| ν47–48 | 1514 | E′ | 16 | ||||
| ν46 | 1466 | A2′ | 0 | ||||
| ν44–45 | 1448 | E′ | 41.4 | ||||
| ν43 | 1347 | A1′ | 0 | 1345 | A1 | 5.7 | 1475 |
| ν42 | 1272 | A2′ | 0 | ||||
| ν40–41 | 1268 | E′ | 0.12 | ||||
| ν39 | 1133 | A1′ | 0 | 1125 | A1 | 0.3 | 1115 |
| ν37–38 | 1130 | E′ | 0.64 | ||||
| ν35–36 | 1035 | E′ | 1.06 | ||||
| ν34 | 1030 | A2′ | 0 | ||||
| ν32–33 | 968 | E′ | 0.12 | ||||
| ν31 | 793 | A2″ | 1.44 | 761 | A1 | 100 | 820 |
| ν29–30 | 758 | E′ | 0 | ||||
| ν27–28 | 745 | E″ | 0 | ||||
| ν26 | 720 | A2″ | 0.1 | ||||
| ν25 | 719 | A1′ | 0 | ||||
| ν23–24 | 697 | E″ | 0 | ||||
| ν22 | 629 | A2′ | 0 | ||||
| ν20–21 | 579 | E″ | 0 | ||||
| ν19 | 578 | A1″ | 0 | ||||
| ν17–18 | 565 | E′ | 0.08 | ||||
| ν16 | 517 | A1′ | 0 | ||||
| ν14–15 | 434 | E′ | 0 | ||||
| ν13 | 380 | A2′ | 0 | ||||
| ν11–12 | 281 | E′ | 0.32 | ||||
| ν10 | 255 | A2″ | 13.2 | ||||
| ν8–9 | 238 | E″ | 0 | ||||
| ν6–7 | 230 | E″ | 0 | ||||
| ν5 | 221 | A2″ | 4.14 | ||||
| ν4 | 208 | A1″ | 0 | ||||
| ν3 | 100 | A2″ | 0.48 | ||||
| ν1–2 | 80 | E″ | 0 | ||||
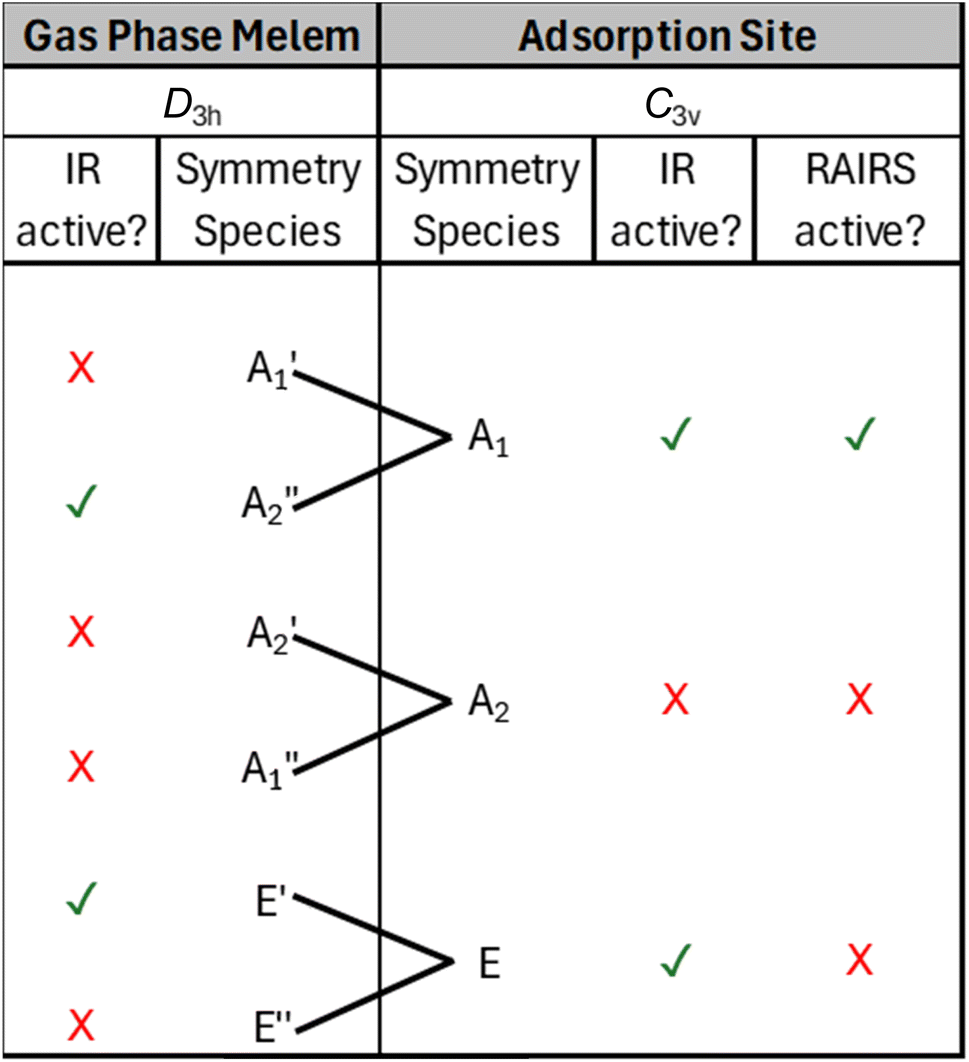
For the D3h point group, only gas phase vibrations belonging to the E′ and A2″ species are infrared active. Upon adsorption in the planar geometry imaged by STM and calculated by DFT, a reduction of symmetry to C3v occurs due to the loss of the molecular σh mirror plane. This results from both the physical presence of the surface, and due to the out-of-plane distortion of the NH2 groups towards the surface, and has a profound effect on the IR activity of the vibrational modes. Table 2 shows the symmetry transformation of the irreducible representations upon descent of symmetry from D3h to C3v and identifies the IR and RAIR active modes. Immediately, it can be seen that the switch of symmetry renders the allowed E′ gas phase modes as RAIRS inactive, while the previously forbidden A1′ modes now become RAIRS active. Thus, the intense out-of-phase NH2 scissoring mode, E′ mode at 1576 cm−1 in the gas phase molecule becomes inactive upon adsorption and is not observed in the RAIR spectrum. Conversely, previously inactive gas phase modes like the A1′ vibrations at 1611 cm−1, 1515 cm−1, 1347 cm−1 and 1133 cm−1 become RAIR active and correspond to the 1591, 1509, 1345 and 1125 cm−1 bands in the calculated RAIR spectrum. The 793 cm−1 A2″ out-of-plane heptazine ring buckling mode is weakly IR active in the gas phase calculation but upon adsorption this mode gives rise to a very strong band at 761 cm−1 in the calculated spectrum and a significant band at 820 cm−1 in the experimental RAIR spectrum.
 |
Unexpected RAIR intensities of formally allowed modes
The symmetry analysis above allows identification of formally allowed and forbidden vibrational modes. However, we note that a mode may be formally allowed but be expected to generate only a small dipole moment change normal to the surface and, therefore, present zero to low intensity RAIR bands. This leads to an interesting point of difference between the simulated and experimental RAIR data, Fig. 3, where there is discrepancy in the relative intensity ratios of the in-plane and out-of-plane vibrations. The calculated RAIR spectrum shows very high intensity of the out-of-plane 761 cm−1 vibration, which is entirely consistent with a planar geometry, Fig. 4. The much lower intensity of the in-plane modes between 1591–1345 cm−1 is also consistent with the major component of the dipole change occurring in the molecular plane. The non-zero intensities in the calculated spectrum may be attributed to the small 7° bend of the N–H bonds towards the surface, Fig. 2b. This is only a slight distortion but the inherently large dipole moment change associated with the NH2 scissoring vibration means that it is sufficient to generate a discernible dipole change normal to the surface. In addition, DFT calculations indicate 0.27 e− exchange between the molecule and surface, likely involving the molecular π system, which would lead the in-plane heptazine ring stretches to generate finite dipole moments normal to the surface.In contrast, the experimental data show a reversal in the relative intensity, with the in-plane NH2 scissoring + heptazine ring stretching modes between 1591–1345 cm−1 displaying significant band-broadening and a greater integrated intensity, while the out-of-plane mode at 820 cm−1 is sharp and possesses a lower integrated intensity. Therefore, it would appear that the experimental system experiences additional perturbations beyond those indicated by DFT calculations of the single adsorbed molecule. There are two main contributing aspects that can be considered. First, it is well established that intermolecular hydrogen bonding has a significant effect on both the vibrational frequencies and its intensity. Commonly, a red shift of the stretching vibration along the X–H direction occurs due to softening of the X–H bond. Concurrently, the bending vibrations can show a slight blueshift to higher frequencies due to stiffening of the bending motion due to H-bonding.50 Hydrogen bonding also induces significant anharmonicity in the vibrational potential energy curve, leading to broadening and large increases in intensity.51–60 STM images in Fig. 1 show that molecules aggregate in small assemblies as coverage increases, attributed to hydrogen bonding between the NH2 groups and the ring nitrogen on the exterior edge of its neighbour. For crystalline melamine61 it has been shown that such interactions lead to significant increases in intensity and broadening of IR bands involving NH2 vibrations, but little perturbation of the inner ring modes. This is consistent with our experimental data for adsorbed melem.
A second effect that may be considered is that melem’s amine hydrogens directly interact with the surface via N–H⋯M interactions that have been observed in transition metal complexes, with descriptions ranging from H-bonding to analogies with agostic and anagostic interactions. In organometallic chemistry, agostic bonds involve C–H⋯M interaction via 3 centre-2 electron bonds, with electron transfer from a C–H σ-bond donor to a vacant metal orbital and back transfer from metal into the antibonding CH σ* orbitals. Agostic interactions significantly increase the intensity and broadening of IR bands, through bond weakening and increased anharmonicity. Similar C–H⋯M interactions have been captured for cyclohexane adsorbed on Cu(111), where significant CH bond softening and intensity increases are observed for the CH stretching modes.62,63 N–H bonds are far more polar than C–H bonds and their interaction with metal surfaces have a larger electrostatic component, leading to anagostic bonds.64,65 Anagostic interactions are weaker than agostic interactions and have larger H…M bond lengths (2.3–2.9 Å) compared to agostic bonds (1.8–2.3 Å).66–69 Our DFT calculations show adsorbed melem is held 3 Å above the surface and its NH2 groups are bent by 7° towards the surface, with the H⋯M bond distance of 2.94 Å to the nearest 2-fold adsorption site verging on the realms of anagostic interactions.64,66–69 Such interactions may also explain why the RAIR spectra display broadening and intensity increases from the lowest coverage, and why extended organised intermolecular H-bonded assemblies are not observed on Cu(111) where, presumably, there is a finer balance between maximising intermolecular H-bonding and optimising molecule–metal interactions. Together, this combination of techniques and data suggests melem adsorption on Cu(111) may lie beyond a simple physisorption description.
Conclusions
The adsorption of melem on Cu(111) at 80 K has been studied using STM, RAIRS, and DFT calculations. STM shows that melem adsorbs with a flat-lying triangular footprint at the surface. The molecules aggregate into small assemblies attributed to H-bonding interactions leading to a stagger along the triangular sides of neighbouring molecules. DFT calculations show that the most stable geometry for a single adsorbed molecule on Cu(111) is flat lying, adsorbed at a height of 3 Å above the surface, which suggests a physisorption state. We posed the question at the outset: what does vibrational spectroscopy add to this already detailed picture? This question is particularly challenging for a physisorbed or weakly adsorbed species, where the effect of the surface is weakly felt and where the combination of STM and DFT calculations would be expected to describe the adsorption state extremely well. Here, we show that vibrational data provides a more sensitive handle on adsorption site symmetry and local perturbations. Melem on Cu(111) is a pertinent example because the major molecular mirror plane switches from being in-plane to out-of-plane upon adsorption. Thus, previously forbidden A1′ modes in the gas phase are now formally allowed in the adsorbed state and, importantly, the gas phase allowed E′ modes now switch off in the adsorbed state. The experimental RAIR spectra deliver further information since they display unexpected intensities and broadening of in-plane vibrational bands. This is attributed to the presence of both intermolecular hydrogen bonding and possible N–H⋯M interactions that lead to increased anharmonicity and significant changes in the frequencies and intensities of the in-plane NH2 scissors + ring stretching vibrational modes. Anharmonicity, which is important in driving molecular behaviours at surfaces, is not easily probed in the STM experiment or by DFT calculations, which work within the simple harmonic oscillator approximation. Overall, the combination of techniques enables a more detailed and nuanced understanding of the system, with STM and DFT calculations providing detailed structural information, while the RAIR information interrogates the adsorption symmetry and signposts the role of molecule–metal and molecule–molecule interactions in driving vibrational bandwidths, intensities and anharmonicity. Specifically, the small charge transfer discerned in the DFT calculations, and the clear vibrational signatures associated with bond softening and anharmonicity provides a sensitive indicator of molecular perturbation that goes beyond a simple physisorption model and delivers a promising handle to gauge the potential reactivity of weakly-adsorbed molecular systems. Finally, although the presence of such effects is indicated in the experimental RAIR spectra, a more detailed theoretical understanding still remains under-explored, especially for complex adsorbed molecules.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available upon request from the corresponding author. Zenodo created with same DOI as paper https://zenodo.org/records/19884778.References
- X. Song, Y. Wu, D. Pan, R. Wei, L. Gao, J. Zhang and G. Xiao, J. CO2 Util., 2018, 24, 287–297 CrossRef CAS.
- H. B. Zheng, W. Chen, H. Gao, Y. Y. Wang, H. Y. Guo, S. Q. Guo, Z. L. Tang and J. Y. Zhang, J. Mater. Chem. C, 2017, 5, 10746–10753 RSC.
- X. Wei, Y. Qiu, W. Duan and Z. Liu, RSC Adv., 2015, 5, 26675–26679 RSC.
- H. Kiuchi, Y. Sonoda, Y. Miyake, F. Kobayashi, J. Tsutsumi, M. Tadokoro and K. Kanai, Phys. Chem. Chem. Phys., 2022, 24, 23602–23611 RSC.
- R. Lei, B. Du, X. Lai, J. Wu, Z. Zhang, S. Liu, R. Wu, X. Li, B. Song and J. Jian, J. Mater. Chem. A, 2019, 7, 13234–13241 RSC.
- M. Bao, X. Wei, L. Cai, Q. Sun, Z. Liu and W. Xu, Phys. Chem. Chem. Phys., 2017, 19, 18704–18708 RSC.
- J. Eichhorn, S. Schlogl, B. V. Lotsch, W. Schnick, W. M. Heckl and M. Lackinger, CrystEngComm, 2011, 13, 5559–5565 RSC.
- S. Uemura, K. Sakata, M. Aono, Y. Nakamura and M. Kunitake, Front. Chem. Sci. Eng., 2016, 10, 294–300 CrossRef CAS.
- L. Wang, H. X. Shi, W. Y. Wang, H. Shi and X. Shao, Acta Phys.–Chim. Sin., 2017, 33, 393–398 CAS.
- S. Uemura, M. Aono, K. Sakata, T. Komatsu and M. Kunitake, J. Phys. Chem. C, 2013, 117, 24815–24821 CrossRef CAS.
- S. Haq, F. Hanke, J. Sharp, M. Persson, D. B. Amabilino and R. Raval, ACS Nano, 2014, 8, 8856–8870 CrossRef CAS.
- M. Clarke, A. Bellamy-Carter, F. Malagreca, J. Hart, S. P. Argent, J. N. O’Shea, D. B. Amabilino and A. Saywell, Mol. Syst. Des. Eng., 2023, 8, 681–689 RSC.
- G. Kuang, S. Z. Chen, W. Wang, T. Lin, K. Chen, X. Shang, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2016, 138, 11140–11143 Search PubMed.
- L. A. Bumm, J. J. Arnold, M. T. Cygan, T. D. Dunbar, T. P. Burgin, L. Jones, D. L. Allara, J. M. Tour and P. S. Weiss, Science, 1996, 271, 1705–1707 CrossRef CAS.
- R. Raval, Faraday Discuss., 2017, 204, 9–33 RSC.
- S. Clair and D. G. De Oteyza, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed.
- F. Hanke, M. S. Dyer, J. Björk and M. Persson, J. Phys. Condens. Matter, 2012, 24, 424217 CrossRef PubMed.
- M. Saeys, M. F. Reyniers, G. B. Marin and M. Neurock, J. Phys. Chem. B, 2002, 106, 7489–7498 CrossRef CAS.
- C. Morin, D. Simon and P. Sautet, J. Phys. Chem. B, 2004, 108, 5653–5665 Search PubMed.
- Y. Jiang, S. Yang, S. Li and W. Liu, Sci. Rep., 2016, 6, 39529 CrossRef CAS PubMed.
- A. Bilić, J. R. Reimers, N. S. Hush, R. C. Hoft and M. J. Ford, J. Chem. Theory Comput., 2006, 2, 1093–1105 CrossRef PubMed.
- M. Sacchi, P. Singh, D. M. Chisnall, D. J. Ward, A. P. Jardine, W. Allison, J. Ellis and H. Hedgeland, Faraday Discuss., 2017, 204, 471–485 RSC.
- J. Schramm and R. Tonner-Zech, J. Phys. Chem. C, 2026, 130, 546–559 CrossRef CAS.
- J. Schramm and R. Tonner-Zech, Chempluschem, 2025, 90, 202400771 CrossRef PubMed.
- X. Shi, R. Q. Zhang, C. Minot, K. Hermann, M. A. Van Hove, W. Wang and N. Lin, J. Phys. Chem. Lett., 2010, 1, 2974–2979 CrossRef CAS.
- X. Q. Shi, Y. Li, M. A. Van Hove and R. Q. Zhang, J. Phys. Chem. C, 2012, 116, 23603–23607 CrossRef CAS.
- B. P. Klein, N. J. Van Der Heijden, S. R. Kachel, M. Franke, C. K. Krug, K. K. Greulich, L. Ruppenthal, P. Müller, P. Rosenow, S. Parhizkar, F. C. Bocquet, M. Schmid, W. Hieringer, R. J. Maurer, R. Tonner, C. Kumpf, I. Swart and J. M. Gottfried, Phys. Rev. X, 2019, 9, 11030 CAS.
- B. P. Klein, J. M. Morbec, M. Franke, K. K. Greulich, M. Sachs, S. Parhizkar, F. C. Bocquet, M. Schmid, S. J. Hall, R. J. Maurer, B. Meyer, R. Tonner, C. Kumpf, P. Kratzer and J. M. Gottfried, J. Phys. Chem. C, 2019, 123, 29219–29230 CrossRef CAS.
- S. Naseri, G. Abbas, G. Johansson, K. Koumpouras, J. D. Baran and J. A. Larsson, J. Phys. Condens. Matter, 2024, 36, 235001 CrossRef CAS.
- M. S. Dyer, A. Robin, S. Haq, R. Raval, M. Persson and J. Klimeŝ, ACS Nano, 2011, 5, 1831–1838 CrossRef CAS PubMed.
- P. Donovan, A. Robin, M. S. Dyer, M. Persson and R. Raval, Chem.–A Eur. J., 2010, 16, 11641–11652 CrossRef CAS PubMed.
- J. Brandhoff, R. K. Berger, F. Otto, M. Schaal, L. Brill, O. T. Hofmann, P. Puschnig, T. Fritz and R. Forker, J. Phys. Chem. C, 2025, 129, 20738–20745 CrossRef CAS PubMed.
- F. M. Chua, Y. Kuk and P. J. Silverman, Phys. Rev. Lett., 1989, 63, 386–389 CrossRef CAS PubMed.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 Search PubMed.
- I. Hamada, Phys. Rev. B:Condens. Matter Mater. Phys., 2014, 89, 121103 CrossRef.
- A. Hjorth Larsen, J. JØrgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. SchiØtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys. Condens. Matter, 2017, 29, 273002 Search PubMed.
- B. Jürgens, E. Irran, J. Senker, P. Kroll, H. Müller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef PubMed.
- X. Yuan, K. Luo, N. Liu, X. Ji, C. Liu, J. He, G. Tian, Y. Zhao and D. Yu, Phys. Chem. Chem. Phys., 2018, 20, 20779–20784 RSC.
- K. Kanai, T. Yamazaki, H. Kiuchi, M. Isobe and Y. Sonoda, ACS Omega, 2025, 10, 16977–16992 CrossRef CAS PubMed.
- K. Tonigold and A. Groß, J. Chem. Phys., 2010, 132, 224701 Search PubMed.
- D. Yang, L. L. Li, G. Xiao and S. Zhang, Appl. Surf. Sci., 2020, 510, 145345 CrossRef CAS.
- T. S. Miller, A. B. Jorge, T. M. Suter, A. Sella, F. Corà and P. F. McMillan, Phys. Chem. Chem. Phys., 2017, 19, 15613–15638 RSC.
- B. V. Lotsch and W. Schnick, Chem.–A Eur. J., 2007, 13, 4956–4968 CrossRef CAS.
- B. V. Lotsch and W. Schnick, Chem. Mater., 2006, 18, 1891–1900 CrossRef CAS.
- L. Li, L. Zhang and C. Zhao, Russ. Chem. Rev., 1964, 33, 400–405 CrossRef.
- H. Meng, M. Kuhn, P. Meng, M. Pfrunder, R. Sun, Z. Liu, X. Zhou, H. Wang, J. McMurtrie and J. Xu, J. Phys. Chem. C, 2025, 129, 13116–13122 CrossRef.
- E. Wirnhier, M. B. Mesch, J. Senker and W. Schnick, Chem.–A Eur. J., 2013, 19, 2041–2049 CrossRef CAS.
- T. Seki, K. Y. Chiang, C. C. Yu, X. Yu, M. Okuno, J. Hunger, Y. Nagata and M. Bonn, J. Phys. Chem. Lett., 2020, 11, 8459–8469 CrossRef CAS PubMed.
- G. C. Pimentel and A. L. McClellan, The Hydrogen Bond, W.H. Freeman and Company, 1960 Search PubMed.
- A. A. Howard, G. S. Tschumper and N. I. Hammer, J. Phys. Chem. A, 2010, 114, 6803–6810 Search PubMed.
- N. S. Myshakina, Z. Ahmed and S. A. Asher, J. Phys. Chem. B, 2008, 112, 11873–11877 CrossRef CAS PubMed.
- X. L. Zhu, Z. Y. Yuan, L. Jiang, K. Zhang, Z. R. Wang, H. W. Luo, Y. Gu, J. W. Cao, X. L. Qin and P. Zhang, New J. Phys., 2019, 21, 043054 Search PubMed.
- T. Fornaro, D. Burini, M. Biczysko and V. Barone, J. Phys. Chem. A, 2015, 119, 4224–4236 Search PubMed.
- W. Qian and S. Krimm, J. Phys. Chem. A, 2002, 106, 6628–6636 Search PubMed.
- A. McNutt, S. Haq and R. Raval, Surf. Sci., 2003, 531, 131–144 Search PubMed.
- C. Clay, S. Haq and A. Hodgson, Phys. Rev. Lett., 2004, 92, 4 Search PubMed.
- J. S. Stevens, S. Coultas, C. Jaye, D. A. Fischer and S. L. M. Schroeder, Phys. Chem. Chem. Phys., 2020, 22, 4916–4923 RSC.
- B. Dereka, Q. Yu, N. H. C. Lewis, W. B. Carpenter, J. M. Bowman and A. Tokmakoff, Science, 2021, 371, 160–164 Search PubMed.
- J. Grabska, K. B. Beć, C. G. Kirchler, Y. Ozaki and C. W. Huck, Molecules, 2019, 24, 1402 CrossRef PubMed.
- R. Raval and M. A. Chesters, Surf. Sci. Lett., 1989, 219, L505–L514 CrossRef CAS.
- R. Raval, S. F. Parker and M. A. Chesters, Surf. Sci., 1993, 289, 227–236 CrossRef CAS.
- D. Braga, F. Grepioni, E. Tedesco, K. Biradha and G. R. Desiraju, Organometallics, 1997, 16, 1846–1856 Search PubMed.
- M. Lein, Coord. Chem. Rev., 2009, 253, 625–634 CrossRef CAS.
- M. Mannarsamy and G. Prabusankar, New J. Chem., 2021, 45, 5933–5938 RSC.
- G. Rajput, V. Singh, A. N. Gupta, M. K. Yadav, V. Kumar, S. K. Singh, A. Prasad, M. G. B. Drew and N. Singh, CrystEngComm, 2013, 15, 4676–4683 RSC.
- E. L. Zins, B. Silvi and M. E. Alikhani, Phys. Chem. Chem. Phys., 2015, 17, 9258–9281 Search PubMed.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |