 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe ever-evolving active site: transformation of single atoms to extended structures during the Rh-catalyzed reverse water–gas shift reaction
Greg Barber†
a,
Xiaobo Chen† bc,
Anastassiya Khand,
Jake Heinleine,
Sabrina M. Gerickeb,
Meng Lib,
Dmitri Zakharovb,
Judith Yangbc,
Ashley R. Headb,
Matteo Cargnelloe,
Robert M. Riouxaf and
Simon R. Bare*d
bc,
Anastassiya Khand,
Jake Heinleine,
Sabrina M. Gerickeb,
Meng Lib,
Dmitri Zakharovb,
Judith Yangbc,
Ashley R. Headb,
Matteo Cargnelloe,
Robert M. Riouxaf and
Simon R. Bare*d
aDepartment of Chemical Engineering, Pennsylvania State University, University Park, Pennsylvania 16802, USA
bCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York 11973, USA
cDepartment of Chemical and Petroleum Engineering, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, USA
dSSRL, SLAC National Accelerator Laboratory, Menlo Park, California 94025, USA. E-mail: srbare@slac.stanford.edu
eDepartment of Chemical Engineering, Stanford University, Stanford, California 94305, USA
fDepartment of Chemistry, Pennsylvania State University, University Park, Pennsylvania 16802, USA
First published on 19th February 2026
Abstract
At low temperatures (<400 °C), single atoms of Rh supported on rutile TiO2 (rTiO2) are responsible for the formation of CO during the reverse water gas shift (RWGS), while methane production is associated with the Rh–TiO2 interface due to the observed correlation between methane formation rates and the volume-averaged Rh nanoparticle diameter. As the temperature is increased to >540 °C, there is a notable increase in CO selectivity as the methane production rates tend towards zero. The time to reach zero depends on the temperature but is independent of the initial Rh structure (single atoms and/or nanoparticles), which is controlled by the catalyst preparation method (wetness impregnation versus colloidal nanoparticles). At 600 °C and >4 h time on stream, the catalytic behaviour becomes completely agnostic to the initial Rh structure as well as weight loading, and the catalysts are highly selective for the RWGS reaction. Post-reaction HR-TEM image analysis confirms Rh nanoparticles crystallize/order during the reaction; at 400 °C, most of the Rh particles are disordered, while at 600 °C, they are more ordered (i.e., there is the development of defined faceting). Infrared spectroscopy of CO adsorption on Rh nanoparticles confirms the appearance of defined facets after annealing in nitrogen at high temperatures. Annealing the Rh/rTiO2 catalysts prior to the RWGS reaction demonstrates the structural transformation of Rh depends only on time and temperature and not on reactant or product fugacity. Sites responsible for stabilizing Rh single atoms are no longer competent at higher temperatures, enabling single atom integration into existent nanoparticles. As the reaction temperature is increased to temperatures >540 °C, the dominant Rh structure for CO production evolves from single atoms to extended surfaces.
1. Introduction
The reverse water gas shift reaction, (RWGS, CO2 + H2 → CO + H2O) is an industrially relevant reaction to produce carbon monoxide (CO) from carbon dioxide (CO2) and hydrogen (H2). This reaction has been discussed as a central component in the so-called power to liquids scenario where water electrolysis is used to make H2, RWGS converts this H2 and CO2 to CO, and then the resulting syngas is converted into liquid fuels using Fischer–Tropsch catalysis.1–3 One of the most significant challenges with the RWGS reaction is selectivity to CO due to the competing Sabatier reaction, CO2 + 4H2 → CH4 + 2H2O. This reaction is heavily exothermic (−165 kJ mol−1), while RWGS is endothermic (41.5 kJ mol−1).Therefore, the RWGS presents fundamental selectivity challenges. If a catalyst is unselective for RWGS versus methanation, then the product distribution is dictated solely by thermodynamics and high temperatures (≥600 °C) are required to suppress CH4 formation and achieve high CO yield. Such high temperatures typically promote sintering of the dispersed active phase of the catalyst. If a catalyst kinetically controls CH4 formation, then substantial CO selectivity is possible at lower temperatures. Most heterogeneous catalysts for RWGS utilize transition metals or oxides as the active species, which are deposited on high surface area oxide supports.4 Interestingly, it has been found that when Ni, Ru, and Rh supported catalysts are synthesized to promote the formation of atomically dispersed or single atom metal species, as opposed to metal nanoparticles (NPs), they are highly selective for RWGS.5–9 These studies support the notion that provided the active metal can be stabilized in the form of single atoms, there are opportunities to perform RWGS at lower temperatures due to the high selectivity of the single atoms at lower temperatures. However, because of RWGS thermodynamics, operation at high temperatures is mandatory. For example, at 400 °C, for 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2/CO2 mixture, the equilibrium conversion is only 38.1%, it increases to 51.9% at 500 °C and is 63.1% at 600 °C (the highest temperature explored in this manuscript). Moreover, the literature teaches that regardless of whether thermodynamics are used to control RWGS selectivity by operating at very high temperatures, or kinetics are used to control selectivity by operating at low temperature using atomically dispersed metal catalysts, sintering of the active metal domains must be suppressed to promote long term catalyst stability.
:1 H2/CO2 mixture, the equilibrium conversion is only 38.1%, it increases to 51.9% at 500 °C and is 63.1% at 600 °C (the highest temperature explored in this manuscript). Moreover, the literature teaches that regardless of whether thermodynamics are used to control RWGS selectivity by operating at very high temperatures, or kinetics are used to control selectivity by operating at low temperature using atomically dispersed metal catalysts, sintering of the active metal domains must be suppressed to promote long term catalyst stability.
As such we have explored the use of Rh/rTiO2 for the RWGS reaction. Earlier work using atomically dispersed Rh/TiO2 (P25) demonstrated isolated atoms and nanoparticles of the same metal on the same support can exhibit uniquely different catalytic selectivity in competing reaction pathways at a reaction temperature of 200 °C and low conversion conditions.5 Moreover, the dispersion of nanoparticles under reaction conditions can play a significant role in controlling stability.5 More recently, under high conversion conditions with Rh/rTiO2, it was demonstrated the product selectivity is governed by the evolving distribution of Rh moieties under reaction conditions: the balance between Rh single atoms and Rh nanoparticle–TiO2 interfacial sites determines CO versus CH4 formation. Moreover, it appeared Rh single atoms are unexpectedly robust over at least 90 h on stream at 500 °C, even at high Rh density, whereas Rh NPs sinter.10 Additionally, we reported the results of a round robin study of Rh/rTiO2 catalysts for RWGS across four laboratories,11 where the time-dependent production rates of CO via RWGS and CH4 via methanation were measured at an appreciable approach to equilibrium. In this study the CH4 formation rate rapidly decreased between 500 °C and 600 °C for higher Rh weight loadings. These studies indicate the reaction temperature plays an important role in the type and stability of the Rh structures present on the titania, motivating the current study.
In this work we report on the evolution of the reactivity and structure of a series of Rh/rTiO2 catalysts under RWGS conditions at high conversions (400–600 °C, 1 atm, 75% H2, 25% CO2) as a function of time on stream (16 h). A series of rutile-supported catalysts were prepared by both wetness impregnation (WI) and colloidal synthesis (CS/NP) with nominal weight loadings ranging from 0.1 to 2 wt% Rh. The catalysts were characterized using transmission electron microscopy (TEM), CO diffuse reflectance infrared spectroscopy (CO DRIFTS), and X-ray photoelectron spectroscopy (XPS). We show that irrespective of both the catalyst preparation method and the Rh weight loading, all catalysts operated at 600 °C exhibit similar selectivity to CO (>98%) after prolonged (16 h) time on stream. TEM analysis after reaction for 16 h shows a similar mean Rh nanoparticle size among all catalysts. We show pre-treatment in an inert gas (annealing) prior to the RWGS reaction significantly improves the initial CO selectivity. We attribute these observations to a change in the ordering/crystallinity of the Rh NPs, which enables the establishment of new active sites for CO production. This work clearly demonstrates that at temperatures >500 °C, evolution of the Rh structure requires only time and temperature; the details of the CO2 hydrogenation atmosphere is not a prerequisite for Rh restructuring.
2. Methods
2.1 Catalyst synthesis
000), glucose (Sigma-Aldrich, 720 mg), and KBr (Sigma-Aldrich, 71.4 mg) were dissolved in 10 mL of water and added to a pressure flask (Ace Glass Inc.). The flask was sealed and heated to 140 °C under magnetic stirring for 3 h. The nanoparticles were then washed in water and separated via precipitation with acetone three times before being dispersed and stored in water. Thermogravimetric analysis (TGA) was performed to determine the Rh NP concentration in the solution.:water ∼ 3:1, v/v). The precipitate was then collected and dried in an oven overnight at 80 °C. The sample was then sieved below 180 µm grain size and calcined at 500 °C for 3 h to remove the organic ligands. Removal of the PVP ligands was ensured by conducting temperature programmed oxidation (Fig. S1) and XPS (Fig. S2). All chemicals were purchased from Sigma-Aldrich and the rTiO2 was purchased from US-Nano (US-Nano, 99.9%, 30 nm particle size). These catalysts are referred to as colloidal synthesis nanoparticle (CS/NP) catalysts, with the designation X wt% Rh/rTiO2 (CS/NP).The reaction environment and thermal profile as a function of time for a representative sample is illustrated in Fig. 1a. The overall protocol for reaction testing was (i) purge reactor tube with 20 sccm of nitrogen (>99.995%) and 100 sccm of 20% O2 in N2 (balance) (>99.995%), (ii) oxidation at 400 °C for 30 min (10 °C min−1 ramp rate) under 20% O2 in N2 (balance) at 32 sccm, (iii) cool catalyst down to <40 °C under same gas flow as in (iii), (iv) purge the system of oxygen with N2 (>99.995%) at 20 sccm for 25 min, (v) switch to 3:1 ratio H2:CO2 at a total flow rate of 32 sccm for 30 min with the sample temperature <40 °C, and (vi) ramp the catalyst to reaction temperature at 10 °C min−1 and hold for 16 h under same gas flow as in (v). This procedure is referred to throughout the manuscript as the normal/standard RWGS protocol (Fig. 1a).11
 | ||
| Fig. 1 Representative reaction environments and thermal profiles for non-annealed samples with the standard protocol (a) and samples that undergo annealing (b) before being exposed to the same 3:1 H2:CO2 environment. In (b), the reduction step occurs at 200 °C under 20% H2 in N2 after the post oxidation purge and prior to annealing under nitrogen and introduction of the RWGS gases. The main processes of purge, oxidation (light orange), reduction (light blue), annealing (light yellow), and time at which the sample is exposed to 3:1 H2:CO2 RWGS environment at reaction temperature (light green) are labelled. | ||
For annealed catalyst samples, the preparation and the reaction protocol was identical through the post oxidation purge (step iv). After this step, the catalyst was reduced and annealed prior to exposure to the 3:1 H2/CO2 mixture. In these experiments, the Rh catalyst was reduced by exposing the sample at <40 °C to 20% H2 in N2 (Linde, >99.995% grade), ramping at 10 °C min−1 to 200 °C and holding for 5 min under at a total flow rate of 25 sccm before purging the reactor with 20 sccm of N2 (Linde, >99.995%) for 25 min. After the post reduction purge, the sample was heated to the annealing temperature at 10 °C min−1 in 20 sccm of N2 for a predetermined amount of time (from 1–2 h) prior to exposure to a 3:1 H2:CO2 mixture. A representative reaction environment and thermal profile for a sample annealed at 500 °C for 2 h is shown in Fig. 1b. In Fig. 1a and b, the main reaction processes of oxidation, reduction, annealing, and being under a RWGS environment at reaction temperature are summarized. The relevant definition of elapsed time and time at reaction temperature are indicated in Fig. 1. Elapsed time is only shown to help guide the reader on the total time for the pre-treatments before collection of the catalytic data using ‘time at reaction temperature.’
The CO2 conversion (XCO2) and CO selectivity (SCO) are defined as follows, where ni denotes the moles of species i leaving the reactor:

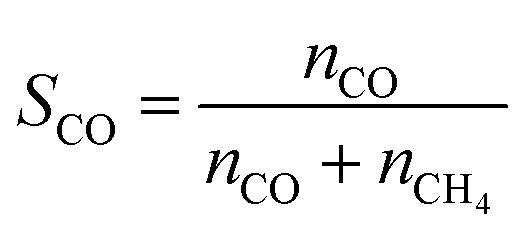
The net production rates are defined as: “CO production rate” (and analogously “CH4 production rate”) denotes CO (or CH4) production rates (rCO/rCH4) normalized per mole of total Rh. These are integral rates, rather than commonly used differential rates for turnover frequency (TOF) or space-time yield (STY) calculations and rate, and ri is defined as  , where Fi is the molar flow rate of CO or CH4 exiting the reactor and molRh is the total moles of Rh in the rTiO2 catalyst.
, where Fi is the molar flow rate of CO or CH4 exiting the reactor and molRh is the total moles of Rh in the rTiO2 catalyst.
2.2 Catalyst characterization
All samples were pretreated in situ in 20% O2/He at 400 °C for 30 min (ramp 10 °C min−1) and then cooled to room temperature in the same O2 flow. After purging with He, the samples were reduced in 20% H2/He at 200 °C for 5 min (ramp 10 °C min−1), followed by a 20 min He purge to remove residual hydrogen. Subsequently, the samples were heated in He to the desired annealing temperature (400, 500, or 575 °C) at a rate of 10 °C min−1 and held for the specified dwell time (30 min, 1 h, or 2 h). The temperature (575 °C) represents the highest temperature the Harrick reactor cell was able to reach. After cooling to room temperature, a background spectrum was collected. The samples were then exposed to 10% CO/Ar (>99.995%, Airgas) at room temperature until the CO adsorption reached saturation coverage, after which gas-phase CO was purged with He for 45 min until all gas phase CO signatures disappeared and the final CO-DRIFTS spectrum was then recorded.
Peak deconvolution was done using OMNIC software with Voigt functions. Peak heights were allowed to be in the range of 0–10 absorbance units, Gaussian full width at half heights were allowed to be in the range of 0–32 cm−1, and Lorentzian widths were allowed to be in the range of 0–450 cm−1. The noise was set to 0.05 and a baseline was not used. The initial number of peaks was determined using the peak finder with low sensitivity to reduce overestimation of the number of peaks. The number of peaks was increased until the fit was constant and the fit parameter no longer decreased.
3. Results and discussion
3.1 Methane selectivity decreases with reaction temperatures >500 °C
We explored the impact of reaction temperature (>500 °C) on CO selectivity for Rh/rTiO2 catalysts produced by two distinct methods (WI and CS/NP) containing both Rh single atoms and nanoparticles (d < 4.5 nm). Fig. 2 summarizes CO and CH4 net production rates at reaction temperatures between 500 and 580 °C for 0.3 wt% Rh/rTiO2 (WI) catalysts over 16 h. In a recent round robin study,11 the 0.3 wt% Rh/rTiO2 had the highest CH4 production yield at 500 °C of all examined Rh/rTiO2; therefore, it was chosen as the initial catalyst to examine the impact of temperature above 500 °C on the production of CH4. Fig. 2a clearly demonstrates CO net production rates increase with temperature and remain relatively constant over 16 h. Duplicate measurements at 540 °C and 560 °C demonstrate reproducible behavior between the experiments conducted on two separate diluted catalyst beds. The fate of CH4 production rates with time on stream follows very different behavior (Fig. 2b). At 500 °C (black data), the CH4 production rate continually decreases over the 16 h experimental window at a rate of CH4 loss of 0.1–0.2 mol molRh−1 s−1 per hour. Similar temporal behavior in the decline of CH4 production rates is observed at 540 °C, and a duplicate catalyst bed demonstrates a similar temporal trend. At 560 °C, the rate of methane production decreases at an average rate of CH4 loss of 0.56 mol molRh−1 s−1 per hour and is <1 mol molRh−1 s−1 at 16 h. At 580 °C, the rate of methane production decreases at ∼1.3 mol molRh−1 s−1 per hour and reaches 1 mol molRh−1 s−1 after 6 h, which corresponds a 60% decrease in time compared with a reaction temperature of 560 °C. The disparate behavior with temperature and time-on-stream between CO and CH4 production rates implies a loss of active sites associated with CH4 formation while the active sites associated with CO formation appear resilient, ultimately suggesting these products are produced on unique sites. These CO-resilient sites could be associated with our prior study where Rh single atoms exhibited stability for the duration of 90 h reactivity measurements. This prior study was conducted at a reaction temperature of 500 °C, which is substantially below the onset of the behavior discussed here. Utilizing the time-on-stream production rates, the CO selectivity is greater than 97% at 580 °C for times at reaction temperature greater than 8 h. | ||
| Fig. 2 CO (a) and CH4 (b) net production rates as a function of temperature for 0.3 wt% Rh/rTiO2 (WI). CO net production rates increase with increasing temperature and are stable with time-on-stream. Initial CH4 production rates do not follow the same trend with temperature and decrease with increasing time on stream. The methane production rate decreases significantly for temperatures greater than 540 °C. The RWGS reaction at 540 and 560 °C were conducted in duplicate (open symbols) to demonstrate reactor reproducibility. | ||
3.2 Methane selectivity at 600 °C is agnostic to the initial Rh structure
To determine if these observations of decreasing CH4 net production rate with temperature in excess of 560 °C were not limited solely to the 0.3 wt% Rh/rTiO2 (WI) catalyst, additional rutile-supported Rh catalysts (0.1 wt% Rh/rTiO2 (WI), 2 wt% Rh/rTiO2 (WI), and 0.3 wt% Rh/rTiO2 (CS/NP)) were tested under the standard RWGS protocol (Fig. 1a) at a temperature of 600 °C for 16 h. Fig. 3 summarizes the CO net production rate (Fig. 3a) and CH4 net production rate (Fig. 3b) for all catalysts tested. The CO net production rate (Fig. 3a) shows a general trend of constant CO production with time greater than 6–8 h on stream after a 0–30% decrease in initial CO production rates over the first several hours. The CH4 production rates in Fig. 3b demonstrate all rutile-supported Rh catalysts exhibit 20–100× lower CH4 production than CO production at 600 °C regardless of Rh weight loading and preparation method. During the first 4 hours on stream at 600 °C, the rapid 20–100× decrease in CH4 production coupled with the more constant CO production yields a substantial increase CO selectivity such that all samples exhibit a maximum in CO selectivity during this time. There is a slow decrease in CO selectivity after the maximum, but this is not due to a decrease in CO production rate but is due to the slow increase in the CH4 production rate (Fig. 3b). This behavior in CH4 net production rates is also seen in Fig. 2b for the 0.3 wt% Rh/rTiO2 (WI) sample at 580 °C suggesting this phenomenon is not limited to a temperature of 600 °C. Several weight loadings and catalyst preparation methods were run in duplicate to demonstrate the reproducibility with time on stream trends across different catalyst beds; all duplicate runs are noted with open symbols in Fig. 3a and b. | ||
| Fig. 3 (a) CO net production rates and (b) CH4 net production rates for all Rh/rTiO2 catalysts exposed to the standard RWGS procedure and a reaction temperature of 600 °C. A general trend of constant CO production rate with time-on-stream regardless of Rh weight loadings and preparation method is observable in (a) while (b) shows all Rh/rTiO2 catalysts have very low CH4 production rates (typically 0.1–0.5 mol molRh−1 s−1) at 600 °C regardless of Rh weight loading and preparation method. Duplicated reaction conditions are represented by open symbols. | ||
3.3 Ex situ TEM characterization of catalysts after reaction displays final particle size distribution
Ex situ TEM characterization was conducted on the post-reaction Rh/rTiO2 catalysts with Rh loadings of 0.1, 0.3, and 2.0 wt% following the standard RWGS reaction conditions at 400 °C, 500 °C, and 600 °C for 16 h. Catalytic performance data for the samples tested at 400 °C and 500 °C are the same as reported previously.11For the 0.1 wt% Rh/rTiO2 (WI) catalyst, a predominantly single atom catalyst in the fresh state (see Fig. S4),10 the TEM image (Fig. 4a) after RWGS reaction at 400 °C reveals the formation of discrete Rh NPs, indicating reaction-induced sintering. The particle-size distribution (Fig. 4b) is relatively uniform, with an average diameter of ∼1.8 nm. The 2.0 wt% Rh/rTiO2 (WI) (Fig. 4c and d) exhibits a similar mean particle size as the 0.1 wt% catalyst even though it has an order of magnitude higher loading of Rh. The comparable size across catalysts with different Rh loadings after RWGS at 400 °C regardless of weight loading suggests thermodynamic factors dominate the reaction-induced restructuring.15,16
 | ||
| Fig. 4 TEM characterization of the post-reaction samples subjected to the standard RWGS procedure at different temperatures. Low-magnification TEM images and corresponding particle size distributions are shown for the 0.1 and 2 wt% Rh/rTiO2 (WI) catalysts after 16 h of RWGS at 400 °C (a)–(d), the 0.1 and 2 wt% Rh catalysts after 16 h at 500 °C (e)–(h), and for the 0.1, 0.3, 2 wt% and 0.3 wt% Rh (CS/NP) catalysts after 16 h at 600 °C (i)–(p). | ||
The 0.1 wt% catalyst (Fig. 4e and f) post reaction at 500 °C exhibits a size distribution comparable to the same catalyst post reaction at 400 °C, whereas the 2.0 wt% catalyst (Fig. 4g and h) shows a broader size distribution but a similar mean size. Furthermore, the 0.1 wt% catalyst post reaction at 600 °C maintains a nearly identical mean particle size (∼1.8 nm) and narrow size distribution (Fig. 4i and j), consistent with large interparticle spacing suppressing particle coalescence at low loading.17 For the higher-loading catalysts (0.3 and 2.0 wt%) post reaction at 600 °C shown in Fig. 4k–n, the mean particle size increases from 1.9 nm to 2.1 nm, respectively, along with a broader size distribution. To further investigate the sintering dynamics of post-reaction samples at different temperatures, the size distributions revealed low-loading samples (0.1 wt% and 0.3 wt%) maintain a narrow particle size distribution without the formation of large particles. In contrast, the 2 wt% post-reaction samples exhibit an obvious increase in the fraction of sintering-induced larger particles (6–10 nm) due to the shorter average distance between Rh species.10 Importantly, the population of large particles (6–10 nm) remains a minor contribution, less than 2% as shown in Fig. 4. For the 600 °C post-reaction, 0.3 wt% Rh/rTiO2 (CS/NP) catalyst, TEM imaging (Fig. 4o and p) reveals unexpected particle shrinkage after reaction, relative to the fresh sample (see Fig. S4), possibly arising from fragmentation during the catalytic reaction.18,19 Surprisingly, the post-reaction particle size distributions after RWGS at 600 °C remain relatively consistent across samples with different loadings (0.1–2.0 wt% Rh) and initial Rh structures (WI or CS/NP), suggesting the final particle size is controlled by a thermodynamic equilibrium between sintering and fragmentation.
3.4 Differences in internal particle order post RWGS reaction
While the number of particles sampled by TEM imaging is small compared to the total number of particles present in each catalyst, the changes in particle size at the different reaction temperatures do not explain the time-dependent trends in CH4 production rate and selectivity. A prior study of the RWGS conducted at 500 °C, concluded that Rh single atoms are responsible for CO formation, and Rh NPs harbour sites (assigned to Rh nanoparticle–TiO2 interfacial sites) responsible for CH4 production.10 Fig. 3b clearly demonstrates the 0.1 wt% Rh/rTiO2 (WI) catalyst produces the least amount of methane on ramp to 600 °C but ultimately has the lowest CO selectivity (albeit still >95%) at 600 °C and 16 h of reaction time. While we previously postulated a population of Rh single atoms on rTiO2 are stable at 500 °C;10 at 600 °C, the appearance of Rh NPs with a mean diameter of ∼1.8 nm suggests these Rh single atoms may no longer be stable. Fig. 3b further demonstrates CO selectivity is >95% for all catalysts, regardless of the Rh weight loading and preparation method at 600 °C, which differs from our recent round robin observations that both a 2 wt% Rh/rTiO2 (WI) or 0.3 wt% Rh/rTiO2 (CS/NP) at 500 °C demonstrated a CH4 selectivity of 15–20%.11 Given our analysis demonstrated similar mean particle sizes after 16 h time-on-stream regardless of temperature (>400 °C), we examined the particles further utilizing HRTEM imaging. Specifically, we sought to determine if structural differences in the supported particles could provide an explanation of the temperature dependent CH4 selectivity.HRTEM images and the corresponding fast Fourier transform (FFT) patterns in Fig. 5 illustrate structural differences between Rh/rTiO2 after reaction at different temperatures. We chose to examine the 2.0 wt% Rh/rTiO2 (WI) since this sample had the highest particle density. HRTEM images of the catalyst post-reaction at 400 °C (Fig. 5a and S5) shows disordered atomic arrangements and irregular nanoparticle morphologies, as further evidenced by the corresponding FFT pattern lacking sharp diffraction spots. HRTEM images of the post-reaction samples (Fig. 5b and S6) at 500 °C contain well-arranged atomic columns, indicating increased structural ordering. In contrast the post-reaction samples at 600 °C exhibit well-defined lattice fringes, faceted morphologies, and enhanced structural ordering, suggesting thermally promoted Rh crystallization under RWGS conditions (Fig. 5c). The crystallized Rh nanoparticles in Fig. 5b and c are the face-centered cubic (FCC) structure of metallic Rh. Fig. 5d quantifies the distribution of ordered and disordered Rh NPs at different temperatures. The results show an increase in the number of ordered Rh NPs with increasing reaction temperature. Most of the Rh NPs are well-ordered after reaction at 600 °C, whereas a vast majority possess defective or disordered structures at 400 °C. It should be noted that the disappearance of lattice fringes in some Rh NPs classified as disordered may arise from slight defocus or off-axis orientation. In this study, we maintain such variations in slight defocus and orientation affect all samples similarly and therefore do not alter the observed temperature-dependent trend. However, the majority of Rh nanoparticles have characteristic sizes of approximately ∼2 nm and therefore primarily govern the observed structural trends. Moreover, the in situ HRTEM images (Fig. S7) reveal dynamic structural evolution between disordered and ordered states during the RWGS reaction at 500 °C. Overall, these temperature-dependent structural variations under RWGS conditions suggest dynamic restructuring processes that impact catalytic performance.
 | ||
| Fig. 5 HRTEM imaging and structural analysis of the post-reaction samples exposed to the standard RWGS procedure at different temperatures. (a)–(c) HRTEM images of the 2 wt% Rh/rTiO2 (WI) catalyst after 16 h of RWGS at 400, 500, and 600 °C, respectively. Insets are the corresponding FFT images of the selected NPs highlighted by the dashed white rectangles. (d) Number of ordered versus disordered structures in the post-reaction samples as a function of reaction temperature. HRTEM imaging of post-reaction Rh/rTiO2 catalysts at 600 °C with different loading: (e) 0.1 wt% Rh (WI), (f) 0.3 wt% Rh (WI), (g) 2 wt% Rh (WI), and (h) 0.3 wt% Rh (CS/NP), showing the well-crystallized and faceted Rh NPs. (i) In situ XPS measurements of the Rh 3d core level of the 2.0 wt% WI catalysts show the fresh catalyst has oxidized Rh and the post-reaction Rh oxidation state is metallic. | ||
Moreover, the Rh NPs are crystallized/faceted with a similar particle size. These are depicted in Fig. 5e–h. Additional HRTEM images are depicted in Fig. S8. This structural consistency across sample preparation methods aligns with the temperature-dependent structural variations after RWGS, providing a potential explanation for the similar catalytic behaviour at 600 °C and >4 h time on stream. These results highlight the critical role of nanoparticle crystallinity in catalytic performance of Rh/rTiO2 (WI) catalysts at 600 °C. Fig. 5i shows Rh 3d XPS data that are consistent with the Rh being metallic post RWGS. The 600 °C condition results in narrower peaks, supporting the more ordered Rh seen in the TEM results (additional discussion in SI). Notably, no strong metal–support interaction (SMSI) overlayer was observed in TEM on any of the samples (400, 500, 600 °C) studied, while Rh–TiO2 interactions may contribute to nanoparticle stabilization to avoid long-distance particle migration.10 The lack of SMSI is further supported by XPS measurements, where the intensity ratio of Rh 3d to Ti 2p peaks is constant for the 400 and 600 °C samples, suggesting no overgrowth and Rh signal attenuation (Fig. S9 and Section S3).
3.5 Pre-reaction annealing of Rh/rTiO2 in an inert atmosphere reduces transient formation of CH4
Given the apparent lack of SMSI, we examined whether the ripening and ordering/crystallization of the Rh is induced by the reactive gases (CO2, H2, and RWGS products – CO and H2O), temperature, time, or a combination of these. First, to gain an understanding of the effect of temperature and time on the observed CO selectivity changes during the first several hours, we examined the impact of an annealing step (i.e., heating the metal catalyst in N2 at a certain temperature and time) prior to the introduction of the 3:1 H2/CO2 mixture to the catalyst. For this investigation we chose the 0.3 wt% Rh/rTiO2 (CS/NP) catalyst, due to the expected structural changes it must undergo to wet the support, and the 2.0 wt% Rh/rTiO2 (WI) Rh, due to the propensity to produce nanoparticles and CH4 in the earlier study.10
In these annealing experiments, after the oxidation pre-treatment, Rh/rTiO2 catalysts were ramped at 10 °C min−1 in 20% H2 in N2 to 200 °C and held for 5 min. Following this reduction step the catalysts were directly heated at 10 °C min−1 in N2 to the stated annealing temperature and held for the different times. After this step the catalyst was heated to the appropriate reaction temperature and the gas composition was changed to the 3:1 H2/CO2 mixture, as depicted in Fig. 1b. It is evident from Fig. 2 and 3 that all major changes are in CH4 production rate while CO production rate is rather constant with time. As a result, changes in CO2 conversion and CO selectivity can be used as indicators of the trends in CH4 production. Fig. 6a and b show the CO2 conversion and CO selectivity for 0.3 wt% Rh/rTiO2 (CS/NP) and Fig. 6c and d are the CO2 conversion and CO selectivity for 2.0 wt% Rh/rTiO2 (WI). Regardless of whether a catalyst was annealed or not, there are no large differences in the final CO2 conversion and CO selectivity after 16 h. However, there are noticeable differences in the initial CO selectivity and the rate at which CO selectivity becomes constant with annealing time (Fig. 6b and d) at temperatures from 400–500 °C. In particular, the 0.3 wt% Rh/rTiO2 (CS/NP) exhibits a rapid increase in initial CO selectivity as the annealing temperature is increased from 400 °C to 500 °C with a further dramatic increase in the initial CO selectivity to >98% for an annealing treatment of 600 °C for 1 h. There is also an increase in CO selectivity with increasing annealing temperature and time for the 2.0 wt% WI catalyst, with the largest increase in CO selectivity being observed as the annealing temperature is again increased from 500 °C to 600 °C for the same 1 h duration. We attribute the different transient behavior in CO selectivity for T < 600 °C for the CS/NP versus WI catalysts to their initial morphology. Particles present on fresh higher weight loading WI catalysts are raft-like (Fig. S4), indicative of favorable wetting behavior to the support, while the colloidal Rh NPs on the Rh/rTiO2 (CS/NP) catalysts are nominally spherical and non-wetting on rTiO2. Fig. 4 and 5 demonstrate after 16 h time on stream, the NPs formed in both catalysts are similar in size and shape. The restructuring of spherical Rh NPs (d ∼ 4.5 nm) requires the movement of many more atoms to form faceted ∼2 nm particles compared to the raft-like particles found in WI catalysts. This temporal difference in catalyst restructuring leads to the dramatic improvement in CO selectivity for CS/NP catalysts.
 | ||
| Fig. 6 (a) and (c) CO2 conversion and (b) and (d) CO selectivity for 0.3 wt% Rh/rTiO2 (CS/NP) (top) and 2 wt% Rh/rTiO2 (WI) (bottom) after annealing in nitrogen for variable times and temperatures. For each catalyst, the total CO2 conversion appears independent of annealing conditions prior to the start of the RWGS reaction at 500 °C under a 3:1 H2:CO2 environment. Annealing the catalyst prior to exposure to RWGS conditions improves the initial CO selectivity with more dramatic improvements observed for the 0.3 wt% CS/NP catalyst than the 2.0 wt% WI catalyst. CO selectivity increases with increasing annealing temperature and time on stream for both catalyst types. For improved clarity, the repeat of the non-annealing reaction condition for the 2.0 wt% WI catalyst is represented by the ‘+’ sign. | ||
3.6 Infrared spectroscopy of CO adsorption confirms defined Rh faceting after annealing
Fig. 7 summarizes the observed CO DRIFTS spectra for three 0.3 wt% Rh/rTiO2 (CS/NP) catalysts after oxidation, reduction, and then annealing under He at various temperature/time combinations prior to exposure to CO at room temperature. The samples were annealed at 400 °C for 30 min (black), 500 °C for 2 h (red), and 575 °C for 2 h (blue). Since catalyst amounts varied between the CO DRIFTS experiments, only relative peak absorption intensities are used to compare results between different annealing conditions. However, peak intensities within each CO DRIFTS experiment can be used to define and compare relative contributions of different adsorbed CO configurations in each spectrum. | ||
| Fig. 7 Infrared spectra of CO adsorbed at room temperature under a He purge on a 0.3 wt% Rh/rTiO2 (CS/NP) catalyst after annealing at 400 °C for 30 min (black), 500 °C for 2 h (red), and 575 °C for 2 h (blue). The spectra were offset vertically. Vertical dashed lines are added to aid the eye and come from peak positions found through spectra deconvolution (see Section S3). All deconvoluted spectra were assigned to specific absorption coverages on Rh crystal facets or for single atoms based on prior literature assignments for CO adsorption. Default fit parameters were used in the software with no forcing of peaks to be at any specific frequency or intensity. | ||
These CO DRIFTS absorption spectra are generally described by assigning the peak in the 1850–1970 cm−1 region to bridge bound CO on a Rh NP, the peaks at 2020 cm−1 and 2090–2100 cm−1 to the asymmetric and symmetric stretches of gem-dicarbonyl on rhodium single atoms and the peak in the 2040–2070 cm−1 region due to linear bound carbonyls on Rh NPs.20–22 We use infrared absorption studies of CO adsorption on Rh single crystal surfaces to provide insight regarding the evolution of the different Rh crystal facets as a function of annealing condition. We emphasize that while Rh single crystal studies are free of interference from gem-dicarbonyl absorption due to their absence on extended low-energy Rh surfaces, the exact CO absorption frequencies and intensities depend on the surface coverage of CO (θCO) on each crystal facet.
The FTIR spectra in Fig. 7 clearly show changes in CO binding configuration as a function of annealing condition. To better understand the influence of annealing temperature each spectrum was deconvoluted. The results of this peak deconvolution for the annealed catalysts in Fig. 7 are shown in Table 1. Full detailed peak deconvolution and fitting parameters are listed in Tables S1–S3 while the fits are shown in SI Fig. S10–S12. The deconvoluted peaks in Table 1 show that overall, the frequencies of the deconvoluted peaks were invariant in frequency and only changed in intensity with annealing conditions further supporting the idea that NPs and their final morphology evolve with time and temperature.
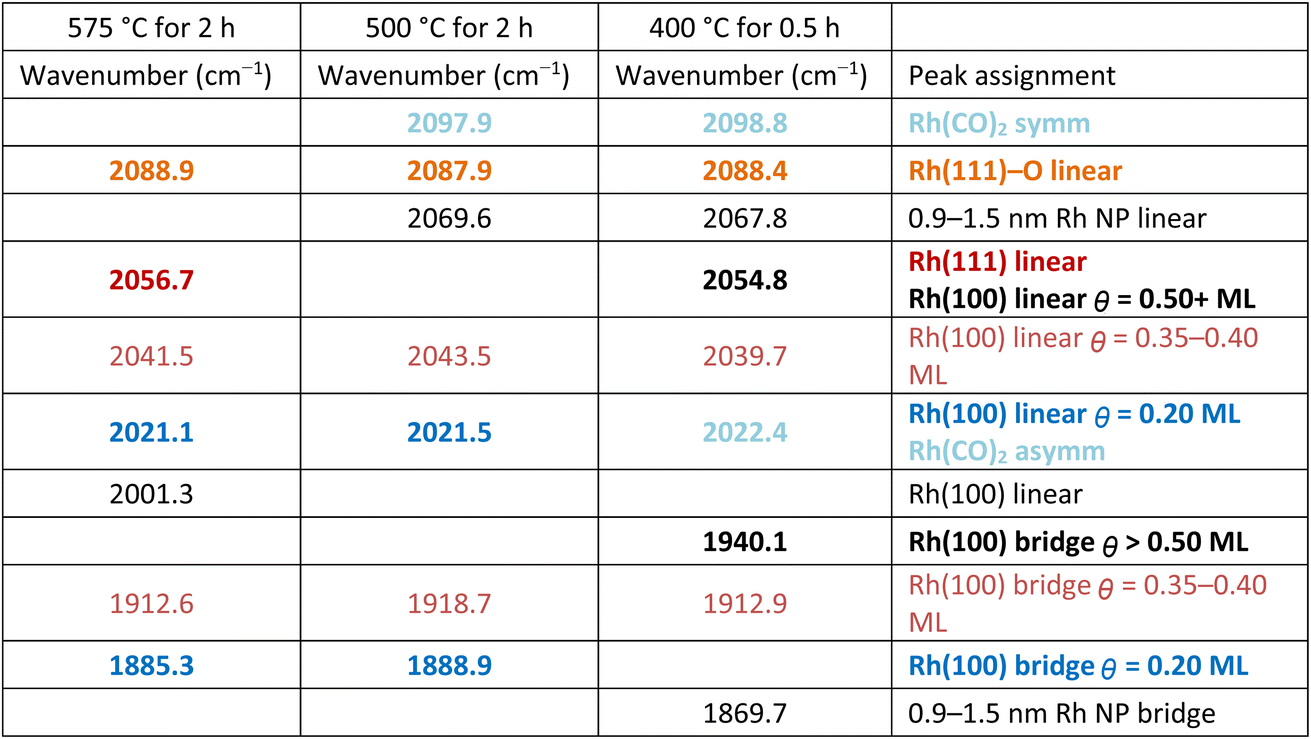 |
The deconvoluted data (Table 1) reveal a total of 11 different absorption peaks can be observed over the 3 different annealing conditions with 4 peaks assignable to CO in a bridging configuration on Rh surfaces (1870–1940 cm−1), 6 peaks assignable to linear CO on Rh surfaces and/or the asymmetric stretch of Rh single atoms (2001–2088 cm−1), and 1 peak associated with the symmetric stretch of Rh single atoms (>2090 cm−1).20–34 (All deconvoluted peaks contained shifts of <±1 cm−1 except the low frequency bridging and the peak at 2041 cm−1 which shifted by ±3 cm−1 and ±2 cm−1 respectively). Of these peaks, only 3 (2021, 2056, and 2088 cm−1) can be interpreted as multiple possible CO configurations belonging to either vibrations on multiple Rh crystal surfaces (2056 cm−1) or Rh surfaces and single atoms (2021 and 2088 cm−1). The bridge and linear frequencies for CO on Rh{100} appear for the same CO surface coverages from the surface science literature without forcing the number of peaks or the peak properties (e.g., center frequency or full width at half maximum) suggesting the sizes of the facets developed during the annealing are energetically equivalent to extended Rh surfaces that can hold up to 0.75 monolayers of CO.23–25
As noted, detailed descriptions of the deconvoluted peaks and their frequencies can be found in Tables S1–S3 and Fig. S10–S12. The most important results of the deconvolutions of the FTIR spectra are the physical changes in Rh–CO binding configurations as a function of annealing temperature. These observed changes and their physical meaning regarding Rh NP development are detailed below.
First, there is a noticeable shift in frequency and an increase in intensity for the infrared bands associated with bridging CO (1850–1970 cm−1) on the Rh{100} and Rh{111} surfaces23–26 as the annealing temperature is increased. Table 1 shows these bridging carbonyls can be assigned to Rh{100} surfaces that accommodate surface coverages of θCO = 0.20 and θCO = 0.35–0.40 ML. Our FTIR results suggest Rh{100} surfaces develop as the annealing temperature is increased, which agrees with the Rh{100} surfaces observable in the post-reaction HRTEM images (Fig. 5, S5, S6 and S8).
Second, all deconvoluted spectra exhibit absorption peaks at 2021 cm−1 and 2041 cm−1. The absorption peak at 2041 cm−1 has been observed on Rh{100} also with a surface coverage of θCO = 0.35–0.40 ML further suggesting the existence of Rh{100} facets. The absorption peak around 2020 cm−1 can be associated with CO bound in a linear configuration on Rh{100} with θCO = 0.20 ML thus matching the same surface coverage for the bridging CO at nominally 1887 cm−1. It should be noted the peak at 2020 cm−1 could also be associated with the asymmetric stretch of CO bound to Rh single atoms implying the Rh CS NPs are fragmented by CO with increasing annealing temperature.
Third, the peak at 2056 cm−1 grows from near zero intensity for the catalyst annealed at 400 °C to a dominant peak for the catalyst annealed at 575 °C while the readily observable 2068 cm−1 peak observable at 400 °C decreases to zero with increasing annealing temperature suggesting these peaks are linked to Rh crystallite development. The 2056 cm−1 peak is most appropriately assigned to linear bound CO on Rh{111}25,26 suggesting Rh{111} facets develop with increasing annealing temperature and support for the existence of these Rh{111} facets can be observed in Fig. 5. The broadening of the 2056 cm−1 peak on the low frequency side is ascribed to linear CO absorption peaks on Rh{100} facets in the 2041 cm−1 region. However, the peak at 2068 cm−1 cannot be assigned to CO bound to low index Rh crystal faces. Instead, a peak at 2068 cm−1 has been reported for CO in a linear configuration on small Rh NPs (0.9–1.5 nm).32 These small Rh NPs also exhibited an absorption peak for bridge bound CO at 1868 cm−1 and a discernible absorption peak is observable at 1869 cm−1 in the spectrum for the catalyst annealed at 400 °C (Fig. S10 and Table S1). Typically, small NPs are highly stepped and the adsorption position of CO on (111) stepped surfaces such as (331) and (755) have not been widely reported. The closest report is for CO in a linear configuration on Rh(331) at 2060 cm−1.33 Therefore, it is reasonable to associate this peak at 2069 cm−1 with highly stepped surfaces of Rh{111}, such as {211} and {311} that would be present in irregularly shaped Rh NPs and observable in the HRTEM images in Fig. 5 of Rh/rTiO2 exposed to the RWGS reaction at temperatures less than 575 °C. As noted above, this peak at 2068 cm−1 associated with Rh NPs (0.9–1.5 nm) and stepped Rh{111} surfaces is noteworthy because of its prominence in Fig. 7 for the catalyst annealed at 400 °C while it is absent in the IR spectra of catalysts annealed at 575 °C. Its presence at low temperatures but disappearance at higher annealing temperatures suggests the development of extended Rh structures with increasing annealing temperature. Rh nanoparticles are >1.5 nm in diameter and/or Rh{111} facets are no longer stepped when the 2068 cm−1 peak disappears. The <10 cm−1 shift in frequency can easily be due to oxygen also bound to the Rh surfaces since CO is known to dissociate on {211},34 {331} and {755} steps35 and B5 and B6 defect sites associated with {111} crystal facets.36 The peak at 2068 cm−1 agrees well with a stepped or ‘roughened’ Rh surface as an asymmetrical shaped CO vibrational peak at 2070 cm−1 was observed on roughened Rh.37
Finally, the peak between 2080 and 2100 cm−1 is comprised of two distinct peaks whose intensities shift depending on the annealing temperature. For the sample annealed at 400 °C, the asymmetry of the peak suggests the higher frequency peak is larger in intensity than the lower frequency peak while for the sample annealed at 575 °C, the opposite is true. Peak deconvolution yields peaks at 2088 cm−1 and 2098 cm−1 for catalysts annealed at 400 and 500 °C with only the 2088 cm−1 peak being present with a slightly larger full-width at half maximum (Tables S1–S3) for the sample annealed at 575 °C. The peak at 2098 cm−1 can be assigned to the symmetric absorption vibration of the rhodium gem-dicarbonyl since CO bound to Rh{111} has not been observed at frequencies higher than 2088.4 cm−1.38 Since Rh{111} facets can be observed from the truncated and non-regularly shaped octahedral Rh NPs in the HRTEM images in Fig. 5 and S8, and since it is well known CO can dissociate at stepped {111} and undercoordinated sites in fcc metals,31–33 it is highly plausible the 2088 cm−1 peak can be assigned to CO in a linear configuration on Rh{111} with O also present on the Rh{111} surface. Beyond the TEM results, support for this can be inferred from the 2098 cm−1:2088 cm−1 peak area ratio which decreased from ∼3:1 for the 0.3 wt% Rh/rTiO2 (CS/NP) catalyst annealed at 400 °C to 0 for the catalyst annealed at 575 °C. The loss of the Rh–CO vibrational signature is most definitively associated with the disappearance of Rh single atoms with increasing annealing temperature. These general observations and their changes with increased annealing temperature suggest Rh is dynamic during the annealing process and evolving with increasing annealing temperature.
3.7 Pre-annealing of Rh/rTiO2 (CS/NP) demonstrates time and temperature determine the agnostic behavior of CO selectivity to initial Rh structure
When heating to the reaction temperature (600 °C) under RWGS reaction conditions (Fig. 3) or with the inclusion of a pre-anneal step prior to exposure to RWGS reactions conditions (Fig. 6), the initial CO selectivity is significantly higher because of the annealing step. There are slight differences in the non-annealing and annealing pre-reaction protocols that we eliminate in a revised reaction protocol. In the revised procedure, two 0.3 wt% Rh/rTiO2 (CS/NP) catalysts were oxidized at 400 °C, reduced at 200 °C, and then annealed at 600 °C for 1 h under N2. However, instead of exposing the catalysts to the RWGS environment as was done for annealed catalysts (in Section 3.5), both catalysts were cooled back down to < 40 °C under N2. One of the catalyst samples was exposed to a 3:1 H2:CO2 gas mixture while the other remained under an inert nitrogen environment before both were simultaneously ramped to 600 °C. Once at a reaction temperature of 600 °C, the sample under N2 was exposed to the 3:1 H2:CO2 gas mixture. The CO2 conversion and CO selectivity as function of elapsed time can be seen in Fig. 8b and c respectively. In Fig. 8a, the reactor temperature, oxidation, reduction, and annealing processing steps are shown using the same color scheme as in Fig. 1.
 | ||
| Fig. 8 Influence of heat to reaction temperature environment on RWGS performance. (a) Pretreatment and reaction environment with corresponding thermal profile. (b) CO2 conversion and (c) CO selectivity for two 0.3 wt% Rh/rTiO2 (CS/NP) as a function of reaction time at 600 °C. Both catalysts were exposed to the same pre-reaction conditions of oxidation, reduction, annealing at 600 °C for 1 h, and cooled back to <40 °C before being ramped to reaction temperature. One catalyst was ramped to a reaction temperature of 600 °C under a 3:1 H2:CO2 environment while the other catalyst was ramped to a reaction temperature of 600 °C under N2 and only exposed to the 3:1 H2:CO2 environment after the catalyst bed reached 600 °C. Both samples exhibit very similar CO2 conversion as a function of time and almost identical CO selectivity as a function of time suggesting the changes in the Rh catalyst occurred prior to being exposed to the RWGS on the ramp to the reaction temperature and the RWGS environment does not affect the Rh catalytic performance on the ramp to the reaction. | ||
The catalysts show very similar CO2 conversion (Fig. 8a) and CO selectivity as a function of time once both were under RWGS reaction conditions at 600 °C. The catalyst heated from <40 °C to 600 °C under N2 prior to the start of the RWGS reaction had a noticeable relative improvement of 10+% in CO2 conversion throughout the entire time-on-stream period. Both samples have the same CO selectivity (Fig. 8b) as a function of time despite one catalyst being ramped to the reaction temperature under N2 and the other under RWGS; the similarity in CO selectivity suggests the Rh catalyst is structurally modified during the annealing step and prior to exposure to RWGS reaction conditions. It is not clear why these catalysts started with an initial CO selectivity of >85% rather than >95% as was previously observed for the sample in Fig. 6b that did not undergo cooling back to <40 °C before being ramped back to 600 °C. Both samples started with a substantially higher CO selectivity than the sample shown in Fig. 6b that underwent a 2 h anneal at 500 °C which only had a starting CO selectivity of about 65%. It is evident the annealing of Rh at higher temperatures (600 °C) improves CO selectivity. Annealing of Rh prior to RWGS reaction at 600 °C for as little as 1 h and/or a RWGS reaction temperature more than 560 °C yields a RWGS catalyst that is highly selective to CO and stable with respect to time.
3.8 Invariance of the apparent activation energy for CO formation with the initial Rh structure demonstrates a common active site
After 16 h time-on-stream, temporal changes in CO2 conversion and CO selectivity are rather small, suggesting the reaction has reached steady-state and/or changes in the catalyst structure are minimal. At this point, the samples were cooled via thermal loss under the 3:1 H2:CO2 mixture. During this cool down, CO and CH4 production rates were calculated as a function of temperature to generate an Arrhenius plot. Fig. 9a and c shows the procedure and the Arrhenius plot for a non-annealed 2.0 wt% Rh/rTiO2 (WI) for both CO and CH4 over a temperature range of 390 °C to 240 °C. The apparent activation energy (Ea) for CO production for the 2.0 wt% Rh/rTiO2 (WI) is 57 kJ mol−1. Duplicate measurements of the “cool down” Ea over the non-annealed 2.0 wt% Rh/rTiO2 (WI) showed good reproducibility.
 | ||
| Fig. 9 CO and CH4 rates for (a) non-annealed 2.0 wt% Rh/rTiO2 (WI) and (b) pre-annealed (600 °C, 1 h) 0.1 wt% Rh/rTiO2 (WI) exposed to 3:1 H2:CO2 mixture for 16 h and their associated cool down activation energy plot data, (c) and (d) respectively, as function of time and temperature. The 2.0 wt% Rh/rTiO2 (WI) sample was run at 500 °C and the 0.1 wt% Rh/rTiO2 (WI) sample was run at 600 °C. The data used for the cool down dynamic activation energy plot can be seen in both image (a) and (b) for times greater than 16 h. The activation energy data plot was fit over a temperature range of nominally 240–390 °C where conversion was 0.1–6%. | ||
The 0.3 wt% Rh/rTiO2 (CS/NP) catalyst annealed at 600 °C for 1 h prior to the RWGS reaction in Fig. 8byields a “heat-up” Ea for CO production of 60.1 kJ mol−1 and a “cool down” Ea for CO production of 60.8 kJ mol−1 both of which agree with the post reaction CS/NP average CO activation energy of 60.5 kJ mol−1 for the non-annealed CS/NP samples that were exposed to RWGS at 500 °C for 16 h. This suggests that catalysts which undergo a high temperature (600 °C) pre-annealing step prior to exposure to RWGS reaction conditions obtain a Rh morphology (size and degree of ordering) similar to catalysts exposed to RWGS reaction conditions at 500 °C for 16 h.
The fresh 0.3 wt% Rh/rTiO2 (CS/NP) contains NPs with a mean spherical particle size of 4.5 nm with little to no single atoms (Fig. S4), while the fresh 2.0 wt% Rh/rTiO2 (WI) contains a mixture of single atoms and raft-like Rh NPs (Fig. S4). Upon reaction under 3:1 H2:CO2 at 600 °C for 16 h, both catalysts contain faceted ∼2 nm Rh NPs. Since these catalysts contained a large fraction of NPs in the fresh state (albeit with different structure), we conducted a 600 °C anneal in N2 for 1 h prior to the RWGS reaction on a single atom catalyst, 0.1 wt% Rh/TiO2 (WI). TEM analysis (Fig. 4, S5, S6 and S8) demonstrates that regardless of reaction temperature, Rh NPs of ∼1.8 nm in diameter are observed. However, the HRTEM analysis was unable to observe Rh single atoms, and we anticipate the population of stable Rh single atoms decreases with reaction temperature. The apparent activation energy on Rh single atoms supported on rutile TiO2 is 101 kJ mol−1,28 while the apparent activation energy on the Rh/rTiO2 (CS/NP) catalyst is 60 kJ mol−1. If the 0.1 wt% Rh/rTiO2 (WI) catalyst still contains single atoms after reaction or pre-annealing, the measured apparent activation energy should reflect a contribution from both the single atoms and ∼1.8 nm NPs. We annealed the 0.1 wt% Rh/TiO2 (WI) catalyst at 600 °C in N2 for 1 h, cooled to <40 °C and heated (10 °C min−1) to 600 °C under RWGS reaction conditions, followed by 16 h time-on-stream, and subsequent measurement of the Ea upon “cool-down”. Fig. 9b represents the time on stream CO and CH4 production rates of a pre-annealed (600 °C, 1 h) 0.1 wt% Rh/rTiO2 (WI) catalyst. Upon completion of 16 h RWGS reaction, the catalyst sample was cooled down via thermal losses from the furnace. Fig. 9d is the Arrhenius plot for the annealed 0.1 wt% Rh/rTiO2 (WI). The value for the Ea for CO production is 57.3 kJ mol−1. A second measurement on a separate bed of the annealed 0.1 wt% Rh/rTiO2 (WI) yielded an apparent Ea of 56 kJ mol−1. The measured Ea during the cool down after 16 h time-on-stream suggests Rh NPs are responsible for CO formation even though the initial Rh structure was dominated by single atoms. The measured apparent activation energy for the RWGS reaction over Rh NP/TiO2 (P25) has been reported to be 55 kJ mol−1 for a 4:1 H2:CO2 ratio.39 The measured apparent activation energy in this work at a 3:1 H2:CO2 ratio is similar, but significantly lower than the value (101 kJ mol−1) reported on rutile-supported single atoms of Rh.28 A value of 55–60 kJ mol−1 for the apparent activation energy appears to be independent of the TiO2 polymorph (rutile versus anatase).39
The apparent Ea for CH4 formation for the 2.0 wt% Rh/rTiO2 (WI) catalyst in a 3:1 H2:CO2 mixture upon cool down is ∼102.8 kJ mol−1 (Fig. 9c), while the Ea upon cool down on the pre-annealed 0.1 wt% Rh/rTiO2 (WI) is 99.7 kJ mol−1 (Fig. 9d). Both experiments were run in duplicate, and the reproducibility between individual catalyst beds was good. These values for the apparent Ea for methane formation agree well with the reported value of Verykios and co-workers of 103 kJ mol−1.39 Reported Ea values for CO2 hydrogenation to CH4 in a 3:1 H2:CO2 mixture over a Rh foil40,41 or an inverse TiO2/Rh foil catalyst42 are in the range of 130–140 kJ mol−1, while a value of 100 kJ mol−1 was reported over an oxidized Rh foil.41 The latter value agrees well with the apparent activation energy for CH4 formation reported here and by Verykios et al.39 The difference in the activation energy reported here compared with those from a Rh foil suggest that undercoordinated sites or tightly bound atomic oxygen at those undercoordinated sites on the Rh nanoparticle surface may be critical for the kinetically relevant CO hydrogenation step.
4. Conclusions
Rh single atoms supported on rutile TiO2 are responsible for CO formation during the reverse water gas shift reaction at low temperatures (<400 °C). As the reaction temperature is increased, the formation of CH4 is observed which is associated with the growth of Rh nanoparticles and the formation of a Rh–TiO2 interface, the presumed location of the active site for CH4 formation. The Rh nanoparticles formed at low temperature are primarily disordered, and CO IR spectroscopy on these catalysts confirms significant Rh speciation (single atoms and nanoparticles). Upon heating to higher temperatures (550–600 °C), the formation of methane decreases significantly even though nanoparticles and their interface with TiO2 persist. At higher temperatures, the particles become more ordered regardless of their initial structure (single atoms or nanoparticles), and IR spectroscopy of adsorbed CO suggests the formation of CO–Rh terrace atom configurations. At a reaction temperature of 600 °C under RWGS conditions (3:1 H2:CO2), the selectivity to CO for all Rh/rTiO2 catalysts is >95%, and the apparent activation energy for multiple Rh/rTiO2 with different initial Rh structures, either pre-annealed at 600 °C or post RWGS (600 °C for 16 h) was 55–60 kJ mol−1, in good agreement with reported activation energies measured on nanoparticle Rh/rTiO2 catalysts. The development of faceted Rh nanoparticles leads to the predominant formation of CO at higher reaction temperatures. Single atoms may persist at these higher temperatures, but the predominance of adsorbed CO associated with extended Rh surfaces and the agreement in measured apparent activation energy on nanoparticle-based Rh/rTiO2 catalysts suggest single atoms are kinetically irrelevant at reaction temperatures of ∼600 °C. This study demonstrates detailed examination of catalyst activation is critical for the prediction of catalyst performance with time on stream.
Author contributions
GB: conceived the initial research idea, catalyst testing, data analysis, original draft writing. XC, AK, JH, SG, LM, DZ: catalyst synthesis, catalyst characterization, discussions, writing and editing. JY, AH, MC, and RR: research supervision, writing and editing. SRB: funding acquisition, project administration, resources, supervision, writing and editing.Conflicts of interest
There are no conflicts of interest to declare.Data availability
All data supporting the findings of this investigation are incorporated within the publication and the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5fd00172b.Acknowledgements
Selin Bac and Seunghwa Hong (UC Santa Barbara) are thanked for catalyst synthesis and helpful discussion and creation of the TOC. Jiyun Hong (SLAC) is acknowledged for help with the references. The work was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, US Department of Energy (DOE) as part of the Accelerate Innovations in Emerging Technologies Initiative, FWP 101064. This research used the Electron Microscopy and Proximal Probes Facility of the Center for Functional Nanomaterials (CFN), which is a U.S. Department of Energy Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704.Notes and references
- J. Xie, P. P. Paalanen, T. W. van Deelen, B. M. Weckhuysen, M. J. Louwerse and K. P. de Jong, Promoted Cobalt Metal Catalysts Suitable for the Production of Lower Olefins from Natural Gas, Nat. Commun., 2019, 10(1), 167, DOI:10.1038/s41467-018-08019-7.
- M. E. Dry, The Fischer–Tropsch Process: 1950–2000, Catal. Today, 2002, 71(3–4), 227–241, DOI:10.1016/S0920-5861(01)00453-9.
- N. Fischer, E. Van Steen and M. Claeys, Structure Sensitivity of the Fischer–Tropsch Activity and Selectivity on Alumina Supported Cobalt Catalysts, J. Catal., 2013, 299, 67–80, DOI:10.1016/J.JCAT.2012.11.013.
- Y. A. Daza and J. N. Kuhn, CO2 Conversion by Reverse Water Gas Shift Catalysis: Comparison of Catalysts, Mechanisms and Their Consequences for CO2 Conversion to Liquid Fuels, RSC Adv., 2016, 6(55), 49675–49691, 10.1039/C6RA05414E.
- J. C. Matsubu, V. N. Yang and P. Christopher, Isolated Metal Active Site Concentration and Stability Control Catalytic CO2 Reduction Selectivity, J. Am. Chem. Soc., 2015, 137(8), 3076–3084, DOI:10.1021/JA5128133.
- H. Guan, J. Lin, B. Qiao, S. Miao, A. Q. Wang, X. Wang and T. Zhang, Enhanced Performance of Rh1/TiO2 Catalyst without Methanation in Water-Gas Shift Reaction, AIChE J., 2017, 63(6), 2081–2088, DOI:10.1002/AIC.15585.
- J. H. Kwak, L. Kovarik and J. Szanyi, CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity, ACS Catal., 2013, 3(11), 2449–2455, DOI:10.1021/CS400381F.
- A. Aitbekova, L. Wu, C. J. Wrasman, A. Boubnov, A. S. Hoffman, E. D. Goodman, S. R. Bare and M. Cargnello, Low-Temperature Restructuring of CeO2-Supported Ru Nanoparticles Determines Selectivity in CO2 Catalytic Reduction, J. Am. Chem. Soc., 2018, 140(42), 13736–13745, DOI:10.1021/JACS.8B07615.
- M. M. Millet, G. Algara-Sille, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. V. Levchenko, R. Schlögl and E. Frei, Ni Single Atom Catalysts for CO2 Activation, J. Am. Chem. Soc., 2019, 141(6), 2451–2461, DOI:10.1021/JACS.8B11729.
- E. K. Schroeder, S. Hong, X. Chen, A. S. Hoffman, Z. Chen, A. Khan, G. D. Barber, S. Bac, R. M. Rioux, J. Yang, C. J. Tassone, S. R. Bare and P. Christopher, Structural Evolution and Stability of Rh/TiO2 Catalysts under CO2 Hydrogenation Conditions: Influence of the Initial Rh Structure, ACS Catal., 2025, 12133–12147, DOI:10.1021/ACSCATAL.5C02205.
- S. Bac, D. Shin, S. Hong, J. Heinlein, A. Khan, G. Barber, Z. Chen, M. M. Albrechtsen, C. Tassone, R. M. Rioux, M. Cargnello, S. R. Bare, K. Winther, P. Christopher and A. S. Hoffman, Quantifying Experimental Uncertainty in Catalyst Deactivation: Round Robin Testing and Implications for Machine-Learned Prediction, 2025, submitted.
- P. H. Chung, A. C. Yang, C. Zhou, J. Oh, M. Homer, C. Lizandara-Pueyo, Y. Li and M. Cargnello, Aqueous-Phase Synthesis of Pt and PGM-Based Nanocrystals with a Controllable Size, Cryst. Growth Des., 2024, 24(24), 10413–10422, DOI:10.1021/ACS.CGD.4C01416.
- L. Li, L. L. Wang, D. D. Johnson, Z. Zhang, S. I. Sanchez, J. H. Kang, R. G. Nuzzo, Q. Wang, A. I. Frenkel, J. Li, J. Ciston, E. A. Stach and J. C. Yang, Noncrystalline-to-Crystalline Transformations in Pt Nanoparticles, J. Am. Chem. Soc., 2013, 135(35), 13062–13072, DOI:10.1021/JA405497P/SUPPL_FILE/JA405497P_SI_002.QT.
- C. N. Eads, J. A. Boscoboinik, A. R. Head, A. Hunt, I. Waluyo, D. J. Stacchiola and S. A. Tenney, Enhanced Catalysis under 2D Silica: A CO Oxidation Study, Angew. Chem., Int. Ed., 2021, 60(19), 10888–10894, DOI:10.1002/ANIE.202013801.
- C. G. Granqvist and R. A. Buhrman, Size Distributions for Supported Metal Catalysts: Coalescence Growth versus Ostwald Ripening, J. Catal., 1976, 42(3), 477–479, DOI:10.1016/0021-9517(76)90125-1.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Sintering of Catalytic Nanoparticles: Particle Migration or Ostwald Ripening?, Acc. Chem. Res., 2013, 46(8), 1720–1730, DOI:10.1021/AR3002427.
- P. Yin, S. Hu, K. Qian, Z. Wei, L. L. Zhang, Y. Lin, W. Huang, H. Xiong, W. X. Li and H. W. Liang, Quantification of Critical Particle Distance for Mitigating Catalyst Sintering, Nat. Commun., 2021, 12(1), 4865, DOI:10.1038/s41467-021-25116-2.
- Y. Li, D. Zakharov, S. Zhao, R. Tappero, U. Jung, A. Elsen, P. Baumann, R. G. Nuzzo, E. A. Stach and A. I. Frenkel, Complex Structural Dynamics of Nanocatalysts Revealed in Operando Conditions by Correlated Imaging and Spectroscopy Probes, Nat. Commun., 2015, 6(1), 7583, DOI:10.1038/ncomms8583.
- W. Yu, S. Yue, M. Yang, M. Hashimoto, P. Liu, L. Zhu, W. Xie, T. Jones, M. Willinger and X. Huang, Operando TEM Study of a Working Copper Catalyst during Ethylene Oxidation, Nat. Commun., 2025, 16(1), 2029, DOI:10.1038/s41467-025-57418-0.
- A. J. Hoffman, C. Asokan, N. Gadinas, P. Kravchenko, A. “Bean” Getsoian, P. Christopher and D. Hibbitts, Theoretical and Experimental Characterization of Adsorbed CO and NO on γ-Al2O3-Supported Rh Nanoparticles, J. Phys. Chem. C, 2021, 125(36), 19733–19755, DOI:10.1021/ACS.JPCC.1C05160.
- A. J. Hoffman, C. Asokan, N. Gadinas, E. Schroeder, G. Zakem, S. V. Nystrom, A. B. Getsoian, P. Christopher and D. Hibbitts, Experimental and Theoretical Characterization of Rh Single Atoms Supported on γ-Al2O3 with Varying Hydroxyl Contents during NO Reduction by CO, ACS Catal., 2022, 12(19), 11697–11715, DOI:10.1021/ACSCATAL.2C02813.
- C. Asokan, H. V. Thang, G. Pacchioni and P. Christopher, Reductant Composition Influences the Coordination of Atomically Dispersed Rh on Anatase TiO2, Catal. Sci. Technol., 2020, 10(6), 1597–1601, 10.1039/D0CY00146E.
- L. W. H. Leung, J. W. He and D. W. Goodman, Adsorption of CO on Rh(100) Studied by Infrared Reflection–Absorption Spectroscopy, J. Chem. Phys., 1990, 93(11), 8328–8336, DOI:10.1063/1.459316.
- A. M. De Jong and J. W. Niemantsverdriet, The Adsorption of CO on Rh(100): Reflection Absorption Infrared Spectroscopy, Low Energy Electron Diffraction, and Thermal Desorption Spectroscopy, J. Chem. Phys., 1994, 101(11), 10126–10133, DOI:10.1063/1.468001.
- R. Linke, D. Curulla, M. J. P. Hopstaken, J. W. Niemantsverdriet, D. Curulla, M. J. P. Hopstaken and J. W. Niemantsverdriet, CO/Rh(111): Vibrational Frequency Shifts and Lateral Interactions in Adsorbate Layers, J. Chem. Phys., 2001, 115(17), 8209–8216, DOI:10.1063/1.1355767.
- G. Krenn, I. Bako and R. Schennach, CO Adsorption and CO and O Coadsorption on Rh(111) Studied by Reflection Absorption Infrared Spectroscopy and Density Functional Theory, J. Chem. Phys., 2006, 124(14), 144703, DOI:10.1063/1.2184308/929455.
- L. H. Dubois and G. A. Somorjai, The Chemisorption of CO and CO2 on Rh(111) Studied by High Resolution Electron Energy Loss Spectroscopy, Surf. Sci., 1980, 91(2–3), 514–532, DOI:10.1016/0039-6028(80)90348-9.
- Y. Tang, C. Asokan, M. Xu, G. W. Graham, X. Pan, P. Christopher, J. Li and P. Sautet, Rh Single Atoms on TiO2 Dynamically Respond to Reaction Conditions by Adapting Their Site, Nat. Commun., 2019, 10(1), 4488, DOI:10.1038/s41467-019-12461-6.
- C. Asokan, M. Xu, S. Dai, X. Pan and P. Christopher, Synthesis of Atomically Dispersed Rh Catalysts on Oxide Supports via Strong Electrostatic Adsorption and Characterization by Cryogenic Infrared Spectroscopy, J. Phys. Chem. C, 2022, 126(44), 18704–18715, DOI:10.1021/ACS.JPCC.2C05426.
- I. Swart, F. M. F. De Groot, B. M. Weckhuysen, D. M. Rayner, G. Meijer and A. Fielicke, The Effect of Charge on CO Binding in Rhodium Carbonyls: From Bridging to Terminal CO, J. Am. Chem. Soc., 2008, 130(7), 2126–2127, DOI:10.1021/JA0772795.
- F. C. Meunier, Relevance of IR Spectroscopy of Adsorbed CO for the Characterization of Heterogeneous Catalysts Containing Isolated Atoms, J. Phys. Chem. C, 2021, 125(40), 21810–21823, DOI:10.1021/ACS.JPCC.1C06784.
- M. A. Albrahim, A. Shrotri, R. R. Unocic, A. S. Hoffman, S. R. Bare and A. M. Karim, Size-Dependent Dispersion of Rhodium Clusters into Isolated Single Atoms at Low Temperature and the Consequences for CO Oxidation Activity, Angew. Chem., Int. Ed., 2023, 62(44), e202308002, DOI:10.1002/ANIE.202308002.
- Z. Wang, X. Pang and R. Wang, Adsorption of CO Molecules on Rh Low Index and (331) Stepped Surfaces, Chin. Sci. Bull., 2004, 49(1), 101012–101019, DOI:10.1360/03wb0127.
- D. Degerman, M. Shipilin, P. Lömker, C. M. Goodwin, S. M. Gericke, U. Hejral, J. Gladh, H. Y. Wang, C. Schlueter, A. Nilsson and P. Amann, Operando Observation of Oxygenated Intermediates during CO Hydrogenation on Rh Single Crystals, J. Am. Chem. Soc., 2022, 144(16), 7038–7042, DOI:10.1021/JACS.2C00300.
- D. G. Castner and G. A. Somorjai, LEED and Thermal Desorption Studies of Small Molecules (H2, O2, CO, CO2, NO, C2H4, C2H2 and C) Chemisorbed on the Stepped Rhodium (755) and (331) Surfaces, Surf. Sci., 1979, 83(1), 60–82, DOI:10.1016/0039-6028(79)90480-1.
- D. A. J. M. Ligthart, I. A. W. Filot, A. A. H. Almutairi and E. J. M. Hensen, Identification of Step-Edge Sites on Rh Nanoparticles for Facile CO Dissociation, Catal. Commun., 2016, 77, 5–8, DOI:10.1016/J.CATCOM.2016.01.006.
- J. W. He, W. K. Kuhn, L. W. H. Leung and D. W. Goodman, Infrared Vibrational Studies of CO Adsorption on Ultrathin Cu Films on a Rh(100) Surface, J. Chem. Phys., 1990, 93(10), 7463–7470, DOI:10.1063/1.459420.
- E. A. Jamka, M. Z. Gillum, C. N. Grytsyshyn-Giger, F. J. Lewis and D. R. Killelea, Temperature-Resolved Surface Infrared Spectroscopy of CO on Rh(111) and (2 × 1)-O/Rh(111), J. Vac. Sci. Technol., A, 2022, 40(4), 43209, DOI:10.1116/6.0001932/2846457.
- Z. Zhang, A. Kladi and X. E. Verykios, Effects of Carrier Doping on Kinetic Parameters of CO2 Hydrogenation on Supported Rhodium Catalysts, J. Catal., 1994, 148(2), 737–747, DOI:10.1006/JCAT.1994.1260.
- B. A. Sexton and G. A. Somorjai, The Hydrogenation of CO and CO2 over Polycrystalline Rhodium: Correlation of Surface Composition, Kinetics and Product Distributions, J. Catal., 1977, 46(2), 167–189, DOI:10.1016/0021-9517(77)90198-1.
- D. G. Castner, R. L. Blackadar and G. A. Somorjai, CO Hydrogenation over Clean and Oxidized Rhodium Foil and Single Crystal Catalysts. Correlations of Catalyst Activity, Selectivity, and Surface Composition, J. Catal., 1980, 66(2), 257–266, DOI:10.1016/0021-9517(80)90030-5.
- K. J. Williams, A. B. Boffa, M. Salmeron, A. T. Bell and G. A. Somorjai, The Kinetics of CO2 Hydrogenation on a Rh Foil Promoted by Titania Overlayers, Catal. Lett., 1991, 9(5–6), 415–426, DOI:10.1007/BF00764834.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2026 |