 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of electrolyte concentration on the depth-dependent structural anisotropy of water at charged interfaces
Álvaro Díaz Duque,
Sarabjeet Kaur,
Martin Wolf ,
Alexander P. Fellows and
Martin Thämer*
,
Alexander P. Fellows and
Martin Thämer*
Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195, Berlin, Germany. E-mail: thaemer@fhi-berlin.mpg.de; Tel: +49 (0)30 8413 5220
First published on 5th February 2026
Abstract
Revealing the structural properties of water at charged interfaces is key for a better understanding of interfacial processes in various fields such as atmospheric chemistry, biology, and electrochemistry. One important aspect of the interfacial water structure in the presence of surface charges is its evolution with depth and how it varies with electrolyte concentration, which is still largely unknown despite various experimental efforts. In this work we investigate the anisotropic water structure in contact with insoluble charged surfactants using our recently developed depth-resolved vibrational spectroscopy which is based on a combination of phase resolved sum- and difference-frequency generation spectroscopy. By probing the line shape of the O–H stretch vibration of water in the first solvation layers of the surface charges and in interfacial regions further away from the phase boundary we obtain detailed insight into the field-induced orientational anisotropy and hydrogen-bonding properties of water molecules inside the electrical double layer. We find that the properties of the hydrogen-bond network in terms of hydrogen-bond strength and connectivity are nearly unaffected by the anisotropic molecular orientation. This is shown to hold throughout the double layer and for all measured electrolyte concentrations. The data, however, reveals significant changes of more than 40% in the amount of orientational anisotropy close to the interface as function of ionic strength, directly opposing a common and crucial assumption made in such investigations.
Introduction
Interfaces between charged surfactants and aqueous electrolytes play a central role in biological, atmospheric, and electro-chemistry.1–6 In the atmosphere, surfactant films on aqueous aerosols regulate the uptake of halide radicals or reactive oxygen species (ROS), while also modulating the interfacial distribution of solvated ions.3,4,7 In biological systems, the ionic distribution at the interface influences membrane potentials and processes such as cell signaling.5,6 At the heart of these phenomena lie the physico-chemical properties of the electrochemical double layer formed at the phase boundary, whose major constituent is water. Numerous studies have shown that interfacial water exhibits a structural organization distinct from that of the bulk, including preferential dipole orientation induced by the presence of the electrostatic field and the formation of specific structural motifs.8–11 These structural phenomena can be accompanied by modifications in strength and connectivity of the interfacial hydrogen-bond network, which not only determines the stabilization of solvated species but also governs the hydration forces that drive the self-assembly of surfactants into specific macrostructures. The extent to which hydrogen-bonding is enhanced or perturbed dictates the stabilization or exclusion of ions and solutes, ultimately controlling interfacial reactivity, transport, and aggregation.12–14 Gaining detailed insight into the structure of water near charged surfactants is therefore essential for understanding the molecular mechanisms that govern aqueous interfacial behavior.As mentioned above, the presence of surface charges makes the water structure in the subsurface region anisotropic with an important aspect of this anisotropy being its evolution with depth. The anisotropy of the molecular structure induced by the charges somehow decays to zero towards the isotropic bulk on a length scale that is largely dictated by the ion concentration of the electrolyte via screening effects.7,11,15 High concentration electrolytes lead to a fast decay of the anisotropy whereas solutions with low salt concentrations can yield decay lengths on the order of 100s of nm. The anisotropy decay at these charged interfaces can generally not be described by a single exponential decay but must typically be separated into two distinct regions: (i) a very thin water layer (ca. 1 nm) that is in direct contact with the surface charges (often labelled as the compact layer, Stern layer, or bonded interfacial layer, BIL) followed by (ii) a potentially much thicker subsurface region with exponentially decaying anisotropy (diffuse layer, DL). The water structure in these two regions can significantly differ with important impact on the mechanistic details of interfacial processes.8,9,11,16,17 It is therefore important to accurately determine the water structure within the electrical double layer as a function of depth.
Among the various experimental techniques to study interfacial water, vibrational sum-frequency generation (SFG) spectroscopy is particularly well suited.11,15,16,18–34 SFG combines high chemical specificity by probing vibrational resonances, with intrinsic sensitivity to molecular orientation which governs the sign of the spectral response. This particularity of second-order nonlinear techniques makes SFG sensitive to structural anisotropy as no sum-frequency signal arises from isotropic (bulk) regions due to cancellation. That way SFG allows for exclusively probing the (anisotropic) interfacial region at aqueous interfaces. Furthermore, analyzing the vibrational line shape of the water response yields valuable information on the hydrogen-bond environment. As the resonance frequency of the O–H stretch vibration is highly sensitive to the number and strength of hydrogen-bonds, any significant variations of the network in the interfacial region become apparent in the spectra.20,35 Consequently, SFG combines selectivity to the anisotropic interfacial region with sensitivity to the hydrogen-bond environment. This capability has established SFG as a powerful tool for probing charged aqueous phase boundaries.
Despite these unique properties of the technique, the elucidation of the depth-dependent water structure solely based on measured SFG spectra from charged aqueous interfaces remains challenging. The reason for this difficulty is the fact that the measured spectra represent the depth-integrated response of the anisotropic region. This makes it difficult to decompose the measured spectra into contributions from the BIL and DL and analyze the spectral responses within these distinct regions separately. To overcome this limitation and to isolate the BIL and DL contributions, several strategies have been developed, either purely spectroscopic or combining spectroscopy with complementary techniques.18,21,36–38 One commonly used approach first identifies the BIL spectrum by performing SFG experiments under conditions of high ionic strength where the DL becomes significantly compressed or collapses. Once the BIL contribution is determined, the DL at different salt concentrations can be determined by simple subtraction of the BIL contribution.36,39,40 Within this method it is assumed that the BIL contribution is independent of the salt concentration of the electrolyte and remains constant for these experiments. An alternative approach uses a reference spectrum of water in the DL that has previously been published in combination with additional experiments (e.g. measurement of the zeta-potential) to model the DL contribution to the measured SFG spectra.21,37,38,41 Subtraction of the obtained DL contribution from the overall SFG spectrum yields also here the desired BIL spectra.
These approaches have proven useful, for instance, in determining the sign of the imaginary part of the DL contribution, thereby revealing trends in the average dipole orientation of water molecules within the BIL and DL regions. Based on such analyses, several impactful conclusions about water structure at charged interfaces have been drawn. For example, it has been shown that the water structures in the BIL and DL differ substantially under pH conditions where surface charging is significant, and that structural anisotropy is predominantly localized in the BIL, while water reorientation within the DL is considerably weaker.18,42 While these results have greatly advanced our understanding of water structure in the electrochemical double layer, it is important to note that the approaches described above rely on assumptions which have so far not been experimentally verified. For the first method to be accurate it is required that SFG contributions from the BIL remain unchanged under varying electrolyte concentrations. While this assumption might seem generally reasonable, several observations from recent studies make its validity questionable. Among these observations are the concentration-dependent contraction of BIL,43 large deviations of the dielectric constant of water close to the phase boundary44 and concentration-dependent dissociation of the surfactants which modulate the surface charge.45 Similarly, the accuracy of the second method described above obviously depends to a large extent on the correct amplitude and line shape of the reference DL spectrum, which has yet to be confirmed. In consequence, it is essential for the validation of past and future SFG studies on charged aqueous interfaces to perform such studies with a method that does not require the above assumptions and/or experimentally test these assumptions and the accuracy of the reference DL spectrum.
Recently, our group developed a depth-resolved nonlinear vibrational spectrometer capable of isolating the BIL and DL spectral contributions to the effective susceptibility from charged aqueous interfaces within a single experiment.42,46,47 In contrast to previous techniques,29,37,48–51 our methodology offers precise depth resolution on the nm scale and enables the direct separation of the BIL and DL responses without altering the chemical composition of the electrolyte (such as ionic strength or pH), thereby preserving the complete physicochemical identity of the sample. As a result, no assumptions are required regarding the invariance of the interfacial structure under different ionic environments. Furthermore, the separation can be done without any modeling of the depth-dependent signal contributions inside the BIL and without the use of a reference DL spectrum which makes the method largely free from model dependent bias and little susceptible to systematic errors. In a previous publication, we demonstrated this capability at a low ionic strength (10−5 M NaCl), allowing for the first time a direct comparison of the BIL and DL spectra under the same experimental conditions.42 In the present contribution, we extend these measurements across a broader range of electrolyte concentrations at a negatively charged surfactant–water interface and study the variation of amplitude and line shape in BIL and DL spectra as a function of ionic strength. Based on these data, we analyze the water structure in these two distinct interfacial regions and obtain a refined picture of the depth-dependent water anisotropy. The results also allow us to verify the validity of the assumptions described above to isolate the BIL spectrum. Furthermore, we determine the intrinsic χ(3) spectrum of interfacial water from our measurements and compare it to the previously reported and widely used reference spectrum. Because of the importance of SFG spectroscopy in the investigation of water structures at various charged interfaces and the clear impact of the results in the field of interfacial water science this study represents an important step towards a more reliable interpretation of measured spectra.
Theory
The induced structural anisotropy of water at charged aqueous interfaces contains two distinct electrostatic regions within the electrical double layer: one arising from the strong chemical and electrostatic interactions of water molecules with the nearby surface charges (BIL), and another from their interaction with a more deeply penetrating screened electric field (DL). The effective susceptibility χ(2)eff that governs the measured SFG response can therefore be expressed as follows:29,42
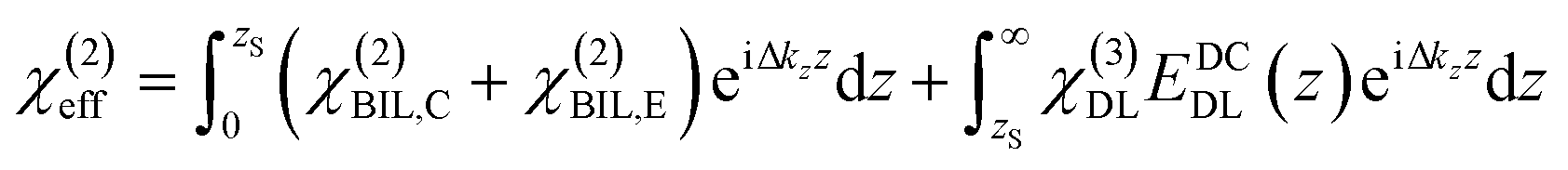 | (1) |
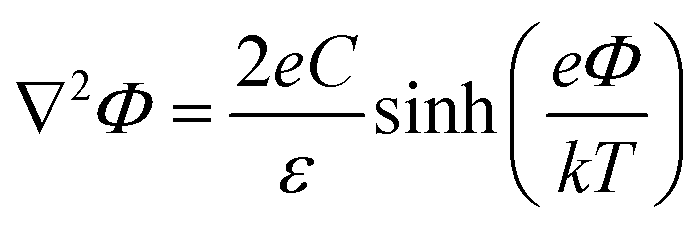 | (2) |
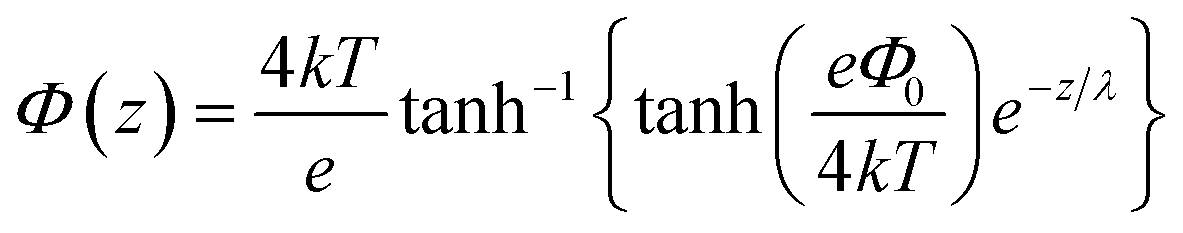 | (3) |
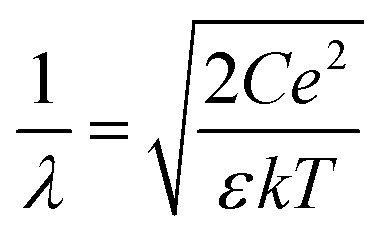 | (4) |
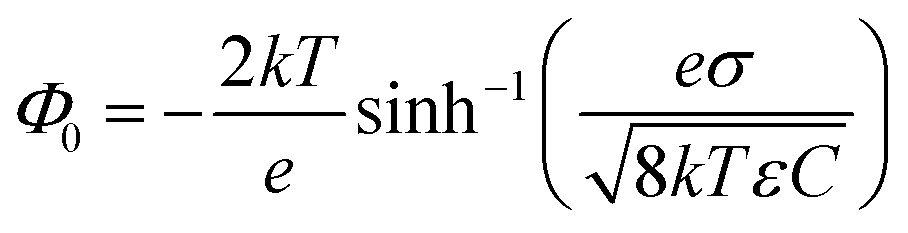 | (5) |
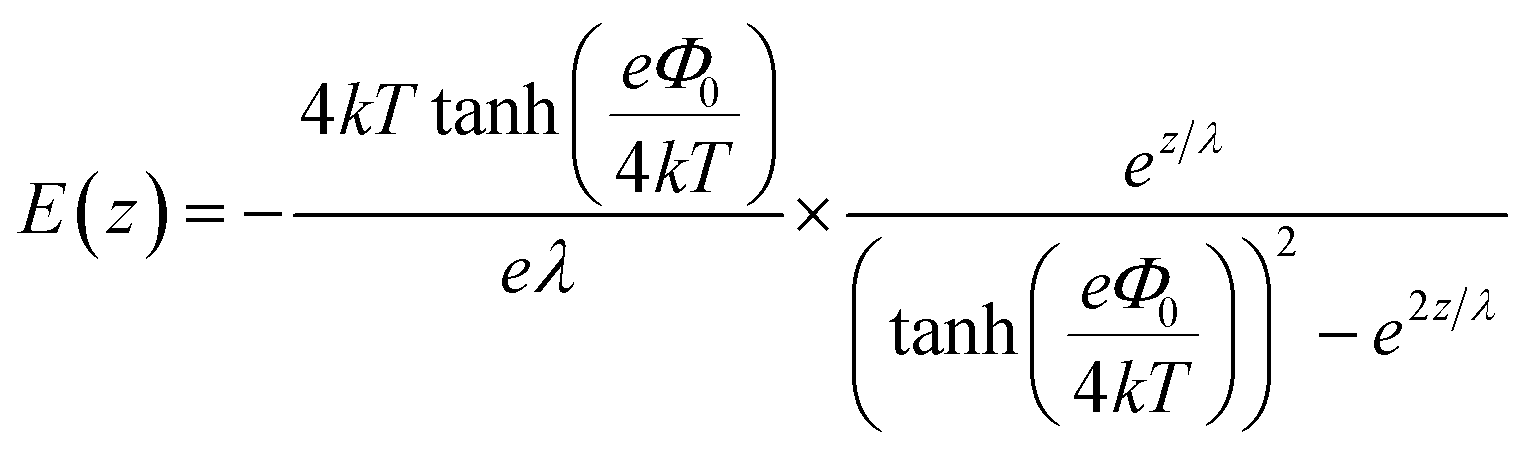 | (6) |
| EDCDL(z,λ) = Ez=zSe−z/λ | (7) |
In contrast, deriving an accurate expression for the SFG contribution from the BIL is much more complex and hardly feasible. This has two main reasons: firstly, as discussed above, an accurate prediction of the evolution of the electrostatic field in the BIL is impossible within the framework of the Poisson–Boltzmann equation, and secondly, even if we precisely knew EDCBIL(z) it would be unclear how to relate the DC field to the nonlinear spectroscopic response. The electrostatic field strengths in the BIL are in the range of 107–108 V m−1, which are so high that it is unreasonable to assume a linear relation between DC field strength and amount of dipolar alignment of water molecules. This already becomes clear by the observation of a strongly decreasing dielectric constant of water within the BIL. As the dielectric constant in water is dominated by the polarization that arises from water reorientation, the dramatic drop of this constant in the BIL shows a strongly reduced tendency of water to further increase its dipolar alignment inside the BIL. In consequence the field-dependent SFG response in the BIL cannot be described by a simple product χ(3)BILEDCBIL(z) which is why we describe it by the effective response χ(2)BIL,E, where the subscript simply indicates the dependency of the response on the DC field without specifying the specific functional relation. Overall, as shown above, predicting the evolution of the SFG signal and thus the anisotropic water structure in the BIL from theory is highly challenging, again underlining the importance of experimental studies.
Experimental insight into the anisotropic water structure in the BIL using SFG spectroscopy requires separation of the contributions from the BIL and DL (eqn (1)). As shown in previous publications, this can be achieved using the SFG/DFG technique.29,42 Eqn (1) shows that contributions from increasing depth acquire a phase shift (approx. 1 degree per nm)47 originating from the propagation phase factor eiΔkzz. Since Δkz differs in sign and magnitude for SFG and DFG responses, these phase shifts have opposite direction yielding the desired depth information.46,47 For the system described above we can safely neglect the phase factor for the contribution in the BIL because of its very small thickness (<1 nm). With 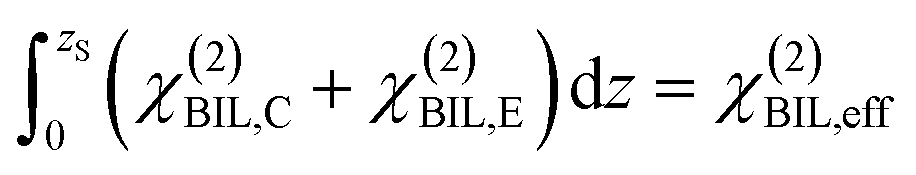 and using the relation
and using the relation 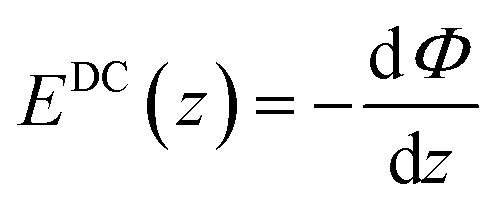 we can obtain following equation
we can obtain following equation
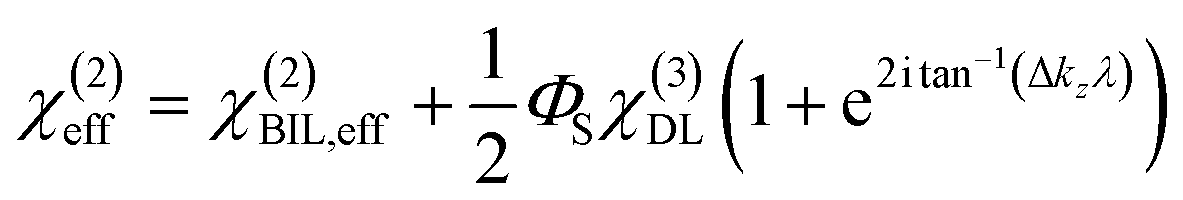 | (8) |
| Δχ(2)eff = ΦSχ(3)DL·G(λ,ΔkSFGz,ΔkDFGz) | (9) |
 | ||
| Fig. 1 SFG/DFG measurements at charged aqueous interfaces: (a) schematic representation of the sample system along with a sketch of the depth-dependent potential induced by the surface charges. The evolution of the potential with depth is divided into two regimes, (i) an exponentially decaying tail at larger depths (purple, DL) and (ii) a much steeper evolution close to the phase boundary (orange, BIL). The exponential decay is given by the Debye length (λ) and is dependent on salt concentration. (b) Argand diagram showing the phase relationship between SFG and DFG. In the BIL region (orange arrow), SFG and DFG contributions are equal in phase and amplitude, whereas in the DL (purple arrow), the additional beam propagation induces phase shifts in opposite directions for SFG (red arrow) and DFG (blue arrow). (c) SFG/DFG spectra at all investigated salt concentrations including the corresponding Debye lengths λ. For decreasing salt concentration (larger λ), the phase difference between the SFG and DFG signals becomes increasingly pronounced. | ||
Results
Fig. 1(c) shows the effective SFG (red) and DFG (blue) spectra (imaginary parts of the second-order susceptibility, χ(2)eff) of a negatively charged aqueous interface measured at different ionic strengths of the electrolyte. The surfactant used in this study is dihexadecyl phosphate (DHP), and the electrolyte is aqueous NaCl (for experimental details, see the SI). In these spectra, the vibrational transitions associated with the O–H stretching modes of interfacial water appear between 3000 and 3650 cm−1, while at lower frequencies, the three sharp features at 2877, 2936, and 2961 cm−1 correspond to the vibrational modes from the terminal CH3 groups of the surfactant tails aliphatic chains of the surfactant tails.42 At high concentrations of the electrolyte (10−1 M, λ = 0.96 nm), the SFG and DFG line shapes nearly overlap, showing that the orientational anisotropy is here confined in a very small thickness. As the ionic strength is decreased, however, deviations arise selectively in the hydrogen-bond stretching region, culminating in markedly dissimilar SFG and DFG profiles at the lowest concentration investigated (10−5.5 M, λ = 171 nm). These divergences in line shape reflect a phase difference between the SFG and DFG signals, indicating that the effective response includes signals from much larger depths. This behavior is a direct consequence of the concentration-dependent decay length of the interfacial electrostatic field into the bulk.7,15 This phase effect is illustrated in the Argand diagram in Fig. 1(b), where the BIL contribution (orange arrow) is indistinguishable between SFG and DFG, whereas the DL contribution (purple arrow) phase-rotates with depth for SFG (red) and DFG (blue). As illustrated in Fig. 1(a), the negatively charged surfactant headgroups form a plane of fixed charge upon proton dissociation, generating an electrostatic field, oriented perpendicular to the surface. This field modulates the anisotropy of interfacial water by aligning molecular dipoles through dipole–field interactions,30 producing the depth-dependent spectral variations observed in SFG and DFG. Hence, probing the depth-dependent water anisotropy allows for deducing the evolution of the electrostatic field with depth, as long as the water anisotropy scales linearly with EDC(z), meaning that the signal is dominated by the χ(3) terms in eqn (1). This condition is fulfilled in the DL as discussed in the previous section.Using the equations shown in the theory section, the presented data is analyzed and decomposed into their different contributions. Fig. 2(a) shows the obtained imaginary spectra of the isolated ΦSχ(3)DL at different ionic strengths, ranging from 10−4 to 10−5.5 M. All spectra exhibit an almost identical line shape, differing only in amplitude, which increases systematically with dilution across the explored concentration range. This similarity becomes even clearer in Fig. 2(b), where all spectra are normalized for direct comparison. These results indicate that variations in electrolyte concentration exert a negligible influence on the overall hydrogen-bond structure of water within the DL across the studied ionic strengths, confirming expectations. This behavior also shows that the influence of the ions on the water structure at the investigated electrolyte concentrations is rather negligible. In Fig. 2(c) the respective isolated BIL spectra are depicted which show excellent overlap of the CH3 stretching modes for the different ionic strengths measured. Also, the vibrational line shape of the water bands is clearly not altered between the different measurements, the water responses only deviate in amplitude. This shows that the hydrogen-bond structure in the BIL seems to be not affected by changes in ion concentration similar to the corresponding observation in the DL.
 | ||
| Fig. 2 Analysis of the measured SFG/DFG data. (a) Isolated vibrational spectra (imaginary parts) from DL and (c) BIL for different ionic strengths. The DL spectra correspond to the term ΦSχ(3)DL and the BIL spectra to χ(2)BIL,eff. For better clarity only the 4 spectra with the lowest concentration are shown. A figure containing all concentrations is shown in the SI. (b) Normalized DL spectra. (d) Comparison of normalized BIL and DL spectra for c(NaCl) = 10−5 mol L−1. (e) Amplitude variations for the symmetric stretch mode of the terminal CH3 group (red) and the O–H stretch mode of water in the BIL (black) as a function of ionic strength. Data is given as deviation in percentage from the reference spectrum (c(NaCl) = 10−5 mol L−1). Calculation of error bars is described in the SI. (f) Line shape comparison between the χ(3) spectrum obtained in this work (black) and the spectrum previously reported in the literature.18 | ||
A particularly instructive observation emerges from the comparison of the absorptive spectra of the DL and BIL (Fig. 2(d)). For better comparability of the line shapes, the spectra are normalized, revealing a remarkable similarity between the water responses in DL and BIL. The main difference between the spectra is the presence of the CH3 modes in the BIL spectrum that are obviously absent in the DL (the surfactants are highly insoluble in water and therefore only decorate the water surface). The near-identical line shapes of the water responses in the BIL and DL shows that the hydrogen-bond structures in both regions must be surprisingly similar. Although there is a vast difference in the amount of preferential molecular orientation in the BIL as compared to the DL,42 this increase in preferential orientation in the BIL does clearly not include any breaking of hydrogen-bonds, which would inevitably lead to marked differences in the vibrational spectrum. Furthermore, the similarity of the BIL and DL spectra also demonstrates that the anisotropy in the BIL must be predominantly governed by field-induced water reorientation because there is no clear spectral signature in the BIL of any structural motifs that are chemically associated with the discontinuity at the interface. In consequence, the χ(2)BIL,C term in eqn (1) must be very small, confirming previous findings.18
The results presented above draw a very clear picture of the water structure within the electrical double layer. Within the reach of the penetrating DC field (including the BIL) the water structure is anisotropic which originates predominantly from preferential dipolar orientation of the water molecules within a hydrogen-bond network that possesses very similar properties in terms of hydrogen-bond strength and connectivity than in bulk water. Nevertheless, it is important to note that this does not mean that the amount of preferential orientation scales linearly with the DC field throughout the double layer. BIL and DL yield almost identical spectra, but this does not include information on the intrinsic amplitude scaling of the SFG response with the electrostatic field in the two regions. It is well possible that the dipolar alignment first increases linearly with increasing field before reaching a plateau that decouples it from any further increase in field strength. This would manifest as a water signal that follows the field well within the DL but then reaches saturation within the BIL where the fields are substantially larger. Such a ‘saturation effect’ in dipolar alignment would then have to be limited by the maximum dipolar alignment that can be reached within the intrinsic rotational degrees of freedom of an otherwise unperturbed hydrogen-bond network. Such a scenario is not only possible but even highly probable because the described ‘saturation’ would indeed lead to a highly decreasing dielectric constant in the BIL as observed.
With this data in hand, we turn to the question if the SFG response from the BIL is independent of the ionic strength as often assumed. In order to do such comparisons, it is essential that the surfactant coverage is reproducible for all measurements. This can be analyzed by comparing the intensity of the CH3 stretch signals which are highly sensitive to molecular density and order. In the upper panel of Fig. 2(e) the intensity of the symmetric CH3 stretch mode (ss-CH3) is shown as a function of NaCl concentration, given as deviation from the reference value. From the plot it becomes evident that the monolayer density is reproducible across all measurements, with a spread of deviations <7%. In contrast, the amplitude of the water band shows significant dependency on the ion concentration, with a spread of deviations larger than 40%. Interestingly, the evolution of the water signal with concentration follows a clear pattern but is not monotonic as it shows a clear minimum for a concentration of 10−4 M NaCl. The reason for these large deviations could be changes in the surface charge density (concentration dependent proton dissociation from the terminal –PO4H2),45 changes in the thickness of the BIL, and/or concentration dependent relations between DC field and amount of water reorientation inside the BIL. The observed non-monotonic concentration dependency of the water signal suggests a complex interplay of multiple contributions. While the exact origin of these changes in the BIL amplitude cannot be determined based on these measurements, the results clearly show the SFG signal from the BIL is not constant for different electrolyte concentrations. In consequence, the method of extracting the DL contribution by subtraction of SFG spectra at different NaCl concentrations will lead to non-negligible errors.
Finally, we compare the line shape of the χ(3) spectrum obtained here with the corresponding spectrum derived by Shen and co-workers18 (see Fig. 2(f)). As mentioned in the introduction, the latter is widely used as a reference for the decomposition of the DL and BIL contributions. We see that the general line shape (in real and imaginary parts) is quite similar, however, closer inspection reveals certain discrepancies. This mainly concerns the low frequency region, where the spectrum derived here shows larger amplitude. While, at first sight, these differences seem minor, they are in fact significant if the spectrum is used for decomposition of SFG spectra. It is at this point not possible to unambiguously say which of the two spectra is more accurate, however, it is important to point out that the spectrum presented in this work is obtained using far fewer assumptions than in the reference work. As stated earlier, for the extraction shown here, the only required assumptions are (i) the electric field in the DL decays exponentially with λ as the decay constant, and (ii) the BIL is very thin such that propagation effects within the BIL contribution can be neglected. In contrast, the assumptions used in the referenced work include that the nonlinear response must scale linearly with the DC field, even in the BIL, and that the evolution of the potential with depth is accurately described by the presented mathematical solution of the (modified) Poisson–Boltzmann equation. Both assumptions are likely to fail in the BIL as discussed above. These uncertainties have particularly large impact on the predicted amplitude of the BIL contribution, thus also influencing the resulting line shape of the obtained DL spectrum. As the method for the determination of the χ(3) spectrum presented here does not require any assumption on the evolution of the dielectric constant at the interface we believe that our result is more accurate.
For the potential use of the χ(3) spectrum reported here it is important to point out that the spectrum is still scaled by the Stern potential (ΦS), which could not be quantified within this study. While this does not affect the line shape, it makes it impossible to simply read-off absolute Stern potentials from the amplitude of the obtained DL spectra. However, our measurements allow for extracting their relative dependency on ionic strength, using the obtained DL spectrum for the 10−5 M NaCl solution as reference. Fig. 3 shows the resulting relative Stern potentials for the different electrolyte concentrations. Except a small kink at a concentration of 10−4 M which is likely originating from measurement inaccuracies, a clear increase in ΦS with increasing Debye length (decreasing salt concentration) can be observed, as expected. The referenced χ(3) spectrum from the literature does report absolute units and, in principle, it would be possible to transform the relative values presented in the figure into absolute potentials. However, given the large uncertainty in the correct scaling of the field amplitudes inside the BIL (as mentioned above) it is unclear to what extent the reported absolute values are accurate.
 | ||
| Fig. 3 Evolution of the relative Stern potential (ΦS) with increasing Debye length (λ). The relative potentials are obtained by determining the ratio between the amplitudes of the extracted DL spectra and the DL spectrum obtained for 10−5 M NaCl. Calculation of error bars is described in the SI. | ||
Conclusion
In this work we have presented a study of the anisotropic water structure at charged interfaces for different electrolyte concentrations. Using the previously introduced SFG/DFG technique we have successfully decomposed the measured data into the spectral contributions from BIL and DL, allowing for a detailed analysis of the corresponding vibrational line shapes. The analysis shows that the line shape of the vibrational responses of the anisotropic water remains largely independent of the ion concentration and is nearly identical throughout the entire electrical double layer. This leads to the conclusion that the structural anisotropy of water in the interfacial region is clearly dominated by DC field-induced preferential molecular orientation for all measured electrolyte concentrations, while the properties of the hydrogen-bond network (hydrogen-bond strength and connectivity) remain largely unaffected by the applied DC field. This implies that the field induced molecular reorientation does not include the breaking of hydrogen-bonds. The variation of salt concentration is found to mainly modulate the evolution of the structural anisotropy with depth which is clearly visible in the measured SFG/DFG raw data.The analysis furthermore reveals that the amplitude of the vibrational water response in the BIL is, in contrast to the line shape, not independent of the electrolyte concentration showing a spread of deviations of more than 40% from the mean value across different ionic strengths. As a commonly used method for decomposing SFG spectra from charged interfaces into their contributions from BIL and DL is based on the assumption of constant BIL signals, these results show that this approach is rather inaccurate and might well lead to misinterpretations of the resulting spectra. A comparison of the DL spectrum obtained in this work with the widely used reference spectrum shows also here small but significant deviations. Together, these findings clearly demonstrate that the methods for analyzing SFG data from charged aqueous interfaces should be refined to avoid inaccurate conclusions. As demonstrated in this work, a clear alternative to these methods is the use of the combined SFG/DFG approach as it allows for the spectral analysis of the vibrational responses from different depths without the need for any far-reaching assumptions.
In summary, the results presented in this work yield deep insight into the anisotropic water structure at charged interfaces and its dependence on electrolyte concentration with somewhat surprising outcome. It will be very interesting to extend these studies to other types of samples such as interfaces to charged oxide materials where very distinct structural motifs of water have been observed in the BIL. Overall, the successful application of the SFG/DFG spectroscopy shown here provides a promising perspective on future investigations of the role of anisotropic water structures in the electrochemical double layer.
Author contributions
A. P. F. and M. T. conceived the project and designed the experiments. A. D. D., S. K. and A. P. F. performed the experiments and analyzed the data. All authors discussed the interpretation of the results. A. D. D. and S. K. drafted the manuscript which was edited by all authors. M. W. and M. T. supervised the work, revised the manuscript and acquired funding.Conflicts of interest
There are no competing interests.Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplementary information (SI). Source data for figures are available from the Zenodo repository at DOI: https://doi.org/10.5281/zenodo.17829584. Supplementary information is available. See DOI: https://doi.org/10.1039/d5fd00155b.Acknowledgements
The authors thank Prof. Julianne Gibbs for providing the reference spectrum shown in Fig. 2(f). Open Access funding provided by the Max Planck Society.References
- R. Vittal, H. Gomathi and K.-J. Kim, Adv. Colloid Interface Sci., 2006, 119, 55–68 CrossRef CAS PubMed.
- M. J. Rosen and J. T. Kunjappu, Surfactants and Interfacial Phenomena, John Wiley & Sons, 2012 Search PubMed.
- S. Lelieveld, J. Wilson, E. Dovrou, A. Mishra, P. S. J. Lakey, M. Shiraiwa, U. Pöschl and T. Berkemeier, Environ. Sci. Technol., 2021, 55, 14069–14079 CrossRef CAS PubMed.
- K. A. Wokosin, E. L. Schell and J. A. Faust, Environ. Sci.: Atmos., 2022, 2, 775–828 Search PubMed.
- R. Friedman, J. Membr. Biol., 2018, 251, 453–460 CrossRef CAS.
- Y. Ma, K. Poole, J. Goyette and K. Gaus, Front. Immunol., 2017, 8, 1513 CrossRef PubMed.
- W. Schmickler and E. Santos, Interfacial Electrochemistry, Springer Science & Business Media, Berlin, Heidelberg, 2010 Search PubMed.
- S. Dewan, V. Carnevale, A. Bankura, A. Eftekhari-Bafrooei, G. Fiorin, M. L. Klein and E. Borguet, Langmuir, 2014, 30, 8056–8065 CrossRef CAS.
- X. Wei, S. C. Hong, A. I. Lvovsky, H. Held and Y. R. Shen, J. Phys. Chem. B, 2000, 104, 3349–3354 CrossRef CAS.
- O. Björneholm, M. H. Hansen, A. Hodgson, L.-M. Liu, D. T. Limmer, A. Michaelides, P. Pedevilla, J. Rossmeisl, H. Shen, G. Tocci, E. Tyrode, M.-M. Walz, J. Werner and H. Bluhm, Chem. Rev., 2016, 116, 7698–7726 CrossRef PubMed.
- G. Gonella, E. H. G. Backus, Y. Nagata, D. J. Bonthuis, P. Loche, A. Schlaich, R. R. Netz, A. Kühnle, I. T. McCrum, M. T. M. Koper, M. Wolf, B. Winter, G. Meijer, R. K. Campen and M. Bonn, Nat. Rev. Chem., 2021, 5, 466–485 CrossRef CAS PubMed.
- M. Li, X. Wang, J. Meng, C. Zuo, B. Wu, C. Li, W. Sun and L. Mai, Adv. Mater., 2024, 36, 2308628 CrossRef CAS PubMed.
- S. Song, Q. Zheng, A. Song and J. Hao, Langmuir, 2012, 28, 219–226 CrossRef CAS PubMed.
- S. Šegota and D. Težak, Adv. Colloid Interface Sci., 2006, 121, 51–75 CrossRef.
- D. K. Hore and K. C. Jena, J. Phys. Chem. C, 2009, 113, 15364–15372 Search PubMed.
- D. K. Hore and E. Tyrode, J. Phys. Chem. C, 2019, 123, 16911–16920 CrossRef CAS.
- J. G. Hedley, H. Berthoumieux and A. A. Kornyshev, J. Phys. Chem. C, 2023, 127, 8429–8447 CrossRef CAS.
- Y.-C. Wen, S. Zha, X. Liu, S. Yang, P. Guo, G. Shi, H. Fang, Y. R. Shen and C. Tian, Phys. Rev. Lett., 2016, 116, 016101 CrossRef PubMed.
- Y. R. Shen and V. Ostroverkhov, Chem. Rev., 2006, 106, 1140–1154 CrossRef CAS PubMed.
- G. L. Richmond, Chem. Rev., 2002, 102, 2693–2724 CrossRef CAS PubMed.
- A. M. Darlington, T. A. Jarisz, E. L. Dewalt-Kerian, S. Roy, S. Kim, M. S. Azam, D. K. Hore and J. M. Gibbs, J. Phys. Chem. C, 2017, 121, 20229–20241 CrossRef CAS.
- S. Kaur, S. Chaudhary, S. T. Khan, D. J. Fairhurst and K. C. Jena, ACS Appl. Mater. Interfaces, 2025, 17, 68566–68578 CrossRef CAS PubMed.
- D. E. Gragson, B. M. McCarty and G. L. Richmond, J. Am. Chem. Soc., 1997, 119, 6144–6152 CrossRef CAS.
- T. Ishiyama, T. Imamura and A. Morita, Chem. Rev., 2014, 114, 8447–8470 CrossRef CAS PubMed.
- A. G. Lambert, P. B. Davies and D. J. Neivandt, Appl. Spectrosc. Rev., 2005, 40, 103–145 CrossRef CAS.
- A. Morita, Theory of Sum Frequency Generation Spectroscopy, Springer Nature Singapore, Singapore, 2018, vol. 97 Search PubMed.
- J. Hunger, J. Schaefer, P. Ober, T. Seki, Y. Wang, L. Prädel, Y. Nagata, M. Bonn, D. J. Bonthuis and E. H. G. Backus, J. Am. Chem. Soc., 2022, 144, 19726–19738 CrossRef CAS PubMed.
- P. B. Miranda and Y. R. Shen, J. Phys. Chem. B, 1999, 103, 3292–3307 CrossRef CAS.
- A. P. Fellows, Á. D. Duque, V. Balos, L. Lehmann, R. R. Netz, M. Wolf and M. Thämer, J. Phys. Chem. C, 2024, 128, 20733–20750 CrossRef CAS.
- A. Montenegro, C. Dutta, M. Mammetkuliev, H. Shi, B. Hou, D. Bhattacharyya, B. Zhao, S. B. Cronin and A. V. Benderskii, Nature, 2021, 594, 62–65 CrossRef CAS PubMed.
- G. Gonella, C. Lütgebaucks, A. G. F. de Beer and S. Roke, J. Phys. Chem. C, 2016, 120, 9165–9173 CrossRef CAS.
- S. Nihonyanagi, S. Yamaguchi and T. Tahara, J. Am. Chem. Soc., 2010, 132, 6867–6869 CrossRef CAS PubMed.
- E. Tyrode, S. Sengupta and A. Sthoer, Nat. Commun., 2020, 11, 493 CrossRef CAS PubMed.
- Y. R. Shen, Fundamentals of Sum-Frequency Spectroscopy, Cambridge University Press, 2016 Search PubMed.
- A. Morita and J. T. Hynes, Chem. Phys., 2000, 258, 371–390 CrossRef CAS.
- N. Garciá Rey, E. Weißenborn, F. Schulze-Zachau, G. Gochev and B. Braunschweig, J. Phys. Chem. C, 2019, 123, 1279–1286 CrossRef PubMed.
- M. M. Uddin and D. K. Hore, Langmuir, 2025, 41, 16634–16640 CrossRef CAS PubMed.
- B. Rehl, E. Ma, S. Parshotam, E. L. DeWalt-Kerian, T. Liu, F. M. Geiger and J. M. Gibbs, J. Am. Chem. Soc., 2022, 144, 16338–16349 CrossRef CAS PubMed.
- Y. Wang, Y. Nagata and M. Bonn, Faraday Discuss., 2024, 249, 303–316 RSC.
- Z. Li, I. Swiderska, L. Dalifoski, S. Lee, N. A. Correa-Rojas, D. Roesel, M. Eremchev, M. Flor, O. B. Tarun, A. Marchioro and S. Roke, Faraday Discuss., 2025, 259, 342–365 RSC.
- B. Rehl and J. M. Gibbs, J. Phys. Chem. Lett., 2021, 12, 2854–2864 CrossRef CAS PubMed.
- Á. Diaz-Duque, V. Balos, M. Wolf, A. P. Fellows and M. Thämer, arXiv, 2025, preprint, arXiv:2508.06912, DOI:10.48550/arXiv.2508.06912.
- M. A. Brown, A. Goel and Z. Abbas, Angew. Chem., 2016, 128, 3854–3858 CrossRef.
- S. V. Kalinin, Science, 2018, 360, 1302 CrossRef PubMed.
- H. Boroudjerdi, Y. Kim, A. Naji, R. Netz, X. Schlagberger and A. Serr, Phys. Rep., 2005, 416, 129–199 CrossRef CAS.
- A. P. Fellows, Á. D. Duque, V. Balos, L. Lehmann, R. R. Netz, M. Wolf and M. Thämer, Langmuir, 2024, 40, 18760–18772 CrossRef CAS PubMed.
- V. Balos, T. Garling, A. D. Duque, B. John, M. Wolf and M. Thämer, J. Phys. Chem. C, 2022, 126, 10818–10832 CrossRef CAS.
- Y. Hsiao, T.-H. Chou, A. Patra and Y.-C. Wen, Sci. Adv., 2023, 9, eadg2823 CrossRef CAS PubMed.
- C. Cai, M. S. Azam and D. K. Hore, J. Phys. Chem. C, 2021, 125, 25307–25315 CrossRef CAS.
- M. M. Uddin, M. S. Azam and D. K. Hore, J. Am. Chem. Soc., 2024, 146, 11756–11763 CrossRef CAS PubMed.
- S. Sun, C. Tian and Y. R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5883–5887 CrossRef CAS PubMed.
- C. G. Gray and P. J. Stiles, Eur. J. Phys., 2018, 39, 053002 CrossRef.
- P. E. Ohno, H. Wang and F. M. Geiger, Nat. Commun., 2017, 8, 1032 CrossRef PubMed.
- M. Sprik, Phys. Rev. E, 2021, 103, 022803 CrossRef CAS PubMed.
- B. D. Storey and M. Z. Bazant, Phys. Rev. E, 2012, 86, 056303 CrossRef PubMed.
- H. Butt, K. Graf and M. Kappl, Physics and Chemistry of Interfaces, Wiley, 2003 Search PubMed.
- R. R. Netz and H. Orland, Eur. Phys. J. E: Soft Matter Biol. Phys., 2000, 1, 203 CrossRef CAS.
- A. G. Moreira and R. R. Netz, Europhys. Lett., 2000, 52, 705–711 CrossRef CAS.
- M. Bischoff, D. Biriukov, M. Předota, S. Roke and A. Marchioro, J. Phys. Chem. C, 2020, 124, 10961–10974 CrossRef CAS PubMed.
- A. P. Fellows, V. Balos, B. John, Á. Díaz Duque, M. Wolf and M. Thämer, J. Chem. Phys., 2023, 159, 164201–164220 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |