DOI:
10.1039/D5FD00148J
(Paper)
Faraday Discuss., 2026, Advance Article
Electrostatic screening in nanotubes: a tubular response function framework
Received
3rd December 2025
, Accepted 4th March 2026
First published on 4th March 2026
Abstract
The structure and transport of electrolytes in nanoscale channels are known to be affected by the electronic properties of the confining walls. This influence is particularly pronounced in quasi-one-dimensional nanotubes, where the high surface-to-volume ratio makes the wall the dominant source of electrostatic screening. For instance, ideal metallic tubes suppress long-range Coulomb interactions between ions exponentially. Yet, there exists no generic framework for evaluating electrostatic interactions in tubular confinement. Here, we introduce tubular response functions – a generalisation of surface response functions that captures how nanotubes with arbitrary electronic properties screen Coulomb interactions. Using this framework, we evaluate the interaction potential of ions confined in a metallic carbon nanotube, treating its long-range electronic properties exactly within a Luttinger liquid model. We demonstrate that the screening characteristic of metallic armchair carbon nanotubes is almost identical to that of an ideal metal, regardless of electron density. We trace the origin of such strong screening to the quantum confinement of electrons around the tube circumference and to the suppression of Friedel oscillations. Our framework opens the way for quantitative descriptions of ionic correlations and charge storage in nanotube-based electrodes, and can be further extended to address confined ion dynamics.
1 Introduction
Ions are at the heart of many nanofluidic applications, from desalination1 to osmotic power generation2 to electrical double-layer supercapacitors.3 These technologies leverage the enormous surface-to-volume ratio of nanoscale channels, exploiting the particularities of confined solid–liquid interfaces, including the surface charge and electric polarisability. Such interfacial effects become particularly pronounced in single-digit nanopores, i.e., channels with a diameter of less than 10 nm, where the surface charge can render the channel impermeable to particular ionic species due to electrostatic repulsion.4,5 Even in the absence of surface charge, the dehydration energy hinders ion entry when the spatial confinement becomes comparable to the ion size. Moreover, the substantial dielectric contrast between water and typical channel materials can lead to a self-energy barrier, reinforcing ion–ion interactions and inhibiting ion entry below a few nanometers of confinement.6–9 This has been identified very early on to have important consequences for ion transport through biological lipid membranes.10
In contrast to the aforementioned ion exclusion phenomena, capacitive ion accumulation in nanoporous carbon materials is most efficient in the smallest accessible pores, where the pore diameter matches the size of the ion.11,12 This striking experimental observation has been attributed to the so-called superionic state,13 promoted by the polarisability of carbon-based nanopores. Kondrat et al. demonstrated a substantial attraction between an ion and its induced image charge in the wall for metallic 2D nanoslits and 1D nanopores.14 Moreover, the image charges in a perfect metal exponentially screen long-range interionic interactions, facilitating the dense packing of like-charge ions.15,18 Such exponential screening is preserved in nanochannels confined by a Thomas–Fermi metal, a model for an imperfect bulk metal, albeit with an increased screening length.15
The dielectric constant of water, as a solvent, is strongly modified under nanoconfinement compared to its isotropic value ∈ ≈ 80 in the bulk. For 2D slits, Fumagalli and co-authors have experimentally measured both the out-of-plane16 and in-plane17 dielectric constant. They find that the out-of-plane dielectric constant drops with decreasing slit height from its bulk value above ∼100 nm down to ∈⊥ ≈ 2 at a few nanometers height. In contrast, the in-plane dielectric constant rises with decreasing slit height from the bulk value above ∼10 nm up to ∈‖ ≈ 1000 at a few nanometers height. For the nanotube geometry, to our knowledge, no experimental data is available. However, molecular dynamics simulations with SPC/E water in frozen and unpolarisable carbon nanotubes give qualitatively similar results, where the radial dielectric constant drops down to ∈r ∼ 1–3 and the axial dielectric constant rises up to ∈z ∼ 103 at the radius R ∼1 nm.18
For two-dimensional nanochannels, the treatment of interionic interactions and electronic screening has recently been generalised to capture arbitrary electronic properties of the confining walls.7 This formalism is based on surface response functions,19 which encode the dielectric response of an arbitrary material across a planar interface. The surface response function can be derived from either microscopic or macroscopic models, and it acts as a reflection coefficient for an evanescent plane-wave potential incident on the interface.7
Focusing on cylindrical nanotubes, early studies have derived the dielectric function and plasmon dispersion for a free electron gas on a cylindrical shell in the random-phase approximation,20,21 as well as the radial and axial polarizability of carbon nanotubes.22 Later, the low-frequency dynamical dielectric function of an arbitrary carbon nanotube (CNT) was analysed.23 CNTs exhibit complex band structures and electronic properties due to their diverse chiralities.24,25 This is because a CNT can be viewed as a sheet of graphene rolled into a cylindrical tube, where the orientation of the tube axis on the graphene lattice is defined by two indices (n, m). These indices fully determine the electronic properties of the CNT: depending on the choice of indices (n, m), the CNT can either be metallic, with an approximately linear band dispersion around the charge-neutrality point, or semiconducting. For quasi-one-dimensional nanotubes, electron–electron interactions are strongly enhanced by geometrical confinement, promoting long-range correlations and power-law response functions characteristic of one-dimensional systems.26 In the long-wavelength, low-energy limit, these interactions can be treated exactly using bosonization, leading to the Tomonaga–Luttinger description of interacting 1D conductors.27–30 This quasi-1D framework is fundamentally different from the Fermi-liquid model, valid in higher dimensions: in a Luttinger liquid, spin and charge degrees of freedom decouple, and fermionic quasi-particles are absent near the Fermi surface, so that the low-energy excitations become collective bosonic modes.26,31 The Luttinger-liquid framework has been explicitly applied to carbon nanotubes, where strong electron–electron interactions lead to the characteristic power-law suppression of the tunnelling density of states.32–36 However, it remains unknown how these intricate electronic properties of CNTs affect interactions between ions confined within the tube.
In this paper, we generalise surface response functions, developed for planar interfaces, to tubular response functions for the cylindrical geometry of nanotubes. We first demonstrate how the tubular response functions emerge from the linear-response properties of a material. We then derive explicit expressions for the tubular response function of various materials, including an insulating, dielectric nanopore, a 1D cylindrical electron gas and a metallic armchair carbon nanotube. For each material, we derive the screened potential of an ion at the centre of the tube, and we deduce the functional form of the decay law at long distances.
2 Evaluation of the confined ion potential
We consider a nanotube of radius R with an ion of charge +Qe placed at its centre (Fig. 1a). We choose cylindrical coordinates (ρ, φ, z), with the z-coordinate aligned along the tube. The tube is filled with water, which we treat as an anisotropic dielectric background where ∈r, ∈φ and ∈z are respectively the radial, azimuthal and axial components of the dielectric tensor ∈ (Fig. 1b). Since we consider quasi-1D nanotubes in this paper, we set the dielectric constants to ∈r = ∈φ = 2 and ∈z = 80. In contrast, for large tube radii we expect them to converge to the values of a flat interface, ∈r ≈ 2 and ∈φ = ∈z ≈ 80.
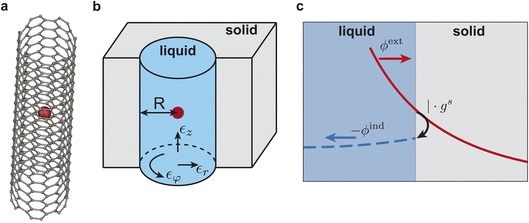 |
| | Fig. 1 (a) Schematic illustration of an ion in a (6,6) armchair carbon nanotube. (b) Schematic of a nanopore of radius R embedded into an infinite dielectric medium. The radial, azimuthal and axial dielectric constants, ∈r, ∈φ and ∈z, are indicated with arrows. (c) Illustration of the tubular response function gs as a reflection coefficient for the external potential ϕext applied to the solid from the liquid side. The external potential polarises the solid, leading to an induced potential ϕind in the liquid. At the solid–liquid interface, the induced potential is ϕind(R) = −gsϕext(R). | |
We aim to compute the screening of the Coulomb potential of the ion inside the “liquid” due to the presence of an arbitrary nanotube material, the “solid”. Phenomenologically, the ion polarises the nanotube, generating an induced potential that acts back on the nanotube's interior. The derivation can be divided into three steps, as illustrated in Fig. 1c: (i) inside the liquid, compute the Coulomb potential of the ion and find the potential at the solid–liquid interface, (ii) inside the solid, compute the dielectric response to the ion potential (decomposed into evanescent cylindrical waves) and find the resulting induced potential at the solid–liquid interface, (iii) compute how the confined liquid screens the potential induced by the solid. Across the solid–liquid interface, the Coulomb potential is continuous. The total potential in the liquid is the sum of the bare potential and the induced potential.
The structure of this problem is generic for any solid–liquid interface geometry, and the formalism for arbitrary electronic properties of the solid has previously been developed for planar surfaces.7,37 For the tubular geometry, we first decompose the Coulomb potential at position (ρ, φ, z) of a point charge Qe at position (ρ′, φ′, z′) into its cylindrical eigenmodes
| |
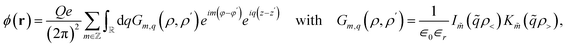 | (1) |
where
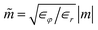
,
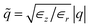
,
ρ< = min{
ρ,
ρ′},
ρ> = max{
ρ,
ρ′}, and
Iν and
Kν are the modified Bessel functions of order
ν. Here,
Gm,q(
ρ,
ρ′) is the Fourier transform with respect to
φ −
φ′ and
z −
z′ of the Coulomb kernel satisfying the Poisson equation ∇
r[
∈·∇
rG(
r,
r′)] = −
δ(
r −
r′). Due to the decomposition in
eqn (1), it is sufficient to study an individual eigenmode (
m,
q) of the ion potential.
For step (i), we apply eqn (1) to an ion at the centre of the tube, yielding the external potential inside the liquid
| |
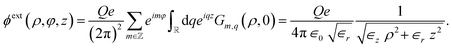 | (2) |
It is worth noting that the potential strength along the tube axis (
ρ = 0) is determined by
∈r. Under ångström-scale confinement,
∈r can be substantially reduced compared to the bulk, leading to an increased Coulomb interaction strength. The eigenmodes of the external potential at the solid–liquid interface are
| |
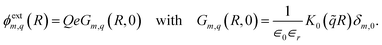 | (3) |
The external potential inside the solid then reads
| |
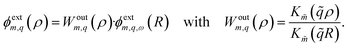 | (4) |
For step (ii), and in analogy with surface response functions for planar interfaces,
7,19 we encode the dielectric response of the nanotube in a tubular response function
gm,q = −
ϕindm,q(
R)/
ϕextm,q(
R). Phenomenologically, the tubular response function acts as a reflection coefficient of the external ion potential impinging on the solid (
Fig. 1c). For step (iii), the potential applied to the liquid by the (polarised) solid is analogous to
eqn (4),
| |
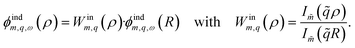 | (5) |
The functions
Winm,q(
ρ) and
Woutm,q(
ρ) respectively describe the radial decay of an evanescent cylindrical wave when going towards or away from the tube axis. The induced potential inside the liquid thus becomes
| |
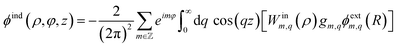 | (6) |
| |
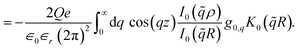 | (7) |
Conceptually, the term in brackets in
eqn (6) is the product of three factors, which represent the steps (i) to (iii) from right to left. The total potential inside the liquid is
ϕtot(
ρ,
φ,
z) =
ϕext(
ρ,
φ,
z) +
ϕind(
ρ,
φ,
z). This formalism is not limited to the description of a single static ion in the centre of the tube. The evanescent electrical potential created by any time-dependent charge distribution inside the tube can be decomposed into cylindrical modes
ϕextm,q,ωWout,sm,q(
ρ)
eimφeiqze−iωt at the interface, and the induced potential is determined by
eqn (6) with a frequency-dependent
gm,q,ω. Moreover, the framework of tubular response functions also applies to charges inside the solid. In this case, the charges polarise the liquid, as encoded in the tubular response function of the liquid,
glm,q,ω. The induced potential inside the solid is then determined by
eqn (6) under the exchange
g →
gl and
Win →
Wout. In the formalism presented so far, the electrostatic response properties of the nanotube are entirely encapsulated into the tubular response function. We now proceed with a derivation of the tubular response function from linear response theory.
3 Tubular response functions
3.1 Dielectric medium
As a first example, we derive the tubular response function of an infinite, purely dielectric medium surrounding a cylindrical pore of radius R (Fig. 1b). For generality, the liquid and solid may have anisotropic dielectric constants. In each region, the electric potential must solve the Poisson equation, where the only charges are the ion in the tube and the induced surface charge at the solid–liquid interface due to the dielectric contrast. Decomposed into cylindrical eigenmodes, the potential must therefore have the form| |
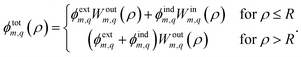 | (8) |
In eqn (8) we have already enforced the continuity of the potential across the interface as well as its asymptotic behaviour for ρ → ∞. For readability, we leave it implicit that the values of ![[m with combining tilde]](https://www.rsc.org/images/entities/i_char_006d_0303.gif) and
and ![[q with combining tilde]](https://www.rsc.org/images/entities/i_char_0071_0303.gif) inside the weight-functions Win/outm,q are generally different for the two material regions. The induced potential amplitude ϕindm,q is determined by the continuity of the radial component of the displacement field
inside the weight-functions Win/outm,q are generally different for the two material regions. The induced potential amplitude ϕindm,q is determined by the continuity of the radial component of the displacement field 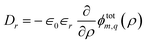 , where ∈r is the radial dielectric constant for the respective material. The general solution is straightforward to obtain, but it is a rather lengthy expression in terms of modified Bessel functions. Here, we focus on an ion at the centre of the tube, which is solely determined by the angular mode m = 0. The resulting tubular response function is
, where ∈r is the radial dielectric constant for the respective material. The general solution is straightforward to obtain, but it is a rather lengthy expression in terms of modified Bessel functions. Here, we focus on an ion at the centre of the tube, which is solely determined by the angular mode m = 0. The resulting tubular response function is| |
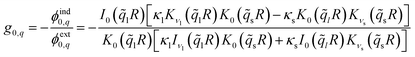 | (9) |
with l and s denoting “liquid” and “solid”, and 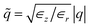 ,
, 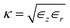 as well as
as well as 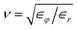 for the respective materials. In the limit R → ∞, eqn (9) reduces to
for the respective materials. In the limit R → ∞, eqn (9) reduces to| |
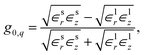 | (10) |
which is independent of the wavevector q and coincides with the surface response function of a planar interface7 for ∈r → ∈⊥ and ∈z → ∈‖. However, for a finite radius R, the tubular response function of a homogeneous dielectric nanopore remains wavevector-dependent. This means that, in contrast to a planar interface, the induced potential is generally not proportional to the external potential in real space.
3.2 Derivation from linear response theory
Microscopically, the dielectric response of a solid-state material stems from the dynamic rearrangement of charges – electrons and nuclei. In linear response theory, this can be characterised by the charge density response function χ(r, r′, t, t′), which, given an external potential ϕext(r′, t′), determines the induced charge density| |
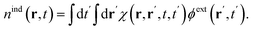 | (11) |
For a nanotube system with translation-invariance along the tube axis, we define the Fourier transform| |
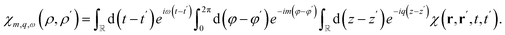 | (12) |
This yields| |
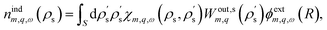 | (13) |
where we explicitly denote the integration over the region S of the solid, and we use 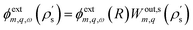 . The induced charge creates an induced potential, which at the interface reads
. The induced charge creates an induced potential, which at the interface reads| |
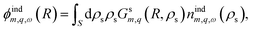 | (14) |
where Gsm,q denotes the Coulomb kernel inside the solid. The resulting expression for the tubular response function reads| |
 | (15) |
Reading the formula from right to left, the external potential propagates from the interface into the solid up to radius 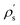 , which, through the density response function χ, induces a charge inside the solid at radius ρs, creating an induced potential at the interface. For completeness, the same reasoning leads to the tubular response function of the liquid, which can be obtained by exchanging the roles of the solid and the liquid:
, which, through the density response function χ, induces a charge inside the solid at radius ρs, creating an induced potential at the interface. For completeness, the same reasoning leads to the tubular response function of the liquid, which can be obtained by exchanging the roles of the solid and the liquid:| |
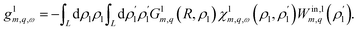 | (16) |
In the derivation of the tubular response functions, eqn (15) and (16), we have formally separated the (possibly anisotropic) dielectric background, which enters in the Coulomb kernel Gm,q, and the dynamic rearrangement of free charges, characterized by the density response function χ. A complete description of the interface then requires a self-consistent treatment of the induced potential due to both the dielectric contrast and the polarisability of mobile charges. We now derive χ from a quantum-mechanical perspective, which will finally enable us to compute the tubular response function of realistic nanotube systems.
3.3 Density response function of tubular systems
For a non-interacting electronic system, the density response function can be obtained from the single-particle eigenenergies Eλ and eigenfunctions ψλ(r) as38| |
 | (17) |
where nF is the Fermi–Dirac distribution and Γ is a phenomenological broadening, accounting for the finite quasi-particle lifetime due to scattering processes. If electron–electron interactions are taken into account at the mean-field level – within the so-called random phase approximation (RPA) – the interacting response function can be obtained in terms of the non-interacting one through an integral Dyson equation:| |
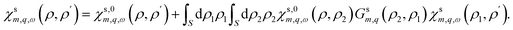 | (18) |
In general, eqn (18) needs to be solved numerically. However, for a nanotube wall with vanishing radial thickness, eqn (18) yields| |
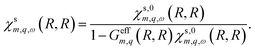 | (19) |
To account for the screening of the electron–electron interactions by the liquid, we use Geffm,q(R,R) = Glm,q(R,R). Additional dielectric screening, for instance due to high-energy excitations in the solid, can be incorporated by renormalising the dielectric constants entering Geffm,q(R,R). Ultimately, the non-interacting tubular response function is given by| | |
gs,0m,q,ω = −Geffm,q(R,R)χs,0m,q,ω(R,R),
| (20) |
and the Dyson equation, eqn (19), translates to| |
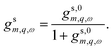 | (21) |
4 Screened potential for model nanotubes
We now proceed with the computation of tubular response functions for different model materials in the static limit ω → 0. We apply the results to an ion at the centre of the nanotube and determine the asymptotic functional form of the potential along the cylinder axis. Based on eqn (2) and (6), the screened potential inside the nanotube is| |
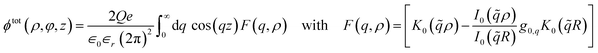 | (22) |
The Bessel function K0(z) is logarithmically divergent for z → 0, so we regularise ϕtot at the tube axis by a small value ρ > 0. The long-range behaviour of the total potential is determined by the asymptotic behaviour of F(q, ρ) in the limit q → 0. For q → 0, and within RPA, the bare density response function of any metallic nanotube converges to minus the density of states, −g(EF), at the Fermi level EF. The asymptotic expansions of the Bessel functions K0(x) and I0(x) for x → 0 are| |
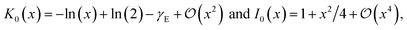 | (23) |
where γE denotes the Euler constant. Thus, for a finite density of states at the Fermi level, the RPA-renormalised tubular response function, eqn (21), converges to 1 for q → 0. In this case, the potential decays faster than 1/|z| as z → ∞. If g0,q < 1 for q → 0, then the asymptotic decay law remains 1/|z|, and the amplitude of the potential is modified by the factor (1 − g0,0). In this case, the nanotube asymptotically acts as a dielectric medium.
In the following, we derive explicit formulas for the tubular response function of various systems and compute the resulting screened ion Coulomb potential. Fig. 2 shows two typical band structures for quasi-1D systems, namely the quasi-1D electron gas and a (6,6) metallic CNT. The resulting tubular response functions and screened ion potentials are depicted in Fig. 3.
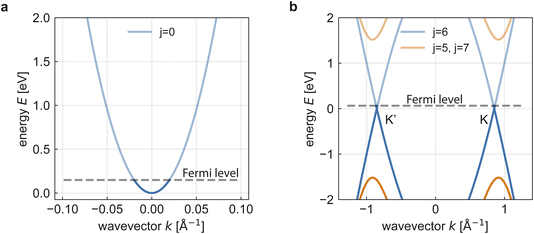 |
| | Fig. 2 (a) Band structure of a quasi-1D electron gas with effective mass 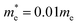 on an an infinite cylinder with radius R = 4 Å. (b) Band structure of a (6,6) armchair CNT with radius R ≈ 4 Å. (a and b) For the chosen electron density n = 5 × 1012 cm−2, only the lowest bands are occupied in both cases. on an an infinite cylinder with radius R = 4 Å. (b) Band structure of a (6,6) armchair CNT with radius R ≈ 4 Å. (a and b) For the chosen electron density n = 5 × 1012 cm−2, only the lowest bands are occupied in both cases. | |
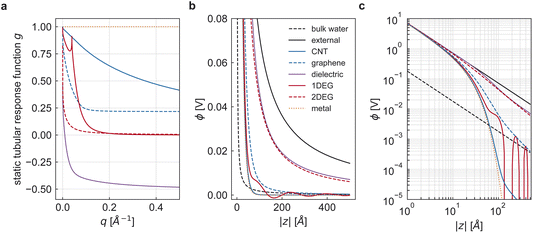 |
| | Fig. 3 Tubular response function and screened ion potential. (a) Tubular response function g0,q for various nanotube systems, defined in the main text, with radius R = 4 Å. The dielectric medium is isotropic with dielectric constants ∈sr,φ,z = 4. The perfect metal has, by definition, g0,q = 1. All other models are based on the same electron density n = 5 × 1012 cm−2. The armchair CNT has the chiral indices (6,6) and the radius R ≈ 4.07 Å. The effective electron mass of the 1D and 2D free electron gas is m∗e = 0.01me. (b) Bare and screened Coulomb potential of an ion at the centre of various nanotubes. The potential is computed along the tube axis with the same parameters as for panel (a). (c) Same data as panel (b), but in double-logarithmic scale. | |
4.1 Homogeneous dielectric medium
The tubular response function g0,q of an infinite, homogeneous, dielectric nanopore from eqn (9) is depicted in Fig. 3a for ∈sr,φ,z = 4. In the long-wavelength limit q → 0, the response function converges to g0,0 = 1 − ∈lr/∈sr < 1. Hence, the long-range amplitude of the bare Coulomb potential is modified by the factor ∈lr/∈sr. Concretely, for ∈lr < ∈sr, the dielectric nanopore provides additional potential screening at long distances, whereas for ∈lr > ∈sr, the nanopore amplifies the Coulomb potential. The latter effect is reminiscent of interaction confinement.7 This phenomenon was initially identified in nanopores with a low dielectric constant filled with a liquid with a high isotropic dielectric constant. In this case, the electric field concentrates within the material with a high dielectric constant, leading to enhanced quasi-1D Coulomb interactions. Accounting for the substantial anisotropy of the dielectric constant of water in ångström-scale nanotubes, Coulomb interactions are again enhanced compared to bulk water (Fig. 3b and c). Here, however, this is an immediate consequence of the anisotropic dielectric background since, according to eqn (2), the Coulomb potential along the cylinder axis is determined by ∈r < ∈z. For ∈lr > ∈sr, the tube walls provide additional interaction enhancement at long distances.
4.2 Ideal metal
An ideal metal can be described as a dielectric in the limit of an infinite dielectric constant, i.e. ∈sr,∈sφ,∈sz → ∞. The tubular response function of an infinite dielectric medium was already derived in eqn (9). The limit yields gm,q = 1, which means that the external potential is fully reflected at all wavelengths. The asymptotic screening law for |z| → ∞ is exponential. For the case of an isotropic dielectric liquid, the screened Coulomb potential in a perfect metallic nanotube has previously been derived.15 For the anisotropic case, we have to consider 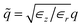 in eqn (22). Rescaling the integration variable q and the coordinate z in eqn (22), we recover the formula for the isotropic case. Thus, the existing result can be generalised to the anisotropic case without explicit calculations. We obtain
in eqn (22). Rescaling the integration variable q and the coordinate z in eqn (22), we recover the formula for the isotropic case. Thus, the existing result can be generalised to the anisotropic case without explicit calculations. We obtain| |
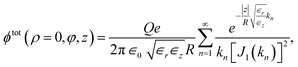 | (24) |
where kn is the n-th zero of the zero-order Bessel function J0, and J1 denotes the first-order Bessel function. Eqn (24) explicitly demonstrates the exponential decay, and at long distances |z| the first term with k1 ≈ 2.40 dominates. Remarkably, the anisotropy of the dielectric constant of water in ångström-scale nanotubes enhances the Coulomb potential so strongly that, for our choice of parameters, the metallic tube walls suppress the Coulomb interaction below the bulk strength only for distances |z| larger than ∼6 nm (Fig. 3b and c).
4.3 Cylindrical quasi-1D electron gas
As a first quantum-mechanical model, we consider a quasi-1D electron gas (1DEG) confined to the surface of a nanotube with radius R. We choose the effective mass 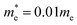 , where me denotes the bare electron mass. The charge density response function of this system has previously been reported.39 However, for completeness, we restate the main findings. Due to the periodic boundary conditions for the wavefunction around the tube circumference, the momentum in this direction is quantised as qj = j/R with
, where me denotes the bare electron mass. The charge density response function of this system has previously been reported.39 However, for completeness, we restate the main findings. Due to the periodic boundary conditions for the wavefunction around the tube circumference, the momentum in this direction is quantised as qj = j/R with  . Thus, the band structure of the electron gas has several bands, indexed by j, with energy
. Thus, the band structure of the electron gas has several bands, indexed by j, with energy 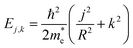 , where k is the wavevector along the nanotube axis (Fig. 2a). The eigenstates λ in eqn (17) can accordingly be labelled as λ = (k, j, σ), with σ denoting spin. Since the eigenfunctions of the electron gas are orthonormal, the non-interacting density response function becomes
, where k is the wavevector along the nanotube axis (Fig. 2a). The eigenstates λ in eqn (17) can accordingly be labelled as λ = (k, j, σ), with σ denoting spin. Since the eigenfunctions of the electron gas are orthonormal, the non-interacting density response function becomes| |
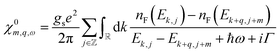 | (25) |
with spin-degeneracy gs = 2.
At room temperature, the thermal energy kBT ≈ 25 meV is much smaller than the Fermi level EF ≈ 150 meV and the subband spacing. We can thus treat the system as if it was at zero temperature, so that the Fermi–Dirac distribution becomes a step function, nF(E) = θ(EF − E), and the integral can be computed analytically. For vanishing broadening Γ = 0, the result is
| |
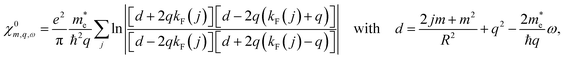 | (26) |
where the sum runs over all partially occupied bands, and
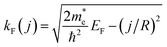
denotes the Fermi wavevector corresponding to band
j. At the electron density under consideration, only the lowest subband contributes. The resulting tubular response function within RPA,
g0,q,ω=0 (see
eqn (19)–(21)), is depicted in
Fig. 3a. In the long-wavelength limit
q → 0, the bare density response function
χ00,q,ω=0 converges to minus the finite density of states at the Fermi level
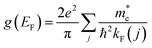
, and therefore the tubular response function converges to 1 (
Fig. 3a). At
q = 2
kF, the tubular response function features a cusp, originating from interbranch transitions from −
kF to +
kF. This gives rise to Friedel oscillations in the screened potential (
Fig. 3b and c).
4.4 Metallic armchair carbon nanotube: Luttinger framework
Many nanofluidic systems are based on carbon nanotubes (CNTs).40 We therefore apply the framework of tubular response functions to the electronic screening of ionic interactions inside CNTs, starting from a quantum-level description of the CNT electronic properties.
To keep the treatment analytical, we start from a nearest-neighbour tight-binding model of the CNT band structure. For details of the model, we refer to the literature.24,25,41 Here, we focus on so-called armchair CNTs with chiral indices m = n. These nanotubes are metallic and at low energies they exhibit the characteristic linear band dispersion in the vicinity of the Dirac points K and K′ (Fig. 2b). As in the quasi-1D electron gas, periodic boundary conditions around the tube circumference give rise to several energy bands. However, for a quasi-1D armchair CNT, the bottom of the second-lowest band has energy E ≈ 10 eV per n and thus lies more than ∼0.6 eV above the charge–neutrality point for nanotubes with radii R < 1 nm, corresponding to n < 15. We therefore consider only the lowest band, whose dispersion is well approximated by the Dirac fermion dispersion Ek = ℏvFk. Here, k is measured from the respective Dirac point, and we use 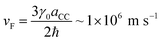 , derived from the tight-binding parameter γ0 = 3.033 eV and the carbon–carbon distance aCC = 1.42 Å.41
, derived from the tight-binding parameter γ0 = 3.033 eV and the carbon–carbon distance aCC = 1.42 Å.41
The response properties of an interacting Dirac fermion system in one dimension can be determined exactly in the long-wavelength limit (q → 0) following the Luttinger liquid framework. This problem has been extensively addressed in the literature,26,28–30 and here we outline only the main ideas. In the long-wavelength limit, the Luttinger framework yields an exact transformation that maps the interacting fermionic Hamiltonian onto a non-interacting bosonic Hamiltonian. The bosonic Hamiltonian splits into two parts: H = Hρ + Hσ, corresponding to charge (ρ) and spin (σ) degrees of freedom. Here, we are interested in the charge excitations, which are governed by26
| |
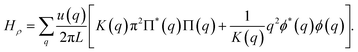 | (27) |
The field
ϕ is related to the charge density
via ρ(
x) = −π
−1∇
ϕ(
x), and Π(
x) is its conjugate momentum. If the fermions are non-interacting, the Fermi velocity reduces to the bare value
u(
q) =
vF, and the Luttinger parameter is
K(
q) = 1. Now, introducing a long-range Coulomb interaction in the initial fermionic Hamiltonian amounts simply to renormalising the parameters of the bosonic one. Accounting for the spin degeneracy
gs = 2 and the valley degeneracy
gv = 2 in the case of CNTs, one obtains
26,31,33| |
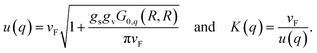 | (28) |
The resulting density response function is
31| |
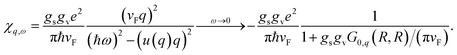 | (29) |
At this point, we remark that the Hamiltonian in
eqn (27) is quadratic in the electronic density: it therefore describes Gaussian charge fluctuations. In Section 2.3 we used the random phase approximation (RPA) for treating electron–electron interactions, and RPA is precisely a Gaussian approximation for charge fluctuations. This implies that the seemingly approximate RPA treatment is in fact exact for the density response function of Dirac fermions in 1D in the long-wavelength limit
q → 0.
26
Going back to eqn (17) for the non-interacting response function, the matrix elements are in general non-trivial due to the sublattice structure of CNTs. However, in armchair CNTs, the matrix element equals 1 if k and k + q are on the same branch, and zero otherwise. Hence, taking into account both valleys in the Brillouin zone, the bare density response function reads42,43
| |
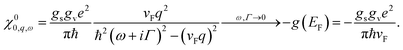 | (30) |
Applying RPA as per
eqn (19), we recover the Luttinger result in
eqn (29). It is worth noting that the result is independent of the CNT electron density. This can be traced back to the fact that the electron density enters the calculation only through the density of states at the Fermi level,
g(
EF), which does not depend on
EF in the linear portion of the Dirac cone (see
Fig. 2b). In
Fig. 3a, we show the tubular response function obtained in the Luttinger/RPA framework. As
q → 0, the tubular response function of armchair CNTs converges to 1 (
Fig. 3a). In contrast to the quasi-1D electron gas, the tubular response function of the armchair CNT does not exhibit a cusp at
q = 2
kF , because of the vanishing inter-branch matrix element. Therefore, Friedel oscillations are suppressed in armchair CNTs, at least at the RPA level. Thus, remarkably, armchair CNTs screen the ion potential almost as if they were a perfect metal (
Fig. 3b and c).
We note that the expressions in eqn (27) and (29) are exact only in the long-wavelength limit q → 0, where the Luttinger–Hamiltonian is indeed quadratic; deviations from these results can be expected around q = 2kF due to non-Gaussian (power law) correlations.26 Further corrections may arise due to the band dispersion not being exactly linear beyond a certain wavevector (|q| ≈ 0.2 Å−1 for a (6,6) CNT) and due to finite-temperature effects. Nevertheless, these corrections are unlikely to affect our conclusions regarding long-range screening determined by the small-q behaviour.
4.5 Rolled-up 2D materials
The very strong screening behaviour that we obtain in the tubular geometry, even with non-ideal metal models, contrasts with the results obtained in the 2D slit geometry, where the shape of the screened ion potential was found to depend significantly on the confining material's electron density (or Thomas–Fermi length).7 We now investigate whether this is an effect of geometry, or rather a quantum effect of dimensionality on electronic properties.
To this end, we introduce a mapping that “rolls up” the density response function of a 2D material χ2D(q) into a tubular geometry with radius R:
| | |
χ1Dm,q = Rχ2D(q) with q = (m/R,q).
| (31) |
In this way, we describe a tube that screens Coulomb interactions exactly like its parent 2D material, save for the geometry. For a 2D electron gas, the charge density is
n2DEG =
kF2/(2π), and the static density response function reads
44| |
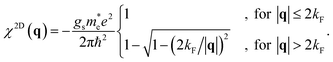 | (32) |
For graphene, the charge density is
ngraph =
kF2/π and the static density response function reads
45| |
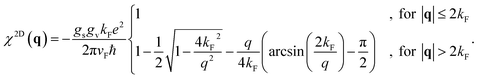 | (33) |
Fig. 3a shows the resulting tubular response functions at the same electron density
n as the quasi-1D models. Both tubular response functions converge to 1 in the long-wavelength limit
q → 0, but at intermediate values of
q they are significantly smaller than their 1D counterparts. Thus, the 2D models screen less effectively than the quasi-1D models (
Fig. 3b and c). We conclude that the predicted qualitative difference in screening between the 1D tube and 2D slit settings is not only a question of geometry: the quantum confinement of electrons along the tube circumference – without which the mapping in
eqn (31) would be exact – is instrumental in the nearly perfect metallic behaviour of armchair CNTs.
5 Self-energy
Until now, we have studied the screened ion potential as a function of distance from the ion along the tube axis. However, an ion also experiences a change in its electrostatic self-energy upon entering a polarisable nanotube, due to two effects. First, the dielectric background provided by water changes from isotropic bulk water with ∈bulk ≈ 80 to anisotropic nanoconfined water, for which we assume ∈z = 80 and ∈φ = ∈r = 2, as in previous sections. We estimate the resulting self-energy by computing the difference in Born energy between the two backgrounds, Σ∈ = Σ1D − Σbulk. Second, the induced potential of the solid acts back on the ion itself according to eqn (6), i.e. ΣNT = ϕind(r = 0). We model the ion as a homogeneously charged ball with radius R0 and total charge Qe. Its charge density is thus n = eQ/V = 3eQ/(4πR03).
In an anisotropic dielectric background, whose dielectric tensor ∈ is diagonal with entries ∈z ≠ ∈r = ∈φ, the Laplace equation for the electrostatic potential ϕ reads ∇[∈∇ϕ(r)] = 0. Rescaling the space coordinates into x′ = x, y′ = y and z′ = αz with 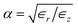 yields an isotropic dielectric constant ∈r. However, it deforms the spherical ion into an ellipsoid with half-axes a = b = R0 and c = αR0, and with charge density n′ = eQ/V′ = n/α, where
yields an isotropic dielectric constant ∈r. However, it deforms the spherical ion into an ellipsoid with half-axes a = b = R0 and c = αR0, and with charge density n′ = eQ/V′ = n/α, where 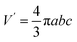 denotes the volume of the ellipsoid. The self-energy is defined as the charge density of the ion interacting with its own potential ϕ(r),
denotes the volume of the ellipsoid. The self-energy is defined as the charge density of the ion interacting with its own potential ϕ(r),
| |
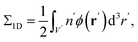 | (34) |
The electrical potential of a homogeneously charged ellipsoid inside its interior is
46| |
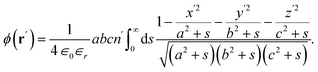 | (35) |
Thus, the integral in
eqn (34) can be evaluated using the formula for the second moments of an ellipsoid. This yields
| |
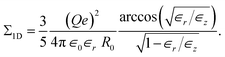 | (36) |
The self-energy in a homogeneous and isotropic dielectric background with dielectric constant
∈ is known to be
| |
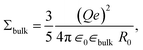 | (37) |
which coincides with
eqn (36) for
∈r →
∈z =
∈bulk.
Fig. 4a shows the water contribution to the self-energy Σ∈ = Σ1D − Σbulk for a monovalent ion (Q = 1) with radius R0 = 1 Å versus the anisotropy factor 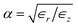 . In ångström-scale nanotubes, there is strong anisotropy,18 corresponding to small values of α and a large self-energy. Upon increasing the tube radius to R ∼10 nm, anisotropy gradually disappears and α converges to its bulk value α = 1, leading to Σ∈ = 0. Fig. 4b shows the nanotube screening contribution ΣNT, for all of our previous nanotube models, with radius R ≈ 4 Å. This contribution can be either positive or negative, depending on the electronic properties of the nanotube.7 Metallic walls yield negative self-energies, thereby reducing the energy cost of an ion entering the nanotube. In contrast, a purely dielectric tube wall can give a positive contribution to the self-energy, further repelling the ion from the nanotube. In both cases, the absolute value decreases with increasing tube radius, as the centrally-placed ion is further away from the wall (Fig. 4c). With the parameters used throughout the paper, R ≈ 4 Å, R0 = 1 Å and α ≈ 0.16, the total self-energy is dominated by the strongly anisotropic dielectric background of water, to which the nanotube polarisability only provides a small correction.
. In ångström-scale nanotubes, there is strong anisotropy,18 corresponding to small values of α and a large self-energy. Upon increasing the tube radius to R ∼10 nm, anisotropy gradually disappears and α converges to its bulk value α = 1, leading to Σ∈ = 0. Fig. 4b shows the nanotube screening contribution ΣNT, for all of our previous nanotube models, with radius R ≈ 4 Å. This contribution can be either positive or negative, depending on the electronic properties of the nanotube.7 Metallic walls yield negative self-energies, thereby reducing the energy cost of an ion entering the nanotube. In contrast, a purely dielectric tube wall can give a positive contribution to the self-energy, further repelling the ion from the nanotube. In both cases, the absolute value decreases with increasing tube radius, as the centrally-placed ion is further away from the wall (Fig. 4c). With the parameters used throughout the paper, R ≈ 4 Å, R0 = 1 Å and α ≈ 0.16, the total self-energy is dominated by the strongly anisotropic dielectric background of water, to which the nanotube polarisability only provides a small correction.
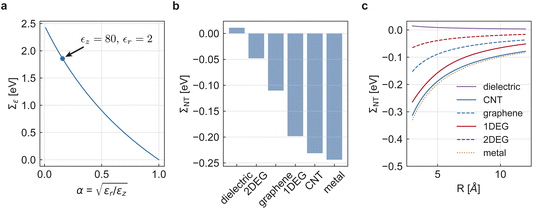 |
| | Fig. 4 Self-energy of a unit-charge ion with radius R0 = 1 Å at the centre of a polarisable nanotube. (a) Self-energy Σ∈ due to the change in dielectric background between bulk and nanoconfined water, as a function of the anisotropy factor 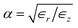 assuming ϵz = ϵbulk. The blue dot corresponds to the choice of dielectric constants used in this paper, ϵz = 80 and ϵr = 2. (b) Nanotube polarisation contribution ΣNT to the self-energy of an ion at the centre of various nanotubes with radius R = 4 Å and identical parameters as in Fig. 3. (c) Same as (b), here depending on the tube radius R. assuming ϵz = ϵbulk. The blue dot corresponds to the choice of dielectric constants used in this paper, ϵz = 80 and ϵr = 2. (b) Nanotube polarisation contribution ΣNT to the self-energy of an ion at the centre of various nanotubes with radius R = 4 Å and identical parameters as in Fig. 3. (c) Same as (b), here depending on the tube radius R. | |
6 Conclusion
In this paper, we have demonstrated that the presence of a polarisable nanotube can fundamentally alter Coulomb interactions in quasi-1D confinement. Due to the substantial anisotropy of the dielectric constant of water in ångström-scale nanotubes, Coulomb interactions are strongly enhanced. A dielectric nanopore material with a low dielectric constant further enhances Coulomb interactions, whereas a metallic nanotube material strongly screens the long-range part of the Coulomb potential. We applied Luttinger liquid theory to show that this behaviour persists under the exact treatment of electron–electron interactions in realistic 1D nanotubes: at intermediate distances, we found that a quasi-1D electron gas and a single-walled carbon nanotube screen the Coulomb potential essentially like a perfect metal. This perfect screening property cannot be reduced to a geometric effect and relies on the quantum confinement of electrons along the tube circumference. At longer distances, the quasi-1D electron gas displays Friedel oscillations, while these are suppressed in a metallic armchair CNT due to the sublattice structure. Accounting for the substantial anisotropy of water in quasi-1D confinement, we found a large self-energy barrier for an ion to enter the nanotube, which is only weakly modified by the dielectric properties of the nanotube walls.
Our approach is based on tubular response functions, which generalise surface response functions to the nanotube, arguably the most widespread geometry in nanofluidics. Our framework can be applied to compute Coulomb interactions in realistic nanotube models, including carbon and boron nitride nanotubes. While the case of a metallic carbon nanotube has been explicitly presented, the 1D-electron gas can serve as a simple model for highly doped nanotubes, where the Fermi level lies within bands with approximately quadratic dispersion. We also expect our findings to qualitatively extend to nanoporous materials, such as carbide-derived carbon, a promising candidate for supercapacitors.47 For large tube radii, our theory converges to the theory of surface response functions for planar interfaces.7
A substantial simplification in our theory is treating water as a dielectric background. This macroscopic model is known to break down in the smallest nanotubes, where the water molecules form a single 1D chain.18 MD-simulations of nano-confined water show that the axial dielectric constant oscillates close to the wall due to layering effects, similar to planar interfaces, and also the radial dielectric constant has a non-monotonous radius-dependence.18 However, even for the smallest possible tubes, uniform effective dielectric constants have been extracted from these simulations and can be used as an input to our theory.18 Short-range electrostatic interactions, in particular the ion self-energy, could be sensitive to the local molecular configuration of the water in the presence of the ion, which is not captured by effective dielectric constants. However, we expect long-range interactions to be largely unaffected, as they are dominated by the polarisability of the solid. More generally, our framework is not restricted to ions in a homogeneous background. It can be applied to any electrolyte or polar liquid whose molecules interact via coulombic forces, such as water itself. Such a treatment could test the assumption of a homogeneous dielectric background and will be left for future investigations.
In this paper, we used the tubular response function of the solid as an input. We also derived a tubular response function of the liquid. In principle, the framework could be generalised to the case in which the solid and the liquid mutually renormalise each other's responses. A dynamical treatment of this effect is closely related to the fluctuation-induced quantum friction force.48 Thus, our framework paves the way for quantitative predictions of electrolyte structure and dynamics in one-dimensional nanofluidic systems.
Author contributions
Conceptualisation: Nikita Kavokine and Peter Gispert. Methodology: Peter Gispert and Nikita Kavokine. Investigation: Peter Gispert. Writing – original draft: Peter Gispert. Writing – review & editing: Peter Gispert and Nikita Kavokine. Supervision: Nikita Kavokine.
Conflicts of interest
There are no conflicts to declare.
Data availability
All data associated with this study is available on Zenodo: https://doi.org/10.5281/zenodo.20128754.
Notes and references
- S. Porada, R. Zhao, A. Van Der Wal, V. Presser and P. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS.
- A. Siria, P. Poncharal, A.-L. Biance, R. Fulcrand, X. Blase, S. T. Purcell and L. Bocquet, Nature, 2013, 494, 455–458 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- R. B. Schoch, J. Han and P. Renaud, Rev. Mod. Phys., 2008, 80, 839–883 CrossRef CAS.
- L. Bocquet and E. Charlaix, Chem. Soc. Rev., 2010, 39, 1073–1095 RSC.
- P. Robin, A. Delahais, L. Bocquet and N. Kavokine, J. Chem. Phys., 2023, 158, 124703 CrossRef CAS PubMed.
- N. Kavokine, P. Robin and L. Bocquet, J. Chem. Phys., 2022, 157, 114703 CrossRef CAS PubMed.
- P. Robin, N. Kavokine and L. Bocquet, Science, 2021, 373, 687–691 CrossRef CAS.
- S. Teber, J. Stat. Mech.:Theory Exp., 2005, 2005, P07001 Search PubMed.
- A. Parsegian, Nature, 1969, 221, 844–846 CrossRef CAS PubMed.
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS.
- A. A. Kornyshev, J. Phys. Chem. B, 2007, 111, 5545–5557 CrossRef CAS PubMed.
- S. Kondrat, G. Feng, F. Bresme, M. Urbakh and A. A. Kornyshev, Chem. Rev., 2023, 123, 6668–6715 CrossRef CAS PubMed.
- C. C. Rochester, A. A. Lee, G. Pruessner and A. A. Kornyshev, ChemPhysChem, 2013, 14, 4121–4125 CrossRef CAS PubMed.
- L. Fumagalli, A. Esfandiar, R. Fabregas, S. Hu, P. Ares, A. Janardanan, Q. Yang, B. Radha, T. Taniguchi, K. Watanabe, G. Gomila, K. S. Novoselov and A. K. Geim, Science, 2018, 360, 1339–1342 CrossRef CAS PubMed.
- R. Wang, M. Souilamas, A. Esfandiar, R. Fabregas, S. Benaglia, H. Nevison-Andrews, Q. Yang, J. Normansell, P. Ares, G. Ferrari, A. Principi, A. K. Geim and L. Fumagalli, Nature, 2025, 646, 606–610 CrossRef CAS PubMed.
- P. Loche, C. Ayaz, A. Schlaich, Y. Uematsu and R. R. Netz, J. Phys. Chem. B, 2019, 123, 10850–10857 CrossRef CAS PubMed.
- J. Pitarke and A. Rivacoba, Surf. Sci., 1997, 377–379, 294–300 CrossRef CAS.
- M. F. Lin and K. W. K. Shung, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 6617–6624 CrossRef CAS PubMed.
- P. Longe and S. M. Bose, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 48, 18239–18243 CrossRef CAS PubMed.
- L. X. Benedict, S. G. Louie and M. L. Cohen, Phys. Rev. B:Condens. Matter Mater. Phys., 1995, 52, 8541–8549 CrossRef CAS PubMed.
- M. Lin and F. Shyu, Phys. B, 2000, 292, 117–126 Search PubMed.
- T. Ando, J. Phys. Soc. Jpn., 2005, 74, 777–817 CrossRef CAS.
- J.-C. Charlier, X. Blase and S. Roche, Rev. Mod. Phys., 2007, 79, 677–732 CrossRef CAS.
- T. Giamarchi, Quantum Physics in One Dimension, Oxford University Press, 2003 Search PubMed.
- F. D. M. Haldane, J. Phys. C: Solid State Phys., 1981, 14, 2585 CrossRef.
- J. M. Luttinger, J. Math. Phys., 1963, 4, 1154–1162 CrossRef CAS.
- S.-I. Tomonaga, Prog. Theor. Phys., 1950, 5, 544–569 CrossRef.
- D. C. Mattis and E. H. Lieb, J. Math. Phys., 1965, 6, 304–312 CrossRef CAS.
- J. Voit, Rep. Prog. Phys., 1995, 58, 977–1116 CrossRef CAS.
- Z. Yao, H. W. C. Postma, L. Balents and C. Dekker, Nature, 1999, 402, 273–276 CrossRef CAS.
- R. Egger and A. O. Gogolin, Phys. Rev. Lett., 1997, 79, 5082–5085 CrossRef CAS.
- R. Egger, Phys. Rev. Lett., 1999, 83, 5547–5550 CrossRef CAS.
- M. Bockrath, D. H. Cobden, J. Lu, A. G. Rinzler, R. E. Smalley, L. Balents and P. L. McEuen, Nature, 1999, 397, 598–601 CrossRef CAS.
- C. Kane, L. Balents and M. P. A. Fisher, Phys. Rev. Lett., 1997, 79, 5086–5089 CrossRef CAS.
- B. Coquinot, M. Becker, R. R. Netz, L. Bocquet and N. Kavokine, Faraday Discuss., 2024, 249, 162–180 RSC.
- J. Rammer, Quantum Field Theory of Non-equilibrium States, Cambridge University Press, Cambridge, 2007 Search PubMed.
- O. Sato, Y. Tanaka, M. Kobayashi and A. Hasegawa, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 48, 1947–1950 CrossRef CAS PubMed.
- Z. Li and A. Noy, Chemical Society Reviews, 2025 Search PubMed.
- R. Saito, G. Dresselhaus and M. S. Dresselhaus, Physical Properties of Carbon Nanotubes, Imperial College Press, London, Repr edn, 2012 Search PubMed.
- A. Thakur, R. Sachdeva and A. Agarwal, J. Phys.: Condens. Matter, 2017, 29, 105701 CrossRef PubMed.
- Q. P. Li, S. Das Sarma and R. Joynt, Phys. Rev. B:Condens. Matter Mater. Phys., 1992, 45, 13713–13716 CrossRef PubMed.
- T. Ando, A. B. Fowler and F. Stern, Rev. Mod. Phys., 1982, 54, 437–672 CrossRef CAS.
- E. H. Hwang and S. Das Sarma, Phys. Rev. B:Condens. Matter Mater. Phys., 2007, 75, 205418 CrossRef.
- S. Chandrasekhar, Ellipsoidal Figures of Equilibrium, Yale University Press, New Haven, 1969 Search PubMed.
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Science, 2010, 328, 480–483 Search PubMed.
- N. Kavokine, M.-L. Bocquet and L. Bocquet, Nature, 2022, 602, 84–90 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Nikita Kavokine
and
Nikita Kavokine

 ,
,  , ρ< = min{ρ, ρ′}, ρ> = max{ρ, ρ′}, and Iν and Kν are the modified Bessel functions of order ν. Here, Gm,q(ρ, ρ′) is the Fourier transform with respect to φ − φ′ and z − z′ of the Coulomb kernel satisfying the Poisson equation ∇r[∈·∇rG(r, r′)] = −δ(r − r′). Due to the decomposition in eqn (1), it is sufficient to study an individual eigenmode (m, q) of the ion potential.
, ρ< = min{ρ, ρ′}, ρ> = max{ρ, ρ′}, and Iν and Kν are the modified Bessel functions of order ν. Here, Gm,q(ρ, ρ′) is the Fourier transform with respect to φ − φ′ and z − z′ of the Coulomb kernel satisfying the Poisson equation ∇r[∈·∇rG(r, r′)] = −δ(r − r′). Due to the decomposition in eqn (1), it is sufficient to study an individual eigenmode (m, q) of the ion potential.







 , where ∈r is the radial dielectric constant for the respective material. The general solution is straightforward to obtain, but it is a rather lengthy expression in terms of modified Bessel functions. Here, we focus on an ion at the centre of the tube, which is solely determined by the angular mode m = 0. The resulting tubular response function is
, where ∈r is the radial dielectric constant for the respective material. The general solution is straightforward to obtain, but it is a rather lengthy expression in terms of modified Bessel functions. Here, we focus on an ion at the centre of the tube, which is solely determined by the angular mode m = 0. The resulting tubular response function is
 ,
,  as well as
as well as  for the respective materials. In the limit R → ∞, eqn (9) reduces to
for the respective materials. In the limit R → ∞, eqn (9) reduces to



 . The induced charge creates an induced potential, which at the interface reads
. The induced charge creates an induced potential, which at the interface reads

 , which, through the density response function χ, induces a charge inside the solid at radius ρs, creating an induced potential at the interface. For completeness, the same reasoning leads to the tubular response function of the liquid, which can be obtained by exchanging the roles of the solid and the liquid:
, which, through the density response function χ, induces a charge inside the solid at radius ρs, creating an induced potential at the interface. For completeness, the same reasoning leads to the tubular response function of the liquid, which can be obtained by exchanging the roles of the solid and the liquid:







 on an an infinite cylinder with radius R = 4 Å. (b) Band structure of a (6,6) armchair CNT with radius R ≈ 4 Å. (a and b) For the chosen electron density n = 5 × 1012 cm−2, only the lowest bands are occupied in both cases.
on an an infinite cylinder with radius R = 4 Å. (b) Band structure of a (6,6) armchair CNT with radius R ≈ 4 Å. (a and b) For the chosen electron density n = 5 × 1012 cm−2, only the lowest bands are occupied in both cases.
 in eqn (22). Rescaling the integration variable q and the coordinate z in eqn (22), we recover the formula for the isotropic case. Thus, the existing result can be generalised to the anisotropic case without explicit calculations. We obtain
in eqn (22). Rescaling the integration variable q and the coordinate z in eqn (22), we recover the formula for the isotropic case. Thus, the existing result can be generalised to the anisotropic case without explicit calculations. We obtain
 , where me denotes the bare electron mass. The charge density response function of this system has previously been reported.39 However, for completeness, we restate the main findings. Due to the periodic boundary conditions for the wavefunction around the tube circumference, the momentum in this direction is quantised as qj = j/R with
, where me denotes the bare electron mass. The charge density response function of this system has previously been reported.39 However, for completeness, we restate the main findings. Due to the periodic boundary conditions for the wavefunction around the tube circumference, the momentum in this direction is quantised as qj = j/R with  . Thus, the band structure of the electron gas has several bands, indexed by j, with energy
. Thus, the band structure of the electron gas has several bands, indexed by j, with energy  , where k is the wavevector along the nanotube axis (Fig. 2a). The eigenstates λ in eqn (17) can accordingly be labelled as λ = (k, j, σ), with σ denoting spin. Since the eigenfunctions of the electron gas are orthonormal, the non-interacting density response function becomes
, where k is the wavevector along the nanotube axis (Fig. 2a). The eigenstates λ in eqn (17) can accordingly be labelled as λ = (k, j, σ), with σ denoting spin. Since the eigenfunctions of the electron gas are orthonormal, the non-interacting density response function becomes

 denotes the Fermi wavevector corresponding to band j. At the electron density under consideration, only the lowest subband contributes. The resulting tubular response function within RPA, g0,q,ω=0 (see eqn (19)–(21)), is depicted in Fig. 3a. In the long-wavelength limit q → 0, the bare density response function χ00,q,ω=0 converges to minus the finite density of states at the Fermi level
denotes the Fermi wavevector corresponding to band j. At the electron density under consideration, only the lowest subband contributes. The resulting tubular response function within RPA, g0,q,ω=0 (see eqn (19)–(21)), is depicted in Fig. 3a. In the long-wavelength limit q → 0, the bare density response function χ00,q,ω=0 converges to minus the finite density of states at the Fermi level  , and therefore the tubular response function converges to 1 (Fig. 3a). At q = 2kF, the tubular response function features a cusp, originating from interbranch transitions from −kF to +kF. This gives rise to Friedel oscillations in the screened potential (Fig. 3b and c).
, and therefore the tubular response function converges to 1 (Fig. 3a). At q = 2kF, the tubular response function features a cusp, originating from interbranch transitions from −kF to +kF. This gives rise to Friedel oscillations in the screened potential (Fig. 3b and c).
 , derived from the tight-binding parameter γ0 = 3.033 eV and the carbon–carbon distance aCC = 1.42 Å.41
, derived from the tight-binding parameter γ0 = 3.033 eV and the carbon–carbon distance aCC = 1.42 Å.41





 yields an isotropic dielectric constant ∈r. However, it deforms the spherical ion into an ellipsoid with half-axes a = b = R0 and c = αR0, and with charge density n′ = eQ/V′ = n/α, where
yields an isotropic dielectric constant ∈r. However, it deforms the spherical ion into an ellipsoid with half-axes a = b = R0 and c = αR0, and with charge density n′ = eQ/V′ = n/α, where  denotes the volume of the ellipsoid. The self-energy is defined as the charge density of the ion interacting with its own potential ϕ(r),
denotes the volume of the ellipsoid. The self-energy is defined as the charge density of the ion interacting with its own potential ϕ(r),



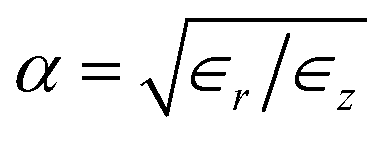 . In ångström-scale nanotubes, there is strong anisotropy,18 corresponding to small values of α and a large self-energy. Upon increasing the tube radius to R ∼10 nm, anisotropy gradually disappears and α converges to its bulk value α = 1, leading to Σ∈ = 0. Fig. 4b shows the nanotube screening contribution ΣNT, for all of our previous nanotube models, with radius R ≈ 4 Å. This contribution can be either positive or negative, depending on the electronic properties of the nanotube.7 Metallic walls yield negative self-energies, thereby reducing the energy cost of an ion entering the nanotube. In contrast, a purely dielectric tube wall can give a positive contribution to the self-energy, further repelling the ion from the nanotube. In both cases, the absolute value decreases with increasing tube radius, as the centrally-placed ion is further away from the wall (Fig. 4c). With the parameters used throughout the paper, R ≈ 4 Å, R0 = 1 Å and α ≈ 0.16, the total self-energy is dominated by the strongly anisotropic dielectric background of water, to which the nanotube polarisability only provides a small correction.
. In ångström-scale nanotubes, there is strong anisotropy,18 corresponding to small values of α and a large self-energy. Upon increasing the tube radius to R ∼10 nm, anisotropy gradually disappears and α converges to its bulk value α = 1, leading to Σ∈ = 0. Fig. 4b shows the nanotube screening contribution ΣNT, for all of our previous nanotube models, with radius R ≈ 4 Å. This contribution can be either positive or negative, depending on the electronic properties of the nanotube.7 Metallic walls yield negative self-energies, thereby reducing the energy cost of an ion entering the nanotube. In contrast, a purely dielectric tube wall can give a positive contribution to the self-energy, further repelling the ion from the nanotube. In both cases, the absolute value decreases with increasing tube radius, as the centrally-placed ion is further away from the wall (Fig. 4c). With the parameters used throughout the paper, R ≈ 4 Å, R0 = 1 Å and α ≈ 0.16, the total self-energy is dominated by the strongly anisotropic dielectric background of water, to which the nanotube polarisability only provides a small correction.
 assuming ϵz = ϵbulk. The blue dot corresponds to the choice of dielectric constants used in this paper, ϵz = 80 and ϵr = 2. (b) Nanotube polarisation contribution ΣNT to the self-energy of an ion at the centre of various nanotubes with radius R = 4 Å and identical parameters as in Fig. 3. (c) Same as (b), here depending on the tube radius R.
assuming ϵz = ϵbulk. The blue dot corresponds to the choice of dielectric constants used in this paper, ϵz = 80 and ϵr = 2. (b) Nanotube polarisation contribution ΣNT to the self-energy of an ion at the centre of various nanotubes with radius R = 4 Å and identical parameters as in Fig. 3. (c) Same as (b), here depending on the tube radius R.