 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionophilicity and transport dynamics of concentrated electrolytes in sub-nanometre graphite confinement
Fulu Zhou *ab and
Paola Carbone*a
*ab and
Paola Carbone*a
aDepartment of Chemistry, University of Manchester, Oxford Rd, Manchester, UK. E-mail: paola.carbone@manchester.ac.uk; fulu.zhou@postgrad.manchester.ac.uk
bDepartment of Chemical Engineering, University of Manchester, Oxford Rd, Manchester, UK
First published on 23rd April 2026
Abstract
Nanoconfined electrolytes exhibit distinct structural and transport properties critical for emerging energy storage and nanofluidic technologies. Graphite nanochannels, with their atomically smooth surfaces and ionophilicity, serve as ideal model systems to explore ion-specific interactions under extreme confinement. To investigate the ionophilicity of graphite, we performed molecular dynamics simulations on four aqueous electrolytes—LiCl, LiTFSI, NaCl, and NaTFSI—confined within 0.89 nm graphite slit pores. For LiCl and LiTFSI, additional simulations across a wide range of concentrations up to the water-in-salt regime were conducted. In agreement with experiments, at low concentrations, the channels show a clear anion selectivity: TFSI−-based electrolytes exhibit strong ion enrichment within the channel, whereas Cl−-based systems display significantly weaker adsorption. This preference is, however, notably reduced at higher concentrations, where ionic distributions become more uniform regardless of anion type. These results are rationalized in terms of capillary pressure and liquid viscosity, which ultimately govern the channel ions uptake. Our findings provide molecular-level insights into how ion-specific interactions regulate transport dynamics in narrow graphite channels, offering guidance for designing efficient nanoconfined electrochemical systems.
Introduction
Ion selectivity—the affinity or repulsion of materials toward specific ions—plays an important role in determining interfacial ion distributions and transport behaviours. The distinct affinities of materials toward particular ions have stimulated extensive research in diverse scientific and industrial fields. For example, in separation and purification technologies, ion selectivity is essential for processes such as seawater desalination and brackish water treatment, which typically rely on reverse osmosis or nanofiltration to selectively remove target ions.1–5 And in electrochemical systems, the ability of solid interfaces to preferentially adsorb or repel specific ions directly governs charge storage, energy conversion, and ion-transport efficiency.6–8 Ion selectivity is not only essential in bulk electrolytes, but also critical under nanoscale confinement.9,10 When electrolytes are confined within nanoscale pores, their structural and dynamic behaviors deviate significantly from the bulk.11–13 The restricted space alters their hydration and screening and magnifies specific ion–surface interactions.9,14,15 Recent studies have reported distinct interfacial phenomena such as ion layering, charge inversion, and concentration-dependent selectivity, which further enable potential applications in nanofluidics and advanced energy storage.16–19 These findings highlight that ionophilicity is vital for understanding the molecular structure and electrochemical properties of confined electrolytes.In recent years, graphite and graphene-based nanochannels have emerged as ideal platforms to investigate such confined ion phenomena.20–23 Graphite slit pores with subnanometer spacing are particularly well suited for probing ion selectivity because their geometry is comparable to the diameter of hydrated ions. A range of experimental approaches has been developed to probe ion behaviour under nanoconfinement: researchers use small-angle X-ray and neutron scattering to capture changes in the concentration of electrolyte ions in micropores during charging;24–26 attenuated total reflection surface-enhanced infrared absorption spectroscopy is used to observe ion electrosorption;27–29 and nuclear magnetic resonance spectroscopy is also used to observe the behavior of adsorbates within carbon micropores.30–32 However, due to the intrinsic structural complexity of nanochannels and the limited spatial resolution at the nanoscale, direct experimental observation of ion behavior within the pores remains challenging.
Theoretical studies have also provided valuable insight into ion behaviour under nanoconfinement, emphasizing the importance of factors such as ion size, pore geometry, and solvation properties in governing adsorption and transport.9,10,13,33,34 Molecular dynamics (MD) simulations offer an approach to observe atomistic details of how specific ions approach and interact with solid surfaces, capturing ion dehydration and local structural rearrangements that are difficult to observe experimentally. Kondrat and collaborators35 used mean-field molecular dynamics simulations to study ion transport in nanochannels and introduced the concept of ionophilicity and ionophobicity, which now have been shown to strongly influence the charge-storage mechanism and kinetics of electrolyte–nanochannel systems.36–38 Anousheh et al.17 used classical molecular dynamic simulation to investigate the ionic structure of different concentrate electrolytes under confinement. Rajput et al.20 performed molecular-dynamics simulations of the ionic liquid [EMIM][NTf2] confined inside charged graphitic slit nanopores, revealing how surface charge governs interfacial ion layering and dynamical asymmetry under confinement. However, many simulation studies have focused on behaviours of ionic liquids or pure water,22,23,39,40 while the ion-specific adsorption of hydrated anions and the concentration dependence of ionophilicity within subnanometer graphite channels remain insufficiently explored, especially at high concentrations.
In a recent experimental study, Bragg et al.41 reported that porous carbon exhibits distinct anion selectivity at low electrolyte concentrations: hydrophobic anions such as bis(trifluoromethanesulfonyl)imide (TFSI−) show strong enrichment within the small pores, while halide anions like Cl− display significantly weaker adsorption. Such behaviour reflects the inherent ion-specific affinity of graphite surfaces, yet it remains unclear how this preference evolves when the electrolyte concentration increases, and electrostatic screening becomes more pronounced. Concentration is a critical but often overlooked parameter governing interfacial ion structure, solvation, and collective correlations. A comprehensive understanding of its influence is essential for linking microscopic ionophilicity to macroscopic transport properties in confined systems.
In this work, we employ molecular dynamics simulations to further investigate the ionophilicity of graphite nanochannels for four aqueous electrolytes—LiCl, NaCl, LiTFSI, and NaTFSI—with a particular focus on the concentration dependence of LiCl and LiTFSI systems. We investigate how different anions at varying concentrations affect ion adsorption, hydration structure, and interfacial density distributions within 0.89 nm slit pores. Our simulations successfully reproduce the clear anion selectivity of porous carbons under dilute conditions reported by Bragg et al.,41 where TFSI−-based electrolytes exhibit strong enrichment near the graphite surface, while Cl−-based systems remain largely excluded. Here we show that this selectivity gradually weakens with increasing electrolyte concentration, eventually leading to nearly identical ionic distributions inside the channel and in the bulk.
Methods
Nanochannel setup
The two-dimensional channel is simulated by two sets of four rigid graphite sheets (each 5 nm × 10 nm), which form the upper and lower boundaries of the channel. The effective channel height, H, is defined as the centre-of-mass distance between the innermost graphene sheets that confine the channel, minus the thickness of a single graphene sheet (∼0.34 nm). In this study, H is fixed at 0.89 nm, consistent with the pore dimensions reported in the experimental work by Bragg et al.41 This specific sub-nanometer height is critical as it accommodates distinct multi-layered water structures while maintaining strong confinement effects.22 Two reservoirs of equal amounts of electrolyte are placed on both sides of the channel to contain the bulk electrolyte. Adjacent to each reservoir, a vacuum region with a volume three times larger than that of the reservoir is added, ensuring that long-range electrostatic interactions between the two reservoirs over periodic images are negligible.42 The entire simulation box is treated with periodic boundary conditions in all directions. A schematic snapshot of the simulation setup is illustrated in Fig. 1. | ||
| Fig. 1 Schematic representation of the simulation system for a 1 m LiTFSI electrolyte confined within a graphite nanochannel. | ||
Graphite contacts with bulk electrolyte
A reference system representing the graphite–electrolyte interface in the bulk state was also constructed to examine the structural properties of the unconfined electrolyte. In this setup, a four-layer graphite plane was placed in direct contact with a cubic electrolyte box measuring 6 nm × 6 nm × 6 nm. On the opposite side of the electrolyte, an 8 nm thick vacuum region was added to prevent long-range electrostatic interactions between periodic images along the surface-normal direction.42 Periodic boundary conditions were applied in the x and y directions, but not in the z direction, which is perpendicular to the graphite surface.Droplet simulation
To further examine the wetting behavior of different electrolytes, additional droplet simulations were performed to determine the contact angle of each electrolyte on graphite. A cylindrical droplet geometry was employed to minimize the influence of line tension.43–46 In these simulations, an equilibrated electrolyte box was initially placed above a four-layer graphite surface, allowing spontaneous spreading and adsorption during the simulation. The contact angle was measured after the system reached equilibrium.In this work, four different salts were investigated: LiCl, NaCl, LiTFSI, and NaTFSI. The TIP4P_2005 model47 was employed to represent water, with the SHAKE constraint algorithm48 applied to fix the lengths and angles of the Ow–H bonds in water. Following our previous study, the Madrid 2019 ion force field was used to simulate LiCl and NaCl and describe their interactions with water,49,50 while LiTFSI and NaTFSI were modelled with the AMBER force field.51,52 The choice of this combination of force field has been extensively validated for aqueous LiCl and LiTFSI electrolytes across a wide concentration range, from dilute solutions to the water-in-salt regime.50 In particular, this parameter set has been shown to reproduce experimental densities, surface tensions, and key structural properties. In the present work, we therefore employ this established force field to investigate the effects of sub-nanometre graphite confinement. Non-bonded interactions were described by the Lennard-Jones (12–6) potential and Coulomb interaction. The real-space cutoff for Lennard-Jones and Coulomb interactions was set to 1.2 nm. The Lennard-Jones (12–6) potential was applied with a switching function from 1.0 to 1.2 nm to smooth the truncation. Long-range electrostatics were treated by the Particle–Particle Particle–Mesh solver.53 The Lorentz–Berthelot combining rule was applied for ion–carbon cross parameters. The cross-interaction energy parameter εC–Ow between carbon and water oxygen was taken from Werder et al., in order to reproduce the experimental contact angle between water and graphite.22,23,54 All simulations were carried out in the NVT ensemble using the Nosé–Hoover thermostat55,56 at 298.5 K, with a relaxation time of 0.5 ps and a timestep of 2 fs. The equilibration time varied depending on the specific setup. For the nanochannel system, simulations were continued until the electrolyte concentration inside the channel reached a steady value. For the reference bulk and droplet systems, the simulations were performed long enough to obtain statistically stable data. The graphite carbon atoms were kept uncharged and fixed throughout all simulations. All simulations were performed by the LAMMPS software package.57
Results and discussion
Ion selectivity under low concentration
In this study, the ion selectivity is quantified by the concentration ratio, α, between the molality of the electrolyte inside the channel (cin) and that in the reservoir (cres), i.e.,
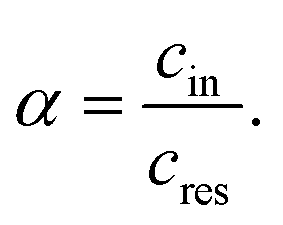 | (1) |
Fig. 2(a) presents the concentration ratio α (specifically, α30 ns) for 1 m LiCl, LiTFSI, NaCl, and NaTFSI confined within the graphite nanochannel after approximately 30 ns of molecular dynamics simulation. A clear anion-dependent selectivity is observed: the graphite channel exhibits pronounced ionophobic behavior toward Cl−, but shows strongly ionophilic behavior toward TFSI−. This observation is consistent with experimental results reported by Bragg et al.41 for 1 M electrolytes. In their work, nuclear magnetic resonance (NMR) spectroscopy was used to quantify the ion populations adsorbed in activated carbon cloth (ACC-20). Crucially, the ACC-20 material possesses a mean pore size of 0.89 nm, matching the geometry of our simulation. Furthermore, the experimental ratios were measured under saturation loading conditions (electrolyte volume exceeding pore volume), ensuring that the in-pore ions were in thermodynamic equilibrium with the bulk reservoir. This enables a meaningful comparison between the experimental cationic molar ratio (χin-pore/χneat) and our calculated concentration ratio α. Results from experiments are also presented in Fig. 2(a) for comparison.
 | ||
| Fig. 2 Ion selectivity and adsorption dynamics of 1 m electrolytes in graphite nanochannels. (a) Equilibrium concentration ratio α at 30 ns for four electrolytes (LiTFSI, NaTFSI, LiCl, NaCl). Simulation results (purple) are compared with available experimental data41 (hatched bars). TFSI−-based electrolytes exhibit strong ionophilic behaviour, whereas Cl−-based electrolytes show pronounced ionophobicity. (b) Time evolution of the concentration ratio α for 1 m LiTFSI and 1 m LiCl. LiTFSI displays continuous enrichment throughout the simulation, while LiCl rapidly reaches a low α plateau. (c) Time evolution of the number of ions and water molecules inside the channel for 1 m LiTFSI. Inset: sustained accumulation of TFSI− and the corresponding increase of Li+, accompanied by gradual water expulsion. (d) Time evolution of species populations inside the channel for 1 m LiCl. Inset: both ions remain at low levels, consistent with the ionophobic character of the channel toward LiCl. | ||
It has been discussed in the literature that ion hydration energy and ion–channel interaction energy play an important role in governing the ion selectivity when the channel is uncharged.41,58–65 Highly charge-dense ions, such as Li+, Na+, and Cl−, generally possess large hydration energies and bind strongly to water molecules. When such ions approach a subnanometer carbon pore, the interaction with the graphite surface is insufficient to compensate for the energetic cost of partial dehydration, resulting in ionophobic behaviour. In contrast, the hydrophobic TFSI− anion exhibits significantly weaker hydration and stronger van der Waals interactions with the graphitic surface, facilitating its adsorption and enrichment within the channel.
Our results indicate that this anion-driven mechanism determines whether the channel's selectivity toward the electrolyte is ionophilic or ionophobic under low-concentration conditions. Although the Li+ cation exhibits a slightly stronger channel affinity than Na+, both species are significantly constrained by their high hydration energies. Although Li+ shows a slightly higher affinity than Na+, both cations are hindered by their high hydration energies, resulting in an overall ionophobic behaviour for both. Consequently, we focus more on LiCl and LiTFSI in the remainder of this study to isolate and highlight the role of the anion.
To further investigate the adsorption process, we monitored the evolution of the concentration ratio, α, in the simulations. As shown in Fig. 2(b), the α value for LiCl reaches a plateau after 20 ns of simulation, indicating that the concentration ratio between the channel and the bulk has reached equilibrium. In contrast, the ratio for LiTFSI continued to increase. To thoroughly investigate this dynamic, the simulation for 1 m LiTFSI was extended until a clear, stable equilibrium in α was observed. As shown in Fig. 2(c), LiTFSI continuously exhibited channel enrichment throughout the 250 ns simulation duration and then reached equilibrium. A further analysis of the evolution of different species within the channel reveals the mechanism: the channel's ionophilic nature towards TFSI− drives a sustained influx of the anions. To maintain electro-neutrality within the channel, the number of Li+ ions consequently increases, effectively following the TFSI− influx. Concurrently, the number of water molecules is observed to decrease after the initial channel filling is complete, reaching the equilibrium. This suggests that the substantial increase in the ratio α for the LiTFSI solution is driven not only by the TFSI− attraction to the channel but also by its hydrophobic character, which facilitates the expulsion of water molecules from the channel. After extending the simulation to 300 ns, it is observed that the concentration ratio between the channel and the bulk, α, for LiTFSI eventually reaches equilibrium at approximately 11.8. In contrast, this behaviour was not observed for the 1 m LiCl solution. As shown in Fig. 2(d), in this case, the number of water molecules quickly reaches equilibrium after entering the channel, and neither Li+ nor Cl− ions showed any significant disparity in their respective counts within the channel.
Ion selectivity under high concentration
Having established the anion-governed selectivity at low concentrations, we next examine how this behavior evolves with increasing electrolyte concentration. To this end, we conducted four distinct concentration simulations for both LiTFSI and LiCl solutions, specifically at 1 m, 3 m, 10 m, and 16 m. Fig. 3(a) shows the equilibrium values of α for LiTFSI solutions at different concentrations. The results demonstrate a clear trend: as the bulk concentration of LiTFSI increases, the equilibrium concentration ratio progressively decreases. This indicates that the ion selectivity of the graphite channel weakens with increasing electrolyte concentration. | ||
| Fig. 3 (a) Equilibrium concentration ratio αeq (purple bars, left axis) and the corresponding in-channel equilibrium molality Ceqin (pink bars, right axis) for TFSI− at different bulk concentrations (1 m, 3 m, 10 m, and 16 m). (b) Time evolution of the number of Li+, TFSI−, and H2O molecules inside the channel for the 16 m LiTFSI system. | ||
Specifically, as the LiTFSI concentration increases from 1 m to 3 m, the equilibrium channel-to-bulk concentration ratio α is observed to decrease to approximately 3.3. By comparing the in-channel TFSI− concentration (Cin) at equilibrium, cequin, we confirm that increasing the initial electrolyte concentration does not significantly enhance the channel's enrichment of TFSI−. In contrast, when the LiTFSI concentration of the reservoir is raised to 10 m or 16 m, the channel's ion selectivity significantly declines, and the internal and external solution concentrations become nearly identical (i.e., α ≈ 1). Furthermore, the phenomenon of water expulsion, which was prominent at low concentrations, also ceases to occur, as shown in Fig. 3(b). These results indicate that the ionophilicity of the graphite channel for TFSI− ions has an upper limit. The channel's enrichment effect is more pronounced for low-concentration LiTFSI solutions, while this ion selectivity virtually disappears when the LiTFSI concentration is raised above a certain threshold.
For the LiCl solutions, a similar concentration-dependent weakening of ion selectivity is also observed. As shown in Fig. 4(a), increasing the initial LiCl concentration in the reservoir leads to a gradual rise in the equilibrium concentration ratio α. This indicates that the channel becomes progressively less ionophobic toward LiCl at higher concentrations. Consistent with the behaviour seen in the LiTFSI systems, when the bulk LiCl concentration in the reservoir is increased to 10 m or 16 m, the channel-to-bulk concentration ratio approaches unity (α ≈ 1), demonstrating that the inherent ionophobic character of the graphite channel toward LiCl effectively disappears under these highly concentrated conditions.
 | ||
| Fig. 4 (a) Equilibrium concentration ratio αeq (purple bars) for LiCl at different bulk concentrations (1 m, 3 m, 10 m, and 16 m). (b) Time evolution of the number of Li+, Cl−, and H2O molecules inside the channel for the 16 m LiCl system. | ||
Structure analysis
To better understand the electrolyte structure within the nanochannel and the concentration-dependent changes in the α ratio, we performed a detailed structural analysis of the confined solution. Our investigation focuses on the normalized density profiles (ñ(z)) and the local solvation environments derived from the radial distribution function (RDF) and coordination number (CN).
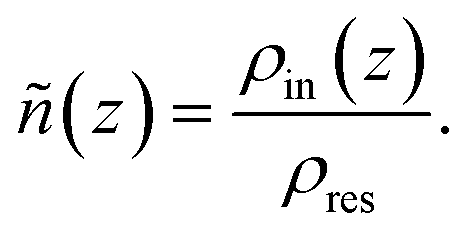 | (2) |
Here, the z-coordinate is defined along the direction normal to the graphite surfaces. The full z-range shown in Fig. 5 and 6 includes both the confined electrolyte region and the thickness of the graphite sheets on either side of the pore. The channel region is defined by the distance between the two innermost graphite layers, which are explicitly marked by vertical dashed lines in the figures.
For the 1 m LiCl solution (Fig. 5(a)), we observe a clear stratification of the confined fluid: water molecules form three distinct layers across the channel, Li+ cations predominantly distribute within two layers adjacent to the walls, while Cl− anions are primarily concentrated in the central layer. This pronounced layering is typical of confined aqueous electrolytes at low concentrations.
 | ||
| Fig. 5 (a and b) Normalized density profiles of H2O, Li+, and Cl− along the surface-normal (z) direction across the graphite nanochannel for (a) 1 m and (b) 16 m LiCl solutions, showing layered structuring near the graphene walls. The vertical dashed lines indicate the positions of the centre of mass of the two innermost graphite layers, which define the 0.89 nm fluid-accessible channel region. (c) Radial distribution functions g(r) between Li+ and O atoms in the confined (channel) and bulk environments at 1 m, indicating similar short-range hydration structures. (d) Li–Ow coordination number as a function of electrolyte concentration for both confined and bulk systems, demonstrating only a slight reduction in hydration with increasing concentration. | ||
For the highly concentrated 16 m solution (Fig. 5(b)), a significant structural transition is observed. While the layered structure of the water molecules remains largely unchanged, the density profiles of Li+ and Cl− exhibit a pronounced overlap within the channel. This overlapping phenomenon aligns with the ion overlap previously reported at the high-concentration LiCl–graphite interface in our previous work.66 This occurs because the high concentration leads to severe solvent scarcity, which compels the strongly hydrated Li+ cations to attract and co-adsorb with Cl− anions via solvent-separated ion pairs, effectively forcing a near 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Li:Cl ratio at the interface. Note that although Fig. 5(b) shows a slight difference in the peak heights of Li+ (red) and Cl− (light green), the total number of the two ions inside the channel remains equal, with this minor difference in normalized peak height resulting from the specific bin size used for density calculation.
:1 Li:Cl ratio at the interface. Note that although Fig. 5(b) shows a slight difference in the peak heights of Li+ (red) and Cl− (light green), the total number of the two ions inside the channel remains equal, with this minor difference in normalized peak height resulting from the specific bin size used for density calculation.
Furthermore, we assessed the local solvation environment of Li+ by calculating the radial distribution function g(r) and coordination number (CN) between the Li+ cation and the oxygen atom of water (Ow). This analysis evaluates the stability of the Li+ hydration shell under nano-confinement. As depicted in Fig. 5(c), the nanoconfinement effect leads to tighter packing of the Li+–Ow correlation in the 1 m case, evidenced by a higher first-peak value in the channel's RDF compared to the bulk solution. Crucially, however, a comparison of the coordination numbers in Fig. 5(d) shows that Li+ maintains its bulk hydration shell structure, indicating that the channel's confinement effect does not significantly perturb the primary solvation layer of the cation.
 | ||
| Fig. 6 (a and b) Normalized density profiles of H2O and Li+ along the surface-normal (z) direction across the graphite nanochannel for LiTFSI solutions at different concentrations, showing persistent layered structuring near the graphite walls. The vertical dashed lines mark the positions of the two innermost graphite layers, which define the 0.89 nm fluid-accessible channel region. (c) Radial distribution functions g(r) between Li+ and O atoms in the confined (channel) and bulk environments at 1 m, indicating similar short-range hydration structures. (d) Li–Ow coordination number as a function of electrolyte concentration for both confined and bulk systems, demonstrating only a slight reduction in hydration with increasing concentration. | ||
A similar trend is observed for the Li+ cation distribution (Fig. 6(b)). As the LiTFSI concentration in the reservoir increases, the enrichment effect on Li+ weakens, evidenced by a progressive decrease in the normalized density peak heights. Despite this concentration-dependent decrease in accumulation, Li+ maintains a similar layered structure within the channel at all concentrations.
Similarly, we calculated the Li+–Ow RDF (Fig. 6(c)) and coordination number for LiTFSI solutions at the four equilibrium concentrations (Fig. 6(d)) to evaluate the stability of the Li+ hydration shell. A higher first-peak value in the channel's RDF compared to the bulk solution is also observed for the 1 m case, and the results of coordination number consistently show that the Li+–Ow coordination number inside the confined channel is identical to that in the bulk solution for all four concentrations. These findings—particularly the layered water structure and the stable Li+ solvation shell—underscore the critical role of solvent in governing the ion selectivity of the channel.
While radial distribution functions are often used to identify structural changes, an equally important observation here is the absence of any significant modification to the primary hydration shell of Li+ under confinement. As shown in Fig. 5(c) and 6(c), the first Li+–Ow peak position and overall short-range structure of g(r) inside the nanochannel closely match those in the bulk solution. A higher first peak is observed in both electrolytes, indicating that nano-confinement does not disrupt the Li+ solvation structure but instead renders the hydration shell more compact. This is further confirmed by the coordination number analysis (Fig. 5(d) and 6(d)), which shows that Li+ retains a bulk-like hydration number across all concentrations investigated.
This structural integrity of the solvation shell has important physical implications. It demonstrates that Li+ does not undergo partial dehydration upon entering the sub-nanometre graphite channel. Consequently, the energetic barrier associated with ion entry at low concentrations originates from the necessity of accommodating a fully hydrated cation within a hydrophobic confinement, rather than from dehydration-induced restructuring. This constraint places a strict upper limit on ion enrichment and directly explains why the channel's selectivity weakens at high concentrations, where solvent availability becomes insufficient to sustain additional hydrated ions.
For LiTFSI solutions, the behaviour observed at low concentrations can be rationalised by considering the balance between ion–surface interactions, hydration constraints, and the availability of water molecules within the nanochannel. At dilute concentrations, the strong attraction of the graphite channel toward TFSI−, together with the hydrophobic character of the anion, drives the sequence of events observed in our simulations: (i) the solution initially enters the channel, (ii) water molecules are partially expelled, and (iii) TFSI− continues to accumulate inside the channel. However, this process cannot proceed indefinitely. Once the number of confined water molecules decreases below a certain threshold, the enrichment ceases. This is because the accompanying Li+ ions possess high hydration energies and require a sufficient number of water molecules to maintain their solvation shells. The repulsion of water by TFSI− is therefore insufficient to disrupt the hydration structure of Li+.
Moreover, although the number of water molecules inside the channel decreases during the enrichment process, the layered water structure within the channel remains largely preserved. As a result, the molality of TFSI− within the channel does not increase indefinitely. Importantly, these structural and solvation constraints do not scale with increasing bulk concentration in the reservoir, which explains why the equilibrium in-channel concentration for the 3 m LiTFSI solution is not significantly higher than that of the 1 m system. When the reservoir concentration is further increased to 10 m, the electrolyte transitions from a dilute solution to a water-in-salt regime. Under these conditions, modifying the concentration inside the channel becomes increasingly difficult: the reduced solvent availability and the strengthened ion–ion correlations prevent the channel from perturbing the bulk-like structure of the solution. Correspondingly, analysis of the confined water population shows that the water-expulsion effect observed at low concentrations is substantially weakened, leading to an equilibrium value of α close to unity. At an even higher reservoir concentration of 16 m, the channel loses its ability to alter the solution structure altogether. In this case, the concentration ratio remains essentially unity throughout the entire filling process, and the water-expulsion mechanism no longer occurs. These observations demonstrate that at extremely high concentrations, the channel no longer exhibits a measurable selectivity toward TFSI−.
For LiCl solutions, a similar concentration-dependent behaviour is observed, yet the underlying mechanism differs from that of LiTFSI due to the strong hydration of both Li+ and Cl−. In dilute solutions, the high hydration energies of these small, highly charge-dense ions prevent them from shedding water molecules when approaching the hydrophobic graphite surface. As a result, the graphite nanochannel exhibits pronounced ionophobic behaviour toward LiCl, reflected by a low equilibrium α value. However, as the bulk concentration increases, the degree of ionophobicity diminishes. In 3 m solution, the number of water molecules per ion decreases, weakening the hydration shells of both Li+ and Cl−. This reduction in hydration strength lowers the energetic penalty associated with partial dehydration, making LiCl less strongly excluded from the channel. Consequently, α increases with concentration. When the LiCl concentration reaches 10 m and 16 m, the system approaches a water-in-salt-like regime in which ion–ion correlations dominate the solution structure. Under these conditions, the graphite channel can no longer significantly perturb the internal organization of the electrolyte. Meanwhile, the high chemical potential of the reservoir drives LiCl into the channel, resulting in nearly identical internal and external concentrations (α ≈ 1). Thus, at sufficiently high concentrations, the intrinsic ionophobicity of the channel toward LiCl effectively disappears.
Channel filling dynamics: wetting properties, capillary pressure and viscosity
Another notable observation in our simulations is the significant variation in the channel filling time (t) among electrolyte solutions of different compositions and concentrations, as summarized in Tables 1 and 2. To understand the origin of this observation, we analyse two key factors that govern the liquid transport in the nanochannel: the capillary pressure (Pcap), which provides the driving force determined by the wetting properties of the electrolyte, and the viscosity (η), which acts as the primary resistive force opposing the transport.| Molality [m] | θ [°] | γlv [mN m−1] | Pc [MPa] | t [ns] |
|---|---|---|---|---|
| 1 | 43.3 ± 1.7 | 60.6 ± 0.3 | 93.4 ± 3.6 | 0.28 ± 0.02 |
| 3 | 23.9 ± 2.5 | 50.4 ± 0.5 | 103.1 ± 4.2 | 0.43 ± 0.03 |
| 10 | 0.0 | 48.4 ± 1.3 | 108.8 ± 2.9 | 2.30 ± 0.07 |
| 16 | 0.0 | 45.5 ± 2.1 | 100.3 ± 4.7 | 78.0 ± 0.05 |
| Molality [m] | θ [°] | γlv [mN m−1] | Pc [MPa] | t [ns] |
|---|---|---|---|---|
| 1 | 65.4 ± 0.5 | 65.1 ± 0.6 | 60.90 ± 1.9 | 0.56 ± 0.03 |
| 3 | 68.4 ± 0.6 | 67.6 ± 0.4 | 55.91 ± 2.1 | 0.84 ± 0.08 |
| 10 | 78.2 ± 0.3 | 76.6 ± 0.8 | 35.21 ± 1.8 | 35.0 ± 0.15 |
| 16 | 81.3 ± 0.4 | 79.4 ± 1.6 | 27.00 ± 2.9 | 128.0 ± 0.25 |
To elucidate the underlying mechanism, we first formalize the capillary pressure (Pcap) for our system. In a quasi-static intrusion process, the pressure difference across a curved liquid–vapour interface is governed by the Young–Laplace equation. The general formal of the Young–Laplace equation is usually written as,
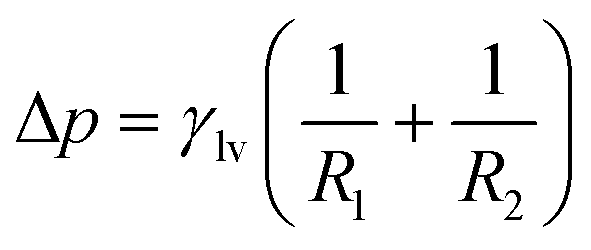 | (3) |
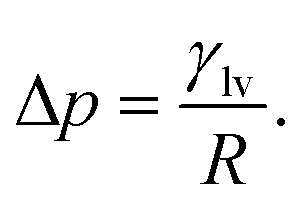 | (4) |
The radius of curvature R is related to the effective channel height H and the static contact angle θ (measured through the liquid) via simple geometry of a circular arc,
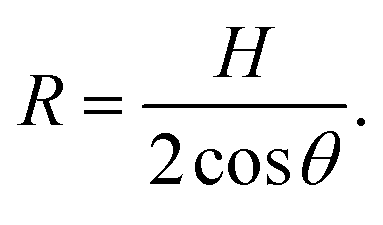 | (5) |
Substituting eqn (5) into eqn (4) yields an explicit expression for the capillary pressure driving liquid intrusion into the two-dimensional graphite slit,
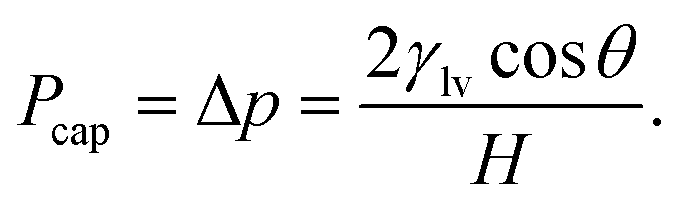 | (6) |
For a given channel height H, the magnitude of Pcap is therefore controlled by the liquid–vapour surface tension and the liquid–graphite contact angle. Both the contact angle (θ) and the liquid–vapour surface tension (γlv) were obtained directly from molecular simulations. The contact angle was determined from separate droplet simulations for each electrolyte and concentration. An equilibrated cylindrical droplet was initially placed above a four-layer graphite slab and allowed to spread spontaneously on the surface. After equilibration, the droplet profile was averaged over time and fitted to a circular arc, from which the macroscopic contact angle θ was extracted and reported in Tables 1 and 2. Specifically, the 10 m and 16 m LiTFSI solutions both exhibited complete wetting on the graphite surface, which is recorded as θ = 0.0° in Table 1. The liquid–vapour surface tension γlv was calculated using the Kirkwood–Buff mechanical route from planar liquid–vapour interface simulations.22,46,67–70 For each electrolyte and concentration, a slab of liquid was placed in equilibrium with its vapour under periodic boundary conditions, and the surface tension γlv is calculated by the equation:
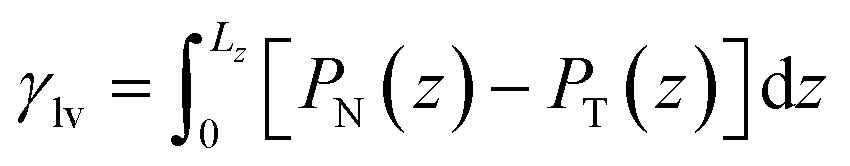 | (7) |
The resistive factor, the electrolyte viscosity (η), is equally important. High viscosity fundamentally slows down mass transport, acting in opposition to the capillary driving force. The experimental viscosity values for LiTFSI and LiCl solutions across different molalities, measured at 25 °C and 1 bar, were sourced from literature and are presented in Table 3. We now analyse the transport dynamics by considering both Pcap and η.
θ) rises significantly. The net result is a strong increase in capillary pressure (Pcap), rising from 93.4 MPa to 108.8 MPa for 10 m, and 100.3 MPa for 16 m. This increasing driving force is highly favorable for filling. Conversely, the LiTFSI viscosity exhibits an even more pronounced increase with concentration than LiCl (e.g., 1.4 mPa s at 1 m to 62.9 mPa s at 20 m), representing a massive increase in resistive force.For 1 m, 3 m and 10 m, despite the rising viscosity, the filling time (t) remains extremely fast (e.g., 0.28 ns to 2.30 ns). This is because the increasing capillary pressure (Pcap) driven by the high wettability and the channel's ionophilic mechanism, provides a dominant force that overcomes the rising viscous resistance. However, at the highest concentration of 16 m, the viscosity (η) becomes the overwhelmingly dominating factor. Although the capillary pressure is still high (Pcap ≈ 100 MPa), the exponential rise in viscous drag means the total filling time dramatically increases from 2.30 ns to 78.0 ns. This shift confirms a transition point where viscous forces suppress the capillary driving mechanism, limiting the rate of dynamic transport.
An additional observation was made during the 16 m LiTFSI droplet simulation. Even though the system eventually reaches an equilibrium contact angle of 0.0°, the droplet takes an exceptionally long time to spread and often maintains its distinct droplet shape on the surface for the majority of the simulation duration. This slow dynamic spreading, despite the thermodynamic driving force (Pcap), provides direct visual confirmation that the extremely high viscosity of the 16 m solution is the sole limiting factor to its transport properties. This observation aligns with early experimental work78 where a poor wetting is observed for 20 m LiTFSI.
Conclusions
In this work, we employed molecular dynamics simulations to systematically investigate the concentration-dependent ion selectivity and transport dynamics of aqueous LiCl and LiTFSI electrolytes confined within sub-nanometer graphite channels. By quantifying selectivity using the concentration ratio α, elucidating mechanisms via structural analysis, and calculating the energetic balance between capillary pressure (Pcap) and viscosity (η), we have provided molecular-level insights into the shift from ion-specific surface affinity to bulk transport limitations.At low concentrations, our simulations confirm a stark, anion-dependent selectivity. TFSI−-based electrolytes exhibit strong ionophilic enrichment driven by the anion's hydrophobic nature and strong van der Waals attraction to the graphite surface, accompanied by water expulsion. Conversely, Cl−-based systems show pronounced ionophobicity due to the high hydration energy of Cl−, which imposes a significant energetic barrier to channel entry. Crucially, we found that for both salts, the intrinsic selectivity of the channel substantially weakens at high concentrations, ultimately leading to a loss of preference where the channel and bulk concentrations are nearly identical (α → 1).
The structural analysis revealed the distinct energetic and structural constraints governing this weakening selectivity. For LiTFSI solutions, the enrichment process is intrinsically limited because the Li+ cation retains its stable, bulk-like hydration shell structure, providing a structural constraint that halts indefinite water expulsion. For LiCl solutions, the ionophobic character is lost in the water-in-salt (WiS) regime because the high concentration leads to solvent scarcity, which weakens the hydration shells of both Li+ and Cl−, effectively lowering the energetic barrier for channel entry. This WiS state also drives the formation of solvent-separated ion pairs (SSIPs), compelling Li+ and Cl− to co-adsorb, which is visually confirmed by the overlap of their density profiles.
Analysis of the channel filling time (t) exposed a critical competition between the capillary driving force (Pcap) and the viscous resistance (η). In the LiTFSI system, the increasing capillary pressure (due to enhanced wettability, θ → 0°) provides a dominant driving force that overcomes the rising viscous drag up to 10 m. The supremacy of this capillary mechanism is demonstrated by the 10 m LiTFSI solution filling the channel orders of magnitude faster than the 10 m LiCl solution, despite LiTFSI having a viscosity nearly four times greater. However, at the highest concentration of 16 m, the exponential increase in viscosity becomes the overwhelmingly dominant factor, suppressing the maximum Pcap and causing a dramatic rise in the filling time (t → 78 ns).
These findings provide crucial molecular-level design rules for nanofluidic and electrochemical systems utilizing concentrated electrolytes. They emphasize that the primary limitation on mass transport in the high-concentration/WiS regime shifts from ion-specific interfacial affinity to bulk fluid viscosity. Future efforts to optimize high-power devices must therefore focus on engineering WiS electrolytes with reduced viscosity, alongside maintaining the thermodynamically favorable wetting properties that maximize capillary driving forces.
Author contributions
Fulu Zhou: methodology, investigation, formal analysis, writing – original draft. Paola Carbone: conceptualization, methodology, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article, including simulation and configuration files, have been uploaded as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5fd00147a.Acknowledgements
P. C. would like to thank the Royal Society for the industrial fellowship IF\R2\242053.Notes and references
- L. F. Greenlee, D. F. Lawler, B. D. Freeman, B. Marrot and P. Moulin, Water Res., 2009, 43, 2317–2348 CrossRef CAS PubMed.
- A. Popova, A. R. D. Verliefde, J. Busch and B. Van der Bruggen, Water Res., 2023, 236, 119880 Search PubMed.
- B. Sawadogo, Y. Konaté, S. K. Sossou, N. F. Ado Saidou, A. W. Nouhou Moussa and H. Karambiri, Water, 2022, 14, 3422 CrossRef CAS.
- A. Abejón, A. Garea and À. Irabien, Chem. Eng. J., 2015, 279, 110–118 Search PubMed.
- B. S. Rathi and R. Kumar, et al., Chem. Eng. J. Adv., 2024, 17, 100626 Search PubMed.
- V. Prabhakaran, B. L. Mehdi, J. J. Ditto, M. H. Engelhard, B. Wang, K. D. D. Gunaratne, D. C. Johnson, N. D. Browning, G. E. Johnson and J. Laskin, Nat. Commun., 2016, 7, 11399 CrossRef CAS PubMed.
- R. P. Misra and D. Blankschtein, J. Phys. Chem. C, 2021, 125, 2143–2153 Search PubMed.
- E. P. Alsaç, D. L. Nelson, S. G. Yoon, K. A. Cavallaro, C. Wang, S. E. Sandoval, U. D. Eze, W. J. Jeong and M. T. McDowell, Chem. Rev., 2025, 125, 2009–2119 CrossRef PubMed.
- S. Kondrat, G. Feng, F. Bresme, M. Urbakh and A. A. Kornyshev, Chem. Rev., 2023, 123, 6668–6715 CrossRef CAS.
- I. Nesterova, N. Kondratyuk, Y. A. Budkov, K. M. Gerke and A. Khlyupin, Adv. Colloid Interface Sci., 2025, 346, 103623 CrossRef CAS PubMed.
- Z. Zhang, C. Li, J. Zhang, M. Eikerling and J. Huang, Nano Lett., 2023, 23, 10703–10709 CrossRef CAS PubMed.
- Y. Wang, F. Tang, X. Yu, K.-Y. Chiang, C.-C. Yu, T. Ohto, Y. Chen, Y. Nagata and M. Bonn, Nat. Commun., 2025, 16, 7288 CrossRef CAS PubMed.
- N. R. Aluru, F. Aydin, M. Z. Bazant, D. Blankschtein, A. H. Brozena, J. P. de Souza, M. Elimelech, S. Faucher, J. T. Fourkas, V. B. Koman, M. Kuehne, H. J. Kulik, H.-K. Li, Y. Li, Z. Li, A. Majumdar, J. Martis, R. P. Misra, A. Noy, T. A. Pham, H. Qu, A. Rayabharam, M. A. Reed, C. L. Ritt, E. Schwegler, Z. Siwy, M. S. Strano, Y. Wang, Y.-C. Yao, C. Zhan and Z. Zhang, Chem. Rev., 2023, 123, 2737–2831 CrossRef CAS PubMed.
- M. L. Barabash, W. A. T. Gibby, C. Guardiani, A. Smolyanitsky, D. G. Luchinsky and P. V. E. McClintock, Commun. Mater., 2021, 2, 65 CrossRef CAS.
- H. Zhu, Y. Wang, Y. Fan, J. Xu and C. Yang, Adv. Theory Simul., 2019, 2, 1900016 CrossRef.
- A. Yaroshchuk, M. L. Bruening and E. Zholkovskiy, Adv. Colloid Interface Sci., 2019, 268, 39–63 CrossRef CAS PubMed.
- N. Anousheh, F. J. Solis and V. Jadhao, AIP Adv., 2020, 10, 125312 CrossRef CAS.
- J. Zeman, S. Kondrat and C. Holm, J. Chem. Phys., 2021, 155, 204501 CrossRef CAS PubMed.
- R. J. E. Reinertsen, S. Kewalramani, F. Jiménez-Ángeles, S. J. Weigand, M. J. Bedzyk and M. O. de la Cruz, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2316537121 CrossRef CAS.
- N. N. Rajput, J. Monk and F. R. Hung, J. Phys. Chem. C, 2012, 116, 14504–14513 CrossRef CAS.
- C.-W. Wang, Y.-W. Kuo, J.-R. Zeng, P.-H. Tang and T.-M. Wu, J. Phys. Chem. B, 2024, 128, 9525–9535 CrossRef CAS PubMed.
- Z. Wei, M. Chiricotto, J. D. Elliott, F. Martelli and P. Carbone, Carbon, 2022, 198, 132–141 CrossRef CAS.
- L. Smith, Z. Wei, C. D. Williams, M. Chiricotto, C. Pereira da Fonte and P. Carbone, ACS Appl. Mater. Interfaces, 2024, 16, 56316–56324 CAS.
- P. W. Ruch, M. Hahn, D. Cericola, A. Menzel, R. Kotz and A. Wokaun, Carbon, 2010, 48, 1880–1888 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 4428–4431 CrossRef CAS.
- C. Prehal, D. Weingarth, E. Perre, R. T. Lechner, H. Amenitsch, O. Paris and V. Presser, Energy Environ. Sci., 2015, 8, 1725–1735 RSC.
- F. W. Richey and Y. A. Elabd, J. Phys. Chem. Lett., 2012, 3, 3297–3301 CrossRef CAS.
- F. W. Richey, B. Dyatkin, Y. Gogotsi and Y. A. Elabd, J. Am. Chem. Soc., 2013, 135, 12818–12826 CrossRef CAS PubMed.
- F. W. Richey, C. Tran, V. Kalra and Y. A. Elabd, J. Phys. Chem. C, 2014, 118, 21846–21855 CrossRef CAS.
- L. Cervini, N. Barrow and J. M. Griffin, Johnson Matthey Technol. Rev., 2020, 64, 152–164 CrossRef CAS.
- J. M. Griffin, A. C. Forse, W.-Y. Tsai, P.-L. Taberna, P. Simon and C. P. Grey, Nat. Mater., 2015, 14, 812–819 CrossRef CAS PubMed.
- I. Hwang, M. Leketas, K. Griffiths, R. Bragg, J. M. Griffin and R. A. W. Dryfe, ChemElectroChem, 2024, 11, e202400099 CrossRef CAS.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed.
- J. D. Elliott, A. A. Papaderakis, R. A. W. Dryfe and P. Carbone, J. Mater. Chem. C, 2022, 10, 15225–15262 RSC.
- S. Kondrat, P. Wu, R. Qiao and A. A. Kornyshev, Nat. Mater., 2014, 13, 387–393 CrossRef CAS PubMed.
- L. Cervini, O. D. Lynes, G. R. Akien, A. Kerridge, N. S. Barrow and J. M. Griffin, Energy Storage Mater., 2019, 21, 335–346 CrossRef.
- Z.-X. Luo, Y.-Z. Xing, Y.-C. Ling, A. Kleinhammess and Y. Wu, Nat. Commun., 2015, 6, 6358 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- T. Dufils, C. Schran, J. Chen, A. K. Geim, L. Fumagalli and A. Michaelides, Chem. Sci., 2024, 15, 516–527 RSC.
- X. Liang, C. Ma and S. Jiao, J. Phys. Chem. B, 2024, 128, 5472–5480 CrossRef CAS.
- R. J. Bragg, K. Griffiths, I. Hwang, M. Leketas, K. Polus, V. Presser, R. A. Dryfe and J. M. Griffin, Carbon, 2024, 229, 119531 CrossRef CAS.
- G. Le Breton and L. Joly, J. Chem. Phys., 2020, 152, 241102 CrossRef CAS PubMed.
- J. H. Weijs, A. Marchand, B. Andreotti, D. Lohse and J. H. Snoeijer, Phys. Fluids, 2011, 23, 022001 CrossRef.
- L. Boruvka and A. W. Neumann, J. Chem. Phys., 1977, 66, 5464–5476 CrossRef CAS.
- F. Leroy and F. Müller-Plathe, J. Chem. Phys., 2010, 133, 044110 CrossRef PubMed.
- F. Zhou, N. Di Pasquale and P. Carbone, J. Chem. Phys., 2025, 162, 054119 CrossRef CAS PubMed.
- J. L. F. Abascal and C. Vega, J. Chem. Phys., 2005, 123, 234505 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- I. M. Zeron, J. L. F. Abascal and C. Vega, J. Chem. Phys., 2019, 151, 134504 CrossRef CAS PubMed.
- H. O. Wood, H. M. Burnett, R. A. W. Dryfe and P. Carbone, Faraday Discuss., 2024, 253, 212–232 RSC.
- Y. Zhang, N. H. C. Lewis, J. Mars, G. Wan, N. J. Weadock, C. J. Takacs, M. R. Lukatskaya, H.-G. Steinrück, M. F. Toney, A. Tokmakoff and E. J. Maginn, J. Phys. Chem. B, 2021, 125, 4501–4513 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- E. L. Pollock and J. Glosli, Comput. Phys. Commun., 1996, 95, 93–110 CrossRef CAS.
- T. Werder, J. H. Walther, R. L. Jaffe, T. Halicioglu and P. Koumoutsakos, J. Phys. Chem. B, 2003, 107, 1345–1352 CrossRef CAS.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef PubMed.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in 't Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- M. H. Abraham and Y. Marcus, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 339–349 RSC.
- A. A. Merdaw, A. O. Sharif and G. A. W. Derwish, Chem. Eng. J., 2011, 168, 229–240 CrossRef CAS.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- T. Osaka and K. Ebina, J. Phys. Chem. B, 1998, 102, 5691–5698 CrossRef.
- H. D. B. Jenkins and Y. Marcus, Chem. Rev., 1995, 95, 2695–2724 CrossRef CAS.
- C. D. Williams, J. Dix, A. Troisi and P. Carbone, J. Phys. Chem. Lett., 2017, 8, 703–708 CrossRef CAS PubMed.
- J. Han, A. Mariani, S. Passerini and A. Varzi, Energy Environ. Sci., 2023, 16, 1480–1501 RSC.
- K. Oununnkai, H. V. Patten, M. Velický, A. K. Farquhar, P. A. Brooksby and J. Downard, et al., Faraday Discuss., 2017, 199, 49–61 RSC.
- H. O. Wood, F. Zhou, J. Dočkal, M. Lísal, F. Moučka, S. Kaewmorakot, R. A. W. Dryfe and P. Carbone, J. Phys. Chem. C, 2026 DOI:10.1021/acs.jpcc.5c08644.
- Z. Wei, J. D. Elliott, A. A. Papaderakis, R. A. Dryfe and P. Carbone, J. Am. Chem. Soc., 2024, 146, 760–772 CrossRef CAS PubMed.
- M. Chiricotto, G. Giunta, H. A. Karimi-Varzaneh and P. Carbone, Soft Mater., 2020, 18, 140–149 CrossRef CAS.
- M. Chiricotto, F. Martelli, G. Giunta and P. Carbone, J. Phys. Chem. C, 2021, 125, 6367–6377 CrossRef CAS.
- N. Di Pasquale, J. Algaba, P. Montero de Hijes, I. Sanchez-Burgos, A. R. Tejedor, S. R. Yeandel, F. J. Blas, R. L. Davidchack, J. R. Espinosa, C. L. Freeman, J. H. Harding, B. B. Laird, E. Sanz, C. Vega and L. Rovigatti, Chem. Rev., 2025, 125, 5003–5053 CrossRef CAS PubMed.
- G. Horwitz, P. Steinberg and H. Corti, J. Chem. Thermodyn., 2021, 158, 106457 CrossRef CAS.
- Y. Sun, L. Kang, Y. Yu, L. Hong and Z. Liu, ChemPhysChem, 2025, 26, e202400981 CrossRef CAS PubMed.
- T. Liang, R. Hou, Q. Dou, H. Zhang and X. Yan, Adv. Funct. Mater., 2021, 31, 2006749 CrossRef CAS.
- I. M. Abdulagatov, A. B. Zeinalova and N. D. Azizov, J. Mol. Liq., 2006, 126, 75–88 CrossRef CAS.
- J. Yue, J. Zhang, Y. Tong, M. Chen, L. Liu, L. Jiang, T. Lv, Y.-S. Hu, H. Li, X. Huang, L. Gu, G. Feng, K. Xu, L. Suo and L. Chen, Nat. Chem., 2021, 13, 1061–1069 CrossRef CAS PubMed.
- M. Paillot, A. Wong, S. A. Denisov, P. Soudan, P. Poizot, B. Montigny, M. Mostafavi, M. Gauthier and S. Le Caër, ChemSusChem, 2023, 16, e202300692 CrossRef CAS PubMed.
- Y. Zhu, S. Zheng, J. Qin, J. Ma, P. Das, F. Zhou and Z.-S. Wu, Fundam. Res., 2024, 4, 307–314 CrossRef CAS PubMed.
- A. A. Papaderakis, A. Ejigu, J. Yang, A. Elgendy, B. Radha, A. Keerthi, A. Juel and R. A. W. Dryfe, J. Am. Chem. Soc., 2023, 145, 8007–8020 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |