 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando X-ray mapping of ion transport and arrangement in a carbon-based supercapacitor electrode
Malina Seyffertitz *ab,
Max Valentin Rauschera,
Sebastian Stocka,
Sylvio Haasc,
Peter Moharitscha and
Oskar Paris*a
*ab,
Max Valentin Rauschera,
Sebastian Stocka,
Sylvio Haasc,
Peter Moharitscha and
Oskar Paris*a
aChair of Physics, Montanuniversitaet Leoben, Austria. E-mail: oskar.paris@unileoben.ac.at
bYusuf Hamied Department of Chemistry, University of Cambridge, UK. E-mail: ms3216@cam.ac.uk
cDeutsches Elektronen Synchrotron DESY, Germany
First published on 16th January 2026
Abstract
Position- and time-resolved X-ray transmission (XRT) and small-angle X-ray scattering (SAXS) are used to map out ion transport and nanoscale ion arrangement in an in-plane carbon-based supercapacitor working electrode with 1 M RbBr aqueous electrolyte. XRT reveals that the charge balancing mechanism depends on the electrode geometry, shifting from ion exchange to predominantly Rb+ transport as the distance between working electrode and counter electrode is increased. SAXS shows that, despite these differences in ion mobilisation, the nanoscale ion arrangement within the smallest carbon nanopores remains essentially unchanged across the electrode and independent of the charging protocol. Small, but systematic differences are observed between sub-nanometre micropores (strong confinement) and larger micropores/mesopores (weaker confinement), the latter showing a distinct time dependence of ion concentration changes attributed to diffusive equilibration processes in these pores. Overall, these findings show that ion transport varies with position on the electrode, whereas the nanoscale ion arrangement does not. The smallest micropores behave uniformly across the electrode, and larger micropores and mesopores likewise show identical local arrangement at all positions, although their additional reservoir-character leads to a slight position dependent ion transport, similar to the overall bulk ion diffusion.
Introduction
The behaviour of ions within charged nanoporous materials defines how electrochemical systems store, convert, and transport charge. In supercapacitors, ions reversibly adsorb and desorb at the electrode–electrolyte interface to form the electric double layer (EDL), which enables electrostatic charge storage. This process is ideally free from any faradaic reactions, and determines the high power-density and exceptional cycle lifetimes of supercapacitors.1,2 Understanding ion behaviour in EDL formation is not only central to improving energy storage performance but also to advancing a broader range of technologies, as similar systems are increasingly applied also in flexible and wearable electronics,3,4 implantable devices for healthcare application,5,6 sensor technology,7 water treatment and desalination,8,9 electrochemical CO2 capture,10 as well as thermo- and chemocapacitive energy harvesting.11–13 Related systems, such as ionic-gating platforms, also depend fundamentally on ion movement and organization at charged interfaces. Across these diverse systems, progress depends on a clear mechanistic understanding of how ions move and arrange under confinement at applied charge.During charging and discharging, the local ion concentration in the electrode can change significantly, by up to 50% for an initial 1 M electrolyte solution.14,15 This concentration change can occur through different charge-balancing mechanisms: co-ion expulsion, where ions of the same charge as the electrode are removed; counter-ion adsorption, where oppositely charged ions are added; or a combination of both, referred to as ion exchange. At low salt concentrations and slow charging speeds, counter ion adsorption typically dominates, whereas higher concentrations and faster charging tend to favour ion exchange in both aqueous16 and organic17 electrolytes in carbon-based supercapacitors. Studies using X-ray and NMR techniques have shown that charging often proceeds in two stages with an initial fast ion exchange followed by a slower equilibration associated with increased counter-ion adsorption.16,18 Simulations further indicate that stepwise or overly rapid charging can cause ion trapping and pore blocking, hindering ion motion and slowing charge redistribution.19,20
The formation of the EDL also involves local rearrangements of ions and solvent molecules, where ions might seek various degrees of confinement in nanopores of the electrode material.21 Ions in particularly strong carbon confinement are often accompanied by desolvation22 and screen charge most effectively, leading to an anomalous increase in capacitance for sub-nanometre pores which are smaller than the hydrated ion size.23–26 While the influence of pore size and other subtle structural features like local disorder27 on overall performance has been extensively studied, much less is known about how the charging behaviour and charge balancing mechanisms vary spatially within the electrode or, in other words, how local ion organization depends on the macroscopic cell design and electrode shape.
In their simplest form, supercapacitors consist of two identical electrodes separated by an ion-permeable membrane and immersed in liquid electrolyte, as used in coin-cell configurations for research and commercial devices. Other formats, such as cylindrically wound and pouch cells, are also available.28 For experimental or application-specific needs, the geometry is often adapted: in situ and operando methods may use asymmetric or laterally arranged electrodes to allow probe access, while flexible and or miniaturized devices may employ interdigitated or in-plane configurations integrated onto substrates. These design variations alter current paths, ion transport distances, and electrical potential distributions, thereby influencing both overall performance and local electrochemical conditions. Previous studies have examined how electrode thickness29,30 and stacking pressure31 affect the macroscopic performance of supercapacitors. Improved power capability has been reported for interdigitated micro-supercapacitors with sharp-edged geometries compared to conventional designs.32 Variations in electrochemical signals observed across different in situ NMR set-ups indicate that electrode geometry and cell design have a strong influence on device response.18 Such studies have primarily focussed on performance changes through the evaluation of overall electrochemical signals. Consequently, spatially resolved mechanistic insights into how geometry affects charge-balancing mechanisms and local ion (re)arrangement remain largely unexplored.
In this work, we use operando position-resolved X-ray transmission (XRT) and small-angle X-ray scattering (SAXS) to map the signal across the activated carbon working electrode of an in-plane supercapacitor with 1 M RbBr aqueous electrolyte. XRT reveals how charge-balancing mechanisms and ion concentrations evolve across the electrode under different charging protocols, while SAXS provides insight into the corresponding nanoscale ion (re-)arrangement within the pores.
Experimental
Materials and electrode preparation
1 M RbBr (aq.) electrolyte was prepared from RbBr salt (99.6% trace metal basis, Sigma-Aldrich) and Milli-Q lab-grade H2O. A porous Whatman GF/A glass fibre separator with a thickness of 200 µm was used. Platinum paper with a thickness of less than 200 nm and a purity greater than 99% served as the current collector.Free-standing activated carbon electrode sheets were fabricated following the procedure described in ref. 21. Briefly, commercially available alkaline-activated carbon powder (MSP-20X, Kansai Coke and Chemicals Co.) was mixed with 5 wt% PTFE binder (60 wt% dispersion in water, Sigma-Aldrich) and ethanol. The resulting mixture was rolled to a thickness of 200 µm using a cold-rolling press equipped with micrometre adjustment screws, yielding thickness variations of less than 5 µm. To remove residual moisture, the electrodes were dried at 110 °C for 24 h in an evacuated oven and subsequently stored in a vacuum furnace until use. Electrode shapes were then punched from the free-standing sheets using custom-machined punch tools. Complementary gas sorption analysis (GSA) of the electrode film using nitrogen at 77 K and carbon dioxide at 273 K was performed with a Quantachrome/Anton Paar AutoSorb iQ (see Fig. S1). The material shows a predominantly microporous character, with most pores smaller than 2 nm, a specific surface area of 1870 m2 g−1, and a specific pore volume of 0.79 cm3 g−1. An associated SEM micrograph showing the particle size and morphology can be seen in Fig. S2.
Cell assembly
Electrochemical cells were assembled using a custom-built design (ref. 33) featuring slits with Kapton windows that allow X-ray access across the electrode stack and are further described in Fig. S3.The working electrode (WE) was circular (diameter: 12 mm), while the counter electrode (CE) was ring-shaped (inner diameter: 14 mm; outer diameter: 34 mm) (Fig. 1a), both punched from the same free electrode film of 200 µm thickness. The WE and CE masses were 13.3 mg and 87.6 mg, respectively, corresponding to a CE/WE oversize factor of approximately 6.6. The cell configuration consisted of a platinum current collector, the circular working electrode, the glass-fibre separator, the ring-shaped counter electrode, and a second platinum current collector. The platinum current collectors cover the full cell area and ensure uniform electronic contact to both electrodes. The in-plane electrode geometry was intentionally chosen to enable spatially resolved investigation of geometry-induced, position-dependent ion transport within the working electrode.
 | ||
| Fig. 1 Mapping of ion transport and charge-balancing mechanisms in an in-plane supercapacitor with operando XRT. (a) Schematic of the electrode geometry showing the circular working electrode (WE), ring-shaped counter electrode (CE), separator, and the horizontal scanning direction of the X-ray beam. (b) XRT profile across the cell stack identifying the positions of the individual components. (c) Schematic illustration of the charge-balancing mechanisms, co-ion expulsion, ion exchange, and counter-ion adsorption. (d) Operando XRT map showing the relative transmission τ/τ0 across the cell during 1 mV s−1 cyclic voltammetry and 1-hour cell voltage holds at ±0.6 V, effectively representing a spatial and temporal map of the charge-balancing mechanism. | ||
480 µL of 1 M RbBr electrolyte was added before sealing the cell, which was then left to equilibrate for 30 min. Prior to operando measurements, the cell was pre-cycled by performing ten cyclic voltammograms (CVs) between ±0.6 V at a scan rate of 50 mV s−1.
X-ray transmission (XRT) and small angle X-ray scattering (SAXS) measurements
Operando X-ray transmission (XRT) and small-angle X-ray scattering (SAXS) measurements were conducted at the P62 SAXSMAT beamline at PETRA III (Deutsches Elektronen-Synchrotron, DESY, Hamburg, Germany).34 The photon energy was set to 11 keV, and the beam was defocused to 0.5 × 0.5 mm to minimize radiation damage. The electrode stack was scanned horizontally in 1 mm steps across 35 positions covering both the central working electrode (WE) and the surrounding counter-electrode (CE) ring (Fig. 1a). The separator is present at all positions. The exposure time at each position was 0.5 s. Including stage movement between positions and the return to the starting point, a complete scan cycle lasted approximately 80 s, providing sufficient temporal resolution to monitor changes at each position throughout the applied voltage protocol.The applied voltage protocol consisted of three CVs at a scan rate of 1 mV s−1, followed by consecutive 1 h chronoamperometry measurements at fixed cell voltages of 0 V → +0.6 V → 0 V → −0.6 V → 0 V → +0.6 V → 0 V → −0.6 V → 0 V, and concluded with another three CVs at 1 mV s−1 (see left part of Fig. 1d).
The recorded 2D-SAXS patterns were azimuthally integrated and corrected for transmission and background according to standard procedures at the beamline. The resulting one-dimensional SAXS profiles are presented as scattered intensity (in arbitrary units) plotted against the magnitude of the scattering vector, 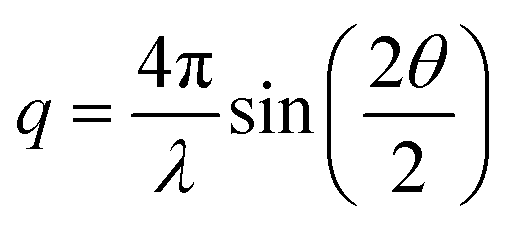 , with 2θ being the scattering angle and λ the photon wavelength.
, with 2θ being the scattering angle and λ the photon wavelength.
Results and discussion
Concentration changes and charge balancing mechanisms from X-ray transmission (XRT)
Fig. 1a shows the electrode set-up with the circular working electrode (WE) surrounded by a ring-shaped counter electrode (CE), and indicates the horizontal scanning direction of the incident X-ray beam across the electrode stack. The corresponding transmission profile in Fig. 1b identifies the positions of the individual components (CE ring, separator, and WE) within the scanned region.Previous studies have shown that the XRT signal can reliably track ion concentration changes in supercapacitors during charging and discharging, allowing the charge-balancing mechanism to be determined.15,16,21,35 The method is based on the direct connection between local ion concentration and X-ray attenuation via the Lambert–Beer law. As the cell is charged, variations in ion concentration change the attenuation and, consequently, the measured transmission. This enables quantitative mapping of concentration changes, assuming that the carbon matrix transmission remains constant and that changes in the water density and concentration during operation of the device have only a minor effect on the total attenuation, as discussed previously.15
For the electrolyte used in this study, 1 M RbBr (aq.), the cation Rb+ and the anion Br− have very similar mass attenuation coefficients (µ/ρ), and comparable sizes, as shown in Table 1. Consequently, during ion exchange, where one counter-ion replaces a co-ion, the overall X-ray transmission τ is expected to remain roughly constant. In contrast, co-ion expulsion would increase the transmitted intensity, while counter-ion adsorption would decrease it. The charge-balancing mechanisms and corresponding changes in ion concentration are schematically illustrated in Fig. 1c.
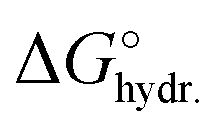 ,39 and number of electrons, e−, for Rb+ and Br−
,39 and number of electrons, e−, for Rb+ and Br−
| Rb+ | Br− | |
|---|---|---|
| µ/ρ at 11 keV (cm2 g−1) | 41.5 | 42.8 |
| dbare (nm) | 0.30 | 0.39 |
| dhydr. (nm) | 0.66 | 0.66 |
| u (10−8 m2 V−1 s−1) | 7.7 | 7.2 |
 |
−275 | −315 |
| e− | 36 | 36 |
To better visualise how the transmission τ changes during operation, the time-dependent transmission signal τ(t) at each scan position was normalised to a baseline function τ0(t). For each position, an individual baseline τ0(t) was obtained by fitting a second-degree polynomial to the τ(t) data at the segments where a cell voltage of 0 V was applied. Examples of the resulting baseline functions together with the corresponding raw transmission data for selected WE positions are shown in the SI (Fig. S4). Using a separate baseline for each position compensates for slow signal drifts and for initial transmission differences across the electrode, which, as visible in Fig. S4, are substantially larger than the subtle changes arising from charging. The resulting values of τ/τ0 therefore represent the transmission relative to the average of all 0 V cell voltage states. A value of 1 corresponds to this baseline level and indicates ion exchange, where one ion replaces the other without changing the overall attenuation. Values greater than 1 indicate higher transmission (co-ion expulsion), and values below 1 indicate lower transmission (counter-ion adsorption). The resulting colour-coded map of τ/τ0 values is shown in Fig. 1d.
From the colour map in Fig. 1d small but systematic variations in the relative transmission τ/τ0 can be observed as the cell voltage is varied. These variations are more distinct in the WE than in the CE and have an overall amplitude of about ±2%. Ion concentration changes are also visible in the separator region between the WE and CE. The trend in this region generally follows that of the WE, although with a slight time delay.
The weaker response in the CE may arise from two factors. First, its behaviour could be dominated by ion exchange, which would cause the transmission signal to remain nearly constant since the two electrolyte ions have almost identical attenuation coefficients. Alternatively, as a supercapacitor is essentially two capacitors in series, the larger size of the CE distributes the same total charge over a much larger electrode surface, leading to smaller local changes in ion concentration and thus weaker detectable transmission variations. In this cell, the CE/WE mass ratio is approximately 6.6, meaning that the concentration changes in the WE are expected to appear about 6.6 times more pronounced. As both effects, i.e. ion exchange dominance and the oversize factor may contribute to the weaker signal in the CE, the CE is not considered in further detail here, and the following discussion focuses on the WE only. It is worth noting that the separator is present in the beam at all positions and its contribution cannot be subtracted, as its response cannot be spatially isolated from that of the electrode. However, the separator contribution at the electrode positions is expected to be small, because in regions containing the electrodes, the separator is very strongly compressed and can be understood as a small additional bulk electrolyte reservoir in close proximity to the carbon, where charge balancing is taking place. This is supported by the CE region, where the separator is likewise included in the beam but shows almost no voltage-dependent variation, indicating that the signal at both electrode positions is dominated by changes within the electrode.
At positive cell voltage, the transmission in the WE increases compared to the average transmission at 0 V, indicating a decrease in the total ion concentration due to co-ion expulsion of Rb+ ions to balance charge. At negative cell voltages the relative transmission decreases, reflecting a higher ion concentration as counter-ion adsorption leads to the accumulation of additional Rb+ ions at the electrode. Consequently, both polarities seem to be dominated by Rb+ mobilisation, although Rb+ and Br− have similar bulk ion mobilities (Table 1).
Overall, the electrode exhibits predominantly homogeneous charge-balancing behaviour, with only slight deviations visible at the edges in the colour map. Since both electrodes are uniformly contacted by the current collectors, the observed centre-edge differences are attributed to geometry-dependent ionic transport rather than to variations in electronic potential. To explore these variations in more detail, centre and edge regions were defined based on the map in Fig. 1d. Positions 14 to 21 correspond to the centre region, while positions 12 to 13 and 22 to 23 represent the edges. The relative transmissions averaged over these regions are shown in Fig. 2 as a function of time and can be regarded as vertical slice averages through the colour map in Fig. 1d.
 | ||
| Fig. 2 Charge balancing behaviour of centre and edge regions on the working electrode. (a) Applied cell voltage (thin grey lines) and averaged relative transmission τ/τ0 for centre (thick black line) and edge (thick dotted line) regions of the working electrode and (b) schematic of the working electrode indicating the corresponding centre and edge positions used for averaging. | ||
From the cyclic voltammetry (CV) segments at the beginning and end of the experiment, where the cell voltage was swept fairly slowly at 1 mV s−1, the centre and edge regions of the working electrode exhibit nearly identical behaviour. Both show the previously discussed Rb+ dominated charge balancing, with co-ion expulsion at positive and counter-ion adsorption at negative cell voltages.
In contrast, during the chronoamperometric (CA) steps, where the voltage was changed stepwise rather than continuously, a difference between edge and centre regions is seen. Particularly at positive voltage holds, the edge regions show considerably smaller changes in transmission, implying corresponding smaller variation of the total ion concentration changes. This behaviour could indicate a larger contribution from ion exchange, which may be favoured near the electrode edges due to shorter diffusion pathways or easier ion access from the separator reservoir and from the counter-electrode. The corresponding transmission signals for the separator and CE are shown in Fig. S5, but will not be further discussed due to the limitations discussed before.
The relative transmission levels at 0 V after each CA step in Fig. 2a differ depending on the polarity of the preceding voltage. Following negative polarisation, the 0 V transmission level is slightly higher (i.e. larger than 1), whereas after positive polarisation it is lower than 1. This indicates a subtle “history” effect, where the cell relaxes back to equilibrium after each step, but the resulting equilibrium state depends on the previous polarisation. This behaviour is consistent with a previous investigation,15 where operando Small-Angle Neutron Scattering (SANS) on the same electrode/electrolyte system and identical electrode geometry was employed. SANS using a neutron beam size that averaged over the entire WE area, revealed a similar dependence of the equilibrium state on charging history. With SANS being particularly sensitive to the solvent water, it was concluded that the water content in the WE was lowest at positive and highest at negative cell voltages. This observation is counterintuitive, as the ion concentration determined here follows the same trend with a minimum in total ion concentration at positive cell voltages due to Rb+ expulsion and a maximum in ion concentration at negative cell voltages due to Rb+ adsorption. If water were to occupy the remaining pore volume, the opposite dependence would be expected. Instead, the results suggest that part of the water may be associated with the ions through hydration and moves together with Rb+ during charging and discharging.
Most interestingly, the difference between edge and centre only appears during stepwise voltage changes, but not during the continuous CV sweeps. This suggests that kinetic effects, such as slower equilibration or transient blocking as predicted by modelling studies19 may play a role. A possible explanation is that during a continuous voltage sweep, charge compensation can proceed primarily via the movement of one preferred ion species, Rb+, which is sufficient to balance the gradually changing potential. During stepwise voltage changes, however, the abrupt potential jump may require a faster charge adjustment that cannot be achieved by the motion of only one ion type. In this situation, ion exchange could become favourable, since the simultaneous expulsion of one ion species and adsorption of the other species effectively transfers two charges in one process.
The in-plane electrode configuration used here represents one extreme of an electrode geometry, where the working and counter electrodes are laterally arranged and do not overlap. In this case, charge compensation is dominated by the movement of Rb+ ions across the electrode, particularly in the central region. At the electrode edges, however, Rb+ adsorption dominates at negative cell voltages, while a trend towards ion exchange emerges at positive polarisation. This behaviour is consistent with the partially overlapping geometry reported previously,15 where an oversized counter electrode (CE) with a central opening allowed the X-ray beam to irradiate a smaller, but partially overlapping working electrode (WE). The edge region of the current in-plane cell, in proximity to the ring-shaped CE, resembles this earlier configuration and therefore shows comparable behaviour. At the opposite limit, an ideally overlapping symmetric supercapacitor with a very small hole in the CE for X-ray access displayed ion-exchange driven charge balancing (Fig. S6 and Supplementary Note 1 in the SI).
In summary, these observations from XRT establish a clear electrode shape trend: as the degree of electrode overlap decreases, the charge balancing mechanism transitions from ion exchange to predominantly Rb+ mediated transport and might be further promoted by stepwise or rapid variations of the cell voltage. This suggests that the charge balancing process and ion concentration changes and mobilisation involve more intricate local effects that are not evident from bulk properties alone. To gain further insight, the local ion rearrangement within the electrode nanopores is examined next.
Local ion re-arrangement in nanoconfinement from small-angle X-ray scattering (SAXS)
The local rearrangement of ions within the nanoconfined pores of the electrode can be probed with small-angle X-ray scattering (SAXS), as demonstrated in ref. 21, 22 and 35. Unlike most spectroscopic techniques, which provide chemical but limited structural information, SAXS is directly sensitive to spatial variations in electron density in the nanometre range, and therefore to structural changes occurring at different length scales and degrees of confinement within the porous carbon.Fig. 3a shows the experimentally obtained SAXS signal of the dry activated carbon electrode, which can be decomposed into three q-dependent contributions: scattering from the micrometre-sized carbon particles (IParticle), scattering from the disordered nanopores (INanopore), and the structure factor (IStructureFactor) as described in ref. 35. The structure factor is proportional to the total number of electrons in the X-ray beam and is assumed to be constant in the SAXS regime, before giving rise to correlation peaks at higher q-values from the atomic scale carbon–carbon interactions. The scattering from the nanopores was divided into two contributions corresponding to different pore sizes with radii of gyration, Rg, of ≈0.7 nm and ≈3 nm, following the fitting of two Debye, Anderson, and Brumberger (DAB)40 model functions as described in ref. 21 and 41. As illustrated by the fitted SAXS curve in Fig. 3a, this model with two pore sizes reproduces the shape of the experimental signal reasonably well. These two distinct average pore sizes are also in good agreement with the pore size distribution from the nitrogen sorption results (Fig. S1a), showing a population of small micropores with a maximum around 0.7 nm and a second broad distribution of pores larger than 1 nm. However, this simplified two-size model should be seen only as a qualitative guide to the underlying length scales.
 | ||
| Fig. 3 Small-angle X-ray scattering (SAXS) profiles of the electrodes. (a) Experimental SAXS profiles of an MSP-20X activated carbon electrode containing 5 wt% PTFE binder, measured in the dry state (thick black line) and after soaking in 1 M RbBr (aq.) (thick grey line). The dry-state curve is decomposed into contributions from particle scattering (thin dash dotted black line), a structure factor term (thin dashed line) and scattering from nanopores, which is divided into two pore-size regimes (thin dotted and full black line) with radii of gyration, Rg, of 3 nm and 0.675 nm respectively. The thin orange line shows the corresponding simulated SAXS profile obtained from the sum of these components. The inset schematically illustrates the SAXS contrast for a dry and an electrolyte-soaked carbon particle. (b) Normalised SAXS intensity I/I0 (I0 being the average SAXS intensity at 0 V) for applied cell voltages during the second cycle of the 1 hour chronoamperometry, shown averaged for centre and edge positions of the working electrode. | ||
Under the assumption of a strict two-phase system consisting of a (homogeneous) matrix and pores, the overall scattering contribution of nanopores, INanopore, is governed by the SAXS contrast, which is proportional to the square of the difference in electron density between these two phases (inset of Fig. 3a). When the electrode is soaked in 1 M RbBr (aq.), the electrolyte fills the previously empty pores. The electron density in the pores increases, and thus the contrast to the carbon matrix is reduced. As seen in Fig. 3a, this leads to a strong decrease in the scattering associated with INanopore. Meanwhile, the structure factor increases after soaking because the electrolyte introduces additional electrons into the irradiated volume, adding the electrolyte structure factor, which is also assumed to be constant in the SAXS regime.
During ion electrosorption, when the cell is charged, the ion concentration and arrangement within the pores are expected to change. As ions enter or leave a pore, the electron density in that pore volume changes, which slightly modifies the SAXS contrast and manifests in a changed scattering signal. Because these changes are small, the measured scattering intensity I is normalised to the averaged scattering signal recorded during segments, where the cell was held at a cell voltage of 0 V, analogous to the normalisation of the transmission values in the previous section. Values of I/I0 greater than 1 indicate an increased scattering contrast relative to the 0 V average scattering curve, while values below 1 indicate a reduced contrast. This normalised scattering intensity, used as a measure of the contrast change, is shown in Fig. 3b for averaged centre and edge positions on the working electrode. Each curve represents the average of all scattering data collected during the 1 hour constant-voltage holds of the second CA cycle. A more quantitative interpretation of the contrast would require detailed knowledge of the electron densities of carbon, solvent, and ions inside pores of different sizes, which is not readily accessible from experiment alone and typically needs dedicated modelling (ref. 22). Therefore, the present analysis focuses on qualitative trends and, in particular, on comparing centre and edge regions rather than determining the exact ion density distribution in the pores.
At high q (>5 nm−1), the scattering is dominated by the structure factor, which should (inversely) mirror the transmission behaviour. As established in the previous section, positive voltages expel Rb+, reducing the number of electrons in the beam, which increases the transmission and correspondingly decreases the structure-factor level. Although this trend is consistent with the transmission results, quantitative interpretation of the structure-factor regime is more complex, as the structure factor in this regime is not necessarily constant (as seen in Fig. 3a by the slight increase in the intensity for the electrolyte-soaked electrode). It may be influenced by ion–ion, ion–carbon, solvent–carbon, and ion–solvent correlations via their partial pair-distribution functions. For this reason, the qualitative consistency of the high-q region in Fig. 3b with the previously analysed transmission is noted, but is not discussed further.
Similarly, the low-q scattering (<0.1 nm−1) arising from large mesopores and macropores (e.g. from the space between the micrometre-scale carbon particles, see Fig. S2) is not further analysed here in detail, as its interpretation is complex and not directly related to the pore-scale ion rearrangement of interest. We note however that there is a positive intensity change (i.e. a contrast increase) for both, positive and negative voltage, as compared to 0 V.
Focusing instead on the confinement and nanopore-relevant range in Fig. 3b, two regions of interest can be identified: one roughly between 0.2 and 0.7 nm−1 and another between 1 and 3 nm−1 with striking different behaviour. We refer to the lower-q region as representing larger nanopores (i.e. large micropores and mesopores) and the higher-q region as representing the smallest nanopores in the system (i.e. small micropores). This distinction is justified by the DAB model fitting shown in Fig. 3a and the pore size distribution (Fig. S1) which shows a class of small micropores <1 nm, and a second peak related to larger micropores and small mesopores.
At higher q values (1–3 nm−1), corresponding to the small micropore regime, both polarities show an increased SAXS contrast compared to the 0 V average state. One possible explanation for this increased contrast is that at 0 V the pores are assumed to be filled mostly with water, and when a voltage is applied, ions enter these small micropores, effectively filling the confined volume and increasing the electron density. Assuming that heavy ions, such as Rb+ or Br−, have a higher electron density than water, and lie further above the carbon electron density than water lies below it, the resulting electron-density difference, and thus the contrast, would increase, as observed in the experimental data. Under positive voltages, the ion present in confinement is expected to be Br−, whereas under negative voltages it is Rb+. There are no observable differences in the contrast variation in the smallest pores between edge and centre regions on the electrode. Interestingly, at positive potential the contrast peak is shifted to higher q values, which could indicate that Br− preferentially occupies more strongly confined environments, despite being the slightly larger ion (Table 1). Together with its slightly reduced bulk ion mobility, this could help explaining why Rb+ is preferentially mobilised during charge balancing. However, further insight into ion-specific behaviour would require systematic studies with different electrolyte salts, which are beyond the scope of the present work.
In the regime associated with the larger pores (with weaker confinement) in the q-range between 0.2 and 0.7 nm−1, the normalised contrast shows a negative contrast change (I/I0 < 1) at positive voltage, and a positive contrast change (I/I0 > 1) at negative cell voltage. Compared to the smaller micropores, a straightforward interpretation of the mesopore regime in terms of contrast is complicated by the hierarchical pore structure.41 For these mesopores, the relevant “matrix” in a two-phase system is not pure carbon, but carbon that contains the smaller micropores. Since the electron density within these micropores changes as discussed above, the effective matrix electron density, as well as the electron density within the mesopores themselves changes as the voltage is varied. This leads to many possible and complex contrast scenarios involving two ion species and solvent water with varying concentrations, making it difficult to assign a reduced or increased contrast to a single physical phenomenon or concentration change. For this reason, the following discussion focuses on qualitative trends and a comparison between edge and centre regions on the electrode rather than attempting a full hierarchical interpretation.
Notably in the mesopore regime, the edge and centre positions show almost no difference at positive cell voltage, and a slight, but significant difference at negative polarisation. This difference is limited to the absolute contrast level and does not involve a shift in q-position. Mechanistically, this indicates that the edge and centre regions respond similarly, with no qualitative difference in how ions rearrange within these larger pores. Only the absolute contrast levels differ slightly, with the edge region showing lower contrast within the space of weak confinement of larger pores.
A similar behaviour is observed at 0 V. As seen in the transmission data in the previous section (Fig. 2a), the signal exhibits a “history” effect, where the response depends on the preceding polarisation. The SAXS data show the same behaviour, but only in the mesopore regime, not in the micropore regime. In the larger pore regime, the edge and centre differ again in the absolute magnitude of the contrast, while the q-positions of the features remain the same, indicating that the underlying ion arrangement is identical and only the total number of ions differs. This suggests that larger pores with weaker confinement contribute to overall charge balancing and ion-concentration changes, but also to the dynamics of the system, in terms of diffusive exchange of ions with the bulk ion reservoir in the macropores and the separator. Obviously, these processes are influenced by electrode shape even in small mesopores and large micropores with weaker confinement, whereas the strong confinement in small micropores results in ion arrangements that are essentially insensitive to electrode shape and macroscopic ion-concentration variations.
The averages at constant cell voltage shown in Fig. 3b are useful for identifying structural differences in the ion rearrangement at applied voltage for different q-regimes, associated with different degree of confinement. However, they represent an average over all scattering curves recorded during the one-hour CA holds. To resolve the time dependence as well, the normalised intensity was evaluated for each individual scattering curve throughout the operando experiment and shown in a colour-coded presentation in Fig. 4a: values greater than 1 (increased contrast relative to the 0 V average) appear in red, whereas values below 1 (reduced contrast) appear in green. We note that the traces shown in Fig. 3b correspond to horizontal averages of these colour maps during the one-hour holds of the second CA cycle. A 3D illustration of how the colour maps were constructed is provided in Fig. S7.
 | ||
| Fig. 4 Operando SAXS contrast during charging and discharging at the edge and centre of the working electrode. (a) Normalised SAXS intensity (I/I0) as a function of scattering vector q and time for the edge and centre regions of the working electrode. Each horizontal line corresponds to one individual SAXS measurement recorded during the operando experiment. The applied cell-voltage protocol is shown on the left. Colours indicate the deviation from the average scattering at 0 V (red: increased contrast; green: decreased contrast). Blue and orange boxes mark the q-ranges used in panels (b) and (c), which show the corresponding evolution of the contrast for the larger-pore and smaller-pore regimes, respectively. | ||
Visually, the colour maps in Fig. 4a for the centre (right) and edge positions (left) appear similar, with no obvious differences in their overall behaviour. There is also qualitatively similar behaviour between CA and CV cycles, and the contrast changes appear very uniform during the voltage holds, with little indication of time-dependent effects. To examine time dependency in more detail, the two length scales identified previously for weak and strong pore confinement, marked by the blue and orange boxes in the colour maps, were averaged. The resulting signals, shown in Fig. 4b and c, are thus vertically averaged cuts through the colour map at these q regions of interest. For consistency, it should be noted that the q-ranges indicated in Fig. 4a are narrower than the broader confinement regimes defined above (0.2–0.7 nm−1 and 1–3 nm−1). These smaller windows were chosen because they capture the regions where the contrast changes are most pronounced, but averaging over the full ranges yields qualitatively identical behaviour.
In strong confinement (panel c), the signals from the edge and centre match perfectly. The contrast also changes nearly instantaneously as the voltage is varied and behaves qualitatively the same during both CV and CA. This indicates that ion rearrangement in small micropores is very fast, independent of whether the voltage is changed stepwise or swept, and essentially unaffected by global ion-concentration changes and thus by the position on the electrode.
In the weaker-confinement regime at lower q, the edge and centre regions also behave similarly, though with some deviations. These deviations are most noticeable at negative cell polarisation, where the centre shows clearly larger quantitative changes, consistent with the behaviour during the constant-voltage holds in Fig. 3b. Examining the time dependence reveals two processes: an initial rapid change followed by a slower, gradual evolution. This likely reflects a fast charge-balancing and rearrangement step, producing the sharp initial rise, and a subsequent slower equilibration of the ion concentration. It is important to emphasise that these changes relate only to the total contrast; that is, to the total number of ions or total electron density within the pore space. As shown earlier and visible in the colour map (Fig. 4a), these variations are not accompanied by shifts in q-position. This indicates that the underlying ion arrangement remains the same at the edge and centre. Instead, the larger pore spaces and more weakly confined regions appear to act as an ion reservoir that accommodates changes in total ion concentration and therefore displays some time dependence, whereas the strongly confined micropores respond almost instantaneously.
Taken together, the voltage-dependent ion arrangement within the porous electrode is largely independent of electrode shape. The centre and edge behave identically for strong pore confinement but show some slight but consistent deviations for larger nanopores, i.e. weaker confinement. Qualitatively, the behaviour does also not depend on whether the voltage is changed stepwise during CA or swept during CV. The data further suggest that Br− ions may tend to occupy more strongly confined pore environments than Rb+. For both ion species, rearrangement in the smallest micropores occurs instantaneously (within the time resolution of the present experiment), whereas larger pores exhibit both a fast, initial response and a slower equilibration of the ion concentration within these pores. This points to an important role of mesopores and large micropores for both, ion storage on the one hand, and ion reservoir for the quick charge compensation in the small micropores.
Conclusions
In this work, operando, position-resolved X-ray transmission (XRT) and small-angle X-ray scattering (SAXS) mapping were used to probe how macroscopic electrode shape influences ion transport and local ion arrangement in a carbon-based in-plane supercapacitor with 1 M RbBr aqueous electrolyte. The in-plane electrode set-up with a circular working electrode and a ring-shaped counter electrode enabled spatially resolved measurements across centre and edge regions of the working electrode.XRT revealed that charge balancing in the central region proceeds predominantly through Rb+ transport, with Rb+ expulsion at positive and Rb+ adsorption at negative polarisation. Near the electrode edge, where diffusion pathways to the counter electrode are shorter, a partial contribution from ion exchange involving both ion species, Rb+ and Br−, appears, particularly during stepwise potential changes. A subtle history effect at 0 V demonstrates that the equilibrium state after each voltage step depends on the preceding polarisation. Together with comparisons to other geometries, a clear trend emerges: larger separation between working and counter electrode favours a transition in the charge balancing mechanism from ion exchange to predominantly Rb+ mediated transport, with this transition most pronounced under rapid stepwise voltage changes.
SAXS mapping complements these findings by resolving the nanoscale ion rearrangement inside the carbon pores. Voltage-dependent changes in SAXS contrast reveal two confinement regimes: (i) a weaker-confinement regime in larger pores that acts as an ion reservoir and shows both, fast and slower equilibration processes, and (ii) a strongly confined small micropore regime that responds almost instantaneously to changes in voltage. In the strongest confinement, no difference is observed between centre and edge positions on the working electrode. In larger pores, slight differences arise but only in the total amount of ions, not in their degree of confinement or local arrangement, underscoring the role of mesopores in charge balancing as well as in serving as an ion reservoir for the rapid compensation in smaller micropores. The data further suggest that Br− ions may occupy more strongly confined pore environments than Rb+, which, together with its slightly lower bulk mobility, offers a possible explanation for the preferential mobilisation of Rb+ during charge balancing.
Taken together, these results demonstrate that ion transport is shaped by the macroscopic electrode geometry, whereas the ion arrangement within the porous carbon is largely unaffected by it. Centre and edge regions on the working electrode behave qualitatively the same across all pore confinement regimes; the only additional nuance is that the larger, more weakly confined pores act as a reservoir and therefore show some slow, diffusion-driven evolution of ion concentration, whereas the strongly confined micropores respond almost immediately. In the studied system, the macroscopic electrode shape dictates which ions move, how fast they respond, and how strongly the overall ion concentration changes, while leaving the nanoscale ion arrangement essentially unaffected. Put simply, electrode shape directs transport, not organisation; an insight that may, quite literally, help reshape the design of future supercapacitor electrodes and other related ion-based devices, enabling control over ion transport without altering their nanoscale distribution inside micropores.
Author contributions
M. S.: conceptualization, methodology, formal analysis, visualization, project administration, investigation, data curation, writing – original draft, writing – review & editing; M. V. R.: investigation, writing – review & editing; S. S.: writing – review & editing; S. H.: data curation, writing – review & editing; P. M.: methodology, investigation; O. P.: conceptualization, methodology, supervision, project administration, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Data availability
All raw and processed data supporting the findings of this study are publicly available on Figshare at: https://doi.org/10.6084/m9.figshare.30747296.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5fd00140d.
Acknowledgements
We acknowledge the Deutsches Elektronen-Synchrotron DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III. XRT and SAXS data were collected at the P62 SAXSMAT beamline operated by Sylvio Haas during the beamtime associated with proposal I-20230206 EC. We thank Andrew Fitch and Catherine Dejoie from the European Synchrotron Radiation Facility (ESRF) for support during the beamtime associated with proposal SC-5511 at the ID22 Beamline for XRF measurements. M. S. also wishes to thank Gerhard Hawranek and Helmut Clemens from the Department Materials Science for recording the SEM images.References
- Supercapacitors – Materials, Systems and Applications, ed. F. Béguin and E. Frąckowiak, Wiley, Weinheim, Germany, 2013 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 Search PubMed.
- D. P. Dubal, N. R. Chodankar, D. H. Kim and P. Gomez-Romero, Chem. Soc. Rev., 2018, 47, 2065–2129 RSC.
- X. Chen, N. S. Villa, Y. Zhuang, L. Chen, T. Wang, Z. Li and T. Kong, Adv. Energy Mater., 2020, 10, 1902769 CrossRef CAS.
- H. J. Sim, C. Choi, D. Y. Lee, H. Kim, J. H. Yun, J. M. Kim, T. M. Kang, R. Ovalle, R. H. Baughman, C. W. Kee and S. J. Kim, Nano Energy, 2018, 47, 385–392 CrossRef CAS.
- N. R. Chodankar, S. V. Karekar, M. Safarkhani, A. M. Patil, P. A. Shinde, R. B. Ambade, J. K. Kim, Y. K. Han, Y. S. Huh, A. al Ghaferi and E. Alhajri, Adv. Funct. Mater., 2024, 34, 2406819 CrossRef CAS.
- N. Wang, W. Dou, S. Hao, Y. Cheng, D. Zhou, X. Huang, C. Jiang and X. Cao, Nano Energy, 2019, 56, 868–874 CrossRef CAS.
- Y. W. Chen, J. F. Chen, C. H. Lin and C. H. Hou, Appl. Energy, 2019, 252, 113417 CrossRef CAS.
- P. Ren, M. Torkamanzadeh, S. Arnold, E. Pameté and V. Presser, Batteries Supercaps, 2024, 7, e202400506 CrossRef CAS.
- B. Kokoszka, N. K. Jarrah, C. Liu, D. T. Moore and K. Landskron, Angew. Chem., Int. Ed., 2014, 53, 3698–3701 Search PubMed.
- A. Härtel, M. Janssen, D. Weingarth, V. Presser and R. Van Roij, Energy Environ. Sci., 2015, 8, 2396–2401 RSC.
- J. S. Teixeira, R. S. Costa, A. L. Pires, A. M. Pereira and C. Pereira, Dalton Trans., 2021, 50, 9983–10013 RSC.
- H. V. M. Hamelers, O. Schaetzle, J. M. Paz-García, P. M. Biesheuvel and C. J. N. Buisman, Environ. Sci. Technol. Lett., 2013, 1, 31–35 CrossRef.
- J. M. Griffin, A. C. Forse, W. Y. Tsai, P. L. Taberna, P. Simon and C. P. Grey, Nat. Mater., 2015, 14, 812–819 CrossRef CAS PubMed.
- M. Seyffertitz, S. Stock, M. V. Rauscher, C. Prehal, S. Haas, L. Porcar and O. Paris, Faraday Discuss., 2024, 249, 363–380 RSC.
- C. Prehal, C. Koczwara, H. Amenitsch, V. Presser and O. Paris, Nat. Commun., 2018, 9, 4145 CrossRef CAS PubMed.
- J. M. Griffin, A. C. Forse, H. Wang, N. M. Trease, P. L. Taberna, P. Simon and C. P. Grey, Faraday Discuss., 2014, 176, 49–68 RSC.
- H. Wang, A. C. Forse, J. M. Griffin, N. M. Trease, L. Trognko, P. L. Taberna, P. Simon and C. P. Grey, J. Am. Chem. Soc., 2013, 135, 18968–18980 CrossRef CAS PubMed.
- K. Breitsprecher, C. Holm and S. Kondrat, ACS Nano, 2018, 12, 9733–9741 CrossRef CAS PubMed.
- K. Breitsprecher, M. Janssen, P. Srimuk, B. L. Mehdi, V. Presser, C. Holm and S. Kondrat, Nat. Commun., 2020, 11, 6085 CrossRef CAS PubMed.
- C. Prehal, D. Weingarth, E. Perre, R. T. Lechner, H. Amenitsch, O. Paris and V. Presser, Energy Environ. Sci., 2015, 8, 1725–1735 RSC.
- C. Prehal, C. Koczwara, N. Jäckel, A. Schreiber, M. Burian, H. Amenitsch, M. A. Hartmann, V. Presser and O. Paris, Nat. Energy, 2017, 2, 16215 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- C. Largeot, C. Portet, J. Chmiola, P. L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- C. Merlet, C. Péan, B. Rotenberg, P. A. Madden, B. Daffos, P. L. Taberna, P. Simon and M. Salanne, Nat. Commun., 2013, 4, 2701 CrossRef CAS PubMed.
- N. Ganfoud, A. Sene, M. Haefele, A. Marin-Laflèche, B. Daffos, P.-L. Taberna, M. Salanne, P. Simon and B. Rotenberg, Energy Storage Mater., 2019, 21, 190–195 Search PubMed.
- X. Liu, D. Lyu, C. Merlet, M. J. A. Leesmith, X. Hua, Z. Xu, C. P. Grey and A. C. Forse, Science, 2024, 384, 321–325 CrossRef CAS PubMed.
- M. E. Şahin, F. Blaabjerg and A. Sangwongwanich, Energies, 2022, 15, 674, DOI:10.3390/en15030674.
- K. C. Tsay, L. Zhang and J. Zhang, Electrochim. Acta, 2012, 60, 428–436 CrossRef CAS.
- S. Kaenket, P. Suktha, K. Kongsawatvoragul, T. Sangsanit, P. Wuamprakhon, R. Songthan, W. Tejangkura, K. Santiyuk, K. Homlamai and M. Sawangphruk, J. Power Sources, 2023, 581, 233512 CrossRef CAS.
- G. Gourdin, A. Meehan, T. Jiang, P. Smith and D. Qu, J. Power Sources, 2011, 196, 523–529 CrossRef CAS.
- J. Kim, S. M. Wi, J. G. Ahn, S. Son, H. Y. Lim, Y. Park, H. J. Eun, J. B. Park, H. Lim, S. Pak, A. R. Jang and Y. W. Lee, Energy Environ. Mater., 2023, 6, e12581 CrossRef CAS.
- M. Seyffertitz, C. J. Balhatchet, M. V. Rauscher, S. Stock, G. Fritz-Popovski, T. Leiner, D. Holec, H. Amenitsch, A. C. Forse and O. Paris, Nat. Commun., 2025, 16, 8649, DOI:10.1038/S41467-025-63772-W.
- S. Haas, X. Sun, A. L. C. Conceição, J. Horbach and S. Pfeffer, J. Synchrotron Rad., 2023, 30(6), 1156–1167 CrossRef CAS PubMed.
- C. Prehal, C. Koczwara, N. Jäckel, H. Amenitsch, V. Presser and O. Paris, Phys. Chem. Chem. Phys., 2017, 19, 15549–15561 RSC.
- C. T. Chantler, K. Olsen, R. A. Dragoset, J. Chang, A. R. Kishore, S. A. Kotochigova and D. S. Zucker, X-Ray Form Factor, Attenuation and Scattering Tables (version 2.1), NIST Standard Reference Database 66, DOI:10.18434/T4HS32, http://physics.nist.gov/ffast.
- E. R. Nightingale, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef CAS.
- S. Koneshan, J. C. Rasaiah, R. M. Lynden-Bell and S. H. Lee, J. Phys. Chem. B, 1998, 102, 4193–4204 CrossRef CAS.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1993, 89, 713–718 RSC.
- P. Debye, H. R. Anderson and H. Brumberger, J. Appl. Phys., 1957, 28, 679–683 CrossRef CAS.
- S. Stock, M. Seyffertitz, N. Kostoglou, M. V. Rauscher, V. Presser, B. Demé, V. Cristiglio, M. Kratzer, S. Rols, C. Mitterer and O. Paris, Carbon, 2024, 221, 118911 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |