 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetic restructuring of catalyst active sites: a MACE-APE study of fluxional Pdn/MgO (n = 3–11) clusters
Patricia Poths *a,
King Chun Laib,
Christoph Scheurera,
Sebastian Materaa and
Karsten Reutera
*a,
King Chun Laib,
Christoph Scheurera,
Sebastian Materaa and
Karsten Reutera
aFritz-Haber-Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: poths@fhi-berlin.mpg.de
bMax Planck Computing and Data Facility, Gießenbachstraße 2, 85748 Garching, Germany
First published on 26th January 2026
Abstract
The role of dynamic restructuring of catalytic interfaces under reaction conditions is of unquestionable importance. Sub-nanocluster catalysts, consisting of just a few atoms on a metal oxide support, are known to be highly fluxional. They are able to populate and interconvert between multiple isomers with similar energies. Computational cost has hitherto restricted first-principles treatment of sub-nanocluster fluxionality to the assumption of thermodynamic equilibrium, where the relative isomer populations are Boltzmann distributed. Here, we exploit the advent of fast and accurate machine-learned interatomic potentials (MLIPs), coupled with an automatic process explorer (APE) approach, to systematically evaluate the kinetics of fluxional behavior of sub-nanoclusters. Specifically, we explore the size-dependent restructuring kinetics of Pd3–Pd11 clusters on a fixed MgO(100) support. Propagation of the constructed comprehensive isomerization networks shows that the timescale for relaxation to thermodynamic equilibrium can vary by several orders of magnitude upon changing the number of Pd atoms in the cluster by only one. There is also no simple correlation between the overall relaxation timescale and the amount of time spent in individual (metastable) isomers. Comparison of the timescales of cluster restructuring to a typical rate-limiting catalytic barrier of 1 eV shows that typical fluxional restructuring occurs on comparable to (much) faster timescales, depending on cluster size and specific isomer geometry. Thus, it becomes clear that explicitly resolving the state-to-state dynamics of fluxional sub-nanoclusters is essential for the development of mechanistic understanding of their catalytic performance.
Introduction
Advances in the capabilities of operando catalyst characterization and first-principles modeling of catalysts has determined beyond a doubt that as-synthesized catalysts generally undergo dramatic restructuring upon exposure to reaction conditions.1–9 In fact, the dynamics of this restructuring has been shown to be key for catalytic processes both thermal9–11 and electrocatalytic.12,13 In the past decades, it has been posited that a “flexible” or dynamic surface may in fact be key to catalytic performance.14,15 The computational treatment of this pre-catalyst activation and restructuring has, however, hitherto primarily focused on treating the restructuring of a catalyst as taking place on timescales entirely separate from its reactivity. One prevalent approach is to explore the stability of restructured surface terminations under different reaction conditions with ab initio thermodynamics,3,16–18 which are then used as a static substrate for further multiscale modeling,5,19–25 for which the reaction mechanism is prepared by hand. Another approach is to explicitly explore the restructuring of a surface during the first part of a loop, after which the reactivity of the surface is evaluated, typically using severe approximations.26,27 Additionally, there has been MD-based exploration of catalyst dynamics, which can provide insight into the short-timescale dynamics of the catalyst,28–31 but cannot access the longer timescales which are critical for long-term operation of catalysts associated with the rate events of overcoming i.e. the rate determining step.3The inclination toward an assumed separation of timescales, as well as the significant approximations made to assess reactivity and restructuring together, arise predominantly from the prohibitive cost that exploring the full range of surface restructuring and associated kinetics with first-principles methods would incur, especially for extended surfaces. Systems which are more computationally accessible are those of cluster catalysis, i.e. where the active material is dispersed in the form of sub-nanoscale metallic clusters consisting of just a few atoms on typically an oxide support. These clusters are known to be highly fluxional, that is, they are able to populate and interconvert between multiple structural motifs, or isomers, with similar energies.32–34 These sub-nanoclusters are known to have size-dependent physical35,36 and catalytic9,37 behavior, and computational work which takes into account the isomeric diversity of structures of these fluxional clusters shows improved agreement with experimental work.37–39 In fact, it has been shown that the observed catalytic performance may even be attributed to metastable isomers, rather than the most stable structure.37–40
Due to the small size of these clusters, first-principles sampling of their structural ensemble, (though often in the gas-phase)41–44 and the catalytic reaction mechanism on multiple structures has been feasible.32,37,38,40 However, up to now explicit treatment of sub-nanoscale cluster catalysts has operated exclusively based on the assumption of thermodynamic equilibrium, assuming that the catalytic performance of a given cluster size can be determined by taking a weighted average of the reactivity of each isomer, based on their relative Boltzmann populations.32,40 A truly comprehensive exploration of the kinetics of the cluster restructuring instead remained largely elusive beyond gas-phase or simple systems,41,42,45 due to both the cost of the involved barrier calculations, and the complexity of ensuring that all kinetic transitions between isomers have been found.46–48 Evaluation of catalytic reaction mechanisms that account for fluxional restructuring have therefore been implicit rather than explicit39 – and they focused on small systems, such as Pt4Hx/Al2O347 or Pt3(H2)/Al2O3,48 or Pt7/Al2O3,46 using hand-prepared reaction networks.
Nevertheless, these works have already shown that even such small and apparently simple systems can exhibit significant kinetic trapping under mild to moderate reaction conditions,47 and that there can be multiple timescales of kinetic restructuring involving the restructuring of the cluster core vs. diffusion of adsorbates.47,48 Most notably, it has also been shown theoretically that just by changing the kinetic barrier for fluxional interconversion between two different cluster active sites, the entire kinetic performance of a cluster catalyst can change.49 In light of these results, it is thus imperative to develop a workflow that can rapidly and thoroughly explore the kinetic restructuring of sub-nanoclusters to get a complete real-time picture of their fluxional behavior, and the resulting impact on their catalytic performance.
Recent advances in fast and accurate machine-learned interatomic potentials (MLIPs), such as GAP50 or MACE,51,52 can reduce the computational cost of such sampling by orders of magnitude. Furthermore, the development of the automatic process explorer (APE)53 provides a powerful tool that can automatically calculate all barriers between restructured states directly from the potential energy surface (PES) of the system. The coupling of MLIPs with APE thus promises a computationally tractable surrogate model for the PES exploration. En route to the application to extended surface catalysis,54 we here demonstrate this for the size-dependent restructuring of Pdn/MgO clusters. Benefiting from the finite size of the dynamically restructuring sites, we exhaustively explore the state-to-state dynamics of these systems. These reveal the kinetic contribution to the fluxionality of sub-nanoclusters. Each cluster size is found to have a unique relaxation behavior to thermal equilibrium, the timescale of which can vary by several orders of magnitude. Furthermore, we find that simply assessing the thermalization timescale of an ensemble is not enough to capture the detailed state-to-state dynamics of the system. Instead, calculated residence times, which identify the amount of time a given structure will remain in a specific structural state before isomerizing, give a better impression of the degree of kinetic restructuring inherent to fluxional sub-nanoscale clusters. While the timescales for relaxation to thermal equilibrium can be quite long, the amount of time spent as a specific structure can be short for most isomers, highlighting the great separation of timescales associated with fluxional behavior. In fact, with the exception of a minority of kinetically-trapped metastable isomers, most isomers’ structures would not persist for long enough to support an entire catalytic cycle, indicating that fluxional restructuring of sub-nanoscale clusters will contribute to their catalytic performance.
Methods
MACE-APE active learning
In order to explore the fluxional behavior of Pdn/MgO clusters, we first train a MACE potential on the region of chemical space in which we are interested. Initial structures are generated using the foundational MACE-MP0 potential,55 taking a fixed MgO(100) support, randomly placing between 3 and 11 Pd atoms on it, then performing (a) local geometry optimization, (b) molecular dynamics, and/or (c) randomly-initiated dimer searches centered on all Pd atoms present. A subset of 400 of these initial structures are selected to perform DFT single-point calculations, and construct the training set of the initial potential. An active-learning loop using APE as the structure generation step is thereafter used to ensure that the MACE potential is ideally suited to identify restructured states, and the barriers between them. The approach here is a slight update to ref. 54 and the corresponding workflow is outlined in Fig. 1. In order to explore the range of compositions of interest to this paper (Pd clusters containing between 3 and 11 atoms on a fixed MgO support), we run APE for each independent composition. After 500 (new) elementary processes per independent APE run have been identified, all corresponding local minimum (LM) and transition state (TS) structures are added to the DFT dataset. Structures are selected as new if they contain atomic environments that cannot be assigned to any of the local environment groups from the existing training set, using the fuzzy classifier DECAF56 with default settings. This is then iterated until no new LM and TS structures are found to add to the training set. For further MACE details and accuracy of the final potential, see SI Note 1. | ||
| Fig. 1 Schematic summarizing the MACE-APE active learning approach. Left panel: active learning loop, starting with DFT data, that is used to train the MACE potential. APE is then run using this MACE potential, until 500 new processes are identified. New local minimum (LM) and transition state (TS) structures are selected from the corresponding APE structures. This is done by scanning the APE structures for local atomic environments that are not represented in the existing DFT training set. The new structures are then added to the DFT training set, and the process is iterated until there are no new APE structures that cannot be represented by the training set. Right panel: summary of the APE workflow. A single input structure is given, of which all local atomic environments are classified. A representative of each environment is added to the to-explore list, which is used to start dimer transition state searches (TSSs). These TSSs yield elementary processes consisting of an initial, transition, and final state, providing the process barrier, which can be converted into a rate constant. Finally, all new structures are subjected to local environment classification, and any structures with new local environments are added to the to-explore list. This is iterated until there are no more local environments to explore. | ||
The training set is constructed from single-point spin-polarized DFT calculations, performed with the Vienna Ab initio Simulation Package (VASP) version 6.3.2,57–59 using the PBE exchange–correlation functional60 with D3-dispersion correction.61 We used a kinetic energy cutoff of 700 eV, and used a (2 × 2 × 1) Γ-centered k-point grid for the (16.71 × 16.71) Å 256-atom MgO(100) supercell, which serves as the support for all Pd cluster sizes explored. The entire MgO support was always kept fixed, to focus solely on the dynamics of the Pdn clusters. SCF energy convergence was set to 10−6 eV. Both DFT energies and forces were included in the training set for MACE. We note that GGAs are known to have issues with the calculation of oxide materials,62 and that physical properties of late transition-metal clusters are known to be functional-dependent.63 However, it has also been shown that GGAs can produce barriers of comparable accuracy to hybrid functionals.64 Thus, we use PBE-D3 to calculate barriers until we can directly validate them with experiment.
The APE algorithm is outlined in detail in ref. 53. Briefly, a single input structure is given, of which all atomic environments are then classified using the DECAF method.56 We then select a representative of each unique atomic environment, and use these to start our single-ended TS searches using the dimer algorithm.65 The dimer algorithm is initiated by randomly displacing the selected atom in the initial state (IS), as well as all atoms within a specified distance cutoff. Subsequently, the final state (FS) structure is obtained via local geometry optimization, starting from the TS structure with atomic positions slightly displaced away from the IS. The IS, TS, and FS structures define the given elementary process. From them, we calculate the activation energy barrier for the forward and reverse processes, which is thereafter converted into a rate constant for the process using harmonic transition state theory,5,66,67
 | (1) |
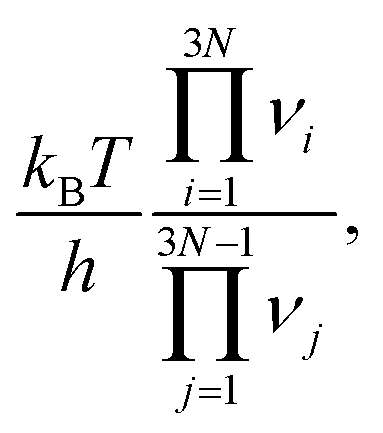 is calculated automatically via APE using the harmonic vibrational modes νi, νj at the IS and TS, respectively, and the activation energy ΔEa is the energy of the TS structure minus the energy of the IS structure.
is calculated automatically via APE using the harmonic vibrational modes νi, νj at the IS and TS, respectively, and the activation energy ΔEa is the energy of the TS structure minus the energy of the IS structure.
All thus identified and defined elementary processes are added to the APE process list, which is a collection of globally unique elementary processes connecting many metastable states. The FS structures of all elementary processes are scanned for new atomic environments. Any structures with new ones are then added to the to-explore list, sorted by ascending energy. APE then iterates until there are no new atomic environments to explore, proceeding in order of the next most stable structure. For a list of APE parameters, see SI Note 2.
For cluster sizes larger than Pd5/MgO, APE did not reach a point where no new local atomic environments were found. For these clusters, we terminated the search once the FS structure that is explored as new IS was >1.5 eV above the most stable IS structure. Based on the ensemble of geometries that we have obtained for each cluster size shown here, we believe that the structural space has been well sampled for most cluster isomerization; however we do not claim that APE is a rigorously complete thermodynamic sampling approach, and it is possible that we would obtain new relevant states if we continued to sample.
Post-processing of APE
The end result of the APE sampling is a large library of elementary steps for each cluster size. While the diversity-driven approach of APE reduces redundant TS searches, it still only relates structures which are globally equivalent. In order to construct the final isomerization network, we need to identify all clusters which are locally equivalent via rotation, translation, or permutation symmetry. This post-processing workflow is outlined in Fig. 2. | ||
| Fig. 2 Schematic summary of the APE post-processing workflow. The APE output is in the format summarized to the left. Each state represents a structure which was used as the initial structure for TS searches. Within each state, there are a number of processes, which are globally unique elementary processes. To convert the APE output into usable form, step (1) involves the clustering and classification of local atomic environments of the Pd atoms within each structure. These are used to reduce the set of globally unique elementary steps into symmetry-equivalent processes, producing a highly inter-connected network, such as is shown in the middle panel. Next, this network undergoes processing step (2), which eliminates all processes which are too fast to be considered stable (aka with barriers <0.1 eV, see text). This involves either merging of multiple states into one, or bypassing one state in the path between two others. For details and examples, please see the SI. This then yields the final isomerization network seen in the right panel, which is used to evaluate the state-to-state dynamics of the system. | ||
As a result of the APE workflow, the library of elementary processes is structured as shown in the left-hand panel of Fig. 2. For the detailed workflow, see Fig. S1. The list of all processes starting from the initial IS is followed by the list of all processes starting from the next IS that was worked on in the to-explore list, and so on and so forth. Each process uniquely connects the IS to a FS; however, there are many more processes than there are connections between states. On the one hand this is due to symmetry redundancies in the IS and FS. To curate this and construct a fully-connected isomerization network, we classify the local atomic environments of all IS and FS structures obtained with APE, and perform fuzzy classification to assign them to equivalence groups using DECAF.56 These groups are used to construct a key, which identifies all clusters which are symmetrically equivalent. These cluster keys are then used to classify processes, which reduces the hundreds to thousands of APE processes to tens to hundreds, depending on the cluster size. This is summarized as step (1), leading to the center panel in Fig. 2. A second reason why there are more processes than connection between states is that APE can identify multiple TS structures between the same two endpoints. We therefore note that each of the processes represented by the double arrows in the center panel of Fig. 2 may actually be a collection of transition states, each associated with a different barrier. We will outline below how this duplicity is handled in the evaluation of the reaction network that results from this post-processing step.
The isomerization networks obtained for the various clusters have elementary processes with barriers ranging from <0.1 eV, to almost 2 eV. To this end, we note that states connected by processes with barriers <0.1 eV are not dynamically stable at catalytically relevant temperatures, as they would only spend times in the state that are comparable to molecular vibrations. The second curation step in the post-processing of the APE library, cf. process (2) in Fig. 2, therefore involves the merging of states which have forward and reverse barriers <0.1 eV into a single state, or bypassing a state which has one exiting barrier <0.1 eV. For the bypassing process, this means that three states that are connected by two processes are reduced into two states connected by a single process. In the SI Note 3, we detail this reduction step for the Pd3/MgO and Pd4/MgO networks for clarity. We note here that we additionally eliminated fragmented clusters from the consideration of cluster networks, as our focus is exclusively on the isomerization dynamics of intact clusters of a given size, rather than cluster migration, Ostwald ripening, or cluster coalescence. Thus, we are excluding the possibility of cluster coalescence at this point in our simulations, though we acknowledge that sub-nanoclusters are particularly sensitive to sintering – though even here, they exhibit size-dependent behavior.36
Kinetics simulations
The minimal isomerization network that results from the post-processing analysis represents all possible transitions between inequivalent states and is the basis for exploration of the state-to-state dynamics of each cluster size. As is common in kinetics, we assume that the state-to-state dynamics can be represented as a Markov jump process.3,68 Thus the vector P of the populations of the different isomers obey the master equation:
 | (2) |
 | (3) |
The construction of the isomerization networks from APE means that the rate matrix representing the state-to-state dynamics is irreducible, i.e. every state can be accessed by every other state by a sequence of elementary steps. As the system is closed and non-driven, the state-to-state dynamics will relax to thermal equilibrium. Therefore, the unique stationary distribution for each cluster size is the Boltzmann distribution, and fulfills detailed balance. G is therefore diagonalizable, with real non-positive eigenvalues λ0, λ1…λN, sorted in descending order, where λ0 = 0 corresponds to an infinite relaxation time, and the corresponding eigenvector v0, when normalized, is the stationary – or Boltzmann – distribution. The relaxation time is then simply given by the slowest non-stationary mode49,68
| τrel = −1/λ1. | (4) |
Equally, we can calculate the residence time of isomer i
 | (5) |
Another approach that we can use to solve the master equation, in order to track the real-time evolution of isomer populations with time is to use the exponential matrix formalism, where we use the rate matrix G,
| P(t) = exp(tG)P(0), | (6) |
We note that the observed cluster population dynamics depend heavily on the accuracy of the barriers in the isomerization network, which are used to construct the rate matrix G. Even a small error (i.e. 0.1 eV) in a given barrier can lead to an order of magnitude difference in rate constants due to the exponential in eqn (1).70,71 While analysis of these errors is beyond the scope of the current work, future work will concern the sensitivity analysis of the kinetic networks for all clusters, to understand which barriers have the most significant impact on the fluxional restructuring of different cluster sizes.72,73
Using the methods outlined above, we identify the isomerization networks for MgO-supported Pd3–Pd11 clusters, and explore their characteristic state-to-state dynamic evolution of isomers. Our goal is (a) to identify whether there is a kinetic contribution to the size-dependent fluxional behavior of these sub-nanoscale clusters, and (b) compare the timescales of kinetic restructuring to typical catalytic timescales, to understand the extent to which fluxional cluster restructuring might play a role in catalytic performance. We define catalytic timescale as the time between two rate-limiting events for a typical catalytic process. For this we assume a barrier of 1 eV.
Results and discussion
Kinetics of the Pd3/MgO system
We begin by exploring the Pd3/MgO system in detail to illustrate how the methodology works on a simple system. The four dynamically stable structural isomers can be seen in Fig. 3(a). These are not the only unique local minima structures identified from the APE output; they are, however, the structures with barriers >0.1 eV exiting them, meaning that they are likely to persist for timescales longer than structural vibrations; two structures which where thusly identified as unstable were removed from the ensemble. For the complete ensemble, including fragmented clusters which were removed as we are only interested in cluster isomerization rather than coalescence at this stage, please see Fig. S3. Global optimization approaches that only identify LM structures would not distinguish between such dynamically stable vs. unstable structures; therefore, APE provides valuable information about the kinetic relevance of a given structure. | ||
| Fig. 3 (a) APE-obtained structural ensemble for Pd3/MgO, and (b) the associated isomerization network, with the lowest barrier between each pair of states shown in eV. Each pathway between the states is also colored with the associated barrier height corresponding to the label. (c) Real-time evolution of the Pd3/MgO ensemble at 300 K, starting from equally populated states (0.25 fractional populations) is shown in the main panel. The dashed vertical line represents the characteristic relaxation time for the isomerization network, calculated from τrel = −1/λ1, where λ1 is the first non-zero eigenvalue of the rate matrix constructed from the network in (b). The right-hand panel shows the Boltzmann populations of each isomer, calculated based on their relative energies. | ||
Fig. 3(b) shows the minimal isomerization network which connects all isomers in (a). The arrow representing the transition between each state is colored by its barrier height, corresponding to the associated label. Only the lowest barrier for each elementary step is shown here; however for almost all processes there were multiple transition states between each pair of endpoints, which were incorporated in the construction of the rate matrix. Fig. S6 shows an example. This isomerization network is then converted into a rate matrix, as described above, which is used to explore the kinetic evolution of states shown in Fig. 3(c) using the matrix exponential approach. The evolution of isomer populations as a function of time is shown, starting from equal populations of all isomers. The relaxation process to the steady-state for even a cluster as simple as Pd3/MgO is not a simple single exponential relaxation; initially, the population of isomer 2 grows rapidly, dramatically overshooting its steady-state (or Boltzmann) population. After this, it decays while the populations of isomers 0 and 1 grow to reach their steady-state populations, which are shown in the right-hand panel of (c). Notably, the time it takes to relax to the steady-state corresponds with the grey vertical dashed line on the plot, which indicates the relaxation time τrel (eqn (4)). The relaxation time calculated from the eigendecomposition of the rate matrix coincides with the kinetics explored using the matrix exponential approach, as is to be expected.
Visual inspection of Fig. 3(b) can provide intuitive insight into the origin of the observed state-to-state dynamics and kinetic trapping outlined above. Isomer 2 clearly has the highest barriers exiting it, and thus will be kinetically trapped compared to the rest of the isomers. This kinetic trapping means that, when starting from equal populations of all isomers, isomer 4 will quickly convert into isomer 2, which will become initially over-populated, after which it slowly decays to populate isomers 0 and 1 to reach thermal equilibrium.
Next, τrel is explored as a function of temperature, from which we can extract an effective barrier that has to be overcome for the system to reach thermal equilibrium using an Arrhenius plot (Fig. 4(a)). Constructing a rate matrix for each temperature, the rate for relaxation is calculated as 1/τrel, of which the natural logarithm is taken and plotted against 1/T. From this, we identify that the effective barrier for the relaxation of the Pd3/MgO ensemble is 0.51 eV. Interestingly, this is higher than the simple numerical average of all lowest barriers used to construct the rate matrix, indicating that the connectivity of the isomerization network also plays a role in determining how the system relaxes.
 | ||
| Fig. 4 (a) The Arrhenius plot for Pd3/MgO. The rates are calculated as 1/τrel, calculated from the rate matrix for temperatures between 300 and 800 K, of which the natural logarithm is taken. From this we can calculate the effective barrier that must be overcome for the system to reach its steady-state population at thermal equilibrium. (b) Plot of the residence times of each Pd3 isomer as a function of temperature. Note that the range of residence times extends over more than 6 orders of magnitude. | ||
Condensing all of the kinetics within even such a simple reaction network as for Pd3/MgO into the single number of an effective barrier for relaxation overlooks a lot of the detailed state-to-state dynamics, however. Beyond just the evolution of the overall populations of different isomers, the amount of time a given isomer might persist for a given cluster, before restructuring into a different isomer, is also of interest. This residence time for isomer i (τres,i), calculated via eqn (5), is shown in Fig. 4(b), showing how the residence time of each isomer changes as a function of temperature.
Residence time is a key factor to consider when evaluating cluster isomers as possible catalytic active sites. Previously, when ensembles of subnanoclusters are considered to provide a variety of catalyst active sites, the activity of the rate determining step is evaluated on each cluster, and scaled by their relative populations.32,40 This assumes that the rate determining step occurs faster than/independently to interconversion between cluster isomers/active sites. Peters has shown for a simplified system that by only changing the rate constant for catalyst active site interconversion, the entire catalytic performance can change.49 By evaluating the residence time of individual metastable isomer structures, we directly evaluate the timescales for active site interconversion to identify their relevance.
Contrary to considering a single barrier for the relaxation to thermal equilibrium, we see that the residence times of each Pd3/MgO isomer can vary by as much as 6 orders of magnitude, depending on the temperature. Therefore, while τrel and associated effective barrier tells us that Pd3/MgO can rapidly reach its steady-state or Boltzmann populations, τres,i tells us that the amount of time spent in each isomer can vary significantly. In fact, the most thermodynamically stable state (isomer 0) does not have the longest residence time; it is isomer 2, which is 0.13 eV less stable, that can persist for much longer. Note that the residence time depends solely on the kinetic barriers required to exit the state, rather than the thermodynamic stability. Fig. S7 shows the histogram of residence times per isomer for Pd3/MgO at 300, 500, and 700 K. Also shown is the rate required to overcome a 1 eV barrier at the given temperature. Regardless of temperature, all isomer lifetimes are shorter than this rate. No isomer would persist long enough to support multiple occurrences of a rate-determining step of 1 eV. However, notably, there can be orders of magnitude differences in the residence time of different isomers. Thus, it becomes clear that most Pd3 isomers will not persist long enough to act as a “static” active site to support multiple consecutive catalytic cycles. Instead, restructuring of Pd3/MgO could occur as part of a catalytic reaction mechanism. This means that, for a system at thermal equilibrium, while the time-average population of each relevant isomer will be the associated Boltzmann population, a single cluster will be converting between isomers. For the case of Pd3/MgO, for instance, while the structural motif associated with isomer 0 will be present more often than isomer 2, the amount of time a structure will remain as isomer 2 before changing into another structure will be longer than for isomer 0. This has significant implications for the evaluation of clusters as catalytic active sites.
Size-dependent kinetics
Now that we have explored the simplest case, we apply the same analysis to larger clusters to explore size-dependent effects, i.e. we systematically explore the dynamic behavior of Pd4/MgO–Pd11/MgO. Using the same APE post-processing approach as above, we construct the minimal isomerization networks for each cluster size. Fig. 5 shows these networks for (a) Pd4/MgO, (b) Pd6/MgO, and (c) Pd7/MgO. For the ensemble of dynamically-stable structures associated with each state (those with barriers >0.1 eV exiting them), please see Fig. S8–S10. The rest of the isomerization networks can be seen in Fig. S11. | ||
| Fig. 5 Minimal isomerization networks for (a) Pd4/MgO, (b) Pd6/MgO, and (c) Pd7/MgO, to illustrate the increasing complexity of the isomerization networks as the number of atoms increases. To see the corresponding structures, see Fig. S8–S10. | ||
As expected, as cluster size increases, so too does the complexity for restructuring. For Pd4 and larger, visual inspection of the network can no longer provide intuitive insight into the state-to-state dynamics of restructuring due to the large number of connections. For the most complex network, Pd11/MgO, we have 108 dynamically stable states, though we had >200 initially, before removing the dynamically unstable states (see Fig. S16), with 386 elementary restructuring processes connecting the states. This level of complexity cannot be treated by hand; APE is essential for the PES sampling and subsequent construction of the isomerization network. As a result, we rely on systematic analysis of the rate matrices to explore the state-to-state cluster dynamics. We note that the isomerization networks for Pd4/MgO and Pd7/MgO in Fig. 5 are more interconnected than those for Pt4Hx/Al2O3 and Pt7/Al2O3,46,47 highlighting the thoroughness of APE sampling in going beyond human intuition in identifying connections between restructured states, and the benefit of MACE-accelerated PES sampling.
We use the networks in Fig. 5 and S11 to construct the rate matrices for the isomerization of each cluster size at temperatures between 300 and 800 K to explore their state-to-state dynamic behavior. Starting from the most coarse-grained level of analysis, we identify the relaxation times τrel to thermal equilibrium for each temperature. These results are summarized in Fig. 6 for all cluster sizes. Note that each column of points contains the data corresponding to the Arrhenius plot for the equivalent cluster size in Fig. S17, from which we can extract the effective relaxation barrier for the isomerization network of each cluster size, which are shown in Table 1.
 | ||
| Fig. 6 Plot of relaxation times vs. cluster size as a function of temperature. Note that the total time required for the system to relax to thermal equilibrium (Boltzmann or steady-state populations) can vary widely based on the cluster size and the given temperature. | ||
| Cluster size | Effective relaxation barrier (eV) | Mean barrier (eV) |
|---|---|---|
| Pd3/MgO | 0.51 | 0.47 |
| Pd4/MgO | 0.77 | 0.71 |
| Pd5/MgO | 0.35 | 0.68 |
| Pd6/MgO | 1.02 | 0.62 |
| Pd7/MgO | 0.93 | 0.63 |
| Pd8/MgO | 0.73 | 0.64 |
| Pd9/MgO | 0.86 | 0.56 |
| Pd10/MgO | 1.00 | 0.56 |
| Pd11/MgO | 0.82 | 0.57 |
Fig. 6 shows the span of relaxation times as a function of temperature for each cluster size; there is no monotonic change in relaxation time with cluster size. Thus, the number of atoms in a cluster is not a descriptor one can use to estimate how long it would take for the system to reach thermal equilibrium. Broadly it takes longer for larger clusters to relax, but this is not a simple linear trend. Instead, the shape of the PES, which dictates the kinetics of cluster restructuring, must be the determining factor; and it is unlikely that this can be captured in such a simple descriptor.
Beyond the highly size-dependent relaxation times for each cluster size, we see a dramatic difference in the timescales required for each cluster size to reach thermal equilibrium. At 300 K, these differences span ∼10 orders of magnitude. As would be expected, they decrease as the temperature is increased, but we still see a span of ∼4 orders of magnitude even at such high a temperature as 800 K. Many cluster sizes (e.g. Pd4, Pd6, Pd10) have relaxation times that reach several seconds to hours even at temperatures as high as 400 K. Any sub-nanocluster catalysts which operate under mild to moderate conditions may thus experience significant kinetic trapping on up to industrial timescales, in agreement with ref. 47.
Next, we consider the effective barriers that have to be overcome for each cluster size to reach thermal equilibrium, compiled in Table 1. Reflecting the wide variation in relaxation times, these barriers vary significantly, from less than 0.4 eV to more than 1 eV. Intriguingly, they are not consistently higher or lower than the numerical mean of all low-barrier processes connecting the networks in Fig. 5 and S11. These mean barriers show less spread than the effective relaxation barriers, clustering between ∼0.6–0.7 eV. For the most part, the mean barriers are somewhat (e.g. for Pd3) to significantly (e.g. Pd10) lower than the effective relaxation barriers. The exception is for Pd5, where the mean barrier is much higher than the effective relaxation barrier. These differences arise from differences in the connectivity of the isomerization networks. None of the established Pd3 to Pd11 networks is fully connected, i.e. there are no single step connections between all states. As illustrated in Fig. S8–S10 and S12–S16, there is instead a partial connectivity, which is in fact higher than might be expected from the structural diversity exhibited in the structural ensembles of the clusters. The effective relaxation barrier results therefore from the dominant pathways for relaxation in these partially connected networks.
As mentioned before in the detailed analysis of Pd3/MgO, reducing the complexity of the cluster isomerization network to a single number per cluster size removes thus a significant amount of detail about how precisely each cluster size relaxes to thermal equilibrium. It is correspondingly essential to move beyond consideration of τrel, and develop an understanding of how the cluster populations evolve, as well as the characteristic residence times of each isomer. From this we can determine how long a given geometry might persist, and whether it makes sense to consider each isomer as a static active site, or if dynamic restructuring is critical.
The evolution of different isomer populations as a function of time for Pd4, Pd6, Pd7, and Pd8 can be seen in Fig. S18. Each cluster size has a unique combination of slow and fast relaxation processes. For all cases, which start with equal populations of all isomers, there are some isomers which initially become over-populated, and then decay to their appropriate steady-state populations, while other isomer populations grow to reach the steady-state. The exact way that the cluster populations relax to equilibrium will depend on the initial populations, however. As this starting point is almost impossible to predict in experimentally-prepared clusters, a more relevant fine-grained feature to explore is the residence time (τrel,i) for each isomer i of a cluster of a given size.
Fig. 7 shows the corresponding per-isomer residence times for (a) Pd4/MgO, (b) Pd6/MgO, and (c) Pd7/MgO associated with the isomerization networks in Fig. 5. Immediately, two things become clear. First, the residence time across all isomers can vary by several orders of magnitude for a given temperature. Second, the residence time of the isomer has no relation to its relative thermodynamic stability. For Pd4/MgO, we see that the structure with the longest residence time is isomer 0, the most stable structure. The next most persistent structure is isomer 10. For Pd6/MgO and Pd7/MgO, however, we see that the multiple most persistent isomers are more or less randomly distributed in energy relative to the most stable isomer. The persistence of a given isomer therefore has no relationship to its thermodynamic stability; it relies only on the energy barrier that must be overcome to exit a given metastable state. This is in agreement with the Pd3/MgO results above, clearly showing that the amount of time a single cluster might reside within a specific state is not related to the stability of the associated structure.
 | ||
| Fig. 7 Residence times of each isomer of (a) Pd4/MgO, (b) Pd6/MgO, and (c) Pd7/MgO. Note that for these clusters, the residence timescales can vary by ∼10 orders of magnitude, depending on the temperature. Furthermore, the thermodynamic stability of the isomer has no correlation to the residence time; it is purely a function of the shape of the PES around each specific metastable state, reflected in the kinetics. | ||
The residence times for the rest of the Pdn/MgO clusters can be seen in Fig. S19–S26. There remains no trend relating the thermodynamic stability of a given isomer to the extent of kinetic trapping that it experiences, with dramatic variation in τrel,i present across all cluster sizes. The temperature dependence of the residence times follows the expected patterns; as temperature increases, the residence times of the longest-persisting structures decrease much faster than the structures which already have a short persistence time. By the time we reach 800 K, while there is still some significant variation between residence times, it is over many fewer orders of magnitude than at 300 K. Notably, even then the residence times can still vary by 2–4 orders of magnitude (Fig. S19–S26). Based on this, while we can conclude that these fluxional clusters undergo highly dynamic restructuring at high temperatures, we cannot truly claim that they have become liquid, which would involve equal populations or residence times across all states. We note that the isomerization networks exclude isomers that have dissociated atoms, meaning that the state-to-state dynamics of the network at the highest temperatures might be somewhat artificially constrained, especially as we neglect cluster coalescence. We would not expect this to have a significant impact on the isomer-specific residence times. Nevertheless, it is very likely that the state-to-state dynamics explored here will impact the catalytic performance of clusters, similarly as has been explored for a gas-phase gold cluster with metadynamics.31 Despite this, the time-averaged population of a given isomer is still dictated by their Boltzmann populations; what is critical here is rather how long a cluster remains as a given isomer before restructuring, and how that timescale compares to that of the rate determining step of a catalytic process. As can be seen in Fig. S27, when comparing the residence times of the Pd4/MgO, Pd6/MgO, and Pd7/MgO isomers to the time required to overcome a 1 eV barrier (a typical value for the rate determining step of a catalytic process), we see that isomer restructuring typically occurs faster than the time between two catalytic events.
Analysis of the state-to-state dynamics of different cluster sizes enables the sorting of cluster sizes into an order of kinetic trapping. There is, however, not a universal order that we can use for kinetic trapping, because it depends on the descriptor that is chosen. For a coarse-grained analysis, we can order the cluster sizes by increasing relaxation time: Pd5 < Pd3 < Pd8 < Pd4 < Pd11 < Pd9 < Pd7 < Pd10 ∼ Pd6. Alternatively we can list the cluster sizes in terms of increasing residence time of the most kinetically trapped isomer at 500 K (see Fig. S19–S26): Pd3 < Pd5 < Pd8 < Pd9 < Pd7 < Pd11 < Pd6 < Pd10 < Pd4. In these two analyses, the order of the clusters changes. Broadly we can say that Pd10 and Pd6 clearly experience the most kinetic trapping, while Pd3, Pd5, and Pd8 experience the fastest state-to-state dynamics, based on both of these metrics. However, other cluster sizes have totally contradicting results, such as Pd4 having a relatively short relaxation time, but a rather long maximum residence time at 500 K. The kinetic behavior of a given cluster size cannot be adequately summarized using a single descriptor; instead, a full analysis of the state-to-state dynamics is essential to properly understand the kinetic contribution to fluxional behavior.
Ultimately, we find that not only do Pdn/MgO (n = 3–11) clusters exhibit size-dependent kinetic behavior, with timescales for relaxation to thermal equilibrium spanning several orders of magnitude, but that they also exhibit isomer-specific kinetic trapping. While the ensemble average proportions of different isomers approach their Boltzmann populations within a given time frame, the amount of time that a cluster might spend in a given structure before isomerization can vary significantly. Many metastable isomers experience this kind of kinetic trapping. Overall, it is clear that all Pdn clusters are highly fluxional, and can readily interconvert on catalytically relevant timescales. We note that this is for clusters in the absence of adsorbates, and that cluster dynamics will inevitably change upon exposure to a reactive gas phase and subsequent adsorption. Adding adsorbates, however, seems unlikely to eliminate all structural dynamics,45,47 especially given the long-standing consensus that catalytic interfaces become increasingly dynamic under reaction conditions compared to UHV surface-science conditions.2,6,14,15
Conclusions
In this work, we used our MACE-APE approach to explore the size-dependent kinetic contribution to the fluxionality of Pdn/MgO (n = 3–11) clusters. We obtained a rich library of elementary processes for the restructuring of each cluster size, which we post-processed to yield a final minimal isomerization network that captures all state-to-state restructuring dynamics. These networks were then converted into rate matrices, from which we extracted information about the real-time kinetic evolution for the isomers of each cluster size.The kinetic behavior of the Pdn/MgO clusters was assessed on multiple levels. On the coarsest level, the state-to-state dynamics of relaxation of the Pdn ensemble to thermal equilibrium could be represented by a relaxation time, with an associated effective barrier. There is no monotonic relationship between cluster size and time required to relax to thermal equilibrium, highlighting that the size-dependent physical behavior of sub-nanoclusters extends to their kinetics. The effective relaxation barrier furthermore is not equivalent to the numerical mean of all kinetic barriers, highlighting the critical component that PES connectivity plays in the relaxation process.
For a finer-grained analysis, we analyzed both the evolution of different isomer populations with time, as well as the amount of time that would be spent in each metastable state. Different cluster isomers have multiple modes of fast and slow relaxation to thermal equilibrium, spanning a wide range of timescales, in agreement with ref. 48. However, this was shown to be highly cluster size dependent. Having multiple relevant timescales for the state-to-state dynamic restructuring of sub-nanoscale clusters appears to be a universal feature of these fluxional species, even in the absence of adsorbates. Furthermore, the complexity of the timescales for interconversion between different active sites aligns nicely with ref. 49, indicating that such state-to-state fluxional dynamics of catalyst active sites would have a consequent impact on the catalytic performance. Calculated residence times for each individual isomer for a given temperature give insight into how often a given cluster would undergo restructuring. Isomer residence times vary by several orders of magnitude, even at the highest temperatures, with significant kinetic trapping occurring for some (metastable) structures. However, the majority of cluster isomers, regardless of their size, will rapidly interconvert with one another at catalytically relevant temperatures. This highly dynamic behavior is reminiscent of studies exploring the “molten” or liquid-like behavior of sub-nanoclusters, which has been used to explain their unique catalytic performance.31,74 We emphasize however that, for the Pdn/MgO clusters studied here, the variation in residence time across the isomers indicates that, while highly dynamic, these clusters cannot be considered to be truly liquid. Furthermore, much of the state-to-state dynamics explored here go well beyond those which can be accessed directly with e.g. molecular dynamics. These results align nicely with the work by Gabor Somorjai, which concluded that more “flexible” or dynamically restructuring surfaces were better substrates for catalytic processes,14,15 with the high dynamic behavior of these clusters perhaps helping to explain their unique behavior compared to their bulk counterparts.
Finally, analysis of the degree to which a given cluster size experiences kinetic hindering can be assessed in one of two ways. For the coarse-grained analysis, the timescale required for the entire ensemble to relax towards equilibrium, can be evaluated. On a finer-grained note, the clusters may be ordered by the length of time the most kinetically hindered metastable isomer will persist. Interestingly, these two analyses do not yield identical results, though Pd10 and Pd6 are shown to exhibit the slowest state-to-state dynamics, and Pd5 and Pd3 exhibit the fastest. Ultimately, fluxional sub-nanoclusters are shown to experience significant – though variable – dynamic restructuring on timescales comparable to or faster than catalysis.
Author contributions
P. P. – conceptualization, project administration, funding acquisition, data curation, methodology, formal analysis, investigation, writing – original draft, review & editing; K. C. L. – methodology; C. S. – conceptualization, supervision; S. M. – conceptualization, supervision, methodology; K. R. – conceptualization, resources, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The MACE potential and code used to post-process the APE libraries is available upon reasonable request.Supplementary information (SI): xyz files of all structures for each cluster size, along with .txt files containing the information about the barriers between isomers for each cluster size used to construct the isomerization networks. See DOI: https://doi.org/10.1039/d5fd00138b.
Acknowledgements
P. P. and K. C. L. gratefully acknowledge the Alexander von Humboldt foundation for postdoctoral research fellowships. P. P. also acknowledges funding from the Fonds der Chemischen Industrie via the Liebig Fellowship. Computing facilities at the Max Planck Computing and Data Facility are gratefully acknowledged.Notes and references
- M. A. Newton, Chem. Soc. Rev., 2008, 37, 2644 RSC.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- K. Reuter, Catal. Lett., 2016, 146, 541–563 CrossRef CAS.
- M. A. van Spronsen, J. W. M. Frenken and I. M. N. Groot, Chem. Soc. Rev., 2017, 46, 4347–4374 RSC.
- A. Bruix, J. T. Margraf, M. Andersen and K. Reuter, Nat. Catal., 2019, 2, 659–670 CrossRef CAS.
- A. Bergmann and B. Roldan Cuenya, ACS Catal., 2019, 9, 10020–10043 CrossRef CAS.
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2020, 121, 882–961 CrossRef PubMed.
- L. Grajciar, C. J. Heard, A. A. Bondarenko, M. V. Polynski, J. Meeprasert, E. A. Pidko and P. Nachtigall, Chem. Soc. Rev., 2018, 47, 8307–8348 RSC.
- X. Li, N. Zhang, D. Wu, Y. Ji, J. Zhang, J. Zhao, Y. Chen, B. C. Gates, J. Liu and C. Xia, J. Am. Chem. Soc., 2025, 147, 39103–39112 CrossRef CAS PubMed.
- S. Nettesheim, A. von Oertzen, H. H. Rotermund and G. Ertl, J. Chem. Phys., 1993, 98, 9977–9985 CrossRef CAS.
- T. Ghosh, J. M. Arce-Ramos, W.-Q. Li, H. Yan, S. W. Chee, A. Genest and U. Mirsaidov, Nat. Commun., 2022, 13, 6176 CrossRef CAS PubMed.
- F. A. Rollier, V. Muravev, A. Parastaev, R. C. J. van de Poll, J. M. J. J. Heinrichs, B. Ligt, J. F. M. Simons, M. C. Figueiredo and E. J. M. Hensen, ACS Catal., 2024, 14, 13246–13259 CrossRef CAS PubMed.
- D. Cheng, K.-L. C. Nguyen, V. Sumaria, Z. Wei, Z. Zhang, W. Gee, Y. Li, C. G. Morales-Guio, M. Heyde, B. Roldan Cuenya, A. N. Alexandrova and P. Sautet, Nat. Commun., 2025, 16, 4064 CrossRef CAS PubMed.
- G. A. Somorjai, Langmuir, 1991, 7, 3176–3182 CrossRef CAS.
- G. A. Somorjai, Catal. Lett., 1992, 12, 17–34 CrossRef CAS.
- K. Reuter and M. Scheffler, Phys. Rev. Lett., 2003, 90, 046103 CrossRef PubMed.
- T. Lee and A. Soon, Nat. Catal., 2024, 7, 4–6 CrossRef.
- F. Riccius, N. Bergmann, H. H. Heenen and K. Reuter, Adv. Sci., 2025, 12, e13878 CrossRef CAS PubMed.
- K. Reuter, D. Frenkel and M. Scheffler, Phys. Rev. Lett., 2004, 93, 116105 CrossRef PubMed.
- K. Reuter and M. Scheffler, Phys. Rev. B, 2006, 73, 045433 CrossRef.
- J. Rogal, K. Reuter and M. Scheffler, Phys. Rev. B, 2008, 77, 155410 CrossRef.
- M. J. Hoffmann and K. Reuter, Top. Catal., 2013, 57, 159–170 CrossRef.
- M. J. Hoffmann, M. Scheffler and K. Reuter, ACS Catal., 2015, 5, 1199–1209 CrossRef CAS.
- Z. Duan and G. Henkelman, ACS Catal., 2014, 4, 3435–3443 CrossRef CAS.
- J. M. Lorenzi, S. Matera and K. Reuter, ACS Catal., 2016, 6, 5191–5197 CrossRef CAS.
- X.-Y. Li, P. Ou, X. Duan, L. Ying, J. Meng, B. Zhu and Y. Gao, JACS Au, 2024, 4, 1892–1900 CrossRef CAS PubMed.
- Y. Yang, A. Hellman and H. Grönbeck, ACS Catal., 2025, 15, 11502–11511 CrossRef CAS.
- O. Abou El Kheir, L. Bonati, M. Parrinello and M. Bernasconi, npj Comput. Mater., 2024, 10, 33 CrossRef CAS.
- S. Perego, L. Bonati, S. Tripathi and M. Parrinello, ACS Catal., 2024, 14, 14652–14664 CrossRef CAS.
- S. Perego, M. Purcel, Y. Baum, S. Chen, A. S. Müller, M. Parrinello, M. Behrens, M. Muhler and L. Bonati, ACS Catal., 2025, 15, 16690–16702 CrossRef CAS PubMed.
- J.-J. Sun and J. Cheng, Nat. Commun., 2019, 10, 5400 CrossRef PubMed.
- H. Zhai and A. N. Alexandrova, ACS Catal., 2017, 7, 1905–1911 CrossRef CAS.
- G. Sun and P. Sautet, Acc. Chem. Res., 2021, 54, 3841–3849 CrossRef CAS PubMed.
- T. Imaoka, T. Toyonaga, M. Morita, N. Haruta and K. Yamamoto, Chem. Commun., 2019, 55, 4753–4756 RSC.
- S. Vajda and M. G. White, ACS Catal., 2015, 5, 7152–7176 CrossRef CAS.
- P. Poths, Z. Hong, G. Li, S. L. Anderson and A. N. Alexandrova, J. Phys. Chem. Lett., 2022, 13, 11044–11050 CrossRef CAS PubMed.
- E. T. Baxter, M.-A. Ha, A. C. Cass, A. N. Alexandrova and S. L. Anderson, ACS Catal., 2017, 7, 3322–3335 CrossRef CAS.
- G. Liu, P. Poths, X. Zhang, Z. Zhu, M. Marshall, M. Blankenhorn, A. N. Alexandrova and K. H. Bowen, J. Am. Chem. Soc., 2020, 142, 7930–7936 CrossRef CAS PubMed.
- P. Poths, G. Li, T. Masubuchi, H. W. T. Morgan, Z. Zhang, A. N. Alexandrova and S. L. Anderson, ACS Catal., 2023, 13, 1533–1544 CrossRef CAS.
- G. Sun and P. Sautet, J. Am. Chem. Soc., 2018, 140, 2812–2820 CrossRef CAS PubMed.
- G. Santarossa, A. Vargas, M. Iannuzzi and A. Baiker, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 174205 CrossRef.
- D. J. Wales, Mol. Phys., 2004, 102, 891–908 CrossRef CAS.
- M. Settem, C. Roncaglia, R. Ferrando and A. Giacomello, J. Chem. Phys., 2023, 159, 094303 CrossRef CAS PubMed.
- L. H. E. Kempen and M. Andersen, npj Comput. Mater., 2025, 11, 8 CrossRef CAS.
- X. Xing, X. Li, B. Yoon, U. Landman and J. H. Parks, Int. J. Mass Spectrom., 2015, 377, 393–402 CrossRef CAS.
- H. Zhai and A. N. Alexandrova, J. Phys. Chem. Lett., 2018, 9, 1696–1702 CrossRef CAS PubMed.
- P. Poths, S. Vargas, P. Sautet and A. N. Alexandrova, ACS Catal., 2024, 14, 5403–5415 CrossRef CAS.
- G. Yan and D. G. Vlachos, ACS Catal., 2023, 13, 10602–10614 CrossRef CAS.
- B. Peters, ACS Catal., 2022, 12, 8038–8047 CrossRef CAS.
- A. P. Bartók, M. C. Payne, R. Kondor and G. Csányi, Phys. Rev. Lett., 2010, 104, 136403 CrossRef PubMed.
- I. Batatia, D. P. Kovacs, G. N. C. Simm, C. Ortner and G. Csanyi, MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields, Advances in Neural Information Processing Systems, 2022 Search PubMed.
- I. Batatia, S. Batzner, D. P. Kovács, A. Musaelian, G. N. C. Simm, R. Drautz, C. Ortner, B. Kozinsky and G. Csányi, The Design Space of E(3)-Equivariant Atom-Centered Interatomic Potentials, arXiv, 2022, preprint, arXiv:2205.06643, DOI:10.48550/arXiv.2205.06643.
- K. C. Lai, P. Poths, S. Matera, C. Scheurer and K. Reuter, Phys. Rev. Lett., 2025, 134, 096201 CrossRef CAS PubMed.
- P. Poths, K. C. Lai, F. Cannizzaro, C. Scheurer, S. Matera and K. Reuter, ACS Catal., 2025, 15, 514–522 CrossRef CAS.
- I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kovács, J. Riebesell, X. R. Advincula, M. Asta, M. Avaylon, W. J. Baldwin, F. Berger, N. Bernstein, A. Bhowmik, F. Bigi, S. M. Blau, V. Cǎrare, M. Ceriotti, S. Chong, J. P. Darby, S. De, F. Della Pia, V. L. Deringer, R. Elijošius, Z. El-Machachi, E. Fako, F. Falcioni, A. C. Ferrari, J. L. A. Gardner, M. J. Gawkowski, A. Genreith-Schriever, J. George, R. E. A. Goodall, J. Grandel, C. P. Grey, P. Grigorev, S. Han, W. Handley, H. H. Heenen, K. Hermansson, C. H. Ho, S. Hofmann, C. Holm, J. Jaafar, K. S. Jakob, H. Jung, V. Kapil, A. D. Kaplan, N. Karimitari, J. R. Kermode, P. Kourtis, N. Kroupa, J. Kullgren, M. C. Kuner, D. Kuryla, G. Liepuoniute, C. Lin, J. T. Margraf, I.-B. Magdǎu, A. Michaelides, J. H. Moore, A. A. Naik, S. P. Niblett, S. W. Norwood, N. O'Neill, C. Ortner, K. A. Persson, K. Reuter, A. S. Rosen, L. A. M. Rosset, L. L. Schaaf, C. Schran, B. X. Shi, E. Sivonxay, T. K. Stenczel, C. Sutton, V. Svahn, T. D. Swinburne, J. Tilly, C. van der Oord, S. Vargas, E. Varga-Umbrich, T. Vegge, M. Vondrák, Y. Wang, W. C. Witt, T. Wolf, F. Zills and G. Csányi, J. Chem. Phys., 2025, 163, 184110 CrossRef CAS PubMed.
- K. C. Lai, S. Matera, C. Scheurer and K. Reuter, J. Chem. Phys., 2023, 159, 024129 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Gerosa, C. E. Bottani, C. Di Valentin, G. Onida and G. Pacchioni, J. Phys.: Condens. Matter, 2017, 30, 044003 CrossRef PubMed.
- T. M. Soini, A. Genest, A. Nikodem and N. Rösch, J. Chem. Theory Comput., 2014, 10, 4408–4416 CrossRef CAS PubMed.
- A. Mahler, B. G. Janesko, S. Moncho and E. N. Brothers, J. Chem. Phys., 2017, 146, 234103 CrossRef PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS.
- B. Peters, in Discrete Stochastic Variables, Elsevier, 2017, p. 363–401 Search PubMed.
- The mpmath development team, mpmath: a Python library for arbitrary-precision floating-point arithmetic, version 1.3.0, 2023 Search PubMed.
- S. Döpking and S. Matera, Chem. Phys. Lett., 2017, 674, 28–32 CrossRef.
- S. Döpking, C. P. Plaisance, D. Strobusch, K. Reuter, C. Scheurer and S. Matera, J. Chem. Phys., 2018, 148, 034102 CrossRef PubMed.
- S. Dortaj and S. Matera, J. Chem. Phys., 2023, 159, 094110 CrossRef CAS PubMed.
- S. Matera, W. F. Schneider, A. Heyden and A. Savara, ACS Catal., 2019, 9, 6624–6647 CrossRef CAS.
- F.-Q. Gong, Y.-P. Liu, Y. Wang, W. E, Z.-Q. Tian and J. Cheng, Angew. Chem., Int. Ed., 2024, 63, e202405379 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |