 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrelating binding energies of adsorbed CO and H on model surfaces with CO/H2 selectivity from co-electrolysis of CO2 and H2O over copper–palladium bimetallic catalysts
Marcus Yu†
 a,
Hong Zhang†b,
William J. Weia,
Ping Liubc and
Jingguang G. Chen*ac
a,
Hong Zhang†b,
William J. Weia,
Ping Liubc and
Jingguang G. Chen*ac
aDepartment of Chemical Engineering, Columbia University, New York, NY 10027, USA. E-mail: jgchen@columbia.edu
bDepartment of Chemistry, State University of New York at Stony Brook, Stony Brook, NY 11794, USA
cChemistry Division, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 29th April 2026
Abstract
Binding energies of adsorbed CO and H are key descriptors governing the activity and selectivity of the co-electrolysis of CO2 and H2O to produce syngas with desired CO/H2 ratios. Palladium hydride (PdH), which forms in situ at negative overpotentials, has been identified as the active Pd phase for CO2 reduction to syngas. Herein, binding energies of CO and H are determined using temperature programmed desorption (TPD) of CO and H2 from Pd(111), PdH/Pd(111), and Cu/PdH/Pd(111) under ultra-high vacuum (UHV) conditions. TPD results reveal that desorption of H2 from subsurface PdH occurs at 460 K, while desorption from surface PdH is more facile at 320 K. CO desorption temperatures shift 20 K lower on PdH/Pd(111) compared to on Pd(111). The presence of 0.7 ML Cu further increases the desorption temperature of H2 by 30 K while simultaneously reducing CO desorption temperatures by 70 K. Density functional theory (DFT) calculations show that CO adsorption onto Pd sites is hindered on the 0.7 ML Cu/PdH/Pd(111) surface while the kinetic barrier for H2 desorption is increased. The trends in the binding energies of CO and H on model surfaces are consistent with electrochemical measurements of CuPd powder catalysts in a membrane electrode assembly (MEA), where H2 evolution is reduced while CO production is enhanced compared to unmodified Pd catalysts. Overall, the results from model surface studies (TPD and DFT) provide a prediction and explanation for the activity and CO/H2 ratios observed in electrochemical experiments. This study also demonstrates that CuPd is a promising catalyst with reduced Pd-loading to produce CO-rich syngas.
1 Introduction
CO2 utilization is a promising method in tackling the rapid increase in atmospheric CO2. Several popular strategies for CO2 utilization are mineralization, syngas production, or as feedstocks for renewable fuels and fertilizer.1–3 Among these, an increasingly attractive avenue for CO2 utilization is the catalytical conversion of CO2 into value-added forms of solid carbon, such as carbon nanofibers (CNFs) or graphene.4–10 However, direct one-pot electrochemical conversion of CO2 to CNFs is thermodynamically challenging and requires high temperatures (>700 °C).11,12 Recently, Xie et al. showed that by coupling an electrochemical reactor for co-electrolysis of CO2 and H2O to syngas with a thermochemical reactor to convert syngas into CNFs, one can produce CNFs at a much lower temperature than was previously demonstrated with either thermochemical or electrochemical reactors alone.4 They identified that the preferred pathway for forming CNFs was the Boudouard reaction (2CO → C(s) + CO2). Based on this, a higher CO production rate from co-electrolysis of CO2 and H2O should lead to more CNF formation by promoting the Boudouard reaction. An ideal electrocatalyst should be highly active and selective for the CO2 reduction reaction (CO2RR) to CO while also producing just enough H2, from the competing hydrogen evolution reaction (HER) to keep the catalyst reduced in the thermochemical reactor. The development of an electrocatalyst that can produce syngas with tunable and desirable CO/H2 ratios is thus necessary to optimize the production of CNFs from the tandem electrochemical–thermochemical reactor scheme.There has been extensive research into CO2RR to syngas, among which precious metals such as Ag, Au, and Pd have been identified as excellent catalysts with high activity, stability, and CO selectivity.13–17 Pd stands out as an interesting CO2RR catalyst due to its ability to dissociate H2 and absorb atomic H to form a Pd hydride (PdH) phase.18 At low overpotentials, Pd remains metallic and converts CO2 to formate selectively.19 However, as potentials are reduced to −0.2 V vs. RHE and below, absorption of atomic H into the bulk Pd lattice occurs, leading to lattice expansion and PdH formation, which coincides with a shift towards CO as the preferred CO2RR product.20
To achieve higher CO/H2 ratios, it is necessary to develop electrocatalysts with moderate CO binding energies. Pd-based catalysts typically suffer from CO poisoning that can reduce activity and selectivity for CO2RR over HER.21,22 As a result, much of the research in using Pd-based catalysts for CO2RR has focused on reducing the CO binding energy to improve CO tolerance using various strategies such as alloying, doping, and facet control.23–26 Several of these studies have identified that the formation of PdH is a key factor in enhancing the CO tolerance of Pd-based catalysts.20,24,27 In the case of alloying, several studies have revealed that the addition of a secondary element such as C or Ag can inhibit PdH formation to control the degree of hydrogenation.28–30 Regarding CO2RR, Ag can be incorporated to hinder PdH formation, leading to enhanced formate selectively, which would be undesirable for the tandem reactor scheme to form CNFs.
In this paper, we experimentally determine activation energies of H2 and CO desorption from a Pd(111) surface under ultra-high vacuum (UHV) with temperature programmed desorption (TPD). Following this, we demonstrate that we can form a subsurface PdH/Pd(111) phase in UHV that is reproducible and stable. A separate, high temperature desorption peak for H2 is observed on the PdH/Pd(111) surface that confirms the successful formation of a PdH phase. Additionally, we measure the effect of sub-monolayer (ML) Cu coverage on the PdH/Pd(111) surface on H2 and CO desorption and reveal that at 0.7 ML, Cu acts to block available Pd sites and significantly hinders H2 desorption while simultaneously reducing CO binding energy. DFT calculations indicate that Cu acts to block available Pd sites, preventing strong CO and H adsorption on Pd hollow sites. Cu significantly increases the diffusion barrier for both surface and subsurface H on PdH/Pd(111) to migrate and desorb, leading to the observed effects in the TPD experiments. We then correlate the results on model surfaces with electrochemical tests to show that trends in CO and H binding energies can be used to tune CO/H2 ratios and guide CO2RR electrocatalyst design.
2 Methods
2.1 Temperature programmed desorption
TPD experiments were carried out in a UHV chamber equipped with a Hiden HAL 3F-RC mass spectrometer for monitoring gas-phase species. The Pd(111) surface was first cleaned using Ne sputtering followed by annealing to 1000 K. Cu was deposited on the surface using physical vapor deposition from heating Cu wires wrapped around a Ta heating lead. Cu was removed and re-deposited before each experiment to ensure each surface was as consistent as possible and to minimize Cu migration into the Pd lattice. An Auger electron spectrometer (AES) was used to characterize the surface elemental composition following the quantification method developed by Cumpson et al.31,32 A deposition curve was developed by monitoring the growth of the Cu (920 eV)/Pd(270 eV) ratio as a function of deposition time (Fig. S1). A sharp break at 8 minutes of deposition time indicated the completion of 1 ML Cu on Pd(111). Additionally, the linear fits suggested that Cu deposition followed the layer-by-layer growth mechanism on the Pd(111) surface. An AES peak ratio of 0.08 Cu (920 eV)/Pd(270 eV) was used to indicate the deposition of 1 ML of Cu on Pd.H2 and CO were dosed into the UHV chamber using a ¼ inch stainless steel tube attached to an external leak valve. Each leak valve was connected to a gas line that was evacuated with a turbopump. Immediately prior to the experiment, the gas-line was sealed from vacuum and filled with H2 or CO respectively. Dosages were measured in Langmuir units (L, 1 L = 1 × 10−6 torr × s). The dosing temperature was 150 K for experiments on Pd(111), and 300 K for experiments on PdH/Pd(111) surfaces. Unless otherwise specified, the heating rate was 2 K s−1.
2.2 Electrochemical experiments
Unless otherwise noted, all chemicals were obtained from Sigma Aldrich. Electrocatalysts with 10 wt% metal loading were synthesized following a previously reported procedure.27 Gas diffusion electrodes were prepared using a standard ink formulation and airbrushing method.4 A zero-gap MEA electrolyzer with a 5 cm2 stainless-steel cathode flow field and titanium anode flow field separated by an anion exchange membrane (Sustainion X37-50 Grade RT, Dioxide Materials) was used to measure CO2RR selectivity. The cathodic flow field was fed a mixture of 10 mL min−1 CO2 and 2 mL min−1 Ar, while the anodic flow field was fed with 2 mL min−1 50 mM KHCO3. Electrochemical measurements were conducted using a BioLogic VSP potentiostat complemented with a Bio Logic VMP3B-20 (20 A/20 V) current booster. The cathode was pre-treated by conducting 20 cyclic voltammetry cycles between −0.2 V and −2.20 V (50 mV s−1). To assess product selectivity, chronoamperometry was conducted at an applied potential of −3.0 V. The cathodic effluent was first passed through a water trap cooled with ice and then directed to the GC, an Agilent 7890B GC equipped with PLOTQ and MOLSIEVE columns and TCD and FID detectors, using an online connection. The CO/H2 ratio was calculated from the molar fractions of CO and H2 detected in the cathodic effluent.2.3 DFT calculations
Spin-polarized DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP).33–35 The Perdew–Burke–Ernzerhof (PBE)36 functional was used to describe electronic exchange and correlation. All structural optimizations were initialized using the Conjugate Gradient ionic relaxation algorithm37 then converged with the RMM-DIIS ionic relaxation algorithm38 with a plane wave cutoff energy of 400 eV. An electronic energy convergence criterion of 1 × 10−6 eV was applied, and ionic relaxation was performed until the Hellmann–Feynman forces on each atom were below 0.02 eV Å−1.A DFT optimized 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 β-PdH FCC unit cell with lattice constant of 4.140 Å was used to construct the PdH(111) surface.39 The 4 × 4 PdH(111) surface consisted of four layers of Pd atoms, whereby the bottom two layers were restrained to represent the bulk structure underneath. The formation energy of the PdH(111) surface was found to be lower when terminated with H (Fig. S5). A 4 × 4 × 1 Monkhorst–Pack grid and first-order Methfessel–Paxton with a smearing width of 0.2 eV were used to integrate over the Brillouin zone. Periodic boundary conditions were applied to extend the surfaces infinitely. A vacuum spacing of 20 Å was added above the surface to ensure at least 15 Å of separation when Cu atoms or intermediate molecules were adsorbed, and dipole corrections were included along the vacuum direction.
:1 β-PdH FCC unit cell with lattice constant of 4.140 Å was used to construct the PdH(111) surface.39 The 4 × 4 PdH(111) surface consisted of four layers of Pd atoms, whereby the bottom two layers were restrained to represent the bulk structure underneath. The formation energy of the PdH(111) surface was found to be lower when terminated with H (Fig. S5). A 4 × 4 × 1 Monkhorst–Pack grid and first-order Methfessel–Paxton with a smearing width of 0.2 eV were used to integrate over the Brillouin zone. Periodic boundary conditions were applied to extend the surfaces infinitely. A vacuum spacing of 20 Å was added above the surface to ensure at least 15 Å of separation when Cu atoms or intermediate molecules were adsorbed, and dipole corrections were included along the vacuum direction.
Different models of Cu supported on PdH(111) with 0.3 ML and 0.7 ML coverage were used to find conformations with the lowest formation energy and minimized Cu–Cu bond strain with respect to the DFT optimized 3.629 Å Cu FCC crystal lattice. Distributed Cu atoms and 2D/3D islands of supported clusters were considered, and the formation energy of Cux/PdH(111) was calculated as:
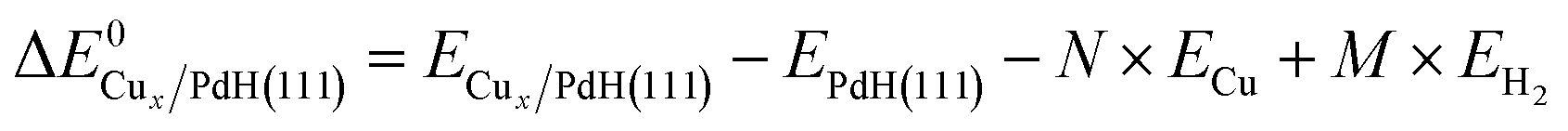 | (1) |
| B.E. = Eadsorbate/surface − Efree molecule − Eclean surface | (2) |
Under a specific temperature used in the TPD experiments, the free energy of adsorption ΔG(T) was calculated as:
| ΔG(T) = Gadsorbate/surface − Eclean surface − µfree molecule | (3) |
| Gadsorbate/surface = Eadsorbate/surface + ZPE − TS | (4) |
 | (5) |
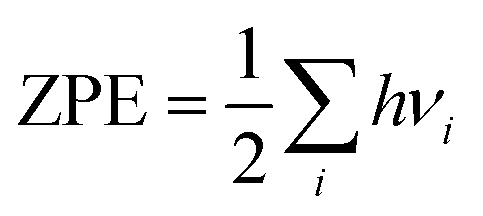 | (6) |
| E0 + ZPE + ΔH(T) − TS | (7) |
The H diffusion rates, whether on the surface or from subsurface PdH, were derived from transition state theory:42
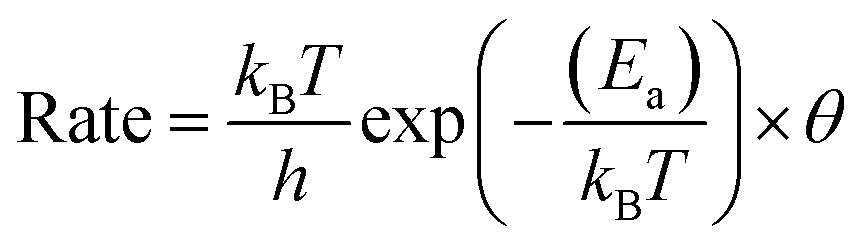 | (8) |
3 Results and discussion
3.1 TPD results of H2 and CO desorption from Pd(111) and PdH/Pd(111)
Binding energies of H and CO on Pd(111) were estimated from a series of TPD experiments with varying heating rates. Following H2 or CO exposure, the Pd(111) surface was then heated at 0.5 K s−1, 1 K s−1, 2 K s−1 and 4 K s−1 and the peak desorption temperatures (TP) were measured (Fig. 1a and b). The linearized Redhead equation (eqn (9)), given below, was then used to determine the activation energy for desorption, Ed, based on the change in Tp as heating rates were varied (Fig. 1c and d).47–49
 | (9) |
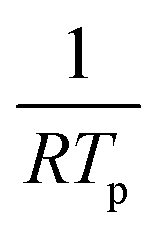 versus
versus  allows the determination of the activation energy for desorption Ed in the units of J mol−1. By assuming that H2 and CO have little to no activation barrier for adsorption, −Ed could be approximated to be the binding energies of H and CO,50,51 which were determined to be −83.6 kJ mol−1 and −161.7 kJ mol−1, or −20.0 kcal mol−1 and −38.6 kcal mol−1, respectively. These values were consistent with previously reported values by Ertl et al., which were −20.8 kcal mol−1 for H and −34 kcal mol−1 for CO on Pd(111).50,52
allows the determination of the activation energy for desorption Ed in the units of J mol−1. By assuming that H2 and CO have little to no activation barrier for adsorption, −Ed could be approximated to be the binding energies of H and CO,50,51 which were determined to be −83.6 kJ mol−1 and −161.7 kJ mol−1, or −20.0 kcal mol−1 and −38.6 kcal mol−1, respectively. These values were consistent with previously reported values by Ertl et al., which were −20.8 kcal mol−1 for H and −34 kcal mol−1 for CO on Pd(111).50,52
 | ||
| Fig. 1 TPD spectra after 10 L exposure of (a) H2 or (b) CO were dosed onto the clean Pd(111) surface at 150 K. Heating rates were varied from 0.5 K s−1 to 4 K s−1. Peak desorption temperatures of (c) H2 and (d) CO at varying heating rates plotted using the linearized Redhead equation. The slope represents the activation energy for desorption in the unit of J mol−1. | ||
Next, H2 and CO were co-adsorbed onto the Pd(111) surface to determine if co-adsorption had any impact on the binding energies. H2 desorption temperatures increased when CO was co-adsorbed on the surface (Fig. 2a). The peak temperature shifted from 318 K to 367 K and 385 K depending on the order of co-adsorption. Changing the order of co-adsorption (H2 first or CO first) significantly impacted the amount of adsorbed H on the surface. Pre-adsorbed CO blocked H2 adsorption, leading to a diminished desorption peak area compared to when H2 was adsorbed first. The desorption peak temperature was highest when CO was dosed first, which may have resulted from the lower coverage of adsorbed H. In comparison, the CO peak desorption temperature was not noticeably affected by the co-adsorption of surface H (Fig. 2b).
 | ||
| Fig. 2 TPD spectra of (a) H2 and (b) CO following the co-adsorption of 10 L H2 and 10 L CO on the Pd(111) surface. The adsorption order was alternated to observe the effect of pre-adsorbed H and CO on H2 and CO binding and desorption. | ||
Previously, it had been determined that a PdH phase was likely the active phase during the electrocatalytic reduction of CO2 to CO.27 Therefore, it was desirable to create a PdH phase under UHV conditions on Pd(111) for further investigation on H2 and CO adsorption. A PdH phase was successfully formed under UHV conditions by increasing the H2 dosage to 600 L and holding the Pd(111) crystal at 300 K for several minutes to allow for H2 dissociation and H migration into the subsurface region of Pd(111) (Fig. 3a). These experimental conditions were adapted from previous studies of H2 adsorption on Pd(111).50,53
 | ||
| Fig. 3 (a) TPD spectra of H2 after 10 L H2 dosage at 150 K and 600 L H2 dosage at 300 K without any CO co-adsorbed on Pd(111). (b) TPD spectra of CO after the co-adsorption of 10 L CO at 250 K onto the 10 L and 600 L H2 pre-dosed surfaces. A broad H2 desorption peak centered at 465 K indicates the formation of a stable, subsurface PdH/Pd(111) phase. | ||
TPD experiments were then performed on the PdH/Pd(111) surface. Following this H2 treatment, a second higher temperature H2 desorption peak at 465 K appeared for PdH/Pd(111). Desorption of H2 at temperatures above 450 K was characteristic of early transition metals, such as Ti, and was typically not observed in noble metals such as Pd or Pt.54–56 Previous studies on Pd single crystal surfaces suggested that adsorption at liquid nitrogen temperatures (80 K) led to a saturation coverage of 1.5 ML H2, with 1 ML on the surface (β-PdH), and another 0.5 ML existing in the subsurface layer (α-PdH).53,57 Typically, this led to two separate desorption regimes, 150–200 K for α-PdH and 300–400 K for β-PdH. Desorption of H2 at temperatures above 450 K suggested successful formation of a separate PdH phase in the Pd(111) subsurface region.
The amount of H2 desorption from the surface PdH phase from 250–350 K after 600 L exposure was substantially reduced compared to H2 desorption after 10 L exposure on clean Pd(111), despite being treated with 60 times more H2. This suggested that the saturation coverage of surface H was reduced on the PdH/Pd(111) surface. The areas of the peaks were approximated using a simple Gaussian fitting (Fig. S2). The H2 desorption peak area from surface PdH/Pd(111) was determined to be approximately half of the desorption peak area from Pd(111). If a saturation coverage of 1 ML of surface H was to be assumed on clean Pd(111), then the coverage of surface H was approximately 0.5 ML on PdH/Pd(111). Additional fitting showed that the desorption area of H from subsurface PdH/Pd(111) above 320 K was equivalent to 2 ML, or twice that of the surface H saturation coverage on Pd(111).
The PdH/Pd(111) surface was cooled to 250 K and 10 L CO was adsorbed onto the surface to determine if PdH formation impacted CO binding energy (Fig. 3b). The peak shape for CO desorption was identical on both Pd(111) and PdH/Pd(111); however, the desorption temperature decreased by about 25 K on PdH/Pd(111). The desorption spectra of H2 from the CO-covered PdH/Pd(111) surface is depicted separately in Fig. 4a. As expected, based on earlier results from the co-adsorption of CO on Pd(111) (Fig. 2), the surface H desorption peak shifted to about 50 K higher with the co-adsorption of CO (Fig. 4) onto PdH/Pd(111). However, CO adsorption on the surface does not seem to influence H desorption from PdH at 465 K. Lastly, AES showed no difference in the C peak before and after CO desorption, suggesting that CO desorbed reversibly on PdH/Pd(111).
 | ||
| Fig. 4 TPD spectra of (a) H2 and (b) CO desorption from CO-covered PdH/Pd(111) and CO-covered Cu-modified PdH/Pd(111) surfaces. The PdH/Pd(111) surface was prepared by 600 L H2 dosage at 300 K. | ||
3.2 TPD results for H2 and CO desorption from Cu/PdH/Pd(111)
Following experiments on clean PdH/Pd(111), Cu was deposited onto the PdH/Pd(111) surface to determine its effect on H and CO binding energies. After exposing the Cu-modified PdH/Pd(111) surface to H2 and CO, two H2 desorption peaks were still observed from the 0.7 ML Cu/PdH surface, suggesting that Cu on the prepared PdH/Pd(111) surface did not disrupt the existing PdH phase. Additionally, when the order of H2 and Cu deposition was reversed, it was demonstrated that exposing a 0.7 ML Cu/Pd(111) surface to the 600 L H2 treatment still led to successful PdH formation, indicating that a sub-ML Cu coverage did not hinder PdH formation (Fig. S3). After depositing 0.7 ML of Cu, the peak desorption temperature of surface H shifted slightly higher to 390 K from 370 K. More notably, the peak desorption temperature of subsurface PdH from the 0.7 ML Cu/PdH surface (Fig. 4a) shifted 30 K higher than that observed on CO-covered PdH/Pd(111) (Fig. 3). Additionally, the subsurface PdH peak became more intense and sharper, with a distinct separation from surface H desorption peak. At lower coverages of Cu (0.3 ML), there was an increase in the intensity and peak temperature of H2 desorption from surface H without significant change in H2 desorption from subsurface PdH. Meanwhile, the CO desorption peak temperature significantly decreased to 380 K from 457 K with 0.7 ML Cu coverage. At 0.3 ML Cu, the CO desorption spectra remained broad and did not result in a sharp peak like it did with 0.7 ML Cu. The CO desorption peak area also decreased by about 30% on 0.3 ML Cu/PdH compared to PdH/Pd(111), which suggested that at low coverages of Cu, CO preferentially bonded to available Pd sites. Overall, TPD results suggest that the presence of 0.7 ML Cu coverage on PdH/Pd(111) significantly reduced CO binding energy while simultaneously increasing H binding energy, which was an unexpected trend as H and CO binding energies often change in tandem instead of in opposite directions, as discussed in DFT calculations next.3.3 DFT results
DFT calculations were carried out to gain a better understanding of the TPD results. Herein, a PdH film supported on Pd(111) was modeled by PdH(111) and two-dimensional Cu clusters adsorbed on PdH(111) were used to describe Cu-modified PdH/Pd(111) (Fig. S5 and S6). The results show that upon exposure to H2, the dissociated hydrogen atom, H, prefers to adsorb at the 3-fold Pd hollow sites on the Pd-terminated PdH(111) surface (Fig. 5a). The surface thermodynamically prefers to be saturated by 1 ML H with an average binding energy (B.E.) of −0.27 eV per surface H atom, which is weaker than that on Pd(111) (B.E. = −0.66 eV per H).58,59 The H monolayer is thermodynamically stable at 300 K and 400 K according to the calculated adsorption free energy (ΔG300K = −0.06 eV; ΔG400K = −0.01 eV) but becomes unstable when the temperature is raised to 500 K (ΔG500K = 0.05 eV). CO adsorption at 0.25 ML of coverage is also substantially weaker at the Pd hollow sites (B.E. = −1.36 eV) on Pd-terminated PdH(111) than that on Pd(111) (B.E. = −2.12 eV).58 Compared to adsorbed H, adsorbed CO is much more stable on Pd-terminated PdH(111), remaining intact up to 500 K (ΔG300K = −0.80 eV; ΔG400K = −0.63 eV; ΔG500K = −0.49 eV). The CO saturation coverage is reached at 0.50 ML (average ΔG300K = −0.75 eV per CO; ΔG400K = −0.57 eV per CO; ΔG500K = −0.47 eV per CO). Upon exposure to a mixture of CO and H2, 50% of Pd sites on Pd-terminated PdH(111) are preferentially saturated by CO, while the rest of the free Pd sites are likely occupied by H, featuring a stable surface configuration of 0.5 ML H + 0.5 ML CO (Fig. 5a). | ||
| Fig. 5 DFT-optimized structures for (a) CO-saturated PdH(111); (b) 0.3 ML Cu/PdH(111); (c) 0.7 ML Cu/PdH(111). (d) DFT calculated B.E. of adsorbed CO at the most stable Cu or Pd site on each surface model. (Grey: Pd; blue: Cu; pink: H; red: CO). | ||
The 0.3 ML Cu/PdH(111) was modeled by a Cu7 cluster supported on Pd-terminated PdH(111) (Fig. 5b). CO preferentially occupies the 2-fold Cu bridge sites at the edge of supported Cu clusters (B.E. = −1.10 eV per CO, Fig. S7a). It saturates at a coverage of 3 CO/Cu7, which is stable up to 500 K (average ΔG300K = −0.53 eV per CO; ΔG400K = −0.37 eV per CO; ΔG500K = −0.24 eV per CO, Fig. 5b). Additional CO adsorption is not preferred (ΔG300K = 0.17 eV per CO; ΔG400K = 0.31 eV per CO; ΔG500K = 0.43 eV per CO). At a low Cu coverage of 0.3 ML, many Pd sites remain exposed at which CO can still adsorb, albeit with slightly weaker binding than that on PdH(111) (B.E. = −1.27 eV, Fig. S7a). Under co-adsorption of CO with H, atomic H does not prefer the Cu7 cluster due to CO saturation, but instead prefers the exposed Pd hollow sites on the surface (Fig. 5b).
The increase in Cu coverage from 0.3 ML to 0.7 ML was described by cluster growth from Cu7 to Cu13 on Pd-terminated PdH(111) (Fig. 5c). Wherein, the 3-fold Cu hollow sites at the edge are the most favorable for CO adsorption (B.E. = −1.17 eV, Fig. S7b), resulting in a saturation coverage of 7 CO/Cu13 with 6 CO at edge Cu hollow sites and 1 CO at the central Cu top site. This CO-saturated 0.7 ML Cu/PdH(111) configuration remains stable when the temperature is raised from 300 K to 500 K (ΔG300K = −0.43 eV; ΔG400K = −0.27 eV; ΔG500K = −0.04 eV, Fig. 5c). At the Cu coverage of 0.7 ML, CO adsorbs only over Cu clusters whereas the very limited Pd sites are only accessible to adsorbed H (Fig. 5c). In this case, atomic H is highly coordinated by both Pd and Cu atoms at the Cu–PdH interface, which increases the stability of adsorbed H by 0.24 eV.
According to the DFT-calculated CO binding energies, increasing Cu coverage from 0 ML to 0.7 ML on PdH progressively weakens CO adsorption, driven primarily by changes in adsorption site preference from Pd hollow sites to Pd + Cu bridge sites and then to Cu only sites (Fig. 5d). This is consistent with the TPD observation of a decrease in CO desorption temperature with increasing Cu coverage (Fig. 4). However, this is not the case for the desorption of atomic H as H2. Specifically, the desorption of H is supposed to be promoted by the presence of CO saturation and Cu deposition on PdH(111), while TPD results show an increase in H2 desorption temperature with the coverage of CO and Cu (Fig. 4a).
Unlike the molecular desorption of CO, H2 desorption requires diffusion of adsorbed H and recombination of two neighboring H atoms. This process is not solely thermodynamically controlled but can also be strongly influenced by kinetics. The kinetic behaviors lie in H diffusion on the surface and from subsurface to surface. The adsorbed CO or deposited Cu atoms on the surface can potentially hinder the H migration along both paths. Indeed, the DFT-calculated diffusion barrier (Ea) for H migration increases when going from adjacent surface sites (Fig. 6a–d) to CO-saturated PdH and to CO-saturated Cu/PdH (Fig. 6a–c). Particularly, on 0.7 ML Cu/PdH, adsorbed H needs to diffuse along a long path that requires overcoming a high barrier of 0.42 eV (Fig. 6d). The Ea for H migration from the subsurface to the surface of Pd-terminated PdH(111) also shows the same trend, with Ea increasing from 0.08 eV on PdH to 0.24 eV on CO-saturated PdH and to 0.39 eV/(0.53 eV) on CO-saturated 0.3 ML/(0.7 ML Cu/PdH) (Fig. 6e–g). Overall, the DFT calculations well describe the peak shifts observed in the TPD results. Compared to Pd, the formation of PdH weakens CO and H adsorption, while Cu deposition progressively reduces CO binding but kinetically hinders H desorption by raising the energetic barrier for diffusion of adsorbed H.
 | ||
| Fig. 6 Surface H diffusion path on (a) PdH(111); (b) 0.5 ML CO saturated PdH(111); (c) CO saturated 0.3 ML Cu/PdH; (d) CO saturated 0.7 ML Cu/PdH. Subsurface H diffusion to surface path on (e) PdH(111); (f) 0.5 ML CO saturated PdH(111); (g) CO saturated 0.3 ML Cu/PdH; (h) CO saturated 0.7 ML Cu/PdH; using Pd-terminated PdH(111) base. (The arrow represents the path of H diffusion). The corresponding barrier Ea is also included. (Grey: Pd; blue: Cu; pink: H; red: CO). | ||
3.4 Co-electrolysis of CO2 and H2O over PdH and CuPdH powder catalysts
To correlate trends in the binding energies of adsorbed CO and H over model surfaces with practical electrocatalysis, electrochemical studies of syngas production from co-electrolysis of CO2 and H2O were performed on Pd and CuPd powder catalysts. The CO/H2 selectivity was evaluated at −3.0 V under enhanced CO2 mass transport conditions by using a zero-gap MEA electrolyzer (Fig. 7a). Under CO2 reduction conditions, the active phase of the catalyst is PdH.27 For the pure Pd/C sample, the observed CO/H2 ratio was 0.9 (Fig. 7b), owing to approximately equal molar production rates of CO and H2. However, the CO/H2 ratio increased to 3.5 and 4.9 as Cu content was subsequently increased in the Cu0.28Pd0.72/C and Cu0.63Pd0.37/C samples, respectively. Compared with previous literature on CO2RR over CuPd electrocatalysts, the observed CO/H2 ratio is relatively high, likely owing to differences in reactor configuration, electrolyte used, and electrocatalyst composition and morphology.27,60 However, the trend in CO/H2 is similar, directly resulting from an increase in selectivity towards CO2RR to CO and a decrease in selectivity towards HER as Cu content was increased. The results from high-angle annular dark field (HAADF) imaging and energy-dispersive X-ray spectroscopy (EDS) mapping of the pristine (Fig. 7c) and spent (Fig. 7d) CuPd electrocatalysts suggest that Cu and Pd remain evenly distributed within nanoparticle clusters before and after CO2RR. Overall, the powder catalyst study supports the TPD and DFT results in that the addition of Cu can be used to progressively weaken CO binding and hinder H2 desorption, allowing one to control the CO/H2 ratio in the syngas product by changing the atomic ratio of Cu/Pd in the bimetallic catalysts. | ||
| Fig. 7 (a) Schematic of the zero-gap MEA electrolyzer. (b) CO/H2 ratios of syngas produced from the co-electrolysis of CO2 and H2O for Pd and CuPd powder catalysts with different Cu/Pd atomic ratios. HAADF-STEM imaging and STEM-EDS mapping of C, Pd, and Cu from the (c) pristine and (d) spent CuPd powder electrocatalysts. Scale bars are 5 nm. | ||
4 Conclusions
CO2 utilization is a necessary technology for closing the carbon cycle and reducing CO2 emissions. Pd catalysts are efficient for the co-electrolysis of CO2 and H2O to produce syngas; however, CO poisoning can represent a challenge for efficient and selective CO2RR to CO. Within this work, experimental and computational studies on Cu/PdH model surfaces reveal key trends in modifying the binding energies of adsorbed CO and H, which are then correlated with electrochemical measurements over the corresponding powder electrocatalysts. TPD results show that the formation of a PdH phase on the Pd(111) surface increases the barrier for H2 desorption and simultaneously slightly reduces the CO desorption temperature. 0.7 ML Cu modification on the PdH/Pd(111) surface further reduces CO binding energy while hindering H2 desorption. DFT results show that at 0.7 ML Cu coverage, there are no available Pd sites for CO to adsorb, forcing CO to adsorb onto Cu–Pd and Cu sites and consequently mitigating CO poisoning. Additionally, the Cu overlayer acts to increase diffusion barriers of adsorbed H, further hindering H2 desorption. When the corresponding electrocatalysts are tested in an MEA electrolyzer, the CO/H2 ratio increased from <1 on pure Pd to as high as 4.9 on CuPd bimetallic catalysts, indicating that alloying Pd with Cu can be used to control effluent CO/H2 ratios while decreasing Pd loading in the electrocatalysts. Overall, results from the current study demonstrate the feasibility of using surface science studies to provide fundamental understanding and design strategies in practical catalysis over the corresponding powder catalysts.Author contributions
MY: investigation, visualization, writing – original draft, writing – review & editing; HZ: investigation, visualization, writing – original draft, writing – review & editing; WW: investigation, visualization, writing – original draft, writing – review & editing; PL: supervision, funding acquisition, writing – review & editing; JGC: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article are available in an Open Science Framework repository. URL: https://osf.io/285yp/overview?view_only=9237a7f4d3874ac798f6308287836e6b.Supplementary information (SI): raw mass spectra from TPD experiments, MEA product concentrations, dataset for all DFT structures and calculation settings. See DOI: https://doi.org/10.1039/d5fd00122f.
Acknowledgements
This work was financially supported by the United States Department of Energy, Office of Basic Energy Sciences, Catalysis Science Program (Grant No. DE-FG02-13ER16381). Marcus Yu acknowledges support by the National Science Foundation Graduate Research Fellowship under Grant No. DGE 2036197. DFT calculations in this work were performed using computational resources at Center for Functional Nanomaterials (CFN), and the Scientific Data and Computing Center, a component of the Computational Science Initiative, at Brookhaven National Laboratory under Contract No. DE-SC0012704, and at the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility, supported by the DOE Office of Science under contract DE-AC02-05CH11231.References
- S. Zhang, M. Wu, Z. Qian, Q. Li, Y. Zhang and H. Zhou, Fuel, 2024, 357, 130087 CrossRef CAS.
- J. Ding, R. Ye, Y. Fu, Y. He, Y. Wu, Y. Zhang, Q. Zhong, H. H. Kung and M. Fan, Nat. Commun., 2023, 14, 4586 CrossRef CAS.
- M. Liu and G. Gadikota, Fuel, 2020, 275, 117887 CrossRef CAS.
- Z. Xie, E. Huang, S. Garg, S. Hwang, P. Liu and J. G. Chen, Nat. Catal., 2024, 7, 98–109 CrossRef CAS.
- Z. Xie, E. Huang, K. K. Turaczy, S. Garg, S. Hwang, P. R. Kasala, P. Liu and J. G. Chen, Nat. Chem. Eng., 2025, 2, 118–129 CrossRef CAS.
- X. Han, K. K. Ostrikov, J. Chen, Y. Zheng and X. Xu, Energy Fuels, 2023, 37, 12665–12684 CrossRef CAS.
- X. Li, X. Wang, X. Hu, C. Xu, W. Shao and K. Wu, J. Magnesium Alloys, 2023, 11, 1206–1212 CrossRef CAS.
- Z. Xie and J. G. Chen, CCS Chem., 2024, 6, 2855–2865 CrossRef CAS.
- Y. Yuan, B. Lamichhane, W. N. Porter, S. Hwang, L. Ma, D. Yang, A. Tayal, N. S. Marinkovic, S. Kattel and J. G. Chen, Chem Catal., 2025, 5, 101428 CAS.
- Z. Xie and J. G. Chen, CCS Chem., 2024, 6, 2855–2865 CrossRef CAS.
- J. Ren, F.-F. Li, J. Lau, L. González-Urbina and S. Licht, Nano Lett., 2015, 15, 6142–6148 CrossRef CAS PubMed.
- S. Licht, A. Douglas, J. Ren, R. Carter, M. Lefler and C. L. Pint, ACS Cent. Sci., 2016, 2, 162–168 CrossRef CAS.
- S. Lu, Y. Shi, N. Meng, S. Lu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2020, 1, 100237 CrossRef.
- T.-W. Jiang, K. Jiang and W.-B. Cai, J. Mater. Chem. A, 2024, 12, 21515–21530 RSC.
- R. T. Rashid, Y. Chen, X. Liu, F. A. Chowdhury, M. Liu, J. Song, Z. Mi and B. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2121174119 CrossRef CAS PubMed.
- B. M. Tackett, J. H. Lee and J. G. Chen, Acc. Chem. Res., 2020, 53, 1535–1544 CrossRef CAS.
- B. Chang, Z. Min, N. Liu, N. Wang, M. Fan, J. Fan and J. Wang, Green Energy Environ., 2024, 9, 1085–1100 CrossRef CAS.
- D. H. Everett and P. Nordon, Proc. R. Soc. London, Ser. A, 1997, 259, 341–360 Search PubMed.
- A. M. Abdellah, F. Ismail, O. W. Siig, J. Yang, C. M. Andrei, L.-A. DiCecco, A. Rakhsha, K. E. Salem, K. Grandfield, N. Bassim, R. Black, G. Kastlunger, L. Soleymani and D. Higgins, Nat. Commun., 2024, 15, 938 CrossRef CAS PubMed.
- W. Sheng, S. Kattel, S. Yao, B. Yan, Z. Liang, C. J. Hawxhurst, Q. Wu and J. G. Chen, Energy Environ. Sci., 2017, 10, 1180–1185 RSC.
- Y. Liu, D. Tian, A. N. Biswas, Z. Xie, S. Hwang, J. H. Lee, H. Meng and J. G. Chen, Angew. Chem., Int. Ed., 2020, 59, 11345–11348 CrossRef CAS PubMed.
- X. Chen, L. P. Granda-Marulanda, I. T. McCrum and M. T. M. Koper, Nat. Commun., 2022, 13, 38 CrossRef CAS PubMed.
- M. Zheng, X. Zhou, Y. Wang, G. Chen and M. Li, Molecules, 2023, 28, 3169 CrossRef CAS PubMed.
- C. Ai, T. Vegge and H. A. Hansen, ChemSusChem, 2022, 15, e202200008 CrossRef CAS.
- B. Xiong, J. Liu, Y. Yang, J. Ding and Z. Hua, New J. Chem., 2022, 46, 1203–1209 RSC.
- H. Dong, L. Zhang, P. Yang, X. Chang, W. Zhu, X. Ren, Z.-J. Zhao and J. Gong, Chem. Eng. Sci., 2019, 194, 29–35 CrossRef CAS.
- J. H. Lee, S. Kattel, Z. Jiang, Z. Xie, S. Yao, B. M. Tackett, W. Xu, N. S. Marinkovic and J. G. Chen, Nat. Commun., 2019, 10, 3724 CrossRef PubMed.
- M. W. Tew, M. Janousch, T. Huthwelker and J. A. van Bokhoven, J. Catal., 2011, 283, 45–54 CrossRef CAS.
- J. Chen, M. Aliasgar, Y. Zhao, F. B. Zamudio, L. Fan, J. Chen, J. Chen, X. Gu, J. Gao, S. M. Kozlov and L. Wang, Nat. Commun., 2025, 16, 10169 CrossRef CAS PubMed.
- K. Hyun, S. Yun and M. Choi, ACS Catal., 2024, 14, 2938–2948 CrossRef CAS.
- P. J. Cumpson and M. P. Seah, Surf. Interface Anal., 1997, 25, 430–446 CrossRef CAS.
- M. P. Humbert and J. G. Chen, J. Catal., 2008, 257, 297–306 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, Numerical Recipes: the Art of Scientific Computing, Cambridge University Press, Cambridge, 3rd edn, 2007 Search PubMed.
- P. Pulay, Chem. Phys. Lett., 1980, 73, 393–398 CrossRef CAS.
- J. E. Schirber and B. Morosin, Phys. Rev. B: Condens. Matter Mater. Phys., 1975, 12, 117–118 CrossRef CAS.
- R. D. Johnson III, NIST 101. Computational Chemistry Comparison and Benchmark Database, https://www.nist.gov/publications/nist-101-computational-chemistry-comparison-and-benchmark-database, accessed October 14, 2025 Search PubMed.
- M. W. Chase, NIST-JANAF Thermochemical Tables, 4th edn, https://www.nist.gov/publications/nist-janaf-thermochemical-tables-4th-edition, accessed October 14, 2025 Search PubMed.
- R. A. Van Santen and J. W. Niemantsverdriet, Chemical Kinetics and Catalysis, Springer, US, Boston, MA, 1995 Search PubMed.
- G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef CAS.
- H. Jónsson, G. Mills and K. W. Jacobsen, in Classical and Quantum Dynamics in Condensed Phase Simulations, World Scientific, 1998, pp. 385–404 Search PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Ertl, M. Neumann and K. M. Streit, Surf. Sci., 1977, 64, 393–410 CrossRef CAS.
- K. K. Turaczy, W. Liao, H. Mou, N. N. Nichols, P. Liu and J. G. Chen, ACS Catal., 2023, 13, 14268–14276 CrossRef CAS.
- A. Noordermeer, G. A. Kok and B. E. Nieuwenhuys, Surf. Sci., 1986, 165, 375–392 CrossRef CAS.
- H. Conrad, G. Ertl and E. E. Latta, Surf. Sci., 1974, 41, 435–446 CrossRef.
- E. Ataman, M. P. Andersson, M. Ceccato, N. Bovet and S. L. S. Stipp, J. Phys. Chem. C, 2016, 120, 16586–16596 CrossRef CAS.
- H. Conrad, G. Ertl, J. Koch and E. E. Latta, Surf. Sci., 1974, 43, 462–480 CrossRef CAS.
- G. E. Gdowski, T. E. Felter and R. H. Stulen, Surf. Sci. Lett., 1987, 181, L147–L155 CrossRef CAS.
- S. Suwarno and V. A. Yartys, Bull. Chem. React. Eng. Catal., 2017, 12, 312–317 CrossRef CAS.
- Y. Hirooka, M. Miyake and T. Sano, J. Nucl. Mater., 1981, 96, 227–232 CrossRef CAS.
- K. Christmann, G. Ertl and T. Pignet, Surf. Sci., 1976, 54, 365–392 CrossRef CAS.
- K. H. Rieder, M. Baumberger and W. Stocker, Phys. Rev. Lett., 1983, 51, 1799–1802 CrossRef CAS.
- H. Zhang and P. Liu, Chem Catal., 2025, 5, 101156 CAS.
- H. Zhang, X. Wang and P. Liu, Phys. Chem. Chem. Phys., 2022, 24, 16997–17003 RSC.
- J. Wang, G. Zhang, H. Liu, Z. Li, L. Wang, J. Tressel and S. Chen, Sep. Purif. Technol., 2023, 320, 124186 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |