 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mode selectivity in electron-promoted vibrational relaxation of chemisorbed hydrogen on molybdenum and tungsten surfaces
Nils Hertl *ab,
Connor L. Boxb and
Reinhard J. Maurer*abc
*ab,
Connor L. Boxb and
Reinhard J. Maurer*abc
aDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: nils.hertl@warwick.ac.uk
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK
cUniversity of Vienna, Faculty of Physics, Kolingasse 14–16, 1090 Vienna, Austria. E-mail: reinhard.maurer@univie.ac.at
First published on 22nd April 2026
Abstract
Electron–phonon coupling in atoms and molecules adsorbed at metal surfaces gives rise to finite vibrational linewidths in infrared or electron energy loss spectra. When it is the dominant contribution to the vibrational lifetime, it manifests itself in the form of a Fano line shape. Here, we report the linewidths of vibrational modes of chemisorbed hydrogen on the (100) and (110) surfaces of molybdenum and tungsten calculated from first-order time-dependent perturbation theory. For those modes with a Fano line shape, our results are in good agreement with the experiment. We further observe that the coupling strength between vibrations and electrons depends on the nature of the mode: for Lorentzian-shaped peaks, the experimental linewidths are always larger than those predicted based on pure electron–phonon coupling. The calculated linewidths exhibit a strong coverage dependence, decreasing towards higher coverages. This finding has important implications for nonadiabatic energy dissipation in hydrogen dynamics at metal surfaces. While electron–hole pair excitation is the dominant energy-transfer mechanism between hydrogen and pristine metal surfaces, other channels for energy dissipation, such as adsorbate–adsorbate interactions, may become more significant on metal surfaces densely covered with hydrogen.
Introduction
Adsorption of hydrogen can induce significant changes in the electronic structure, leading to enhanced electron–phonon coupling in two-dimensional materials,1–3 or at metal surfaces such as tungsten.4–6 Electron–phonon coupling is pivotal to driving phenomena in hydrogen–metal systems, ranging from the Jahn–Teller effect,7,8 and superconductivity,9,10 to vibrational relaxation at surfaces.11–13 Hydrogen adsorption is also a crucial step in the formation of metal hydrides14 and in heterogeneously catalysed reactions at surfaces, such as the Haber–Bosch process15 or the Fischer–Tropsch process.16 Moreover, adsorption of atomic hydrogen on tungsten surfaces is an elementary step that contributes to the macroscopic recycling coefficient of plasma fuel in a fusion reactor.17 Yet, incoming hydrogen will only adsorb when the excess energy is efficiently dissipated. At metal surfaces, the dissipation of the excess energy released during H2 bond dissociation is governed to a large extent by its interaction with the electrons of the substrate. In this scenario, electron–phonon coupling enables energy dissipation via excitation of electron–hole pairs (EHPs) triggered by the motion of the adsorbed atom.11–13 Other dissipation mechanisms, such as coupling of the hydrogen-atom motion to the surface phonons of the substrate, are considered to be inefficient due to the large mass mismatch between the adsorbed hydrogen's mass and the atomic mass of the substrate.18,19Vibrational spectra of atoms and molecules on surfaces can reveal much about the nature and effectiveness of energy exchange between adsorbate and substrate, because the dissipation rate contributes intrinsically to the overall linewidth of individual spectral signals. However, isolating the different broadening contributions is challenging, as inhomogeneous broadening, pure dephasing, and instrumental (Gaussian) broadening all contribute to the overall linewidth.12,20 Sum-frequency generation experiments provide a means to directly measure the depopulation time and thus decipher the kinetics of energy relaxation from the broadening caused by pure dephasing.20 Yet the depopulation time, T1, is the inverse of the relaxation rate of all mechanisms that dampen the vibration. Thus, experiments have often been performed in conjunction with theoretical support, allowing for an estimate of the effectiveness of electron–phonon-mediated dissipation in comparison to phonon–phonon-mediated dissipation.21–26
From a theoretical standpoint, EHP-mediated vibrational relaxation is commonly described within the framework of electronic friction.27,28 In this framework, the coupling between the vibrations and the electrons of the substrate is assumed to be weak and can therefore be derived with time-dependent perturbation theory, which yields a Fermi's golden rule-type expression for the dampening rate.11,22,28,29 Recent developments have connected this formalism to full-potential Kohn–Sham density functional theory within the numeric atomic orbital software FHI-aims,30 enabling efficient ab initio calculations of mode-resolved linewidths and the corresponding friction tensors for adsorbates on metal surfaces.31–33
In addition to the linewidth, the shape of the spectral line can also contain information about the broadening mechanism. Langreth showed that a vibrational signature has a line shape of the Fano-type when its relaxation is dominated by an EHP dampening mechanism.34,35 In fact, Fano line shapes have been observed in infrared (IR) reflection experiments on H/Mo(100),36 H/W(100),36,37 and H/Cu(111),38 as well as for CO/Cu(100).39 Electron energy loss spectroscopy (EELS) has likewise revealed Fano-type asymmetry for H/Mo(110)40 and H/W(110).41 More recently, asymmetric line shapes have also been reported for polyatomic molecules on coinage-metal surfaces.42,43 Aside from IR and EELS experiments, Fano line shapes have also been observed in inelastic electron tunnelling spectroscopy (IETS) measurements for chemisorbed hydrogen on Cu(100).44 These line shapes have been characterised theoretically using the theory developed by Lorente and Persson.45–47 The line-shape formula derived by Langreth has been used to characterise these experimental observations, but, to the best of our knowledge, no first-principles characterisation of the experimental IR and EELS experiments on full monolayer coverages of H/metal surfaces for Mo and W has been performed.
In this work, we leverage the time-dependent perturbation theory framework to compute vibrational linewidths for the different modes of hydrogen and deuterium adsorbed on the (100) and (110) surface facets of molybdenum and tungsten. Our calculations reproduce the linewidths of those vibrational modes that exhibit Fano line shapes in experiment, supporting the interpretation that these modes are predominantly broadened by EHP excitations. In contrast, vibrations on the (100) surfaces that display Lorentzian profiles in IR and EELS spectra show significantly broader experimental linewidths than predicted by our calculations, indicating that additional broadening mechanisms beyond EHP-mediated relaxation contribute strongly in this case. This also allows for a realistic assessment of the extent to which EHP excitations contribute to the overall linewidth of the remaining modes. Finally, we analyse the influence of hydrogen coverage on the vibrational linewidth and electronic-friction magnitude. We find that high coverages lead to markedly reduced linewidths (decreased electronic friction). We discuss the implications of these findings for molecular dynamics with electronic friction.28 Essentially, electronicfriction frameworks that rely in practice on the electronic density of the pristine metal48,49 might overestimate electronically non-adiabatic effects when used to model processes at high-coverage hydrogen–metal surfaces.50–52
Methods
Computational details
All calculations were performed using density functional theory (DFT) with the PBE exchange–correlation functional53,54 as implemented in the software package (version 20251007).30,55–58 Scalar-relativistic corrections were included via the atomic ZORA approach.59 Our employed convergence criteria for the energy, eigenvalues, density, and forces are 10−6 eV, 10−3 eV, 10−5 e/a03, and 10−4 eV Å−1, respectively. For all structural optimisation calculations, we used the default “tight” basis set definition. For all calculations, we used a Gaussian occupation smearing width of 0.1 eV. For the lattice constant optimisation, the Brillouin zone was sampled by a 36 × 36 × 36 k-point mesh with sampling method proposed by Monkhorst and Pack.60 The optimised lattice constants for Mo and W are 3.16 and 3.18 Å, respectively, and are in good agreement with previous calculations employing the same functional61,62 as well as experimental values.63
software package (version 20251007).30,55–58 Scalar-relativistic corrections were included via the atomic ZORA approach.59 Our employed convergence criteria for the energy, eigenvalues, density, and forces are 10−6 eV, 10−3 eV, 10−5 e/a03, and 10−4 eV Å−1, respectively. For all structural optimisation calculations, we used the default “tight” basis set definition. For all calculations, we used a Gaussian occupation smearing width of 0.1 eV. For the lattice constant optimisation, the Brillouin zone was sampled by a 36 × 36 × 36 k-point mesh with sampling method proposed by Monkhorst and Pack.60 The optimised lattice constants for Mo and W are 3.16 and 3.18 Å, respectively, and are in good agreement with previous calculations employing the same functional61,62 as well as experimental values.63
All H/metal surfaces were modelled as periodically repeated slabs consisting of six layers, of which the two lowest layers remained fixed in the geometry optimisation. The forces have been minimised until the convergence criterion of 10−3 eV Å−1 was met. For the primitive p(1 × 1)H/metal cells, we placed two hydrogen atoms on the bridge site in the case of the (100) surface, and one hydrogen on the three-fold hollow site for the (110) surface, which yields a hydrogen coverage, Θ, of 1 ML. The structures are represented by the black, opaque areas in Fig. 1. For the primitive cells used in the geometry optimisation, the Brillouin zone was sampled by a 36 × 36 × 1 k-point mesh. Lower coverages for the H/metal(110) surfaces were realised by placing one hydrogen on n-supercells. To ensure the same k-grid density throughout the different n × n × 1 supercells, we divided the k-point mesh by the factor of n.
 | ||
| Fig. 1 Orthographic view on the p(1 × 1)H/bcc(100) surface shown in panel (a) and the p(1 × 1)H/bcc(110) surface shown in panel (b). The black arrows define the Cartesian coordinates parallel to the surface. The black and white opaque areas define the primitive cell and a 3 × 3 × 1 supercell of the H/metal surfaces, respectively. | ||
Electron–phonon coupling and linewidths
Harmonic vibrational frequencies and normal modes were calculated from the dynamical matrix, constructed with finite difference displacement of 10−3 Å with respect to the relaxed atomic positions. The calculations of the frequency-dependent electron–phonon rates are based on the electronic friction formalism reported by Box et al.,64 which are implemented in version 20251007. For both the vibrational frequencies ω and rate tensor Λ(ħω), we omitted the degrees of freedom of the metal atoms. While this is in line with previous calculations of Λ(ħω) for small adsorbates on metal surfaces,32,64,65 it is also well justified for the vibrational modes of hydrogen due to the large mass mismatch between H and Mo and W, which leads to energetically well-separated phonon bands.66 Consequently, the coupling between the hydrogen phonon bands and the metal is weak.
version 20251007. For both the vibrational frequencies ω and rate tensor Λ(ħω), we omitted the degrees of freedom of the metal atoms. While this is in line with previous calculations of Λ(ħω) for small adsorbates on metal surfaces,32,64,65 it is also well justified for the vibrational modes of hydrogen due to the large mass mismatch between H and Mo and W, which leads to energetically well-separated phonon bands.66 Consequently, the coupling between the hydrogen phonon bands and the metal is weak.
The frequency-dependent electron–phonon relaxation rate tensor in Cartesian coordinates Λ(ħω) was evaluated using first-order time-dependent perturbation theory (TDPT) for phonons at the Γ-point, i.e., q = 0:
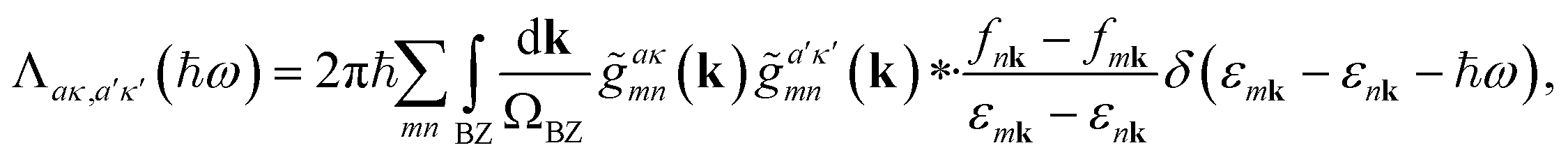 | (1) |
![[g with combining tilde]](https://www.rsc.org/images/entities/i_char_0067_0303.gif) aκmn(k) couple pairs of occupied and unoccupied Kohn–Sham states that differ in energy by ħω, corresponding to the absorption or emission of a vibrational quantum (with momentum q = 0). In practice, the integral was evaluated on a 4 × 4 six-layer slab of the p(1 × 1) H/metal(hkl) systems using a 9 × 9 × 1 of the Brillouin zone as described above, and the δ-function was replaced by a normalised Gaussian of width 0.075 eV to ensure convergence of the frequency-resolved coupling matrix. For the surface structures with different hydrogen coverages, Θ, which were modelled using supercells of varying lateral size, we evaluated eqn (1) using k-point grids adjusted to maintain a constant k-grid density. For each structure, the k-point grid employed to compute the friction tensor was identical to the one used in the geometry optimisation of the corresponding slab, as described above. As previous benchmarks65 for H on metals show a weak influence of the chosen basis set on the TDPT calculations, we used a light basis set, but with a dense integration grid for converged forces. The temperature in the Fermi-distribution fnk is set to 100 K, which is close to the experimental conditions.36,40 The factor of two in eqn (1) arises from ignoring the explicit sum over spin channels, which is suitable for H atoms close to a metallic surface.33
aκmn(k) couple pairs of occupied and unoccupied Kohn–Sham states that differ in energy by ħω, corresponding to the absorption or emission of a vibrational quantum (with momentum q = 0). In practice, the integral was evaluated on a 4 × 4 six-layer slab of the p(1 × 1) H/metal(hkl) systems using a 9 × 9 × 1 of the Brillouin zone as described above, and the δ-function was replaced by a normalised Gaussian of width 0.075 eV to ensure convergence of the frequency-resolved coupling matrix. For the surface structures with different hydrogen coverages, Θ, which were modelled using supercells of varying lateral size, we evaluated eqn (1) using k-point grids adjusted to maintain a constant k-grid density. For each structure, the k-point grid employed to compute the friction tensor was identical to the one used in the geometry optimisation of the corresponding slab, as described above. As previous benchmarks65 for H on metals show a weak influence of the chosen basis set on the TDPT calculations, we used a light basis set, but with a dense integration grid for converged forces. The temperature in the Fermi-distribution fnk is set to 100 K, which is close to the experimental conditions.36,40 The factor of two in eqn (1) arises from ignoring the explicit sum over spin channels, which is suitable for H atoms close to a metallic surface.33
Projection of Λ onto a mass-weighted, normal mode displacement vector of mode ν, ũν, (from the same simulation cell) yields the mode-resolved linewidth γν, corresponding to the full width at half maximum (FWHM), and corresponding lifetime due to electron–hole pair dissipation, τν:
| γν = ħũνΛ(ħων)ũTν, | (2) |
| τν = ħ/γν. | (3) |
Since the phonon bands we consider in this work are monoisotopic, we can reformulate the expression on the right-hand side of eqn (2) as
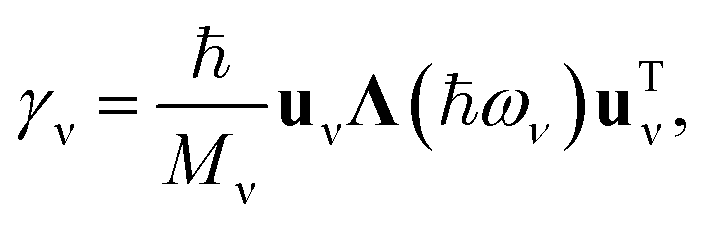 | (4) |
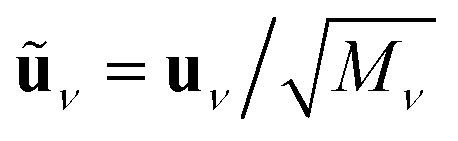 . From the 1/M dependence, it can also be inferred that the contributions from neighbouring metal atoms to the linewidth of the hydrogen vibrations are small.
. From the 1/M dependence, it can also be inferred that the contributions from neighbouring metal atoms to the linewidth of the hydrogen vibrations are small.
Identification of coherent and averaged modes
Although the implementation operates strictly at q = 0, we employed larger real-space supercells (nx × ny × nz), such that band folding provides us with access to phonon modes with q ≠ 0. As a result, a single supercell Γ-point calculation contains a dense manifold of modes representing different phase relations between primitive-cell repeats.In a finite real-space supercell, all vibrational modes are formally labelled as Γ-point modes of that supercell. However, these supercell Γ-point modes do not all correspond to the true q = 0 modes of the primitive cell. Enlarging the real-space cell reduces the size of the Brillouin zone, such that phonons with finite wave-vectors q in the primitive cell are folded back to the Γ-point of the supercell. Each such folded mode appears as a distinct eigenmode with the same local adsorbate motion (e.g. wagging or stretching), but with different phase relations between neighbouring primitive cells.
Among the supercell modes, one displacement pattern corresponds to the coherent q = 0 motion, where all adsorbate images move in phase across the supercell. As the wavelength of light used is typically much larger than the lattice constant, this mode predominantly gets excited in spectroscopic experiments before it dephases into modes with finite wave-vectors q. The q = 0 mode is straightforward to identify visually in the eigenvector animations and was used to obtain the “coherent” linewidth γ0 reported in Table 2.
The remaining modes with the same vibrational character but different phase relations represent finite-q vibrations of the primitive cell that have been folded to the supercell Γ-point. Since the employed implementation operates strictly at the supercell Γ-point, evaluating linewidths for these folded modes provides a practical route to accessing electron–phonon dissipation for finite-q vibrations without explicitly performing q-dependent calculations.
To get an upper limit for the linewidths, we assume that the light-excited coherent mode ν at q = 0 dephases instantaneously into a localised vibration that can be represented as a linear combination of mode ν at all possible wave-vectors q. As detailed by Box et al.,64 the linewidth of phonon mode ν in this limit of instantaneous dephasing can be written as:
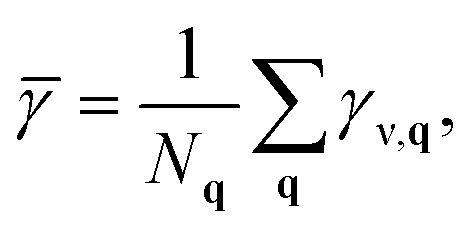 | (5) |
![[small gamma, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c4.gif) , represents the depopulation rate of an incoherent ensemble of locally similar vibrations.64
, represents the depopulation rate of an incoherent ensemble of locally similar vibrations.64
Together, γ0 and provide lower and upper bounds on the first-order electron–phonon contribution to the linewidth: γ0 corresponds to the perfectly coherent limit where all adsorbates oscillate in phase, while represents the instantaneously dephased limit in which neighbouring adsorbates move with arbitrary phase relations.
Results and discussion
Mode characterisation
We now discuss the modes of the hydrogen vibrations on the (100) and (110) surfaces for both molybdenum and tungsten. The structures of the p(1 × 1)H/metal(100) and H/metal(110) surfaces are shown in Fig. 1. The p(1 × 1)H/bcc(100) structure for molybdenum and tungsten consists of two H atoms per unit cell adsorbed on the bridge sites between two metal atoms.67–69 The H atoms in the p(1 × 1)H/M(110) phase for molybdenum and tungsten are located in the centre of the triangle formed by the metal atoms, usually referred to as a three-fold site.40,41,67,70,71Both p(1 × 1)H/Mo(100) and p(1 × 1)H/W(100) have an in-plane wagging mode, an out-of-plane symmetric stretch mode, and an in-plane asymmetric stretch mode.67,68,72,73 For both systems, the in-plane wagging mode is the lowest-energy mode, followed by the out-of-plane mode, while the planar asymmetric stretch mode is the highest energetic mode.36,72–74 The hydrogen vibrations in both the p(1 × 1)H/Mo(110) and p(1 × 1)H/W(110) structures can be characterised as a planar wagging mode with no normal component, a rocking mode, and a symmetric stretch mode.40,41 The vibrations are displayed in Fig. 2. The corresponding computed frequencies of these modes are in good agreement with experimental EELS data (Table 1).
 | ||
| Fig. 2 Adsorbate vibrational modes for the p(1 × 1) phases of hydrogen adsorbed on molybdenum and tungsten. Panels (a)–(c) display the vibrations on the (100) facet, whereas panels (d)–(f) shows the vibrations of the (110) facet. Note that the wagging mode in panel (a) and the asymmetric stretch mode in panel (c) have a two-fold degeneracy. | ||
| Surface facet | Mode | ωDFT/cm−1 | ωlit/cm−1 |
|---|---|---|---|
| Mo(100) | Wagging | 566 | 555 (ref. 72) |
| Sym. stretch | 1034 | 1025 (ref. 72) | |
| Asym. stretch† | 1342 | 1260 (ref. 72) | |
| W(100) | Wagging | 665 | 645 (ref. 68) |
| Sym. stretch | 981 | 1048 (ref. 68) | |
| Asym. stretch† | 1282 | 1290 (ref. 68) | |
| Mo(110) | Wagging | 785 | 742 (ref. 40) |
| Rocking† | 853 | 802 (ref. 40) | |
| Sym. stretch | 1261 | 1229 (ref. 40) | |
| W(110) | Wagging | 686 | 655 (ref. 41) |
| Rocking | 858 | 809 (ref. 41) | |
| Sym. stretch | 1338 | 1300 (ref. 41) |
In infrared absorption spectroscopy experiments conducted by Chabal37 on p(1 × 1)H/W(100), two absorption signals were observed: one at 1070 cm−1 with a symmetric Lorentzian line shape and one at 1270 cm−1 with an asymmetric line shape of the Fano type. Reutt et al.36 made similar observations for p(1 × 1)H/Mo(100): they found a symmetric signal with a Lorentzian line shape at 1016 cm−1 and an asymmetric signal at 1300 cm−1. Both studies assigned the signal with the Lorentzian line shape to the symmetric stretch mode, and the signal with the Fano line shape to the overtone of the wagging mode.36,37 However, given that the overtone of the wagging mode and the fundamental of the asymmetric stretch mode are nearly isoenergetic for H/W(100), this interpretation was questioned.75 With the help of angle-resolved photoelectron spectroscopy (ARPES) measurements and symmetry arguments, the Fano-type line observed in the IR experiment on H/W(100) could be unambiguously assigned to the asymmetric stretch mode.76 For H/Mo(100), however, ARPES measurements remained inconclusive about the assignment of the mode, and it was argued that both modes may in fact contribute to the signal observed in the IR experiments.73 Yet, with 555 cm−1, the frequency for the fundamental of the wagging mode measured with EELS by Zaera et al.72 is not half the size of the fundamental of the asymmetric stretch mode (1260 cm−1). In fact, the overtone would be expected to be in the area of ∼1100 cm−1. Therefore, it is more natural to assign the asymmetric signal at 1300 cm−1 in the IR spectrum for H/Mo(100) to the asymmetric stretch mode rather than the first overtone of the wagging mode. Asymmetric line shapes of the Fano type have also been observed in EELS measurements for p(1 × 1)H/Mo(110), which could be unambiguously assigned to the rocking mode.40 For H/W(110), a continuum of signals starting at about 810 cm−1 was reported, which the authors interpreted as a 2D liquid phase.41
Vibrational linewidths for the p(1 × 1)H/metal surfaces
In this section, we discuss the linewidths due to the EHP-excitation mechanism for the three different hydrogen vibration modes of the four H/metal surfaces and compare them to experimental findings reported in ref. 36 and 40. Adsorbate vibrations with an asymmetric line shape in spectroscopic experiments are of particular interest because Fano-type line shapes are fingerprints of non-adiabatic effects in vibrational energy dissipation.34 The expression for the line shape of such a phenomenon is
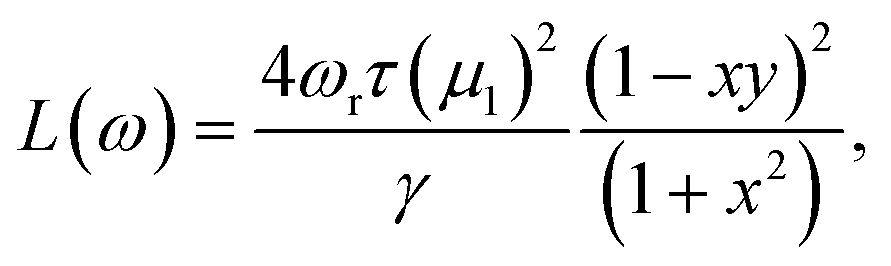 | (6) |
The results from our calculations for the coherent vibrational linewidth, γ0, and the linewidth of the q-averaged mode, γ, are given in Table 2 for both hydrogen and deuterium. In all reported cases, γ0 is smaller than the γ values, which is consistent with previous observations for vibrational lifetimes of c(2 × 2)CO/Cu(100) computed with the same framework as in this work.32,64 Novko et al. further showed that the q > 0 modes have larger vibrational linewidths for the internal stretch vibration of CO adsorbed on Cu(100).79,80 We make the same observations for all modes in the (1 × 1) hydrogen-covered phases on molybdenum and tungsten surfaces. The coherent modes have linewidths below 10 cm−1, except for the H/Mo(100) symmetric stretch mode and the H/Mo(110) wagging mode, which exhibit linewidths of 13 cm−1 and 12 cm−1, respectively. The averaged modes have linewidths up to 30 cm−1. When comparing linewidths between Mo and W for the same surface facet, the linewidths on Mo are generally larger. For the coherent modes, γ0 values on the (100) facets follow the same ordering: the linewidth of the symmetric stretch mode is the largest, followed by the asymmetric stretch mode and then the wagging mode. For the (110) surfaces, the wagging mode is the largest one for both elements, but otherwise, the order for the other modes is swapped. In general, γ0 values are smaller and more sensitive to details of the electronic structure description, making systematic trends difficult to establish. For the averaged linewidths , clearer differences emerge. Hydrogen vibrations on Mo(100) exhibit noticeably broader values than those on W(100), indicating stronger electronic coupling on the molybdenum surface. On the (110) facets, the averaged linewidths for molybdenum and tungsten are of comparable magnitude, except for the wagging mode, which is significantly broader for Mo(110). For deuterium, linewidths are typically about half those of the corresponding hydrogen modes, consistent with the expected inverse mass scaling, as discussed more in the following section.
. All values are obtained from calculations using a (4 × 4 × 6) slab with a Θ = 1 ML coverage. The reported experimental linewidths γlit for those modes labelled with a † symbol in the table are values resulting from a fit of the Fano-type line shape given in eqn (6) to the experimental data. All other experimental linewidths are FWHMs of vibrational modes with a Lorentzian line shape
| Surface facet | Mode | Isotope | γ0/cm−1 | /cm−1 |
γlit/cm−1 |
|---|---|---|---|---|---|
| Mo(100) | Wagging | H | 3.5 | 9 | — |
| D | 1.3 | 4 | — | ||
| Sym. stretch | H | 12.0 | 29 | 65 (ref. 36) | |
| D | 9.6 | 18 | — | ||
| Asym. stretch† | H | 8.0 | 30 | 12 (ref. 36) | |
| D | 2.9 | 22 | — | ||
| W(100) | Wagging | H | 3.2 | 2 | — |
| D | 1.4 | 1 | — | ||
| Sym. stretch | H | 3.1 | 11 | 103 (ref. 36) | |
| D | 1.9 | 6 | 50 (ref. 36) | ||
| Asym. stretch† | H | 4.7 | 15 | 22 (ref. 36) | |
| D | 1.8 | 6 | 25 (ref. 36) | ||
| Mo(110) | Wagging | H | 13.0 | 30 | — |
| D | 4.5 | 13 | — | ||
| Rocking† | H | 2.1 | 20 | 35 (ref. 40) | |
| D | 0.9 | 9 | 16 (ref. 40) | ||
| Sym. stretch | H | 3.2 | 10 | 27 (ref. 40) | |
| D | 1.3 | 5 | 18 (ref. 40) | ||
| W(110) | Wagging | H | 3.9 | 11 | — |
| D | 1.7 | 5 | — | ||
| Rocking | H | 2.3 | 18 | — | |
| D | 0.9 | 8 | — | ||
| Sym. stretch | H | 1.4 | 6 | — | |
| D | 0.6 | 3 | — |
Our computed γ0 values of the Fano-type modes are smaller than the experimental values γlit. The same observation was made for static TDPT calculations of vibrational lifetimes for CO on copper surfaces.32,64 This finding can be easily rationalised if one recalls that the γ0 values assume no dephasing and are therefore the lower bound of the vibrational linewidths of the adsorbates. However, the averaged linewidths are in better agreement with the experimental values as the differences are up to 18 cm−1. Translated to vibrational lifetimes, the differences between the experimental and our computed averaged values are within ±0.1 ps. This confirms the assumption that all other dissipation channels for the asymmetric modes are of ancillary importance. The lifetimes deduced from the computed values and experimental linewidths of the modes with the Fano line shape range from 0.1 ps to 0.3 ps and are significantly lower than the estimated vibrational lifetimes due to EHP excitations for in-plane vibrations of H on Cu(111)38 and Ni(111)81 with values of 0.7 ps and 1 ps, respectively.
For those lines which exhibit a Lorentzian line shape in the experiments, we find that our computed vibrational linewidths—both γ0 and —are always smaller than the experimental ones. The linewidths of the symmetric stretch mode of H/Mo(100) and H/W(100) measured by Reutt et al.36 are 65 cm−1 and 103 cm−1, respectively, which are significantly larger than the values we predict with our approach. For the symmetric stretch modes of the H/metal(100) systems, EHP excitations alone cannot account for the finite linewidth. Hence, other broadening mechanisms, such as adsorbate–adsorbate interactions or inhomogeneous broadening, have to be taken into account too. This is further corroborated by a pronounced temperature dependence observed for the linewidths of the symmetric stretch mode of H/W(100).36,37 The temperature dependence would be small if the vibrational relaxation is governed by EHP excitations,11,32,82 which was, in fact, observed for the asymmetric stretch mode.36 For the symmetric stretch mode of H/Mo(110), the situation is different: the difference between our calculated linewidth and the reported linewidth is similar to the difference we have seen for the asymmetric rocking mode of H/Mo(110). Combined with the fact that this linewidth was observed to be barely affected by temperature,40 our findings suggest that the main relaxation channel of the symmetric stretch might also be due to electron–phonon coupling.
Isotope effect
If the coupling between the electrons and the nuclei is weak, then the linewidths that are caused by EHP excitations show an inverse dependence on the adsorbate mass M.11,12 We quantify the kinetic isotope effect (KIE) as the H/D linewidth ratio KIE = γH/γD for a given mode and facet. In the weak-coupling limit of first-order TDPT, eqn (4) implies γ ∝ 1/M at fixed perturbation energy, so KIE ≈ 2 is the canonical expectation. If we were to evaluate this coupling matrix for both hydrogen and deuterium at the same energy, the linewidths for the deuterium modes would be exactly half the rates for the corresponding H vibrations. However, since the Λ(ħω) is evaluated at the respective frequencies of the H(D) modes, the frequencies change and therefore deviations from this expected factor of two arise. Since deuterium vibrates at a lower frequency than hydrogen (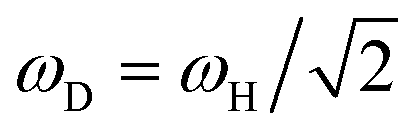 , in the harmonic approximation), H and D modes sample different parts of Λ(ħω). For most vibrations, we observe that the ratio of the averaged linewidths for H and D vibrations is slightly larger than 2, which can be seen by comparing the H and D values of all modes given in Table 2.
, in the harmonic approximation), H and D modes sample different parts of Λ(ħω). For most vibrations, we observe that the ratio of the averaged linewidths for H and D vibrations is slightly larger than 2, which can be seen by comparing the H and D values of all modes given in Table 2.
The isotope effect is mode- and momentum-dependent. In particular, KIE for the coherent mode is generally larger and more dispersed than KIE for the averaged manifold. This reflects that (a) the coherent mode samples a very narrow portion of the electron–hole continuum and is therefore highly sensitive to the precise perturbation energy and numerical convergence (k mesh density and smearing width), while (b) the averaged mode integrates over backfolded finite-q patterns and thereby averages out some frequency- and phase-relation sensitivities. Consequently, the KIE clusters around the weak-coupling expectation (∼2, here spanning 1.4–2.5), whereas KIEs for the coherent mode span broader values (in the range 2–5).
Since we have established that the averaged linewidths show a better agreement with the experimental linewidths, we will focus on them in the discussion of the isotope effect (Table 3). When comparing to experimental findings, we see that our approach correctly reproduces the isotope effect observed for the rocking mode and the symmetric stretch mode of p(1 × 1)H(D)/Mo(110). Note that this is another indicator that the relaxation of the excited symmetric stretch mode of H(D)/Mo(110) is driven by the EHP-excitation mechanism. However, our calculations cannot capture the absent isotope effect of the lifetime for the asymmetric mode of p(1 × 1)D/W(100) observed in the experiment.36,37 The weak isotope dependence of this mode was subject to investigation by Zhang and Langreth,75 who showed that the vibrational energy relaxation, when mediated by strong non-adiabatic effects, has a smaller dependence on the mass of the adsorbate. This interpretation would not only explain the discrepancy in the strength of the isotope effect between our calculations and the experiment for this mode, but also the slightly lower value for our computed averaged linewidths of the asymmetric stretch mode of H/W(100) in comparison to the experimental lifetime.
from Table 2. Fano-type (EHP-dominated) modes are indicated by †
Influence of the hydrogen coverage
Thus far, we have discussed H/metal systems with a full surface coverage Θ = 1 ML. We will investigate coverage dependence for the bcc(110) surfaces as Mo(100) and W(100) undergo different surface reconstructions at different hydrogen coverages.83 The relaxation rates for the coherent modes, γ0, of hydrogen on both bcc metal(110) surfaces are shown in Fig. 3.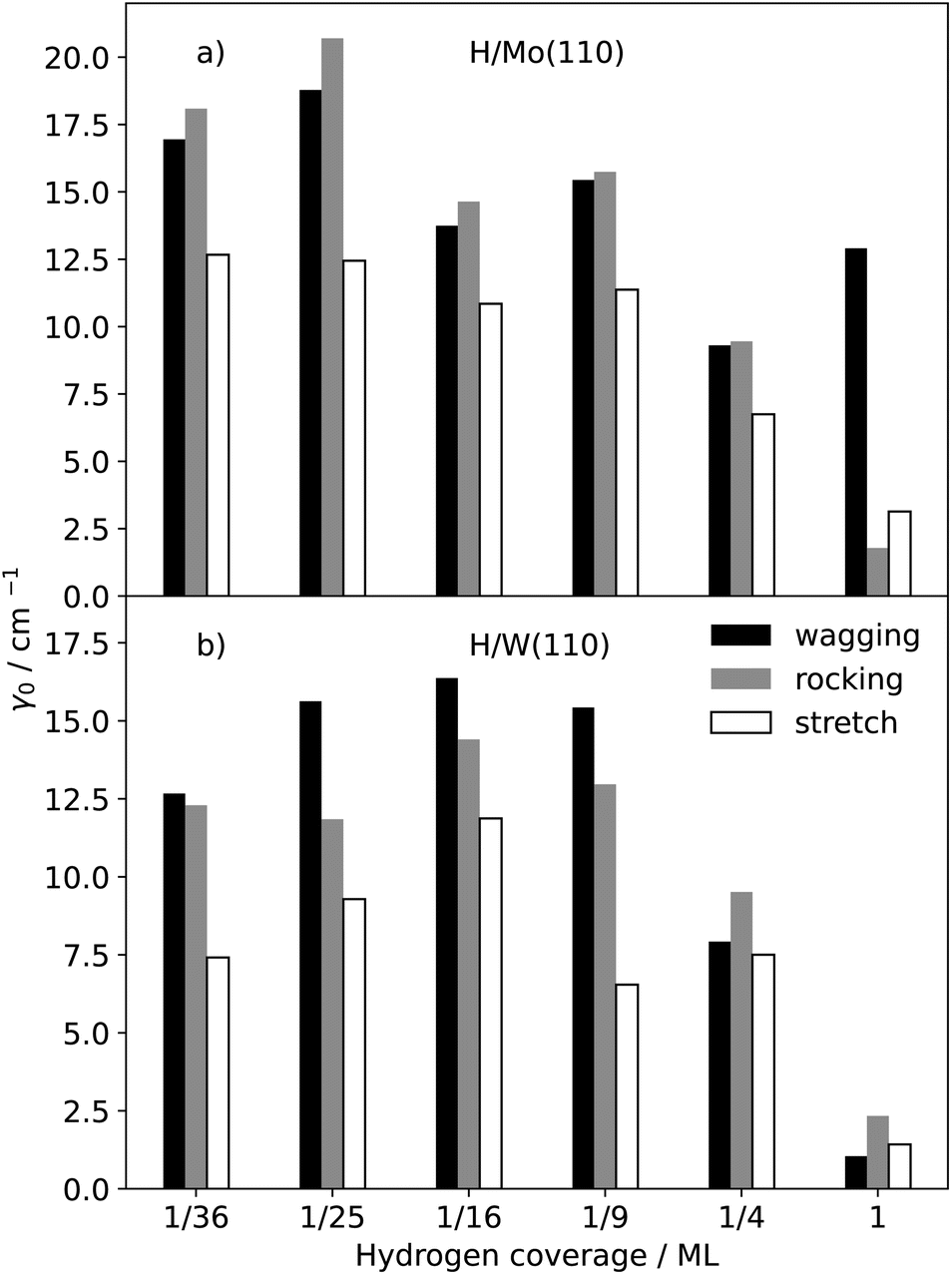 | ||
| Fig. 3 Electron–phonon-coupling-induced vibrational linewidths of the three different collective modes of hydrogen adsorbed on Mo(110), shown in panel (a), and W(110), shown in panel (b), as a function of hydrogen coverage Θ. The 1/n2 coverages were realised by placing one hydrogen on the three-fold side of a p(n × n) surface slab. | ||
The EHP-induced linewidths increase markedly as the hydrogen coverage decreases, with enhancements of up to a factor of eight across the different substrates and vibrational branches. Although we do not compute the density of states explicitly here, high adsorbate coverages are expected to alter the surface electronic structure and therefore the density of available electronic excitations that contribute to eqn (1). Such coverage-related changes in the local electronic environment offer a plausible explanation for the weaker relaxation of the coherent modes at full monolayer coverage. For H/Mo(110) systems, the relaxation rates for the vibrations start to plateau once the coverage has reached 1/9 ML. For H/W(110), the linewidths rise until they reach 1/16 ML coverage and then show a trend of decline with decreasing coverage. Furthermore, we also observe that the strength of the coverage dependence of the relaxation rates is also dependent on the mode. The wagging mode of H/Mo(110) shows the weakest dependence on coverage among all modes, whereas the corresponding mode of H/W(110) exhibits a noticeably stronger one. We attribute this difference to a more pronounced coverage dependence of the non-adiabatic couplings in tungsten than in molybdenum, as discussed in more detail below. The symmetric stretch mode of hydrogen on both metals shows a smaller dependence in comparison to the rocking mode. Both modes have similar linewidths for Θ = 1 ML, but when the coverage is decreased, the rocking mode increases more strongly than the asymmetric stretch mode.
In Table 4, the entries of the non-adiabatic coupling matrix in the zero-frequency limit, Λ(ħω → 0), are presented. In the zero-frequency limit, this quantity can be interpreted as a Markovian electronic friction tensor.28,32 The xx-component of the friction tensor, Λxx, differs significantly between the tungsten and molybdenum surfaces. For Mo(110), this entity is insensitive to changes in the coverage, whereas it shows a strong coverage dependence in the case of W(110). This is the reason why the calculated γ0 values for the wagging mode of H/Mo(110) do not increase when hydrogen coverage at the surface drops. All other entries of the friction tensor of both systems increase with decreasing coverage and start to plateau around a coverage of Θ = 1/9 ML. Furthermore, the diagonal elements of the friction tensor are clearly different for H/Mo(110) at Θ = 1 ML. The friction tensor for H/W(110) at this coverage, on the other hand, is nearly isotropic. At low coverage, on the other hand, the Λzz component is the smallest friction entry on the diagonal, whereas Λxx and Λyy are comparatively similar. Hence, the friction tensors for hydrogen atoms close to the surfaces are anisotropic. For both systems, we find that the off-diagonal elements are small compared to the diagonal elements. For both H/Mo(110) and H/W(110), the yz-component of the friction tensor, Λyz, is the only significant off-diagonal element that reflects the geometry of the surface as the yz direction points directly towards a metal atom. Whilst at high coverages, Λyz is almost insignificant, it is almost three or four times smaller than Λzz at small coverages and thence we conclude that off-diagonals cannot be omitted, per se, if one wishes to study hydrogen diffusion on metal surfaces or diffusion into the interior of the substrate.
| System | Cell size | Θ/ML | Λxx/ps−1 | Λyy/ps−1 | Λzz/ps−1 | Λyz/ps−1 | Λiso/ps−1 |
|---|---|---|---|---|---|---|---|
| H/Mo(110) | p(1 × 1) | 1 | 1.64 | 0.31 | 0.39 | 0.04 | 0.78 |
| p(2 × 2) | 1/4 | 1.17 | 1.04 | 0.79 | 0.26 | 1.00 | |
| p(3 × 3) | 1/9 | 1.63 | 1.61 | 1.13 | 0.28 | 1.46 | |
| p(4 × 4) | 1/16 | 1.51 | 1.47 | 1.18 | 0.26 | 1.38 | |
| p(5 × 5) | 1/25 | 1.87 | 1.96 | 1.22 | 0.34 | 1.68 | |
| p(6 × 6) | 1/36 | 1.79 | 1.79 | 1.31 | 0.32 | 1.63 | |
| H/W(110) | p(1 × 1) | 1 | 0.29 | 0.23 | 0.27 | 0.09 | 0.26 |
| p(2 × 2) | 1/4 | 0.87 | 0.84 | 0.71 | 0.23 | 0.81 | |
| p(3 × 3) | 1/9 | 1.64 | 1.25 | 0.93 | 0.38 | 1.27 | |
| p(4 × 4) | 1/16 | 1.66 | 1.45 | 1.09 | 0.15 | 1.40 | |
| p(5 × 5) | 1/25 | 1.69 | 1.41 | 0.89 | 0.23 | 1.33 | |
| p(6 × 6) | 1/36 | 1.44 | 1.21 | 0.95 | 0.31 | 1.20 |
Our findings have implications for molecular dynamics with electronic friction (MDEF): an established technique in theoretical surface science for including non-adiabatic effects into molecular dynamics at metal surfaces.28,48,84 This technique has proven highly successful in describing the energy transfer dynamics observed in H-atom scattering experiments on pristine metal surfaces85–87 and hot-H-atom dissipation,88,89 but was shown to fail for adsorbate-covered surfaces.90 Note that in these studies, friction was treated as a single coefficient dependent on the background electronic density of the pristine metal surface, an approach commonly referred to as the local density friction approximation (LDFA).48,49,91 The LDFA and TDPT approaches give similar results for hydrogen dynamics at metal surfaces provided that the coverage is low.33,65,92,93 In light of the coverage dependence of the friction tensor and the agreement of the here-computed linewidths with spectroscopic experiments, we conclude this agreement between LDFA and TDPT might not generally hold true at high coverages. Inelastic and reactive hydrogen-atom scattering from densely hydrogen-covered tungsten surfaces was modelled with LDFA-MDEF, and in those studies, the couplings between the electrons and the nuclei were found to be the main dissipation channel, whereas coupling to vibrations was weaker.51,52,94 Close to the surface, LDFA-based friction coefficients for hydrogen were shown to have values of ∼6 ps−1 for a variety of transition-metal surfaces,33,49,95 including molybdenum and tungsten.61,96 This might also affect computed laser-induced desorption simulations on densely hydrogen-covered Ru(0001).50 We introduce the isotropic friction tensor,
| Λiso = (Λxx + Λyy + Λzz)/3, | (7) |
Conclusions
In this work, we carried out first-principles electron–phonon coupling calculations for hydrogen adsorbed on the (100) and (110) surfaces of molybdenum and tungsten, enabling us to determine EHP-induced vibrational linewidths for all adsorbate modes. The collective vibrational linewidths are generally smaller than the corresponding averaged values, reaching an order of magnitude difference in some cases. For the modes that exhibit Fano line shapes in experiment, the collective-mode linewidths alone do not reproduce the measured widths, with the possible exception of the asymmetric stretch of hydrogen on molybdenum(100).Under the assumption of instantaneous dephasing, our calculated q-averaged linewidths show good overall agreement with the experimentally fitted FWHMs for Fano-type modes, consistent with rapid electron-mediated dephasing of the collective vibration. The averaged linewidths typically agree within a factor of two with experiment for all modes that display Fano line shapes. A notable exception is the asymmetric stretch of hydrogen on molybdenum(100), where our value is roughly twice as large as the experiment.
For Lorentzian line shapes, the situation is more varied. The symmetric stretch modes of hydrogen and deuterium on W(100) are underpredicted by about an order of magnitude, indicating that other dissipation mechanisms may be dominant, such as phonon–phonon coupling. Other recent studies have compared the magnitude of these effects.97 In contrast, the symmetric stretch modes of hydrogen on Mo(100) and Mo(110) yield averaged linewidths that lie within a factor of two of the experimental values. For the symmetric stretch on Mo(110), the similarity between its deviation and that of the asymmetric rocking mode, combined with its negligible temperature dependence reported in experiment,40 suggests that its relaxation pathway may also be dominated by coupling with EHP excitations.
By explicitly calculating the coverage-dependent linewidths and electronic friction tensors for hydrogen on Mo(110) and W(110), we find a significant and systematic coverage dependence in both quantities. The coherent vibrational linewidths remain relatively consistent at low coverages but decrease noticeably at the highest coverages (Θ = 1/4 and 1 ML). A notable exception is the wagging mode on Mo(110) at one monolayer, which shows an unusually large linewidth caused by strong mode anisotropy. This trend is closely reflected in the friction tensor. At full monolayer coverage, the hydrogen–molybdenum(110) system becomes strongly anisotropic, whereas hydrogen–tungsten(110) shows a similar reduction in friction at high coverage but retains a comparatively isotropic character in its diagonal elements. At sparse coverages, among all directions, motion along the surface normal experiences significantly reduced electronic friction compared with motion parallel to the surface, indicating pronounced anisotropy.
These findings have direct implications for molecular dynamics with electronic friction. Approaches that rely on isotropic electronic friction approximations, such as the LDFA approach, may be suitable at low coverages but likely overestimate electronically non-adiabatic effects when applied to densely covered surfaces.
Appendix
We checked the influence of the chosen exchange–correlation functional on the friction tensor Λ(0) at the level of the local density approximation and different functionals within the generalised gradient approximation. We find that the choice of the exchange–correlation functional does not have significant impact on the friction tensor—see Table 5.| Functional | Λxx/ps−1 | Λyy/ps−1 | Λzz/ps−1 | Λyz/ps−1 |
|---|---|---|---|---|
| LDA | 1.68 | 0.31 | 0.42 | 0.05 |
| PBE | 1.64 | 0.31 | 0.39 | 0.04 |
| PW91 | 1.72 | 0.32 | 0.38 | 0.04 |
| RPBE | 1.63 | 0.31 | 0.38 | 0.04 |
Author contributions
N. H. conceptualised the project. R. J. M. supervised the project. N. H. performed the calculations and analysed and curated the data. N. H., C. L. B., and R. J. M. discussed and interpreted the findings. N. H. and C. L. B. wrote the original draft. N. H., C. L. B., and R. J. M. edited and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
All linewidth and electronic friction calculations discussed in this manuscript are available within the NOMAD repository (https://doi.org/10.17172/NOMAD/2025.11.17-1).Acknowledgements
Funding is acknowledged from the UKRI Future Leaders Fellowship programme (MR/X023109/1), a UKRI frontier research grant (EP/X014088/1), and an MSCA postdoctoral fellowship (EP/Z001498/1). High-performance computing resources were provided via the Scientific Computing Research Technology Platform of the University of Warwick, the EPSRC-funded Materials Chemistry Consortium (EP/R029431/1, EP/X035859/1), and the UK Car-Parrinello consortium (EP/X035891/1) for the ARCHER2 UK National Supercomputing Service, and the EPSRC-funded HPC Midlands+ computing centre for access to Sulis (EP/P020232/1).Notes and references
- J. Bekaert, M. Petrov, A. Aperis, P. M. Oppeneer and M. V. Milošević, Phys. Rev. Lett., 2019, 123, 077001 CrossRef CAS PubMed.
- X. Yan, S. Ding, X. Zhang, A. Bergara, Y. Liu, Y. Wang, X.-F. Zhou and G. Yang, Phys. Rev. B, 2022, 106, 014514 CrossRef CAS.
- B. N. Soskic, J. Bekaert, C. Sevik and M. V. Milosevic, Nano Lett., 2024, 24, 12650–12657 CrossRef CAS PubMed.
- E. Rotenberg and S. D. Kevan, Phys. Rev. Lett., 1998, 80, 2905–2908 CrossRef CAS.
- E. Rotenberg, J. Schaefer and S. D. Kevan, Phys. Rev. Lett., 2000, 84, 2925–2928 CrossRef CAS PubMed.
- E. Rotenberg and S. Kevan, J. Electron Spectrosc. Relat. Phenom., 2002, 126, 125–132 CrossRef CAS.
- H. A. Jahn, E. Teller and F. G. Donnan, Proc. R. Soc. A, 1937, 161, 220–235 CAS.
- I. B. Bersuker, Chem. Rev., 2021, 121, 1463–1512 CrossRef CAS PubMed.
- D. Duan, Y. Liu, Y. Ma, Z. Shao, B. Liu and T. Cui, Natl. Sci. Rev., 2017, 4, 121–135 CrossRef CAS.
- D. V. Semenok, I. A. Kruglov, I. A. Savkin, A. G. Kvashnin and A. R. Oganov, Curr. Opin. Solid State Mater. Sci., 2020, 24, 100808 CrossRef CAS.
- M. Persson and B. Hellsing, Phys. Rev. Lett., 1982, 49, 662–665 CrossRef CAS.
- J. Gadzuk and A. Luntz, Surf. Sci., 1984, 144, 429–450 CrossRef CAS.
- J. C. Tully, Annu. Rev. Phys. Chem., 2000, 51, 153–178 CrossRef CAS PubMed.
- J. Bloch and M. H. Mintz, J. Alloys Compd., 1997, 253, 529–541 CrossRef.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainability Res., 2021, 2, 2000043 CrossRef CAS.
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS.
- R. A. Causey, J. Nucl. Mater., 2002, 300, 91–117 CrossRef CAS.
- J. Nørskov and B. Lundqvist, Surf. Sci., 1979, 89, 251–261 CrossRef.
- E. K. Grimmelmann, J. C. Tully and M. J. Cardillo, J. Chem. Phys., 1980, 72, 1039–1043 CrossRef CAS.
- P. Guyot-Sionnest and A. Harris, Laser Spectroscopy and Photochemistry on Metal Surfaces: Part 1, World Scientific, 1995 Search PubMed.
- R. Ryberg, Chem. Phys. Lett., 1981, 83, 423–426 CrossRef CAS.
- B. Persson and R. Ryberg, Phys. Rev. Lett., 1982, 48, 549 CrossRef CAS.
- M. Morin, N. Levinos and A. Harris, J. Chem. Phys., 1992, 96, 3950–3956 CrossRef CAS.
- M. Head-Gordon and J. C. Tully, J. Chem. Phys., 1992, 96, 3939–3949 CrossRef CAS.
- J. C. Tully, M. Gomez and M. Head-Gordon, J. Vac. Sci. Technol., A, 1993, 11, 1914–1920 CrossRef CAS.
- B. N. J. Persson and R. Ryberg, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 10273–10281 CrossRef CAS PubMed.
- E. G. d'Agliano, P. Kumar, W. Schaich and H. Suhl, Phys. Rev. B: Condens. Matter Mater. Phys., 1975, 11, 2122–2143 CrossRef.
- M. Head-Gordon and J. C. Tully, J. Chem. Phys., 1995, 103, 10137–10145 CrossRef CAS.
- B. Hellsing and M. Persson, Phys. Scr., 1984, 29, 360–371 CrossRef CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- M. Askerka, R. J. Maurer, V. S. Batista and J. C. Tully, Phys. Rev. Lett., 2016, 116, 217601 CrossRef PubMed.
- R. J. Maurer, M. Askerka, V. S. Batista and J. C. Tully, Phys. Rev. B, 2016, 94, 115432 CrossRef.
- C. L. Box, N. Hertl, W. G. Stark and R. J. Maurer, J. Phys. Chem. Lett., 2024, 15, 12520–12525 CrossRef CAS PubMed.
- D. C. Langreth, Phys. Rev. Lett., 1985, 54, 126–129 CrossRef CAS PubMed.
- D. C. Langreth, Phys. Scr., 1987, 35, 185 CrossRef CAS.
- J. E. Reutt, Y. J. Chabal and S. B. Christman, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 3112–3132 CrossRef CAS PubMed.
- Y. J. Chabal, Phys. Rev. Lett., 1985, 55, 845–848 CrossRef CAS PubMed.
- C. L. A. Lamont, B. N. J. Persson and G. P. Williams, Chem. Phys. Lett., 1995, 243, 429–434 CrossRef CAS.
- C. Hirschmugl, G. Williams, F. Hoffmann and Y. Chabal, J. Electron Spectrosc. Relat. Phenom., 1990, 54, 109–114 CrossRef.
- J. Kröger, S. Lehwald and H. Ibach, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10895–10904 CrossRef.
- M. Balden, S. Lehwald and H. Ibach, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 7479–7491 CrossRef CAS PubMed.
- P. Jakob and S. Thussing, Phys. Rev. Lett., 2021, 126, 116801 CrossRef CAS PubMed.
- R. Priya and P. Jakob, J. Phys. Chem. C, 2025, 129, 918–926 CrossRef CAS.
- L. J. Lauhon and W. Ho, Phys. Rev. Lett., 2000, 85, 4566–4569 CrossRef CAS PubMed.
- N. Lorente and M. Persson, Faraday Discuss., 2000, 117, 277–290 RSC.
- N. Lorente and M. Persson, Phys. Rev. Lett., 2000, 85, 2997–3000 CrossRef CAS PubMed.
- S. Paavilainen and M. Persson, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 085417 CrossRef.
- Y. Li and G. Wahnström, Phys. Rev. Lett., 1992, 68, 3444–3447 CrossRef CAS PubMed.
- J. I. Juaristi, M. Alducin, R. Díez Muiño, H. F. Busnengo and A. Salin, Phys. Rev. Lett., 2008, 100, 116102 CrossRef CAS PubMed.
- J. Juaristi, M. Alducin and P. Saalfrank, Phys. Rev. B, 2017, 95, 125439 CrossRef.
- R. Martin-Barrios, O. Galparsoro, A. Martínez Mesa, L. Uranga-Piña, C. Crespos and P. Larregaray, J. Phys. Chem. C, 2021, 125, 14075–14081 CrossRef CAS.
- R. Martin Barrios, N. Omar, O. Galparsoro, C. Crespos and P. Larregaray, J. Phys. Chem. C, 2024, 128, 13333–13338 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- V. Havu, V. Blum, P. Havu and M. Scheffler, J. Comput. Phys., 2009, 228, 8367–8379 CrossRef CAS.
- X. Ren, P. Rinke, V. Blum, J. Wieferink, A. Tkatchenko, A. Sanfilippo, K. Reuter and M. Scheffler, New J. Phys., 2012, 14, 053020 CrossRef.
- A. Marek, V. Blum, R. Johanni, V. Havu, B. Lang, T. Auckenthaler, A. Heinecke, H.-J. Bungartz and H. Lederer, J. Phys.: Condens. Matter, 2014, 26, 213201 CrossRef CAS PubMed.
- S. V. Levchenko, X. Ren, J. Wieferink, R. Johanni, P. Rinke, V. Blum and M. Scheffler, Comput. Phys. Commun., 2015, 192, 60–69 CrossRef CAS.
- E. Van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- N. Hertl, A. Kandratsenka and A. M. Wodtke, Phys. Chem. Chem. Phys., 2022, 24, 8738–8748 RSC.
- S. Ghosh, Eur. Phys. J. B, 2023, 96, 57 CrossRef CAS.
- R. W. G. Wyckoff and R. W. Wyckoff, Crystal Structures, Interscience publishers, New York, 1963, vol. 1 Search PubMed.
- C. L. Box, W. G. Stark and R. J. Maurer, Electron. Struct., 2023, 5, 035005 CrossRef CAS.
- W. G. Stark, C. L. Box, M. Sachs, N. Hertl and R. J. Maurer, Phys. Rev. B, 2025, 112, 035422 CrossRef CAS.
- A. Eiguren and C. Ambrosch-Draxl, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 045124 CrossRef.
- R. F. Willis, Surf. Sci., 1979, 89, 457–466 CrossRef CAS.
- W. Ho, R. F. Willis and E. W. Plummer, Phys. Rev. Lett., 1978, 40, 1463–1466 CrossRef CAS.
- R. Biswas and D. R. Hamann, Phys. Rev. Lett., 1986, 56, 2291–2294 CrossRef CAS PubMed.
- K. Kwak, M. Chou and N. Troullier, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 13734 CrossRef CAS PubMed.
- B. Kohler, P. Ruggerone and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 13503–13518 CrossRef CAS.
- F. Zaera, E. Kollin and J. Gland, Surf. Sci. Lett., 1986, 166, L149–L154 CrossRef CAS.
- K. E. Smith and S. D. Kevan, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1831–1834 CrossRef CAS PubMed.
- L. Lou, D. Langreth and P. Nordlander, Surf. Sci., 1990, 234, 412–420 CrossRef CAS.
- Z. Y. Zhang and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 10028–10046 CrossRef PubMed.
- K. E. Smith and S. D. Kevan, Phys. Rev. Lett., 1990, 64, 567 CrossRef CAS PubMed.
- U. Fano, Phys. Rev., 1961, 124, 1866 CrossRef CAS.
- R. S. Sorbello, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 6294 CrossRef CAS PubMed.
- D. Novko, M. Alducin and J. I. Juaristi, Phys. Rev. Lett., 2018, 120, 156804 CrossRef CAS PubMed.
- D. Novko, J. C. Tremblay, M. Alducin and J. I. Juaristi, Phys. Rev. Lett., 2019, 122, 016806 CrossRef CAS PubMed.
- B. N. J. Persson, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 3277–3296 CrossRef CAS PubMed.
- D. C. Langreth and M. Persson, Laser Spectroscopy and Photochemistry onMetal Surfaces: Part 1, World Scientific, 1995, pp. 498–541 Search PubMed.
- J. A. Prybyla, P. J. Estrup, S. C. Ying, Y. J. Chabal and S. B. Christman, Phys. Rev. Lett., 1987, 58, 1877–1880 CrossRef CAS PubMed.
- D. J. Auerbach, J. C. Tully and A. M. Wodtke, Nat. Sci., 2021, 1, e10005 CrossRef CAS.
- O. Bünermann, H. Jiang, Y. Dorenkamp, A. Kandratsenka, S. M. Janke, D. J. Auerbach and A. M. Wodtke, Science, 2015, 350, 1346–1349 CrossRef PubMed.
- Y. Dorenkamp, H. Jiang, H. Köckert, N. Hertl, M. Kammler, S. M. Janke, A. Kandratsenka, A. M. Wodtke and O. Bünermann, J. Chem. Phys., 2018, 148, 034706 CrossRef PubMed.
- N. Hertl, K. Krüger and O. Bünermann, Langmuir, 2022, 38, 14162–14171 CrossRef CAS PubMed.
- M. Blanco-Rey, J. I. Juaristi, R. D. Muino, H. F. Busnengo, G. J. Kroes and M. Alducin, Phys. Rev. Lett., 2014, 112, 103203 CrossRef CAS PubMed.
- O. Galparsoro, H. F. Busnengo, A. E. Martinez, J. I. Juaristi, M. Alducin and P. Larregaray, Phys. Chem. Chem. Phys., 2018, 20, 21334–21344 RSC.
- L. Lecroart, N. Hertl, Y. Dorenkamp, H. Jiang, T. N. Kitsopoulos, A. Kandratsenka, O. Bünermann and A. M. Wodtke, J. Chem. Phys., 2021, 155, 034702 CrossRef CAS PubMed.
- Y. Li and G. Wahnström, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 14528–14542 CrossRef CAS PubMed.
- P. Spiering and J. Meyer, J. Phys. Chem. Lett., 2018, 9, 1803–1808 CrossRef CAS PubMed.
- P. Spiering, K. Shakouri, J. Behler, G. J. Kroes and J. Meyer, J. Phys. Chem. Lett., 2019, 10, 2957–2962 CrossRef CAS PubMed.
- O. Galparsoro, R. Martin-Barrios, P. E. Ibañez-Almaguer, M. Márquez-Mijares, J. D. Cremé, Y. Silva-Solis, J. Rubayo-Soneira, C. Crespos and P. Larregaray, Phys. Chem. Chem. Phys., 2025, 27, 3052–3060 RSC.
- S. M. Janke, D. J. Auerbach, A. M. Wodtke and A. Kandratsenka, J. Chem. Phys., 2015, 143, 124708 CrossRef PubMed.
- R. Martin-Barrios, N. Hertl, O. Galparsoro, A. Kandratsenka, A. M. Wodtke and P. Larrégaray, Phys. Chem. Chem. Phys., 2022, 24, 20813–20819 RSC.
- L. Hörmann and R. J. Maurer, JACS Au, 2025, 5, 5027–5038 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2026 |