
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metallocenium salts as tunable dopants for enhanced efficiency of perovskite solar cells
Thomas
Webb†
ab,
Francesco
Vanin†
 ac,
Danpeng
Gao
c,
Lei
Zhu
d,
William D. J.
Tremlett
a,
Amanz
Azaden
ab,
Alice
Rodgers
ab,
Polina
Jacoutot
ab,
Andrew J. P.
White
a,
M. Saiful
Islam
d,
Nicholas J.
Long
a,
Zonglong
Zhu
*c and
Saif A.
Haque
*ab
ac,
Danpeng
Gao
c,
Lei
Zhu
d,
William D. J.
Tremlett
a,
Amanz
Azaden
ab,
Alice
Rodgers
ab,
Polina
Jacoutot
ab,
Andrew J. P.
White
a,
M. Saiful
Islam
d,
Nicholas J.
Long
a,
Zonglong
Zhu
*c and
Saif A.
Haque
*ab
aDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, London, W12 0BZ, UK. E-mail: s.a.haque@imperial.ac.uk
bCentre for Processable Electronics, Molecular Sciences Research Hub, Imperial College London, London, W12 0BZ, UK
cDepartment of Chemistry, City University of Hong Kong, Kowloon, 999077, Hong Kong. E-mail: zonglzhu@cityu.edu.hk
dDepartment of Materials, University of Oxford, Oxford, OX1 3PH, UK
First published on 5th December 2025
Abstract
The generation of free carriers through extrinsic doping is essential for transforming the electronic properties of organic semiconductors (OSCs). Doped OSCs play a crucial role in the successful operation of a wide range of electrical and optoelectronic devices, but challenges remain associated with dopant design, such as processability, stability and efficacy. Herein, we introduce a class of versatile p-type dopants based on metallocenium salts with the general formula ([M(C10H10−n)(X)n]+[Y]−) that meet these requirements. Critical to this approach is the ability to independently tune the cation via the redox-active metal cation (M) and the functionality (X) on the cyclopentadiene ring, allowing control over the oxidation strength. Simultaneously, the ability to tune the counter-anion (Y) allows control over the doping efficacy and stability of the resultant doped OSC+ salt. In this study, we systematically investigate the effect of cation and anion structures on the doping of OSCs and elucidate structure–property relationships for dopant design. We unravel the doping mechanism and demonstrate that such dopants can be used to enhance the hole extraction yield by 45% at perovskite/OSC heterojunctions. Perovskite/OSC photoactive layers using metallocenium dopants show significantly increased tolerance to moisture induced degradation as compared to films using conventional LiTFSI based dopants. Finally, we showcase the use of our optimised ferrocenium dopant in n–i–p configuration perovskite solar cells, demonstrating LiTFSI-free and additive-free devices with impressive solar-light to electrical power conversion efficiencies reaching 25.30%.
Broader contextOrganic semiconductors (OSCs) are critical components of next-generation electronics, enabling the development of low-cost, lightweight devices that surpass current technologies in fields spanning light-emitting diodes, field-effect transistors, thermoelectrics, and photovoltaics. The key to realising the beneficial electronic properties of OSCs is the use of chemical dopants, making the doping method critical for device performance and commercialisation. However, issues with dopant design remain, including poor processability, limited tunability, and chemical instability, restricting performance and stability. Herein, we introduce a design strategy for molecular dopants based on metallocenium salts, elucidating structure–property relationships to guide independent tuning of redox potential and counter-anion interactions. This approach provides precise control over doping strength and stability, establishing design rules for doping OSCs. As a case study, we apply these air-stable dopants to perovskite solar cells (PSCs), replacing the hygroscopic and volatile lithium-based dopants typically used in the organic hole transport layer (HTL). Perovskite/HTL films containing our metallocenium dopants show enhanced resistance to environmental degradation and improved interfacial charge separation. We report lithium-free and additive-free devices with efficiencies exceeding 25%, demonstrated using an archetypal HTL, namely spiro-OMeTAD. This work addresses the universal challenge of controlled and stable doping, providing a platform for durable, high-performance OSC devices across next-generation optoelectronics. |
Introduction
Chemical doping plays a critical role in tuning the intrinsic carrier conduction properties of organic semiconductors (OSCs).1–3 The increased carrier density in doped OSCs results in improved charge carrier mobility and enhanced conductivity. The use of doping as a means to improve the electronic properties has enabled OSCs to be incorporated within a wide range of applications, most notably light-emitting diodes (LEDs),1,4 field effect transistors (FETs),5,6 thermoelectrics (TE),7,8 and photovoltaics (PV), including perovskites.9–11 Despite recent advances in the field of doped OSCs, major challenges remain, including: (i) the dopant's and doped OSC's chemical stability, (ii) precise tuning and matching of the redox potential between the dopant and ionisation potential of the OSC, (iii) control over the coulombic binding between the dopant and OSC, and (iv) the ability to produce mild dopants with high doping efficacy.12–15To facilitate p-type doping of OSCs, the oxidation potential of the dopant must exceed the oxidation potential of the OSC such that free holes are generated in the semiconductor. The use of strong oxidisers often compromises the chemical stability of the OSC, thus creating a preference for sufficiently mild oxidising dopants that maintain a high doping efficacy. Additionally, the incorporation of excessively strongly oxidising dopants within OSCs can lead to morphological defects and phase-segregation, introducing additional challenges such as hygroscopicity.16–19 As such, a one-size-fits-all approach in dopant design that relies on a selection of universal dopants is sub-optimal when used across a range of OSCs. These challenges are further compounded by the often expensive and complex dopant synthesis procedures or the use of thermal activation to generate carriers, which can further impair device stability.15,20–22
Herein, we introduce a new strategy for doping hole-transporting OSCs (Fig. 1). Our approach is based on the use of metallocenium salts, which allows tuning and optimisation of both the oxidiser strength and the oxidation efficacy. This tunability is enabled by the synthetic and chemical design flexibility afforded using metallocene compounds. The dopants have a general structural formula of [(M)(C10H10−n)(X)n]+[Y]−, where M is a charge transition metal ion, X corresponds to the inclusion of n functional group(s) on the cyclopentadienyl (Cp) ring, and Y− is a counter anion (Fig. 1). In this system, the metallocenium cation dictates the oxidation potential of the dopant, allowing the doping of a range of OSCs with varying ionisation potentials. For this work, the ferrocenium ion (M = Fe) is predominantly used owing to its ideal redox proximity to the ionisation energy of spiro-OMeTAD amongst other hole transporting OSCs. The ability to modify the Cp ring functionality (X) with electron-donating or withdrawing groups provides an additional degree of freedom to fine-tune the redox properties of the dopant (Fig. 1).23 Simultaneously, the choice of counter-anion influences the ionic interaction and binding strength of the oxidised OSC salt, allowing control over the chemical stability of the charged ion-pair and dictating the doping efficacy.
 | ||
| Fig. 1 Design freedom afforded by metallocenium-based dopants for organic semiconductors (OSCs). The [M(C10H10−n)(X)n]+[Y]− dopants can be tuned via the (M) metal, (X) functional groups and (Y) anion choice. The redox properties can be tuned via the design of the metallocenium cation, whereas the chemical stability of the dopant is dictated by the anion group. The ability to independently tune these parameters creates a 3D design space of possible structures for different applications. | ||
Chemical doping of OSCs is a long-standing challenge in the field of perovskite solar cells (PSCs). PSCs have achieved reported efficiencies as high as 27%, which has been enabled by the integration and optimisation of OSCs.24 Of these OSCs, doped 2,2′,7,7′-tetrakis[N,N-di(4-methoxyphenyl)amino]-9,9′-spirobifluorene or spiro-OMeTAD has been particularly influential as a hole transport layer (HTL) in n–i–p architecture devices, enabling power conversion efficiencies (PCEs) exceeding 26%.25–27 Nevertheless, PSCs prepared by conventional high-performance spiro-OMeTAD doping suffer poor chemical stability, as a consequence of the hygroscopic nature of alkali metal-based ions, in this case, lithium bis(trifluoromethylsulfonimide) (LiTFSI). This has led to frequent substitution of the HTL with more stable alternatives when assessing device stability.25,28–31 To date, strategies to mitigate the hygroscopic LiTFSI dopant have heavily relied on the use of alternative metal cations or a handful of molecular cations.32–34 However, these dopants have yet to achieve the high performance of PSCs prepared using LiTFSI. This is, in part, a consequence of the difficulty in tailoring the doping characteristics to replicate the high-performing LiTFSI and Tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)cobalt(III) tris(bis(trifluoromethylsulfonyl)imide) (FK209) co-dopants,35 using a selection of cation alternatives with limited tunability.32–34,36
In this study, we report the application of ferrocenium salts as oxidative dopants for the HTL in n–i–p PSCs. Through a combination of chemical design, spectroscopy, ab initio modelling and photovoltaic device studies, we investigate the influence of the molecular structure of the ferrocenium salts on the doping strength and efficacy. This allows us to elucidate key structure–property relationships and design criteria that can be used to guide the doping of OSCs. Specifically, we investigate the effect of the metal cation, counter-anion, and Cp ring functionality on the doping performance of spiro-OMeTAD. We rationalise how the inclusion of electron-withdrawing and -donating groups can modify the oxidation potential of the metallocene core. Similarly, we show how softer, more polarisable anions provide stronger coulombic binding energies to spiro-OMeTAD, yielding higher doping efficacy. We prepare nickelocenium and cobaltocenium TFSI, to demonstrate the effect of metal substitution on redox potential. To demonstrate the versatility of our approach, we extend this concept to doping of other widely used OSCs, namely polytriarylamine (PTAA) and polythiophene (P3HT). We showcase the excellent air stability and chemical stability of the ferrocenium dopants, observing significantly reduced dopant-induced chemical instability at the perovskite/doped-HTL interface. Furthermore, the hydrophobicity of the metallocene salts provides improved tolerance of the HTL to moisture and humidity, leading to retention of hole extraction yield under high humidity conditions. Transient absorption spectroscopy studies reveal that ferrocenium dopants can be used to optimize and enhance the hole extraction yield by 45% at perovskite/OSC heterojunctions. Finally, we demonstrate that tailored metallocenium [(M)(C10H10−n)(X)n]+[Y]− dopants can effectively replace the commonly used hygroscopic LiTFSI and FK209 co-dopants in perovskite solar cells, leading to solar light electrical power conversion efficiencies exceeding 25%.
Results and discussion
Optimisation of Y− anion choice
A range of ferrocenium salts with different anions were synthesised (Fig. 2a) using simple ferrocene oxidation with Ag salts of the target anions (see Methods).37 Ferrocenium salts with TFSI−, BF4−, OTf−, and PF6− anions were prepared to study the effect of the anion structure on the doping yield and performance in doped HTLs. To confirm the oxidation of the ferrocene component to ferrocenium, NMR was used to probe the chemical shift of the Cp ring protons. The spectra show a downfield shift from 4.1 ppm to 32.8 ppm and broadening, indicating successful one-electron oxidation to the respective paramagnetic ferrocenium compounds (SI, Fig. S1 and S2).37 Likewise, in the case of the TFSI anion, a single peak in 19F NMR spectra attributed to the CF3 groups experiences an upfield shift from −76 to −88 ppm, consistent with displacement onto the ferrocenium ion. The exchange of the counter anion was further confirmed through the ATR-FTIR vibrational modes; [BF4] 1028 & 1000 cm−1 (B–F stretch),38 [OTf] 1255 cm−1 (SO3 stretch) and 1151 cm−1 (CF3 stretch),39 [PF6] 811 cm−1 (P–F antisymmetric stretch),38 [TFSI] 1343 & 1048 cm−1 (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches), and 1185 cm−1 (CF3 stretch) (SI, Fig. S3). The changes in the recorded IR spectra post-oxidation are accompanied by red-shifted absorbance onset at 690 nm, an indicator of ferrocenium formation (SI, Fig. S4). We confirm the composition of [Fc][TFSI] using single-crystal X-ray diffraction (XRD), where we observe a 1
O stretches), and 1185 cm−1 (CF3 stretch) (SI, Fig. S3). The changes in the recorded IR spectra post-oxidation are accompanied by red-shifted absorbance onset at 690 nm, an indicator of ferrocenium formation (SI, Fig. S4). We confirm the composition of [Fc][TFSI] using single-crystal X-ray diffraction (XRD), where we observe a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ferrocenium to anion ratio (crystallographic parameters are detailed in SI, Methods, Fig. S5 and S6, and Table S1).
:1 ferrocenium to anion ratio (crystallographic parameters are detailed in SI, Methods, Fig. S5 and S6, and Table S1).| Fc+Y− + spiro-OMeTAD → Fc + spiro-OMeTAD+Y− | (1) |
 | ||
| Fig. 2 Optimisation of the anion (Y−) of [M(C10H10−n)(X)n]+[Y]− dopants. (a) Chemical structures of anions from left to right: bis(trifluoromethylsulfonimide) [TFSI−], hexafluorophosphate [PF6−], trifluoromethylsulfimide [OTf−], and tetrafluoroborate [BF4−]. (b) 19F NMR showing shifts in the anion chemical environment before and after addition to spiro-OMeTAD. (c) 1H NMR of pre-doped spiro-OMeTAD (yellow) with the reformation of the ferrocene (orange) Cp singlet upon addition of [Fc][TFSI] (blue) to spiro-OMeTAD, forming [spiro-OMeTAD+˙][TFSI−](red). (d) EPR data of spiro-OMeTAD doped with different ferrocenium salts and (e) correlation between EPR oxidation yield and absorbance of spiro-OMeTAD+˙ polarons at 1400 nm. (f) Key photoinduced charge transfer reactions at the TiO2/perovskite/spiro-OMeTAD heterojunction. Schematic of electron (red) and hole (blue) processes in a typical transient absorbance spectroscopy (TAS) experiment, (1) excitation of the perovskite sample to form electrons and holes. (2) Hole injection into spiro-OMeTAD, where the yield is directly proportional to ΔODmax. (3) Electron injection into an electron transport layer (TiO2). (4) Interfacial recombination between electrons in the conduction bands of perovskite and TiO2 and holes injected into spiro-OMeTAD. (g) TAS of TiO2/MAPbI3/doped spiro-OMeTAD films. The yield of hole transfer at the MAPbI3/doped spiro-OMeTAD is proportional to the change in optical density ΔOD (mΔOD = ΔOD (×10−3)). In this study, the spiro-OMeTAD was doped with ferroceniums with different anions. (h) Comparison between the choice of ferrocenium anion species and the performance of a spiro-OMeTAD OSC. | ||
In order to perform the doping of the HTL, the solid ferrocenium salt was directly added to a solution of spiro-OMeTAD, in line with previously reported procedures.40,41 This resulted in a colour change from yellow to red, consistent with the oxidation of spiro-OMeTAD (SI, Fig. S7).40,41 We used 19F NMR to examine the chemical structure of the counter in the [spiro-OMeTAD+˙][Y−] product formed after the doping reaction (eqn (1)). The 19F NMR spectra are presented in Fig. 2b and demonstrate that the exchange of anions is independent of the anion choice, resulting in the formation of a range of attainable [spiro-OMeTAD+˙][Y−] salts. The ability to tune the doped OSC anion is crucial, enabling control over key properties such as ionic binding energy (vide infra), chemical stability, solubility and electronic properties.35,42,43 In the 19F NMR spectrum of [Fc][TFSI], the –CF3 fluorine shows a single chemical shift at −79 ppm, consistent with spiro-OMeTAD doped with LiTFSI and FK209, demonstrating that the same doped [spiro-OMeTAD+˙][TFSI] species is formed (SI, Fig. S8). To confirm the reduction of ferrocenium, an additional 1H NMR spectrum was collected on [Fc][TFSI] doped solutions, which revealed the formation of neutral ferrocene (Fig. 2c).29 Matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI) further confirmed eqn (1) as the primary doping mechanism, where the peak at 1505 is attributed to [spiro-OMeTAD+˙][TFSI] (SI, Fig. S9).
We now turn to the relationship between the structure of the counter anion (Y) and the doping efficacy and performance of doped spiro-OMeTAD as an HTL. Electronic paramagnetic resonance (EPR) enables the determination of the concentration of unpaired electrons through the magnitude of perturbation caused by an applied magnetic field.44 EPR signals of ferrocenium compounds are not observed at the experimental temperature of 298 K owing to fast spin–lattice relaxations (SI, Fig. S10).45 This simplification allows for correlation of the EPR double integral intensity as an estimate of the molar percentage of spiro-OMeTAD oxidation reported in Fig. 2d and e. From these data, we observe oxidation efficacies ranging from 0.77% in the BF4 salt to 3.50% with the TFSI anion. These findings are in agreement with the UV-vis absorbance intensity of the spiro-OMeTAD+˙ polaron (λ = 1400 nm) (SI, Fig. S11). From the EPR and UV-vis studies, we establish a trend in anion doping efficacy following the order BF4 < OTf < PF6 < TFSI, shown in Fig. 2e.
Transient absorption spectroscopy (TAS) was used to probe the photoinduced hole transfer reaction at the MAPbI3 perovskite/doped spiro-OMeTAD heterojunction.29,46–48 We determined the kinetics of charge recombination between the photoexcited electrons in the perovskite and holes in the spiro-OMeTAD by monitoring the decay of the spiro-OMeTAD+˙ polarons at 1600 nm following pulsed laser excitation at 532 nm (Fig. 2f). Fig. 2g shows typical decay traces of hole polarons within spiro-OMeTAD as a function of dopant anion. We note that the TAS decays for the PF6 and TFSI anions exhibit negative change in optical density (ΔOD) signals on millisecond timescales; the origin and optimisation of these kinetics are discussed in greater detail below (vide infra). The ΔOD is directly related to the concentration of photogenerated spiro-OMeTAD+˙ and is therefore a measure of the yield of hole transfer. SI, Fig. S12 presents the relative hole transfer yield ΔODmax (defined as ΔOD at 1 µs) for the different dopant anions. Photoluminescence (PL) quenching measurements (SI, Fig. S13) agree with the transient absorption data; samples with greater yield of PL quenching exhibit larger ΔODmax values.49 In summary, Fig. 2h presents a direct correlation between the hole transfer yield, anion type, and the extent of doping.
We used density functional theory (DFT) to obtain further atomistic insights into the relationship between the chemical structure of the anion and doping efficacy. Electronic surface potential (ESP) maps of the spiro-OMeTAD+˙ optimised geometry when doped with a set of TFSI, PF6 or OTf anions reveal an increase in electronic potential localised on the anion in the order OTf > PF6 > TFSI (SI, Fig. S14). This trend is further observed upon inspection of the charge distribution across the isolated anions and reflects the polarizability of the anions (SI, Fig. S15). The variation in the distribution of charge across the anion directly influences the binding energy (ΔGb) between the anions and spiro-OMeTAD+˙, yielding binding energies of 2.87, 2.96 and 3.30 eV, for OTf, PF6 and TFSI, respectively. As such, the polarizability of the anion dictates the doping efficacy and subsequent performance.50,51 The tendency of polarisable anions to produce high-performance OSC dopants is reflected in the prevalence of large, conjugated counter-anions TFSI, F4-CTNQ and poly(styrenesulfonate) (PSS) within the literature.13,51 Finally, we predict the thermodynamic driving force for the ion-exchange doping reaction given in eqn (1) (ΔGr (eqn (1))). Free energies of eqn (1) of −0.87, −0.99 and −1.11 eV were calculated for OTf, PF6 and TFSI dopants, respectively, and demonstrate a strong correlation with the molar spiro-OMeTAD oxidation and performance as an HTL (SI, Fig. S16). As such, ferroceniums with polarizable counter anions show greater yields of both doping and hole transfer, originating from a smaller binding energy with spiro-OMeTAD+˙ and a greater thermodynamic driving force for doping.
Optimisation of the [(M)(C10H10−n)(X)n]+ cation
We now consider the role of the ferrocenium cation in the oxidative doping of OSCs. We prepared a range of ferrocenium TFSI salts with different functional groups (electron-donating to electron-withdrawing) substituted onto the Cp ring (Fig. 3a). Microscope images of the resultant ferrocenium crystals are provided in SI, Fig. S17. The ATR-FTIR spectrum for each compound showed distinctive TFSI SO stretches at 1344 and 1047 cm−1 and a CF3 stretch at 1176 cm−1. In the ferrocenium salt containing a COOH group ([Fc(COOH)][TFSI]), stretches at 1698 and 1292 cm−1 correspond to the CO and C–O stretching modes, respectively (SI, Fig. S18). A red shift in the absorbance spectra was also observed in all ferrocene derivatives upon oxidation, shown in SI, Fig. S19. Metallocenium salts with alternative metal centres, nickelocenium ([Nc]) and cobaltocenium ([Cc]) bis(trifluoromethylsulfonimide) salts, were also prepared using the same process as that of the ferrocenium derivatives. In the case of [Nc][TFSI], brown crystals were obtained, identified through the characteristic TFSI FTIR stretching modes at 1344 and 1047 cm−1 and the presence of a singlet peak at 76 ppm in the 19F NMR spectrum (SI, Fig. S20 and S21). [Cc][TFSI] formed a black solid and was characterised using 1H NMR (SI, Fig. S21), from which a downfield shift from −17.5 to 5.7 ppm of the Cp ring was recorded following oxidation. The 19F spectrum showed a TFSI peak at 78 ppm (SI, Fig. S21).
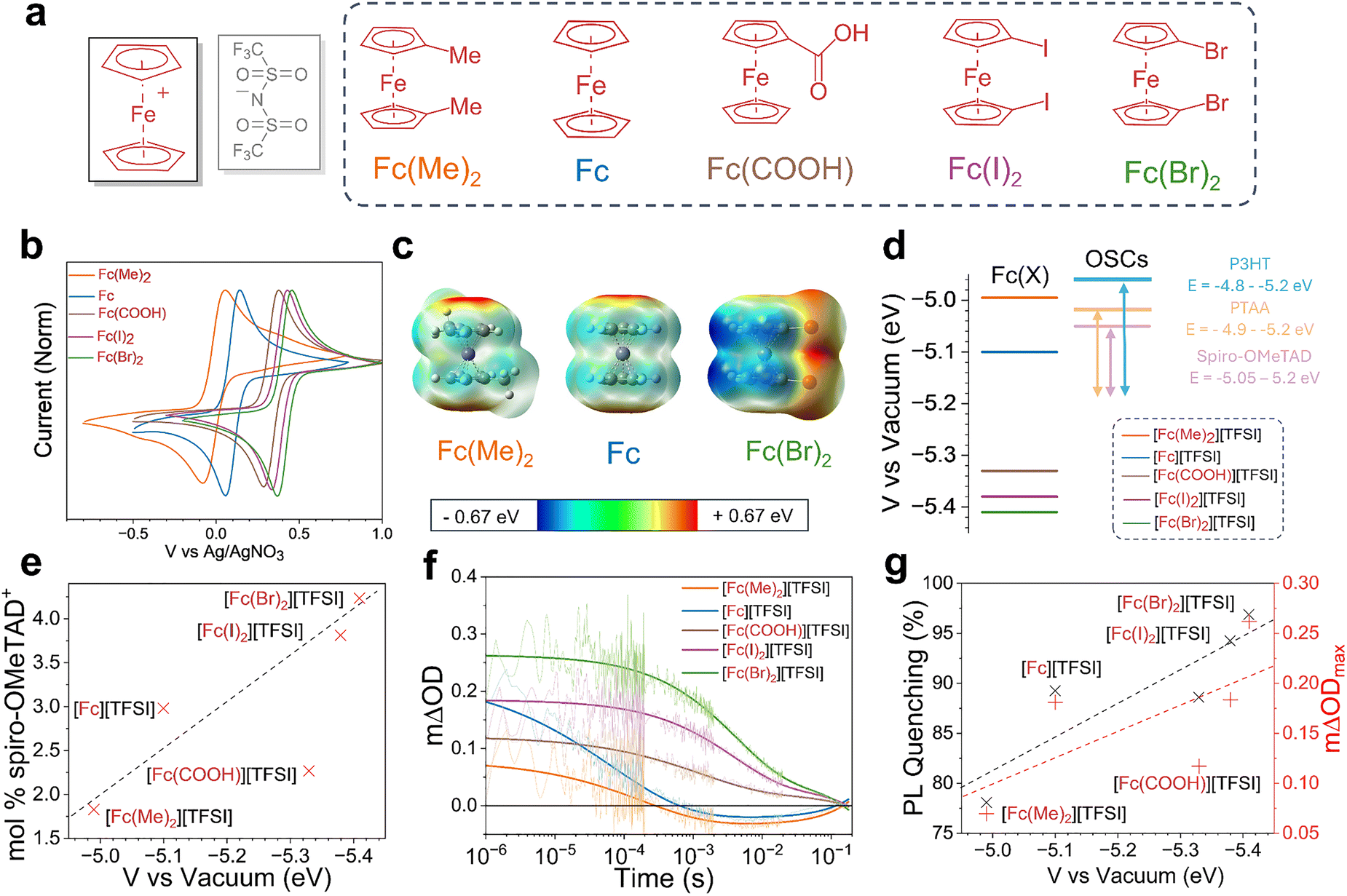 | ||
| Fig. 3 Optimisation of the cation functionality (X) of [(M)(C5H5−n)2(X)]+[Y]− dopants. (a) Chemical structures of ferrocene derivatives used to produce ferrocenium dopants. (b) Cyclic voltammetry (CV) measurements of ferrocene derivatives at 2 mM in 0.1 M [NBuPF6] ACN electrolyte against an Ag/AgNO3 reference electrode. (c) DFT electronic surface potential (EPS) of the most electron-donating functionalised derivative [Fc(Me)2], ferrocene, and the most electron-withdrawing functionalised derivative [Fc(Br)2], red areas indicate areas of electronegativity. (d) Comparison of oxidation potential between ferrocene-derivatives and common OSC materials. (e) Comparison between the molar oxidation percentage of spiro-OMeTAD calculated from EPR spectroscopy against the vacuum potential of ferrocene-derivatives. (f) Transient absorption spectroscopy (TAS) studies of TiO2/MAPbI3/doped spiro-OMeTAD films. TAS data showing changes in the hole extraction efficiency in spiro-OMeTAD doped with different ferrocenium cations. The change in optical density ΔOD is proportional to the yield of hole transfer (note that mΔOD = ΔOD × 10−3). (g) Correlation between the performance of spiro-OMeTAD as an HTL and the oxidation potential of the ferrocenium cation used to dope. | ||
The functions of the anion and cation in the metallocenium salts are distinct, allowing independent optimisation of multiple properties. Whilst the anion is primarily involved in stabilisation of the spiro-OMeTAD+˙ polaron, the cation design determines the oxidation strength of the dopant. Oxidative doping of OSCs is expected to occur when the redox potential (EOx) of the metallocenium cation is greater than the redox potential of the OSC such that there is an overall decrease in the potential of the electron and ETot is positive (eqn (2) and (3)). Cyclic voltammetry (CV) was used to evaluate the oxidation potentials of the different metallocene derivatives (Fig. 3b). Taking the half-wave potential between the anodic and cathodic waves gives a redox potential for the Fc/Fc+ half-cell of 0.10 V w.r.t Ag/Ag+. This value can then be converted to a vacuum potential of −5.10 eV, using previously reported methodology.40,52 Comparing this value to the first oxidation potential of spiro-OMeTAD (EOx = 5.05 eV, SI, Fig. S22) rationalises our observations to conclude that the ferrocenium cation is sufficiently oxidising to dope spiro-OMeTAD.40 Addition of a methyl group to each ring lowers EOx to 5.01 eV, producing a milder oxidant. The electron-donating effect of the methyl substituents on the electronic surface potential is shown in Fig. 3c, whereby electron density is directed onto the Cp rings. Accordingly, EPR spectroscopy shows a lower spiro-OMeTAD doping yield of 1.8 mol% when prepared with Fc(Me)2. The effect of EOx was further demonstrated through lowering the potential to −4.78 eV by replacement of the Fe3+ centre with Ni3+ in [Nc][TFSI] and lower still to 3.96 eV using Co3+ [Cc][TFSI] (SI, Fig. S23). As a result of the lower redox potential, [Nc][TFSI] is too mild to oxidise the spiro-OMeTAD system, yielding no [spiro-OMeTAD][TFSI] formation (SI, Fig. S24).
| ETot = −(Eox([(M)(C10H10−x)n(x)n]+[Y]−) − Eox(OSC)) > 0 | (2) |
| ΔG = −nFETot | (3) |
Stronger oxidising dopants can be produced through the inclusion of electron-withdrawing functionality, such as a carboxyl group or halogen atom. CV was used to evaluate the redox potentials of ferrocene-derivatives containing a carboxylic acid group (Fc(COOH); Eox = −5.30 eV), diiodoferrocene (Fc(I)2; Eox = −5.38 eV) and dibromoferrocene (Fc(Br)2; Eox = −5.40 eV). In these compounds, electron density is withdrawn from the redox-active metal centre (Fig. 3c). The relative Eox potentials of the different ferrocenium cations are presented in Fig. 3d, alongside common OSCs. In the case of Fc(I)2 and Fc(Br)2, we find that the observed Eox values also exceed the second oxidation potential of spiro-OMeTAD (Eox = −5.25 eV), allowing opportunities to access higher oxidation states through control of the molecular design. EPR measurements on spiro-OMeTAD doped with ferroceniums with electron-withdrawing functionality are presented in Fig. 3e. As expected, a strong correlation is observed between Eox and mol% of doping, reaching up to 4.3 mol% when doped with 2.5 mM [Fc(Br)2][TFSI], the most oxidising dopant tested herein. We note that [Fc(COOH)][TFSI] exhibited poor solubility in chlorobenzene, which may rationalise the lower mol% oxidation observed.
We next investigate the influence of the cation functional groups on the performance of the doped OSC when used as an HTL. First, TAS was used to compare the yield of hole extraction from a perovskite active layer, providing a comparison between the performances of the different cations when used to dope spiro-OMeTAD. From the decays presented in Fig. 3f, the yield of hole transfer (mΔODmax) was found to follow the same trend as observed in the EPR measurements, where the inclusion of electron-withdrawing substituents improved the hole extraction yield. Using [Fc(Br)2]+, we observe that the yield of hole transfer can be enhanced by 45%. In contrast, replacing the [Fc]+ cation with the less oxidising [Fc(Me)2]+ cation decreased the hole transfer yield by 62%. These findings were further supported using PL spectroscopy (Fig. 3g and SI, Fig. S25), whereby greater quenching of the perovskite active layer was achieved using dopants with more electron-withdrawing ring substituents. As such, the design of the functionality on the Cp ring is critical in determining the performance of the doped OSC when used as a hole extraction layer (Fig. 3g).
We note the presence of a negative transient feature on the order of 10−2 s in spiro-OMeTAD in samples prepared with [Fc(Me)2] and [Fc] cations. We further note that this feature was also observed in [Fc][PF6] doped samples (Fig. 2f). We attribute this feature to a temporary de-doping effect induced by the close energetic proximity between the Fc/Fc+ redox potential and the first oxidation of spiro-OMeTAD combined with the reversible nature of the Fc/Fc+ (<0.1 eV) (SI, Note S1). This transient effect is not observed using cations with a larger offset compared to the OSC, owing to the formation of a large energetic barrier to de-doping. Notably, while [Fc(COOH)][TFSI] doped spiro-OMeTAD exhibits lower yields of hole transfer owing to poor solubility in chlorobenzene, the effect is still present, confirming that the process is driven by energetics.
Finally, we consider the influence of the neutral ferrocene that forms as a by-product of eqn (1). Formation of neutral ferrocene within the HTL can (i) improve the performance of the HTL and (ii) provide a hydrophobic alternative to hygroscopic metal cations, such as Li+.29,53 In synergy with the experimental work, we used DFT and ab initio molecular dynamics methods to gain insights into ferrocene-perovskite surface interactions, which are not fully understood on the atomic scale. For such a detailed modelling analysis, ferrocene provides a simple representative system, allowing us to consider the behaviour of the dopant by-product at the perovskite interface. The simulations examined the atomistic effects at the (001) PbI2-terminated surfaces of MAPbI3, as it is one of the most stable perovskite facets (computational details are provided in the Methods section).
Four important results emerged. First, the ferrocene molecule adsorbs favourably on the MAPbI3 surface, with a binding energy of −0.69 eV. For this configuration, Fig. 4a shows the changes in the charge-density profile across the ferrocene–perovskite interface, suggesting that both the Fe metal centre and the Cp rings engage in significant electronic interactions with undercoordinated Pb. Second, adsorption of the ferrocene molecule results in the reduction of the work function by 1.01 eV (SI, Fig. S26 and Table S2), suggesting band bending at the perovskite surface, which agrees well with our previous experimental findings using Kelvin probe force microscopy.23,29 This is also in accord with the TAS results (Fig. 3f), which reveal longer hole polaron lifetimes in spiro-OMeTAD doped with strongly oxidising ferroceniums (Fig. S27 and Supplementary Note S1, SI).
 | ||
| Fig. 4 DFT modelling of the impact of residual ferrocene, the by-product of ferrocenium doping. (a) Side view of the simulated lowest-energy configuration of ferrocene on the (001) PbI2-terminated MAPbI3 surface and charge density profile (with charge density gain and loss coloured in yellow and blue, respectively). (b) Calculated electronic density of states (DOS) for the MAPbI3 surface with and without Fc doping. DOS are projected onto atoms in the top three layers of the surface slab (blue) and onto ferrocene atoms (orange). Top panel: surface without ferrocene; middle and bottom panels: surfaces with the ferrocene molecule relaxed near the surface, showing additional hybridized states not found in the undoped system. All DOS plots are aligned to the surface VBM, set to zero on the energy axis. The black dashed line indicates the position of the highest occupied level at the surface. (c) Schematic energy level diagram illustrating the proposed mechanism of hole transport at the interface between the perovskite (PVK) layer and the HTL without (left) and with (right) Fc doping. | ||
Third, the simulated electronic density of states (DOS) (Fig. 4b) indicates that the neutral ferrocene modifies the electronic structure of the surface by introducing additional energy states within the band gap, which do not appear in the bulk DOS (Fig. S28). These results suggest that ferrocene forms hybridized states with undercoordinated surface Pb2+, consistent with studies pertaining to interaction of neutral Fc with perovskite surfaces.23,29 Also, a small blue shift (0.03 eV) of the surface band gap was found, which indicates possible passivation of the perovskite surface, mitigating non-radiative recombination centres. Finally, the ferrocene-surface hybridized states elevate the highest occupied energy level above the valence band maximum (VBM), which helps to account for the work function shift at the surface. As a result, these hybridized states serve as intermediate channels between the HOMO level of the Fc-doped spiro-OMeTAD layer and the VBM level of the perovskite, which would facilitate efficient hole extraction (illustrated schematically in Fig. 4c).
In summary, we have shown that the chemical structure of the metallocenium cation can be easily modified to tune its oxidation potential and strength through (i) substitution of the metal or (ii) fine-tuning of the Cp ring functionality. Our ab initio simulations suggest that the neutral ferrocene by-products and their influence on the perovskite (e.g. band bending, surface hybridized states, and passivation) collectively contribute to the enhanced hole extraction at the interface between the perovskite and the HTL layer as observed from the TAS experiments.
Application of ferrocenium dopants in n–i–p PSCs
To this point, we have demonstrated the flexibility afforded by ferrocenium dopants using spiro-OMeTAD as a technologically relevant molecular OSC. We next investigate the ferrocenium salt doping on PTAA and P3HT, archetypal examples of poly(triarylamine) and poly(thiophene) classes of OSCs, respectively (Fig. 5a). It has been reported that both PTAA and P3HT have similar values of Eox(OSC) to spiro-OMeTAD, allowing both OSCs to be doped upon addition of [Fc][TFSI], leading to a change in colour shown in Fig. 5b (inset) and the absorption profile (SI, Fig. S29).11,54,55 Confirmation of doping for both OSCs using [Fc][TFSI] was made using EPR spectroscopy, presented in Fig. 5b and distinct changes in the 1H NMR spectra arising from the presence of paramagnetic effects (SI, Fig. S30). 19F NMR spectroscopy further confirmed the formation of both PTAA and P3HT TFSI salts, demonstrating that eqn (1) holds for different OSCs, provided Eox of the ferrocenium centre is larger than the ionisation potential of the OSC (SI, Fig. S31). In all three OSCs tested, we observe improvements to the electrical conductivity (σ) upon doping with [Fc][TFSI], increasing from 1.6 × 10−10 to 2.3 × 10−5 S cm−1, 3.7 × 10−4 to 7.2 × 10−4 S cm−1 and 2.4 × 10−5 to 4.8 × 10−5 S cm−1 for spiro-OMeTAD, PTAA and P3HT, respectively (Fig. 5c).56 Similarly, TAS and time-resolved photoluminescence (TRPL) measurements on perovskite heterojunctions prepared with PTAA or P3HT HTLs show improved yields of hole injection post-doping, indicating enhanced OSC performance of the HTL (Fig. 5d and SI, Fig. S32). | ||
| Fig. 5 Applicability of ferrocenium dopants in alternative OSCs and PSCs. (a) Chemical structures of poly(triarylamine) (PTAA) and poly(3-hexylthiophene) (P3HT). (b) EPR spectra of PTAA (above) and P3HT (below) with and without [Fc][TFSI] doping. (Inset) Photographs showing the colour change of PTAA and P3HT pre-doping (left) and post-doping (right). (c) J–V characteristics of the ohmic region of spiro-OMeTAD (top), PTAA (centre), and P3HT (bottom) using an ITO/PEDOT:PSS/OSC/Au architecture with and without [Fc][TFSI] doping. Conductivity σ calculated using I = σAVd−1.56 (d) µs-TAS decays of the perovskite/HTL interface prepared with and without [Fc][TFSI] in PTAA (green) and P3HT (purple). (e) Absorbance spectra of perovskite substrates after interfacial contact with LiTFSI or [Fc][TFSI] for a period of 2 minutes. (inset) Microscope photograph of LiTFSI-induced cation washout in the perovskite, forming yellow PbI2 regions. (f) Absorbance spectra of perovskite substrates prepared with spiro-OMeTAD doped using LiTFSI + FK209 or [Fc][TFSI] under high humidity conditions (85% RH, 25 °C). (g) Device architecture schematic of n–i–p PSCs used to demonstrate ferrocenium doping of spiro-OMeTAD as an HTL. (h) Champion J–V curves and (i) PCE statistical distribution of PSCs prepared using LiTFSI, [Fc][TFSI] or [Fc(Br)2][TFSI]. | ||
To evaluate the stability of ferrocenium-based dopants against conventional LiTFSI, solid forms of both dopants were deposited on exposed perovskite substrates and the change in absorbance was measured (Fig. 5e). The hygroscopic LiTFSI samples quickly condensed water in as little as 120 seconds, forming visible water droplets on the perovskite and producing yellow regions in the film (SI, Fig. S33). In these regions, the perovskite cation is removed, leaving behind PbI2 as confirmed by absorbance spectroscopy.57 In contrast, negligible changes occur to the perovskite upon contact with [Fc][TFSI], owing to the excellent air-stability of the ferrocenium salts when in the solid-state and the Cp rings increasing the hydrophobicity at the interface. We next consider the stability of MAPbI3/doped spiro-OMeTAD photoactive layers following ageing under ISOS-D3 (RH = 85%) damp storage conditions. In these experiments, we observed a significant loss in the perovskite absorbance in the LiTFSI-doped films following 1 hour of ageing. In contrast, we observed only a minor change in the UV-vis spectra of [Fc][TFSI] doped samples under the same ageing conditions (Fig. 5f).58 TAS experiments were further used to probe the yield of hole transfer in perovskite/doped spiro-OMeTAD films during storage under ISOS-D3 conditions. Comparing the evolution of ΔODmax, we report a retention of 56% of the initial hole extraction yield after 96 hours in the [Fc][TFSI] doped films. In contrast, samples prepared with LiTFSI presented no measurable hole extraction after 72 hours (SI, Fig. S34). Moreover, contact-angle measurements (SI, Fig 35, Table S3) show that LiTFSI-doped spiro-OMeTAD films rapidly become hydrophilic (decreasing from 50.9° to 13.0° within fifteen minutes), consistent with hygroscopic wetting. In contrast, [Fc(Br)2][TFSI] doped films retain a stable contact angle of ∼72°, confirming that our ferrocene-core TFSI salts significantly enhance short-term moisture resistance, consistent with optical microscopy observations (SI, Fig 36). Beyond moisture stability, TGA also showed high thermal stability and a sublimation temperature of 277 °C (SI, Fig. S37).
Finally, PSC devices were prepared to demonstrate the performance of ferrocenium dopants when used within a technologically relevant application. Devices were prepared using an n–i–p architecture comprising ITO/SnO2/Perovskite/spiro-OMeTAD/MoO3/Ag shown in Fig. 5g. The devices prepared using the conventional LiTFSI and FK209 co-dopants achieved a benchmark PCE of 24.59% as shown in Fig. 5h. Both LiTFSI and FK209 co-dopants were replaced with the [Fc][TFSI] dopant and PSCs yielded a similar PCE of 24.26%. Additional devices were prepared using [Fc(Br)2][TFSI] and achieved an impressive PCE of 25.30%, surpassing that of the conventional Li-based cells. These findings are reflected within the statistical analysis presented in Fig. 5i and are in accord with the design rules established herein; the introduction of electron-withdrawing functionality to the dopant enhances both oxidation strength (Fig. 3e) and hole extraction characteristics (Fig. 3g). Moreover, in all cases, PSCs showed excellent hysteresis between the forward and reverse scans (SI, Fig. S38). To validate the current density curves collected and measured by the solar simulator, additional external quantum efficiency (EQE) was collected, giving values of 25.03 and 25.22 mA cm−2 for [Fc][TFSI] and [Fc(Br)2][TFSI], respectively, within a 5% tolerance to the J–V measurements (SI, Fig. S39). Additionally, stabilised output point tracking was measured over a 300 s duration and showed no immediate instability under operation (SI, Fig. S40). Overall, the fabrication of devices demonstrates that the use of ferrocenium-derived dopants can exceed the current state-of-the-art LiTFSI-based cells and that optimizing the redox potential and counterion of the metallocenium dopant is critical in optimising the performance. This improved performance obtained through appropriately designed single-component metallocenium dopants is attributed to three main factors: (i) low doping density with clearly-defined neutral ferrocene side-products as opposed to large LiTFSI and tert-butyl pyridine (TBP) dopant loadings, (ii) solubility of dopants in chlorobenzene as opposed to acetonitrile and (iii) strong oxidation potential that does not require oxygen exposure for activation.
Conclusions
In this work, we have demonstrated the application of metallocenium salts [M(C10H10−n)(X)]+[Y]− as a tunable framework suitable for doping organic semiconductors (OSCs) that enhance the efficiency of PSCs. These dopants can be prepared simply via oxidising functionalized ferrocene species with silver salts, affording air-stable crystals. These silver salts can be synthesised through salt metathesis, allowing a wide range of flexibility and seamless integration within existing technologies.59,60 We show that ferrocenium compounds can be used to dope spiro-OMeTAD, a high-performance HTL typically used in PSCs. The performance of these dopants is influenced by two key factors: (i) the polarizability of the anion (Y−) and (ii) the oxidation potential of the metallocenium cation, modified through either changing the metal (M) or the incorporation of electron-donating or -withdrawing substituents (X) on the Cp rings. It should also be stressed that post-doping, the ability to influence the perovskite surface properties via the ferrocene by-products provides an additional advantage over the use of other cations, enhancing hole extraction. Beyond spiro-OMeTAD, we demonstrate the doping of other OSCs, namely PTAA and P3HT, showing that the doping mechanism is widely applicable, provided the design of the metallocenium dopant has a larger redox potential than the OSC. Finally, we report the use of these dopants in n–i–p perovskite solar cells, where structural tuning facilitated the production of LiTFSI-free devices with PCEs reaching 25.30%. We envisage that the tunability of metallocenium dopants, combined with their chemical and air stability, makes these compounds highly suitable for a range of applications such as OFETs, LEDs and PVs, including perovskite devices.Methods
Sample preparation
To prepare PTAA, 10 mg mL−1 of PTAA (Osilla) was dissolved in CB, stirred and filtered. The solution was doped using the same 2.5 mM concentration of metallocenium dopant affording a red solution. The solution was filtered using a 0.2 µm PTFE filter and spin-coated at 4000 rpm.
For P3HT (Sigma-Aldrich, Mn = 54–74000), 10 mg mL−1 was dissolved in CB. The solution was filtered affording an orange solution. 2.5 mM of metallocenium dopant was added to the P3HT solution, changing the colour of the solution to purple. The solution was spin-coated at 2000 rpm.
The (FA0.98MA0.02)0.95Cs0.05Pb(I0.98Br0.02)3 perovskite precursor solution (1.55 M) was prepared by dissolving CsI, FAI, MABr, PbI2, and PbBr2 in 1 ml of mixed DMF:DMSO (4:1 v/v) with the addition of 12.6 mol% MACl and 7 mol% excess of PbI2 in a N2-filled glovebox. Prior to device fabrication, 0.04 mol% of ODADI was added to the precursor solution and allowed to stir for 2 h; the precursor was filtered through a 0.22 µm PTFE filter. Deposition of the perovskite layer was performed by spin-coating 50 µL of the as-prepared precursor on the glass/ITO/SnO2 substrate at 1000 rpm for 10 s and subsequently 5000 rpm for 40 s; 350 µl of CB was dripped onto the centre of the film 10 s before the end of the spin-coating procedure. The substrates were then directly annealed at 100 °C for 30 min. The cooled perovskite layers were then treated with a passivation layer consisting of 1 mg ml−1 of PDADI dissolved in 150:1 IPA:DMF at 5000 rpm for 30 s and annealed at 100 °C for 5 minutes. All procedures were conducted in a N2-filled glovebox with a controlled temperature between 18 and 23 °C by the integrated conditioner, and water and oxygen levels were maintained <5 ppm.
Doped spiro-OMeTAD solutions, prepared as described above, were dynamically spin-coated onto the cooled perovskite layers at 3000 rpm. No further annealing was performed.
Finally, 6 nm of MoO3 was evaporated at a rate of 0.5 Å s−1 followed by the 100 nm silver electrode (evaporated at a rate of 0.05 Å s−1 for the first 10 nm and 2 Å s−1 for the remaining 90 nm) under high vacuum (<4 × 10−6 Torr). A 125 nm thick magnesium fluoride layer was evaporated at a rate of 1 Å s−1 on the back of the ITO substrate to reduce reflective losses.
Characterisation
:2 v/w, deposited using a spin speed of 5000 rpm and annealed using an annealing temperature of 500 °C. MAPbI3 was used as an archetypal and comparable absorber layer. The doped and undoped HTLs were prepared using the methods given in the Sample preparation section.
Samples were pulsed using an Opotek (Opolette UX06230) wavelength tunable OPO laser at a wavelength of 532 nm at a power of ∼100 µJ cm−2 at a repetition rate of 4 Hz and across a sample area of approximately 1 × 1 cm2. A Bentham 100 W Tungsten lamp was used as a probe source on an orthogonal axis to the pump beam. The probe wavelength was set to 1600 nm using a Bentham monochromator, corresponding to the maximum spiro-OMeTAD hole polaron yield. The probe beam was detected following transmission through the sample using an InGaAs photodiode (Hamatsu Photonics), amplified (Costronics Electronics) and collected in Labview via a digital oscilloscope (Tekronics DPO3012). Tas decays were fitted using a stretched exponential function: ΔOD = exp(−t/τ)a. The maximum yield of hole transfer (ΔODmax) (corresponding to the largest deviation in absorbance) was collected at t = 1 µs.
| Fc+Y− + spiro-OMeTAD → Fc + spiro-OMeTAD+Y− | (1) |
| ΔGr(eqn (1)) = [ΔG(spiro-OMeTAD+Y−) + ΔG(Fc)] − [ΔG(spiro-OMeTAD) + ΔG(Fc+Y−)] |
To calculate the ionic binding energy between the spiro-OMeTAD+˙ the following method was used:
| ΔGr(spiro-OMeTAD+Y−) = [ΔG(spiro-OMeTAD+Y−)] − [ΔG(spiro-OMeTAD+) + ΔG(Y−)] |
For the bulk MAPbI3 structure, ab initio calculations were performed using a plane-wave/pseudopotential DFT approach as implemented in the Vienna Ab initio Simulation Package (VASP), with valence electron configurations explicitly defined as: 1s1 (H), 2s22p2 (C), 2s22p3 (N), 2s22p4 (O), 3s23p63d74s1 (Fe), 5s25p5 (I), and 5d106s26p2 (Pb). The generalized gradient approximation (GGA) exchange–correlation functionals of Perdew–Burke–Ernzerhof (PBE) was used. The Grimme's DFT-D3 dispersion correction term was added to all of our total energy calculations (apart from the validation simulations using r2SCAN and spin–orbital coupling). A kinetic energy cut-off of 520 eV and a Gaussian smearing of 0.01 were implemented for the self-consistent field (SCF) method calculations, with a convergence criterion of 1 × 10−6 eV. Spin-polarization was considered due to the presence of the iron atom. For geometry optimizations, a k-point grid of 4 × 4 × 4 was used for the bulk system (1 × 1 × 1 for surface slabs). For electronic structure calculations, a finer 6 × 6 × 6 k-point grid was used for the bulk system (2 × 2 × 1 for surface slabs). To obtain an accurate bulk tetragonal-phase MAPbI3 structure, a 2 × 2 × 2 supercell of the MAPbI3 structure with random orientations of MA cations was generated using ab initio molecular dynamics (AIMD) with an on-the-fly machine-learning interatomic potential under NVT ensemble conditions at 300 K in VASP. The final configuration was fully relaxed from the dynamic structure and selected based on agreement with experimentally reported values for lattice constants (a = b = 8.76 Å, c = 12.71 Å), mean Pb–I bond length (3.19 Å), and band gap energy (1.57 eV). The structural relaxation was terminated when all forces relaxed smaller than 1 × 10−2. Such methods have been used successfully in recent related studies of perovskite halides.63–65
Our PbI2-terminated (001) surface slab was constructed from the fully optimized 2 × 2 × 2 tetragonal-MAPbI3 supercell. A 2 × 2 × 3 slab structure was constructed with a vacuum region of 20 Å on top to avoid any spurious interactions between the periodic images. Starting from the DFT-optimized molecule-surface structure, AIMD simulations were carried out at 300 K for at least 10 ps under the NVT ensemble. To accurately describe the C–H and N–H vibrations, a time step of 0.5 fs was employed. To reduce the computational cost at each step, a reduced SCF convergence criterion of 1 × 10−4 eV and a Gaussian smearing width of 0.05 were used in the electronic calculations. Dipole moment correction was applied on both energies and forces for all surface simulations, with the dipole centre set at the midpoint of the slab. The binding energies were calculated as: EBinding = ES+MTot − ESTot − EMTot, where ES+MTot, ESTot, and EMTot are the total energies of surface-molecule, surface-only, and molecule-only in the same simulation box respectively. The ferrocene-surface hybridized states also appeared using the meta-GGA level functional r2SCAN and spin–orbital coupling, suggesting that the hybridized states were not artefacts from the GGA-level functional.
Conflicts of interest
The authors declare no other competing interests.Data availability
Datasets are available from the corresponding author upon reasonable request.The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. see DOI: https://doi.org/10.1039/d5ee05482f.
CCDC 2479149 contains the supplementary crystallographic data for this paper.66
Acknowledgements
S. A. H. and M. S. I. gratefully acknowledge funding from the Engineering and Physical Sciences Research Council (EPSRC, EP/X012344/1 and EP/X012484/1). T. W. thanks the Engineering and Physical Sciences Research Council (EPSRC, DTP EP/T51780X/1) for funding. Z. L. gratefully acknowledges funding from the Research Grants Council of Hong Kong, Innovation and Technology Fund (ITS/147/22FP, MHP/079/23), N. J. L. acknowledges funding from the Imperial College Sir Edward Frankland BP Endowment, the Imperial College Deep Tech Funding Scheme and the UKRI IAA project on “Organometallic-enhanced perovskite solar cells”.References
- J. Blochwitz, M. Pfeiffer, T. Fritz and K. Leo, Low voltage organic light emitting diodes featuring doped phthalocyanine as hole transport material, Appl. Phys. Lett., 1998, 73, 729–731 CrossRef CAS.
- J. J. Andre, et al., Molecular semiconductors and junction formation: Phthalocyanine derivatives, Synth. Met., 1987, 18, 683–688 CrossRef.
- B. Maennig, et al., Controlled p-type doping of polycrystalline and amorphous organic layers: Self-consistent description of conductivity and field-effect mobility by a microscopic percolation model, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 195208 CrossRef.
- M. Pfeiffer, et al., Doped organic semiconductors: Physics and application in light emitting diodes, Org. Electron., 2003, 4, 89–103 CrossRef.
- M. Ahles, R. Schmechel and H. von Seggern, n-type organic field-effect transistor based on interface-doped pentacene, Appl. Phys. Lett., 2004, 85, 4499–4501 Search PubMed.
- Y. Hu, et al., Doping Polymer Semiconductors by Organic Salts: Toward High-Performance Solution-Processed Organic Field-Effect Transistors, ACS Nano, 2018, 12, 3938–3946 CrossRef PubMed.
- G.-H. Kim, L. Shao, K. Zhang and K. P. Pipe, Engineered doping of organic semiconductors for enhanced thermoelectric efficiency, Nat. Mater, 2013, 12, 719–723 CrossRef.
- W. Zhao, J. Ding, Y. Zou, C. Di and D. Zhu, Chemical doping of organic semiconductors for thermoelectric applications, Chem. Soc. Rev., 2020, 49, 7210–7228 Search PubMed.
- Y. Xiong, et al., Revealing the Impact of F4-TCNQ as Additive on Morphology and Performance of High-Efficiency Nonfullerene Organic Solar Cells, Adv. Funct. Mater., 2019, 29, 1806262 CrossRef.
- Y. Dong, et al., Dopant-induced interactions in spiro-OMeTAD: Advancing hole transport for perovskite solar cells, Mater. Sci. Eng., R, 2025, 162, 100875 CrossRef.
- F. M. Rombach, S. A. Haque and T. J. Macdonald, Lessons learned from spiro-OMeTAD and PTAA in perovskite solar cells, Energy Environ. Sci., 2021, 14, 5161–5190 RSC.
- M. Jha, et al., Stability Study of Molecularly Doped Semiconducting Polymers, J. Phys. Chem. C, 2024, 128, 1258–1266 CrossRef.
- Y. Yamashita, et al., Efficient molecular doping of polymeric semiconductors driven by anion exchange, Nature, 2019, 572, 634–638 CrossRef PubMed.
- J. Pan, et al., Efficient molecular doping of polymeric semiconductors improved by coupled reaction, Nat. Commun., 2024, 15, 5854 CrossRef PubMed.
- M. Schwarze, et al., Molecular parameters responsible for thermally activated transport in doped organic semiconductors, Nat. Mater., 2019, 18, 242–248 CrossRef PubMed.
- J. Meng, I. E. Castelli and Z. Lan, Heavy Doping-Induced Phase Segregation and Heterojunction Formation, ACS Mater. Lett., 2025, 7, 891–897 CrossRef.
- F. Deschler, et al., Imaging of morphological changes and phase segregation in doped polymeric semiconductors, Synth. Met., 2015, 199, 381–387 CrossRef.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Molecular Electrical Doping of Organic Semiconductors: Fundamental Mechanisms and Emerging Dopant Design Rules, Acc. Chem. Res., 2016, 49, 370–378 CrossRef PubMed.
- G. Zhu, et al., Unveiling the Critical Role of Oxidants and Additives in Doped Spiro-OMeTAD toward Stable and Efficient Perovskite Solar Cells, ACS Appl. Energy Mater., 2022, 5, 3595–3604 CrossRef.
- J. E. Rainbolt, et al., Synthesis and characterization of p-type conductivity dopant 2-(3-(adamantan-1-yl)propyl)-3,5,6-trifluoro-7,7,8,8-tetracyanoquinodimethane, J. Mater. Chem. C, 2013, 1, 1876–1884 RSC.
- P. K. Koech, et al., Synthesis and Application of 1,3,4,5,7,8-Hexafluorotetracyanonaphthoquinodimethane (F6-TNAP): A Conductivity Dopant for Organic Light-Emitting Devices, Chem. Mater., 2010, 22, 3926–3932 CrossRef CAS.
- Y. Karpov, et al., Hexacyano-[3]-radialene anion-radical salts: a promising family of highly soluble p-dopants, Chem. Commun., 2018, 54, 307–310 RSC.
- F. Vanin, et al., Modulating Perovskite Surface Energetics Through Tuneable Ferrocene Interlayers for High-Performance Perovskite Solar Cells, Angew. Chem., Int. Ed., 2025, 64, e202424041 CrossRef CAS.
- Best Research-Cell Efficiency Chart | Photovoltaic Research | NREL. https://www.nrel.gov/pv/cell-efficiency.
- L. Shen, et al., Ultrathin polymer membrane for improved hole extraction and ion blocking in perovskite solar cells, Nat. Commun., 2024, 15, 10908 CrossRef.
- J. Park, et al., Controlled growth of perovskite layers with volatile alkylammonium chlorides, Nature, 2023, 616, 724–730 CrossRef CAS PubMed.
- Y. Shen, et al., Strain regulation retards natural operation decay of perovskite solar cells, Nature, 2024, 635, 882–889 CrossRef CAS PubMed.
- Q. Jiang, et al., Surface passivation of perovskite film for efficient solar cells, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- T. Webb, et al., A Multifaceted Ferrocene Interlayer for Highly Stable and Efficient Lithium Doped Spiro-OMeTAD-based Perovskite Solar Cells, Adv. Energy Mater., 2022, 12, 2200666 CrossRef CAS.
- B. Wen, et al., Robust chelated lead octahedron surface for efficient and stable perovskite solar cells, Nat. Commun., 2024, 15, 7720 CrossRef CAS PubMed.
- J. Suo, et al., Multifunctional sulfonium-based treatment for perovskite solar cells with less than 1% efficiency loss over 4500-h operational stability tests, Nat. Energy, 2024, 9, 172–183 CrossRef CAS PubMed.
- J.-Y. Seo, et al., Dopant Engineering for Spiro-OMeTAD Hole-Transporting Materials towards Efficient Perovskite Solar Cells, Adv. Funct. Mater., 2021, 31, 2102124 CrossRef CAS.
- S.-M. Bang, et al., Defect-Tolerant Sodium-Based Dopant in Charge Transport Layers for Highly Efficient and Stable Perovskite Solar Cells, ACS Energy Lett., 2020, 5, 1198–1205 CrossRef CAS.
- G. Ren, et al., Strategies of modifying spiro-OMeTAD materials for perovskite solar cells: a review, J. Mater. Chem. A, 2021, 9, 4589–4625 RSC.
- J. Burschka, F. Kessler, M. K. Nazeeruddin and M. Grätzel, Co(III) Complexes as p-Dopants in Solid-State Dye-Sensitized Solar Cells, Chem. Mater., 2013, 25, 2986–2990 CrossRef CAS.
- Y.-N. Kim, J.-H. Jo, J. Kim, H.-S. Kim and W. I. Lee, Outstanding Thermal Stability of Perovskite Solar Cells Based on Zn(TFSI)2-Doped Spiro-MeOTAD, ACS Appl. Energy Mater., 2023, 6, 10225–10232 CrossRef CAS.
- F. S. Tuglak Khan, A. L. Waldbusser, M. C. Carrasco, H. Pourhadi and S. Hematian, Synthetic, spectroscopic, structural, and electrochemical investigations of ferricenium derivatives with weakly coordinating anions: ion pairing, substituent, and solvent effects, Dalton Trans., 2021, 50, 7433–7455 RSC.
- T. Buffeteau, J. Grondin and J.-C. Lassègues, Infrared Spectroscopy of Ionic Liquids: Quantitative Aspects and Determination of Optical Constants, Appl. Spectrosc., 2010, 64, 112–119 CrossRef CAS PubMed.
- D. H. Johnston and D. F. Shriver, Vibrational study of the trifluoromethanesulfonate anion: unambiguous assignment of the asymmetric stretching modes, Inorg. Chem., 1993, 32, 1045–1047 CrossRef CAS.
- W. H. Nguyen, C. D. Bailie, E. L. Unger and M. D. McGehee, Enhancing the Hole-Conductivity of Spiro-OMeTAD without Oxygen or Lithium Salts by Using Spiro(TFSI)2 in Perovskite and Dye-Sensitized Solar Cells, J. Am. Chem. Soc., 2014, 136, 10996–11001 CrossRef CAS.
- L. Ye, et al., Superoxide radical derived metal-free spiro-OMeTAD for highly stable perovskite solar cells, Nat. Commun., 2024, 15, 7889 CrossRef CAS PubMed.
- U. B. Cappel, T. Daeneke and U. Bach, Oxygen-Induced Doping of Spiro-MeOTAD in Solid-State Dye-Sensitized Solar Cells and Its Impact on Device Performance, Nano Lett., 2012, 12, 4925–4931 CrossRef CAS.
- H. Yang, et al., Iodonium Initiators: Paving the Air-free Oxidation of Spiro-OMeTAD for Efficient and Stable Perovskite Solar Cells, Angew. Chem., Int. Ed., 2024, 63, e202316183 CrossRef CAS.
- E. Clifford, J. Wright, A. Collauto, J. Hirst and M. Roessler, Characterization of a semiquinone radical bound in the active site of respiratory complex I by hyperfine EPR spectroscopy, ( 2025), preprint at https://doi.org/10.26434/chemrxiv-2025-br3z5.
- R. Prins and F. J. Reinders, Electron spin resonance of the cation of ferrocene, J. Am. Chem. Soc., 1969, 91, 4929–4931 CrossRef CAS.
- F. T. F. O’Mahony, et al., Improved environmental stability of organic lead trihalide perovskite-based photoactive-layers in the presence of mesoporous TiO2, J. Mater. Chem. A, 2015, 3, 7219–7223 RSC.
- R. J. E. Westbrook, et al., 2D Phase Purity Determines Charge-Transfer Yield at 3D/2D Lead Halide Perovskite Heterojunctions, J. Phys. Chem. Lett., 2021, 12(13), 3312–3320 CrossRef CAS PubMed.
- R. J. E. Westbrook, Dr. I. Sanchez-Molina, Dr. J. Manuel Marin-Beloqui, Dr. H. Bronstein and Dr. S. A. Haque, Effect of Interfacial Energetics on Charge Transfer from Lead Halide Perovskite to Organic Hole Conductors, J. Phys. Chem. C, 2018, 122, 1326–1332 CrossRef CAS.
- A. Azaden, et al., Understanding and Overcoming the Fundamental Chemical and Electronic Challenges of SnBr4 Impurities in Tin Perovskite Solar Cells, Small Sci., 2025, e202500426 CrossRef PubMed.
- C. Choi, et al., Hard-Cation-Soft-Anion Ionic Liquids for PEDOT:PSS Treatment, J. Phys. Chem. B, 2022, 126, 1615–1624 CrossRef CAS PubMed.
- W. H. K. Perera, et al., 23.2% efficient low band gap perovskite solar cells with cyanogen management, Energy Environ. Sci., 2025, 18, 439–453 RSC.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- R. He, et al., Ferrocene Derivatives as Redox-Active Antisolvent Additives Enabling Defect Elimination and Residual Stress Mitigation in Perovskite Solar Cells, ACS Photonics, 2025, 12(8), 4745–4756 CrossRef CAS.
- D. L. Cheung, D. P. McMahon and A. Troisi, Computational Study of the Structure and Charge-Transfer Parameters in Low-Molecular-Mass P3HT, J. Phys. Chem. B, 2009, 113, 9393–9401 CrossRef CAS.
- M. Böckmann, et al., Structure of P3HT crystals, thin films, and solutions by UV/Vis spectral analysis, Phys. Chem. Chem. Phys., 2015, 17, 28616–28625 RSC.
- T. J. Macdonald, et al., Phosphorene Nanoribbon-Augmented Optoelectronics for Enhanced Hole Extraction, J. Am. Chem. Soc., 2021, 143, 21549–21559 CrossRef CAS PubMed.
- P. K. Bhushan, M. Jamshidi and J. M. Gardner, The impact of moisture on the stability and degradation of perovskites in solar cells, Mater. Adv., 2024, 5, 2200–2217 RSC.
- M. V. Khenkin, et al., Consensus statement for stability assessment and reporting for perovskite photovoltaics based on ISOS procedures, Nat. Energy, 2020, 5, 35–49 CrossRef.
- K. Ogawa, T. Kitagawa, S. Ishida and K. Komatsu, Synthesis and Structure of a New Tetrakis(pentafluorophenyl)borate Salt of the Silver(I) Cation with Novel Trigonal Planar Tris(benzene) Coordination, Organometallics, 2005, 24, 4842–4844 CrossRef CAS.
- S. Lukin, et al., Mechanochemical Metathesis between AgNO3 and NaX (X = Cl, Br, I) and Ag2XNO3 Double-Salt Formation, Inorg. Chem., 2020, 59, 12200–12208 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- G. García, M. Atilhan and S. Aparicio, Assessment of DFT methods for studying acid gas capture by ionic liquids, Phys. Chem. Chem. Phys., 2015, 17, 26875–26891 RSC.
- Z. Jia, et al., Film-forming polymer nanoparticle strategy for improving the passivation and stability of perovskite solar cells, Energy Environ. Sci., 2024, 17, 7221–7233 RSC.
- A. N. Arber, Vikram, F. C. Mocanu and M. S. Islam, Ion Migration and Dopant Effects in the Gamma-CsPbI3 Perovskite Photovoltaic Material: Atomistic Insights through Ab Initio and Machine Learning Methods, Chem. Mater., 2025, 37, 4416–4424 CrossRef CAS.
- Y.-H. Lin, et al., Bandgap-universal passivation enables stable perovskite solar cells with low photovoltage loss, Science, 2024, 384, 767–775 CrossRef CAS PubMed.
- CCDC 2479149: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2p6rk7.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |