
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable perovskite–organic tandem solar cells enabled by chloride-doped evaporated wide-bandgap perovskites
Xiao
Guo†
 ab,
Zhenrong
Jia†
ab,
Zijing
Dong†
ab,
Nikhil
Kalasariya
c,
Jingcong
Hu
ab,
Zhuojie
Shi
ab,
Julian A.
Steele
de,
Yi-Hsun
Chen
e,
Gordon
Ochsner
f,
Jinxi
Chen
ab,
Xiangkun
Jia
ab,
Yu-Duan
Wang
ab,
Ran
Luo
ab,
Ling Kai
Lee
b,
Tao
Wang
ab,
Shunchang
Liu
ab,
Chao
Luo
ab,
Jia
Li
b,
Martin
Stolterfoht
c and
Yi
Hou
*ab
ab,
Zhenrong
Jia†
ab,
Zijing
Dong†
ab,
Nikhil
Kalasariya
c,
Jingcong
Hu
ab,
Zhuojie
Shi
ab,
Julian A.
Steele
de,
Yi-Hsun
Chen
e,
Gordon
Ochsner
f,
Jinxi
Chen
ab,
Xiangkun
Jia
ab,
Yu-Duan
Wang
ab,
Ran
Luo
ab,
Ling Kai
Lee
b,
Tao
Wang
ab,
Shunchang
Liu
ab,
Chao
Luo
ab,
Jia
Li
b,
Martin
Stolterfoht
c and
Yi
Hou
*ab
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Singapore, 117585, Singapore. E-mail: yi.hou@nus.edu.sg
bSolar Energy Research Institute of Singapore (SERIS), National University of Singapore, Singapore, 117574, Singapore
cElectronic Engineering Department, The Chinese University of Hong Kong, Sha Tin N.T., Hong Kong SAR, China
dAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia
eSchool of Mathematics and Physics, The University of Queensland, Brisbane, QLD 4072, Australia
fSchool of Chemical Engineering, The University of Queensland, Brisbane, 4067, Queensland, Australia
First published on 18th March 2026
Abstract
Wide-bandgap perovskites are critical for achieving high-efficiency perovskite-based tandem solar cells, yet their practical deployment remains limited by stability challenges such as ion migration and halide phase segregation. Herein, we demonstrate that introducing ∼1% PbCl2 as a vapor-phase additive during thermal evaporation effectively stabilizes wide-bandgap perovskite devices. Detailed characterization studies reveal strong chemical interactions between chloride anions and formamidinium cations, resulting in improved crystallinity and enhanced photoluminescence quantum yield. The Cl-doped wide-bandgap perovskite films exhibit suppressed halide phase segregation and mitigated ionic losses. As a result, a 1.75 eV single-junction perovskite cell achieves a T80 lifetime of 1639 hours under continuous maximum power point tracking, along with improved efficiency and open-circuit voltage. When integrated with an organic subcell (PCE-10:P2EH-2V), the resulting perovskite–organic tandem solar cell achieves a power conversion efficiency of 24.86% and a maximum T80 of 1979 hours, surpassing the stability of all the previously reported perovskite–organic and perovskite–perovskite tandem devices.
Broader contextIn the context of global transition to cleaner energy sources, perovskites are revolutionizing photovoltaic technologies due to their remarkable properties. The broad bandgap tunability of such materials enables perovskite-based tandem solar cells with efficiency threshold beyond the theoretical limit. Particularly, wide-bandgap perovskites are essential for state-of-the-art perovskite-based tandem cells. However, their practical deployment is bottlenecked by multiple stability and efficiency challenges. Herein, we demonstrate that introducing ∼1% PbCl2 as a vapor-phase additive during thermal evaporation effectively stabilizes wide-bandgap perovskites. Characterization studies reveal strong chemical interactions between chloride and formamidinium cations, resulting in improved crystallinity and enhanced photoluminescence intensity. The Cl-doped wide-bandgap perovskites exhibit suppressed halide phase segregation and mitigated ionic losses. As a result, a 1.75 eV single-junction perovskite cell achieves a T80 lifetime of 1639 hours under maximum power point tracking. When integrated with an organic subcell, the perovskite–organic tandem achieves 24.86% efficiency and a T80 lifespan of 1979 hours, surpassing all of the reported perovskite-based thin-film tandem cells. This work underscores the significance of stabilizing wide-bandgap perovskites through a one-stop additive strategy, and pushes the boundary of operational stability towards an unprecedented 2000 hours for perovskite-based thin-film tandem photovoltaics. |
Introduction
Perovskite solar cells (PSCs) have emerged as the most promising next-generation photovoltaic technologies, achieving rapid progress in power conversion efficiency (PCE), with the current record reaching 26.95%.1 Despite this remarkable achievement, further closing the gap between the PCE and the Shockley–Queisser limit2 remains increasingly challenging within the constraints of single-junction device architecture. In this context, tandem solar cell configurations are considered the most practical pathway to overcome the theoretical efficiency limit of single-junction cells, offering the potential to achieve PCEs exceeding 30%.Due to excellent semiconductor properties3–6 and broad bandgap tunability7,8 of metal halide perovskites, numerous types of perovskite-based tandem solar cells (TSCs) have already demonstrated remarkable PCE values.9–11 However, the common and essential wide-bandgap perovskite subcell of these TSCs typically suffers from more severe stability and efficiency losses compared to its narrower-bandgap counterparts,12 bottlenecking further optimizations of the device performance of perovskite-based TSCs. Actually, this fact is associated with multiple harmful processes that occur inside wide-bandgap perovskites. Hoke et al. first discovered a halide phase segregation phenomenon of wide-bandgap perovskites with Br contents more than 20%,13 and subsequent studies confirm its adverse impact on device stability.14,15 Mahesh et al. pointed out the key role that the defect-assisted non-radiative recombination plays on the open-circuit voltage (VOC) loss of the device.16 More recently, Thiesbrummel et al. correlated the operational stability loss with the mobile ion-induced field screening effect, which is created by the generation and migration of mobile ion species during aging.17 Shah et al. further studied the influence of such effect on the stability of perovskite-based TSCs.18
Given the complex origins of stability and efficiency losses in wide-bandgap perovskites, identifying the fundamental mechanisms or dominant species responsible remains a significant challenge. Cs–formamidinium (FA+) mixed perovskites are widely recognized for their enhanced thermal stability and photostability against halide phase segregation and device operational stability advantages, compared to their methylammonium (MA+)-containing counterparts.19–22 In such Cs–FA mixed perovskites, the FA+ cation has been identified as a key contributor to instability through ion migration and loss processes.23 Furthermore, volatile FABr escape from the perovskite material is found to be responsible for thermal instabilities of perovskites.24 Meanwhile, the incorporation of Cl-based additives into Br–I mixed-halide perovskites has been shown to suppress halide phase segregation25 and reduce non-radiative recombination losses in solution-processed wide-bandgap perovskites.26 However, the controlled incorporation of Cl into an evaporated Cs-rich, low-Br wide-bandgap perovskite—with simultaneously reduced FA+ and Br− contents for mitigated mobile ion-related losses and minimized halide phase segregation—to collaboratively address both stability and efficiency losses has yet to be realized.
Herein, in this work, we initially developed a Cs-rich (∼70% Cs), low-Br (∼20% Br), and MA+-free wide-bandgap perovskite enabled by thermal evaporation of PbI2, CsBr and FAI to minimize negative impacts brought by FA+ and Br− on device stability as mentioned above. As a step further, we innovatively incorporated a relatively limited amount (∼1%) of PbCl2 with respect to PbI2 in molar ratio as an additive to the vapor. Results from various characterization studies lead to the conclusion that Cl− remains in the perovskite and influences crystal and electronic structures of the perovskite, indicating the successful doping of Cl into the perovskite. More in-depth analyses suggest that the incorporated Cl− manifests chemical interactions with FA+ and enhances crystallinity of the perovskite, suppresses halide phase segregation, reduces non-radiative recombination, and mitigates ionic losses of the PSC device. With these advantages, we demonstrate a PCE of 18.17% for the 1.75 eV Cl-doped evaporated perovskite single-junction device with enhanced VOC. The T80 lifetime of such PSCs reaches 1639 hours under continuous maximum power point (MPP) tracking. When integrating this subcell with a PCE-10:P2EH-2V organic photovoltaic (OPV) subcell, we realize a PCE of 24.86% for the perovskite-OPV tandem with a T80 of 1979 hours under the same MPP aging conditions. To the best of our knowledge, the achieved stability performance of our tandem device surpasses previous reports on perovskite-based thin-film TSCs, i.e., perovskite–OPV and perovskite–perovskite tandem devices.
Results and discussion
Chloride-doping in evaporated perovskites and its impact on material properties
We begin with confirming the successful doping of Cl− and determining the specific chemical compositions of our deposited perovskite thin-films by thermal evaporation. Based on a Cs-rich, low-Br, and MA-free recipe as a control that involves sublimation of PbI2, CsBr, and FAI precursor salts, we introduced another source of PbCl2 as an additive to the vapor during the deposition process (Fig. S1, SI). The evaporation rate of PbCl2 is controlled at 5% of the rate of PbI2 according to quartz crystal microbalance (QCM) sensors. We then performed X-ray photoelectron spectroscopy (XPS) to analyze the chemical compositions of control and Cl-treated perovskite thin-films. As illustrated in Fig. 1a, we found clear signals corresponding to Cl 2p in PbCl2-incorporated thin-film confirming the existence of chlorine elements remaining in the evaporated perovskite, while the control sample shows no signs of peaks within this specific binding energy range. This observation of the presence of Cl in the perovskite is particularly interesting, considering that the majority of previous studies employing chloride salts into the bulk of perovskite do not yield detectable Cl signals in XPS measurements.27–34 Soto-Montero et al. pointed out that those other works reporting the existence of Cl in such cases,35,36 the observations of Cl signals in XPS are always accompanied by other phases, such as CsPbCl3 or MAPbCl3.29 In fact, in our study, we did not observe such phases in subsequent X-ray diffraction (XRD) results. Considering all of these previous reports, we infer that it is our unique evaporated Cs-rich, low-Br, and MA-free perovskite that leads to the detectable retention of Cl in perovskite without the co-existence of other Cl-containing phases, rather than possible Cl losses in the form of volatile MACl.37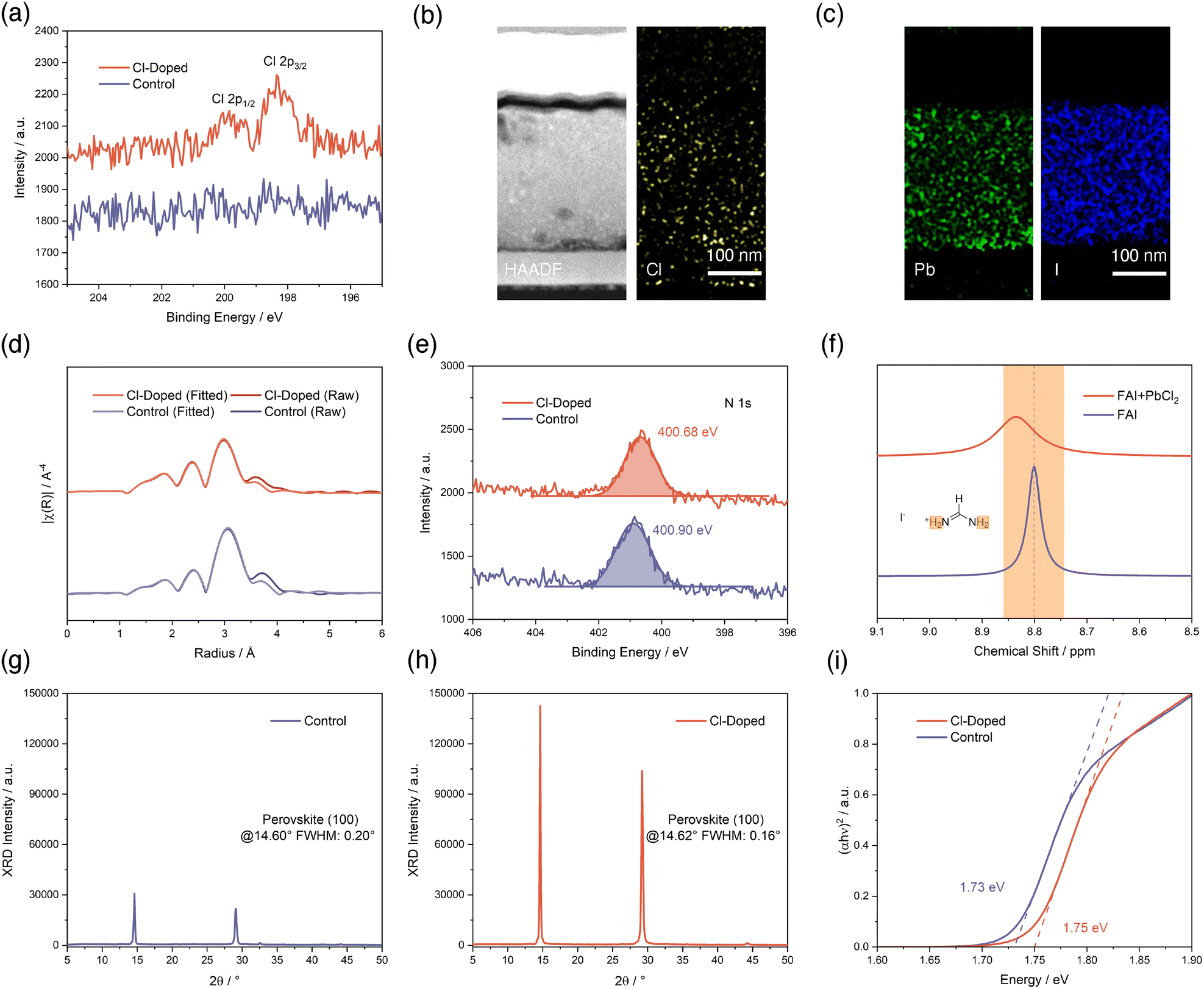 | ||
| Fig. 1 (a) Cl 2p XPS results of perovskites without and with Cl-doping. (b) HAADF-STEM image of FIB-cut cross-section of a Cl-doped PSC and STEM-EDX mapping of Cl element, respectively. (c) STEM-EDX mappings of Pb and I elements, respectively. (d) EXAFS spectra plotted in R-space with phase correction of perovskites without and with Cl-doping measured in transmission mode. (e) N 1s XPS results of perovskites without and with Cl-doping. The signals are fitted by a Gaussian function and the peak positions marked are determined accordingly. (f) 1H-NMR results of FAI without and with PbCl2 mixed in DMSO-d6 solvent. XRD diffraction patterns, perovskite (100) peak positions and FWHMs extracted of perovskites without (g) and with Cl-doping (h). (i) Tauc-plots from UV-vis measurements showing optical bandgaps of perovskites without and with Cl-doping. | ||
To determine the specific chemical compositions of evaporated perovskites, we captured high-angle annular dark-field (HAADF) images using scanning transmission electron microscopy (STEM) for a focused ion beam (FIB)-cut PSC with PbCl2 introduced. As shown in Fig. 1b and c and Fig. S2 (SI), the cross-section of the PSC device is fully exposed after FIB cut, and we extracted energy dispersive X-ray spectroscopy (EDX) mappings for multiple elements. The revealed distribution of Cl elements further confirms the successful introduction of Cl to the perovskite layer. The atomic ratios of Pb, I, and Cl elements are determined from EDX mappings and listed in Table S1 (SI). Specifically, the Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb ratio is calculated as 2.39%. According to these ratios, we managed to confirm that the perovskite composition with PbCl2 added is approximately Cs0.66FA0.34Pb(Cl0.01Br0.22I0.77)3, given the condition that the incorporation of Cl into the lattice is complete. As a result, the composition for control perovskite should be between Cs0.69FA0.31Pb(Br0.23I0.77)3 to Cs0.66FA0.34Pb(Br0.22I0.78)3. The specific PbCl2 amount incorporated with the amount of PbI2 as a 100% reference is calculated to be 1.21% in molar ratio according to a Cl:Pb ratio of 2.39% (Note S1, SI). We notice that this value is lower than the PbCl2 to PbI2 rate ratio of 5% according to QCM sensors, nevertheless, we believe that the value determined from STEM-EDX rather than QCM sensors is more realistic to reflect the actual Cl amount incorporated into the perovskite layer.
:Pb ratio is calculated as 2.39%. According to these ratios, we managed to confirm that the perovskite composition with PbCl2 added is approximately Cs0.66FA0.34Pb(Cl0.01Br0.22I0.77)3, given the condition that the incorporation of Cl into the lattice is complete. As a result, the composition for control perovskite should be between Cs0.69FA0.31Pb(Br0.23I0.77)3 to Cs0.66FA0.34Pb(Br0.22I0.78)3. The specific PbCl2 amount incorporated with the amount of PbI2 as a 100% reference is calculated to be 1.21% in molar ratio according to a Cl:Pb ratio of 2.39% (Note S1, SI). We notice that this value is lower than the PbCl2 to PbI2 rate ratio of 5% according to QCM sensors, nevertheless, we believe that the value determined from STEM-EDX rather than QCM sensors is more realistic to reflect the actual Cl amount incorporated into the perovskite layer.
Next, we conducted various characterizations to investigate the role of introduced Cl in the perovskite from a microscopic perspective. We start with analyzing the extended X-ray absorption fine structure (EXAFS) measurement results for the Pb L3-edge of perovskites without and with Cl-incorporation (Fig. S3, SI). The EXAFS Fourier transform plotted in R-space is exhibited in Fig. 1d. The signals are phase corrected and fitted according to the EXAFS equation using ARTEMIS, part of the IFEFFIT software package (SI).38Via the fits, it emerged that the derived Pb–I bond length is reduced from 3.16 Å to 3.12 Å for perovskite after Cl-treatment, implicating a spatial distortion of PbX6 (X can be Cl, Br, and I) octahedron unit of perovskite induced by Cl-introduction, given the smaller radius of Cl compared to Br and I. This observation also suggests a potentially stronger Pb–I bonding strength. Then, we proceed to investigate the chemical interactions between introduced Cl with perovskite compositions. We firstly analyze XPS results for perovskites without and with Cl-treatment, as shown in Fig. 1e and Fig. S4 (SI). Among all of the elements from the perovskite, the sharpest contrast between the two conditions appears in N 1s XPS results. The N 1s peak for the control perovskite at 400.90 eV obviously shifted to 400.68 eV for the Cl-incorporated perovskite. This binding energy change is in accordance with the electron-donating effect brought by Cl− to FA+, and visualizing the chemical interactions between Cl− and FA+. The 1H-nuclear magnetic resonance (NMR) study further substantiates this claim (Fig. 1f and Fig. S5, SI). The signal from the hydrogen atoms of FA+ at 8.80 ppm clearly moves towards a higher chemical shift of 8.84 ppm upon addition of PbCl2, confirming the formation of hydrogen bonding between Cl− and FA+. This kind of chemical interaction formed is also beneficial for suppressing FA+ migration and loss processes.39 Additionally, we also observed the influences of such interactions from Fourier-transform infrared spectroscopy (FTIR) measurements plotted in Fig. S6 (SI).
Subsequently, we used XRD to reveal the difference in crystal properties brought by incorporating Cl into perovskite. As shown in Fig. 1g, the control perovskite shows a peak intensity of around 30000 counts with a peak position at 14.60° with a full width at half maximum (FWHM) of 0.20°. In sharp contrast, the Cl-treated perovskite exhibits a significantly enhanced peak intensity of about 140000 counts, over four times as high as the control, with a slightly enlarged 2θ of peak position to 14.62° (Fig. 1h). The peak is also obviously narrowed with a FWHM of 0.16°. These features first indicate a drastic enhancement in the crystallinity of the perovskite with the introduction of Cl. Additionally, the slightly shifted peak position to a larger 2θ direction reflects the crystal lattice changes of the perovskite induced by Cl incorporation, which is consistent with our observations in the EXAFS study above. Furthermore, the Tauc-plots in Fig. 1i created using UV-visible absorption (UV-vis) measurements shown in Fig. S7 (SI) confirm that the introduction of Cl into the perovskite also affects the electronic structure of the perovskite. The optical bandgap of the perovskite determined from the Tauc-plots is widened from 1.73 eV to 1.75 eV upon the incorporation of Cl, and these values are in line with previous reports with similar Cs-rich perovskite compositions.40,41
Summarizing all of our findings above, we conclude that the Cl is successfully introduced into the bulk of the perovskite, interacting with a critical cation species FA+ and doping the perovskite, considering the observed crystal and electronic structure changes of the perovskite. Consequently, we mark the target condition of the perovskite with PbCl2 as “Cl-doped” for precisely describing the role of Cl throughout this work.
Photoluminescence-related enhancements by Cl-doping
Next, we performed a series of photoluminescence (PL)-based characterization to further investigate the effects that Cl-doping brought on the PL emission properties of perovskite thin-films. It is worth noting that the configuration of perovskite thin-films we fabricated in this section is indium tin oxide (ITO)/perovskite.We begin with steady-state PL measurements for both conditions of perovskite thin-films, as illustrated in Fig. 2a and Fig. S8 (SI). It is found that the PL peak intensity of the perovskite reveals a significant enhancement after Cl-doping, compared with control. Furthermore, we also observed a peak position difference between both conditions, which is consistent with the optical bandgap changes we reported in Fig. 1i. The PL emission peak for control perovskite is centred at 716.2 nm, while the Cl-doped perovskite reveals a peak with a shifted position towards a lower wavelength of 706.8 nm. Then, we proceed to fit the quasi-Fermi-level splitting (QFLS) and calculate the photoluminescence quantum yield (PLQY) accordingly for both perovskite thin-films following the established method in previous reports (Fig. 2b and Fig. S9, SI).11,42 It turned out that the fitted QFLS value for Cl-doped perovskite exhibits an obvious increment of 41 meV, from 1.193 eV of the control to 1.234 eV. We note that the improved QFLS can be attributed to both an enlarged bandgap and the enhanced PLQY: the determined PLQY values are 0.027% and 0.076%, respectively for the two conditions. This nearly three-fold enhancement of PLQY suggests that Cl-doping is beneficial in terms of reducing VOC loss and suppressing non-radiative recombination processes, marking the improved quality of the perovskite.
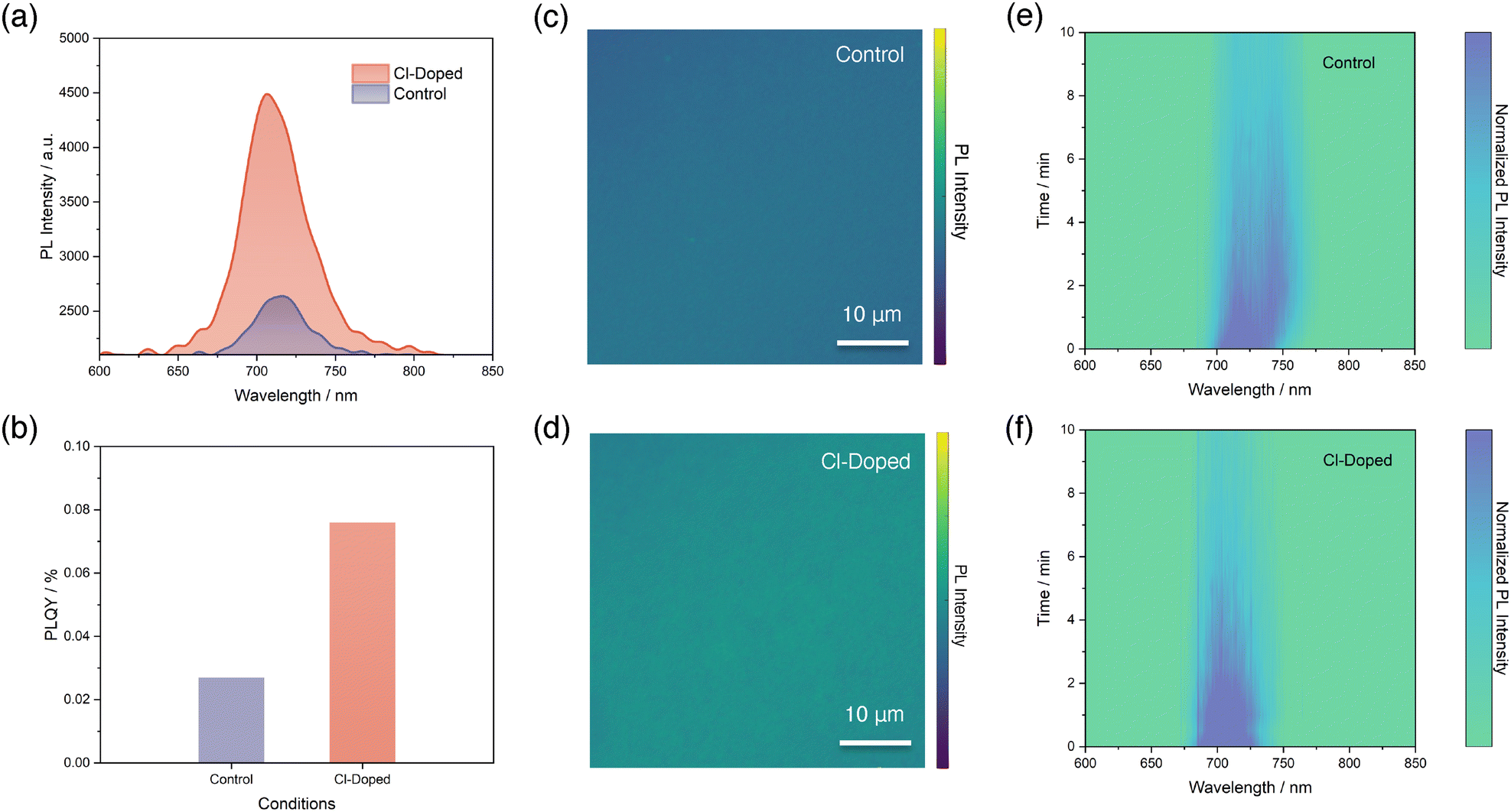 | ||
| Fig. 2 (a) Steady-state PL spectra of perovskite thin-films without and with Cl-doping. Both curves are smoothed by a FFT filter method with a point of window of 15. (b) Calculated PLQY values for perovskite thin-films without and with Cl-doping. Confocal PL mappings of perovskite thin-films without (c) and with (d) Cl-doping. Contour plots extracted from PL evolution study for perovskite thin-films without (e) and with (f) Cl-doping under continuous 100 mW cm−2 532 nm laser excitation. The configuration of all perovskite thin-films in this section is ITO/perovskite. | ||
To provide spatial insight of PL emissions of perovskite thin-films, we captured confocal PL mapping images for both conditions, as shown in Fig. 2c and d. First of all, we noted that both control and Cl-doped perovskite thin-films show very uniform PL emission intensities under confocal PL mappings, aligning well with previous reports of exceptional homogeneity of perovskites deposited by thermal evaporation.43,44 Next, we also observed distinct contrasts in terms of PL emission intensity for the two samples. The Cl-doped perovskite shows obviously enhanced PL intensity, confirming again the benefits gained by Cl-doping of the perovskite.
Lastly, to investigate the effects brought by Cl-doping on suppressing halide phase segregation, we performed a PL evolution study, where the perovskite thin-films were exposed to 10 minutes of continuous 532 nm laser excitations with a power density of 100 mW cm−2 and the PL spectra were recorded every 30 seconds. In Fig. 2e and f, we use contour plots to show the evolutions of PL peaks of both conditions. For the control perovskite thin-film, shown in Fig. 2e, a new emerging PL peak is observed at a longer wavelength position around 740 nm compared to the original PL peak, right after the beginning of exposure to laser excitation. This phenomenon indicates the formation of an iodide-rich segregated phase from the original well-mixed phase and suggests the occurrence of halide phase segregation.13 It is common to expect and observe such a process for a wide-bandgap perovskite with Br:I ratio higher than 20%.13 However, in stark contrast, the halide phase segregation process is significantly suppressed and becomes negligible in Cl-doped perovskite, as depicted in Fig. 2f. This leads to the conclusion that the Cl-doping of our control wide-bandgap perovskite is helpful in regulating the halide phase segregation process25 and brings an advantage to the stability of wide-bandgap perovskites.45
Performances of single-junction wide-bandgap PSCs
Leveraging the above-mentioned benefits of Cl-doping on wide-bandgap perovskite, we fabricated single-junction wide-bandgap PSCs utilizing an inverted p–i–n structure, which is ITO/[4-(3,6-dimethyl-9H-carbazol-9-yl)butyl]phosphonic acid (Me-4PACz)/evaporated wide-bandgap perovskite/C60/SnOx/Ag from the bottom to the top, as revealed in the cross-sectional scanning electron microscopy (SEM) image in Fig. 3a. In Fig. 3b and Fig. S10 (SI), we display the champion current–voltage (J–V) characteristics of fabricated single-junction wide-bandgap PSCs for control and Cl-doped conditions. The champion J–V performance of PSCs with Cl-doping exhibits a drastic improvement, with the PCE from 16.75% to 18.17% and comprehensively enhanced VOC, which is greater than the bandgap increment, short-circuit current density (JSC), and fill factor (FF), despite the enlarged bandgap of the Cl-doped perovskite. We also provide statistics of these parameters in Fig. S11 (SI) to further confirm the reproducibility of advantageous effects from Cl-doping in optimizing J–V performances of single-junction wide-bandgap PSCs. Results of external quantum efficiency (EQE) measurements depicted in Fig. 3c substantiate an enhanced photovoltaic response for the Cl-doped PSC device and reveal the effect of bandgap-broadening induced by Cl-doping again. The photovoltaic bandgaps of PSCs are determined from the inflection points of EQE curves and displayed in Fig. S12 (SI), agreeing well with the optical bandgaps derived from UV-vis results (Fig. 1i).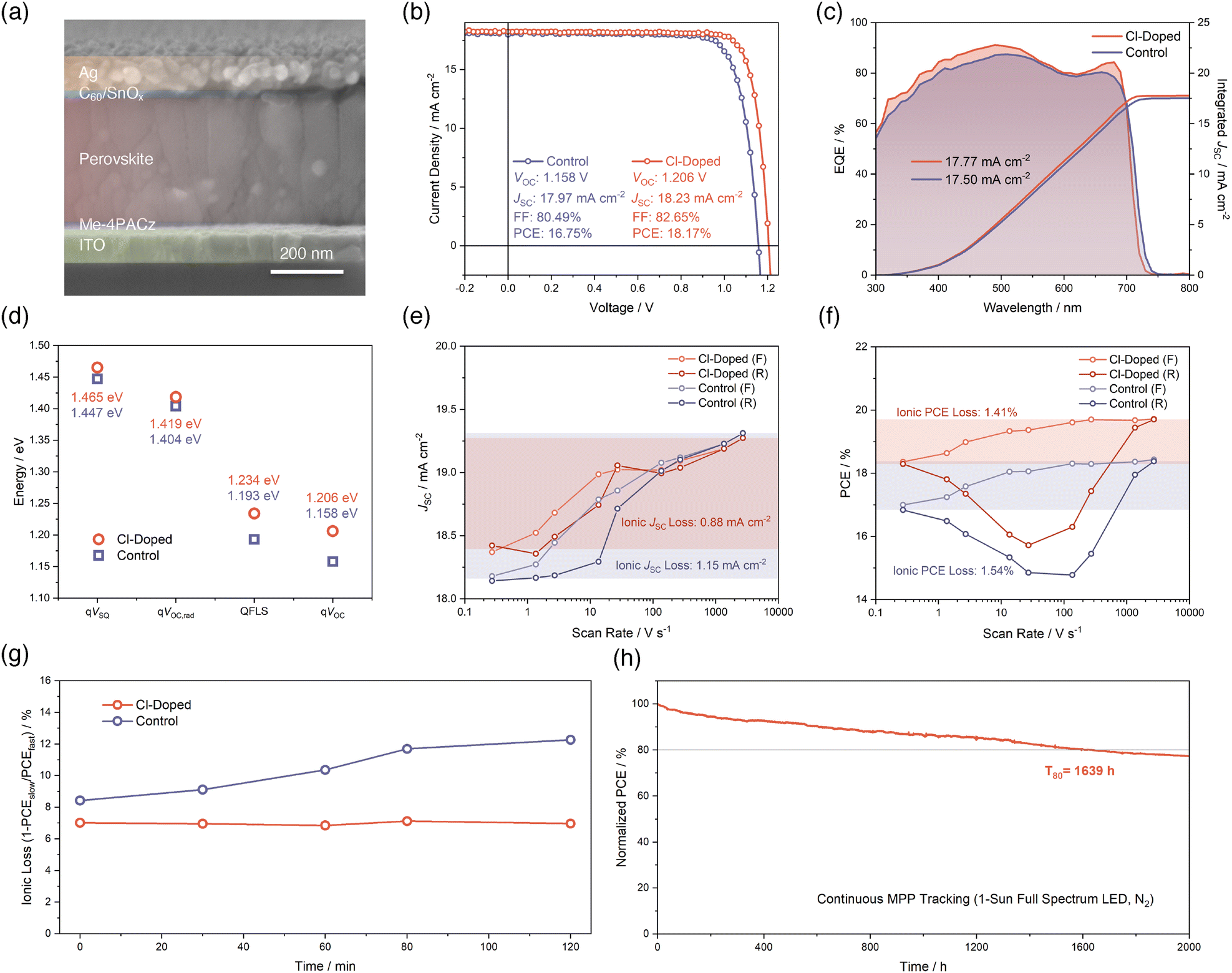 | ||
| Fig. 3 (a) Cross-sectional SEM image of single-junction wide-bandgap PSCs. (b) Champion J–V curves and photovoltaic parameters in reverse scan directions of single-junction wide-bandgap PSCs. (c) EQE curves and integrated JSC values for single-junction wide-bandgap PSCs. (d) VOC loss analysis for single-junction wide-bandgap PSCs. JSC (e) and PCE (f) measured at different J–V scan speeds in both of the reverse and forward scan directions for wide-bandgap PSCs. “F” denotes forward scan direction while “R” denotes reverse scan direction. (g) The evolution of ionic loss for wide-bandgap PSCs plotted against MPP aging time. (h) Long-term MPP stability assessment for single-junction wide-bandgap PSC. The device is unencapsulated and there is no active temperature control, which leads to an actual temperature of about 45 °C. The full spectrum refers to a spectrum from 350 to 900 nm for the white LED light. | ||
Besides, regarding the obviously suppressed VOC loss of the Cl-doped wide-bandgap PSC devices, we conducted a VOC loss analysis to elucidate the origins of mitigated VOC loss. To begin with, we performed high-resolution external quantum efficiency (HREQE) characterization, which allows us to extract the radiative-limit VOC (VOC,rad) and calculate the Urbach energy of the PSCs (Fig. S13 and S14, SI), following previously reported methods.11,46,47 It turned out that the fitted Urbach energies is 37.01 meV and 36.73 meV for control and Cl-doped PSC, respectively. This reduced Urbach energy for Cl-doped PSC indicates that the incorporation of Cl to form a triple-halide perovskite reduced the sub-bandgap electronic state density. This is consistent with previous observations that an appropriately small amount of Cl can help decrease trap state density and Urbach energy.48 Together with the fitted QFLS (Fig. S9, SI) from steady-state PL results, measured VOC of champion PSCs and the Shockley–Queisser limit VOC (VSQ), we visualized the VOC loss mechanism of control and Cl-doped PSCs, in Fig. 3d. It can be concluded that apart from about 20 meV of bandgap difference, the remaining 28 mV improvement of device VOC is majorly from the alleviated loss between qVOC,rad and QFLS, which is 185 meV for Cl-doped compared to 211 meV for control. This sharp QFLS contrast supports the claim that the Cl-doping of the control wide-bandgap perovskite enhances the quality of the perovskite and leads to a better device VOC. Considering the relatively more severe VOC deficits for evaporated-based PSCs compared to solution-processed ones,49 this result is particularly encouraging.
Subsequently, we performed fast hysteresis J–V measurements17,50 to reveal the advantages of Cl-doping in mitigating ion migration-related losses and instabilities for our single-junction wide-bandgap PSCs. This methodology is based on changing the sweeping speed of J–V scans in a broad range to determine the ‘steady-state’ and ‘ion-freeze’ J–V characteristics of PSCs under slow and fast scan rates, respectively. When the scan speed is significantly faster than the ion diffusion rate, the ions will be effectively immobilized and their contributions to device performance will be excluded. Therefore, the gap that can be observed in J–V curves between ‘steady-state’ and ‘ion-freeze’ states corresponds to the influences of ion migration on photovoltaic parameters of PSCs, namely the ionic losses. Given that the JSC of PSCs is typically the most sensitive to mobile ion-induced losses rather than others,17 we first analyzed the JSC values of control and Cl-doped PSCs derived from different J–V scan rates, as shown in Fig. 3e. The JSC losses due to ion migrations are visualized and calculated based on slow and fast scan speeds of 0.272 V s−1 and 2720 V s−1, respectively. The result proves that the Cl-doping can help reduce the ionic JSC loss from 1.15 mA cm−2 to 0.88 mA cm−2, suggesting an alleviated ionic loss of the PSC devices. Indeed, we also included the PCE values obtained from various scan rates as well (Fig. 3f), and the outcome confirms again the conclusion that the Cl-doping brings positive effects for mitigating performance losses induced by mobile ions. This is further substantiated by bias-assisted charge extraction (BACE) measurement51 results in Fig. S15 (SI). The calculated mobile ion concentrations for control and Cl-doped PSCs are 2.61 × 1017 and 4.68 × 1016 cm−3, respectively, further supporting the conclusion above.
Besides the static comparisons between ionic JSC and PCE losses of control and Cl-doped PSCs, as shown in Fig. 3e and f, the fast hysteresis characterization can also assist in monitoring the evolution of ionic losses during the aging of PSC devices. This dynamic process was reported to be correlated with the generation of mobile ions and their accumulation in perovskite during aging.17 Therefore, the result is strongly correlated with the operational stability of the PSC devices. Here, in Fig. 3g and Fig. S16 (SI), we summarize the evolution of ionic loss quantified in the form of 1-PCEslow/PCEfast within 120 minutes of aging time. During this period, the PSCs are aged by exposure to continuous 100 mW cm−2 light illumination and under MPP tracking. It can be seen that the Cl-doped PSC exhibits an obviously lower, and much more stable ionic loss of about 7% throughout the whole aging period. However, the control PSC shows a larger ionic loss of nearly 8.5% at the beginning of aging, and the ionic loss keeps increasing to an elevated level of more than 12% at the end of the aging. This comparison supports that Cl-doping can not only reduce the ionic loss compared to the control, but also suppress the development of enhanced ionic losses during continuous operation of PSCs. This highlights another significant advantage gained by Cl-doping for the enhancement of operational stability of PSCs based on Cl-doping.
Furthermore, as shown in Fig. 3h, we tracked the long-term stability of a Cl-doped unencapsulated single-junction wide-bandgap PSC, which continuously operated at its maximum power point under 1-sun full spectrum (350–900 nm) white LED illumination in a N2-filled glovebox. The temperature of the device is not actively controlled and can be up to 45 °C. Under such an environment, the Cl-doped PSC can maintain 80% of its initial PCE for as long as 1639 hours (T80). This outstanding performance enabled by Cl-doping is among the best stabilities achieved so far for PSCs with a wide bandgap of 1.75 eV.
Transferring Cl-doped wide-bandgap PSCs to stable perovskite–OPV TSCs
Based on the above-discovered enhancements of the performance of single-junction wide-bandgap PSCs enabled by Cl-doping, we integrated this optimized subcell to the fabrication of monolithic perovskite–OPV TSCs. To realize a balanced current-matching condition considering the 1.75 eV bandgap of Cl-doped perovskite and maximize the operational stability of perovskite–OPV TSC, we selected a narrow-bandgap non-fullerene acceptor P2EH-2V (Fig. S17, SI) with an optical bandgap of 1.20 eV,52 mixed with a donor polymer of poly([2,6′-4,8-di(5-ethylhexylthienyl)benzo[1,2-b;3,3-b]dithiophene]{3-fluoro-2[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl}) (PCE-10) to form a tailored OPV subcell of PCE-10:P2EH-2V (Fig. S18 and S19, SI). Fig. 4a reveals the cross-sectional device structure of the fabricated perovskite–OPV TSC, which is ITO/Me-4PACz/perovskite/C60/SnOx/ITO/MoOx/OPV/2,9-bis[3-[[3-(dimethylamino)propyl]amino]propyl]-anthra[2,1,9-def:6,5,10-d′e′f′]diisoquinoline-1,3,8,10(2H,9H)-tetrone (PDINN)/Ag from the bottom to the top. We summarized the champion J–V characteristics and photovoltaic parameters in both forward and reverse scan directions in Fig. 4b. The champion perovskite–OPV TSC reached a PCE of 24.86% with a VOC of 1.813 V, a JSC of 16.69 mA cm−2, and a FF of 82.16% in reverse scan. This is the first demonstration of a perovskite–OPV TSC based on a thermally evaporated wide-bandgap perovskite subcell. We then performed EQE characterization for each subcell and showed the results in Fig. 4c. The integrated JSC values for the two subcells are 16.26 and 16.08 mA cm−2, respectively. These values indicate a balanced current-matching achieved in our perovskite–OPV TSC. Next, to check the reproducibility of our perovskite–OPV TSC, 18 cells are included in forming statistics of PCEs (Fig. 4d). It turned out that the distribution of PCEs generally follows a normal distribution centered at about 24.51%.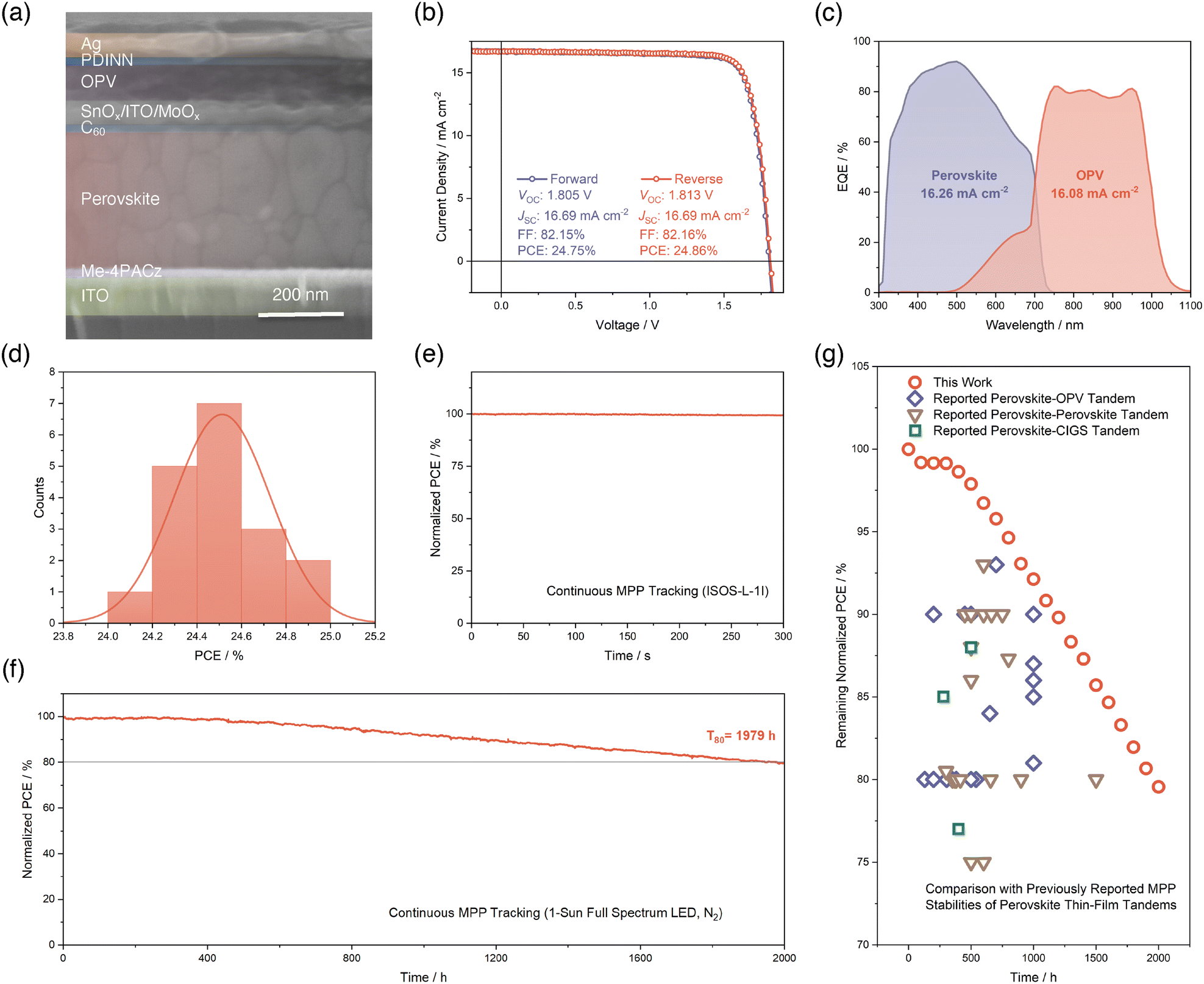 | ||
| Fig. 4 (a) Cross-sectional SEM image of perovskite–OPV TSC. (b) Champion J–V curves and photovoltaic parameters in forward and reverse scan directions of perovskite–OPV TSC. (c) EQE curves and integrated JSC values for perovskite and OPV subcells in perovskite–OPV TSC. (d) Statistical distribution of PCEs from 18 perovskite–OPV TSCs. The red line represents the fitted distribution by a Gaussian function. (e) Short-term MPP tracking result of perovskite–OPV TSC within 300 seconds following ISOS-L-1I protocol. (f) Long-term MPP stability assessment for perovskite–OPV TSC. The device is unencapsulated without active temperature control, which leads to an actual temperature of about 45 °C. The full spectrum refers to a spectrum from 350 to 900 nm for the white LED light. (g) Comparison between our results and previously reported MPP stabilities of perovskite-based thin-film TSCs. The red circles represent our MPP stability result of perovskite–OPV TSC in (f) with a time interval of 100 hours. | ||
To comprehensively disclose the stability of our fabricated perovskite–OPV PSCs, we initially begin with a short-term MPP tracking of the TSC in a N2-filled glovebox and at a room temperature of 25 °C under 1-sun simulated air mass 1.5 global (AM1.5G) light illumination, following the ISOS-L-1I protocol.53 As depicted in Fig. 4e, within the 300 seconds of continuous tracking, we did not find any signs of noteworthy PCE degradation of our perovskite–OPV TSC. Furthermore, when shifted to long-term MPP stability assessment in a N2-filled glovebox with an actual temperature of ∼45 °C under 1-sun full spectrum (350–900 nm with UV) white LED light illumination, our TSC device manifests an outstanding performance where the T80 lifespan can be as long as 1979 hours (Fig. 4f). To the best of our knowledge, this operational stability lifetime is the best among all of the reported perovskite–OPV TSCs. What's more, we also elevated the testing temperature to 65 °C and applied active temperature control to maintain such a high temperature, in accordance with ISOS-L-2I protocol.53 This is the first time that a perovskite–OPV TSC has been subjected to such a high aging temperature continuously. This strict thermal stress, together with UV-containing illumination, poses a challenge for the durability of our TSC device. However, under such harsh conditions, the T80 lifetime for our perovskite–OPV TSC can still reach 195 hours, as illustrated in Fig. S20 (SI). Finally, in Fig. 4g, we summarized previous reports of MPP stability of perovskite–OPV TSCs, and included perovskite–perovskite and perovskite–copper indium gallium selenide (CIGS) TSCs to complete a comparison of our result with dual-junction perovskite-based thin-film TSCs. The plot shows the remaining PCEs versus time elapsed of different TSCs at different testing conditions (Table S2, SI). We also include our MPP stability result as red circles with a time interval of 100 hours from Fig. 4f in the same plot for ease of comparison. It can be clearly seen that our perovskite–OPV TSC based on Cl-doping exhibits remarkable MPP stability, outcompeting previously reported stabilities of perovskite–OPV and perovskite–perovskite TSCs. This observation suggests the stability benefits gained by applying our Cl-doping strategy to evaporated wide-bandgap perovskite and significantly advances the device MPP stability beyond the previous reports of dual-junction perovskite-based TSCs in literature, paving the way towards further development and commercialization of such kind of TSCs.
Conclusions
In summary, targeting the enhancement of the durability of wide-bandgap PSCs and perovskite–OPV TSCs, we intentionally incorporate ∼1% PbCl2 as a vapor additive to a Cs-rich, low-Br, and MA-free evaporated wide-bandgap perovskite. It is observed that the introduced Cl− interacts with FA+ in the perovskite, enlarges the bandgap of the perovskite, improves its crystallinity and suppresses non-radiative recombination in the perovskite. The resulting Cl-doped perovskite thin-films and devices show resistance to halide phase segregation and ionic losses, leading to a T80 lifetime of 1639 hours with optimized PCE and VOC for a 1.75 eV single-junction PSC. When paired with a tailored OPV subcell with suitable photovoltaic properties, we demonstrate a champion PCE of 24.86% with an extended T80 lifetime of 1979 hours under continuous MPP tracking. This result is the best stability performance among reported perovskite–OPV and perovskite–perovskite TSCs and sheds light on designing effective strategies to advance the stability of perovskite–OPV TSCs towards further development and commercialization in the future.Author contributions
Conceptualization, X. G., Z. J., Z. D., and Y. H.; methodology, X. G., Z. J., Z. D., and Y. H.; formal analysis, X. G., Z. J., Z. D., and Y. H.; investigation, X. G., Z. J., Z. D., N. K., J. H., Z. S., J. A. S., Y.-H. C., G. O., J. C., X. J., Y.-D. W., R.L., L. K. L., T. W., S. L., C. L., J. L., M. S., and Y. H.; resources, J. A. S., J. L., M. S., and Y. H.; writing – original draft, X. G.; writing – review & editing, X. G., Z. J., Z. D., and Y. H.; visualization, X. G., Z. J., Z. D., and Y. H.; supervision, Y. H.; project administration, Y. H.; funding acquisition, Y. H.Conflicts of interest
Y. H. is the founder of Singfilm Solar, a company commercializing perovskite photovoltaics. The remaining authors declare no conflicts of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ee04477d.Acknowledgements
Y. H. acknowledges the support from the Agency for Science, Technology and Research (A*STAR) under its MTC IRG Grant (M23M6c0108). The authors of this paper are affiliated with the Solar Energy Research Institute of Singapore (SERIS), a research institute at the National University of Singapore. SERIS is supported by the National University of Singapore, the National Research Foundation Singapore, the Energy Market Authority of Singapore, and the Singapore Economic Development Board. M. S. acknowledges the Vice-Chancellor Early Career Professorship Scheme from CUHK for funding. J. A. S. acknowledges financial support from the Australian Research Council (DE230100173). Part of this work was conducted on the XAS beamline at the Australian Synchrotron, part of ANSTO.References
- Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html, accessed July 2025.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- W.-J. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104, 063903 CrossRef.
- A. Walsh and A. Zunger, Nat. Mater., 2017, 16, 964–967 CrossRef CAS PubMed.
- B. Zhao, M. Abdi-Jalebi, M. Tabachnyk, H. Glass, V. S. Kamboj, W. Nie, A. J. Pearson, Y. Puttisong, K. C. Gödel, H. E. Beere, D. A. Ritchie, A. D. Mohite, S. E. Dutton, R. H. Friend and A. Sadhanala, Adv. Mater., 2017, 29, 1604744 CrossRef PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- J. Liu, Y. He, L. Ding, H. Zhang, Q. Li, L. Jia, J. Yu, T. W. Lau, M. Li, Y. Qin, X. Gu, F. Zhang, Q. Li, Y. Yang, S. Zhao, X. Wu, J. Liu, T. Liu, Y. Gao, Y. Wang, X. Dong, H. Chen, P. Li, T. Zhou, M. Yang, X. Ru, F. Peng, S. Yin, M. Qu, D. Zhao, Z. Zhao, M. Li, P. Guo, H. Yan, C. Xiao, P. Xiao, J. Yin, X. Zhang, Z. Li, B. He and X. Xu, Nature, 2024, 635, 596–603 CrossRef CAS PubMed.
- Z. Liu, R. Lin, M. Wei, M. Yin, P. Wu, M. Li, L. Li, Y. Wang, G. Chen, V. Carnevali, L. Agosta, V. Slama, N. Lempesis, Z. Wang, M. Wang, Y. Deng, H. Luo, H. Gao, U. Rothlisberger, S. M. Zakeeruddin, X. Luo, Y. Liu, M. Grätzel and H. Tan, Nat. Mater., 2025, 24, 252–259 CrossRef CAS PubMed.
- X. Guo, Z. Jia, S. Liu, R. Guo, F. Jiang, Y. Shi, Z. Dong, R. Luo, Y.-D. Wang, Z. Shi, J. Li, J. Chen, L. K. Lee, P. Müller-Buschbaum, D. S. Ginger, D. J. Paterson and Y. Hou, Joule, 2024, 8, 2554–2569 CrossRef CAS.
- K. O. Brinkmann, P. Wang, F. Lang, W. Li, X. Guo, F. Zimmermann, S. Olthof, D. Neher, Y. Hou, M. Stolterfoht, T. Wang, A. B. Djurišić and T. Riedl, Nat. Rev. Mater., 2024, 9, 202–217 CrossRef CAS.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- S. G. Motti, J. B. Patel, R. D. Oliver, H. J. Snaith, M. B. Johnston and L. M. Herz, Nat. Commun., 2021, 12, 6955 CrossRef CAS PubMed.
- Y. Guo, C. Zhang, L. Wang, X. Yin, B. Sun, C. Wei, X. Luo, S. Yang, L. Sun and B. Xu, Energy Environ. Sci., 2025, 18, 2308 RSC.
- S. Mahesh, J. M. Ball, R. D. Oliver, D. P. McMeekin, P. K. Nayak, M. B. Johnston and H. J. Snaith, Energy Environ. Sci., 2020, 13, 258–267 RSC.
- J. Thiesbrummel, S. Shah, E. Gutierrez-Partida, F. Zu, F. Peña-Camargo, S. Zeiske, J. Diekmann, F. Ye, K. P. Peters, K. O. Brinkmann, P. Caprioglio, A. Dasgupta, S. Seo, F. A. Adeleye, J. Warby, Q. Jeangros, F. Lang, S. Zhang, S. Albrecht, T. Riedl, A. Armin, D. Neher, N. Koch, Y. Wu and M. Stolterfoht, Nat. Energy, 2024, 9, 664–676 CrossRef CAS.
- S. Shah, F. Yang, E. Köhnen, E. Ugur, M. Khenkin, J. Thiesbrummel, B. Li, L. Holte, S. Berwig, F. Scherler, P. Forozi, J. Diekmann, F. Peña-Camargo, M. Remec, N. Kalasariya, E. Aydin, F. Lang, H. Snaith, D. Neher, S. De Wolf, C. Ulbrich, S. Albrecht and M. Stolterfoht, Adv. Energy Mater., 2024, 14, 2400720 CrossRef CAS.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- R. D. J. Oliver, P. Caprioglio, F. Peña-Camargo, L. R. V. Buizza, F. Zu, A. J. Ramadan, S. G. Motti, S. Mahesh, M. M. McCarthy, J. H. Warby, Y.-H. Lin, N. Koch, S. Albrecht, L. M. Herz, M. B. Johnston, D. Neher, M. Stolterfoht and H. J. Snaith, Energy Environ. Sci., 2022, 15, 714–726 RSC.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D’Haen, L. D’Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. De Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- Q. Wang, N. Phung, D. Di Girolamo, P. Vivo and A. Abate, Energy Environ. Sci., 2019, 12, 865–886 RSC.
- W. Li, M. Hao, A. Baktash, L. Wang and J. Etheridge, Nat. Commun., 2023, 14, 8523 CrossRef CAS PubMed.
- Z. Dong, J. Hu, X. Guo, Z. Shi, H. Chen, Y. Wang, R. Luo, J. A. Steele, Z. Degnan, E. Solano, Q. Zhou, N. Kalasariya, N. Li, T. Wang, J. Chen, L. K. Lee, Y. Wang, J. Li, M. Stolterfoht, M. Sui, Y. Lu and Y. Hou, Nat. Mater., 2025 DOI:10.1038/s41563-025-02375-8.
- J. Xu, C. C. Boyd, Z. J. Yu, A. F. Palmstrom, D. J. Witter, B. W. Larson, R. M. France, J. Werner, S. P. Harvey, E. J. Wolf, W. Weigand, S. Manzoor, M. F. A. M. Van Hest, J. J. Berry, J. M. Luther, Z. C. Holman and M. D. McGehee, Science, 2020, 367, 1097–1104 CrossRef CAS PubMed.
- X. Shen, B. M. Gallant, P. Holzhey, J. A. Smith, K. A. Elmestekawy, Z. Yuan, P. V. G. M. Rathnayake, S. Bernardi, A. Dasgupta, E. Kasparavicius, T. Malinauskas, P. Caprioglio, O. Shargaieva, Y.-H. Lin, M. M. McCarthy, E. Unger, V. Getautis, A. Widmer-Cooper, L. M. Herz and H. J. Snaith, Adv. Mater., 2023, 35, 2211742 CrossRef CAS PubMed.
- A. Ren, H. Lai, X. Hao, Z. Tang, H. Xu, B. M. F. Y. Jeco, K. Watanabe, L. Wu, J. Zhang, M. Sugiyama, J. Wu and D. Zhao, Joule, 2020, 4, 1263–1277 CrossRef CAS.
- J. Zhou, L. Tan, Y. Liu, H. Li, X. Liu, M. Li, S. Wang, Y. Zhang, C. Jiang, R. Hua, W. Tress, S. Meloni and C. Yi, Joule, 2024, 8, 1691–1706 CrossRef CAS.
- T. Soto-Montero, S. Kralj, R. Azmi, M. A. Reus, J. S. Solomon, D. M. Cunha, W. Soltanpoor, D. S. Utomo, E. Ugur, B. Vishal, M. Ledinsky, P. Müller-Buschbaum, F. Babbe, D. K. Lee, C. M. Sutter-Fella, E. Aydin, S. De Wolf and M. Morales-Masis, Joule, 2024, 8, 3412–3425 CrossRef CAS.
- B. Philippe, B.-W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
- T. G. Kim, S. W. Seo, H. Kwon, J. Hahn and J. W. Kim, Phys. Chem. Chem. Phys., 2015, 17, 24342–24348 RSC.
- M. Kim, G.-H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Joule, 2019, 3, 2179–2192 CrossRef CAS.
- H. Yu, F. Wang, F. Xie, W. Li, J. Chen and N. Zhao, Adv. Funct. Mater., 2014, 24, 7102–7108 CrossRef CAS.
- K. H. Stone, A. Gold-Parker, V. L. Pool, E. L. Unger, A. R. Bowring, M. D. McGehee, M. F. Toney and C. J. Tassone, Nat. Commun., 2018, 9, 3458 CrossRef PubMed.
- K. B. Lohmann, S. G. Motti, R. D. J. Olive, A. J. Ramadan, H. C. Sansom, Q. Yuan, K. A. Elmestekawy, J. B. Patel, J. M. Ball, L. M. Herz, H. J. Snaith and M. B. Johnston, ACS Energy Lett., 2022, 7, 1903–1911 CrossRef CAS PubMed.
- A. Babaei, W. Soltanpoor, M. A. Tesa-Serrate, S. Yerci, M. Sessolo and H. J. Bolink, Energy Tech., 2019, 8, 1900784 CrossRef.
- V. L. Pool, A. Gold-Parker, M. D. McGehee and M. F. Toney, Chem. Mater., 2015, 27, 7240–7243 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- W. Yang, K. Zhang, W. Yuan, L. Zhang, C. Qin and H. Wang, Adv. Mater., 2024, 36, 2313461 CrossRef CAS PubMed.
- J. Kim, J. Park, G. Kim, W. Xu, S. D. Stranks, H. Min and S. I. Seok, Small, 2025, 21, 2500197 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, N. Tian, Z. Huang, S. Duan, J. Wang, D. Yao, G. Zheng, Y. Yang, B. Zhou and F. Long, Sol. Energy Mater. Sol. Cells, 2024, 275, 113038 CrossRef CAS.
- T. Kirchartz, J. A. Márquez, M. Stolterfoht and T. Unold, Adv. Energy Mater., 2020, 10, 1904134 CrossRef CAS.
- J. Ávila, C. Momblona, P. P. Boix, M. Sessolo and H. J. Bolink, Joule, 2017, 1, 431–442 CrossRef.
- J. Li, H. Wang, X. Y. Chin, H. A. Dewi, K. Vergeer, T. W. Goh, J. W. M. Lim, J. H. Lew, K. P. Loh, C. Soci, T. C. Sum, H. J. Bolink, N. Mathews, S. Mhaisalkar and A. Bruno, Joule, 2020, 4, 1035–1053 CrossRef CAS.
- J. Hieulle, X. Wang, C. Stecker, D.-Y. Son, L. Qiu, R. Ohmann, L. K. Ono, A. Mugarza, Y. Yan and Y. Qi, J. Am. Chem. Soc., 2019, 141, 3515–3523 CrossRef CAS PubMed.
- L. Krückemeier, U. Rau, M. Stolterfoht and T. Kirchartz, Adv. Energy Mater., 2019, 10, 1902573 CrossRef.
- F. Urbach, Phys. Rev., 1953, 92, 1324 CrossRef CAS.
- D. Zhao, C. Chen, C. Wang, M. M. Junda, Z. Song, C. R. Grice, Y. Yu, C. Li, B. Subedi, N. J. Podraza, X. Zhao, G. Fang, R.-G. Xiong, K. Zhu and Y. Yan, Nat. Energy, 2018, 3, 1093–1100 CrossRef CAS.
- T. Abzieher, D. T. Moore, M. Roß, S. Albrecht, J. Silvia, H. Tan, Q. Jeangros, C. Ballif, M. T. Hoerantner, B.-S. Kim, H. J. Bolink, P. Pistor, J. C. Goldschmidt, Y.-H. Chiang, S. D. Stranks, J. Borchert, M. D. McGehee, M. Morales-Masis, J. B. Patel, A. Bruno and U. W. Paetzold, Energy Environ. Sci., 2024, 17, 1645–1663 RSC.
- V. M. Le Corre, J. Diekmann, F. Peña-Camargo, J. Thiesbrummel, N. Tokmoldin, E. Gutierrez-Partida, K. P. Peters, L. Perdigón-Toro, M. H. Futscher, F. Lang, J. Warby, H. J. Snaith, D. Neher and M. Stolterfoht, Sol. RRL, 2021, 6, 2100772 CrossRef.
- N. Kalasariya, P. F. Sowmeeh, F. Pena-Camargo, F. Vanin, T. Lukas, Y. Dong, Q. Feng, Z. Liu, W. A. Memon, D. Gao, J. Gong, X. Wu, A. F. C. Mendez, J. Hagenberg, Z. Abadi, T. Hultzsch, X. Zhao, S. Shah, H. Yu, V. Srivastava, J. Xu, N. Zhao, F. Lang, Z. Zhu and M. Stolterfoht, Adv. Energy Mater., 2026, e03866 CrossRef.
- Z. Jia, X. Guo, X. Yin, M. Sun, J. Qiao, X. Jiang, X. Wang, Y. Wang, Z. Dong, Z. Shi, C.-H. Kuan, J. Hu, Q. Zhou, X. Jia, J. Chen, Z. Wei, S. Liu, H. Liang, N. Li, L. K. Lee, R. Guo, S. V. Roth, P. Müller-Buschbaum, X. Hao, X. Du and Y. Hou, Nature, 2025, 643, 104–110 CrossRef CAS PubMed.
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame, A. Di Carlo, R. Cheacharoen, Y.-B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan, D. Di Girolamo, M. Grätzel, A. Hagfeldt, E. von Hauff, H. Hoppe, J. Kettle, H. Köbler, M. S. Leite, S. Liu, Y.-L. Loo, J. M. Luther, C.-Q. Ma, M. Madsen, M. Manceau, M. Matheron, M. McGehee, R. Meitzner, M. K. Nazeeruddin, A. F. Nogueira, Ç. Odaba şı, A. Osherov, N.-G. Park, M. O. Reese, F. De Rossi, M. Saliba, U. S. Schubert, H. J. Snaith, S. D. Stranks, W. Tress, P. A. Troshin, V. Turkovic, S. Veenstra, I. Visoly-Fisher, A. Walsh, T. Watson, H. Xie, R. Yıldırım, S. M. Zakeeruddin, K. Zhu and M. Lira-Cantu, Nat. Energy, 2020, 5, 35–49 CrossRef.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |