
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing EDL-driven reduction mechanisms in binary, ternary, and quaternary fluorinated electrolytes via an integrated MD–DFT–ML framework
Md Jamil Hossain†
 a,
Tilas Kabengele†ad,
Qisheng Wua,
Patrick J. Barryef,
Mia S. Mishaaneh,
Shan Yanfg,
Xiao Tongi,
Amy C. Marschilokefgh,
Kenneth J. Takeuchiefgh,
Esther S. Takeuchiefgh,
Brenda M. Rubensteinbcd and
Yue Qi*a
a,
Tilas Kabengele†ad,
Qisheng Wua,
Patrick J. Barryef,
Mia S. Mishaaneh,
Shan Yanfg,
Xiao Tongi,
Amy C. Marschilokefgh,
Kenneth J. Takeuchiefgh,
Esther S. Takeuchiefgh,
Brenda M. Rubensteinbcd and
Yue Qi*a
aSchool of Engineering, Brown University, Providence, RI 02912, USA. E-mail: yueqi@brown.edu
bDepartment of Physics, Brown University, Providence, RI 02912, USA
cData Science Institute, Brown University, Providence, RI 02912, USA
dDepartment of Chemistry, Brown University, Providence, RI 02912, USA
eInstitute of Sustainability, Electrification, and Energy (I:SEE), Stony Brook University, Stony Brook, NY 11794, USA
fDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, USA
gInterdisciplinary Science Department, Brookhaven National Laboratory, Upton, NY 11973, USA
hDepartment of Materials Science and Chemical Engineering, Stony Brook University, Stony Brook, NY 11794, USA
iCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 23rd April 2026
Abstract
Accurately predicting solid electrolyte interphase (SEI) formation requires explicitly resolving the electric double layer (EDL) structure, which deviates significantly from that of the bulk electrolyte. Although an established molecular dynamics (MD) and Density Functional Theory (DFT) framework can model SEI formation by evaluating reduction reactions of local clusters in the EDL, it suffers from a combinatorial computational bottleneck. To overcome this limitation, we introduce a machine-learning-accelerated simulation workflow (MD–DFT–ML), integrating a gradient-boosted regression model trained on EDL composition data to efficiently predict reduction potentials. We apply this framework to seven fluorinated electrolytes comprising fluorinated anions, a fluorinated ester solvent, two types of diluent (ion-solvating ester vs. non-solvating ether), and an FEC additive. The analysis shows that the EDL selectively accumulates cation-binding species; consequently, the non–cation-binding ether diluent rarely enters the EDL and makes minimal contributions to SEI formation. DFT calculations on statistically representative EDL clusters provide reduction potentials and fluorine-release pathways, while the ML model, which substantially reduces the DFT workload, predicts cluster reduction energies with a mean absolute error of 0.1 eV. The combined MD–DFT–ML approach also quantifies contributions from different sources to LiF formation in the SEI. This methodology establishes a generalizable route for multiscale modeling electrolyte and interphase design for next-generation electrochemical energy-storage systems.
Broader contextHigh-energy rechargeable batteries, essential for electrified transportation and renewable energy storage, operate under strongly charged conditions during cycling. Near these charged electrode surfaces, the local electrolyte environment differs dramatically from the bulk liquid. Designing electrolytes that enable stable solid electrolyte interphase (SEI) formation, therefore, requires resolving electrical double layer (EDL) structures rather than relying solely on bulk measurements or isolated molecular calculations. Here, we integrate molecular dynamics, density functional theory, and machine learning into a unified DFT–MD–ML framework to explicitly resolve EDL structures and statistics in complex electrolytes and quantify how different species contribute to SEI formation. Applying this approach to emerging multicomponent fluorinated electrolytes containing fluorinated anions, solvents, diluents, and additives, we show that interfacial partitioning, rather than bulk composition, controls electrolyte reduction processes and LiF generation. This predictive, electrochemical interface framework moves beyond bulk-centric electrolyte formulation toward rational interphase engineering, advancing safer and longer-lasting energy storage technologies. |
1. Introduction
Electrolytes are critical components of any battery system, as they govern ion transport, charge transfer kinetics, interfacial stability, and ultimately, the performance, safety, and longevity of the device.1–6 To achieve this multifunctionality, electrolytes must support the formation of a stable solid electrolyte interphase(SEI)—a thin passivation layer formed on the anode surface—in the first few battery cycles. The SEI is arguably the most important component at the electrode/electrolyte interface, as it protects the electrode from parasitic redox reactions of the electrolyte while still maintaining high cation permeability.7,8 However, controlling and promoting SEI formation in battery design is extremely difficult due to our limited understanding of the morphology and reaction mechanisms involved in SEI formation.8–12Although bulk electrolyte properties, such as ionic conductivity, viscosity, and solvation structure, are relatively easy to obtain experimentally and computationally,13 accurately predicting which electrolyte formulations and electrochemical compositions will yield a stable SEI remains exceptionally challenging. This difficulty arises from the chemical complexity and multicomponent nature of modern electrolytes, as well as the inaccessibility of the interface to experimental characterization during battery cycling. Conventional electrolytes, consisting of salts, multiple solvents, and additives, are low-concentration (e.g., 1M) electrolytes with homogeneous liquid structures. However, the recently developed localized high concentration electrolytes (LHCEs)14–16 exhibit micelle-like heterogeneous nanoscale structures17,18 and cannot be treated as homogeneous solutions. Structural inhomogeneities in such complex electrolytes fundamentally alter interfacial composition19 and require specialized experimental and computational techniques that can capture resolutions beyond those needed to model the bulk electrolyte.
Quantum chemistry-based methods, including Density Functional Theory (DFT)20–26 and Ab Initio Molecular Dynamics (AIMD),27–29 have been used to illustrate the initial SEI formation mechanisms due to electrolyte reduction and decomposition, supporting experimental observations.3,30 However, due to limitations on the system size and timescale of DFT calculations, the electrolyte in these studies is often simplified to a few ions in a few solvent molecules, which is not able to represent the electrolyte composition accurately. Previous simulation efforts have primarily focused on the reduction reactions of simple solvent or Li+-coordinated single-solvent molecules and neglected the actual electrical double layer (EDL) structure under an electric field.8,22–25,31–33 Thus, the effects of electrode charge and long-range diffusion are often ignored.34–37 Multiscale modeling approaches aim to capture different time and length scales and provide a more complete physical picture of the kinetics and interfacial processes. Such models include phase-field models and hierarchical, scale-bridging modeling frameworks whose goal is to resolve different time or length scales.8,38–41
Considering the facts that electrochemical reactions occur within a few nanometers of the electrified interface and that the EDL composition deviates from that of the bulk electrolyte, it is extremely important to incorporate EDL structures into SEI formation simulations. Recently, Wu and co-workers developed an MD–DFT computational modeling framework to address a key question: Which species accumulate at the negatively-charged electrode surface in a complex, multicomponent electrolyte? In the absence of multiscale modeling to track the long-term SEI formation and growth process (kinetics), this approach made a thermodynamic assumption that all species in the EDL may eventually be reduced according to the reduction voltages of the local structures. The statistical distribution of local structures in the EDL was obtained from classical MD simulations and was used as input for DFT calculations of reduction potentials. This MD–DFT model was applied to investigate several key electrolytes, including: a prototypical LHCE composed of lithium bis(fluorosulfonyl)imide (LiFSI) salt, dimethoxyethane (DME) solvent, and tris(2,2,2-trifluoroethyl) orthoformate (TFEO) diluent;19 1 M lithium hexafluorophosphate (LiPF6) in a mixed ethylene carbonate (EC) and ethyl methyl carbonate (EMC) solvent system; and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in mixed 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME) ether-based electrolytes, with and without fluoroethylene carbonate (FEC) additive. At a fraction of the cost of AIMD simulations, the MD–DFT model provided conclusions well-aligned with experimental observations.42 More importantly, this EDL and thermodynamics-focused DFT–MD model revealed the temperature-sensitive changes in EDL structure and the resulting SEI compositions, in agreement with experimental observations, which could not be captured by considering only bulk electrolyte solvation structures.42
With an increasing number of components, especially advanced additives, in the electrolyte design, the number of DFT evaluations grows combinatorially with species count (N) and cluster size (M), scaling as  . This means, for a typical electrolyte with more than 10 components, more than 105 combinations may be generated.43 These factors pose a computational bottleneck and hinder the rapid screening of candidate electrolyte components and EDL structures in the SEI-guided electrolyte design workflow. This work presents a machine learning (ML) extension to the previously proposed MD–DFT workflow to address its drawbacks associated with the combinatorial increase in the number of EDL clusters. Given the critical role the electrolyte plays in battery systems and the vastness of potential electrolyte candidates, it remains unrealistic, at least in the near future, to rely solely on quantum chemistry methods. In a recent review, Xu and co-workers likened the underexplored chemical space of potential electrolyte candidates to the Universe.44 ML and AI approaches are thus needed to streamline and accelerate the discovery of new electrolyte materials. While previous ML studies in electrochemistry have mainly focused on machine learning force fields of bulk electrolytes rather than predicting localized interfacial properties,37,39,45–48 our work integrates an ML scheme based on gradient-boosting regression into the MD–DFT workflow.
. This means, for a typical electrolyte with more than 10 components, more than 105 combinations may be generated.43 These factors pose a computational bottleneck and hinder the rapid screening of candidate electrolyte components and EDL structures in the SEI-guided electrolyte design workflow. This work presents a machine learning (ML) extension to the previously proposed MD–DFT workflow to address its drawbacks associated with the combinatorial increase in the number of EDL clusters. Given the critical role the electrolyte plays in battery systems and the vastness of potential electrolyte candidates, it remains unrealistic, at least in the near future, to rely solely on quantum chemistry methods. In a recent review, Xu and co-workers likened the underexplored chemical space of potential electrolyte candidates to the Universe.44 ML and AI approaches are thus needed to streamline and accelerate the discovery of new electrolyte materials. While previous ML studies in electrochemistry have mainly focused on machine learning force fields of bulk electrolytes rather than predicting localized interfacial properties,37,39,45–48 our work integrates an ML scheme based on gradient-boosting regression into the MD–DFT workflow.
We demonstrate this approach in the design of fluorinated electrolyte systems with heterogeneous structures. The electrolyte system is based on recent experimental work by Quilty and co-workers49 and Hossain et al.50 Fluorinated solvents, additives, and diluents are chosen, as they were proposed to increase the presence of fluorine in the SEI, whether as simple inorganic fluorides (LiF) or organofluoro groups, for enhanced interface stability.14,51–54 Additional advantages of fluorinated electrolytes include enhanced oxidative stability due to fluorine's electron-withdrawing properties and low freezing temperature.52 Adding low solubility diluents further increases the salt anion fraction in the Li-ion inner solvation shell.55–59 Our recent joint experimental and computational study showed that this set of fluorinated ternary electrolytes with salt, solvent, and diluent enabled operation under extreme conditions of high voltage, fast charge, and low and high temperature and demonstrated improved capacity retention compared to carbonate-based electrolytes.49 To further increase the fluoride content in the SEI, FEC is often added.60–68 However, it was found to be more efficient in increasing F-containing SEI for low-concentration carbonate-based electrolytes than ether-based electrolytes at room temperature.42,69 This is due to whether F-containing anions can enter the EDL or not. Therefore, adding FEC does not necessarily increase F-containing species in the SEI, requiring detailed EDL simulations. Since all four components, namely anions, solvent, diluent, and FEC, are fluoridated, their contributions to the EDL and reduction potentials, computed from MD and DFT respectively, jointly impact the LiF content in the SEI. Several hypotheses related to the LiF content in the SEI will be quantified with the newly developed MD–DFT–ML modeling framework.
The remainder of the paper is structured as follows: section 2 outlines the electrolyte systems studied in this work and presents the computational details and design of our MD–DFT–ML modeling framework. Section 3 presents and discusses the results obtained from our model, starting with the impact of diluent and solvent concentrations on the EDL structures. We show using the EDL model how accounting for interfacial local composition and structures alters the reduction behavior of the electrolyte species and drives the formation of the SEI. The ML model based on simple electrolyte cluster compositions to predict their reduction potentials achieves a mean absolute error of 0.1 eV on the reduction potential of electrolyte species compared to pure DFT calculations, and also shows the sensitivity of the reduction potential to different electrolyte components. Overall, our work demonstrates how ML techniques can be incorporated into ab initio calculations and used to accelerate the modeling and design of interfacial battery properties.
2. Methods
2.1. Numerical design of electrolyte system
Our numerical design followed a typical electrolyte virtual lab scenario. Seven electrolyte systems, with their compositions listed in Table 1, were picked based on five species (components), with their molecular structures shown in Fig. 1a. The electrolyte design started from dissolving LiFSI salt in fluorinated ester methyl 3,3,3-triflouropropionate (MTFP) solvent (E1). Two chemically different diluents, namely the ion-solvating fluorinated ester 2,2,3,3-tetrafluoropropyl trifluoroacetate (TFPTFA) and the non-ion-solvating fluorinated ether 1,1,2,2-tetrafluoroethyl 2,2,2-trifluoroethyl ether (TFETFE) were added at two different compositions, denoted as E2, E3 and E4, E5. To investigate how an FEC additive plays a role in accumulation in the EDL and subsequently as LiF in SEI, FEC was added to E1, generating E6; and to E4, generating E7. Given the repeated molecular components, it is natural to ask if the MD–DFT–ML workflow can retain the memory of previously tested electrolytes. | ||
| Fig. 1 (a) The molecular structures of the components that form the electrolytes. (b) The ternary representation of the complex electrolytes. (c) The computational workflow. | ||
| Name | Mixing | Electrolyte components and concentration | Number of molecules in the simulation cell | ||||
|---|---|---|---|---|---|---|---|
| LiFSI | MTFP | TFPTFA | TFETFE | FEC | |||
| E1 | — | LiFSI–3.2M TFP | 480 | 1536 | — | — | — |
| E2 | E1 + TFPTFA | LiFSI–3.2M TFP–1.6M TFPTFA | 360 | 1152 | 576 | — | — |
| E3 | E1 + TFPTFA | LiFSI–3.2M TFP–3.2M TFPTFA | 240 | 768 | 768 | — | — |
| E4 | E1 + TFETFE | LiFSI–3.2M TFP–1.6M TFETFE | 360 | 1152 | — | 576 | — |
| E5 | E1 + TFETFE | LiFSI–3.2M TFP–3.2M TFETFE | 240 | 768 | — | 768 | — |
| E6 | E1 + FEC | LiFSI–3.2M TFP–0.68M FEC | 480 | 1536 | — | — | 324 |
| E7 | E4 + FEC | LiFSI–3.2M TFP–1.6M TFETFE–1.03M FEC | 345 | 1104 | — | 552 | 354 |
2.2. Computational workflow
Fig. 1c illustrates the workflow for the combined MD and DFT calculations. In this workflow, classical molecular dynamics (MD) simulations were used to simulate the bulk and EDL structures. Then, representative building blocks of the EDL, including solvation clusters of Li+ and free solvents, were captured and input into DFT calculations to obtain their reduction voltage and kinetics. Finally, the DFT-computed reduction voltages and the statistics of the solvation clusters and free solvents were combined to predict the probabilities of different reduced species that contribute to SEI formation. Meanwhile, the DFT-computed reduction potentials generate a dataset on which to train a machine learning (ML) model, which can be used to directly predict the reduction potentials based on cluster compositions. Since most of the computational details and justifications for MD and DFT calculations can be found in our previous work, we will describe these calculations briefly while focusing on new details related to the ML model.Bulk electrolytes, especially heterogeneous electrolytes, were built following the approaches described in our previous publications,18,50 specifically by constructing different initial salt-solvent cluster configurations to accelerate the equilibration time. The interfacial models were built from the bulk electrolyte structures by expanding the cell in one direction, large enough (150 Å) to ensure bulk electrolyte behavior in the center region,72 and confining it between two graphene electrodes separated by a ∼200 Å thick vacuum layer. The x- and y-dimensions of the simulation cells are 51.12 Å × 54.12 Å. To accelerate the equilibration time for heterogeneous interface structures, two different initial configurations were used for each electrolyte by expanding the already equilibrated bulk electrolyte into different directions before being confined by the graphene electrodes. The configuration that leads to the lower averaged energy was used for data analysis. The number of electrolyte constituents for each interface model is given in Table 1.
For EDL calculations,42 constant charges, σ = ±0.6 e nm−2 corresponding to the surface charge at the Li+/Li deposition potential,73 were applied to the two graphene electrodes.42,74 The liquid density (the distance between the two graphene electrodes) was determined under uncharged conditions with a 2.0 ns NVT simulation. The graphene electrodes were then fixed for 32.0 ns under charged conditions, as it takes a longer time to equilibrate under charged conditions. Additional 8.0 ns simulations for each charge density were conducted and used for statistical analysis. The timestep was set to 2.0 fs with fixed-length hydrogen bonds, and the temperature was set to 298 K for all simulations.
The use of a non-polarizable force field such as COMPASS III enables access to larger system sizes and longer simulation times, which are necessary to capture the mesoscale features of the EDL. We therefore expect the qualitative conclusions and comparative trends reported here to remain robust, while acknowledging that explicit polarization may be required for quantitatively accurate interfacial properties. More advanced polarizable force field and constant potential methods can be used to further improve the accuracy of MD simulations of the EDL structures.75 The improvement may be necessary for the dynamics of EDL but less significant for steady-state EDL structures.76
The Gradient Boosting Regressor (GBR), initialized with a Linear Regression (LR) model, was employed to directly predict ΔGR based on compositions using the Python scikit-learn library85 following the residual-boosting framework introduced by Friedman.86–89 Unlike constant initializers, which have no predictive performance, or nonlinear initializers such as decision trees, which are prone to overfitting, the linear base learner improves the extrapolation performance of the model and prevents GBR from overfitting by capturing only the underlying linear trends in the data.90,91 The LR prediction is then incrementally enhanced and regularized in subsequent GBR iterations using decision trees and hyperparameter tuning. A grid search approach was used to automatically select the optimal hyperparameters in the GBR model and circumvent overfitting. Our ML algorithm follows a residual-boosting technique, where simpler models are sequentially added to learn the residuals from previous models, effectively correcting previous errors, and enhancing predictive performance. To maximize model transferability and predictive performance across different electrolyte datasets, hyperparameter tuning was conducted iteratively, updating the parameters with each new dataset version. At each iteration, a 5-fold cross-validation strategy was applied to evaluate model performance and select the optimal combination of hyperparameters.85,92 The hyperparameter grid considered at each iteration was as follows: number of estimators = [20, 40, 60]; learning rate = [0.05, 0.1]; maximum tree depth = [2, 3, 4]; minimum samples per leaf = [1, 2, 3].
Electrolyte preparation, electrode preparation, cell assembly, cell testing, sample recovery, XPS measurements, and data analysis. Electrolytes were prepared matching compositions of E1, E6, and E7 as described in Table 1. The salts were used as received and the solvents were dried prior to use. Two electrode cells with lithium metal and graphite electrodes were prepared and cycled for two (dis)charge cycles at 30 °C in a 0.010–1.8 V window at a C/10 rate. Cells were disassembled within argon-filled gloveboxes and the graphite electrodes were recovered and washed with dimethylcarbonate (DMC) solvent. X-ray photoelectron spectroscopy (XPS) measurements were collected on the recovered graphite anodes after formation cycling. Measurements were collected at the Center for Functional Nanomaterials (CFN) at Brookhaven National Laboratory. Measurements were performed under a base pressure of 5 × 10−6 Pa in an ultra-high vacuum chamber. The X-ray source and detector were a non-monochromatized Al Kα (hν = 1486.6 eV, V = 15 kV, I = 20 mA) source and a SPECS Phoibos 100 MCD analyzer, respectively. An inert transfer suitcase was used to transfer all samples from an argon-filled glovebox to the XPS instrument. A step size of 0.05 eV and an Epass of 20 eV were used for the measurement of the C 1s, O 1s, and F 1s spectral regions. All peak fitting of the XPS spectra was performed with CasaXPS software. A Shirley background function was used and the line shape was Gaussian–Lorentzian.
3. Results and discussion
3.1. Reduction reactions of electrolyte components w/wo Li-ion association
As the focus is on electrolyte reduction reactions and if they can contribute to LiF formation in the SEI, individual components that form the electrolytes are first computed. Table 2 reports the single-electron equilibrium reduction potentials and energy barriers of reduction reactions of different electrolyte components without and with Li+ coordination, respectively. Fig. S2–S6 show the reduction pathways of various electrolyte molecules with and without Li+ coordination. Typically, associating with Li+ will increase the reduction potential of a species, as confirmed by the results in Table 2. The greater the value, the greater the tendency the species has for reduction to occur. Table 2 shows the possibility of reduction for Li+-coordinated species is ordered as follows: FSI− > TFPTFA > FEC > TFETFE > MTFP. From Table 2, it can be seen that different components may contribute to LiF formation after reduction decomposition. The free MTFP solvent is electrochemically stable against Li–metal, indicated by its lower voltage than Li+/Li. But, Li+ associated MTFP can be reduced (Fig. S3). In other words, the MTFP molecules in the salt-solvent clusters can be reduced. For the common F-containing additive, FEC, it was found that Li+-coordinated FEC can form LiF when reduced, but the free FEC ring opening reaction is more favored than the F-dissociation reaction for the one-electron reduction reaction. A second electron reduction could lead to defluorination from ring-opened FEC (Fig. S6). Two diluent components, TFPTFA and TFETFE, are compared. Since most diluent molecules have no or very small Li–salt solubility, they do not enter the Li+ solvation shell. Therefore, the reduction results for free species are of interest. Both can be reduced even without association with Li+ and contribute to F-dissociation. TFPTFA shows a higher reduction potential without defluorination reactions (0.49 V for defluorination vs. 0.73 V for no defluorination). The energy barriers for the reduction reactions of all the species studied are low and of a similar order of magnitude, suggesting that they can all be easily reduced if they enter the EDL and receive electrons from the electrode. Therefore, the rest of the modeling work will focus on the thermodynamics of the reduction reactions of the EDL. This assumption assumes any species within the EDL will be reduced if their reduction voltage is above the Li-reduction potential, corresponding to final SEI composition rather than time-dependent SEI evolution, which can be treated with other multiscale modeling approaches, e.g., DFT and phase-field modeling.41| Species | Pathway | Free species | Li+-coordinated species | ||||
|---|---|---|---|---|---|---|---|
| Reduction potential (V) | Energy barrier (eV) | F− dissociation? | Reduction potential (V) | Energy barrier (eV) | Li–F formation? | ||
| FSI− (trans) anion | — | 0.80 | 0.045 | Y | 2.75 | 0.026 | Y |
| MTFP solvent | — | −0.01 | 0.312 | N | 1.80 | 0.002 | Y |
| TFPTFA diluent | F-dissociation | 0.49 | 0.122 | Y | 2.44 | 0.075 | Y |
| No F-dissociation | 0.73 | 0.058 | N | — | — | — | |
| TFETFE diluent | — | 0.25 | 0.247 | Y | 2.15 | 0.004 | Y |
| FEC | F-dissociation | 0.39 | 0.102 | Y | 2.18 | 0.051 | Y |
| Ring opening | 0.84 | 0.045 | N | 2.06 | 0.058 | N | |
3.2. Electrolyte structures in the bulk and EDL
Mixing these components creates a large chemical and compositional design space for the electrolyte. Fig. 1b represents the bulk electrolyte compositions and solubility range. The binary salt and solvent mixed electrolytes are on the left edge in Fig. 1b. Mixing the salt-solvent-diluent into a ternary electrolyte system is represented in the green area in Fig. 1b. Previous literature often calls the salt-solvent-diluent electrolytes as “localized high concentration electrolytes” (LHCEs), without specifying the concentrations. High concentration electrolyte (HCE) is used to refer to electrolytes at salt concentrations at which no free solvent is available. However, not every salt-solvent system can reach such high concentrations (limited by solubility). In our example, the fluorinated MTFP solvent can dissolve LiFSI salt up to 2.25 M,50 while a stronger DME solvent can dissolve LiFSI up to 4 M.18 The 2.25 M LiFSI in MTFP leaves many free solvent molecules (not coordinated to Li+) in the solution.50 To be more accurate, we will refer to the LiFSI–MTFP saturated electrolyte as a medium concentration electrolyte (MCE), to show its difference from a low concentration electrolyte (LCE) at 1M and a high concentration electrolyte (HCE) at 4 M. The corresponding ternary MCE-diluent mixed system is referred to as a “localized-MCE” (LMCE). In the ternary phase diagram representation (Fig. 1b), each corner (component) can contain multiple species, such as multiple salts, multiple solvents (referred to as cosolvents), as well as multiple diluents. “Additives”, species with small concentrations, can be categorized into any one of the components based on their interactions with the ions (e.g., FEC can be considered a cosolvent, as it participates in the solvation shell of Li+ due to strong interactions with Li+).The reduction reaction of the complex electrolyte strongly depends on the electrolyte structures near the interface, typically nanometers when electron tunneling occurs. We analyzed the EDL structures for the seven electrolytes sandwiched between two electrodes, following the approach described in the Methods section. The final frames of each interface model at a charge density of σ = ±0.6 e nm−2 are shown in Fig. 2a. Fig. 2b shows the charge density profile within 1.5 nm of the negatively-charged surface, averaged parallel to the electrode, along the direction perpendicular to the electrode (z-axis) for all electrolytes studied. The oscillatory pattern near the electrode surface indicates ion layering in the EDL and is similar among all of the electrolyte formulations studied here. The EDL structures consist of an adsorbed cation layer with solvated anions, solvents, and diluents, followed by a diffuse layer of ions, solvents, and diluents. We will refer to the first 5 Å adsorbed layer as ‘the inner layer’ (Helmholtz layer) and the 5–13 Å diffuse layer as ‘the outer layer’ of the EDL. The charge density becomes relatively flat, representing the bulk phase density of the electrolyte beyond 13 Å from the negatively-charged electrodes for all of the electrolytes.
 | ||
| Fig. 2 (a) Final frames of the MD trajectories for the 7 electrolytes at an electrode charge density of σ = ±0.6 e nm−2 and (b) the charge density profile next to the negatively-charged surface, averaged over time and parallel to the electrode. (c) Typical salt clusters (solvents and diluents are removed for ease of visualization) in bulk electrolyte from MD simulations. The FSI− anions are color-coded according to the number of Li+ cations coordinated. The presence of 1 to 2 Li+ cations coordinated to each FSI− indicates chain-like clusters. The presence of 4 to 6 Li+ coordinated to each FSI− indicates aggregated clusters. (d) The schematics of the ELD near the negatively-charged electrode as they extended to the bulk phase. | ||
Before discussing EDL structures, we will first discuss the bulk electrolyte characteristics, as they play a role in the corresponding EDL structures. The electrolytes return to their bulk compositions and structures in the center of the interface model. E1 and E6 can be considered to be homogeneous electrolytes with salt and solvent (Fig. 2a). The additive FEC can be considered to be a cosolvent, as it participates in the solvation shell of Li+ due to strong interactions with Li+. E2–E5 and E7 have nano-scale heterogeneous structures due to the diluent, which does not dissolve the salt (ideally) but forms solutions with the solvent. (Fig. 2a) The difference between the two diluents has been extensively studied in our previous MD simulations.50 We found that two chemically different diluents (TFPTFA or TFETFE) result in different solvation structures and nano-scale salt-solvent clusters because TFPTFA can enter into the salt-solvation clusters, whereas TFETFE minimally participates in these clusters. Fig. 2c shows the typical salt clusters (solvents and diluents are removed for ease of visualization) from MD simulations of the bulk electrolytes based on the cation (Li+) to anion (FSI−) coordination numbers (CNs). The CNs reveal the local salt concentration in the salt-solvent clusters and compare well with the Raman spectra results. In a dilute electrolyte, one would expect the cation–anion CN to be 0 or 1, as isolated ions or ion pairs, respectively. The chain-like regions are dominated by each FSI− anion coordinated to 1 to 2 Li+ cations, due to the close to 2.25 M salt concentration. Thus, the salt clusters in the E1 are formed by anions and cations consecutively arranging in a chain-like network that percolates in three dimensions with regions consisting of aggregates (Fig. 2c). Adding TFPTFA-diluent into E1 results in similar chain-like characteristics and similar Li+–FSI− coordination numbers. In contrast, the addition of TFETFE-diluent to E1 further increases the salt concentration, showing 4 to 5 Li+ cations coordinated to each FSI− anion (Fig. 2c). Although TFETFE-diluent does not enter the solvation shell, it removes solvent molecules from the salt-solvation cluster, resulting in higher Li+–FSI− CNs. These are other examples of micelle-like structures with increased local salt concentrations due to solvent–diluent interactions, resulting in more complicated nano-scale heterogeneous structures.
As seen in Fig. 2a, for E2 E5, E7, we observe distinct heterogeneous regions in the EDL. Due to the larger fraction of free solvent molecules available in the MCE, the corresponding LMCE has cation-solvents in the inner adsorption layer (indicated by the MTFP number density in Table 3). This is drastically different from the LHCE, in which diluent enters the inner adsorption layer since insufficient solvent is available.19 Fig. S7 shows front views of the EDL structures for all of the electrolytes. The salt clusters in the TFPTFA-based LMCEs appear to be scattered throughout the xy-plane, while the TFETFE-based LMCEs show localized clustering of the salt.
| Electrolyte | Inner adsorbed layer (0–5 Å) | Total EDL (0–13 Å) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Li+ | FSI− | MTFP–Li+ | Free MTFP | Diluent | FEC | Li+ | FSI− | MTFP–Li+ | Free MTFP | Diluent | FEC | |
| E1 | 1.42 | 0.44 | 1.51 | 0.09 | — | — | 0.98 | 0.47 | 0.99 | 1.02 | — | — |
| E2 | 1.99 | 0.74 | 1.94 | 0.11 | 0.62 | — | 1.54 | 0.78 | 1.40 | 0.79 | 0.66 | — |
| E3 | 2.59 | 0.65 | 2.70 | 0.26 | 0.68 | — | 1.59 | 0.56 | 1.43 | 0.82 | 0.86 | — |
| E4 | 2.41 | 0.62 | 2.07 | 0.47 | 0.05 | — | 1.67 | 0.71 | 1.12 | 1.25 | 0.59 | — |
| E5 | 2.56 | 0.71 | 2.70 | 0.86 | 0.07 | — | 1.88 | 0.90 | 1.41 | 1.39 | 0.51 | — |
| E6 | 1.47 | 0.49 | 1.13 | 0.08 | — | 2.05 | 1.23 | 0.72 | 0.90 | 0.84 | — | 1.36 |
| E7 | 2.08 | 0.62 | 1.59 | 0.39 | 0.02 | 2.28 | 1.45 | 0.65 | 1.00 | 1.16 | 0.50 | 1.55 |
Based on our MD simulation results for the different interface models and the CN counting, we developed schematics as shown in Fig. 2d to better describe the electrolyte structures in the bulk as well as in the negative electrode/electrolyte interfacial region. Near the negatively-charged interface, due to repulsion between the negatively-charged electrode and salt anions, fewer anions are present, leading excess cations to form solvation shells with ion-solvating species (e.g. MTFP, FEC, TFPTFA). An Li+ adsorption layer is formed with anions, solvents, and diluents coordinating with Li+ to form half-solvation shells that screen the electrode charge. This adsorbed layer is followed by a diffuse layer of salt, solvent, and diluent molecules. The cation-rich EDL region contains chain-like salt clusters, which connect to the 3D branched salt network in the bulk electrolyte in E1. Like bulk E1, only a few aggregates can be found (Fig. 2a). Diluent modulates these chain structures in the EDL, as it does in the bulk electrolyte. Adding an ion-solvating diluent (TFPTFA) into E1 creates chains of alternatingly connected cations and anions, and some ion aggregates, all interconnected through a 3D branched network. The mobility of these chain-like clusters is restricted by these branched networks. Conversely, a non-ion solvating diluent (TFETFE) in E1 leads to more ion aggregates with increasing TFETFE. More isolated island-like clusters, which possess greater mobility, were found in E4 and E5.
3.3. Reduction of the EDL
The reduction of complex electrolytes requires a realistic representation of EDL structures and their corresponding reduction potentials. Table 1 only shows which species have a higher chance of reducing without considering their local environment in the electrolyte, especially in the EDL, which has a different composition than the bulk electrolyte.To quantitatively show that the electrolyte concentration in the EDL is different from that in the bulk electrolyte, the number density of each species was normalized to the bulk values and listed in Table 3. With increasing cations (>1) and decreasing anions (<1) in the EDL (especially in the inner adsorbed layer), cation binding species, such as FEC, increased; and non-binding species, such as diluent, decreased in the EDL. Among the two diluents, ion-nonbinding TFETFE cannot be found in the inner adsorption layer, suggesting fewer contributions to the SEI formation. Diluent modulates the anion distribution in the EDL as well. Comparing electrolytes E2 and E3, as the amount of ion-solvating TFPTFA diluent is increased, the amount of FSI− is decreased in the EDL due to the overall decreased salt concentration in Table 3. Conversely, comparing E4 and E5, despite a decrease in overall salt concentration in the electrolyte with increased dilution, the non-ion-solvating TFETFE-diluent allowed for higher salt concentration and an increased amount of FSI− in the EDL region (opposite to the case of TFPTFA). This trend holds in both the inner adsorbed layer and the total EDL region.
Thus, we obtained the statistics of the possible Li+ solvation structures in the EDL for various electrolytes and calculated the reduction potentials of these individual structures that can lead to LiF formation (section 3.5). Due to the large number of cluster combinations, only the local structures with an occurrence that accounts for 90% of the EDL are generally included in the DFT calculations. The rest of 10% of clusters are chemically similar major cation-solvent clusters (shown in Fig. 3), therefore they can be omitted from the DFT calculations. With this simplification, 30 clusters are generated for DFT calculations in E6 and E7 (Fig. S8).
 | ||
| Fig. 3 Tree maps of various Li+ centric first solvation shells and free species in the EDL of electrolytes. Color coded according to the reduction potentials for reduction reactions. | ||
From each frame of the 8 ns MD simulation trajectory, the total number of each of the possible Li+ solvation structures and the number of free (not Li-ion associate) species in the EDL is counted and averaged over all frames to calculate the probability of occurrence of each Li solvation cluster and free species. Fig. 3 shows the probability of occurrence of major Li+ solvation clusters along with the free species in a tree map color-coded with their respective reduction potentials vs. Li+/Li. The relative size of a nested rectangle compared to the total area of the tree map for a given electrolyte indicates the fraction of the overall probability (normalized to 1) assigned to that cluster. Apart from free species, only the ten most probable Li+ solvation clusters are shown in the tree maps, while the rest of the clusters are grouped as ‘others’ and color-coded according to their corresponding weighted average reduction potential. This weighted average reduction potential for the ‘others’ group is calculated by summing the products of the occurrence probability of each cluster in this group and its respective reduction potential vs. Li+/Li. Regions with darker blue correspond to higher reduction potentials as shown in the reduction potential color spectrum. Smaller solvation clusters (fewer species coordinated to Li+) tend to show higher reduction potentials. In a CV scan, clusters with higher reduction voltages will be reduced first.
The most noticeable change is the contribution of free MTFP solvent, which has a reduction −0.01 V vs. Li+/Li. They are counted as zero reduction potential in the tree maps, as well as in the average reduction potential calculations (shown in SI Table S1). A large portion of free solvents (light colored regions) is seen for all electrolytes. For E1, the free MTFP solvent corresponds to 55% of the reducing species in the EDL (Fig. 3a). Adding FEC additive to E1 brings free solvents down to 45% by replacing some portions with beneficial free FEC additives (Fig. 3b). Addition of TFPTFA diluent in E1 decreases free MTFP solvents to 33–37% (Fig. 3c and d). TFETFE-based LMCEs have a higher portion of free MTFP solvents (37–45%) than TFPTFA-based LMCEs (Fig. 3e and f) due to TFETFE's non-ion-solvating nature. Adding FEC to E4 to form E7 (Fig. 3g) retains the free MTFP solvent portion while increasing the free FEC contribution to 6%.
Considering the appearance of each species and their association with Li+ in the EDL, the reduction sequence of individual species that contribute to F− generation can be obtained from Table 2, as FSI− (Li+) > TFPTFA(Li+) > FEC(Li+) > MTFP(Li+) > FEC (free) > TFPTFA(free) > TFETFE (free). A more comprehensive understanding of the features (e.g., species in the clusters) and the DFT-computed reduction energy, ΔGR, were obtained by a feature correlation analysis using the Pearson Correlation coefficient, rXY, defined as:
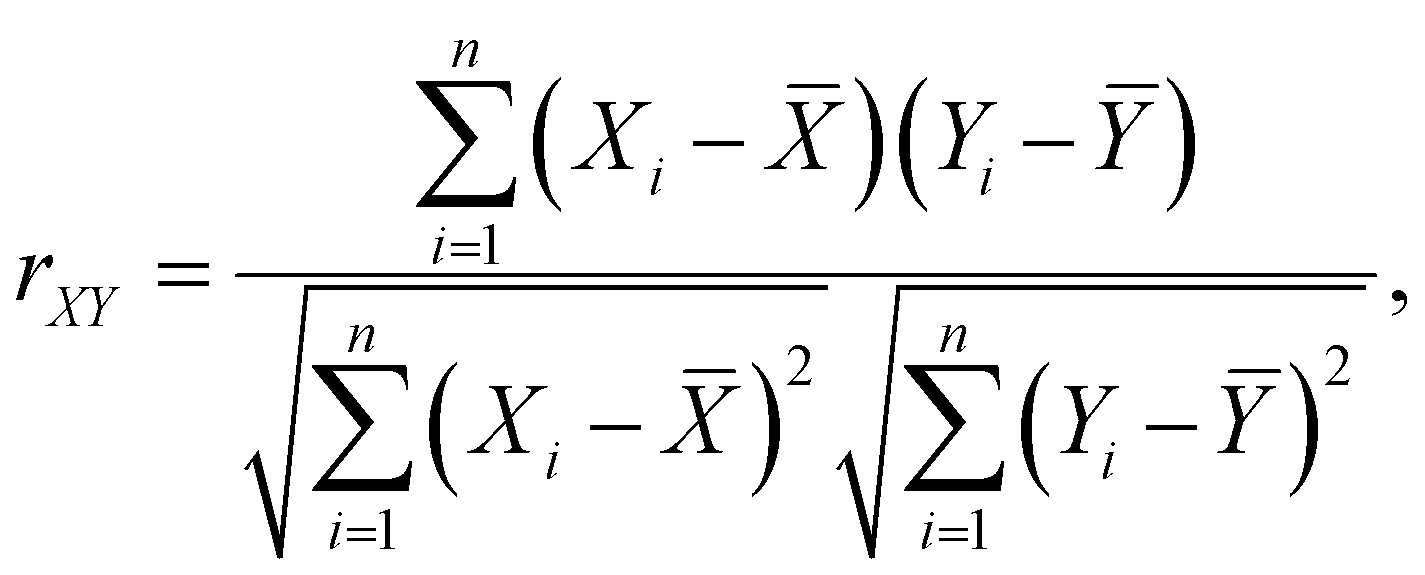 | (1) |
![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) and Ȳ are their respective means.
and Ȳ are their respective means.
The features are the species in the solvation shell. Since FEC can bind with Li+ in two ways – with its carbonyl oxygen or with its fluorine atom, these configurations are denoted as FEC and fFEC, respectively. TFETFE is excluded from the analysis as it does not coordinate significantly with Li+ and was not explicitly represented as a feature in the dataset. As shown in Fig. 4, the pairwise correlation matrix (left panel) reveals no strong correlations among the input features, justifying their inclusion in the model. The ranked list of feature-target correlations (right panel) shows that Li+ and FSI− are most strongly correlated with ΔGR, followed by the fluorinated additives TFPTFA, fFEC, and FEC. While Li+ decreases ΔGR, or increases the reduction potential, FSI− decreases the reduction potential. FEC increases its reduction potential if it is associated with Li+ via the fluorine atom. These results reinforce chemical intuition. Li+ and FSI− play a dominant electrostatic role in the stability of the cluster and, hence, in the reduction potential. TFPTFA and fFEC exhibit strong correlations due to their ability to enter the first solvation shell and modulate the electronic environment around Li+. In contrast, MTFP shows weak correlation with ΔGR, consistent with its role as a relatively inert solvent. These correlation results greatly enhanced our understanding based on chemical intuition or single component calculations.
 | ||
| Fig. 4 Feature correlation and ranking of input variables based on their linear association with the reduction potential (ΔG). Left: Pairwise correlation heatmap. Right: Sorted correlation coefficients with respect to ΔG. | ||
3.4. ML accelerated reduction voltage prediction and correlation
Due to the common species in these clusters, ML becomes efficient in accelerating the reduction voltage calculations. As shown in Fig. 5, the training by electrolyte (TBC) protocol begins with E1 and progresses sequentially through E6. In each step, the model is tested on the next unseen electrolyte, with performance evaluated using Mean Absolute Error (MAE) and the coefficient of determination R2. Table S2 reports the complete evaluation results. The model initially trained on E1 performs moderately when tested on E2 (MAE = 0.207 eV, R2 = 0.659). The addition of E2 data improves generalization significantly, yielding an MAE of 0.083 eV and R2 of 0.890 when tested on E3. This pattern continues as more electrolytes are included: testing on E4 after training on E1–E3 produces an MAE of 0.037 eV, while testing on E5 gives a remarkably low MAE of 0.026 eV and an R2 of 0.998. A slight performance drop is observed when moving to the E6 test step (MAE = 0.176 eV; R2 = 0.798), which is chemically distinct due to the presence of the FEC additive. Nonetheless, the model retains strong predictive power. The final test on E7, which is unseen during training, yields an MAE of 0.113 eV and R2 of 0.911. These results demonstrate robust generalization across electrolytes with increasingly complex chemical compositions. The ML model has effectively identified and weighted the chemically relevant features that govern the reduction potential in Li+-coordinated clusters.Alternatively, one can first pretrain the ML model with the initial DFT-computed ΔGR from the individual species (w/wo Li+ association) in Table 2, and then test and retrain with different electrolytes. This training protocol is named train by component (TBC) and the results are listed in Fig. S9. However, training from the components did not lead to a satisfactory prediction for the clusters in the E1 EDL (MAE = 0.826 eV, R2 = 2.842), confirming that component-level information alone is insufficient for predicting complex EDL cluster behavior. However, when training on E1 and testing on E2, the TBC strategy outperforms the TBE strategy, with a lower MAE (0.158 vs. 0.207 eV) and higher R2 (0.751 vs. 0.659). This advantage continues through later steps. Later, both strategies achieve similar performance, but TCB is generally slightly better at unseen electrolyte data. The ML resting results in Fig. 5 and S9 suggest that training with different electrolyte components (chemistry) is necessary, and the ML model trained on different clusters can be effectively extended to electrolytes with different compositions and different mixtures.
 | ||
| Fig. 5 Iterative steps of the Train by Electrolyte protocol. The ML model was trained (yellow panels) on cumulative electrolyte data from E1 to Ei, thus labeled as “Train(E1…Ei), Test(Ei)”. The ML model was tested (green panels) on the next unseen electrolyte at each step, thus labeled as “Without Retraining, Test(Ei + 1)”. Final validation was performed on E7. | ||
It may be surprising to see the level of performance based on such a small dataset and simple features used in the electrolyte reduction energy/voltage prediction model. However, this performance is supported by the strong correlations with the features in Fig. 4b, where it shows the cation and anion (Li+ and FSI−) are most strongly correlated with ΔGR. While the impact of Li+ on electrolyte reduction potential is well known, the solvent reduction potentials are often computed with Li-ion, as in the procedure for Table 2. The impact of an anion on electrolyte reduction was not often considered. This suggests ML can accelerate the DFT-based reduction voltage calculations, while the remaining important information on electrolyte reduction reactions and their contributions to the SEI relies on accurately predicting the EDL structure and compositions.
3.5. EDL composition and LiF in the SEI
We are particularly interested in the components that can generate Li–F species in the SEI due to reduction reactions. The results in Table 2 and their occurrence in the EDL indicate that FSI− (Li+), TFPTFA (Li+), FEC (Li+), MTFP (Li+), TFPTFA (free), FEC (free), and TFETFE (free) can contribute to LiF components in the SEI.We first show the distributions of these species in the EDL. Fig. 6 shows representative distributions of the number density of the Li and F atoms in the EDL along the z-direction (perpendicular to the electrode) normalized by the number density in the bulk region. Addition of FEC in E6 and E7 shows a spike in the number density of the F atoms from FEC in the EDL region, confirming that FEC accumulates in the EDL, and this is due to its higher polarity than other solvents and diluents in the electrolyte.
 | ||
| Fig. 6 Distribution of number density of atoms in the EDL along the z-direction (perpendicular direction to the electrode) normalized with respect to the bulk number density, along with a simplified image of the inner adsorbed layer with different possible orientations of FSI−, MTFP, and TFPTFA. (a) Number density plot, (b) view of inner adsorbed layer from the yz-plane, and (c) distribution of number density of Li/F atoms along the z-direction normalized with respect to the bulk number density for E1–E7. | ||
We analyzed the atom number fraction of possible LiF generated by EDL reduction reactions. LiF content is predicted based on the average number of de-fluorinated fluorine atoms from reduction and combination with Li. Assuming each reducible and LiF-forming species in the EDL contributes one fluorine atom, the total number of fluorine atoms is summed. According to our simulations, this number is larger than the number of lithium atoms in the EDL. Thus, as neutral LiF is formed, the charge imbalance due to excess negative fluorine would require more lithium atoms from the bulk to accumulate in the EDL to screen the charge. Taking this additional number of lithium atoms into account, the total number of lithium and previously counted fluorine atoms is summed up and divided by the total number of atoms in the EDL to obtain the LiF atom fraction as follows:
 | (2) |
Note, free MTFP does not contribute to LiF formation since it has a negative reduction potential vs. Li+/Li. MTFP contributes to LiF only when coordinated to Li+. The rest of the electrolyte components all reduce with or without Li-coordination and contribute to de-fluorination and LiF formation.
We analyzed the atom fraction of possible LiF formation from different contributors in the EDL structures and reported them in Table 4. If regenerated LiF-containing SEI from the liquid electrolyte plays a more critical role in protecting the Li-surface,93 adding FEC will increase LiF content. According to Table 4, adding the FEC to the E1 increased the LiF atomic percentage (by 2%). Similarly, the LiF atomic percentage increases from 13.03% in E4 to 14.81% in E7 after adding FEC. Others proposed that the anion reduction leads to a more stable inorganic-rich SEI with higher LiF content.93 Due to the limited salt solubility in the fluorinated MTFP solvent, the anion contribution is relatively low in E1, a MCE, compared to other HCEs with higher salt concentrations.19 In this measure, E6 and E3 have the highest and lowest anion contributions, respectively. Recently, it was also argued that heterogeneous nano-grained LiF in the SEI is desired.94 In this case, multiple LiF sources may generate more heterogeneous nano-grained LiF phases in the SEI. Therefore, E7 may stand out as all anions, FEC-additive, TFETFE-diluent, and MTFP-solvent all contribute to LiF content.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :FEC:diluent:MTFP) in the Li+-coordinated environment
:FEC:diluent:MTFP) in the Li+-coordinated environment
| Electrolyte | Molar ratio in simulations | LiF % atom | Species ratio (anion:FEC:diluent:MTFP) in Li+-coordinated environment |
|---|---|---|---|
| E1 | LiFSI–3.2MTFP | 14.02% | 1:0.0:0.0:4.35 (MTFP and anions) |
| E2 | LiFSI–3.2MTFP–1.6TFPTFA | 14.38% | 1:0.0:1.36:2.99 (MTFP, diluent, anions) |
| E3 | LiFSI–3.2MTFP–3.2TFPTFA | 13.11% | 1:0.0:4.92:3.46 (less anion contribution) |
| E4 | LiFSI–3.2MTFP–1.6TFETFE | 13.03% | 1:0.0:1.80:3.82 (less anion contribution) |
| E5 | LiFSI–3.2MTFP–3.2TFETFE | 13.62% | 1:0.0:1.82:2.70 (MTFP, diluent, anions) |
| E6 | LiFSI–3.2MTFP–0.68FEC | 16.05% | 1:1.20:0:2.21 (increased anion contributions) |
| E7 | LiFSI–3.2MTFP–1.6TFETFE–1.03FEC | 14.81% | 1:2.44:1.22:2.42 (more FEC contributions) |
Experimental data were collected on delithiated graphite electrodes recovered from lithium/graphite half cells containing electrolytes E1, E6, and E7, Fig. S10. X-ray photoelectron spectroscopy (XPS) results are in Fig. S11 and S12. Interestingly, electrolyte E1 shows a higher LiF atomic percentage than electrolytes E6 and E7 that both contain FEC and may preferentially react at the negative electrode surface, forming a more effective passivation layer to mitigate surface solvent reactivity. Thus, adding FEC may not always increase LiF in the SEI. Furthermore, E1 exhibits more first-cycle capacity loss than E6 and E7, suggesting the source of F species may be more important for SEI formation.
4. Conclusion
In this work, we developed and demonstrated a machine-learning-accelerated molecular simulation workflow that integrates classical molecular dynamics (MD), Density Functional Theory (DFT), and gradient-boosted regression (GBR) models to efficiently investigate solid electrolyte interphase (SEI) formation while explicitly accounting for the electric double layer (EDL) structure. By combining atomistic molecular simulations with data-driven regression models, we established a multiscale modeling framework that quantitatively links local solvation environments, EDL composition, and electrochemical reduction behavior. The inclusion of EDL-resolved statistical descriptors derived from MD trajectories into the gradient-boosted regression model enabled accurate prediction of reduction potentials, achieving a mean absolute error of approximately 0.1 eV relative to DFT calculations, while reducing the computational burden associated with exhaustive quantum-mechanical sampling.Our results reveal that the local EDL environment significantly alters the reduction behavior of electrolyte species and therefore governs the composition and morphology of the SEI. In particular, fluorinated solvents, diluents, and additives such as TFPTFA, TFETFE, and FEC exhibit varying propensities to contribute fluorine-containing fragments (e.g., LiF) depending on their position within the EDL and their coordination with Li+. The model captures the shift in reducible cluster distributions upon compositional tuning, revealing that dilution with weakly solvating fluorinated cosolvents promotes selective reduction pathways. These findings highlight the mechanistic importance of EDL heterogeneity in determining the early stages of SEI formation and interfacial stability in Li-ion batteries. However, it is still inconclusive in terms of the factors that lead to a more stable SEI based on LiF contributions. For future studies, it is necessary to combine quantitative modeling with experiments to test various hypotheses in the SEI structure–property relationship.
Beyond the specific systems studied here, the proposed MD–DFT–ML framework offers a generalizable and computationally tractable approach for exploring electrochemical interfaces across a broad chemical space. By coupling interfacial modeling with data-driven learning, this methodology enables rapid screening and rational design of electrolyte formulations optimized for targeted interfacial chemistries. This framework may pave the way toward predictive and interpretable design principles for next-generation electrolytes and interphases that underpin high-energy-density and long-life electrochemical energy storage systems.
Author contributions
MJH and TK performed the modeling and data analysis. MJH, TK, and YQ prepared the initial manuscript draft. QW contributed to the computational design protocol as well as manuscript writing and editing. PB, MM, SY, XT, AM, KT, and ET carried out all the supporting experimental work and electrolyte design. BMR and YQ provided guidance on data interpretation and analysis, contributed to writing and editing, provided mentorship, and supervised the project.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available in the online repository https://doi.org/10.5281/zenodo.19051119.Supplementary information is available. See DOI: https://doi.org/10.1039/d6eb00085a.
Acknowledgements
This work was initially supported by the U. S. Department of Energy Office of Energy Efficiency and Renewable Energy under the Vehicle Technologies program, award DE-EE0009643. The workflow design was also supported by DOE DE-SC0026248. We also acknowledge the Center for Computation and Visualization at Brown University for providing computational resources and the Brown Open Graduate Education program for supporting T. K. This research used XPS resources of the Center for Functional Nanomaterials (CFN), which is a U.S. Department of Energy Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704. E. S. T. acknowledges support as the William and Jane Knapp Chair in Energy and the Environment.References
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS.
- E. Markervich, G. Salitra, M. Levi and D. Aurbach, J. Power Sources, 2005, 146, 146–150 CrossRef CAS.
- D. Aurbach, M. D. Levi, E. Levi and A. Schechter, J. Phys. Chem. B, 1997, 101, 2195–2206 CrossRef CAS.
- D. Aurbach, B. Markovsky, M. Levi, E. Levi, A. Schechter, M. Moshkovich and Y. Cohen, J. Power Sources, 1999, 81–82, 95–111 Search PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, NPJ Comput. Mater., 2018, 4, 15 CrossRef.
- J. Lee, Y. Park and J. W. Choi, Nano Converg., 2025, 12, 25 CrossRef CAS PubMed.
- Y. Xu, K. Dong, Y. Jie, P. Adelhelm, Y. Chen, L. Xu, P. Yu, J. Kim, Z. Kochovski, Z. Yu, W. Li, J. LeBeau, Y. Shao-Horn, R. Cao, S. Jiao, T. Cheng, I. Manke and Y. Lu, Adv. Energy Mater., 2022, 12, 2200398 CrossRef.
- L. von Kolzenberg, A. Latz and B. Horstmann, ChemSusChem, 2020, 13, 3901–3910 CrossRef CAS PubMed.
- J. Wu, M. Ihsan-Ul-Haq, Y. Chen and J.-K. Kim, Nano Energy, 2021, 89, 106489 CrossRef CAS.
- R. Atwi, M. Bliss, M. Makeev and N. N. Rajput, Sci. Rep., 2022, 12, 15760 CrossRef CAS PubMed.
- X. Zhang, L. Zou, Y. Xu, X. Cao, M. H. Engelhard, B. E. Matthews, L. Zhong, H. Wu, H. Jia, X. Ren, P. Gao, Z. Chen, Y. Qin, C. Kompella, B. W. Arey, J. Li, D. Wang, C. Wang, J. Zhang and W. Xu, Adv. Energy Mater., 2020, 10, 2070098 CrossRef CAS.
- L. Jiang, C. Yan, Y. Yao, W. Cai, J. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2020, 60, 3402–3406 CrossRef PubMed.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- G. A. Giffin, Nat. Commun., 2022, 13, 5250 CrossRef CAS PubMed.
- C. M. Efaw, Q. Wu, N. Gao, Y. Zhang, H. Zhu, K. Gering, M. F. Hurley, H. Xiong, E. Hu, X. Cao, W. Xu, J.-G. Zhang, E. J. Dufek, J. Xiao, X.-Q. Yang, J. Liu, Y. Qi and B. Li, Nat. Mater., 2023, 22, 1531–1539 CrossRef CAS PubMed.
- Q. Wu and Y. Qi, Energy Environ. Sci., 2025, 18, 3036–3046 RSC.
- T. Li and P. B. Balbuena, Chem. Phys. Lett., 2000, 317, 421–429 CrossRef CAS.
- Y. Wang, S. Nakamura, K. Tasaki and P. B. Balbuena, J. Am. Chem. Soc., 2002, 124, 4408–4421 CrossRef CAS PubMed.
- Y. Wang, S. Nakamura, M. Ue and P. B. Balbuena, J. Am. Chem. Soc., 2001, 123, 11708–11718 CrossRef CAS PubMed.
- O. Borodin, M. Olguin, C. E. Spear, K. W. Leiter and J. Knap, Nanotechnology, 2015, 26, 354003 CrossRef PubMed.
- S. A. Delp, O. Borodin, M. Olguin, C. G. Eisner, J. L. Allen and T. R. Jow, Electrochim. Acta, 2016, 209, 498–510 CrossRef CAS.
- K. Leung, Chem. Phys. Lett., 2013, 568–569, 1–8 CrossRef CAS.
- O. Borodin, M. Olguin, C. Spear, K. Leiter, J. Knap, G. Yushin, A. Childs and K. Xu, ECS Trans., 2015, 69, 113–123 CrossRef CAS.
- K. Leung and J. L. Budzien, Phys. Chem. Chem. Phys., 2010, 12, 6583 RSC.
- K. Leung, Y. Qi, K. R. Zavadil, Y. S. Jung, A. C. Dillon, A. S. Cavanagh, S.-H. Lee and S. M. George, J. Am. Chem. Soc., 2011, 133, 14741–14754 CrossRef CAS PubMed.
- L. E. Camacho-Forero, T. W. Smith, S. Bertolini and P. B. Balbuena, J. Phys. Chem. C, 2015, 119, 26828–26839 CrossRef CAS.
- Y. Jin, N.-J. H. Kneusels, P. C. M. M. Magusin, G. Kim, E. Castillo-Martínez, L. E. Marbella, R. N. Kerber, D. J. Howe, S. Paul, T. Liu and C. P. Grey, J. Am. Chem. Soc., 2017, 139, 14992–15004 CrossRef CAS PubMed.
- M. D. Halls and K. Tasaki, J. Power Sources, 2010, 195, 1472–1478 CrossRef CAS.
- Y.-K. Han, J. Jung, S. Yu and H. Lee, J. Power Sources, 2009, 187, 581–585 CrossRef CAS.
- O. Borodin, W. Behl and T. R. Jow, J. Phys. Chem. C, 2013, 117, 8661–8682 CrossRef CAS.
- F. Chen, Curr. Opin. Electrochem., 2022, 35, 101086 CrossRef CAS.
- J. Vatamanu and O. Borodin, J. Phys. Chem. Lett., 2017, 8, 4362–4367 CrossRef CAS PubMed.
- O. Borodin, X. Ren, J. Vatamanu, A. von Wald Cresce, J. Knap and K. Xu, Acc. Chem. Res., 2017, 50, 2886–2894 CrossRef CAS PubMed.
- N. Yao, X. Chen, Z.-H. Fu and Q. Zhang, Chem. Rev., 2022, 122, 10970–11021 CrossRef CAS PubMed.
- B. Horstmann, F. Single and A. Latz, Curr. Opin. Electrochem., 2019, 13, 61–69 CrossRef CAS.
- D. Diddens, W. A. Appiah, Y. Mabrouk, A. Heuer, T. Vegge and A. Bhowmik, Adv. Mater. Interfaces, 2022, 9, 2101734 CrossRef CAS.
- J. Wagner-Henke, D. Kuai, M. Gerasimov, F. Röder, P. B. Balbuena and U. Krewer, Nat. Commun., 2023, 14, 6823 CrossRef CAS PubMed.
- K. Zhang, Y. Ji, Q. Wu, S. A. Nabavizadeh, Y. Qi and L.-Q. Chen, Energy Environ. Sci., 2025, 18, 7541–7554 RSC.
- Q. Wu, M. T. McDowell and Y. Qi, J. Am. Chem. Soc., 2023, 145, 2473–2484 CrossRef CAS PubMed.
- J. Scholl, P. Scharpmann, A. Bagheri, J. Lisec, E. Meiers, C. Jaeger, R. Leonhardt, H. Haase and M. Koch, J. Power Sources, 2026, 673, 239739 CrossRef CAS.
- D. Hannah, Y. Zhang, X. Li, D. Dong, J. Han, G. Park, H. Gan, B. Liu, K. Liu, Q. Hu and K. Xu, Electrochem. Soc. Interface, 2025, 34, 35 CrossRef CAS.
- S. Gong, Y. Zhang, Z. Mu, Z. Pu, H. Wang, X. Han, Z. Yu, M. Chen, T. Zheng and Z. Wang, Nat. Mach. Intell., 2025, 1–10 CAS.
- R. Zhou, K. Luo, S. W. Martin and Q. An, ACS Appl. Mater. Interfaces, 2024, 16, 18874–18887 CrossRef CAS PubMed.
- Y. Shao, L. Knijff, F. M. Dietrich, K. Hermansson and C. Zhang, Batteries Supercaps, 2021, 4, 585–595 CrossRef CAS.
- M. Zohair, V. Sharma, E. A. Soares, K. Nguyen, M. Giammona, L. Sundberg, A. Tek, E. Vital Brazil and Y.-H. La, NPJ Comput. Mater., 2025, 11, 283 CrossRef CAS.
- C. D. Quilty, E. J. Marin Bernardez, A. Nicoll, M. J. Hossain, A. Kingan, D. J. Arnot, H. A. Mohamed, C. L. O'Connor, X. Tong, C. Jaye, D. A. Fischer, L. Wang, Y. Qi, E. S. Takeuchi, A. C. Marschilok, S. Yan, D. C. Bock and K. J. Takeuchi, RSC Appl. Interfaces, 2024, 1, 1077–1092 RSC.
- M. J. Hossain, Q. Wu, E. J. Marin Bernardez, C. D. Quilty, A. C. Marschilok, E. S. Takeuchi, D. C. Bock, K. J. Takeuchi and Y. Qi, J. Phys. Chem. Lett., 2023, 14, 7718–7731 CrossRef CAS PubMed.
- C. Wang, Y. S. Meng and K. Xu, J. Electrochem. Soc., 2018, 166, A5184–A5186 CrossRef.
- Y. Yang, P. Li, N. Wang, Z. Fang, C. Wang, X. Dong and Y. Xia, Chem. Commun., 2020, 56, 9640–9643 RSC.
- J. Holoubek, M. Yu, S. Yu, M. Li, Z. Wu, D. Xia, P. Bhaladhare, M. S. Gonzalez, T. A. Pascal, P. Liu and Z. Chen, ACS Energy Lett., 2020, 5, 1438–1447 CrossRef CAS.
- W. Zhang, T. Yang, X. Liao, Y. Song and Y. Zhao, Energy Storage Mater., 2023, 57, 249–259 CrossRef.
- Y. Chen, Z. Yu, P. Rudnicki, H. Gong, Z. Huang, S. C. Kim, J.-C. Lai, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2021, 143, 18703–18713 CrossRef CAS PubMed.
- J. Zhang, Q. Li, Y. Zeng, Z. Tang, D. Sun, D. Huang, Y. Tang and H. Wang, ACS Energy Lett., 2023, 8, 1752–1761 CrossRef CAS.
- E. Park, J. Park, K. Lee, Y. Zhao, T. Zhou, G. Park, M.-G. Jeong, M. Choi, D.-J. Yoo, H.-G. Jung, A. Coskun and J. W. Choi, ACS Energy Lett., 2023, 8, 179–188 CrossRef CAS.
- W. Cai, Y. Deng, Z. Deng, Y. Jia, Z. Li, X. Zhang, C. Xu, X. Zhang, Y. Zhang and Q. Zhang, Adv. Energy Mater., 2023, 13, 2301396 CrossRef CAS.
- D. Penley, H. Gerber, M. N. Garaga, N. P. Wickramasinghe, S. G. Greenbaum, E. J. Maginn, Y. Zhang and B. Gurkan, J. Power Sources, 2024, 593, 233984 CrossRef CAS.
- E. R. Logan, D. S. Hall, M. M. E. Cormier, T. Taskovic, M. Bauer, I. Hamam, H. Hebecker, L. Molino and J. R. Dahn, J. Phys. Chem. C, 2020, 124, 12269–12280 CrossRef CAS.
- J. Holoubek, Y. Yin, M. Li, M. Yu, Y. S. Meng, P. Liu and Z. Chen, Angew. Chem., 2019, 131, 19068–19073 CrossRef.
- X. Dong, Y. Lin, P. Li, Y. Ma, J. Huang, D. Bin, Y. Wang, Y. Qi and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 5623–5627 CrossRef CAS PubMed.
- X. Ma, J. Li, S. L. Glazier, L. Ma, K. L. Gering and J. Dahn, Electrochim. Acta, 2018, 270, 215–223 CrossRef CAS.
- Q. Q. Liu, D. J. Xiong, R. Petibon, C. Y. Du and J. R. Dahn, J. Electrochem. Soc., 2016, 163, A3010–A3015 CrossRef CAS.
- X. Lin, G. Zhou, J. Liu, J. Yu, M. B. Effat, J. Wu and F. Ciucci, Adv. Energy Mater., 2020, 10, 2001235 CrossRef CAS.
- L. Chen, X. Fan, E. Hu, X. Ji, J. Chen, S. Hou, T. Deng, J. Li, D. Su, X. Yang and C. Wang, Chem, 2019, 5, 896–912 CAS.
- X. Zhang, X. Chen, X. Cheng, B. Li, X. Shen, C. Yan, J. Huang and Q. Zhang, Angew. Chem., 2018, 130, 5399–5403 CrossRef.
- D. Weintz, S. P. Kühn, M. Winter and I. Cekic-Laskovic, ACS Appl. Mater. Interfaces, 2023, 15, 53526–53532 CrossRef CAS PubMed.
- A. C. Thenuwara, P. P. Shetty, N. Kondekar, S. E. Sandoval, K. Cavallaro, R. May, C.-T. Yang, L. E. Marbella, Y. Qi and M. T. McDowell, ACS Energy Lett., 2020, 5, 2411–2420 CrossRef CAS.
- BIOVIA, Materials Studio, 2020 Search PubMed.
- R. L. C. Akkermans, N. A. Spenley and S. H. Robertson, Mol. Simul., 2020, 47, 540–551 CrossRef.
- D. Rakov, M. Hasanpoor, A. Baskin, J. W. Lawson, F. Chen, P. V. Cherepanov, A. N. Simonov, P. C. Howlett and M. Forsyth, Chem. Mater., 2022, 34, 165–177 CrossRef CAS.
- Y. Li and Y. Qi, Energy Environ. Sci., 2019, 12, 1286–1295 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Vatamanu, D. Bedrov and O. Borodin, Mol. Simul., 2017, 43, 838–849 CrossRef CAS.
- L. Zeng, T. Wu, T. Ye, T. Mo, R. Qiao and G. Feng, Nat. Comput. Sci., 2021, 1, 725–731 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, Gaussian 09, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Wohlfarth, Static Dielectric Constants of Pure Liquids and Binary Liquid Mixtures, Springer, Berlin Heidelberg, 2015, pp. 1–2 Search PubMed.
- L. E. Roy, E. Jakubikova, M. G. Guthrie and E. R. Batista, J. Phys. Chem. A, 2009, 113, 6745–6750 CrossRef CAS PubMed.
- R. A. Marcus, Pure Appl. Chem., 1997, 69, 13–30 CrossRef CAS.
- E. W. C. Spotte-Smith, R. L. Kam, D. Barter, X. Xie, T. Hou, S. Dwaraknath, S. M. Blau and K. A. Persson, ACS Energy Lett., 2022, 7, 1446–1453 CrossRef CAS.
- S. F. Nelsen, S. C. Blackstock and Y. Kim, J. Am. Chem. Soc., 1987, 109, 677–682 CrossRef CAS.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss and V. Dubourg, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- J. Friedman, T. Hastie and R. Tibshirani, Ann. Stat., 2000, 28, 337–407 Search PubMed.
- J. H. Friedman, Ann. Stat., 2001, 1189–1232 Search PubMed.
- J. H. Friedman, Comput. Stat. Data Anal., 2002, 38, 367–378 CrossRef.
- T. Hastie, R. Tibshirani and J. Friedman, The Elements of Statistical Learning: Data Mining, Inference, and Prediction, Springer, New York, 2nd edn, 2009 Search PubMed.
- A. Natekin and A. Knoll, Front. Neurorobot., 2013, 7, 21 Search PubMed.
- Z.-H. Zhou, Ensemble methods: foundations and algorithms, CRC press, 2025 Search PubMed.
- L. Ward, A. Agrawal, A. Choudhary and C. Wolverton, NPJ Comput. Mater., 2016, 2, 1–7 Search PubMed.
- M. He, R. Guo, G. M. Hobold, H. Gao and B. M. Gallant, Proc. Natl. Acad. Sci. U. S. A., 2019, 117, 73–79 CrossRef PubMed.
- A. Svirinovsky-Arbeli, M. Juelsholt, R. May, Y. Kwon and L. E. Marbella, Joule, 2024, 8, 1919–1935 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |